PredLnc-GFStack: A Global Sequence Feature Based on a Stacked Ensemble Learning Method for Predicting lncRNAs from Transcripts

 and
and
Abstract
:1. Introduction
2. Datasets and Method
2.1. Datasets
2.2. Features Extraction
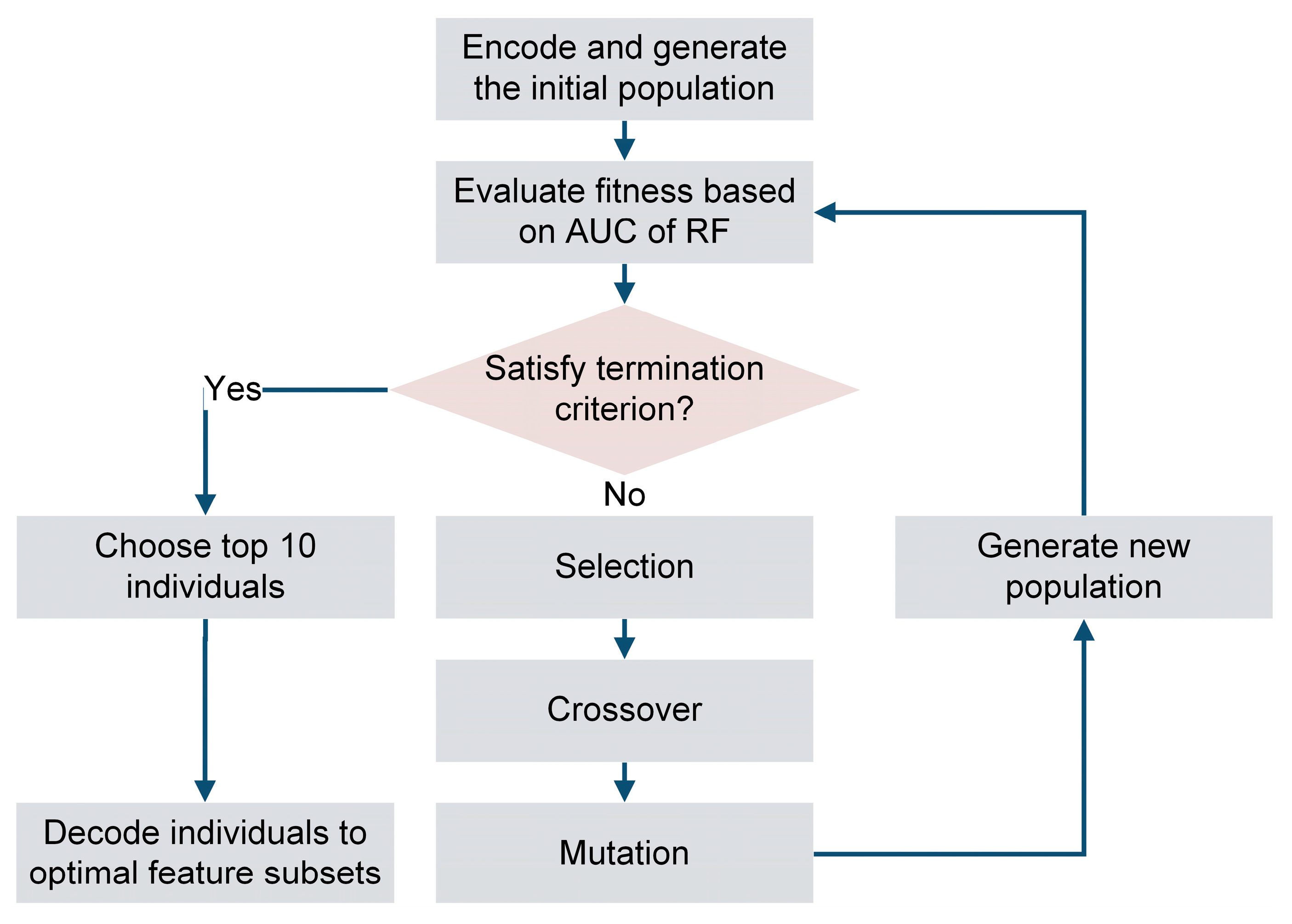
2.3. Feature Selection by Genetic Algorithm and Random Forest (GA-RF)
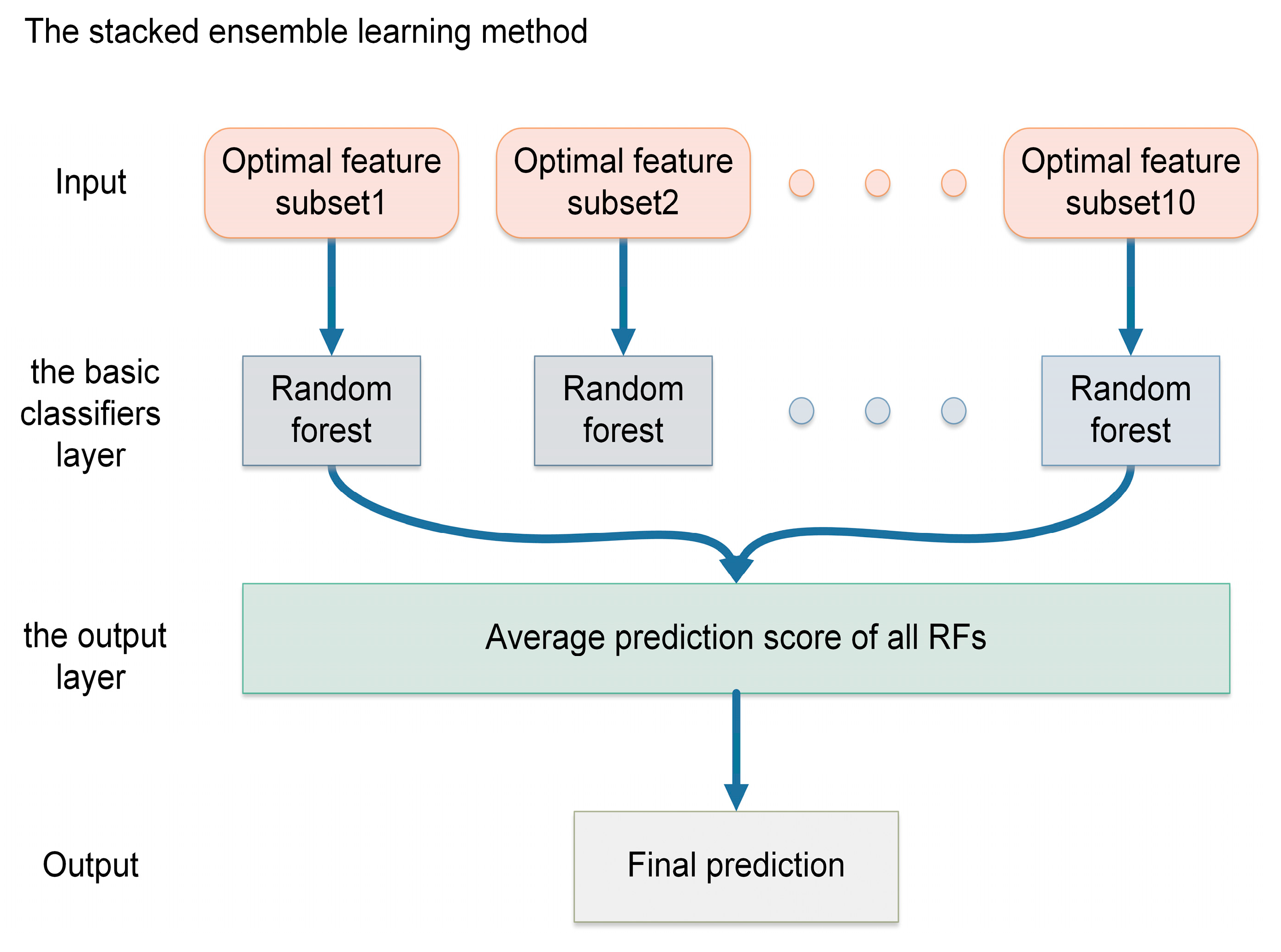
2.4. Stacked Ensemble Learning in PredLnc-GFStack
3. Results and Discussion
3.1. Performance Evaluation
| (1) | |
| (2) | |
| (3) | |
| (4) | |
| (5) |
3.2. Evaluation of the Optimal Feature Subsets
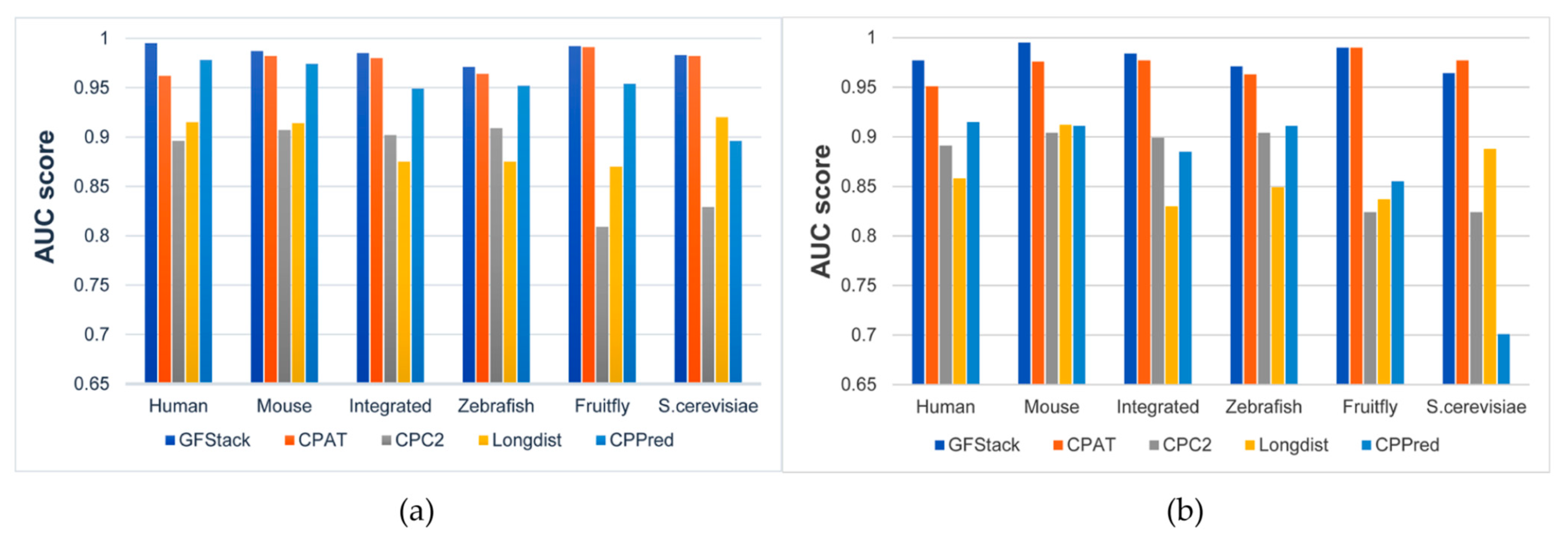
3.3. Evaluation of PredLnc-GFStack on Different Datasets
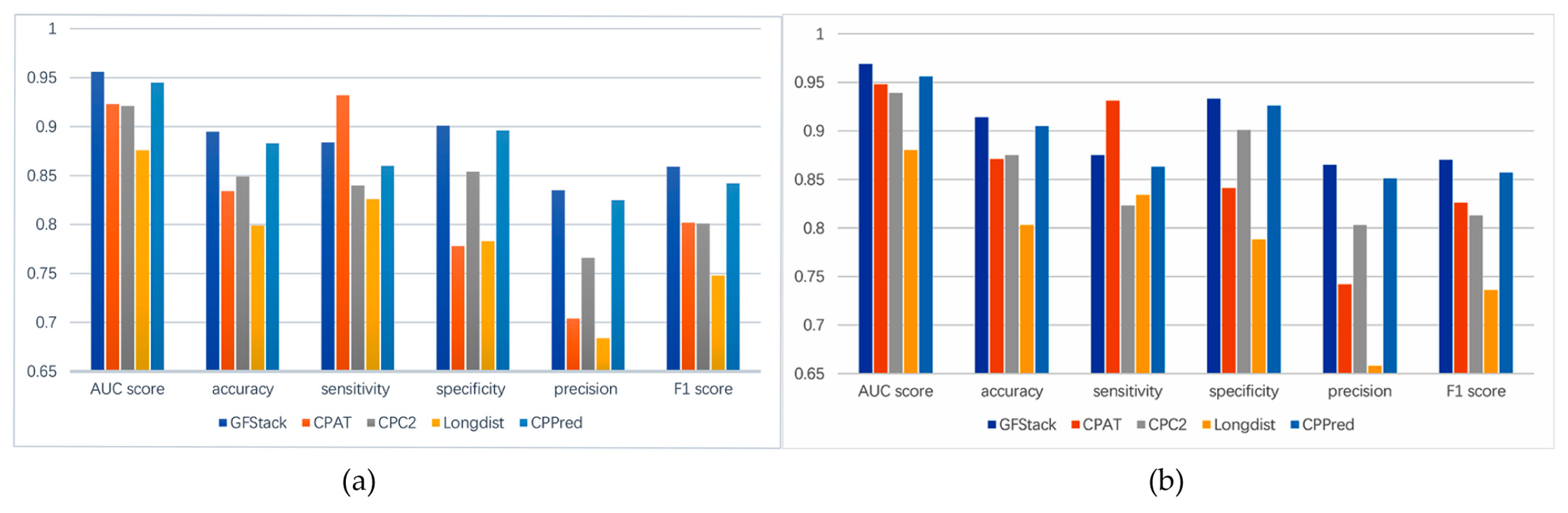
3.4. Comparison with Other Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrich, J.A.; Kugel, J.F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol. 2006, 7, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Elsner, T.; Gou, D.; Kremmer, E.; Sauer, F. Noncoding RNAs of trithorax response elements recruit Drosophila Ash1 to Ultrabithorax. Science 2006, 311, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.; Handley, P.; Wong, L.; McLachlan, D.C. BC200 RNA in normal human neocortex, non-Alzheimer dementia (NAD), and senile dementia of the Alzheimer type (AD). Neurochem. Res. 1992, 17, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Ravindranath, L.; Tran, N.; Petrovics, G.; Srivastava, S. Regulation of apoptosis by a prostate-specific and prostate cancer-associated noncoding gene, PCGEM1. Dna Cell Biol. 2006, 25, 135–141. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef]
- Li, D.; Chen, G.; Yang, J.; Fan, X.; Gong, Y.; Xu, G.; Cui, Q.; Geng, B. Transcriptome analysis reveals distinct patterns of long noncoding RNAs in heart and plasma of mice with heart failure. PLoS ONE 2013, 8, e77938. [Google Scholar] [CrossRef]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.-Y.; Yedavalli, V.S.R.K.; Jeang, K.-T. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. MBio 2013, 4, e00596-12. [Google Scholar] [CrossRef] [PubMed]
- Jathar, S.; Kumar, V.; Srivastava, J.; Tripathi, V. Technological developments in lncRNA biology. In Long Non Coding RNA Biology; Rao, M.R.S., Ed.; Springer Singapore: Singapore, 2017; pp. 283–323. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Garcia, J.T.; Hung, T.; Flynn, R.A.; Shen, Y.; Qu, K.; Payumo, A.Y.; Peres-da-Silva, A.; Broz, D.K.; Baum, R.; et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat. Genet. 2016, 48, 1370. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, C. Coding or noncoding, the converging concepts of RNAs. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Li, M.; Zhao, K.; Liu, J.; Wu, F.-X.; Pan, Y.; Wang, J. LDAP: A web server for lncRNA-disease association prediction. Bioinformatics 2016, 33, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qu, Q.; Zhang, Y.; Wang, W. The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 2018, 273, 526–534. [Google Scholar] [CrossRef]
- Zhang, W.; Yue, X.; Tang, G.; Wu, W.; Huang, F.; Zhang, X. SFPEL-LPI: Sequence-based feature projection ensemble learning for predicting LncRNA-protein interactions. PLoS Comput. Biol. 2018, 14, e1006616. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Akhtar, A.; Barlow, D.P.; Bird, A.P.; Brockdorff, N.; Duboule, D.; Ephrussi, A.; Ferguson-Smith, A.C.; Gingeras, T.R.; Haerty, W.; et al. Considerations when investigating lncRNA function in vivo. eLife 2014, 3. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Sun, L.; Liu, H.; Zhang, L.; Meng, J. lncRScan-SVM: A tool for predicting long non-coding RNAs using support vector machine. PLoS ONE 2015, 10, e0139654. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.W.; Raiol, T.; Brigido, M.M.; Walter, M.; Stadler, P.F. A support vector machine based method to distinguish long non-coding RNAs from protein coding transcripts. BMC Genom. 2017, 18, 804. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Liu, S. CPPred: Coding potential prediction based on the global description of RNA sequence. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forest. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Achawanantakun, R.; Chen, J.; Sun, Y.; Zhang, Y. LncRNA-ID: Long non-coding RNA IDentification using balanced random forests. Bioinformatics 2015, 31, 3897–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Xu, Z.; Hu, B.; Lu, Z.J. COME: A robust coding potential calculation tool for lncRNA identification and characterization based on multiple features. Nucleic Acids Res. 2017, 45, e2. [Google Scholar] [CrossRef] [PubMed]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, F.; Veltri, P.; Prosperi, M.; Tradigo, G. On the identification of long non-coding rnas from RNA-Seq. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 1103–1106. [Google Scholar]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-N.; Zhang, S.-W. LncRNA-MFDL: Identification of human long non-coding RNAs by fusing multiple features and using deep learning. Mol. Biosyst. 2015, 11, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Lee, B.; Kwon, S.; Yoon, S. LncRNAnet: Long non-coding RNA identification using deep learning. Bioinformatics 2018, 34, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, L.; Zhou, M.; Xie, H.; Zhang, C.; Wang, M.D.; Zhu, H. LncADeep: An ab initio lncRNA identification and functional annotation tool based on deep learning. Bioinformatics 2018, 34, 3825–3834. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Andrews, B. Distinguishing long non-coding RNAs from mRNAs using a two-layer structured classifier. In Proceedings of the 2017 IEEE 7th International Conference on Computational Advances in Bio and Medical Sciences (ICCABS), Orlando, FL, USA, 19–21 October 2017; pp. 1–5. [Google Scholar]
- Simopoulos, C.M.A.; Weretilnyk, E.A.; Golding, G.B. Prediction of plant lncRNA by ensemble machine learning classifiers. BMC Genom. 2018, 19, 316. [Google Scholar] [CrossRef] [PubMed]
- Pian, C.; Zhang, G.; Chen, Z.; Chen, Y.; Zhang, J.; Yang, T.; Zhang, L. LncRNApred: Classification of long non-coding RNAs and protein-coding transcripts by the ensemble algorithm with a new hybrid feature. PLoS ONE 2016, 11, e0154567. [Google Scholar] [CrossRef] [PubMed]
- Ventola, G.M.; Noviello, T.M.; D’Aniello, S.; Spagnuolo, A.; Ceccarelli, M.; Cerulo, L. Identification of long non-coding transcripts with feature selection: A comparative study. BMC Bioinform. 2017, 18, 187. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S. GENCODE: The reference human genome annotation for the ENCODE project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Curwen, V.; Eyras, E.; Andrews, T.D.; Clarke, L.; Mongin, E.; Searle, S.M.J.; Clamp, M. The ensembl automatic gene annotation system. Genome Res. 2004, 14, 942–950. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Vilela, C.; McCarthy, J.E. Regulation of fungal gene expression via short open reading frames in the mRNA 5′ untranslated region. Mol. Microbiol. 2003, 49, 859–867. [Google Scholar] [CrossRef]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.-H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef] [PubMed]
- Davis, L. Handbook of Genetic Algorithms; Van Nostrand Reinhold: New York, NY, USA, 1991. [Google Scholar]
- Blickle, T.; Thiele, L. A Mathematical analysis of tournament selection. In Proceedings of the ICGA, San Francisco, CA, USA, 1995; pp. 9–16. [Google Scholar]
- Dietterich, T.G. Ensemble learning. In The Handbook of Brain Theory and Neural Networks; MIT Press: Cambridge, MA, USA, 2002; Volume 2, pp. 110–125. [Google Scholar]
- Perez-Ortiz, M.; Gutierrez, P.A.; Hervas-Martinez, C. Projection-based ensemble learning for ordinal regression. IEEE Trans. Cybern. 2014, 44, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jing, K.; Huang, F.; Chen, Y.; Li, B.; Li, J.; Gong, J. SFLLN: A sparse feature learning ensemble method with linear neighborhood regularization for predicting drug–drug interactions. Inf. Sci. 2019, 497, 189–201. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, X.; Fu, Y.; Tsuji, J.; Weng, Z. Predicting human splicing branchpoints by combining sequence-derived features and multi-label learning methods. BMC Bioinform. 2017, 18, 464. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, D.; Zhang, W.; Tu, S.; Zhu, X.; Tian, G. Accurate prediction of transposon-derived piRNAs by integrating various sequential and physicochemical features. PLoS ONE 2016, 11, e0153268. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, L.; Zhang, W.; Liu, F.; Luo, F. A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinform. 2016, 17, 329. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef] [Green Version]
- Kearns, M. Thoughts on hypothesis boosting. Unpubl. Manuscr. 1988, 45, 105. [Google Scholar]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, J.; Zhao, M.; Li, Q. Predicting linear B-cell epitopes by using sequence-derived structural and physicochemical features. Int. J. Data Min. Bioinform. 2012, 6, 557–569. [Google Scholar] [CrossRef]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Zhang, W.; Niu, Y.; Zou, H.; Luo, L.; Liu, Q.; Wu, W. Accurate prediction of immunogenic T-cell epitopes from epitope sequences using the genetic algorithm-based ensemble learning. PLoS ONE 2015, 10, e0128194. [Google Scholar] [CrossRef] [PubMed]
- Bühlmann, P.; Yu, B. Analyzing bagging. Ann. Stat. 2002, 30, 927–961. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Sources | Name | Coding RNAs | NcRNAs |
|---|---|---|---|
| GENCODE | Human-Main | 35760 | 20299 |
| Human-Independent | 1500 | 1500 | |
| Mouse-Main | 23987 | 11746 | |
| Mouse-Independent | 1500 | 1500 | |
| preprocess CPPred | Human-Testing | 8557 | 8241 |
| Mouse-Testing | 31102 | 19930 | |
| Zebrafish-Testing | 15594 | 10662 | |
| Fruit-fly-Testing | 17400 | 4098 | |
| S.cerevisiae-Testing | 6713 | 413 | |
| Integrate-Testing | 13903 | 13903 |
| Types | Features (Dimension) |
|---|---|
| codon-related features | stop codon count (1), stop codon frequency (1), stop codon frame score (1), stop codon frequency frame score (1), nucleotide position frequencies (4), Fickett TESTCODE score (1) |
| Open reading frame (ORF)-related features | the first ORF length (1), the longest ORF length (1), the ORF coverage (2), the ORF integrity (1), ORF frame score (1), the entropy density profiles (EDP) of ORF (16) |
| GC-related features | GC (1), GC1 (1), GC2 (1), GC3 (1), GC frame score (1), UTR GC content (2) |
| coding sequence-related features | Coding sequence (CDS) length (1), CDS percentage (1), coding potential of the transcripts (CDS score) (1) |
| transcript-related features | transcript length (1), k-mer (168), CTD (20), Hexamer score (1), Signal to noise ratio (SNR) (1), untranslated region (UTR) coverage (2), EDP (20) |
| structure-related features | Molecular weight (Mw) (1), isoelectric point (pI) (1), pI/Mw (1), pI/Mw frame score (1), Gravy (1), Instability index (1) |
| Optimal Feature Subset No. | Human | Mouse | ||
|---|---|---|---|---|
| AUC | Number of Features | AUC | Number of Features | |
| 1 | 0.94979 | 134 | 0.96382 | 118 |
| 2 | 0.94946 | 137 | 0.96350 | 125 |
| 3 | 0.94940 | 131 | 0.96343 | 127 |
| 4 | 0.94934 | 136 | 0.96334 | 123 |
| 5 | 0.94929 | 138 | 0.96327 | 114 |
| 6 | 0.94929 | 134 | 0.96324 | 123 |
| 7 | 0.94923 | 129 | 0.96323 | 115 |
| 8 | 0.94916 | 127 | 0.96323 | 121 |
| 9 | 0.94913 | 137 | 0.96322 | 122 |
| 10 | 0.94910 | 128 | 0.96322 | 119 |
| Dataset | AUC | ACC | SN | SP | PRE | F1 |
|---|---|---|---|---|---|---|
| Human | 0.956 | 0.895 | 0.884 | 0.901 | 0.835 | 0.859 |
| Mouse | 0.969 | 0.914 | 0.875 | 0.933 | 0.865 | 0.870 |
| Training Dataset | Testing Dataset | AUC | ACC | SN | SP | PRE | F1 |
|---|---|---|---|---|---|---|---|
| Human-Main | Human-Testing | 0.995 | 0.968 | 0.962 | 0.974 | 0.973 | 0.967 |
| Mouse-Testing | 0.987 | 0.941 | 0.879 | 0.981 | 0.968 | 0.921 | |
| Integrated-Testing | 0.985 | 0.907 | 0.831 | 0.982 | 0.979 | 0.899 | |
| Zebrafish-Testing | 0.971 | 0.901 | 0.772 | 0.989 | 0.980 | 0.863 | |
| Fruit-fly-Testing | 0.992 | 0.940 | 0.714 | 0.993 | 0.962 | 0.819 | |
| S.cerevisiae-Testing | 0.983 | 0.960 | 0.828 | 0.969 | 0.621 | 0.710 | |
| Mouse-Main | Human-Testing | 0.977 | 0.887 | 0.807 | 0.964 | 0.955 | 0.875 |
| Mouse-Testing | 0.995 | 0.944 | 0.869 | 0.992 | 0.985 | 0.924 | |
| Integrated-Testing | 0.984 | 0.871 | 0.757 | 0.985 | 0.981 | 0.855 | |
| Zebrafish-Testing | 0.971 | 0.843 | 0.626 | 0.991 | 0.979 | 0.764 | |
| Fruit-fly-Testing | 0.990 | 0.917 | 0.593 | 0.994 | 0.957 | 0.733 | |
| S.cerevisiae-Testing | 0.964 | 0.942 | 0.382 | 0.976 | 0.500 | 0.433 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Zhao, X.; Zhang, G.; Li, W.; Liu, F.; Liu, S.; Zhang, W. PredLnc-GFStack: A Global Sequence Feature Based on a Stacked Ensemble Learning Method for Predicting lncRNAs from Transcripts. Genes 2019, 10, 672. https://doi.org/10.3390/genes10090672
Liu S, Zhao X, Zhang G, Li W, Liu F, Liu S, Zhang W. PredLnc-GFStack: A Global Sequence Feature Based on a Stacked Ensemble Learning Method for Predicting lncRNAs from Transcripts. Genes. 2019; 10(9):672. https://doi.org/10.3390/genes10090672
Chicago/Turabian StyleLiu, Shuai, Xiaohan Zhao, Guangyan Zhang, Weiyang Li, Feng Liu, Shichao Liu, and Wen Zhang. 2019. "PredLnc-GFStack: A Global Sequence Feature Based on a Stacked Ensemble Learning Method for Predicting lncRNAs from Transcripts" Genes 10, no. 9: 672. https://doi.org/10.3390/genes10090672