A Comparative Transcriptome Analysis Reveals Physiological Maturation Properties of Mycelia in Pleurotus tuoliensis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Pleurotus Tuoliensis Mycelia
2.2. Library Preparation and RNA-Seq
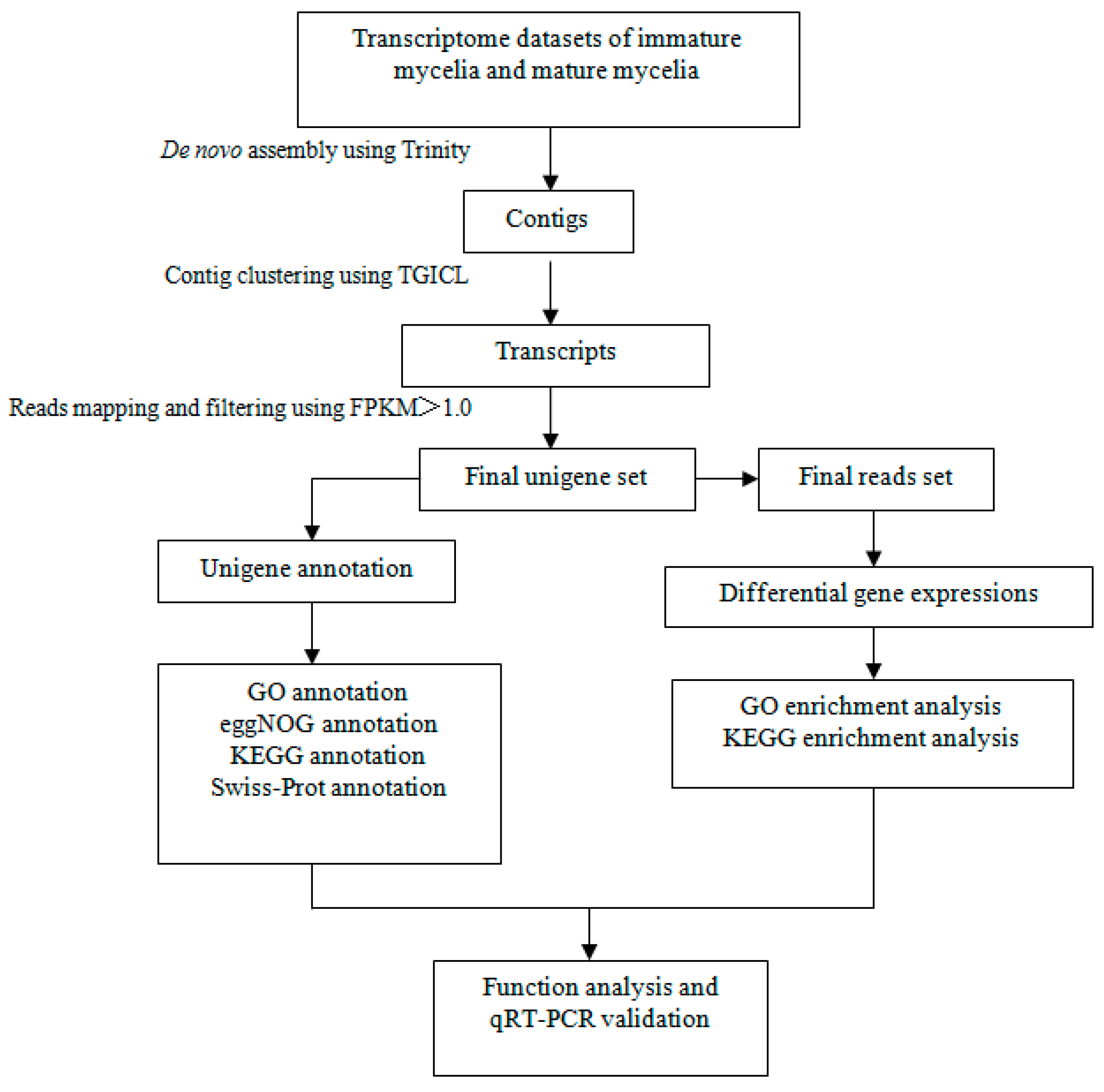
2.3. De Novo Transcriptome Assembly and Annotation
2.4. Identification and Functional Analysis of Differentially Expressed Genes (DEGs)
2.5. Reverse Transcription-Quantitative PCR (RT-qPCR) Analysis
3. Results
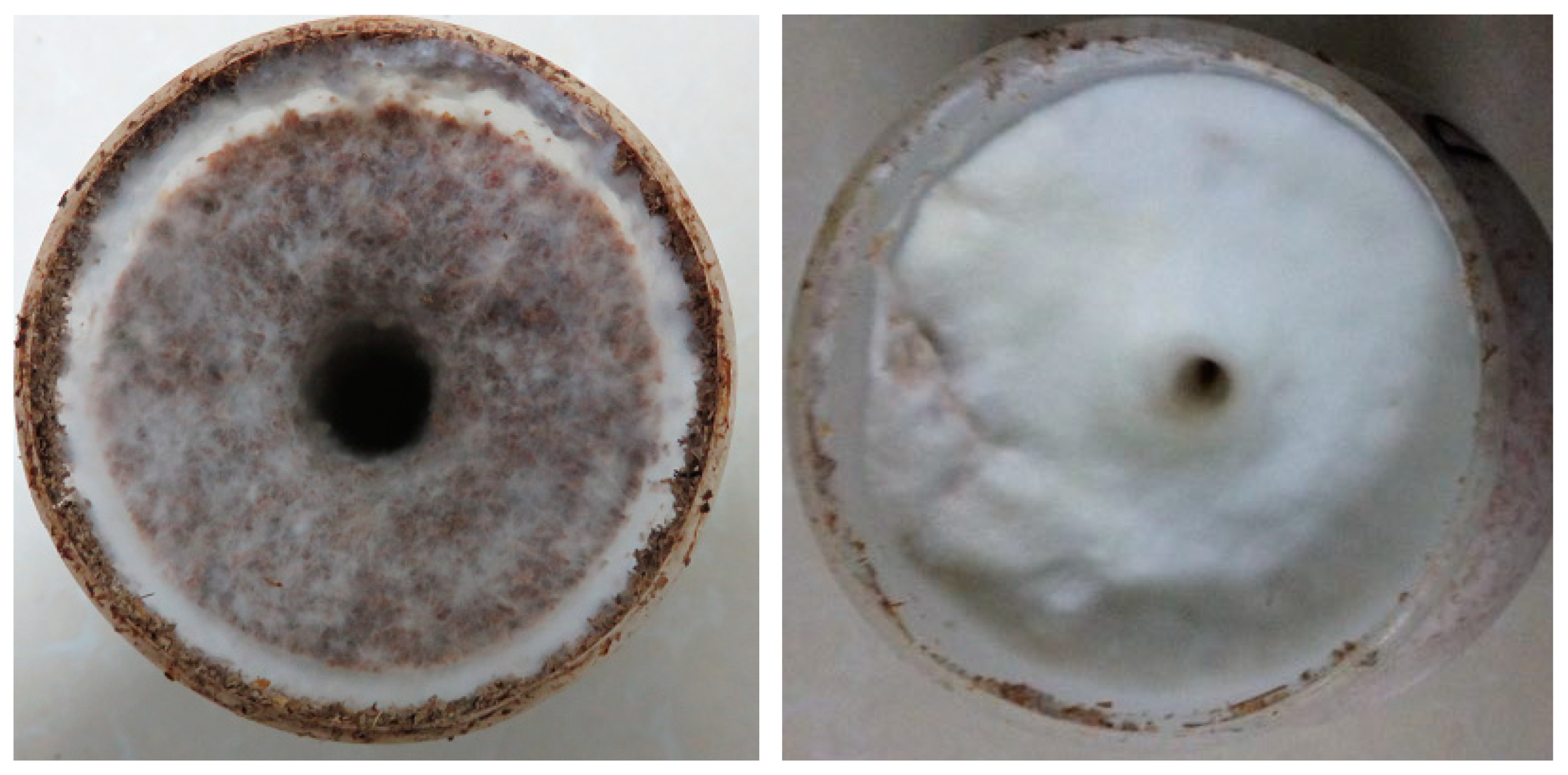
3.1. Growth of P. Tuoliensis Mycelia
3.2. Sequencing and Gene Annotation
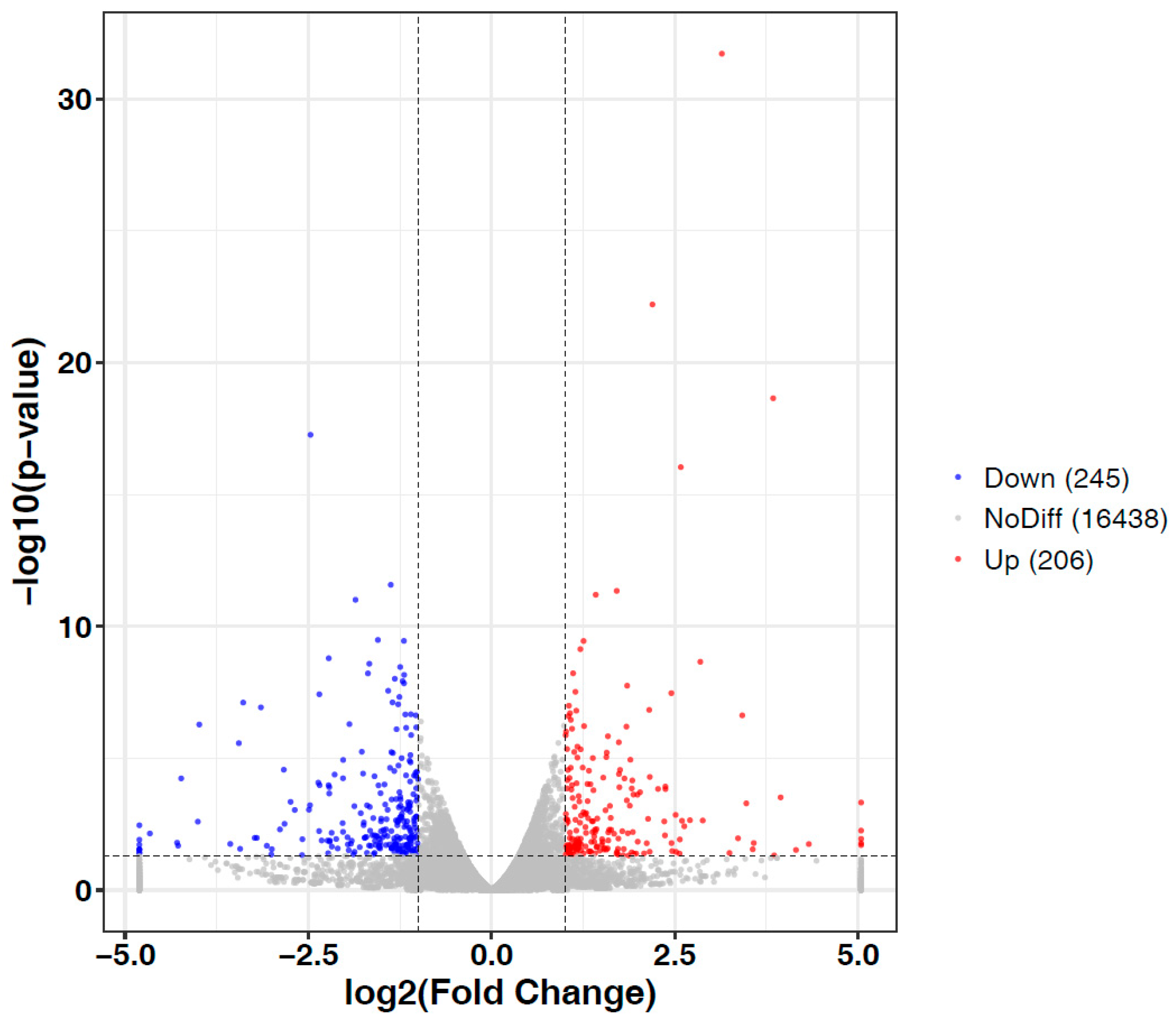
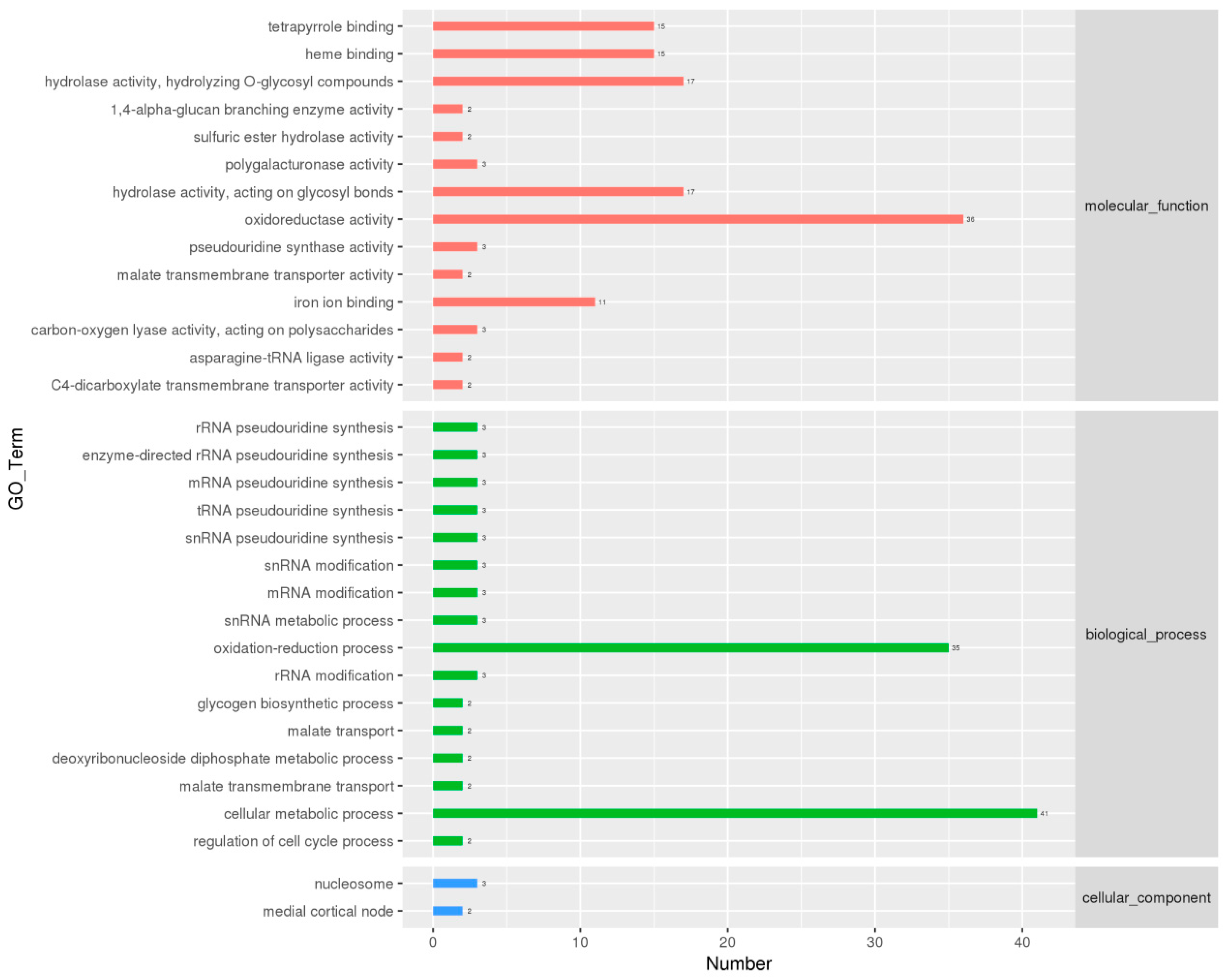
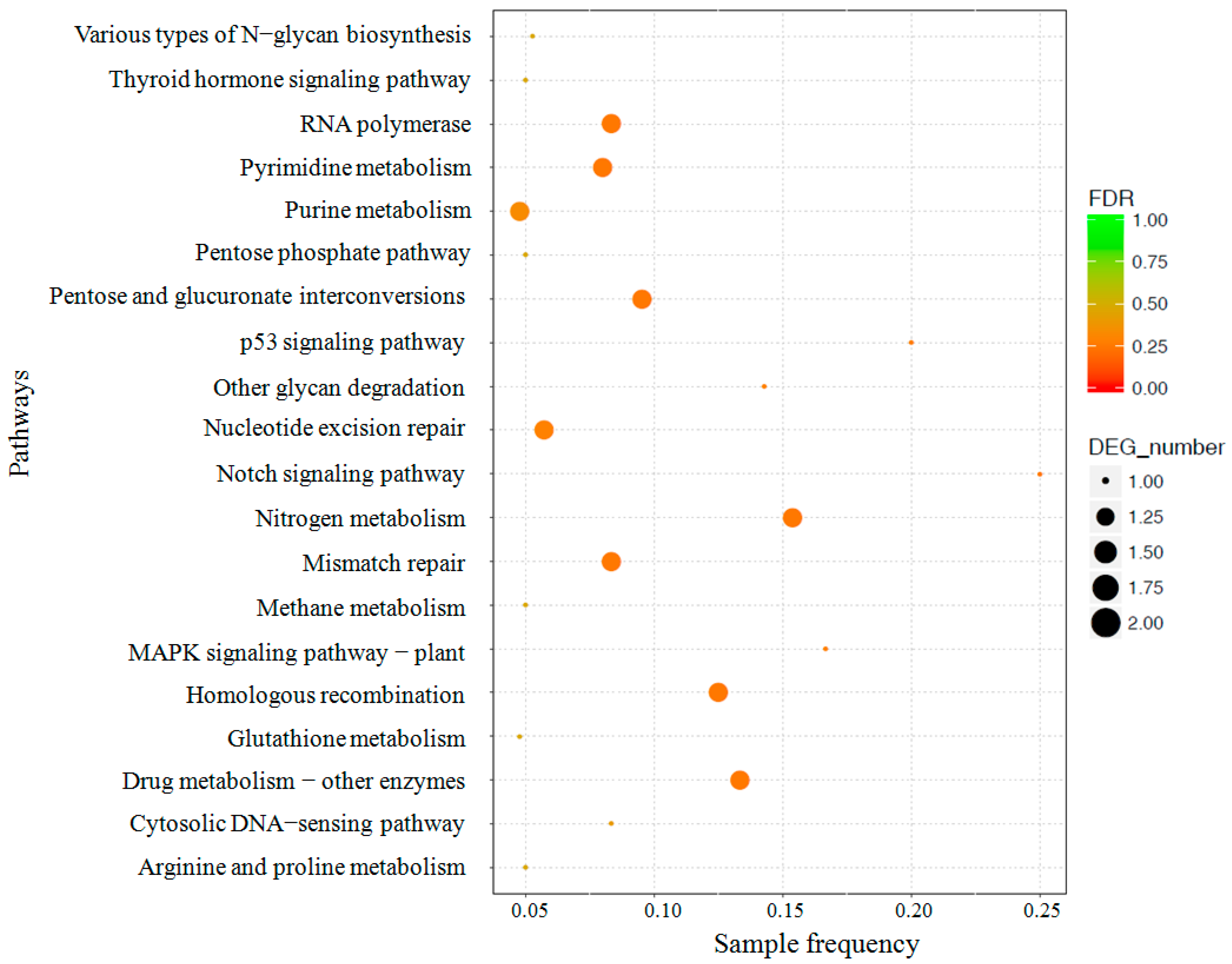
3.3. Identification and Functional Enrichment Analysis of Differentially Expressed Genes (DEGs)
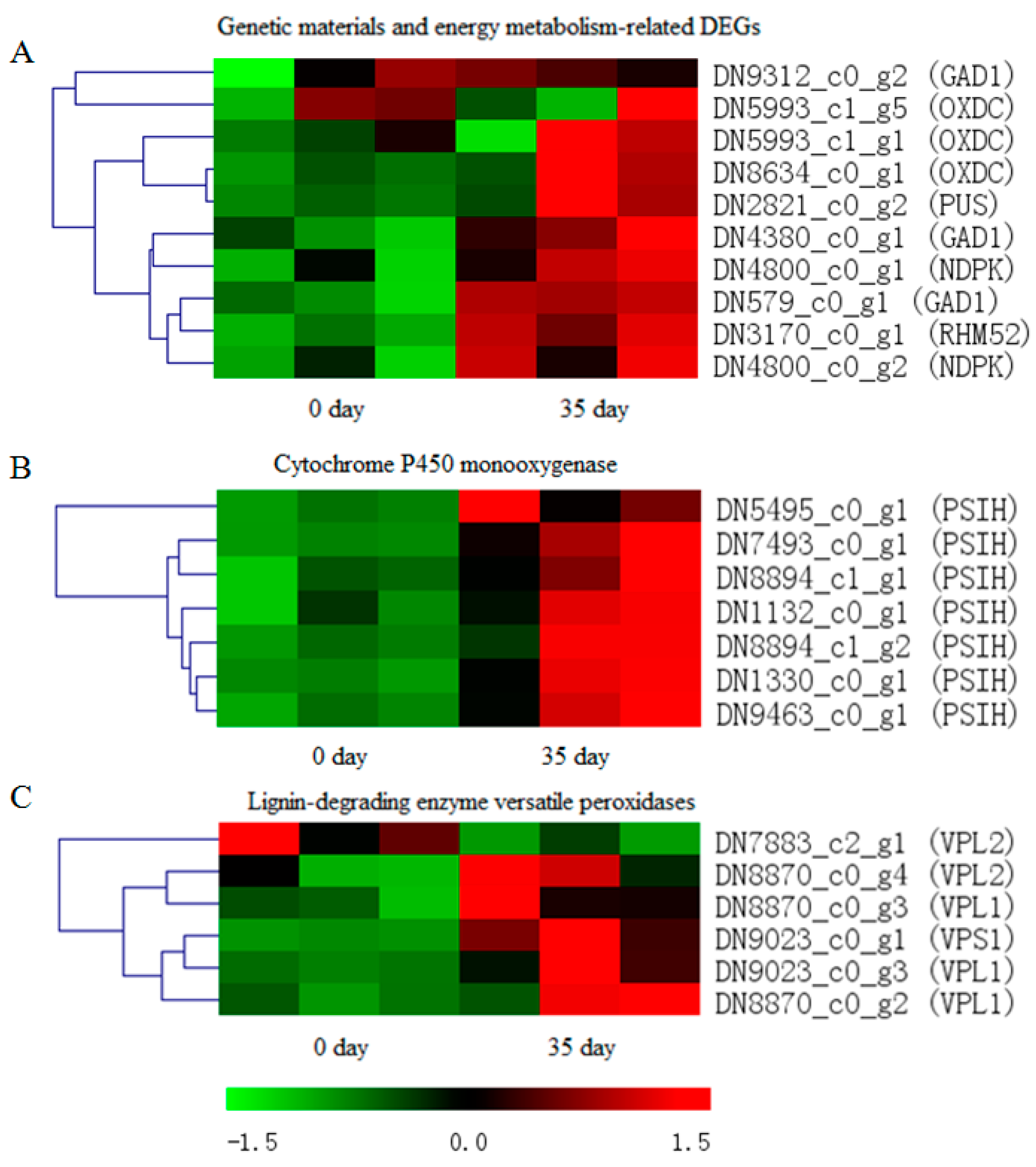
3.4. Identification of DEGs Related to Physiological Maturation of Mycelia
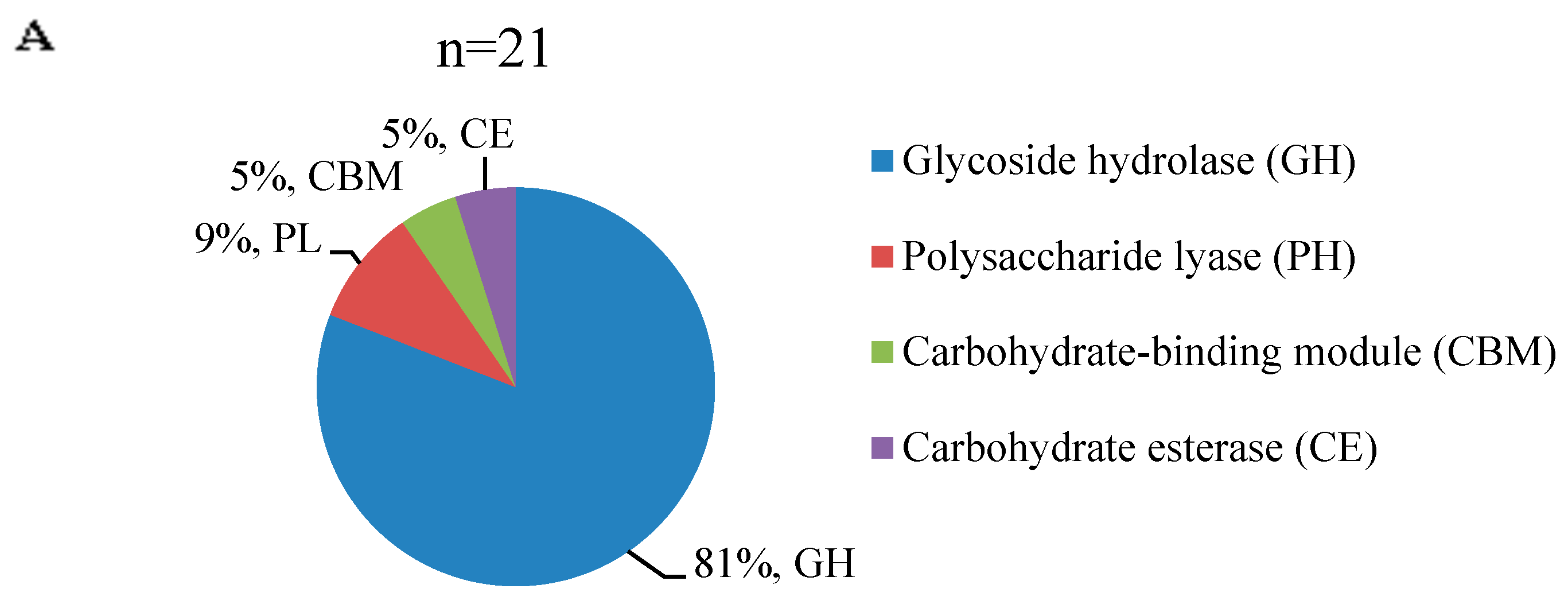
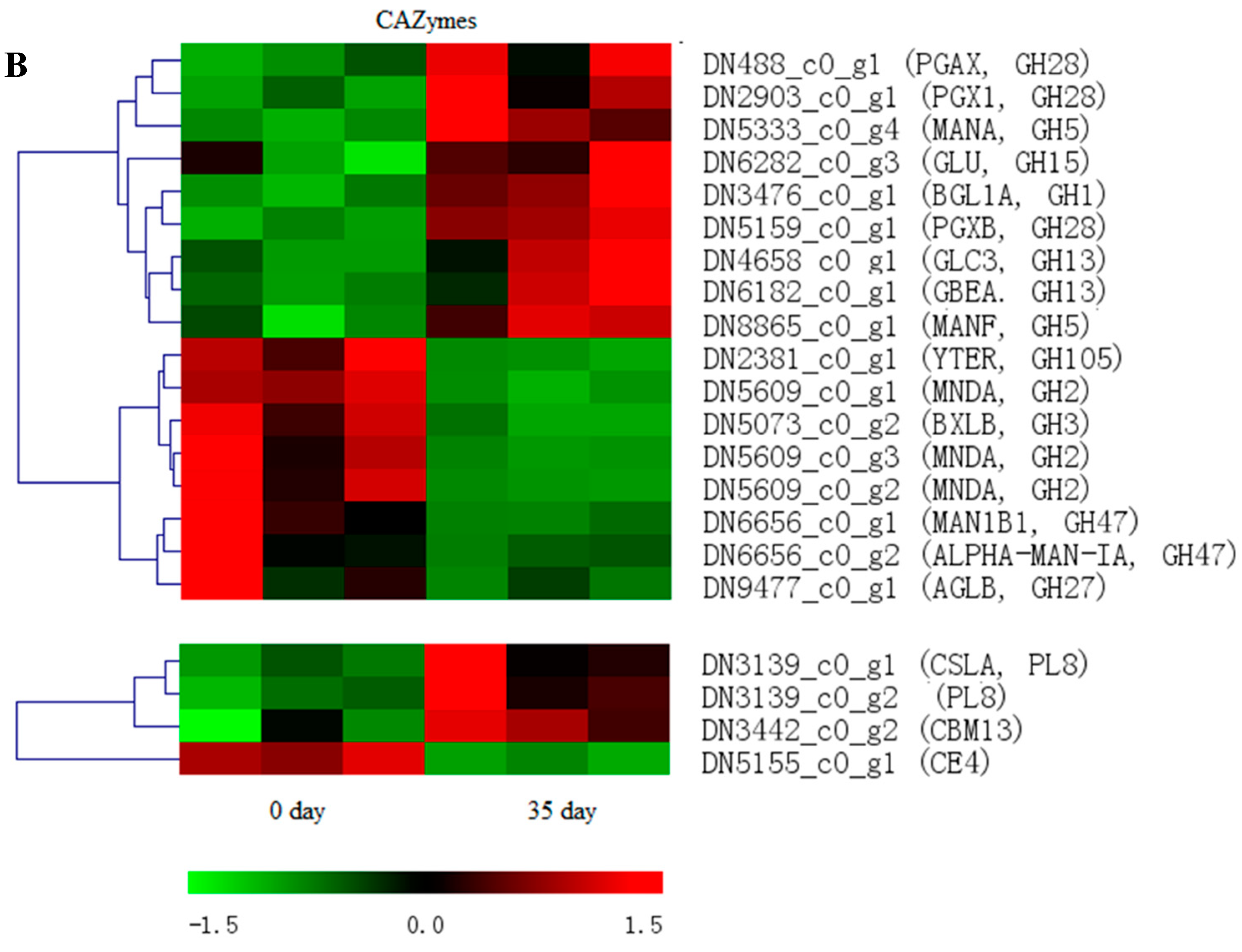
3.5. Distinct Distribution of Carbohydrate-Active Enzymes during Mycelial Maturation
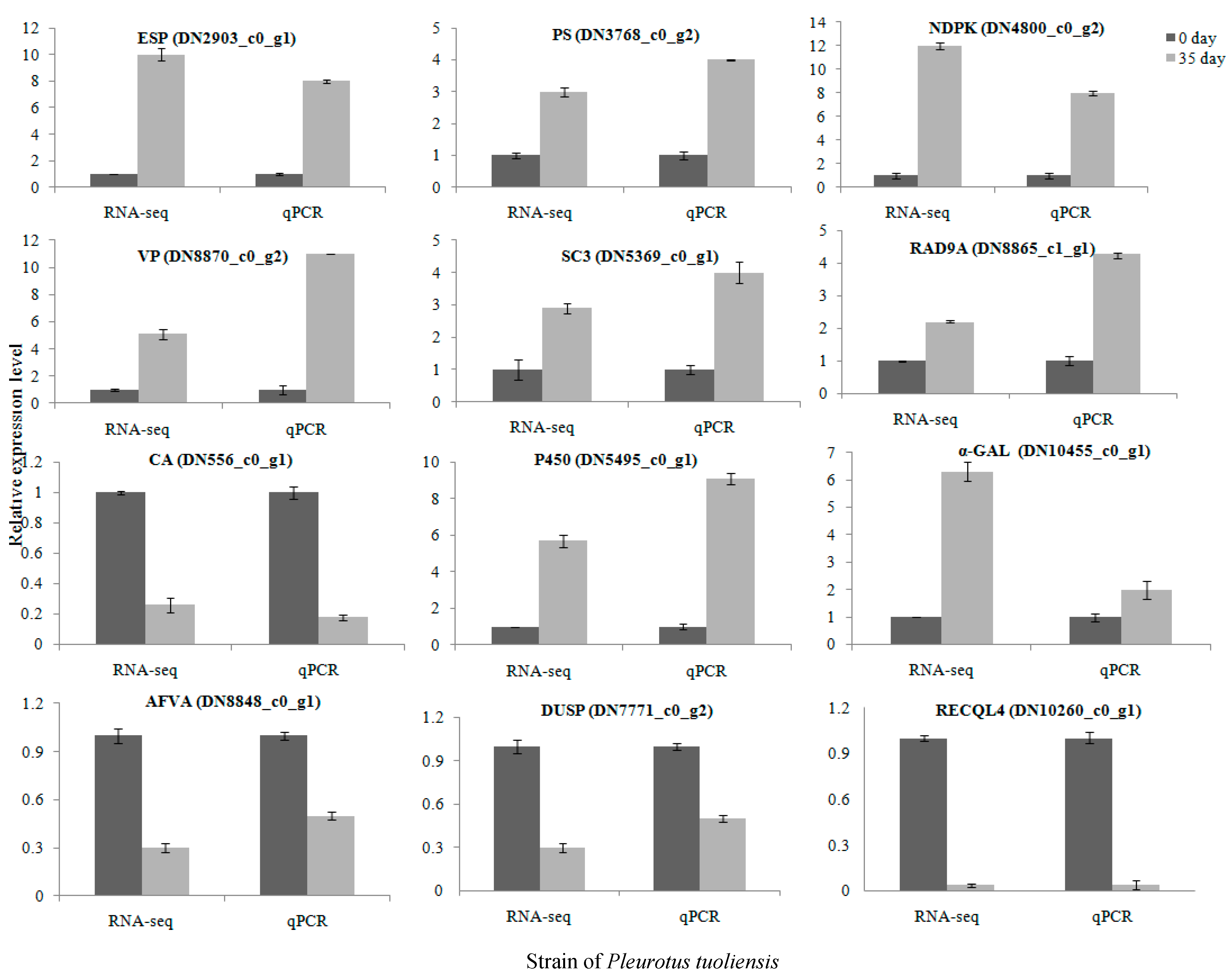
3.6. Validation of DEGs by RT-qPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, M.R.; Zhang, J.X.; Chen, Q.; Wu, X.L.; Gao, W.; Deng, W.Q.; Huang, C.Y. The famous cultivated mushroom Bailinggu is a separate species of the Pleurotus eryngii species complex. Sci. Rep. 2016, 6, 33066. [Google Scholar] [CrossRef] [PubMed]
- Kawai, G.; Babasaki, K.; Neda, H. Taxonomic position of a Chinese Pleurotus “Bai-Ling-Gu”: It belongs to Pleurotus eryngii (DC.: Fr.) Quél. and evolved independently in China. Mycoscience 2008, 49, 75. [Google Scholar] [CrossRef]
- Wang, C.L.; Cui, H.Y.; Wang, Y.R.; Wang, Z.F.; Li, Z.J.; Chen, M.H.; Li, F.J. Bidirectional immunomodulatory activities of polysaccharides purified from Pleurotus nebrodensis. Inflammation 2014, 37, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.X.; Zhao, S.; Huang, Z.X.; Yin, L.M.; Hu, J.; Li, J.H.; Liu, Y.; Rong, C.B. Development of a highly productive strain of Pleurotus tuoliensis for commercial cultivation by crossbreeding. Sci. Hortic. 2018, 234, 110–115. [Google Scholar] [CrossRef]
- Gou, Y.P.; He, Y.W.; Mao, Z.Y. Effect of after-riping time on the fruitng and yield of Pleurotus tuoliensis. Mushroom 2003, 25, 33–34. [Google Scholar]
- Fu, Y.P.; Liang, Y.; Dai, Y.T.; Yang, C.T.; Duan, M.Z.; Zhang, Z.; Hu, S.N.; Zhang, Z.W.; Li, Y. De novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar] [CrossRef]
- Du, F.; Zou, Y.J.; Hu, Q.X.; Jing, Y.G.; Yang, X.H. Metabolic profiling of Pleurotus tuoliensis during mycelium physiological maturation and exploration on a potential indicator of mycelial maturation. Front. Microbiol. 2019, 9, 3274. [Google Scholar] [CrossRef]
- Wong, M.M.; Cannon, C.H.; Wickneswari, R. Identification of lignin genes and regulatory sequences involved in secondary cell wall formation in Acacia auriculiformis and Acacia mangium via de novo transcriptome sequencing. BMC Genom. 2011, 12, 342. [Google Scholar] [CrossRef]
- Xie, C.L.; Gong, W.B.; Zhu, Z.H.; Yan, L.; Hu, Z.X.; Peng, Y.D. Comparative transcriptomics of Pleurotus eryngii reveals blue-light regulation of carbohydrate-active enzymes (CAZymes) expression at primordium differentiated into fruiting body stage. Genomics 2018, 110, 201–209. [Google Scholar] [CrossRef]
- Yin, J.; Xin, X.D.; Weng, Y.J.; Gui, Z.Z. Transcriptome-wide analysis reveals the progress of Cordyceps militaris subculture degeneration. PLoS ONE 2017, 12, e0186279. [Google Scholar] [CrossRef]
- Yu, G.J.; Wang, M.; Huang, J.; Yin, Y.L.; Chen, Y.J.; Jiang, S.; Jin, Y.X.; Lan, X.Q.; Wong, B.H.C.; Liang, Y. Deep insight into the Ganoderma lucidum by comprehensive analysis of its transcriptome. PLoS ONE 2012, 7, e44031. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Dai, Y.T.; Yang, C.T.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.W.; Li, Y. Comparative transcriptome analysis identified candidate genes related to Bailinggu mushroom formation and genetic markers for genetic analyses and breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Moran, Y.; Levin, J.Z.; Thompson, D.A.; Ido, A.; Xian, A.; Lin, F.; Raktima, R.; Qiandong, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Huang, X.Q.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.D.; White, J.; Cheung, F.; Parvizi, B. TIGR gene indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Stefan, G.T.; Juan Miguel, G.G.; Javier, T.; Williams, T.D.; Nagaraj, S.H.; María José, N.; Montserrat, R.; Manuel, T.; Joaquín, D.; Ana, C. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar]
- Yuki, M.; Masumi, I.; Shujiro, O.; Yoshizawa, A.C.; Minoru, K. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and, dispersion for RNA-seq data with DESeq. 2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Corinne, R.; Thomas, B.; Vincent, L.; Bernard, H. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233. [Google Scholar] [CrossRef]
- Tang, L.H.; Tan, Q.; Bao, D.P.; Zhang, X.H.; Jian, H.H.; Li, Y.; Yang, R.H.; Wang, Y. Comparative proteomic analysis of light-induced mycelial brown film formation in Lentinula edodes. BioMed Res. Int. 2016, 2016, 5837293. [Google Scholar] [CrossRef]
- Hetmann, A.; Kowalczyk, S. Nucleoside diphosphate kinase isoforms regulated by phytochrome A isolated from oat coleoptiles. Acta Biochim. Pol. 2009, 56, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Hasunuma, K.; Yabe, N.; Yoshida, Y.; Ogura, Y.; Hamada, T. Putative functions of nucleoside diphosphate kinase in plants and fungi. J. Bioenerg. Biomembr. 2003, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Yoshida, Y.; Hasunuma, K. Nucleoside diphosphate kinase-1 regulates hyphal development via the transcriptional regulation of catalase in Neurospora crassa. FEBS Lett. 2009, 583, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- André, I.; Potocki-Véronèse, G.; Barbe, S.; Moulis, C.; Remaud-Siméon, M. CAZyme discovery and design for sweet dreams. Curr. Opin. Chem. Biol. 2014, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Sathya, T.A.; Khan, M. Diversity of glycosyl hydrolase enzymes from metagenome and their application in food industry. J. Food Sci. 2014, 79, R2149–R2156. [Google Scholar] [CrossRef] [PubMed]
- Couturier, M.; Roussel, A.; Rosengren, A.; Leone, P.; Stalbrand, H.; Berrin, J.G. Structural and biochemical analyses of glycoside hydrolase families 5 and 26 β-(1,4)-mannanases from Podospora anserina reveal differences upon manno-oligosaccharide catalysis. J. Biol. Chem. 2013, 288, 14624–14635. [Google Scholar] [CrossRef] [PubMed]
- Sathya, T.A.; Jacob, A.M.; Khan, M. Cloning and molecular modelling of pectin degrading glycosyl hydrolase of family 28 from soil metagenomic library. Mol. Biol. Rep. 2014, 41, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Giardina, P.; Palmieri, G.; Fontanella, B.; Rivieccio, V.; Sannia, G. Manganese peroxidase isoenzymes produced by Pleurotus ostreatus grown on wood sawdust. Arch. Biochem. Biophys. 2000, 376, 171–179. [Google Scholar] [CrossRef]
- Camarero, S.; Sarkar, S.; Ruiz-Duenas, F.J.; Martinez, M.J.; Martinez, A.T. Description of a versatile peroxidase involved in the natural degradation of lignin that has both manganese peroxidase and lignin peroxidase substrate interaction sites. J. Biol. Chem. 1999, 274, 10324–10330. [Google Scholar] [CrossRef]
- Rico, A.; Rencoret, J.; Del Río, J.C.; Martínez, A.T.; Gutiérrez, A. Pretreatment with laccase and a phenolic mediator degrades lignin and enhances saccharification of Eucalyptus feedstock. Biotechnol. Biofuels 2014, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome Biol. 2000, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Joh, J.H.; Lee, S.H.; Lee, J.S.; Kim, K.H.; Jeong, S.J.; Youn, W.H.; Kim, N.K.; Son, E.S.; Cho, Y.S.; Yoo, Y.B.; et al. Isolation of genes expressed during the developmental stages of the oyster mushroom, Pleurotus ostreatus, using expressed sequence tags. FEMS Microbiol. Lett. 2007, 276, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jolr, J.H.; Lee, J.S.; Lim, J.H.; Kim, K.Y.; Yoo, Y.B.; Lee, C.S.; Kim, B.G. Isolation of genes specifically expressed in different developmental stages of Pleurotus ostreatus using macroarray analysis. Mycobiology 2009, 37, 230–237. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Annotation | Primer |
|---|---|---|
| DN2903_c0_g1 | Exopolygalacturonase (EPS) | F:GTGAGTTGGCTTGCTTCT R:CGATTCCTATCTGTCTTGGT |
| DN3768_c0_g2 | Pseudouridine synthase (PS) | F:TCGTCTACCTCCATATTGTC R: CTCTCAGCACCATACCTAG |
| DN4800_c0_g2 | Nucleoside diphosphate kinase (NDPK) | F: GCTGTTGGACGCAATATC R:CACACTTCCTAAGACTTCAC |
| DN8870_c0_g2 | Versatile peroxidase (VP) | F:GCAATGATGAAGACACAGAC R: GCAACTACGCAGCCTAAT |
| DN5369_c0_g1 | Fruiting body protein SC3 (SC3) | F: CTTCATCGCCTATCTTCACT R: CGCACCAATAACAGTAACG |
| DN5495_c0_g1 | Cytochrome P450 monooxygenase (P450) | F: TCCTTCGTATTCGTCTTCAG R: CGTCGCTACCTTACATCAA |
| DN8865_c1_g1 | Cell cycle checkpoint control protein RAD9A (RAD9A) | F: GCATCACCGTCCATCATT R: CTTCCGCAATCTCGTCTT |
| DN556_c0_g1 | Carbonic anhydrase (CA) | F:CGTCTCATCTCTGTCACTAAT R: CCGAGGTGTTCAGGAATC |
| DN10455_c0_g1 | α-galactosidase (α-GAL) | F: CAGAATACGACCGCATGG R: AAGCACCACCAACCAATG |
| DN8848_c0_g1 | NADPH dehydrogenase afvA (AFVA) | F: CCTTCCTATCGTCTTCTCTG R: CCACTTCAACTGCCTCTC |
| DN7771_c0_g2 | Dual specificity protein phosphatase (DUSP) | F: AGGAACGACTGAACTGAG R: GGTATGGTGGAGGTGAAG |
| DN10260_c0_g1 | ATP-dependent DNA helicase Q4 (RECQL4) | F: GTCTGTTGCTGCTTGTCT R: CTCTATCCATTGCTTGATGTC |
| Samples | PE01 | PE02 | PE03 | PE351 | PE352 | PE353 |
|---|---|---|---|---|---|---|
| Raw reads | 41,410,374 | 46,175,662 | 43,409,086 | 45,505,956 | 48,825,340 | 48,802,066 |
| Clean reads | 41,222,978 | 45,964,590 | 43,208,980 | 45,306,508 | 48,616,554 | 48,565,980 |
| Percentage of clean reads (%) a | 99.54 | 99.54 | 99.53 | 99.56 | 99.57 | 99.51 |
| Assembled statistics | ||||||
| Number of assembled transcripts | 60,330 | |||||
| Mean length of assembled transcripts (bp) | 1897 | |||||
| Number of assembled unigene | 17,030 | |||||
| Total assembled unigene size (bp) | 21,120,061 | |||||
| Mean length of assembled unigene (bp) | 1240 | |||||
| N50 (bp) b | 1891 | |||||
| The number of N50 transcripts | 3412 | |||||
| Annotation | ||||||
| NR | 13,611 | |||||
| GO | 8804 | |||||
| KEGG | 2942 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, F.; Ya Li mai mai ti, N.E.z.y.; Hu, Q.; Zou, Y.; Ye, D.; Zhang, H. A Comparative Transcriptome Analysis Reveals Physiological Maturation Properties of Mycelia in Pleurotus tuoliensis. Genes 2019, 10, 703. https://doi.org/10.3390/genes10090703
Du F, Ya Li mai mai ti NEzy, Hu Q, Zou Y, Ye D, Zhang H. A Comparative Transcriptome Analysis Reveals Physiological Maturation Properties of Mycelia in Pleurotus tuoliensis. Genes. 2019; 10(9):703. https://doi.org/10.3390/genes10090703
Chicago/Turabian StyleDu, Fang, Nu Er zi ya Ya Li mai mai ti, Qingxiu Hu, Yajie Zou, Dou Ye, and Haijun Zhang. 2019. "A Comparative Transcriptome Analysis Reveals Physiological Maturation Properties of Mycelia in Pleurotus tuoliensis" Genes 10, no. 9: 703. https://doi.org/10.3390/genes10090703