siRNAs Trigger Efficient Silencing of a Parasitism Gene in Plant Parasitic Root-Knot Nematodes

Abstract
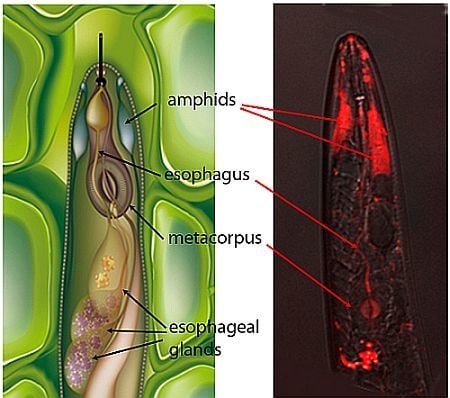
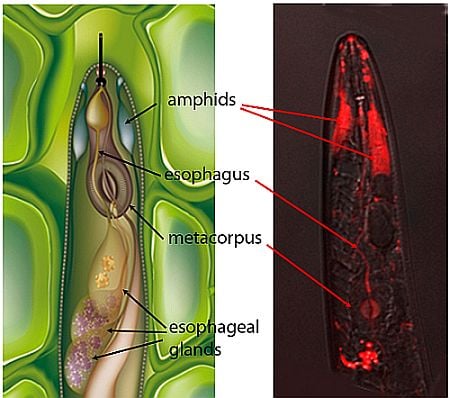
:
1. Introduction
2. Results and Discussion
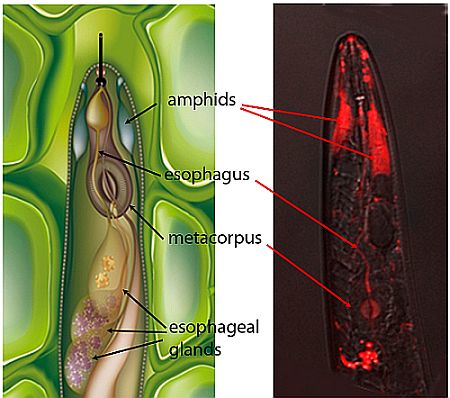
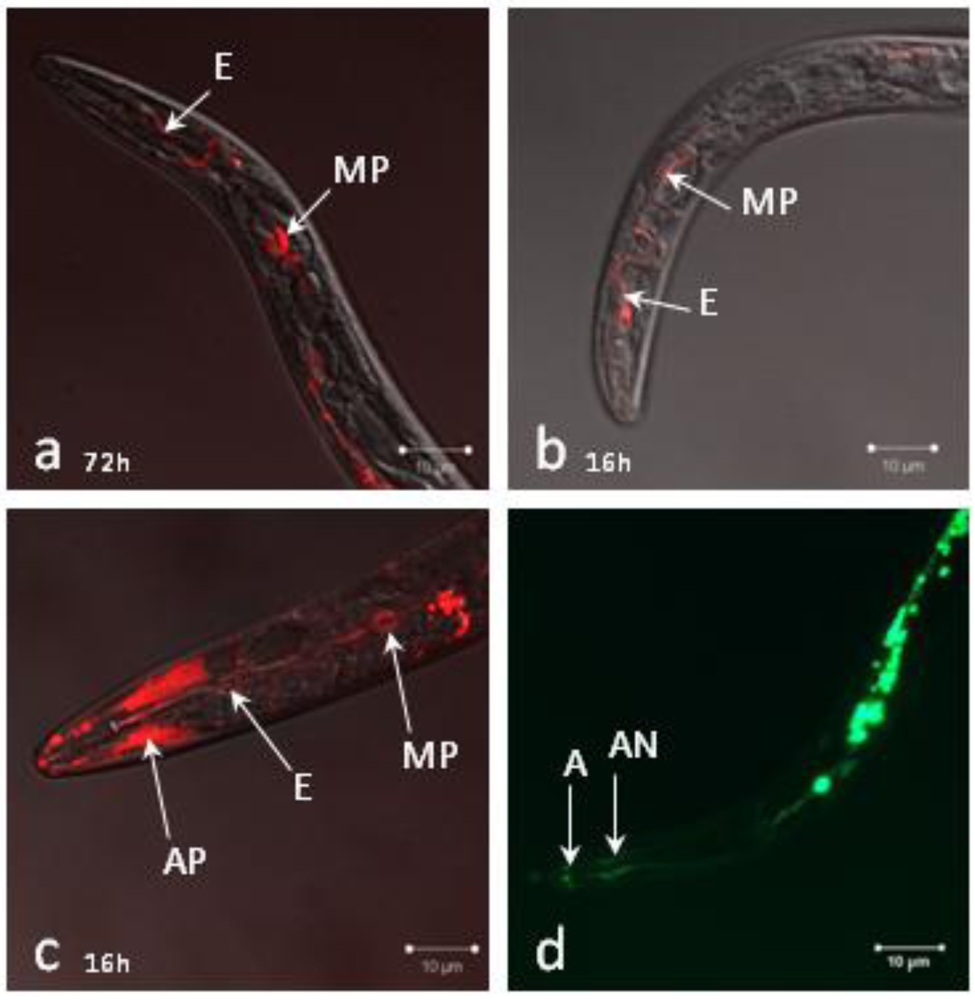
2.1. Localization of siRNAs within Soaked Nematodes
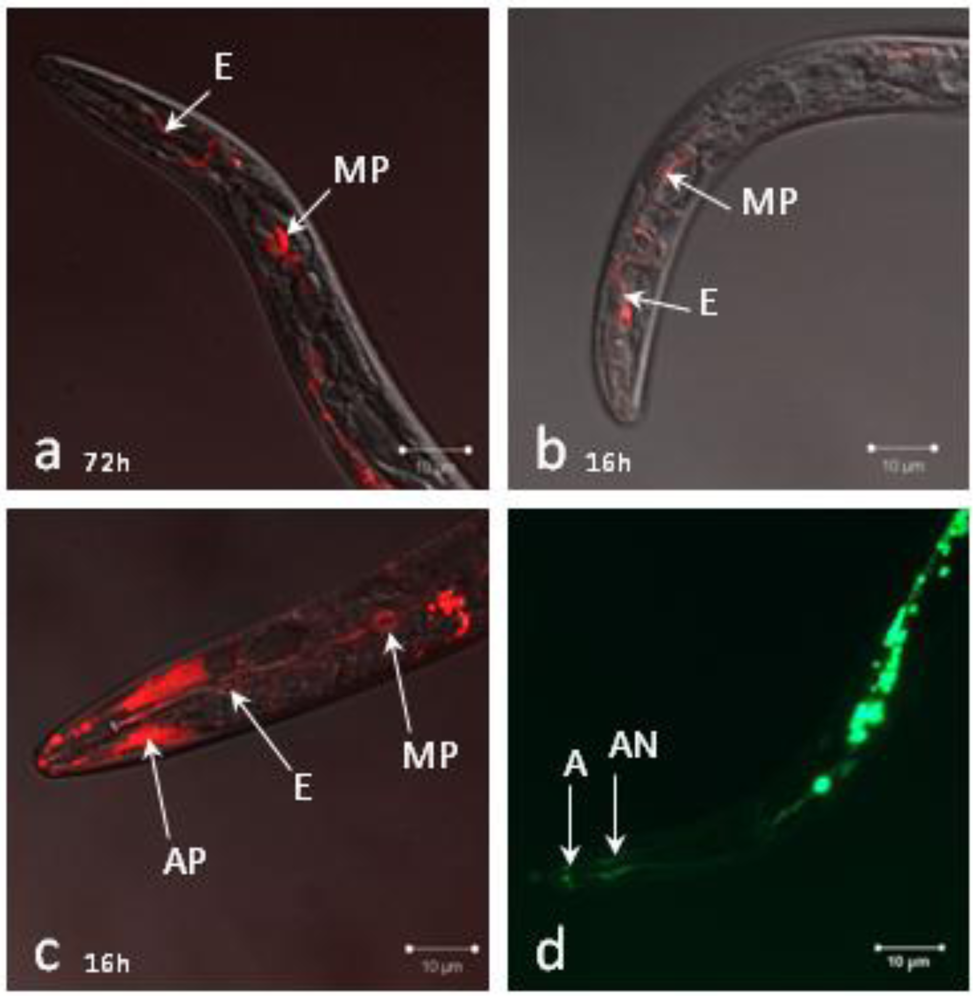
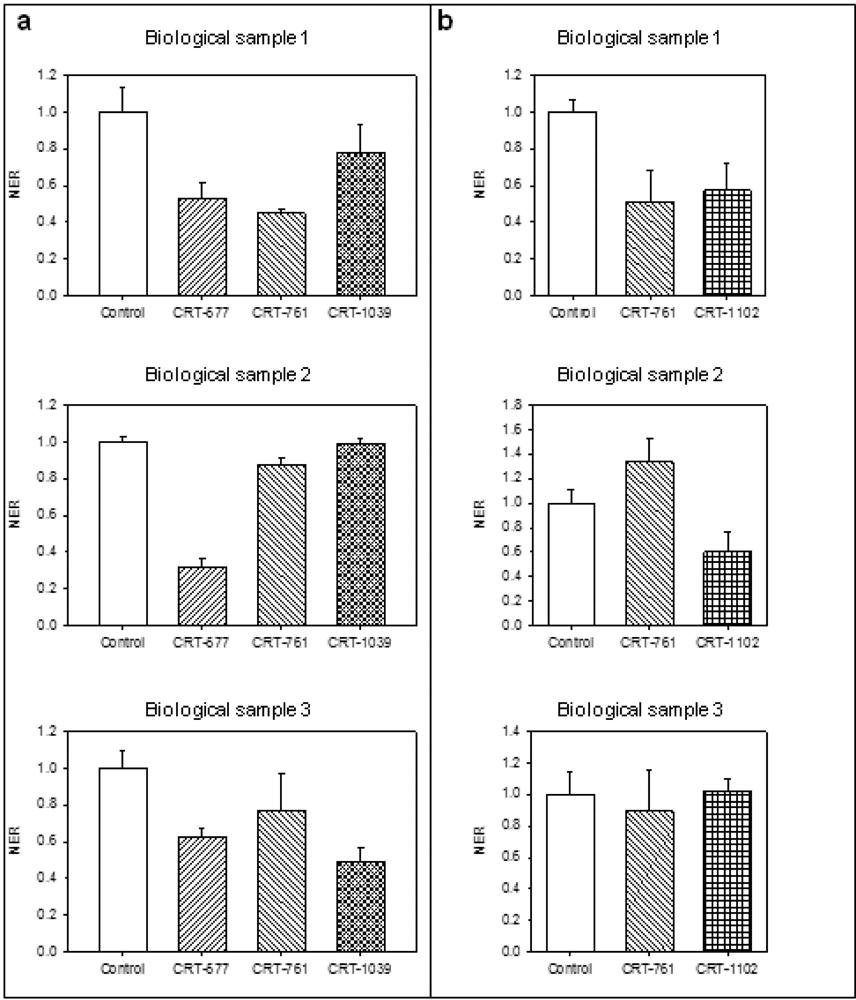
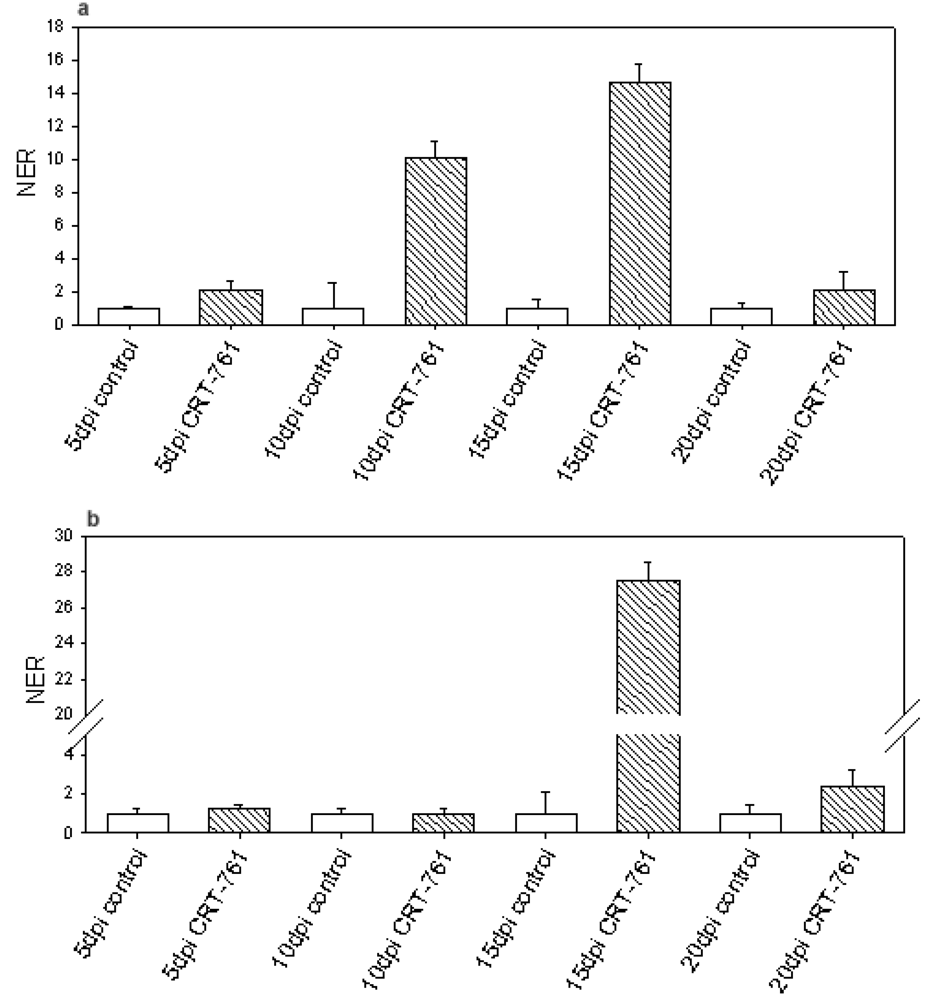
2.2. siRNAs Trigger RNAi in Infective Juveniles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
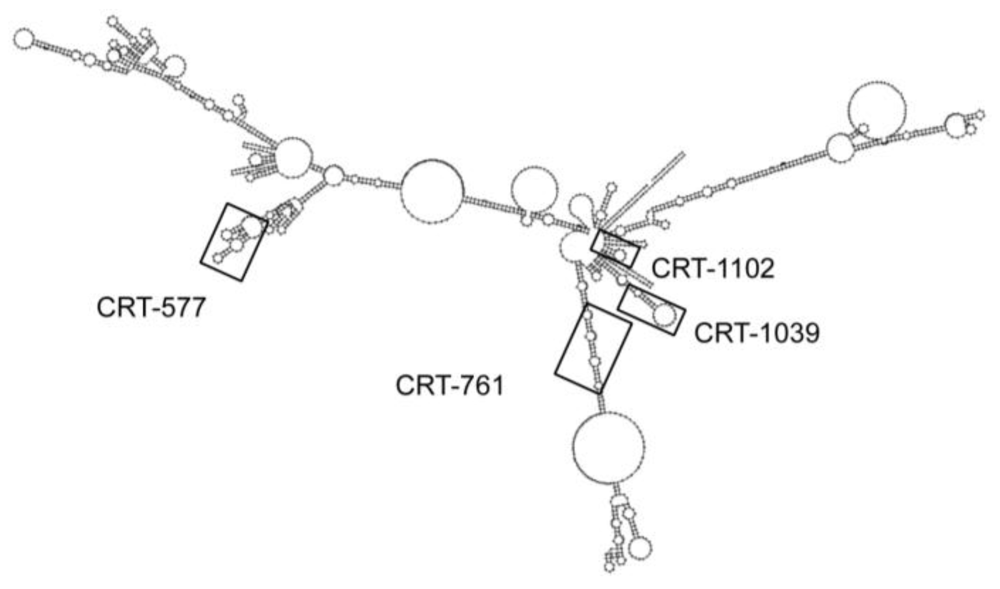
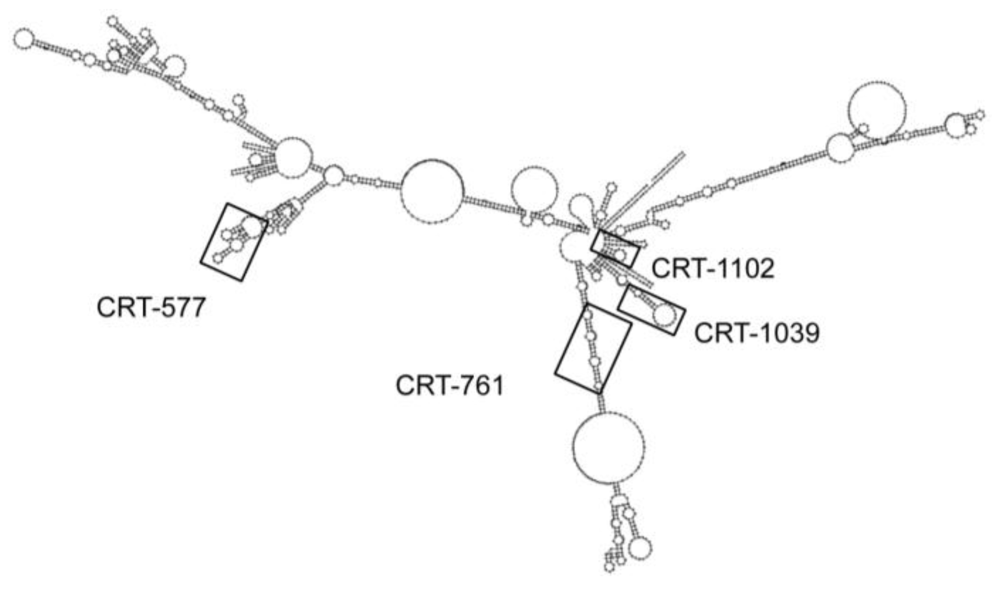
| siRNA | Sequence 5'→3' | Target Matching Region | |
|---|---|---|---|
| Size (bp) | GC (%) | ||
| CRT-577 | F: AAGGCAGAAAGTGGAGAGCTTCCTGTCTC | 21 | 47.61 |
| R: AAAAGCTCTCCACTTTCTGCCCCTGTCTC | |||
| CRT-761 | F: AAGACTGGGATGATGATATGGCCTGTCTC | 21 | 42.85 |
| R: AACCATATCATCATCCCAGTCCCTGTCTC | |||
| CRT-1039 | F: AAGGAAACATTTGAGCCATTGCCTGTCTC | 21 | 38.09 |
| R: AACAATGGCTCAAATGTTTCCCCTGTCTC | |||
| CRT-1102 | F: AACGCAAGAAGTTTGACGAGGCCTGTCTC | 21 | 47.61 |
| R: AACCTCGTCAAACTTCTTGCGCCTGTCTC | |||
| Control siRNA | F: AAGTACATTTAACGCCAGCCTCCTGTCTC | 21 | 42.85 |
| R: AAAGGCTGGCGTTAAATGTACCCTGTCTC | |||
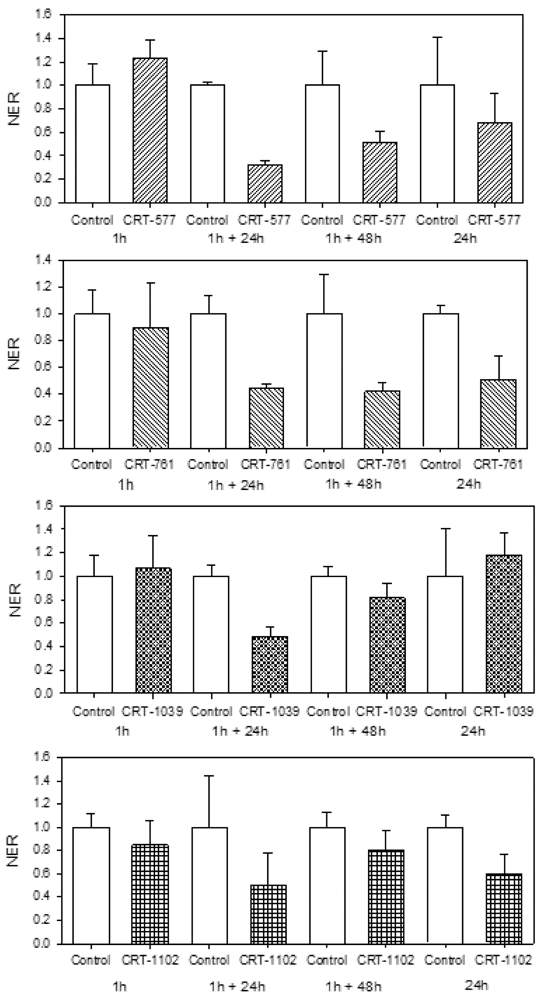
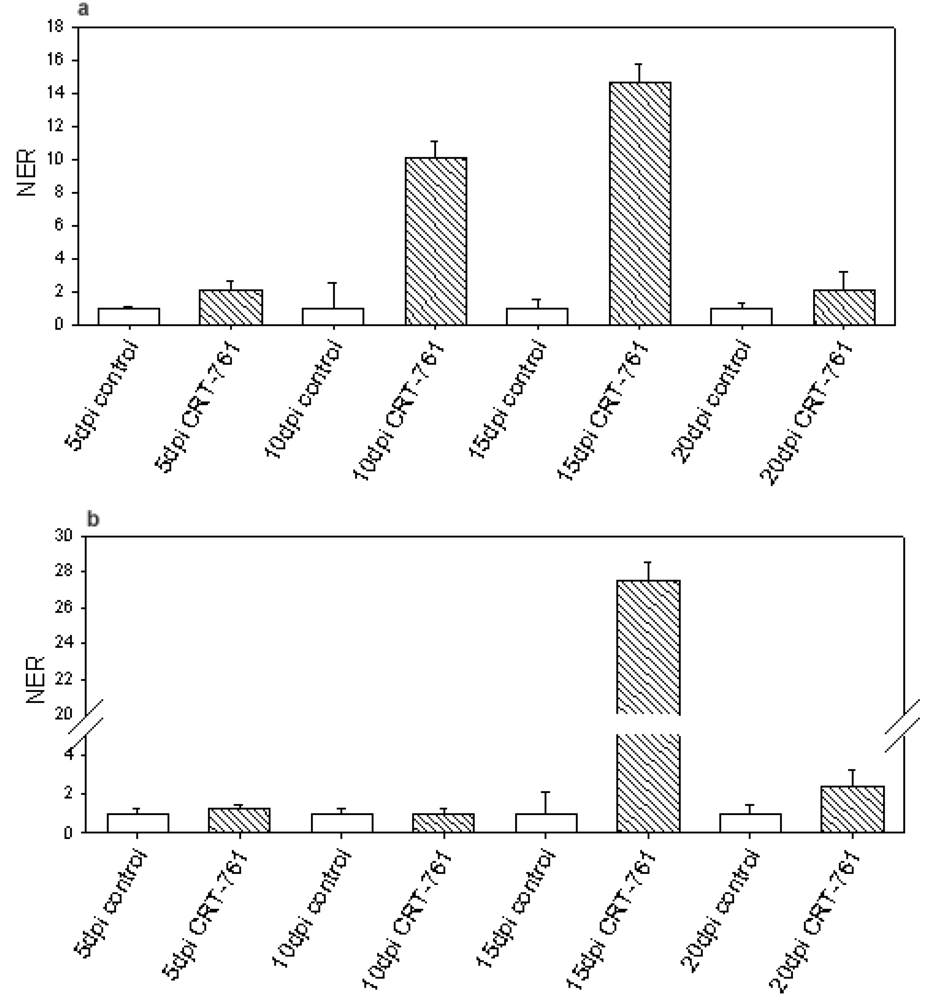
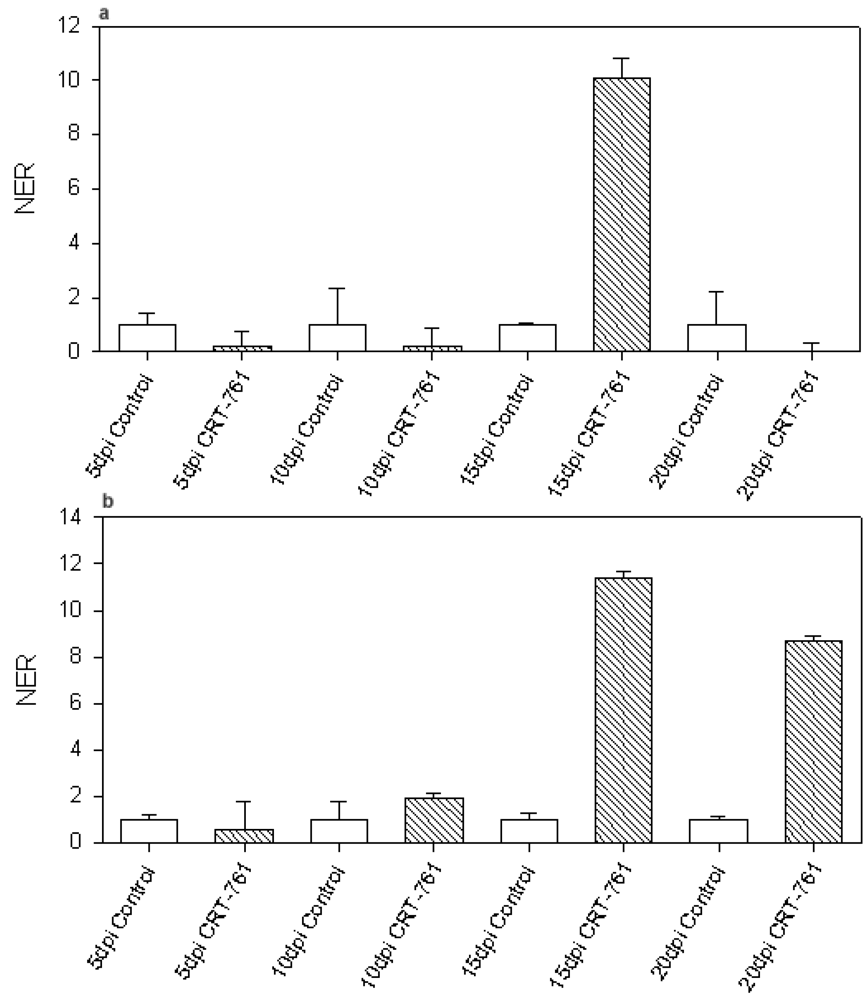
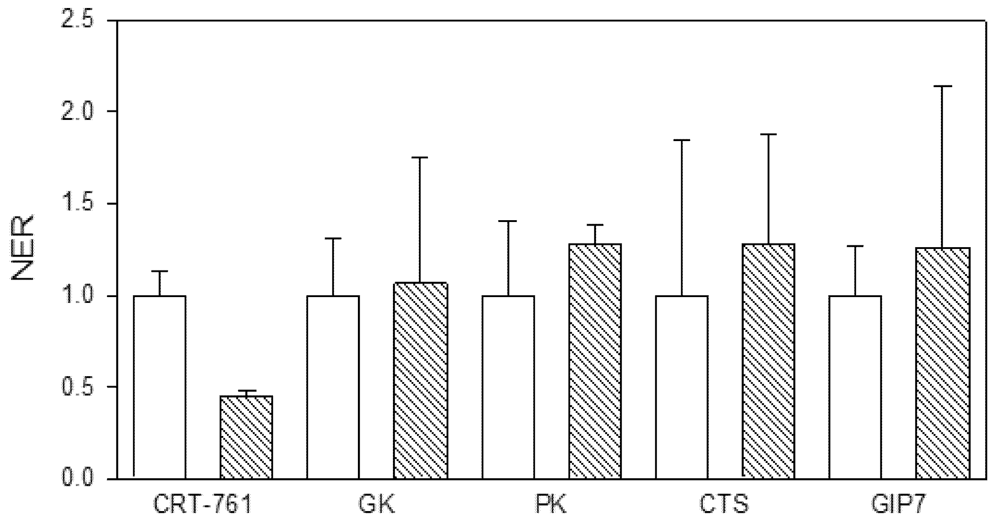
2.3. Analysis of Gene-Silencing in Parasitic Stages
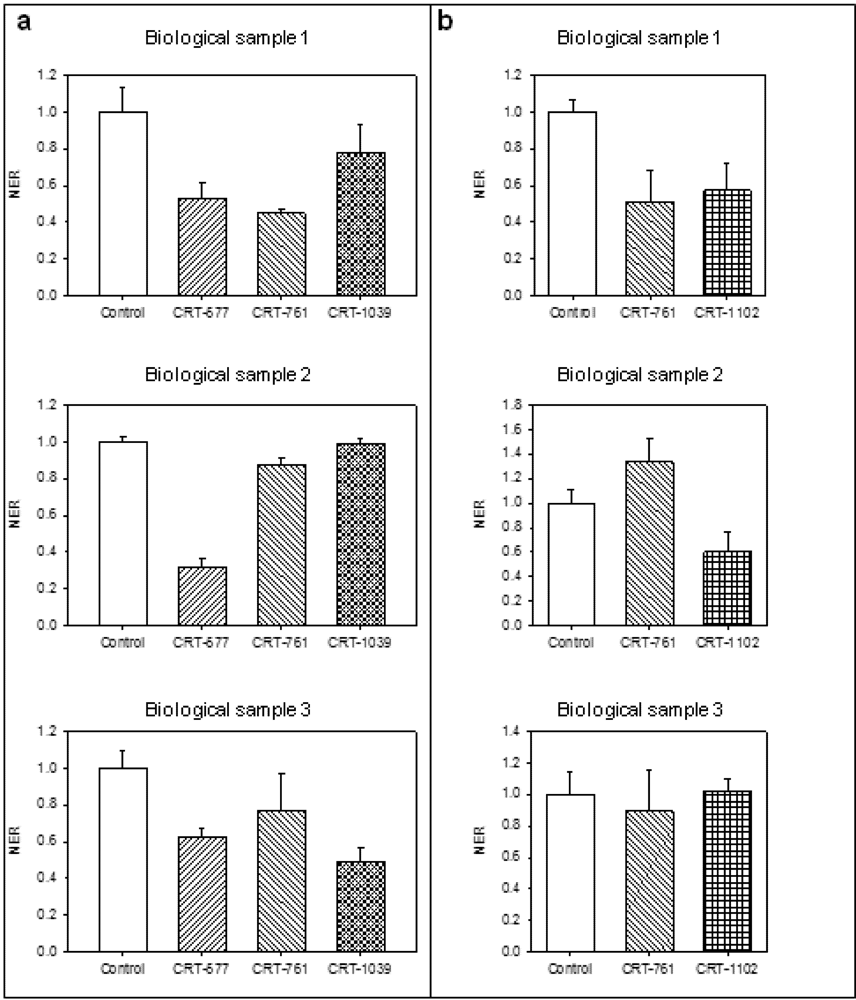
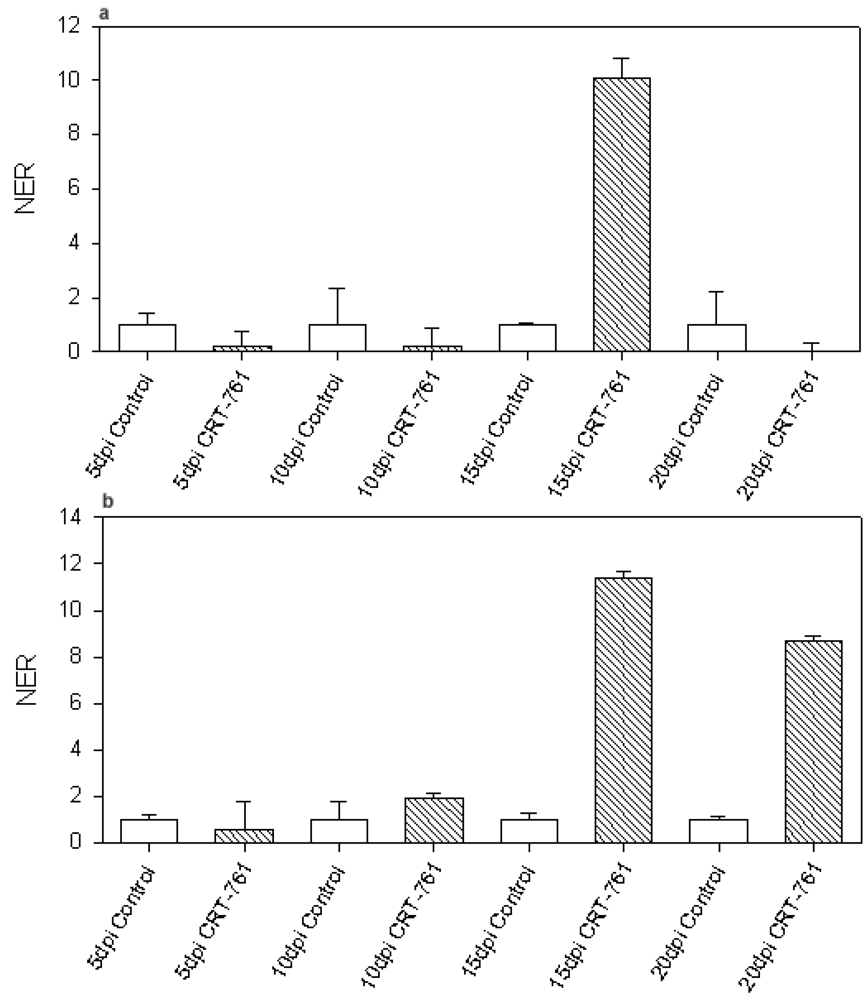
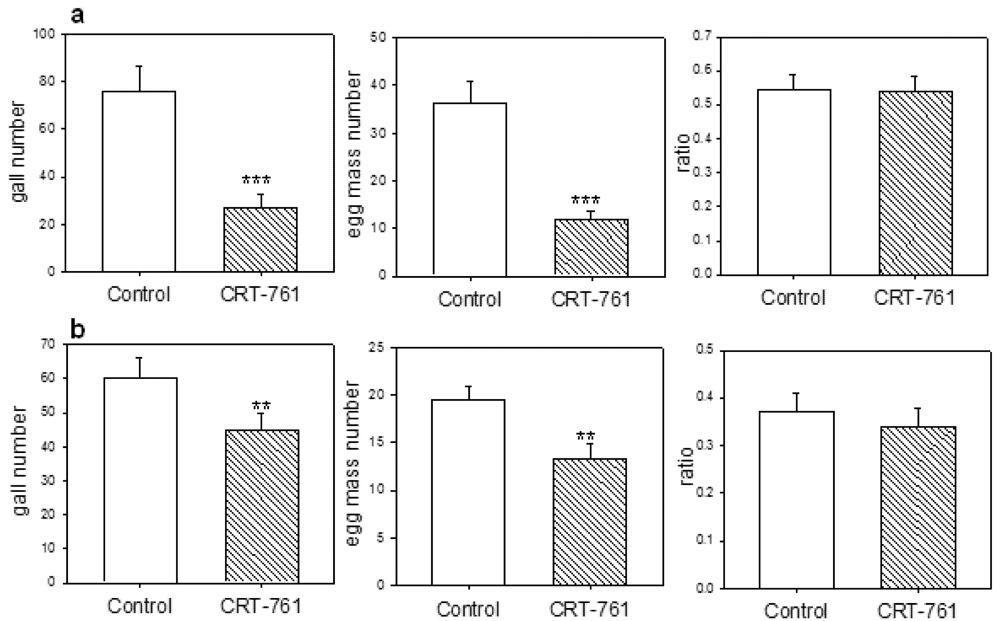
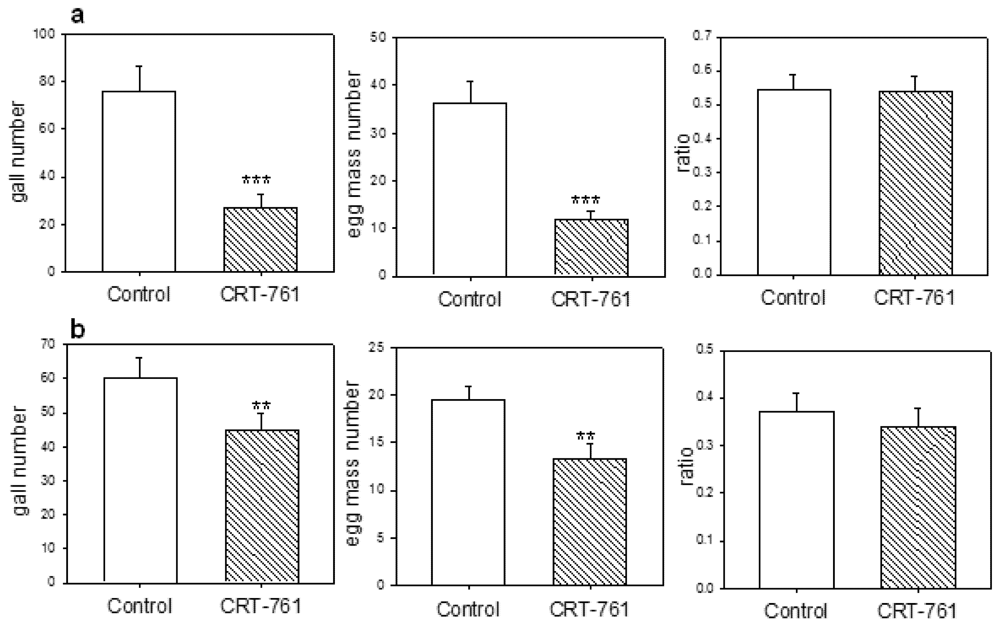
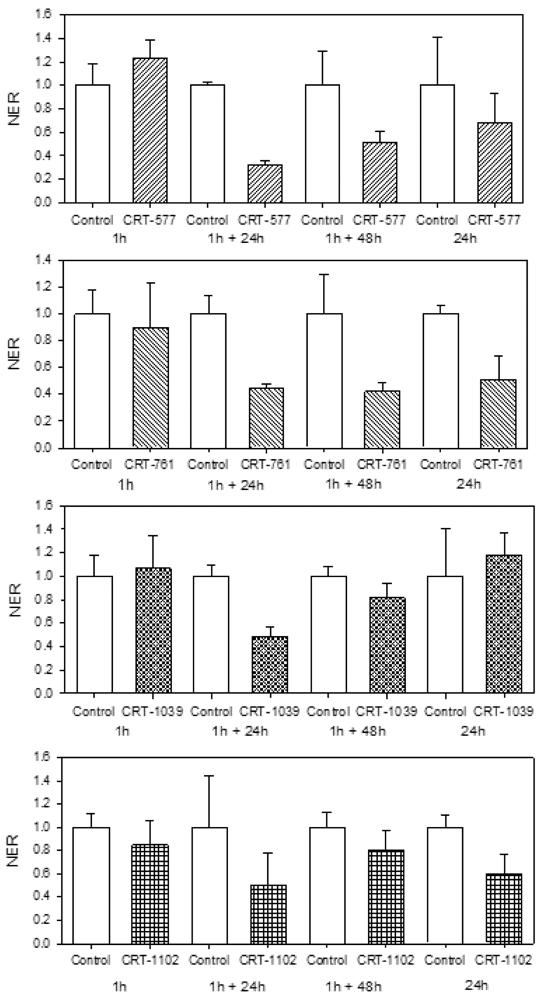
2.4. Nematode Viability after siRNA-Mediated RNAi
| Gene | name | primer sequence (5'→ 3') | size | T° |
|---|---|---|---|---|
| Target gene: | ||||
| Mi-CRT | CRT9 | GCCTGAGGATTGGGATGAG | 19 | 60 |
| CRT10 | TTTGGTGGCTCCCATTCTC | 19 | 60 | |
| Reference genes: | ||||
| ACT | ACT23 | AAGACGAAGCAGCTGTAGCC | 20 | 60 |
| ACT24 | GGTGTTACGCACACAGTTCC | 20 | 60 | |
| GAPDH | GAP17 | GGTTATCTCAGCTCCGTCTGC | 21 | 60 |
| GAP18 | TAACCTTCGCAAGAGGAGCA | 20 | 60 | |
| Mi-eri-1: | ||||
| Qp2F | CTGAATATCCGGCGTTCATT | 20 | 58 | |
| Qp2R | AGCGTCATCCATTCCACAAT | 20 | 58 | |
| Metabolism genes: | ||||
| ACDH | ACD19 | GCTCTGATGTTTCCGGACTT | 20 | 60 |
| ACD20 | AGCATCACCATCCACAACAA | 20 | 60 | |
| GK | GK30 | AGCATGGAATTGAGCCTGAG | 20 | 60 |
| GK31 | ACGTTATCCAGAAGCCAACG | 20 | 60 | |
| PK | PK38 | GCTGAGGGAAGTGATGTTGC | 20 | 60 |
| PK39 | ACACGAAATAGCCGCAGAAG | 20 | 60 | |
| ACO-C | ACO44 | GGAGAGGGCTCTTCTCGTG | 19 | 60 |
| ACO45 | CTGGAGCAAGAGTGTTGAGC | 20 | 60 | |
| CTS | CTS21 | TCGTGAAGGAACATCTGTGG | 20 | 60 |
| CTS22 | ATTGCCGCAGAGAAAGAGAG | 20 | 60 | |
| GIP7 | GIP46 | TGGTACAGGCTGTGTTCCAT | 20 | 60 |
| GIP47 | TACAACAGATCCGGCACGTA | 20 | 60 | |
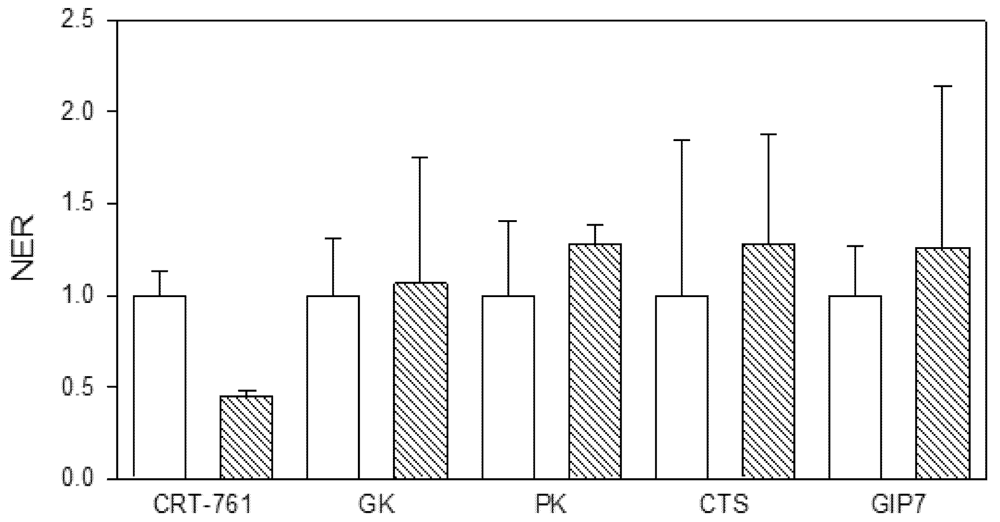
3. Experimental Section
4. Conclusions
Acknowledgments
References
- Abad, P.; Favery, B.; Rosso, M.-N.; Castagnone-Sereno, P. Root-knot nematode parasitism and host response: Molecular basis of a sophisticated interaction. Mol. Plant Pathol. 2003, 4, 217–224. [Google Scholar] [CrossRef]
- Djian-Caporalino, C. Root-knot nematodes (meloidogyne spp.), a growing problem in french vegetable crops. OEPP/EPPO Bull. 2012, 42, 1–12. [Google Scholar]
- Blok, V.C.; Jones, J.T.; Phillips, M.S.; Trudgill, D.L. Parasitism genes and host range disparities in biotrophic nematodes: The conundrum of polyphagy versus specialisation. BioEssays 2008, 30, 249–259. [Google Scholar] [CrossRef]
- Rosso, M.-N.; Hussey, R.S.; Davis, E.L.; Smant, G.; Baum, T.J.; Abad, P.; Mitchum, M.G. Nematode Effector Proteins: Targets and Functions in Plant Parasitism. In Effectors in Plant-Microbe Interactions; Wiley-Blackwell: Hoboken, NJ, USA, 2011; pp. 327–354. [Google Scholar]
- Rosso, M.N.; Jones, J.T.; Abad, P. Rnai and Functional genomics in plant parasitic nematodes. In Annual Review of Phytopathology; Annual Reviews: Palo Alto, CA, USA, 2009; Volume 47, pp. 207–232. [Google Scholar]
- Gheysen, G.; Vanholme, B. Rnai from plants to nematodes. Trends Biotechnol. 2007, 25, 89–92. [Google Scholar] [CrossRef]
- Lilley, C.J.; Davies, L.J.; Urwin, P.E. Rna interference in plant parasitic nematodes: A summary of the current status. Parasitology 2012, 139, 1–11. [Google Scholar] [CrossRef]
- Mello, C.C.; Conte, D. Revealing the world of rna interference. Nature 2004, 431, 338–342. [Google Scholar] [CrossRef]
- Sijen, T.; Fleenor, J.; Simmer, F.; Thijssen, K.L.; Parrish, S.; Timmons, L.; Plasterk, R.H.A.; Fire, A. On the Role of RNA Amplification in dsRNA-Triggered Gene Silencing. Cell 2001, 107, 465–476. [Google Scholar] [CrossRef]
- Abad, P.; Gouzy, J.; Aury, J.M.; Castagnone-Sereno, P.; Danchin, E.G.J.; Deleury, E.; Perfus-Barbeoch, L.; Anthouard, V.; Artiguenave, F.; Blok, V.C.; et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nature Biotechnology 2008, 26, 909–915. [Google Scholar]
- Dalzell, J.J.; McVeigh, P.; Warnock, N.D.; Mitreva, M.; Bird, D.M.; Abad, P.; Fleming, C.C.; Day, T.A.; Mousley, A.; Marks, N.J.; Maule, A.G. RNAi Effector Diversity in Nematodes. Plos Neglected Tropical Diseases 2011, 5. [Google Scholar]
- Dalzell, J.J.; Warnock, N.D.; Stevenson, M.A.; Mousley, A.; Fleming, C.C.; Maule, A.G. Short interfering RNA-mediated knockdown of drosha and pasha in undifferentiated Meloidogyne incognita eggs leads to irregular growth and embryonic lethality. International Journal for Parasitology 2010, 40, 1303–1310. [Google Scholar] [CrossRef]
- Huang, G.Z.; Allen, R.; Davis, E.L.; Baum, T.J.; Hussey, R.S. Engineering broad root-knot resistance in transgenic plants by rnai silencing of a conserved and essential root-knot nematode parasitism gene. Proc. Natl. Acad. Sci. USA 2006, 103, 14302–14306. [Google Scholar]
- Yadav, B.C.; Veluthambi, K.; Subramaniam, K. Host-generated double stranded rna induces rnai in plant-parasitic nematodes and protects the host from infection. Mol. Biochem. Parasitol. 2006, 148, 219–222. [Google Scholar] [CrossRef]
- Fairbairn, D.J.; Cavallaro, A.S.; Bernard, M.; Mahalinga-Iyer, J.; Graham, M.W.; Botella, J.R. Host-delivered rnai: An effective strategy to silence genes in plant parasitic nematodes. Planta 2007, 226, 1525–1533. [Google Scholar] [CrossRef]
- Dubreuil, G.; Magliano, M.; Dubrana, M.P.; Lozano, J.; Lecomte, P.; Favery, B.; Abad, P.; Rosso, M.N. Tobacco rattle virus mediates gene silencing in a plant parasitic root-knot nematode. J. Exp. Bot. 2009, 60, 4041–4050. [Google Scholar] [CrossRef]
- Valentine, T.A.; Randall, E.; Wypijewski, K.; Chapman, S.; Jones, J.; Oparka, K.J. Delivery of macromolecules to plant parasitic nematodes using a tobacco rattle virus vector. Plant Biotechnol. J. 2007, 5, 827–834. [Google Scholar] [CrossRef]
- Dalzell, J.J.; McMaster, S.; Johnston, M.J.; Kerr, R.; Fleming, C.C.; Maule, A.G. Non-nematode-derived double-stranded rnas induce profound phenotypic changes in meloidogyne incognita and globodera pallida infective juveniles. Int. J. Parasitol. 2009, 39, 1503–1516. [Google Scholar] [CrossRef]
- Dalzell, J.J.; McMaster, S.; Fleming, C.C.; Maule, A.G. Short interfering rna-mediated gene silencing in globodera pallida and meloidogyne incognita infective stage juveniles. Int. J. Parasitol. 2010, 40, 91–100. [Google Scholar] [CrossRef]
- Vieira, P.; Danchin, E.G.J.; Neveu, C.; Crozat, C.; Jaubert, S.; Hussey, R.S.; Engler, G.; Abad, P.; de Almeida-Engler, J.; Castagnone-Sereno, P.; et al. The plant apoplasm is an important recipient compartment for nematode secreted proteins. J. Exp. Bot. 2011, 62, 1241–1253. [Google Scholar]
- Luedtke, S.; O'Connor, V.; Holden-Dye, L.; Walker, R.J. The regulation of feeding and metabolism in response to food deprivation in caenorhabditis elegans. Invertebr. Neurosci. 2010, 10, 63–76. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, H.; Jiang, X.J.; Zhou, W.; Zhu, B.; Zeng, Y.; Yao, K.T.; Ren, C.P. Lipofectamine rnaimax: An efficient sirna transfection reagent in human embryonic stem cells. Mol. Biotechnol. 2008, 40, 19–26. [Google Scholar] [CrossRef]
- Meloidogyne Incognita Resource. Available online: http://www.inra.fr/meloidogyne_incognita/ (Accessed on January 2011).
- RNAfold WebServer. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cg (Accessed on January 2011).
- Melillo, M.T.; Leonetti, P.; Bongiovanni, M.; Castagnone-Sereno, P.; Bleve-Zacheo, T. Modulation of reactive oxygen species activities and h2o2 accumulation during compatible and incompatible tomato–root-knot nematode interactions. New Phytol. 2006, 170, 501–512. [Google Scholar] [CrossRef]
- Inoh, Y.; Furuno, T.; Hirashima, N.; Kitamoto, D.; Nakanishi, M. Rapid delivery of small interfering rna by biosurfactant mel-a-containing liposomes. Biochem. Biophys. Res. Commun. 2011, 414, 635–640. [Google Scholar] [CrossRef]
- Dubreuil, G.; Magliano, M.; Deleury, E.; Abad, P.; Rosso, M.N. Transcriptome analysis of root-knot nematode functions induced in the early stages of parasitism*. New Phytol. 2007, 176, 426–436. [Google Scholar] [CrossRef]
- Bakhetia, M.; Urwin, P.E.; Atkinson, H.J. Characterisation by rnai of pioneer genes expressed in the dorsal pharyngeal gland cell of heterodera glycines and the effects of combinatorial rnai. Int. J. Parasitol. 2008, 38, 1589–1597. [Google Scholar] [CrossRef]
- Bakhetia, M.; Urwin, P.E.; Atkinson, H.J. Qpcr analysis and rnai define pharyngeal gland cell-expressed genes of heterodera glycines required for initial interactions with the host. Mol. Plant Microbe Interact 2007, 20, 306–312. [Google Scholar] [CrossRef]
- Jaubert, S.; Milac, A.L.; Petrescu, A.J.; de Almelda-Engler, J.; Abad, P.; Rosso, M.N. In planta secretion of a calreticulin by migratory and sedentary stages of root-knot nematode. Mol. Plant Microbe Interact. 2005, 18, 1277–1284. [Google Scholar] [CrossRef]
- Rosso, M.-N.L.; Favery, B.; Piotte, C.; Arthaud, L.; De Boer, J.M.; Hussey, R.S.; Bakker, J.; Baum, T.J.; Abad, P. Isolation of a cdna encoding a î²-1,4-endoglucanase in the root-knot nematode meloidogyne incognita and expression analysis during plant parasitism. Mol. Plant Microbe Interact. 1999, 12, 585–591. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. The single-step method of rna isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time rt-pcr. Nucleic Acids Res. 2001, 29, E45. [Google Scholar] [CrossRef]
- Hong, J.; Qian, Z.K.; Shen, S.Y.; Min, T.S.; Tan, C.; Xu, J.F.; Zhao, Y.C.; Huang, W.D. High doses of sirnas induce eri-1 and adar-1 gene expression and reduce the efficiency of rna interference in the mouse. Biochem. J. 2005, 390, 675–679. [Google Scholar] [CrossRef]
- Melnyk, C.W.; Molnar, A.; Baulcombe, D.C. Intercellular and systemic movement of rna silencing signals. EMBO J. 2011, 30, 3553–3563. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arguel, M.-J.; Jaouannet, M.; Magliano, M.; Abad, P.; Rosso, M.-N. siRNAs Trigger Efficient Silencing of a Parasitism Gene in Plant Parasitic Root-Knot Nematodes. Genes 2012, 3, 391-408. https://doi.org/10.3390/genes3030391
Arguel M-J, Jaouannet M, Magliano M, Abad P, Rosso M-N. siRNAs Trigger Efficient Silencing of a Parasitism Gene in Plant Parasitic Root-Knot Nematodes. Genes. 2012; 3(3):391-408. https://doi.org/10.3390/genes3030391
Chicago/Turabian StyleArguel, Marie-Jeanne, Maëlle Jaouannet, Marc Magliano, Pierre Abad, and Marie-Noëlle Rosso. 2012. "siRNAs Trigger Efficient Silencing of a Parasitism Gene in Plant Parasitic Root-Knot Nematodes" Genes 3, no. 3: 391-408. https://doi.org/10.3390/genes3030391