Mixing Genetically and Morphologically Distinct Populations in Translocations: Asymmetrical Introgression in A Newly Established Population of the Boodie (Bettongia lesueur)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Translocation Site and History
2.2. Sampling and DNA Extraction
2.3. mtDNA Control Region Sequences
2.4. Microsatellites
2.5. Data Analysis
3. Results
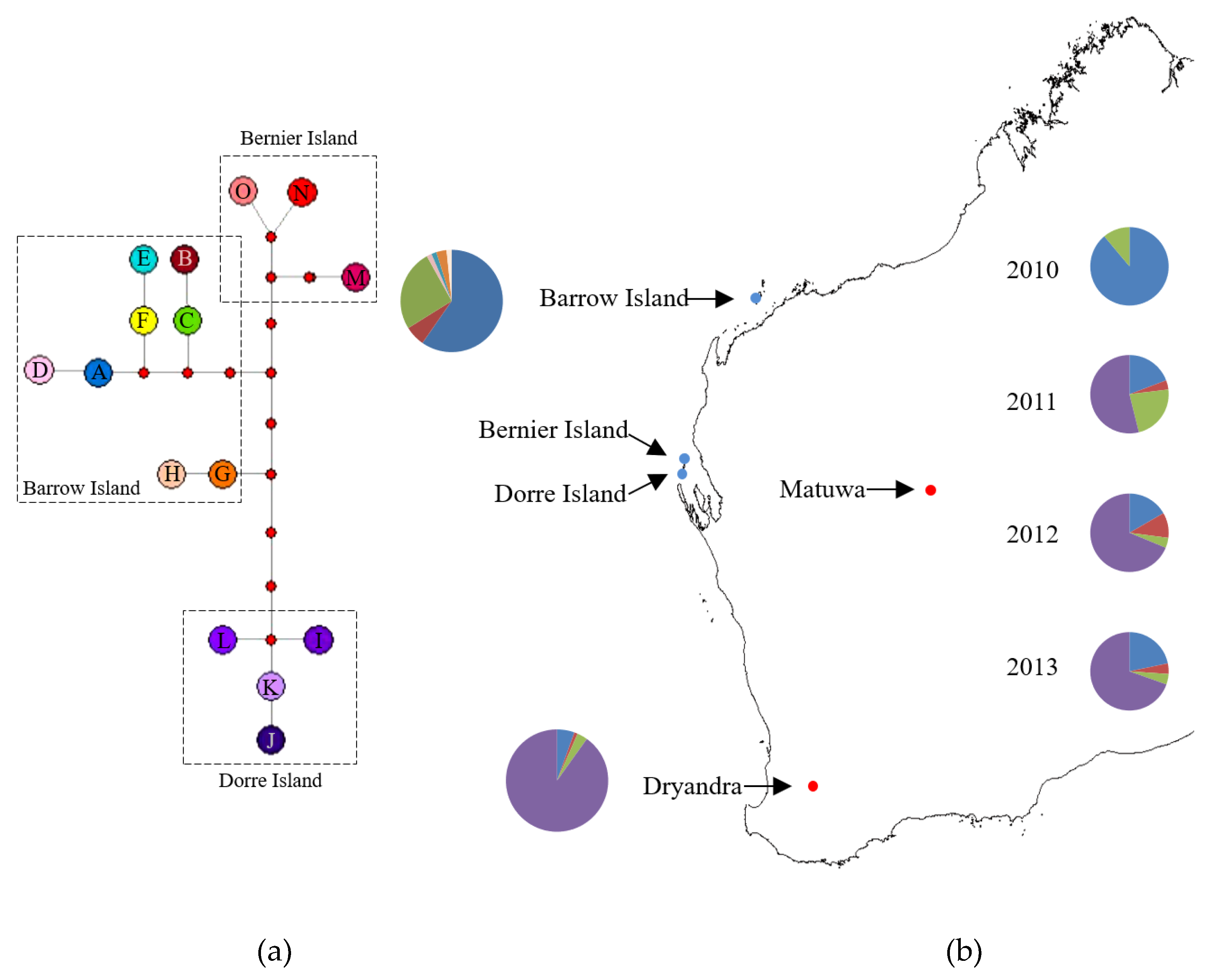
3.1. Mitochondrial DNA Variation
3.2. Microsatellite Variation
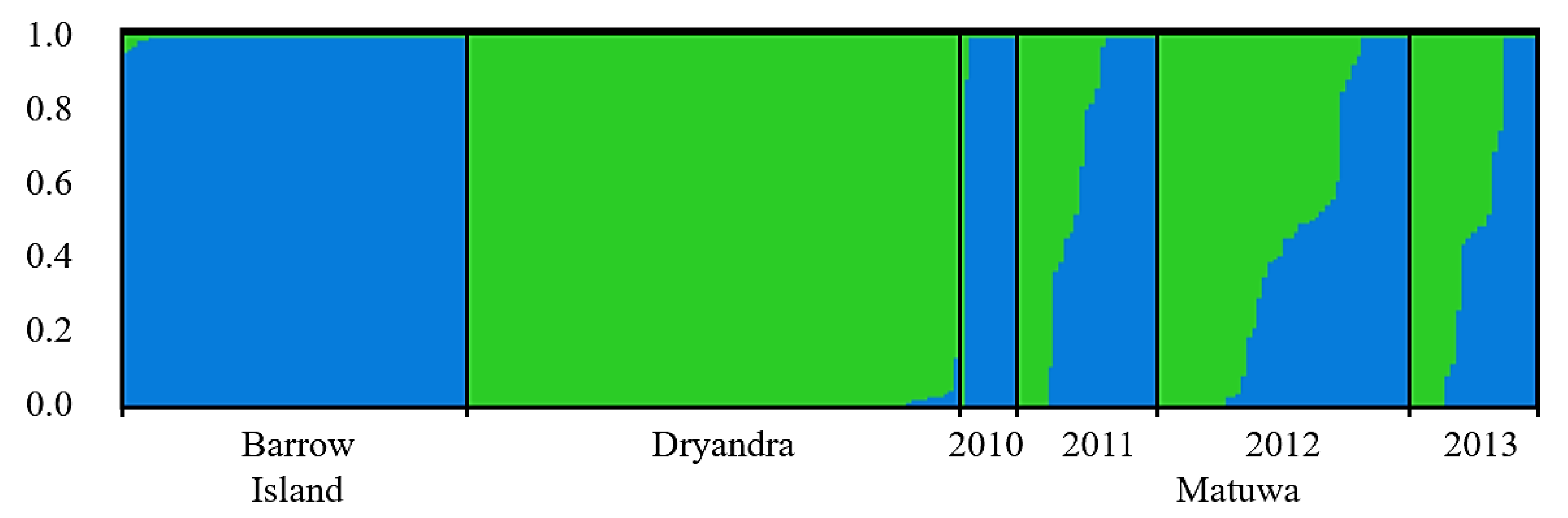
3.3. Population Structure and Genetic Mixing
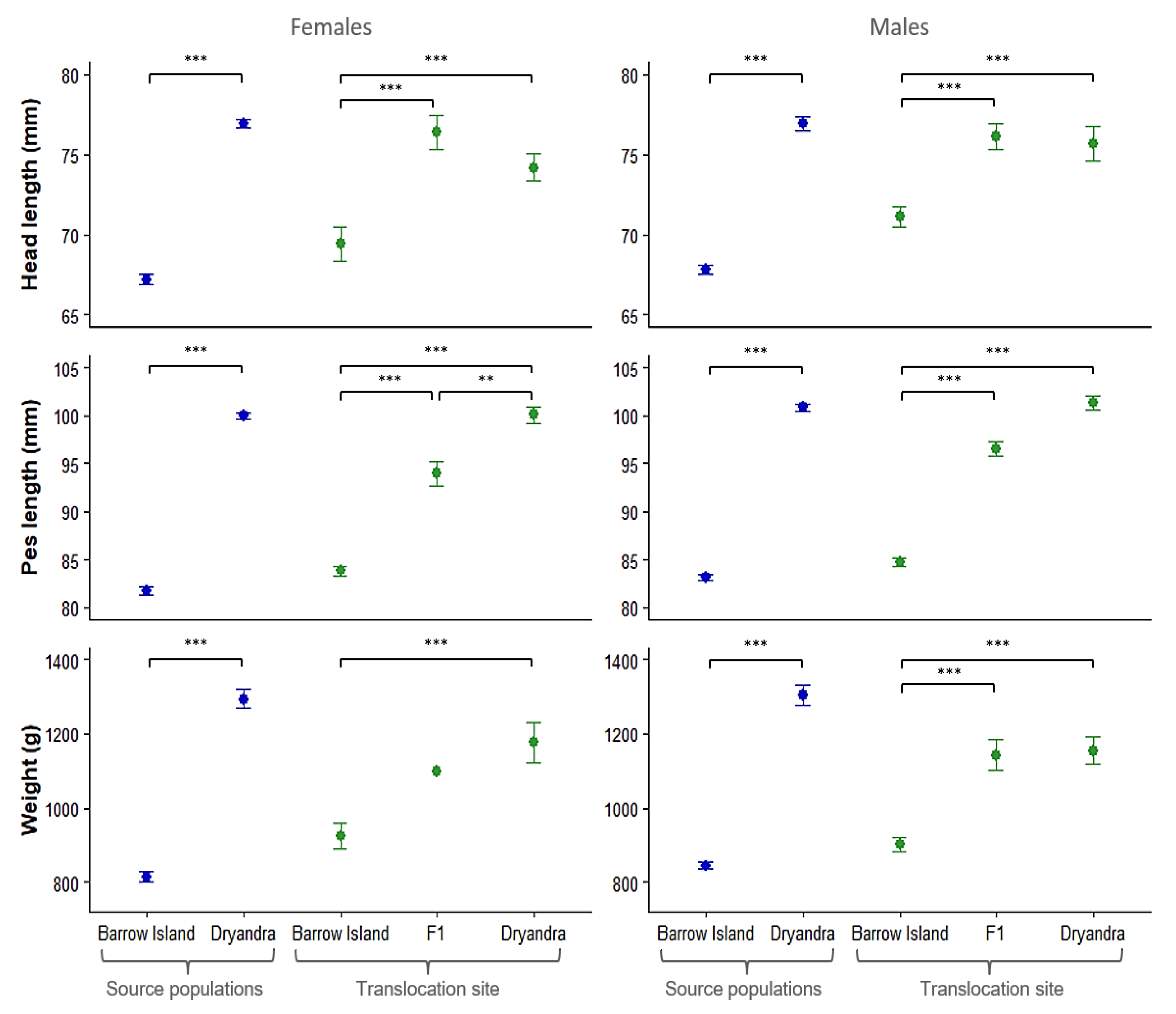
3.4. Variation in Body Size Among Parental and Admixed Generations
4. Discussion
4.1. Phenotypic and Genetic Differentiation Between Island Populations
4.2. Non-Random Mating and Asymmetrical Introgression Between Source Population Lineages
4.3. Increased Genetic Diversity in a Newly Established Population
4.4. Future Direction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoffmann, A.A.; Sgrò, C.M.; Kristensen, T.N. Revisiting adaptive potential, population size, and conservation. Trends Ecol. Evol. 2017, 32, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Garcia-Dorado, A. Understanding inbreeding depression, purging, and genetic rescue. Trends Ecol. Evol. 2016, 31, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Willi, Y.; Van Buskirk, J.; Hoffmann, A.A. Limits to the adaptive potential of small populations. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 433–458. [Google Scholar] [CrossRef]
- Wayne, R.K.; Bruford, M.W.; Girman, B.D.; Rebholz, W.E.R.; Sunnucks, P.; Taylor, A.C. Molecular genetics of endangered species. In Creative Conservation: Interactive Management of Wild and Captive Animals; Olney, P.J.S., Mace, G.M., Feistner, A.T.C., Eds.; Chapman & Hall: London, UK, 1994; pp. 92–117. [Google Scholar]
- Weeks, A.R.; Stoklosa, J.; Hoffmann, A.A. Conservation of genetic uniqueness of populations may increase extinction likelihood of endangered species: The case of australian mammals. Front. Zool. 2016, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C. Conservation units and translocations: Strategies for conserving evolutionary processes. Hereditas 1999, 130, 217–228. [Google Scholar] [CrossRef]
- Ransler, F.A.; Quinn, T.W.; Oyler-McCance, S.J. Genetic consequences of trumpeter swan (Cygnus buccinator) reintroductions. Conserv. Genet. 2011, 12, 257–268. [Google Scholar] [CrossRef]
- Frankham, R. Genetic rescue of small inbred populations: Meta-analysis reveals large and consistent benefits of gene flow. Mol. Ecol. 2015, 24, 2610–2618. [Google Scholar] [CrossRef]
- Weeks, A.R.; Heinze, D.; Perrin, L.; Stoklosa, J.; Hoffmann, A.A.; van Rooyen, A.; Kelly, T.; Mansergh, I. Genetic rescue increases fitness and aids rapid recovery of an endangered marsupial population. Nat. Commun. 2017, 8, 1071. [Google Scholar] [CrossRef]
- Edmands, S.; Timmerman, C.C. Modeling factors affecting the severity of outbreeding depression. Conserv. Biol. 2003, 17, 883–892. [Google Scholar] [CrossRef]
- Johnson, W.E.; Onorato, D.P.; Roelke, M.E.; Land, E.D.; Cunningham, M.; Belden, R.C.; McBride, R.; Jansen, D.; Lotz, M.; Shindle, D.; et al. Genetic restoration of the florida panther. Science 2010, 329, 1641–1645. [Google Scholar] [CrossRef]
- Pimm, S.L.; Dollar, L.; Bass, O.L. The genetic rescue of the florida panther. Anim. Conserv. 2006, 9, 115–122. [Google Scholar] [CrossRef]
- Hostetler, J.A.; Onorato, D.P.; Nichols, J.D.; Johnson, W.E.; Roelke, M.E.; O’Brien, S.J.; Jansen, D.; Oli, M.K. Genetic introgression and the survival of florida panther kittens. Biol. Conserv. 2010, 143, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.B.; King, J.M.; Loupis, A.K.; Spencer, P.B.S.; Taylor, A.C.; Pope, L.C.; Hall, G.P. Unprecedented low levels of genetic variation and inbreeding depression in an island population of the black-footed rock-wallaby. Conserv. Biol. 1999, 13, 531–541. [Google Scholar] [CrossRef]
- Thompson, S.L.; Lamothe, M.; Meirmans, P.G.; Perinet, P.; Isabel, N. Repeated unidirectional introgression towards Populus balsamifera in contact zones of exotic and native poplars. Mol. Ecol. 2010, 19, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B.; Boughman, J.W.; Groenenboom, M.; Sniatynski, M.; Schluter, D.; Gow, J.L. Speciation in reverse: Morphological and genetic evidence of the collapse of a three-spined stickleback (Gasterosteus aculeatus) species pair. Mol. Ecol. 2006, 15, 343–355. [Google Scholar] [CrossRef]
- Miller, J.M.; Poissant, J.; Hogg, J.T.; Coltman, D.W. Genomic consequences of genetic rescue in an insular population of bighorn sheep (Ovis canadensis). Mol. Ecol. 2012, 21, 1583–1596. [Google Scholar] [CrossRef]
- Kennington, W.J.; Hevroy, T.H.; Johnson, M.S. Long-term genetic monitoring reveals contrasting changes in the genetic composition of newly established populations of the intertidal snail Bembicium vittatum. Mol. Ecol. 2012, 21, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.R.; Vucetich, L.M.; Hedrick, P.W.; Peterson, R.O.; Vucetich, J.A. Genomic sweep and potential genetic rescue during limiting environmental conditions in an isolated wolf population. Proc. R. Soc. Lond. B Biol. Sci. 2011, 278, 3336–3344. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R. Genetic rescue benefits persist to at least the F3 generation, based on a meta-analysis. Biol. Conserv. 2016, 195, 33–36. [Google Scholar] [CrossRef]
- Binks, R.M.; Kennington, J.W.; Johnson, M.S. Rapid evolutionary responses in a translocated population of intertidal snail (Bembicium vittatum) utilise variation from different source populations. Conserv. Genet. 2007, 8, 1421–1429. [Google Scholar] [CrossRef]
- Edmands, S. Between a rock and a hard place: Evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 2007, 16, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Tallmon, D.A.; Luikart, G.; Waples, R.S. The alluring simplicity and complex reality of genetic rescue. Trends Ecol. Evol. 2004, 19, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Brodie, E.D. Correlational selection for color pattern and antipredator behavior in the garter snake Thamnophis ordinoides. Evolution 1992, 46, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Fishman, L.; Willis, J.H. Evidence for dobzhansky-muller incompatibilites contributing to the sterility of hybrids between Mimulus guttatus and M. nasutus. Evolution 2001, 55, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Gharrett, A.J.; Smoker, W.W.; Reisenbichler, R.R.; Taylor, S.G. Outbreeding depression in hybrids between odd- and even-broodyear pink salmon. Aquaculture 1999, 173, 117–129. [Google Scholar] [CrossRef]
- Huff, D.D.; Miller, L.M.; Chizinski, C.J.; Vondracek, B. Mixed-source reintroductions lead to outbreeding depression in second-generation descendents of a native North American fish. Mol. Ecol. 2011, 20, 4246–4258. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.L.; Bradshaw, C.J.A.; Goldsworthy, S.D.; Sunnucks, P. Lower reproductive success in hybrid fur seal males indicates fitness costs to hybridization. Mol. Ecol. 2007, 16, 3187–3197. [Google Scholar] [CrossRef]
- Alexandrino, J.; Baird, S.J.E.; Lawson, L.; Macey, J.R.; Moritz, C.; Wake, D.B. Strong selection against hybrids at a hybrid zone in the Ensatina ring species complex and its evolutionary implications. Evolution 2005, 59, 1334–1347. [Google Scholar] [CrossRef]
- Latch, E.K.; Harveson, L.A.; King, J.S.; Hobson, M.D.; Rhodes, O.E. Assessing hybridization in wildlife populations using molecular markers: A case study in wild turkeys. J. Wildl. Manag. 2006, 70, 485–492. [Google Scholar] [CrossRef]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer Association: Sunderland, MA, USA, 2004. [Google Scholar]
- Hedrick, P.W.; Robinson, J.A.; Peterson, R.O.; Vucetich, J.A. Genetics and extinction and the example of isle royale wolves. Anim. Conserv. 2019, 22, 302–309. [Google Scholar] [CrossRef]
- Edmands, S. Does parental divergence predict reproductive compatibility? Trends Ecol. Evol. 2002, 17, 520–527. [Google Scholar] [CrossRef]
- Lynch, M. The genetic interpretation of inbreeding depression and outbreeding depression. Evolution 1991, 45, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Weeks, A.R.; Sgro, C.M.; Young, A.G.; Frankham, R.; Mitchell, N.J.; Miller, K.A.; Byrne, M.; Coates, D.J.; Eldridge, M.D.B.; Sunnucks, P.; et al. Assessing the benefits and risks of translocations in changing environments: A genetic perspective. Evol. Appl. 2011, 4, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Close, R.L.; Lowry, P.S. Hybrids in marsupial research. Aust. J. Zool. 1989, 37, 259–267. [Google Scholar] [CrossRef]
- Burbidge, A.A.; Short, J.C. Burrowing bettong. In The mammals of Australia; Strahan, R., Ed.; Australian Museum and Reed Books: Chatswood, NSW, Australia, 1995. [Google Scholar]
- Short, J.; Turner, B. Ecology of burrowing bettongs, Bettongia lesueur (marsupialia: Potoroidae), on Dorre and Bernier islands, Western Australia. Wildl. Res. 1999, 26, 651–669. [Google Scholar]
- Donaldson, F.; Bencini, R.; Morris, K.; Teale, R.; Wale, C.H.; How, R.A.; Schmitt, L.H. The burrowing bettongs of barrow island: Demographic and genetic insights into a threatened macropod. Aust. J. Zool. 2018, 65, 257–272. [Google Scholar] [CrossRef]
- Sander, U.; Short, J.; Turner, B. Social organisation and warren use of the burrowing bettong, Bettongia lesueur (macropodoidea: Potoroidae). Wildl. Res. 1997, 24, 143–157. [Google Scholar] [CrossRef]
- Jarman, P.J. Social behavior and organization in the macropodoidea. In Advances in the Study of Behavior; Slater, P.J.B., Rosenblatt, J.S., Beer, C., Milinski, M., Eds.; Academic Press: Cambridge, MA, USA, 1991; Volume 20, pp. 1–50. [Google Scholar]
- Woinarski, J.; Burbidge, A.A. Bettongia lesueur. In The IUCN Red List of Threatened Species; The IUCN Red List of Threatened Species: Cambridge, UK, 2019. [Google Scholar]
- Short, J.; Turner, B. The distribution and abundance of the burrowing bettong (marsupialia: Macropodoidea). Wildl. Res. 1993, 20, 525–534. [Google Scholar] [CrossRef]
- Richards, J.D. Western Barred Bandicoot Perameles bougainville, Burrowing Bettong Bettongia lesueur and Banded Hare-wallaby Lagostrophus fasciatus National Recovery Plan; Department of Environment and Conservation: Perth, Australia, 2012. [Google Scholar]
- Dortch, C.E.; Morse, K. Prehistoric stone artefacts on some offshore islands in Western Australia. Aust. Archaeol. 1984, 19, 31–47. [Google Scholar]
- Sims, C.; Morris, K.; Blythman, M. Rangeland Restoration: Fauna Recovery at Matuwa (Lorna Glen), Western Australia; Annual Report 2016; Department of Parks and Wildlife: Perth, Australia, 2017. [Google Scholar]
- Frankham, R.; Ballou, J.D.; Eldridge, M.D.B.; Lacy, R.C.; Ralls, K.; Dudash, M.R.; Fenster, C.B. Predicting the probability of outbreeding depression. Conserv. Biol. 2011, 25, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Sunnucks, P.; Hales, D.F. Numerous transposed sequences of mitochondrial cytochrome oxidase i-ii in aphids of the genus Sitobion (hemiptera: Aphididae). Mol. Biol. Evol. 1996, 13, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, L.; Pope, L.C.; Taberlet, P.; Moritz, C. Versatile primers for the amplification of the mitochondrial DNA control region in marsupials. Mol. Ecol. 1997, 6, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The clustal_x windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Donaldson, F.R.; Vercoe, P.E. Cross-family amplification: Microsatellites isolated from macropodidae are polymorphic in potoroidae. Mol. Ecol. Resour. 2008, 8, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.B.S.; Odorico, D.M.; Jones, S.J.; Marsh, H.D.; Miller, D.J. Highly variable microsatellites in isolated colonies of the rock-wallaby (Petrogale assimilis). Mol. Ecol. 1995, 4, 523–525. [Google Scholar] [CrossRef]
- Zenger, K.R.; McKenzie, L.M.; Cooper, D.W. The first comprehensive genetic linkage map of a marsupial: The tammar wallaby (Macropus eugenii). Genetics 2002, 162, 321–330. [Google Scholar]
- Pope, L.C.; Sharp, A.; Moritz, C. Population structure of the yellow-footed rock-wallaby Petrogale xanthopus (gray, 1854) inferred from mtDNA sequences and microsatellite loci. Mol. Ecol. 1996, 5, 629–640. [Google Scholar] [CrossRef]
- Luikart, G.; Painter, J.; Crozier, R.H.; Westerman, M.; Sherwin, W.B. Characterization of microsatellite loci in the endangered long-footed potoroo Potorous longipes. Mol. Ecol. 1997, 6, 497–498. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. Dnasp v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Polzin, T.; Daneshmand, S.V. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003, 31, 12–20. [Google Scholar] [CrossRef]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.; Derek, P.; Wills, M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Goudet, J. Fstat, a Program to Estimate and Test Gene Diversities and Fixation Indices. Version 2.9.3. 2001. Available online: http://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 4 November 2013).
- Weir, B.S.; Cockerham, C.C. Estimating f-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Anderson, E.C.; Thompson, E.A. A model-based method for identifying species hybrids using multilocus genetic data. Genetics 2002, 160, 1217–1229. [Google Scholar] [PubMed]
- Woinarski, J.C.Z.; Burbidge, A.A.; Harrison, P.; Kelly, J. The Action Plan for Australian Mammals 2012; CSIRO Publishing: Collingwood, Australia, 2014. [Google Scholar]
- Arntzen, J.W.; Jehle, R.; Bardakci, F.; Burke, T.; Wallis, G.P. Asymmetric viability of reciprocal-cross hybrids between crested and marbled newts (Triturus cristatus and T. marmoratus). Evolution 2009, 63, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, D.; Garcia-Vazquez, E. Maintenance of asymmetric hybridization between atlantic salmon (Salmo salar) and brown trout (Salmo trutta) via postzygotic barriers and paternal effects. Can. J. Fish. Aquat. Sci. 2011, 68, 593–602. [Google Scholar] [CrossRef]
- Freegard, C.J.; Calver, M.C.; Richards, J.D.; Bradley, S. Age estimation of pouch young and growth of the burrowing bettong Bettongia lesueur (marsupialia: Potoroidae) on Heirisson prong, Shark bay, Western Australia. Aust. Mammal. 2008, 29, 157–167. [Google Scholar] [CrossRef]
- Teixeira, C.P.; de Azevedo, C.S.; Mendl, M.; Cipreste, C.F.; Young, R.J. Revisiting translocation and reintroduction programmes: The importance of considering stress. Anim. Behav. 2007, 73, 13. [Google Scholar] [CrossRef]
- Moseby, K.E.; Read, J.L.; Paton, D.C.; Copley, P.; Hill, B.M.; Crisp, H.A. Predation determines the outcome of 10 reintroduction attempts in arid south australia. Biol. Conserv. 2011, 144, 2863–2872. [Google Scholar] [CrossRef]
- Biebach, I.; Keller, L.F. Genetic variation depends more on admixture than number of founders in reintroduced alpine ibex populations. Biol. Conserv. 2012, 147, 197–203. [Google Scholar] [CrossRef]
- Nelson, N.J.; Keall, S.N.; Brown, D.; Daugherty, C.H. Establishing a new wild population of tuatara (Sphenodon guntheri). Conserv. Biol. 2002, 16, 887–894. [Google Scholar] [CrossRef]
- Holleley, C.E.; Dickman, C.R.; Crowther, M.S.; Oldroyd, B.P. Size breeds success: Multiple paternity, multivariate selection and male semelparity in a small marsupial, Antechinus stuartii. Mol. Ecol. 2006, 15, 3439–3448. [Google Scholar] [CrossRef]
- Kraaijeveld-Smit, F.J.L.; Ward, S.J.; Temple-Smith, P.D.; Paetkau, D. Factors influencing paternity success in Antechinus agilis: Last-male sperm precedence, timing of mating and genetic compatibility. J. Evol. Biol. 2002, 15, 100–107. [Google Scholar] [CrossRef]
- Parrott, M.L.; Ward, S.J.; Temple-Smith, P.D.; Selwood, L. Females choose mates based on genetic relatedness in a small dasyurid marsupial, the agile antechinus (Antechinus agilis). PLoS ONE 2015, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.S.C.; Mezger, M.; Kolar, M.; Michler, F.-U.; Sommer, S. The best smellers make the best choosers: Mate choice is affected by female chemosensory receptor gene diversity in a mammal. Proc. R. Soc. Lond. B Biol. Sci. 2018, 285, 20182426. [Google Scholar] [CrossRef]
- Lipshutz, S.E.; Meier, J.I.; Derryberry, G.E.; Miller, M.J.; Seehausen, O.; Derryberry, E.P. Differential introgression of a female competitive trait in a hybrid zone between sex-role reversed species. Evolution 2019, 73, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Huff, D.D.; Miller, L.M.; Vondracek, B. Patterns of ancestry and genetic diversity in reintroduced populations of the slimy sculpin: Implications for conservation. Conserv. Genet. 2010, 11, 2379–2391. [Google Scholar] [CrossRef]
- White, L.C.; Moseby, K.E.; Thomson, V.A.; Donnellan, S.C.; Austin, J.J. Long-term genetic consequences of mammal reintroductions into an Australian conservation reserve. Biol. Conserv. 2018, 219, 11. [Google Scholar] [CrossRef]
- Miller, K.A.; Nelson, N.J.; Smith, H.G.; Moore, J.A. How do reproductive skew and founder group size affect genetic diversity in reintroduced populations? Mol. Ecol. 2009, 18, 3792–3802. [Google Scholar] [CrossRef]
- Sigg, D.P. Reduced genetic diversity and significant genetic differentiation after translocation: Comparison of the remnant and translocated populations of bridled nailtail wallabies (Onychogalea fraenata). Conserv. Genet. 2006, 7, 577–589. [Google Scholar] [CrossRef]
- Bolnick, D.I.; Turelli, M.; López-Fernández, H.; Wainwright, P.C.; Near, T.J. Accelerated mitochondrial evolution and “darwin’s corollary”: Asymmetric viability of reciprocal F1 hybrids in centrarchid fishes. Genetics 2008, 178, 1037–1048. [Google Scholar] [CrossRef]
- Rick, K.; Ottewell, K.; Lohr, C.; Thavornkanlapachai, R.; Byrne, M.; Kennington, W.J. Population genomics of Bettongia lesueur: admixing increases genetic diversity with no evidence of outbreeding depression. Genes submitted. 2019. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Microsatellites | Mitochondrial DNA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Allelic Richness | H | FIS | Pairs of Loci in GD | N | Number of Haplotypes | H | Nucleotide Diversity | |
| Barrow Island | 66 | 3.8 (0.5) | 0.56 (0.07) | 0.02 | 0 | 62 | 7 | 0.58 (0.01) | 0.008 (0.0003) |
| Dryandra | 95 | 2.8 (0.2) | 0.47 (0.07) | −0.03 | 2 | 91 | 4 | 0.19 (0.01) | 0.004 (0.0001) |
| Matuwa 2010 | 11 | 4.4 (0.5) | 0.61 (0.05) | 0.18 * | 0 | 9 | 2 | 0.22 (0.06) | 0.001 (0.0004) |
| Matuwa 2011 | 27 | 4.9 (0.5) | 0.69 (0.04) | 0.19 * | 16 | 26 | 4 | 0.64 (0.01) | 0.012 (0.0002) |
| Matuwa 2012 | 48 | 5.0 (0.5) | 0.69 (0.05) | 0.11 * | 24 | 48 | 4 | 0.50 (0.01) | 0.010 (0.0002) |
| Matuwa 2013 | 24 | 4.9 (0.5) | 0.71 (0.04) | 0.19 * | 3 | 24 | 4 | 0.47 (0.02) | 0.009 (0.0004) |
| Matuwa overall | 113 | 5.1 (0.5) | 0.70 (0.04) | 0.17 * | 64 | 110 | 4 | 0.59 (0.00) | 0.011 (0.0001) |
| Barrow Island | Dryandra | 2010 | 2011 | 2012 | 2013 | |
|---|---|---|---|---|---|---|
| Barrow Island | – | 0.72 * | −0.02 | 0.35 * | 0.49 * | 0.50 * |
| Dryandra | 0.42 * | – | 0.75 * | 0.30 * | 0.11 * | 0.10 |
| 2010 | 0.01 | 0.38 * | – | 0.26 * | 0.43 * | 0.44 * |
| 2011 | 0.09 * | 0.20 * | 0.04 | – | 0.02 | 0.02 |
| 2012 | 0.18 * | 0.10 * | 0.11 * | 0.02 | – | –0.03 |
| 2013 | 0.17 * | 0.13 * | 0.10 * | 0.02 | 0.00 | – |
| Sample | N | Observed | Expected | χ2 | P | ||
|---|---|---|---|---|---|---|---|
| BWI | DRY | BWI | DRY | ||||
| 2010 | 11 | 0.895 | 0.105 | 0.468 | 0.532 | – | – |
| 2011 | 27 | 0.609 | 0.391 | 0.468 | 0.532 | 2.1 | 0.143 |
| 2012 | 48 | 0.431 | 0.569 | 0.468 | 0.532 | 0.3 | 0.605 |
| 2013 | 24 | 0.450 | 0.550 | 0.468 | 0.532 | 0.0 | 0.858 |
| Overall | 113 | 0.537 | 0.463 | 0.468 | 0.532 | 2.1 | 0.143 |
| Sample | Haplotype | χ2 | P | ||||
|---|---|---|---|---|---|---|---|
| A | B | C | I | Others | |||
| Pure Barrow Island | 23 (26.7) | 6 (1.9) | 11 (9.5) | – | 0 (1.9) | 1.99 | 0.158 |
| Pure Dryandra | 1 (1.2) | 0 (0.0) | 0 (1.2) | 30 (28.5) | – | 0.96 | 0.327 |
| F1 hybrid | 1 (5.7) | 1 (0.4) | 0 (2.4) | 18 (11.2) | 0 (0.4) | 9.48 | 0.002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thavornkanlapachai, R.; Mills, H.R.; Ottewell, K.; Dunlop, J.; Sims, C.; Morris, K.; Donaldson, F.; Kennington, W.J. Mixing Genetically and Morphologically Distinct Populations in Translocations: Asymmetrical Introgression in A Newly Established Population of the Boodie (Bettongia lesueur). Genes 2019, 10, 729. https://doi.org/10.3390/genes10090729
Thavornkanlapachai R, Mills HR, Ottewell K, Dunlop J, Sims C, Morris K, Donaldson F, Kennington WJ. Mixing Genetically and Morphologically Distinct Populations in Translocations: Asymmetrical Introgression in A Newly Established Population of the Boodie (Bettongia lesueur). Genes. 2019; 10(9):729. https://doi.org/10.3390/genes10090729
Chicago/Turabian StyleThavornkanlapachai, Rujiporn, Harriet R. Mills, Kym Ottewell, Judy Dunlop, Colleen Sims, Keith Morris, Felicity Donaldson, and W. Jason Kennington. 2019. "Mixing Genetically and Morphologically Distinct Populations in Translocations: Asymmetrical Introgression in A Newly Established Population of the Boodie (Bettongia lesueur)" Genes 10, no. 9: 729. https://doi.org/10.3390/genes10090729