Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations


1
Faculty of Health and Medical Sciences, UWA Medical School, The University of Western Australia, Crawley, WA 6009, Australia
2
Department of Molecular Life Science, Division of Basic Medical Science and Molecular Medicine, Tokai University School of Medicine, Isehara 259-1193, Japan
Cells 2019, 8(5), 480; https://doi.org/10.3390/cells8050480
Submission received: 5 May 2019
/
Revised: 16 May 2019
/
Accepted: 17 May 2019
/
Published: 20 May 2019
(This article belongs to the Special Issue Major Histocompatibility Complex (MHC) in Health and Disease)
Abstract
:The HCP5 RNA gene (NCBI ID: 10866) is located centromeric of the HLA-B gene and between the MICA and MICB genes within the major histocompatibility complex (MHC) class I region. It is a human species-specific gene that codes for a long noncoding RNA (lncRNA), composed mostly of an ancient ancestral endogenous antisense 3′ long terminal repeat (LTR, and part of the internal pol antisense sequence of endogenous retrovirus (ERV) type 16 linked to a human leukocyte antigen (HLA) class I promoter and leader sequence at the 5′-end. Since its discovery in 1993, many disease association and gene expression studies have shown that HCP5 is a regulatory lncRNA involved in adaptive and innate immune responses and associated with the promotion of some autoimmune diseases and cancers. The gene sequence acts as a genomic anchor point for binding transcription factors, enhancers, and chromatin remodeling enzymes in the regulation of transcription and chromatin folding. The HCP5 antisense retroviral transcript also interacts with regulatory microRNA and immune and cellular checkpoints in cancers suggesting its potential as a drug target for novel antitumor therapeutics.
1. Introduction
The human major histocompatibility complex (MHC), also known as the human leukocyte antigen (HLA), covers 0.13% of the human genome and spans ~4 Mbp on the short arm of chromosome six at position 6p21 within a region that contains more than 250 annotated genes and pseudogenes [1,2]. The classical class I and class II regions within the MHC have extensive patterns of linkage disequilibrium (LD), and a high degree of single nucleotide polymorphisms (SNPs) at the HLA genes can differentiate worldwide populations [1,3,4,5]. HLA polymorphisms are a crucial determinant of the adaptive immune response to infectious agents, allograft success, or rejection and self/nonself immune recognition that can contribute to more autoimmune diseases than any other region of the genome [1,2,6,7,8]. Apart from the adaptive immune response, MHC class I molecules have a role in brain development, synaptic plasticity, axonal regeneration, and immune-mediated neurodegeneration [9,10,11,12]. At least half of the molecules encoded by this highly polymorphic locus are involved in antigen processing and presentation, inflammation regulation, the complement system, and the innate and adaptive immune responses, highlighting the importance of the MHC in immune-mediated autoimmune and infectious diseases [1,2]. Polymorphisms expressed by the MHC genomic region influence many critical biological traits and individuals’ susceptibility to the development of chronic autoimmune diseases such as type I diabetes, rheumatoid arthritis, celiac disease, psoriasis, ankylosing spondylitis, multiple sclerosis, Graves’ disease, schizophrenia, bipolar disorder, inflammatory bowel disease, and dermatomyositis [2,6,7,8]. Furthermore, different viral infections and cancers are associated strongly with the suppression of MHC genomic expression activity, particularly in the region of the MHC class I and class II loci [6,13,14,15].
There are tens of thousands of genomic loci that express microRNA (miRNA) [16] and lncRNA [17,18,19,20], but only about 50 have been investigated in any great detail with respect to their role in the regulation of the immune system and disease [21,22,23,24]. Although there are many miRNA and lncRNA loci within the MHC genomic region, they have been ignored largely in favor of studies on polymorphisms of the HLA class I and class II gene loci in health, disease, and transplantation cell/tissue/organ typing [25,26]. This review focuses on the structure and function of only one of these HLA lncRNA, the HCP5 lncRNA, which is located between the MICA and MICB genes and ~105 kb centromeric of the HLA-B gene.
In 1993, Vernet et al. [27] discovered a novel coding sequence belonging to a new multicopy pseudogene family P5 that they mapped within the HLA class I region and named P5-1 (alias for HCP5). They found that it expressed a 2.5-kb transcript in human B-cells, phytohemagglutinin-activated lymphocytes, a natural killer-like cell line, normal spleen, hepatocellular carcinoma, neuroblastoma, and other non-lymphoid tissue, but not in T-cells. HCP5 (P5-1) appeared to be a hybrid sequence created by nonhomologous recombination between two pseudogenes or nonmobile genetic elements that possibly produced a protein comprising 219 amino acids (aa’s) [28]. A few years later, the HCP5 (P5-1) gene was mapped precisely to a region between the MICA and MICB genes and downstream, at the centromeric end, of the two classical HLA class I genes HLA-B and HLA-C (Figure 1) [29]. In 1999, Kulski and Dawkins [30] used the computer programs Censor and RepeatMasker and dot-plot DNA and RNA sequence analyses to demonstrate that the HCP5 gene sequence and its transcripts were composed mainly of the 3′LTR and pol sequences of an ancient HERV16 insertion, which was a member of the HERVL or class III category of endogenous retroviruses (ERVs) in the human and mammalian genomes [31,32].
Because HCP5 expressed an antisense transcript that was complementary to retrovirus pol mRNA sequences and a 3′LTR, Kulski and Dawkins [30] suggested that it might have a role in immunity to retrovirus infection. They considered that the lncRNA of HCP5 might hybridize with retroviral sense mRNA sequences to suppress viral transcription, translation, and transport. Eight years later, a single-nucleotide polymorphism (rs2395029) in the HCP5 gene was associated with HLA-B*57:01 and correlated with a lower HIV-1 viral set point [33], indicating that these two alleles within a particular haplotype may have a role in viral control [34]. However, when Yoon et al. [35] tested the antisense/antiviral hypothesis for HCP5 by infecting TZM-bl cells in vitro with HIV-1 and plasmids expressing high levels of HCP5 transcripts, they observed no restriction with infectivity throughout the viral life cycle. They concluded from their findings that the HCP5 gene had no direct antiviral effect, and that the association of an HCP5 variant with viral control most likely was due to an HLA-B*57:01-related effect or other functional variants in the haplotype or both. In fact, it appears that the role of HCP5 in immunity and human disease is far more complex than previously envisioned, and that its antiviral affects might occur by way of some secondary mechanisms such as the possible involvement of miRNA inhibition rather than by hybridization of the HCP5 transcript with the complementary viral pol transcripts.
During the last two decades, HCP5 SNPs have been associated with many different diseases in genome-wide association studies (GWASs), gene expression studies, and cancer studies investigating tissue and cellular biomarkers of tumor progression and inhibition. To better understand the genetics, molecular biology, and functions of HCP5, this paper reviewed the available data and literature on the genomic organization, structure, and function of the HCP5 gene (HLA complex P5 (non-protein coding), HGNC:21659) in health and disease (MIM:604676), particularly its association with autoimmune diseases, cancer, and infections by way of its endogenous interactions with miRNA and various gene targets. Table S1 lists the online databases and repositories that were searched and interrogated to find the available data on HCP5. Table S2 lists a summary of the downloaded functional associations in all of the datasets linked with Harmonizome, which is an integrated knowledge base connecting big data with a collection of information about genes and proteins from 114 datasets provided by 66 online resources (Table S1).
2. HCP5 Genomic Organization, Gene Structure, and the HERV16 Antisense Transcript
2.1. Human MHC Class I Genomic Structure as Duplication Blocks and HCP5 Location within an HERV16 Duplicated Sequence
The HLA class I genomic region within chromosome six is composed of highly polymorphic frozen blocks [36] that were generated by a series of duplication, deletion, and various genomic rearrangement events during mammalian and primate evolution. Numerous crossover events of a tripartite gene combination (duplicon) of MIC, HERV16, and HLA class I generated the duplication blocks and the HLA class I structural organization of the HLA class I and MIC genes within the beta and alpha blocks of the MHC genomic region in an unidentified ancestor [37]. The ancient endogenous retroviral sequence HERV16 is repeated at least twelve times, along with the HLA class I coding and noncoding sequences within the alpha and beta block of the MHC class I region, and it appears, along with other retroelements, to have been a recombination site for many of the duplication events involving unequal crossovers [37,38]. The beta-block of approximately 362 kb between the POU5F1 and MCCD1 genes harbors the MICA, MICB, HLA-B, and the HLA-C genes as well as the HCP5 gene that is centromeric of a short, fragmented HLA-X pseudogene (645 bp) and telomeric of the HCG26 long noncoding RNA gene (Figure 1). The HCP5 gene is located between the MICA and MICB genes at the centromeric end of the HLA beta block, and it also has a neighboring 3538 bp HERV16 fragmented duplication (alias P5.8) at chr6:31383746-31387283 between the HLA-S (alias HLA-17) pseudogene (919 bp) and the MICA-AS1 RNA gene [30,38]. The HCP5 gene is different from all the other duplicated HERV16 sequences in the MHC in that the ancestral endogenous antisense 3′LTR and part of the internal pol antisense sequence of HERV16 (exon 2) is linked to an HLA class I promoter and leader sequence at the 5′-end (exon 1) that regulates its expression (see Section 2.5).
There are at least four HCP5 gene sequence variants in the genomic databases to consider: HCP5-1 with 2630 bp (or 2432 to 2658 bp); HCP5-2 of 23 kb; HCP5-3 of 575 bp; and HCP5-4 of 465 bp ((HGNC: 21659, Entrez Gene: 10866, Ensembl: ENSG00000206337, OMIM: 604676, UniProtKB: Q6MZN7, Vega: OTTHUMG00000031282, and GCID: GC06P031400). The original P5.1 sequence (GenBank L06175) reported by Vernet et al. [27] was 2535 bp. According to the online Ensembl database (February 2019), HCP5 has four transcript or splice variants and one gene allele that is associated with 64 phenotypes. In this review, the HCP5 gene of 2,630 bp is defined by the NCBI Gene ID: 10,866 (updated 31 January 2019) and by its location on the GRCh38.p12 assembly (annotation release 109 in March 2018) at chr6:31,463,180–31,465,809 (NC_000006.12) with a transcript length of 2547 bp, including eight adenines of the RNA polyA tail (NCBI ncRNA reference sequence NR_040662.1). It spans 2630 bp of a 5153 bp (nt positions from 717 to 3346) extended HCP5 gene reference with 5′ and 3′ noncoding regions (GenBank AB088109.1) and the chromosome six reference sequence NC_000006.12. DNA sequence alignment of NR_040662.1 and AB088109.1 or NC_000006.12 revealed that the HCP5 gene is composed of two exons: a 100 bp exon 1 and a 91 bp intron and 2355 bp exon 2. The HERV16 sequence begins at nucleotide position 150 within exon 2 of HCP5 and occupies most of exon 2 of the HCP5 RNA sequence NR_040662.1.
Instead of simply annotating the HCP5 gene at positions 31,463,180–31,465,809 on chromosome six, the Ensembl human genome assembly GRCh38.p10 that is linked to various other gene and genomic databases (OMIM, UniProtKB, HGNC, Vega, and GCID) complicated the genomic location by placing the variant (Ensembl: ENSG00000206337) at the extended genomic coordinate of 6:31,400,702 to 31,477,506 (76,805 nt). This extended genomic coordinate for the HCP5 large variant is confusing because the 5′-end is within the MICA gene sequence ~76,000 bp from the transcription start site and overlaps MICA by 60% rather than within the 5′-UTR promoter region closer to the transcription start site of the 2630 bp HCP5 variant (Figure 1, Table 1). Furthermore, these extended genomic coordinates for the large HCP5 variant can be misleading with respect to the location of SNPs within the HCP5 gene and the transcription factor regulatory elements within the 5′-UTR of the HCP5 gene. For example, the HCP5 SNP rs6938467 (T/G) that is associated significantly with Kawasaki disease in Korean children [39] is positioned at chr6:31,440,139 (GRCh38.p12) well outside the HCP5 RNA reference sequence (NR_040662.1) and upstream of the LINC01149 locus starting within the MICA gene in the genomic location of chr6:31,400,702–31,465,809 and ENSG00000206337. Therefore, this expanded version of the large HCP5 variant sequence (ENSG00000206337) with a genomic length of 65,108 to 76,000 bp has been excluded from this review except for occasionally noting whether or not the published HCP5 SNPs in disease association studies lie inside or outside the 2630 bp version of the HCP5 gene sequence.
2.2. HCP5 RNA, a Complementary Sequence of ERV16 (an ERV Class III (ERVL))
The gene structure of HCP5 is unusual because it is a hybrid sequence of an HLA class I gene fragment in exon 1 and the fragmented portions of the 3′LTR and internal sequence of ERV16 in exon 2 [30], annotated by RepeatMasker as 3′LTR16B and complementary (minus strand) ERV3-16A3 (Table 1). The entire ERV16 sequence from 5′LTR16B to 3′LTR16B with the internal ERV3-16A3 is located on the reverse strand between chr6:31,469,592 to 31,463,420 (6173 bp), and it is 2.4 times longer than the HCP5 gene sequence of 2630 bp (Figure 1). The internal ERV3-16A3 sequence contained between the flanking LTRs has about 60% similarity to HERVL, mainly within the pol gene region and with only a slight identity to parts of the gag gene. An assumed primer binding sequence (5′ -TGGCTTCAGGAGTGGTCC-3′) for leucine-tRNA is present two nucleotides downstream from the 3′ end of the 5′-LTR16B sequence, which has identity with 15 of 18 nucleotides of HERVL [30]. Thus, the HCP5 2547 bp transcript of the reference sequence NR_040662.1 terminates about a third of the way from the 3′LTR16B to the 5′LTR16B2 of the ERV16 sequence, with the two LTR16B sequences possibly acting as promoters and/or enhancers for HCP5 and various other expressed sequences within the vicinity [30]. The ERV16 sequence downstream of HCP5 is also interrupted with insertions from other retroelements including AluSp, THE1B, and AluSx (Table 1), indicating that this ERV16 sequence is at least as old as the wave of AluSp/x insertions that occurred in primates about 37 million years ago [40].
2.3. HCP5 and the Human Papillomavirus (HPV) Minor Structural Protein Interacting Protein (PMSP) Gene
A surprising finding during a BLAST search of HCP5 sequences in GenBank was that a portion of the HCP5 RNA (NR_040662.1, 1844–2011 bp) shares 99% identity with the 186 bp sequence of the papillomavirus minor structural protein interacting protein (PMSP) gene sequence (AJ437509.2) (Figure 2) that is only 7% of the length of the entire HCP5 sequence. PMSP codes for a 61 aa peptide (UniProtKB-Q8TCT4) that interacts with the capsid L2 protein of HPV1, HPV11, and HPV16 in the cytoplasm, and both proteins are transported into cellular nuclear dots for viral assembly [41]. In this regard, the 7% of HCP5 appears to be the solitary genomic locus for PMSP gene expression. Alternatively, the PMSP gene might be found at a different genomic locus that is still to be identified. The start of the PMSP gene (1 to 22 bp) has 100% identity with HCP5 (NR_040662.1) at nt positions 37 to 58, and the remainder of the PMSP gene has 98% identity with 164 bp of HCP5 (NR_040662.1) at nt positions 1847 to 2012 located in the ERV16 portion of HCP5 (Figure 2). Thus, both the PMSP gene and the HCP5 gene of the human genome share the same part of the ERV16 sequence, possibly generated as a hybrid at some time before or early in primate history. The 164 bp portion of the PMSP sequence within HCP5 is also present in macaque monkeys with 91% identity, but not in the chimpanzee or gorilla where the HCP5 sequence between the two MIC genes has been deleted in available sequences [42]. The PMSP gene also has about 88% identity for 141 bp within the multicopy Homo sapiens zinc ribbon domain containing one antisense pseudogene (ZNRD1ASP) transcript variant 1, noncoding RNA (2450 bp) at three locations within the class I region of the MHC on chr6:31,465,114–31,465,281 in MICB; 30,001,688–30,001,549 between HLA-J and ZNRD1; and 29,713,247–29,713,129 between HLA-F and the IFITM4P noncoding RNA gene. The PMSP portion of the HCP5 transcript might be upregulated during warts infections in humans; plantar warts are associated significantly with the SNP rs9267257 that is located at 6:31488485 within the LOC102725068, an intergenetic variant position between HCP5 and MICB [43]. Conversely, Karim et al. [44] showed that episomal copies of HPV16 and HPV18 in undifferentiated keratinocytes downregulated the expression of HCP5 and the HLA-A, -B, -C, and -G gene products that were involved with the antigen presenting pathway in order to allow persistence of latent infections. However, expression of the HCP5 gene that was downregulated substantially by human papillomavirus (HPV) infection was again upregulated 2.2-fold in HPV-infected keratinocytes after 24 h of treatment with polyI:C compared to greater than three-fold upregulation in HPV-uninfected cells during the same period [44]. Further studies are needed to confirm the possible association between HCP5, PMSP, and the assembly of the HPV capsids in various papillomaviral infections.
2.4. HLA Class I Leader Sequence and Cytomegalovirus (CMV) Signal Peptides
The HCP5 RNA sequence has an open reading frame (ORF) that may code for a peptide of 132 aa (Q6M2N7.1) with a domain resembling HLA class I signal peptides [28,30]. It has another ORF with the first 24 of 52 aa of the putative HCP5 peptide starting from methionine and sharing identity with at least 18 of 24 aa of the leader peptide of HLA-A, -B, and -C, whereas the remaining 28 aa have no sequence similarity to any known protein [30]. The first 23 aa, RVMESRTLLLLFSGAVALIOTWA, of the putative 52 aa HCP5 peptide sequence shares at least 65% (15/23 aa) identity and up to 78.3% (18/23 aa) identity with the leader peptides of HLA-A, -B, and -C. However, it is only the MESRTLL portion of the sequence that is found at the start of the 61 aa peptide that is encoded by the PMSP gene (Figure 2). Also, the HCP5 peptide sequence VMESRTLLL shares 74% identity with the VMAPRTLIL sequence within the signal peptides of HLA-A and the UL40 protein of the human cytomegalovirus (HCMV), which binds and stabilizes cell surface expression of both the endogenous HLA-E and the HCMV-encoded MHC-I homologue gpUL18 to regulate differentially two distinct natural killer cell evasion pathways [45,46]. HLA-E and gpUL18 bind a restricted subset of peptides derived from the leader peptides of other class I molecules, but whether they bind the putative HCP5 leader peptide or the PMSP leader peptide is not known.
2.5. HLA-Class I Gene Promoters and Enhancers within HCP5
The HCP5 genomic sequence has a putative HLA-class I gene promoter for transcriptional activation of the P5-1 mRNA (Figure 1 and Table S3) that is linked to a 159 bp sequence homologous to exon 1 (signal peptide) and part of intron 1 of classical class I genes [28,30]. Table S3 shows the Clustal W alignment of 360 bp of sequence, including the promoter regions of the HCP5 and HLA-A genes, approximately 120 bp of the start of the HCP5 RNA sequence, and 159 bp of the start of the HLA-A mRNA sequence. The 5′ nonretroviral promoter region of the HCP5 gene has at least 88% identity with the promoter regions of various class I HLA genes including HLA-A, -B, -C, -E, -F, and -G [44] (Table S3). There is also 84% to 87% identity between the HCP5 sequence and the first ~159 bp of the HLA class I transcripts.
The HCP5 gene promoter and the associated exon and intron 1 of MHC class I genes are located within a 1075 bp genomic region between the MER4C and HERV sequence (Table 1). The putative gene promoter and the peptide-coding region are linked to the 3′LTR16B fragment and are in reverse orientation to the HLA-X pseudogene (Figure 1) that is positioned between MER4C and MER21B fragment within the HCP5 genomic sequence [30]. The HLA-class I gene promoter of HCP5 has an intact TATA box, CAAT box, and regulatory response sequences for interferon, kappa B1, and kappa B2 that have been described for the regulatory complex region of class I promoters [28,30,47]. Numerous copies of a motif (AGAAA) for heat shock transcription factor are distributed in forward and reverse orientation within the HCP5 gene promoter region, and a potential cap signal sequence (5′-GACTCAGATTC-3′) for transcription initiation is located 26 nt downstream from the TATA box [30].
Although few experiments have been performed directly on the HCP5 promoter, the high sequence similarity with the HLA class I promoters suggests that it is regulated and expressed similarly to HLA-A, -B, -C, -G, and -F but with differences dependent on polymorphisms and mutations providing sequence variants [47] (Table S3) and tissue-specific effects on expression [48]. Regulatory elements and transcription factors associated with the MHC class I promoter region were reviewed recently [47,48,49,50] and highlighted that the MHC class I promoter region with the MHC class I NF-κB enhanceosome, together with the RFX-complex, ATF1/CREB, and the NFY-complex (binding the SXY-module), activated MHC class I transcription. Additionally, type II interferons (IFN-γ) can activate MHC class I transcription by upregulation of IRF1, which bind to the interferon-sensitive response elements (ISREs) of the MHC class I promoter [51,52]. Gene Expression Omnibus (GEO) expression profiles reveal that HCP5 gene expression is regulated by IFN-γ (Table 2) along with other MHC class I genes [53,54,55]. Recently, TGFbeta was shown to induce over-expression of the HCP5 gene and activity of the S-mothers against decapentaplegic homolog 3 (SMAD3) protein complex in adenocarcinomas [56]. Also, the ubiquitous transcription factor SP1, which is known to regulate MHC class I gene expression both constitutively and in a tissue-specific manner [48], was shown to upregulate HCP5 expression and induce the development of osteosarcoma [57]. Moreover, the osteosarcoma study also found that the transcription factors STAT3, E2F1, and NF-κB1 had no effect on HCP5 expression. In contrast, NF-κB binding to Enhancer A is necessary for both constitutive and induced MHC class I expression [58], and NF-κB-induced MHC class I expression is most prominent for the HLA-A locus, which contains two NF-κB binding sites in its Enhancer A region [49,59]. Figure 3 shows the comparison and strong correlation between HCP5 and HLA-B expression in the same 27 tissues that were the results of the NCBI BioProject PREJEB4337. In general, HLA-B transcription is expressed at much higher levels with a range of 1 to 590 reads per kilobase of transcript per million mapped reads (RPKM ) than HCP5 transcription with a range of 1 to 39 RPKM.
The class I transactivator (CITA) NLRC5 (alias NOD4, NOD27, and CLR16.1) induces genes involved in the MHC class I-dependent antigen processing and presentation pathway; it activates the promoters of MHC class I and related genes by co-binding within the SXY module of MHC class I to form a CITA enhanceosome and induces the expression of classical MHC class I, nonclassical class I, the beta-2 microglobulin, immunoproteasome component (PSMB9), peptide transporter (TAP1) [61,62,63], and HCP5 [64].
The HCP5 RNA also contains the 3′LTR16B2 and a substantial portion of the internal ERV16 sequence (Table 1). It should be noted however that there are ~860 ERV16 copies of LTR16B2 with the ERV16 internal regions and ~18,100 copies of the solitary LTR16B within the human genome [31]. These ERVs provide an enormous reservoir of autonomous gene regulatory modules, some of which play important roles in normal regulation of genes and gene networks and influence the transcription of host genes. At least one of these 5′LTR16B sequences is known to act as a promoter for the activation of the isoform anaplastic lymphoma kinase (ALK) of the receptor tyrosine kinase (RTK) in ~11% of skin cutaneous melanomas [65]. The regulatory elements of HERV/LTRs tend to locate near to the genes involved in immune responses in order to interact with them, indicating that these regulatory elements play an important role in controlling the immune regulatory network [66]. Therefore, the possibility exists that the 3′LTR16B2 antisense sequence within the lncRNA of HCP5 might interact with one or other LTR16B2 sense sequences distributed across the human genome to regulate the expression of LTR16B2-regulated genes outside the MHC genomic region.
2.6. HCP5-CCCTC-Binding Factor (CTCF) Connection and Chromatin Structural Regulation
There are two high-probability CCCTC-binding factor (CTCF) binding sites within the body of the HCP5 genomic sequence that demarcate the likely position of open and closed chromatin (Figure 4). The CTCF gene (ID: 10664) encodes the transcriptional repressor CTCF, also known as 11-zinc protein or CCCTC-binding factor, which is involved in many cellular processes including transcriptional regulation, insulator activity, regulation of interferon-gamma (IFNg) induction of MHC class I and class II gene expression [49,66,67], V(D)J recombination in B and T cells [68], and regulation of chromatin architecture [69]. The primary role of CTCF is in the regulation of the 3D structure of chromatin by binding together strands of DNA to form chromatin loops and anchor DNA to the nuclear lamina [70], but it also has an essential role in blocking the interaction between enhancers and promoters to prevent oncogenesis [71]. This nuclear protein is able to use different combinations of the zinc finger domains to bind different DNA target sequences and proteins, and depending upon the context of the site, the protein can interact with a histone acetyltransferase (HAT)-containing complex as a transcriptional activator or a histone deacetylase (HDAC)-containing complex as a transcriptional repressor. If the protein is bound to a transcriptional insulator element, it can block communication between enhancers and upstream promoters, thereby regulating imprinted expression.
The insulator factor CTCF controls MHC class I and II gene expression and is required for the formation of long-distance chromatin interactions [49,66,67]. A search of the ENCODE database revealed that the proximal end of the HCP5 sequence had two CTCF binding sites in addition to peak clusters of DNase I hypersensitivity, H3K4me3, and H3K27ac with Z scores >1.8 (Figure 4). In this regard, the first 100 nucleotides of HCP5 exon 1 has a DNase I hypersensitivity region of CpG island DNA (Figure 1f, Table 1) that has 100% sequence homology with CpG island DNA on chr 22 (NCBI, AJ236677.1) [72]. A number of published studies have shown that there are differential methylation sites such as CTCF within the HCP5 gene, and that hypomethylation is associated strongly with the regulation of a number of phenotypes and diseases (see Section 4, HCP5 methylomics). The two experimentally unverified CTCF binding sites in the Ensembl database were at the HCP5 core positions ENSR0000078786 and ENSR00000195544. Of the 123 cells studied for position ENSR0000078786, 2 were active, 28 inactive, 3 poised, 6 repressed, and the regulatory activity was unknown for 84 cell types. The cells in the active or poised positions for ENSR0000078786 were DND-41 (a human leukemic T-cell line with a p53 mutation) and MCF7, and those for ENSR00000787587 were A549 (an epithelial lung carcinoma), a keratinocyte, and MCF7. The presence of CTCF binding sites at the HCP5 locus (Figure 4) is noteworthy given the importance of the CTCF transcription factor in diverse genomic regulatory functions including regulating chromatin structure, gene expression, and development of various acute and chronic diseases. IFNg probably activates the CTCF binding sites at the HCP5 locus [51,52,53,54,55], but this requires experimental confirmation.
2.7. Structural Deletion of HCP5 and MICA Genes
Examination of the HCP5 locus in the Database of Genomic Variants (Table S1) revealed that this gene region is the site of copy number variants (CNVs) with multiple reported deletions (losses) and duplications (gains). The deletion of this locus is associated mostly with a null haplotype for the MICA and MICB genes and LD with HLA-B48:01—reported initially to occur in 3.7% of East Asian individuals [42,73]. Subsequently, the HLA-B48:01 allele was genotyped at a frequency of 26% or higher in some ethnic groups such as the Ami of Taiwan [74] or the Angaite Amerindian community in Paraguay [75]. In a cohort of 2026 disease-free Caucasians and African Americans, 38 deletions and four duplications of the HCP5 locus were detected [76]—29 of the deletions had the same apparent breakpoints and seven had different breakpoints. For one of the reported sample sets of 65% Caucasians and 34% African Americans, deletions were detected in 31 African Americans and only seven Caucasians [76]. In another study of 1854 samples that represented 790 Europeans and 1064 individuals from 51 different populations, HCP5 locus deletions were detected in 34 ethnically diverse individuals (e.g., Chinese, Japanese, Palestinian, Pakistani, and Yoruban) and in only three Europeans [77]. The unforeseen deletion of HCP5 might interfere with the HCP5 genotyping assay for abacavir hypersensitivity [78]. However, an alternative and simple proxy assay for HLA-B*57 and HLA-B*48:01 could be the structurally polymorphic AluMICB, which is a lineage indel within intron 1 of the MICB gene [79].
The HCP5 genomic region has been deleted in gorillas and chimpanzees because of recombination and fusion between the MICB and MICA genes [80,81,82]. On the other hand, the transcriptional activity and function of the P5.1.1/HCP5-like ortholog in the Macaque monkey is not known, although its structure has diverged substantially from that of HCP5 in the human. The human lineages studied to date carry two functional MIC genes, MICA and MICB, and a number of unprocessed pseudogenes (MICC, D, E, F, and G) [1,2]. MICA and MICB appear to have diverged from each other 33–44 million years ago [83,84], before the divergence of chimpanzee and human. Nevertheless, deletion of HCP5 and some forms of the MIC genes from humans, gorillas, and chimpanzees highlights the plasticity of this intergenic genomic region in different primate species.
In Japan, the HLA-B*48:01 haplotype was estimated to have a 3.2% frequency with 62% of these haplotypes carrying the MIC/HCP5 null configuration. Based on the current size of the Japanese population and a homozygote rate of 0.1025%, more than 80,000 individuals could be expected to have the homozygous HLA-B*48:01 haplotype [85]. Apparently, the MIC/HCP5-deficient individuals have no overt clinical symptoms of endangered health, unlike those without MHC class I or class II molecules who suffer from mild to severe immune deficiency syndromes and diseases. In an unpublished study, homozygous HLA-B*48:01 individuals were reported to have no immunological defect in T cell or B cell populations, no change in T cell or B cell receptor repertoire, no obvious change in immunological signals, and no known susceptibility to any diseases [85]. However, ethnic Thais with the HLA-B48 haplotypes might be susceptible to developing secondary dengue hemorrhagic fever [86], and deletion between the two MIC genes was associated with nasopharyngeal carcinoma in Malaysian Chinese [87] but not Southern Chinese Han [88].
3. HCP5 Single Nucleotide Variant (SNV) Associations with Disease
Many reviews are available that outline the enormous advancements made during the last twenty years to improve our understanding of the MHC locus and genetic susceptibility to autoimmune and infectious diseases because of the availability of dense genotyping platforms and hybridization chips such the custom-made Illumina Infinium SNP chip (Immunochip) [8,89]. The online Genome-Wide Association Studies (GWAS) Catalog [90] revealed that HCP5 has 131 associations in 86 studies (86 variants) with most severe consequences associated with SNP in the regulatory and transcript region from chromosomal position 31,462,917 to 31,466,072. Some of the different diseases and phenotypes that were associated with the internal and 5′ or 3′UTR single nucleotide variants (SNVs) of the HCP5 gene, using dense genotyping platforms such as Illumina’s HumanHap550 BeadChips or candidate genotyping by PCR, are listed in Table 3. However, many of these associations need to be considered with caution because they are likely to be part of extended haplotypes or in LD with other identified or unidentified genes in the MHC. In most of these association studies, the HCP5 SNV was in strong LD with SNV from other MHC gene loci. This is not surprising given the relatively large genetic diversity within the HLA class I genomic blocks observed between different HLA haplotypes [5], which has resulted in the formation of ancestral haplotypes with relatively frozen polymorphic blocks at the HLA class I and class II loci [36].
3.1. HIV and AIDS
Fellay et al. [33,34] were the first to find a significant association of the HCP5 gene variant rs2395029 (T>G) with disease nonprogression in a cohort of HIV-infected individuals. The association was particularly strong with 10% of the variation among individuals during the viral asymptomatic set point (stabilized viral load) period of infection with the minor C/G-allele being associated with a lower viral load. Their hypothesis was that the association observed for the variation in HIV-1 set point was due mainly to HLA-B*57:01, although they acknowledged the possible effect of the HCP5 variation because it was an endogenous retroviral element (ERV) with sequence homology to retroviral pol genes, and that its transcripts were known to be expressed in lymphocytes with the possible production of predicted short protein antigens with an amino acid substitution at the SNP rs2395029 [28,30]. A model in which a combined haplotypic effect of HCP5, HLA-B*57:01, and the SNV rs9264942 located in the 5′ region of the HLA-C gene, 35 kb away from transcription initiation and 156 kb telomeric of the HCP5 gene on the HIV-1 set point, was consistent with the observation that suppression of viremia can be maintained in B*57:01 patients with undetectable viral load, even if HIV-1 undergoes mutations that allow escape from cytotoxic T lymphocyte (CTL)–mediated restriction. Therefore, HCP5 seemed to be a good candidate to interact with HIV. Furthermore, Fellay et al. [33] found that the strongest association with progression to AIDS included a set of seven polymorphisms located in and near to the ring finger protein 39 (RNF39) and zinc ribbon domain-containing 1 (ZNRD1) genes in the MHC regions.
The association of the HCP5 polymorphism at rs2395029-G with HLA-B*57:01 and HIV nonprogression to AIDS was confirmed in follow-up studies [93,95]. Limou et al. [95] used their own Genomics of Resistance to Immunodeficiency Virus (GRIV) cohort and the AIDS GWAS of the Euro-CHAVI (Center for HIV/AIDS Vaccine Immunology) cohort and replicated the results of Fellay et al. [33]. As expected, they obtained the strongest association with the HCP5 rs2395029 minor SNP and the second strongest association with the C6orf48 rs9368699 SNP, which was in LD with HCP5 and with several SNPs located in the MHC class I and class III region including HLA-B, MICB, PSORS1C1 (class I region), TNXB, TNF, LTB, BAT1, BAT2, BAT3, and RDBP (class III region). Their study suggested an independent role for the ZNRD1 gene in disease progression. Of the 50 best signals found in their meta-analysis, 46 originated from the HLA locus, emphasizing the critical role of the MHC in the control of HIV-1 replication and delayed disease progression.
A GWAS of a multiethnic cohort of HIV-1 controllers and progressors [93] obtained similar results as in the previous two studies [33,95] but with the added novelty that (1) the nature of the HLA-viral peptide interaction was the major factor modulating durable control of HIV infection, (2) HCP5 rs2395029-G was a proxy not only for HLA-B*57:01 but also for many protective and risk HLA alleles (predominantly at HLA-B), and (3) with an independent effect on HLA-C gene expression that together differentially affected the response to HIV and delayed progression to AIDS [106,107]. However, HCP5 rs2395029 was not associated with viral load at set point in African populations [108], instead, the viral load was associated with the HLA-B*57:03 allele [92] suggesting a difference in individuals of African and European ancestry. The rs2395029-G polymorphism is missing from the Yoruban population of the Niger-Congo, and they have the HLA-B*57:03 allele instead of the HLA-B*57:01 of Europeans [108,109].
The Catano et al. [110] study of mainly European Americans was noteworthy because they showed in their univariate analysis that the HCP5 minor allele was associated with a slow disease course and lower viral loads, whereas in the multivariate models, after partitioning out the protective effects of HLA-B*57, the HCP5 minor allele was associated with disease acceleration and enhanced viral replication. These contradictory associations for HCP5 are generally obscured, possibly because of the very strong LD between this allele and a subset of protective HLA-B*57 alleles. Furthermore, they found that HCP5 and HLA-C alleles stratified the HLA-B*57-containing genotypes into those that associated with either disease retardation or progression, “providing one explanation for the long-standing conundrum of why some HLA-B*57-carrying individuals are long-term non-progressors, whereas others exhibit progressive disease.” Their study highlighted the strong dependence of genotype–phenotype relationships upon cohort design, phenotype selection, LD patterns, and populations studied. However, in a more recent study [109], the minor alleles of HCP5 rs2395029, HLA-C rs9264942, and ZNRD1 rs3689068 were associated strongly with lower viral load among antiretroviral-naïve individuals who had a shorter time to first viral load of less than 51 copies/mL during combination antiretroviral therapy, even after adjustment for viral load before combination antiretroviral therapy. The authors of a 2016 study [109] concluded that more studies are needed to elucidate the function and mechanisms of these SNVs in relation to HIV disease progression and disease course as well as to clarify whether these are functional SNV or whether they simply reflect a strong LD with other SNVs that are actually functional but, as yet, unidentified.
In 2010, Yoon et al. [35] infected TZM-bl cells in vitro with HIV-1 and plasmids expressing high levels of HCP5 transcripts; they showed that the HCP5 gene had no direct antiviral effect. This implied that the association of an HCP5 variant with viral control more likely was due to various other complex interactions and epistatic factors. Thus, HCP5 rs2395029 was deemed more as a useful genetic marker or proxy for HLA-B*57:01 in Europeans; and it was used as such by other researchers to show that hypersensitivity in HIV patients to abacavir treatment was associated with HLA-B*57:01 and HIV [97,110,111]. Abacavir is a potent nucleoside reverse transcriptase inhibitor, and it is an integral part of antiretroviral therapy in combination with other antivirals with good efficacy and a favorable long-term toxicity profile in the treatment of HIV. However, serious hypersensitivity to abcavir is recognized as a prohibitive and life-threatening treatment in approximately 8% of Caucasians and 3% of African Americans. Rodriguez-Novoa et al. [97] concluded from their analysis of 245 HIV patients that “the use of HCP5 rs2395029 testing could be as useful as HLA-B*57:01 typing to prevent the abacavir hypersensitivity reaction. Given that HCP5 testing is cheaper, less time-consuming and easier to perform than HLA typing, it may confidently replace the latter in clinical settings.” However, in 2012, when Melis et al. [78] genotyped both the HCP5 SNP and HLA-B*57:01 in a set of 1888 samples, they found a good correlation, but they also found that one HLA-B*57:01-positive sample tested negative for the HCP5 SNP, and that HCP5 could not be amplified in two samples as a consequence of a homozygous deletion of the HCP5 gene. They concluded that copy number variation and incomplete LD interfered with the HCP5 genotyping assay for abacavir hypersensitivity, and that ethnicities should be considered when using the HCP5 SNP as a surrogate marker for HLA-B*57:01. Discovering the absence of HCP5 SNPs in two individuals was not a surprising result because the HCP5 gene was known to be deleted along with the MICA gene in a number of Asian HLA haplotypes including the HLA-B*48 haplotype in Japanese [42,73]. Despite the occasional deletion or structural mutation, genotyping for the HCP5 rs2395029 minor allele is still a quick and practical method for assessing the possibility of abacavir hypersensitivity associated with HLA-B*57:01 in order to avoid potentially fatal consequences [112]. Also, the structurally polymorphic AluMICB, which is a lineage indel within intron 1 of the MICB gene, possibly could be used as an alternative and simple proxy assay for HLA-B*57 and HLA-B*48:01 [79]. Ultimately, genotyping for HLA-B*57 is essential because the most widely accepted hypothesis proposes that abacavir alters the repertoire of peptides that are able to bind to MHC, allowing for presentation of novel self-peptides, which in the absence of abacavir would not bind to HLA-B*57:01 [113]. These altered peptides are perceived as foreign by T cells, and a cytotoxic response is triggered that can result in the abacavir hypersensitivity reaction [114].
3.2. Herpes Zoster
Crosslin et al. [99] reported that MHC genomic regions of HCP5, especially at SNPs rs116062713 (now rs75640364) and rs114864815 (now rs77349273), were strongly associated with susceptibility to herpes zoster (shingles) caused by the varicella zoster virus. The HCP5 SNP marker rs75640364 is in the 3′UTR of HCP5 at chr6:31,465,789 and SNP rs77349273 is a 2 kb upstream variant of HCP5 located between MICA and HLA-X.
3.3. Autoimmune Disease and Drug Hypersensitivity
Apart from HIV infection, AIDS, and abacavir hypersensitivity, the HCP5 [rs2395029] SNVs also were associated with other adverse drug reactions, autoimmune diseases, and abnormal phenotypes. The same HCP5 and HLA-B genotypes were associated with psoriasis (PS) and psoriatic arthritis (PSA) [96,115] and found to be a major determinant of flucloxacillin-induced liver injury [94]. Liu et al. [96] found that the HCP5 G2V polymorphism at rs2395029 had the highest odds ratio with both psoriasis (PS) and psoriatic arthritis (PSA) for 223 Caucasian individuals, and its effect was independent (not in significant LD) of the most highly associated SNP rs10484554 that was 34.7 kb upstream of HLA-C. The MHC, in particular the HLA class I region, is the only genomic region that has been shown to be consistently associated with PS. The nine top-ranking SNPs in the Liu et al. [96] study were from the MHC, and seven were significant, even when statistically adjusted for multiple testing. The authors also noted that the same significant HCP5 SNV in psoriasis and HIV infections is not surprising since psoriasis can be triggered by infection with HIV and other viruses. Hence, it is possible that HCP5-C carriers mount a strong immune reaction to viral infection, and when psoriasis is associated with other genes such as corneodesmosin, POU5F1, MICA, and HLA-C in the MHC and genes outside the MHC in genetically susceptible individuals [116], then this reaction might lead to excessive inflammation in skin and joints.
The antimicrobial agent flucloxacillin is a common cause of drug-induced liver injury (DILI), but the genetic basis for susceptibility remains unclear. A study of 51 cases of flucloxacillin DILI, along with a replication study of another 23 cases, found that rs2395029(G) carriers were at highly increased risk (odds ratio 80, p = 8.7 × 10−33) [94]. The rare homozygote rs2395029(G;G) increases the odds of flucloxacillin -induced liver injury by 45×.
Many associations have been attributed to HCP5 SNVs outside the gene locus, either upstream in the HLA-X pseudogene or beyond the HCP5 3′UTR, positioned almost up to the locus for the HCG lncRNA. For example, SNP rs3099844 was associated with nevirapine-induced Steven–Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN) drug reactions [117], systemic lupus erythematosus (SLE), anti-Ro/SSA [118], Sjogren Syndrome, leukopenia, and lymphoma [119]. Thus, SNV was referred to incorrectly as being in the HCP5 gene when it was actually positioned in LOC102725068 (chr6:31,479,918–31,494,794 of GRCh38/hg38), which was the MICB-DT gene that coded for the 14,877 bp MICB divergent transcript. The MICB-DT lncRNA is interesting in its own right because it immediately neighbors the MICB gene, has many transcription factor binding sites, and is associated with various diseases or phenotypes such as myositis, asthma, mumps, plantar warts, blood protein measures, MICB protein levels, and lymphocyte and monocyte counts [90].
In a recent genome-wide association analysis of a total of 915 children with Kawasaki disease and 4553 controls in the Korean population, the susceptibility locus for the disease was identified by Kim et al. [39] to be NMNAT2 on chromosome 1q25.3 (rs2078087) and the human leukocyte antigen (HLA) region on chromosome 6p21.3 (HLA-C, HLA-B, MICA, and HCP5 with rs9380242, rs9378199, rs9266669, and rs6938467, respectively). The HCP5 rs6938467 (T/G) is positioned at chr6:31,440,139 (GRCh38.p12) well outside the HCP5 RNA reference sequence (NR_040662.1) and upstream of the LINC01149 locus. However, this is within the alternative genomic location of the old Hugo Gene Nomenclature Committee (HGNC definition) of HCP5 (HGNC ID:21659) using Gencode Gene ENSG00000206337.10 and the Gencode Transcript ENST00000414046.2 starting within the MICA gene in the genomic location of chr6:31,400,702–31,465,809, which provided a transcript, including UTRs, of 65,108 bp in sequence length.
3.4. Transplantation
HCP5 upstream and downstream SNVs were associated with disease relapse in 53 patients with unrelated cord blood transplantation (UCBT) when compared to HLA-matched unrelated donors [105]. The diagnosed diseases before transplantation were transfusion-dependent thalassemia (19), genetic diseases (10), anemias (8), leukemias (11), and other neoplastic diseases (5). Of the 58 SNVs that were analyzed by genotyping, seven SNVs were associated with the risk of relapse, and two of these SNVs, rs2523675 and rs2518028, were located ~2.5 kb downstream of HCP5 but still within the tailing ERV16 (ERV3-16A3 and 5′LTR16B2) sequence of ~4.6 kb. The other SNVs were in the MICD gene and the HLA-DOA gene. Hematological disease relapses after UCBT were defined as recurrence of malignancy and/or relapse of nonmalignant hematological disorders defined by conversion to partial or complete nonresponse. Although the two HCP5 SNVs resulted in 2.75 and 4.52 times greater risk of relapse for the recipients than the donors, a possible molecular mechanism for the relapse was not provided.
3.5. Cancer
A number of different SNVs located in the MICA and MICB genes and in the genomic region between them have been associated with cancer. In a recent analysis of eight GWAS datasets with 17,153 cases and 239,337 controls by Yuan et al. [92], at least six HCP5 SNVs (including rs3130907 within the HCP5 sequence) were associated significantly with lung cancer susceptibility along with the novel risk SNV rs114020893 in the lncRNA NEXN-AS1 region at 1p31.1. The authors noted that lung cancer risk-related loci (6p21 and 15q25) were enriched in lncRNAs, such as HCP5, RP11-650L12.2, XXbac-BPG27H4.8, and HCG17, and that using noncoding regions in GWAS and gene-based and pathway-based analyses should be complementary to protein coding-related approaches [92].
4. HCP5 Methylomics
The study of differentially methylated sites, differential gene expression, and epigenetic mechanisms represent a complementary method to genetic association studies for the identification of molecular and biological pathways that contribute to good health during a normal life cycle and to clinical heterogeneity of autoimmune and chronic diseases and cancer [120]. Recent studies have identified differentially methylated sites within, or neighboring, the HCP5 gene sequence associated with epigenetic regulation of various disease phenotypes (obesity, SLE) and in response to fetal development, aging, HIV infection, and vaccination (Table 4).
Hypomethylation of HCP5 was associated with autoantibody production against dsDNA, Sjogren’s syndrome-related antigen A (SSA), Smith (Sm) antigen, and ribonucleoprotein (RNP) in SLE [122,126], with gene expression and humoral immune response to influenza [124], with hypomethylated PSORS1C1-associated allopurinol-induced severe cutaneous adverse reactions in Han Chinese [127], accelerated aging in chronic HIV infection [123], endometrial receptivity [125], sexual bias in the human placental sexome [128,129], age-related monocyte and T cell gene expression [121], lung adenocarcinoma [130], and with hypomethylated POU5F1-associated ankylosing spondylitis [131]. In contrast to the more common observation of hypomethylation, HCP5 hypermethylation was associated with obesity and BMI in an epigenome-wide association study of adiposity in Ghanaian African migrants using whole blood to measure DNA methylation [64].
Age-related HCP5 DNA methylation was associated with gene expression in human monocytes and T cells [121], and the expressed genes that linked to potentially functional age-related methylation sites were enriched with antigen processing and presentation MHC class I and class II genes that were implicated in ‘parainflammation’ and the development of age-related chronic inflammatory diseases and autoimmune diseases. The total effects of age on gene expression (which increased with age) were significant (FDR < 0.05) for seven MHC genes—HLA-B, -E, -DPA1, -DPB1, TAP2, TAPBP, and HCP5—with hypomethylation within and/or near to all of those genes. On the other hand, Gross et al. [123] found an HCP5 CpG DNA methylation signature in blood cells of patients with chronic, well-controlled HIV infection that correlated with accelerated aging, and that it also was independently associated with HLA expression and corresponding HIV control. The level of methylation at HCP5 was correlated with a patient’s CD4+/CD8+ T cell ratio to provide further evidence that the observed changes were functional [123]. Chronic HIV infection, even when viral loads were kept below the level of detection, is associated with early onset of diseases linked to aging, including cardiovascular disease, kidney disease, cancer, and premature death. Highly active antiretroviral therapy (HAART) controls the burden of HIV, without curing the infection, enabling HIV-infected patients to live for many decades, provided they continue their medications. The increased methylation changes in HIV-infected patients found beyond their chronological age suggested about a five-year increase in aging compared to healthy controls [123].
Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disease of unknown etiology that can affect most organs and is characterized by the development of autoantibodies associated with specific clinical manifestations implicated in the pathogenesis of lupus nephritis and decreased survival [122,126]. The genetic risk factors suggested for SLE include alleles in IRF5, STAT4, BLK, TNFAIP3, TNIP1, FCGR2B, and other genes [132]. Genome-wide DNA methylation analysis of SLE revealed persistent hypomethylation of interferon genes and compositional changes to CD4+ T cell populations. For example, Chung et al. [122] characterized the methylation status of 467,314 CpG sites in 326 women with SLE DNA methylation profiling, performed using the Infinium HumanMethylation450 BeadChip (Illumina), and they identified and replicated significant associations between anti-dsDNA autoantibody production and the methylation status of 16 CpG sites in 11 genes. Differential methylation for these CpG sites was also associated with anti-SSA, anti-Sm, and anti-RNP autoantibody production. Overall, associated CpG sites were hypomethylated in autoantibody-positive samples compared to autoantibody-negative cases. In the discovery/replication analysis, associations with hypomethylated CpG sites were within genes (IFIT1, IFI44L, MX1, RSAD2, OAS1, and EIF2AK2) that were either induced by type 1 interferon or that regulated type 1 interferon signaling (NLRC5). Except for hypomethylation at HCP5 and the PSMB8 gene in the class III region, differential methylation of CpG sites within the MHC was not strongly associated with autoantibody production. Thus, hypomethylation of CpG sites within HCP5 and other genes from different pathways that could not be explained by DNA sequence variation were associated strongly with anti-dsDNA, anti-SSA, anti-Sm, and anti-RNP production in SLE.
In a system-wide association study between DNA methylation, gene expression, and humoral immune response to influenza [124], a cohort of 158 individuals who were 50 to 70 years old showed that HCP5 along with HLA-B and HLA-DQB2 had an important role in methylation expression, particularly when the humoral immune response to influenza was measured by a hemagglutination inhibition assay (HAI). Only two genes showed association in all three independent analyses: ADARB2, an inhibitor of adenosine deaminase activity (RNA editing), and SPEG, a kinase with a known function in myocyte development. The small number of genes, including HCP5, that overlapped across two or more methods and at multiple time points were HLA-B, HLA-DQB2, the histone deacetylase HDAC4, RWDD2B, PTPRN2 that (de)phosphorylates phosphoinositols in an insulin regulatory role, DNAH2, HCP5, FAM24B, LOC399815, and the transcription factor genes PAX7 and PAX9. Many genes (~640 genes) that were identified in one of these analyses had direct protein–protein interacting genes identified in the other analyses, revealing that the impact of methylation on humoral immunity is complex and highly dependent upon the immune outcome. Zimmermann et al. [124] reported that methylation levels of a CpG within the gene body of HLA-B (hypermethylation) were strongly associated with HAI and had an opposite trend to that of HCP5 (hypomethylation).
Human and animal studies have identified that the placenta expresses select transcripts in a sexually dimorphic manner [129]. A microarray-based study identified sex-dependent differences in the placental transcriptomic profile in males and females (sexome) with isolated cells derived from human placental villi [128]. The four cell types examined included cytotrophoblasts, synctiotrophoblasts, and arterial and venous endothelial cells. For sex-dependent differences, the males demonstrated enrichment of signaling pathways previously reported to mediate graft versus host disease and transcripts involved in immune function and inflammation such as HLA-DQB1 (syncytiotrophoblast), HLA-DQA1 (syncytiotrophoblast and cytotrophoblast), HCP5 (cytotrophoblast), NOS1 (cytotrophoblast), FSTL3, PAPPA, SPARCL, FCGR2C (trophoblast epithelium), CD34 (cytotrophoblast), HLA-F (cytotrophoblast), and BCL2 (syncytiotrophoblast). Males demonstrated a greater in utero vulnerability, and the findings of Cvitic et al. [128] suggested that these effects may partially be due to reduced maternal–fetal compatibility for males who were then required to up-regulate immune-associated transcripts in an attempt to combat an attack by the maternal immune system.
In a genome-wide methylome analysis of endometrial biopsies collected from 17 healthy fertile-aged women from prereceptive and receptive phases of a menstrual cycle, Kukushkina et al. [125] found that extracellular matrix organization and immune response were the pathways most affected by methylation changes during the transition from prereceptive to receptive phase. The overall methylome remained relatively stable during the two time points of the menstrual cycle with small-scale changes affecting 5% of the studied CpG sites (22,272 out of 437,022 CpGs, FDR < 0.05). The study confirmed that the differential methylation of KRTAP17-1, CASP8, RANBP3L, WT1, MPP7, PTPRN2, and HCP5 between the early and midsecretory phases were similar to those observed in the previous studies [133,134]. The differential methylation of PTPRN2 and HCP5 in the endometrium is an interesting connection, given that they both were differentially methylated in a system-wide association study between DNA methylation, gene expression, and humoral immune response to influenza [134]. The PTPRN2 (NCBI gene: 5799) gene product (de)phosphorylates phosphoinositols, has an insulin regulatory role, and it may be an autoantigen in insulin-dependent diabetes mellitus, but its actual function as a methylation site in the endometrium or human monocytes and T cells is not known.
In an epigenome-wide association study using whole blood measures of adiposity in 547 Ghanaian African migrants, Meeks et al. [64] found that obesity and body mass index (BMI) were related to HCP5 hypermethylation and 18 differentially methylated positions (DMPs) for BMI, 23 for waist circumference, and three for obesity. Fourteen DMP overlapped between BMI and waist circumference. The two epigenome-wide loci that were significantly hypomethylated for both general adiposity and abdominal adiposity were CPT1A (carnitine palmitoyltransferase 1A) and BCAT1 (branched chain amino acid transaminase 1), whereas NLRC5 ((NLR family CARD domain containing 5) and most other DMPs including those for six HLA genes—HCP5, HLA-B, TAP1, TAP2, PSMB8, and HLA-E—were hypermethylated. The hypermethylation of NLRC5 was highly significant, and this gene is known to regulate the expression of MHC class I genes and to limit the activation of inflammatory pathways [54,55,56]. Thus, the results of Meeks et al. [64] suggested that obesity might suppress the adaptive immune response and induce inflammation that could also result in insulin resistance.
Coit et al. [131] identified a total of 68 differentially methylated sites between ankylosing spondylitis (AS) patients and osteoarthritis controls. HCP5 and POU5F1 were both hypomethylated in HLA-B*27-positive compared to HLA-B*27-negative AS patients. They suggested that HLA-B*27 might play a role in AS in part through epigenetic linkage disequilibrium-inducing epigenetic dysregulation. The POU5F1 gene (alias OCT4) is located at the telomeric end of the MHC beta-block, ~98 kb upstream of the HLA-C gene, and its role in methylation is well described [120].
HCP5 is known to be involved in lung cancer [130], and Yuan et al. [92] described at least six HCP5 SNVs, including rs3130907, that were associated significantly with lung cancer susceptibility (Table 3). Previously, Orvis et al. [135] showed that inactivation of the BRG1 gene, also known as SMARCA4, which encodes the ATPase subunits of the SW1/SNF chromatin remodeling complex, contributed to non-small cell lung cancer aggressiveness by altering nucleosome positioning in a wide range of genes as well as by downregulating the expression of HCP5 and all of the classical and nonclassical HLA class I genes.
Presumably, hypomethylation of HCP5 leads to added interactions and connectivity with proteins and other RNA sequences, especially with miRNA regulators. Studies on hypermethylation of HCP5 are still lacking, and such studies might provide a more contrasting view of the action of methylation on the function of HCP5 in health and disease. However, the overall absence of hypermethylation data for HCP5 might be related to the DNA methylation paradox, whereby methylation of the transcribed region and the region of transcription initiation have opposite effects on gene expression [120]. Although methylation can affect gene expression in both directions depending on the genomic region, there are more negative correlations in the 5′ UTR, while positive correlations are more common in the gene body region. While this was the case for HLA-B, the reverse was observed for HCP5 in humoral immune response to influenza [124].
5. HCP5, Gene Targets, and Transcription Factors in Interaction Networks
Many hundreds of different transcription factors (TFs) are believed to target the HCP5 sequence and regulate its expression. Although only a few experiments have examined the relationship between particular transcription factors and the expression of HCP5 and its neighboring genes in the region between MICA and MICB, a variety of datasets have predicted connections between HCP5 and many known TFs. Table S4 shows five Internet databases sourced from Harmonizome and GeneCards (Table S1) that associated TFs with HCP5. For example, the dataset of JASPAR Predicted Transcription Factor Targets predicted that 57 transcription factors were associated with regulating the expression of HCP5, whereas the MotifMap Predicted Transcription Factor Targets dataset predicted only seven associations: alpha-CP1, E2A, ETS2, MAFA, NF-kB, NF-Y, and TEF-1. On the other hand, the TRANSFAC curated dataset only listed PTF1A as a TF, interacting with the HCP5 gene in low- or high-throughput transcription factor functional studies. PTF1A is the pancreas-specific transcription factor 1a, with a role in mammalian pancreatic development and in determining whether cells allocated to the pancreatic buds continue towards pancreatic organogenesis or revert back to duodenal fates. Also, HCP5 is only one of 233 target genes for the PTF1A transcription factor. In contrast, the TRANSFAC dataset predicted that 13 TFs regulated the expression of the HCP5 gene: ATF2, ELF3, ETS1, HINFP, JDP2, LEF1, LTF, MYC, NFE2L2, RUNX1, SMAD4, SMARCA2, and SPI1. Most of these predicted transcription factors also targeted many other genes as part of interaction networks, cascades, or divergent pathways.
In a study of interactions between HCP5 and transcription factors, Warner et al. [136] showed that HCP5, together with NOD2 and IL-8, was associated strongly with decreased viability of cells in a study of the inactivation of the NF-kB1 gene by knockout. In their datafile, the other MHC genes that were strongly associated with decreased viability were HLA-A, -DQA1, -DRB1, -DRB4, -DOA, and -DOB but not HLA-B, -E, -G, -F, -DRB5, -DMA, -DRA –DQA2, -DQB1, -DRB3, -DMB, -DPB1, -DPA1, MICA, MICB, TNF, LTA, LTB, C4A, and C4B in the HEK293 cell line. Thus, NF-kB1 (located on chr 4) regulates HCP5 and the gene expression of some other MHC genes as well as a wide variety of biological functions, including inappropriate activation associated with inflammatory diseases, inappropriate immune cell development, and delayed cell growth. This study [136] also demonstrated that although HCP5 was often in LD with many genes in the MHC, it could be activated or suppressed independently from most of them.
Coit et al. [131] identified a total of 68 differentially methylated sites in a study of ankylosing spondylitis (AS) patients and osteoarthritis controls; HCP5 and POU5F1 were both hypomethylated in HLA-B*27-positive compared to HLA-B*27-negative AS patients. This predicted cis relationship between the transcription factor POU5F1 and HCP5 is interesting, given that both genes are located in the beta block of the HLA class I region [1,2]. Also, Meeks et al. [64] in an epigenome-wide association study of measures of adiposity among Ghanaians showed that NLRC5 and HCP5, HLA-B, TAP1, TAP2, PSMB8, and HLA-E were all significantly hypermethylated for both general adiposity and abdominal adiposity.
The interactive website Pathwaynet [137] predicts both the presence of a functional association and the most likely interaction type among human genes or their protein products on a whole-genome scale. It is based on a large compendium of refined regulatory interactions within 77 tissues, with their curated pathways taken from primary experimental datasets such as 690 ChIP-Seq datasets, numerous mass spectrometry of metabolites, protein–protein interactions, disease samples, etc., in order to capture the interaction networks. Figure 5 shows the top 15 genes that interacted with HCP5, as predicted by Pathwaynet [137], with a high relationship confidence of between 0.9247 (BTN3A3 and IRF9) and 0.9523 (HLA-F). All of the genes were within the MHC region except for UBE2L6 (chr11q12.1), TRIM22 (chr11p15.4), IRF1 (chr5q31.1), CASP1 (chr11q22.3), and IRF9 (chr14q12). Pathwaynet did not specify the type of functional relationships between HCP5 and these 15 genes or gene products; therefore, the predicted gene interactions should be considered with considerable caution. However, it is evident that the non-HCP5 genes have roles in antigen processing and presentation, the proteasome, graft versus host disease, allograft rejection, autoimmune disease, response to type 1 interferon or interferon gamma, regulation of viral reproduction, IL-6- and IL-12-mediated and NOD-like signaling pathways, and signal transduction by the p53 class I mediator. This connection is supported to a large degree by the top 52 genes that were positively associated with HCP5 gene expression in the Comparative Toxicogenomics Database (CTD) datasets (Table 5). The HCP5 interaction and regulation is probably by way of the methylome and the competitive endogenous RNA regulatory networks, although this premise needs to be investigated further in both in vivo and in vitro experiments and association studies.
6. HCP5 Gene Expression and Gene Interactions
In 1993, Vernet et al. [27] originally reported that HCP5 expressed a 2.5 kb transcript in human B cells, phytohemagglutinin-activated lymphocytes, a natural killer-like cell line, normal spleen, hepatocellular carcinoma, neuroblastoma, and other nonlymphoid tissue but not in T cells. Since then, numerous studies of genome-wide gene expression using dense Affymetrix expression arrays were published, but the findings rarely reported directly on the expression of HCP5. To identify the expression profile of HCP5 in various scenarios, the databanks needed to be investigated and interrogated separately. In this way, it was possible to find the particular pattern of HCP5 expression especially in comparison to the other class I and class II genes. HCP5 is widely expressed at low levels, but it is primarily expressed at higher levels in cells of the immune system such as spleen, blood, and thymus (http://smd-www.stanford.edu/), consistent with potential roles in autoimmunity and cancer.
Harmonizome (Table S1) was a good starting point to find data about HCP5 expression under various experimental conditions [138]. The gene expression results of HCP5 in 53 tissues from 8555 samples (570 donors) were sourced from GTEx RNA-seq using the University of California, Santa Cruz (UCSC) online browser (Table S1). Also, interrogation of the online NCBI Gene Expression Omnibus (GEO) with the keyword “HCP5” produced 1771 results to review. Eighty-nine results were related to up and down differential expression, 6 results to the keyword ‘immunity’, 375 results to ‘cancer’, 28 to ‘HIV’, 50 to ‘virus’, 36 to ‘interferon’, 42 to ‘host defense’, and 4 to ‘MHC’. These results were browsed with a visual profile of the effects of treatments and experiments on the gene expression of HCP5 and/or other genes of investigative choice. Nine studies in GEO confirmed that IFN and IL28B upregulated HCP5 RNA in some cell types, whereas IL10 downregulated HCP5 RNA in peripheral blood mononuclear cells (PBMCs) (Table 2). There were little or no significant data for other cytokine-positive or -negative effects on HCP5.
In comparison, Table S5 shows the ~37 drugs and chemicals that induce or suppress HCP5 expression with effects on inferred diseases, and that were identified in the Comparative Toxicogenomics Database (CTD) [139]. Based on the data in Table S5, HCP5 gene expression seems to be decreased by various immunosuppressants and neurotoxins. This includes the immunosuppressive aflatoxin B1 that increased methylation of the HCP5 gene [140]. In addition, the CTD database [139] revealed that HCP5 reacted with 714 different genes in various gene expression studies. The top 52 genes that HCP5 interacted with most often included PTGS2, TNF, IL1B, PTGS1, and CASP3 (Table 5). The summary information provided by NCBI RefSeq for each of these five genes was the following: PTGS1 and PTGS2 are prostaglandin-endoperoxide synthases or cyclooxygenases, key enzymes in the biosynthesis of prostaglandin that are regulated by specific stimulatory events involved in inflammation and mitogenesis. The tumor necrosis factor (TNF) gene that is located downstream of HCP5 in the MHC class III region encodes a multifunctional, proinflammatory cytokine that is a member of the tumor necrosis factor (TNF) superfamily. This cytokine is mainly secreted by macrophages. It is involved in the regulation of a wide spectrum of biological processes including cell proliferation, differentiation, apoptosis, lipid metabolism, and coagulation. It has been implicated in a variety of diseases including autoimmune diseases, insulin resistance, and cancer. Knockout studies in mice suggested that TNF also has a neuroprotective function. IL1B is a cytokine expressed on chromosome two and produced by activated macrophages as a proprotein, which is proteolytically processed to an active form by caspase 1 (CASP1/ICE). This cytokine is an important mediator of the inflammatory response and is involved in a variety of cellular activities including cell proliferation, differentiation, and apoptosis. IL1B induces PTGS2/COX2 in the central nervous system and contributes to inflammatory pain hypersensitivity. CASP3 or caspase 3 is a protease with a central role in the execution phase of cell apoptosis. It inactivates poly(ADP-ribose) polymerase, while it cleaves and activates sterol regulatory element binding proteins as well as caspases 6, 7, and 9. Also, it is the predominant caspase involved in the cleavage of amyloid-beta 4A precursor protein, which is associated with neuronal death in Alzheimer’s disease. Therefore, it is evident that HCP5 RNA is strongly associated with the inflammatory innate immune response as well as adaptive immune responses as indicated by its coexpression with various class I genes in expression studies in the databases (e.g., GEO) and the published literature.
6.1. HCP5 Expression in HIV-Infected Cells
Given that HCP5 has been associated strongly with viral suppression in HIV-infected cells in GWAS (Table 3), it is surprising that so few papers have specifically addressed the correlation between HCP5 gene expression levels and HIV levels or response to HIV infection [35]. However, there are a few studies in the GEO database to suggest that HCP5 expression in response to HIV is induced, suppressed, or unaffected in some cell types. For example, HCP5 RNA was significantly lower in the three HIV-negative controls than in three samples of jejunal mucosal cells from HIV patients on highly active antiviral therapy [141]. Also, HCP5 transcription activity was high in three mononuclear cell samples, but it was low or absent in three T cell samples and three fibroblast samples with HIV DNA integration sites [142]. Similarly, HCP5 RNA was higher in the brains of 10 of 26 patients receiving antiretroviral therapy for HIV-associated neurocognitive disorder than in nine uninfected controls [143]. However, in some studies, there was no significant effect of HIV on HCP5 RNA levels. For example, there was no difference in the HCP5 RNA levels of 23 infected and 12 noninfected peripheral blood mononuclear cell samples [144], little difference between eight infected and eight uninfected macrophage samples [145], and little or no difference between five uninfected and 15 HIV-infected CD4+ samples and 15 CD8+ T cell samples [146]. Alternatively, HCP5 RNA was low or absent in three T cell samples infected with HIV-based vector or three samples treated with TNF-alpha, but it was relatively higher in the three untreated T cell samples [147]. Unfortunately, in the one study on RUNX1 in the regulation of HIV, no data were provided about HCP5 RNA levels. Thus, based on these limited analyses, the role of HCP5 in HIV infection and AIDS remains unclear.
6.2. HCP5 Expression in Cancer
HCP5 has been found upregulated or downregulated in a number of different cancers. The interactions between HCP5 and three transcription factors with potential antioncogenic functions are noteworthy. HCP5 was confirmed as one of the KAT8 (alias hMOF) downregulated genes by qPCR and ChIP in the hMOF siRNA knockdown HeLa cells and 20 of 28 clinically diagnosed ovarian cancer tissues [148]. KAT8 (lysine acetyltransferase 8) encodes a member of the MYST histone acetylase protein family involved with the p53 pathway and chromatin organization as well as with the suppression of epithelial to mesenchymal transition and tumor progression [149]. In contrast, Teng et al. [150] demonstrated that the HCP5 transcribed sequence interacted with an miRNA sequence and the runt-related transcriptional regulator RUNX1 in a feedback loop to regulate the malignant behavior of glioma cells of the brain. Another noteworthy interaction was between HCP5 and SATB1 (special AT-rich sequence binding protein 1), which is a nuclear matrix-associated DNA binding protein that functions as a chromatin organizer. SATB1 is highly expressed in aggressive breast cancer cells and promotes growth and metastasis by reprogramming gene expression [151]. It also enhanced HCP5 epigenetically and suppressed the oncogenic long noncoding RNA urothelial carcinoma-associated 1 (UCA1) in breast cancer cells. Recently, Zhao and Li [57] showed that transcription factor SP1 induced upregulation of HCP5, which in turn promoted the development of osteosarcoma, whereas inhibition of HCP5 expression reversed cell invasion and epithelial–mesenchymal transition. In addition, HCP5 is overexpressed in tumor tissues of patients with lung adenocarcinoma, and it is positively correlated with poor prognosis specifically in patients who are smokers with EGFR and KRAS mutations [56]. HCP5 also was overexpressed in lymph node metastasis of small cell lung cancer [130,152], glioma tissue [150], colorectal cancerous tissue [153], and cancers of the colon [154], thyroid [155], cervix [156], and breast [151,157].
Interrogation of the TCNG Cancer Network Galaxy Database (Table S1) produced 206 networks for genes regulating or regulated by the HCP5 gene as estimated from publicly available cancer gene expression data. There are ~1010 genes that were predicted to interact with HCP5 either as a child node (regulated gene) or parent node (regulating gene). In about 590 interactions, HCP5 was the parent or regulatory node, and in the remainder (420 nodes) HCP5 was the child or regulated node. For example, HLA-A, -B, -G, -H, and –J were ranked as child nodes downstream of the HCP5 parent node in four experimental arrays on breast cancer and one experiment on colon cancer. That is, HCP5 was predicted to regulate the HLA-class I genes in those experiments. On the other hand, HCP5 was predicted to be the regulated or child node with NLRC5, a member of the NOD-like receptor family that acts as a transcriptional activator of MHC class I genes [61,62,63], that was the parent or regulatory node in some gene expression experiments such as between adenocarcinoma and squamous cell carcinoma in non-small-cell lung carcinoma, breast cancer cell line profiles, non-Hodgkins lymphoma cell lines, complex genetic sarcomas, meningiomas, prostate cancer, and uveal melanoma primary tumors. Since there were far too many data to review here, further information on the activation or suppression of HCP5 gene expression in many different cancers can be obtained by interrogating the TCNG Cancer Network Galaxy Database with ‘HCP5’ as the search query and following the links including those to the expression arrays at GEO. A more detailed account of HCP5 RNA interaction in micro RNA regulatory networks in cancer is provided in the following section.
7. HCP5 lncRNA Interactions with Regulatory miRNA in Cancer
In recent years, an increasing number of lncRNA, including HCP5, were found to have potential functions in cancer [158,159,160]. The oncogenic lncRNA appear to regulate the transcription and translation of neighboring and distant genes by cis and trans-regulatory functions in a series of biological steps involving dosage compensation, genomic imprinting, and cell cycle dysregulation leading to cancer and its progression [158,159,160]. A particularly important mechanism to emerge from many of these studies is the role of lncRNAs to bind with regulatory miRNA that control antioncogenic or oncogenic pathways. This three-way binding interaction between lncRNA, miRNA, and regulatory protein coding genes, such as those coding for regulatory transcription factors, has become known as the competing endogenous RNA (ceRNA) mechanism/network [161,162]. In this regard, association studies (Table 3), expression data analysis, and knockdown experiments (Table 2, Table 4, Table 5 and Table 6, Tables S4 and S5) have shown that HCP5 can promote or suppress cancers depending on the HCP5 allelic form and the cancer type. Since 2016, at least ten different cancer types were found to occur and/or progress by way of the HCP5–miRNA–gene regulator interactions or the ceRNA mechanism (Table 6).
In an integrated analysis on the dosage effect of lncRNAs in lung adenocarcinoma, Wei et al. [130] found that the protein coding genes CTSS, FGL2, and PDCD1LG2 (alias PDL2) competed with HCP5 (ENSG00000206337) and formed a single regulatory subnet with the miRNAs miR-106b-5p and miR-17-5b. This in part confirmed a previous study that found that HCP5 was involved in the process of lung cancer by competing with PDL2, an immune checkpoint gene, and FGL2, a therapeutic target to suppress carcinogenesis [92]. This also was consistent with their finding that at least six HCP5 SNPs, including rs3130907, were associated significantly with lung cancer susceptibility along with the novel risk SNP rs114020893 in the lncRNA NEXN-AS1 region at 1p31.1.
HCP5 expression was positively correlated with the oncogenesis of a pathological grade of glioma tissues, and knockdown of HCP5 exerted tumor-suppressive effects in human glioma cells by allowing an increase in expression of the miRNA tumor suppressor miR-139 [150]. The malignant behavior of glioma cells in the brain appears to be regulated by an HCP5–miR-139–RUNX1 feedback loop, whereby RUNX1 increased the promoter activities and expression of HCP5 that binds to the tumor suppressor miRNA-139 (miR-139) and, therefore, acts as an oncogene. HCP5 absorbed the tumor suppressor miR-139 to downregulate its expression. Upregulated RUNX1 also inhibited apoptosis in glioma cell-lines U87 and U251. RUNX1 down-regulation by knockdown using miR-139 as an inhibitor had the opposite effect on apoptosis and exerted tumor-suppressive effects in human glioma cells. MiR-139 inhibited RUNX1 expression by targeting the 3′-UTR, and HCP5 knockdown suppressed RUNX1 expression by allowing miR-139 overexpression [150]. Thus, it was concluded that HCP5 promotes cell proliferation, cell migration, and invasion (motility) and inhibits apoptosis, as do many other lncRNAs participating in glioma phenotypes [165].
RUNX1 also is known to suppress HIV reactivation in T cells, resulting in a negative correlation between RUNX1 expression and viral load. The pharmacologic inhibition of RUNX1 by a small molecule inhibitor, Ro5-3335, synergized with the histone deacetylase (HDAC) inhibitor SAHA (Vorinostat), enhanced the activation of latent HIV-1 in cell lines and PBMCs from patients [166]. However, the effect of HCP5 on RUNX1 expression in HIV-infected cells is not known, although this effect might occur by way of epigenetic or transcriptomic regulation.
Liang et al. [155] found that lncRNA HCP5 promoted follicular thyroid carcinoma (FTC) progression as a competing endogenous RNA (ceRNA) sponge for miR-22-3p, mi-186-5p, and miR-216a-5p and activated alpha-2,6-sialyltransferase 2 (ST6GAL2). Functional experiments showed that HCP5 promoted ST6GAL2, which in turn mediated the proliferation, migration, invasiveness, and angiogenic ability of FTC cells. In comparison, Yu et al. [156] showed that overexpressed HCP5 promoted the development of cervical cancer by absorbing miRNA-15a to promote expression of the MET transcription regulator MACC1 (MACC1). MiRNA-15a overexpression in vitro inhibited MACC1 expression and suppressed the proliferation of cervical cancer cells. In contrast, miRNA-15a knockdown experiments or absorption by HCP5 allowed increased MACC1 expression and the proliferation of cervical cancer cells. Furthermore, HCP5 and MACC1 were overexpressed in cervical cancer tissues compared to paracancerous tissue, and the survival rate of patients with cervical cancer was negatively correlated to HCP5 expression and positively correlated to miRNA-15a. Luciferase reporter gene assay also showed that miRNA-15a bound directly to either HCP5 or MACC1. Because HPV16 and HPV18 are associated strongly with cervical cancer progression, the question arises whether the PMSP gene [38] of HCP5 is also expressed and what role it might have with the increased proliferation of cervical cancer cells.
Both MACC1 and HCP5 are expressed in gastric cancer, and their expression may be regulated by miRNAs. In a metabolic network analysis, Mo et al. [163] found that HCP5 was coexpressed with 34 metabolic-related protein-coding genes and five lncRNAs, and regulated by three miRNAs, miR-128, miR-101, and miR-103a, that were downregulated in gastric cancer. The TOPORS-AS1 lncRNA and the NADH ubiquinone oxireductase subunit B6 (NDUFB6) coding gene that were associated with HCP5 in the network analysis were downregulated in gastric cancer samples [163]. In some cancers, TGFbeta might induce HCP5 transcription via the activity of SMAD3 [56], and increased levels of HCP5 RNA might either directly or indirectly affect GSR, ASCL1, MET, GRM8, DACHI [152], ETN3A1, ETN3A3, CCDC50, HERC6, TAP1, and PSMB9 [164] as well as many other genes (Table 6).
Based on a data expression analysis, Olgun et al. [157] found that HCP5 was one of seven lncRNA that interacted with miRNA in breast cancer. HCP5 bound to miR-155 at the hub of the ceRNA network of interactions in the basal subtype of breast cancer. This basal subtype in breast cancer was characterized by a positive correlation between immune cell infiltration and aggressiveness with a key role for interferon signaling and the induction of cell proliferation by the complement cascade. Olgun et al. [157] detected C2, C3, C3AR1, C4A, and C7 complement genes in the basal ceRNA interactions, suggesting that the complement cascade pathway may be significant for progression of the basal subtype.
In an analysis of differentially expressed profiles of lncRNAs and mRNA in ceRNA networks during transformation of diffuse large B cell lymphoma (DLBCL), Tian et al. [164] identified HCP5 as a key regulator interacting with many miRNA and protein coding genes associated with transcription (KLF2), cell adhesion and proliferation (CD47), lipid metabolism (BTN3A1), and the adaptive immune response (TAP1, PSMB9). However, they concluded that the molecular function of HCP5 remained unknown.
HCP5 has been associated with various other cancers including cutaneous melanoma [167], HPV-infected head and neck squamous cell carcinoma [168], squamous cell carcinoma cells [169], HCV-induced liver cancer [170], and upper tract urothelial carcinoma [171] in which ceRNA cross-talk has yet to be tested. The ceRNA network and cross-talk mechanism also might have a role in autoimmune diseases such as idiopathic thrombocytopenia [172], viral infections, and HCP5-associated phenotypes (Table 3, Table 4 and Table 6) that, as yet, have not been examined for interactions with miRNA and with the other lncRNA regulators and protein coding genes.
8. Perspective
Since the discovery of HCP5 in 1993 [27], a large amount of data has been gathered about its expression, function, and disease associations. Much of this data, however, is buried in large datasets that require considerable effort to locate, analyze, validate, and interpret [89,90,137,138,139] (Table S1). Nevertheless, sufficient amounts of published and unpublished data that were retrieved from online public databanks for this review revealed that HCP5 had important roles in health and disease, particularly with respect to its role as a ceRNA regulator and biomarker in autoimmune diseases and cancer. GWAS indicated that the HCP5 SNV rs2395029 was a potential marker for abacavir-induced hypersensitivity, a marker for HLA-B*57:01 in populations of mainly Caucasian or Hispanic descent, as well as flucloxacillin drug liver injury, HIV control, and psoriasis and psoriatic arthritis (Table 3). Other SNVs within the HCP5 gene or within 2.5 kb of the 5′ or 3′ UTR region are useful association markers for various diseases including myositis, herpes infection, cancer, and risk of relapse after transplantation (Table 3). Methylomic studies have associated HCP5 strongly with HIV progression, SLE, ankylosing spondylitis (AS) and obesity. Various expression studies have shown that HCP5 is a useful biomarker for interferon and IL28-related inflammatory response, monocyte response to influenza A infection, and various cancers (Table 4, Table 6, and Table S5). Many of the HCP5-associated diseases such as SLE, AS, psoriasis and psoriatic arthritis, myositis, obesity, and cancer are associated also with accelerated aging, morbidity, and mortality.
The HCP5 gene within the MHC class I genomic region has evolved by exaptation from an ancient endogenous retroviral and gained a new regulatory function by sequestering the MHC promoter and enhancer region from a fragmented/deleted ancient HLA class I gene. It appears to have generated the PMSP sequence that codes for a 61 aa peptide that binds to the HPV L2 capsid protein for viral assembly [41]. The function of the PMSP peptide in different cell types other than its interaction with HPV in keratinocytes still needs to be determined and whether the PMSP gene is regulated or continuously expressed along with the HCP5 transcript. The HCP5 promoter also contains a 22 nt RUNX1 sequence (Table S3) that might contribute to the interaction between the HCP5 transcript and the Runx transcription factor in glioma and in the monocytes of HIV patients. Because the HCP5 promoter has many of the canonical TF binding sites of the HLA class I promoter (Table S3), HCP5 is often expressed concomitantly with classical and nonclassical class I genes, unless they are differentially separated from each other by epigenomic or ceRNA regulatory processes. For example, in some instances, HCP5 might be upregulated, whereas class I HLA genes are downregulated as exemplified in the study of humoral immune response to influenza [124]. While it is notable that HCP5 is often in LD with the HLA class I genes and that their expression is often coordinated in response to various stimulators or suppressors, functional interactions between the products of these genes suggest that HCP5 has an associated role in antigen processing and presentation, the proteasome, graft versus host disease, allograft rejection, autoimmune disease, response to type 1 interferon or interferon gamma, and in the regulation of viral reproduction, especially in HIV restriction [6,14,24,50,52,107]. The mechanisms by which HLA class I genes and the lncRNA genes HCP5 and ZNRD1 might interact with HIV also are worth investigating further because these lncRNAs could be exploited therapeutically using small RNA inhibitors [109,110]. Similarly, the interaction of HCP5, microRNA, and protein coding genes in cancer (Table 6) suggests that HCP5 could be targeted for knockdown or knockout in antitumor therapeutics.
It is evident from the results and reports presented in this review that HCP5 contributes to regulating viral and autoimmune diseases and cancer, and it can be upregulated or downregulated depending on its response to various exogenous or endogenous stimulators or suppressors. One shared feature of the investigated cancerous and noncancerous diseases is hypomethylation of the HCP5 gene and upregulation of its transcript, which highlights the enormous potential of this lncRNA as a diagnostic biomarker in these pathologies. The HCP5 gene sequence is a differentially methylated site associated with the epigenetic regulation of some disease phenotypes, such as obesity [64] and SLE [132], and it may also act in response to fetal development [128], aging [121], HIV [123] or influenza infection, and vaccination [124]. Presumably, hypomethylation of HCP5 leads to added interactions and connectivity with protein and other RNA sequences, especially with microRNA regulators. Although increased HCP5 levels seem to be a common event upon viral infection and in response to interferon stimulation and some cancers, the consequences of HCP5 upregulation are diverse. The mode of HCP5 action, whether acting as a promoter or suppressor, probably depends on the occurring downstream events. From the mechanistic point of view, HCP5 either acts as a differentially methylated site involved in epigenetic regulation, or it is involved in transcriptional regulation of protein coding genes by sequestering miRNA as reported for a variety of different cancers (Table 6). Although transcriptional regulation by sequestration of transcription factors or regulatory miRNA seems to be the predominant mode of action by HCP5 in cancerous diseases, it might also act as a specific or nonspecific ‘sponge’ for miRNAs in noncancerous diseases. The involvement of HCP5 lncRNA in various human diseases and cancers underscores the importance of understanding its functions in the ceRNA networks as an important step towards future drug development. Perturbations in cellular regulatory functions due to interactions between HCP5 and transcription factors probably contribute to some malignancies. For instance, recent studies of the interaction between HCP5 and Runx family proteins suggests that they may play key roles in stem cell biology, particularly in regulating apoptosis and the G0/G1 transition by way of the ceRNA networks [150]. Runx1 increases the promoter activities and expression of HCP5, and it is also known to suppress HIV reactivation in T cells perhaps by way of HCP5 mediation [166]. In many of the recent studies, the function of the HCP5 gene is described as a defense-response gene often acting in unison with the inflammatory, innate, and adaptive immune response systems. However, HCP5 also has disease progression and oncogenic effects that suggest that it may act like a double-edged sword to defend and to attack depending on other endogenous and exogenous regulators in the pathway.
The absence of the HCP5 gene from the MHC genomic region of chimpanzees and gorillas and some human haplotypes that carry the HLA-B*48:01 allele appears not to be deleterious or life-threatening [85]. Great apes and humans without the HCP5 gene seem to live in relatively good health, which suggests that the deletion, if not harmful, might even confer some selective advantage [173]. Nevertheless, it is evident from an exploration of the recent literature and accumulating public databases that HCP5 has many regulatory or associated functions at the genetic, molecular, and physiological levels. It appears to interact with numerous other genes and/or their products by way of intermediaries such as transcription factors and miRNA, LTR16B2 genomic repeats, and as an enhancer or superenhancer in the regulation of other genes and in chromatid structural changes. There is still much to learn about the ceRNA actions of HCP5, and many of the future surprises about this hybrid MHC class I endogenous retroviral gene undoubtedly will arise from knockdown, knockout, and knockin gene expression studies. The human homologous HLA-B*48:01 haplotype is a naturally occurring HCP5-MICB deletion or knockout haplotype in the human population that warrants genetic and epidemiological analysis to help elucidate the importance of these genes for most humans who still have the intact functional versions. Although the large databanks, datasets, and available publications have provided important insights into the functions of HCP5, much work still remains in order to elucidate the actual mechanisms and role of this intriguing MHC class I hybrid retroelement in immunity, health, and disease. More detailed studies are needed, particularly applying knockdown, knockout, and knockup studies, to find out more about its function as a regulator of various molecular and biological processes related to health and disease and the MHC.
9. Conclusions
HCP5 is a unique human-specific gene within the MHC class I genomic region that encodes a hybrid HLA class I endogenous retroviral lncRNA with peptide coding potential. It has functional relationships with many other genes within or outside the MHC genomic region that are involved with antigen processing and presentation, the interferon regulatory pathway, and epigenomic and ceRNA networks; however, many of these functional interactions between multiple genetic variants are still poorly understood. HCP5 gene SNVs and neighboring upstream and downstream SNVs have been associated with HIV viral load, HPV infection, autoimmune diseases, disease relapse after transplantation, and various cancers. Much still needs to be determined about the defensive and pathological functions of HCP5 and its structural and functional role in RNA editing and signaling to enable epigenetic plasticity and immune response pathways. Judging from the recent new findings about the possible oncogenic role of HCP5 as a ‘sponge’ for sequestering regulatory miRNA in cancer, new insights about its diverse mechanisms and functions in health and disease undoubtedly will continue to emerge and surprise in the near future.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/8/5/480/s1. Table S1, Databases and Repositories (docx). Table S2, At least 1899 HCP5 functional associations with biological entities found within the Harmonizome datasets (xls). Table S3, Clustal W(1.83) alignments of the transcription factor binding motifs and proximal promoter region of HCP5, HLA-A, -B, -C, -G, and -F (xls). Table S4, Internet databases with predicted HCP5-associated transcription factors (TFs) (docx). Table S5, Chemical and drug effects on HCP5 expression and inferred diseases (xls).
Funding
This research received no external funding.
Acknowledgments
I thank the staff of Cells’ editorial office, especially Daniela Wang for her assistance with the Special Issue on the MHC in Health and Disease. I also thank my assistant guest editors Hans Djikstra and Takashi Shiina for their invaluable contribution and all the authors who submitted and published their papers in this Special Issue. It was hard and relentless work, but ultimately satisfying and rewarding with an erudite outcome.
Conflicts of Interest
The author declares no conflict of interest.
References
- Shiina, T.; Inoko, H.; Kulski, J.K. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens 2004, 64, 631–649. [Google Scholar] [CrossRef]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef]
- Alter, I.; Gragert, L.; Fingerson, S.; Maiers, M.; Louzoun, Y. HLA class I haplotype diversity is consistent with selection for frequent existing haplotypes. PLoS Comput. Biol. 2017, 13, e1005693. [Google Scholar] [CrossRef]
- Meyer, D.; Single, R.M.; Mack, S.J.; Erlich, H.A.; Thomson, G. Signatures of Demographic History and Natural Selection in the Human Major Histocompatibility Complex Loci. Genetics 2006, 173, 2121–2142. [Google Scholar] [CrossRef] [Green Version]
- Shiina, T.; Ota, M.; Shimizu, S.; Katsuyama, Y.; Hashimoto, N.; Takasu, M.; Anzai, T.; Kulski, J.K.; Kikkawa, E.; Naruse, T.; et al. Rapid Evolution of Major Histocompatibility Complex Class I Genes in Primates Generates New Disease Alleles in Humans via Hitchhiking Diversity. Genetics 2006, 173, 1555–1570. [Google Scholar] [CrossRef] [Green Version]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosaad, Y.M. Clinical Role of Human Leukocyte Antigen in Health and Disease. Scand. J. Immunol. 2015, 82, 283–306. [Google Scholar] [CrossRef]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Cabarrocas, J.; Bauer, J.; Piaggio, E.; Liblau, R.; Lassmann, H. Effective and selective immune surveillance of the brain by MHC class I-restricted cytotoxic T lymphocytes. Eur. J. Immunol. 2003, 33, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Lazarczyk, M.J.; Kemmler, J.E.; Eyford, B.A.; Short, J.A.; Varghese, M.; Sowa, A.; Dickstein, D.R.; Yuk, F.J.; Puri, R.; Biron, K.E.; et al. Major Histocompatibility Complex class I proteins are critical for maintaining neuronal structural complexity in the aging brain. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Nelson, P.A.; Sage, J.R.; Wood, S.C.; Davenport, C.M.; Anagnostaras, S.G.; Boulanger, L.M. MHC class I immune proteins are critical for hippocampus-dependent memory and gate NMDAR-dependent hippocampal long-term depression. Learn. Mem. 2013, 20, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, M.; Inoko, H.; Kulski, J.K.; Yoshimura, S. Major histocompatibility complex (Mhc) class Ib gene duplications, organization and expression patterns in mouse strain C57BL/6. BMC Genom. 2008, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Prugnolle, F.; Manica, A.; Charpentier, M.; Guégan, J.F.; Guernier, V.; Balloux, F. Pathogen-Driven Selection and Worldwide HLA Class I Diversity. Curr. Biol. 2005, 15, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Aptsiauri, N.; Ruiz-Cabello, F. The Escape of Cancer from T Cell-Mediated Immune Surveillance: HLA Class I Loss and Tumor Tissue Architecture. Vaccines 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.-A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elem. 2011, 1, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Kraus, W.L. From Discovery to Function: The Expanding Roles of Long NonCoding RNAs in Physiology and Disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Mattick, J.S. The State of Long Non-Coding RNA Biology. Non-Coding RNA 2018, 4, 17. [Google Scholar] [CrossRef]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355, eaah7111. [Google Scholar] [CrossRef]
- Sigdel, K.R.; Cheng, A.; Wang, Y.; Duan, L.; Zhang, Y. The Emerging Functions of Long Noncoding RNA in Immune Cells: Autoimmune Diseases. J. Immunol. Res. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Geng, H.; Tan, X.-D. Functional diversity of long non-coding RNAs in immune regulation. Genes Dis. 2016, 3, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Spurlock, C.F.; Crooke, P.S.; Aune, T.M. Biogenesis and Transcriptional Regulation of Long Noncoding RNAs in the Human Immune System. J. Immunol. 2016, 197, 4509–4517. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Satpathy, A.T.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.M.; Chitnis, N.; Shieh, M.; Kamoun, M.; Johnson, F.B.; Monos, D. Novel and Haplotype Specific MicroRNAs Encoded by the Major Histocompatibility Complex. Sci. Rep. 2018, 8, 3832. [Google Scholar] [CrossRef] [PubMed]
- Gensterblum-Miller, E.; Wu, W.; Sawalha, A.H. Novel Transcriptional Activity and Extensive Allelic Imbalance in the Human MHC Region. J. Immunol. 2018, 200, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Vernet, C.; Ribouchon, M.-T.; Chimini, G.; Jouanolle, A.-M.; Sidibe, I.; Pontarotti, P. A novel coding sequence belonging to a new multicopy gene family mapping within the human MHC class I region. Immunogenetics 1993, 38, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Avoustin, P.; Ribouchon, M.T.; Vernet, C.; N’Guyen, B.; Crouau-Roy, B.; Pontarotti, P. Non-homologous recombination within the Major Histocompatibility Complex creates a transcribed hybrid sequence. Mamm. Genome 1994, 5, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Tamiya, G.; Oka, A.; Yamagata, T.; Yamagata, N.; Kikkawa, E.; Goto, K.; Mizuki, N.; Watanabe, K.; Fukuzumi, Y.; et al. Nucleotide Sequencing Analysis of the 146-Kilobase Segment around the IkBL and MICA Genes at the Centromeric End of the HLA Class I Region. Genomics 1998, 47, 372–382. [Google Scholar] [CrossRef]
- Kulski, J.K.; Dawkins, R.L. The P5 multicopy gene family in the MHC is related in sequence to human endogenous retroviruses HERV-L and HERV-16. Immunogenetics 1999, 49, 404–412. [Google Scholar] [CrossRef]
- Babaian, A.; Mager, D.L. Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLos Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef]
- Fellay, J.; Shianna, K.V.; Ge, D.; Colombo, S.; Ledergerber, B.; Weale, M.; Zhang, K.; Gumbs, C.; Castagna, A.; Cossarizza, A.; et al. A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. Science 2007, 317, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Fellay, J.; Ge, D.; Shianna, K.V.; Colombo, S.; Ledergerber, B.; Cirulli, E.T.; Urban, T.J.; Zhang, K.; Gumbs, C.E.; Smith, J.P.; et al. Common Genetic Variation and the Control of HIV-1 in Humans. PLoS Genet. 2009, 5, e1000791. [Google Scholar] [CrossRef]
- Yoon, W.; Ma, B.-J.; Fellay, J.; Huang, W.; Xia, S.-M.; Zhang, R.; Shianna, K.V.; Liao, H.-X.; Haynes, B.F.; Goldstein, D.B. A polymorphism in the HCP5 gene associated with HLA-B*5701 does not restrict HIV-1 in vitro. AIDS 2010, 24, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Dawkins, R.; Leelayuwat, C.; Gaudieri, S.; Tay, G.; Hui, J.; Cattley, S.; Martinez, P.; Kulski, J. Genomics of the major histocompatibility complex: Haplotypes, duplication, retroviruses and disease. Immunol. Rev. 1999, 167, 275–304. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K.; Gaudieri, S.; Martin, A.; Dawkins, R.L. Coevolution of PERB11 (MIC) and HLA Class I Genes with HERV-16 and Retroelements by Extended Genomic Duplication. J. Mol. Evol. 1999, 49, 84–97. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Inoko, H.; Dawkins, R.L. Comparison Between Two Human Endogenous Retrovirus (HERV)-Rich Regions Within the Major Histocompatibility Complex. J. Mol. Evol. 1999, 48, 675–683. [Google Scholar] [CrossRef]
- Kim, J.-J.; Yun, S.W.; Yu, J.J.; Yoon, K.L.; Lee, K.-Y.; Kil, H.-R.; Kim, G.B.; Han, M.-K.; Song, M.S.; Lee, H.D.; et al. A genome-wide association analysis identifies NMNAT2 and HCP5 as susceptibility loci for Kawasaki disease. J. Hum. Genet. 2017, 62, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.; Jurka, J. The age of Alu subfamilies. J. Mol. Evol. 1996, 42, 59–65. [Google Scholar] [CrossRef]
- Görnemann, J.; Hofmann, T.G.; Will, H.; Müller, M. Interaction of Human Papillomavirus Type 16 L2 with Cellular Proteins: Identification of Novel Nuclear Body-Associated Proteins. Virology 2002, 303, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Komatsu-Wakui, M.; Tokunaga, K.; Ishikawa, Y.; Kashiwase, K.; Moriyama, S.; Tsuchiya, N.; Ando, H.; Shiina, T.; Geraghty, D.E.; Inoko, H.; et al. MIC-A polymorphism in Japanese and a MIC-A-MIC-B null haplotype. Immunogenetics 1999, 49, 620–628. [Google Scholar] [CrossRef]
- Tian, C.; Hinds, D.A.; Hromatka, B.S.; Kiefer, A.K.; Eriksson, N.; Tung, J.Y. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. BioRxiv 2016, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J.M.; van der Burg, S.H.; Boer, J.M. Human Papillomavirus Deregulates the Response of a Cellular Network Comprising of Chemotactic and Proinflammatory Genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef] [PubMed]
- Prod’homme, V.; Tomasec, P.; Cunningham, C.; Lemberg, M.K.; Stanton, R.J.; McSharry, B.P.; Wang, E.C.Y.; Cuff, S.; Martoglio, B.; Davison, A.J.; et al. Human Cytomegalovirus UL40 Signal Peptide Regulates Cell Surface Expression of the NK Cell Ligands HLA-E and gpUL18. J. Immunol. 2012, 188, 2794–2804. [Google Scholar] [CrossRef] [PubMed]
- Halenius, A.; Gerke, C.; Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: So many targets—But how many arrows in the quiver? Cell. Mol. Immunol. 2015, 12, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Ramsuran, V.; Hernández-Sanchez, P.G.; O’hUigin, C.; Sharma, G.; Spence, N.; Augusto, D.G.; Gao, X.; García-Sepúlveda, C.A.; Kaur, G.; Mehra, N.K.; et al. Sequence and Phylogenetic Analysis of the Untranslated Promoter Regions for HLA Class I Genes. J. Immunol. 2017, 198, 2320–2329. [Google Scholar] [CrossRef]
- Barbash, Z.S.; Weissman, J.D.; Campbell, J.A.; Mu, J.; Singer, D.S. Major Histocompatibility Complex Class I Core Promoter Elements Are Not Essential for Transcription in vivo. Mol. Cell. Biol. 2013, 33, 4395–4407. [Google Scholar] [CrossRef] [Green Version]
- van den Elsen, P.J. Expression Regulation of Major Histocompatibility Complex Class I and Class II Encoding Genes. Front. Immunol. 2011, 2, 48. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2017. [Google Scholar] [CrossRef]
- Gobin, S.J.P.; Peijnenburg, A.; Keijsers, V.; van den Elsen, P.J. Site alpha Is Crucial for Two Routes of IFNgamma-Induced MHC Class I Transactivation: The ISRE-Mediated Route and a Novel Pathway Involving CIITA. Immunity 1997, 6, 601–61111. [Google Scholar] [CrossRef]
- Zhou, F. Molecular Mechanisms of IFN-γ to Up-Regulate MHC Class I Antigen Processing and Presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Indraccolo, S.; Pfeffer, U.; Minuzzo, S.; Esposito, G.; Roni, V.; Mandruzzato, S.; Ferrari, N.; Anfosso, L.; Dell’Eva, R.; Noonan, D.M.; et al. Identification of genes selectively regulated by IFNs in endothelial cells. J. Immunol. 2007, 178, 1122–1135. [Google Scholar] [CrossRef]
- Johnson-Huang, L.M.; Suárez-Fariñas, M.; Pierson, K.C.; Fuentes-Duculan, J.; Cueto, I.; Lentini, T.; Sullivan-Whalen, M.; Gilleaudeau, P.; Krueger, J.G.; Haider, A.S.; et al. A Single Intradermal Injection of IFN-γ Induces an Inflammatory State in Both Non-Lesional Psoriatic and Healthy Skin. J. Invest. Dermatol. 2012, 132, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Rock, R.B.; Hu, S.; Deshpande, A.; Munir, S.; May, B.J.; Baker, C.A.; Peterson, P.K.; Kapur, V. Transcriptional response of human microglial cells to interferon-γ. Genes Immun. 2005, 6, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Wang, R.; Fang, L.; Ge, X.; Chen, L.; Zhou, M.; Zhou, Y.; Xiong, W.; Hu, Y.; Tang, X.; et al. HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics 2019, 9, 2460–2474. [Google Scholar] [CrossRef]
- Zhao, W.; Li, L. SP1-induced upregulation of long non-coding RNA HCP5 promotes the development of osteosarcoma. Pathol. Res. Pract. 2019, 215, 439–445. [Google Scholar] [CrossRef]
- Tilburgs, T.; Meissner, T.B.; Ferreira, L.M.R.; Mulder, A.; Musunuru, K.; Ye, J.; Strominger, J.L. NLRP2 is a suppressor of NF-ƙB signaling and HLA-C expression in human trophoblasts. Biol. Reprod. 2017, 96, 831–842. [Google Scholar] [CrossRef]
- Gobin, S.J.P.; Keijsers, V.; van Zutphen, M.; van den Elsen, P.J. The Role of Enhancer A in the Locus-Specific Transactivation of Classical and Nonclassical HLA Class I Genes by Nuclear Factor κB. J. Immunol. 1998, 161, 2276–2283. [Google Scholar]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Downs, I.; Vijayan, S.; Sidiq, T.; Kobayashi, K.S. CITA/NLRC5: A critical transcriptional regulator of MHC class I gene expression: CITA/NLRC5. BioFactors 2016, 42, 349–357. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef]
- Vijayan, S.; Sidiq, T.; Yousuf, S.; van den Elsen, P.J.; Kobayashi, K.S. Class I transactivator, NLRC5: A central player in the MHC class I pathway and cancer immune surveillance. Immunogenetics 2019, 71, 273–282. [Google Scholar] [CrossRef]
- Meeks, K.A.C.; Henneman, P.; Venema, A.; Burr, T.; Galbete, C.; Danquah, I.; Schulze, M.B.; Mockenhaupt, F.P.; Owusu-Dabo, E.; Rotimi, C.N.; et al. An epigenome-wide association study in whole blood of measures of adiposity among Ghanaians: The RODAM study. Clin. Epigenetics 2017, 9, 103. [Google Scholar] [CrossRef]
- Wiesner, T.; Lee, W.; Obenauf, A.C.; Ran, L.; Murali, R.; Zhang, Q.F.; Wong, E.W.P.; Hu, W.; Scott, S.N.; Shah, R.H.; et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015, 526, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Majumder, P.; Boss, J.M. CTCF Controls Expression and Chromatin Architecture of the Human Major Histocompatibility Complex Class II Locus. Mol. Cell. Biol. 2010, 30, 4211–4223. [Google Scholar] [CrossRef]
- Ottaviani, D.; Lever, E.; Mao, S.; Christova, R.; Ogunkolade, B.W.; Jones, T.A.; Szary, J.; Aarum, J.; Mumin, M.A.; Pieri, C.A.; et al. CTCF binds to sites in the major histocompatibility complex that are rapidly reconfigured in response to interferon-gamma. Nucleic Acids Res. 2012, 40, 5262–5270. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, J.; Skok, J.A. The role of CTCF in regulating V(D)J recombination. Curr. Opin. Immunol. 2012, 24, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.E.; Corces, V.G. CTCF: Master Weaver of the Genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [Green Version]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Bailey, C.; Metierre, C.; Feng, Y.; Baidya, K.; Filippova, G.; Loukinov, D.; Lobanenkov, V.; Semaan, C.; Rasko, J. CTCF Expression is Essential for Somatic Cell Viability and Protection Against Cancer. Int. J. Mol. Sci. 2018, 19, 3832. [Google Scholar] [CrossRef]
- Cross, S.H.; Clark, V.H.; Simmen, M.W.; Bickmore, W.A.; Maroon, H.; Langford, C.F.; Carter, N.P.; Bird, A.P. CpG island libraries from human chromosomes 18 and 22: Landmarks for novel genes. Mamm. Genome 2000, 11, 373–383. [Google Scholar] [CrossRef]
- Ota, M.; Bahram, S.; Katsuyama, Y.; Saito, S.; Nose, Y.; Sada, M.; Ando, H.; Inoko, H. On the MICA deleted-MICB null, HLA-B*4801 haplotype. Tissue Antigens 2000, 56, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Middleton, D.; Menchaca, L.; Rood, H.; Komerofsky, R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens 2003, 61, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Aida, K.; Russomando, G.; Kikuchi, M.; Candia, N.; Franco, L.; Almiron, M.; Ubalee, R.; Hirayama, K. High frequency of MIC null haplotype (HLA-B48-MICA-del-MICB*0107 N) in the Angaite Amerindian community in Paraguay. Immunogenetics 2002, 54, 439–441. [Google Scholar] [CrossRef]
- Shaikh, T.H.; Gai, X.; Perin, J.C.; Glessner, J.T.; Xie, H.; Murphy, K.; O’Hara, R.; Casalunovo, T.; Conlin, L.K.; D’Arcy, M.; et al. High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res. 2009, 19, 1682–1690. [Google Scholar] [CrossRef] [Green Version]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melis, R.; Lewis, T.; Millson, A.; Lyon, E.; McMillin, G.A.; Slev, P.R.; Swensen, J. Copy Number Variation and Incomplete Linkage Disequilibrium Interfere with the HCP5 Genotyping Assay for Abacavir Hypersensitivity. Genet. Test. Mol. Biomark. 2012, 16, 1111–1114. [Google Scholar] [CrossRef]
- Dunn, D.; Ota, M.; Inoko, H.; Kulski, J.K. Association of MHC dimorphic Alu insertions with HLA class I and MIC genes in Japanese HLA-B48 haplotypes. Tissue Antigens 2003, 62, 259–262. [Google Scholar] [CrossRef]
- Anzai, T.; Shiina, T.; Kimura, N.; Yanagiya, K.; Kohara, S.; Shigenari, A.; Yamagata, T.; Kulski, J.K.; Naruse, T.K.; Fujimori, Y.; et al. Comparative sequencing of human and chimpanzee MHC class I regions unveils insertions/deletions as the major path to genomic divergence. Proc. Natl. Acad. Sci. 2003, 100, 7708–7713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulski, J.K.; Shiina, T.; Anzai, T.; Kohara, S.; Inoko, H. Comparative genomic analysis of the MHC: The evolution of class I duplication blocks, diversity and complexity from shark to man. Immunol. Rev. 2002, 190, 95–122. [Google Scholar] [CrossRef] [PubMed]
- Wilming, L.G.; Hart, E.A.; Coggill, P.C.; Horton, R.; Gilbert, J.G.R.; Clee, C.; Jones, M.; Lloyd, C.; Palmer, S.; Sims, S.; et al. Sequencing and comparative analysis of the gorilla MHC genomic sequence. Database 2013, 2013, bat011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudieri, S.; Giles, K.M.; Kulski, J.K.; Dawkins, R.L. Duplication and polymorphism in the MHC: Alu generated diversity and polymorphism within the PERB11 gene family. Hereditas 1997, 127, 37–46. [Google Scholar] [CrossRef]
- Hughes, A.L.; Yeager, M.; Ten Elshof, A.E.; Chorney, M.J. A new taxonomy of mammalian MHC class I molecules. Immunol. Today 1999, 20, 22–26. [Google Scholar] [CrossRef]
- Bahram, S. MIC genes: From genetics to biology. Adv. Immunol. 2000, 76, 1–60. [Google Scholar] [PubMed]
- Vejbaesya, S.; Luangtrakool, P.; Luangtrakool, K.; Kalayanarooj, S.; Vaughn, D.W.; Endy, T.P.; Mammen, M.P.; Green, S.; Libraty, D.H.; Ennis, F.A.; et al. TNF and LTA Gene, Allele, and Extended HLA Haplotype Associations with Severe Dengue Virus Infection in Ethnic Thais. J. Infect. Dis. 2009, 199, 1442–1448. [Google Scholar] [CrossRef]
- Low, J.S.Y.; Chin, Y.M.; Mushiroda, T.; Kubo, M.; Govindasamy, G.K.; Pua, K.C.; Yap, Y.Y.; Yap, L.F.; Subramaniam, S.K.; Ong, C.A.; et al. A Genome Wide Study of Copy Number Variation Associated with Nasopharyngeal Carcinoma in Malaysian Chinese Identifies CNVs at 11q14.3 and 6p21.3 as Candidate Loci. PLoS ONE 2016, 11, e0145774. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tian, W.; Zhu, F.; Li, L.; Cai, J.; Wang, F.; Liu, K.; Jin, H.; Wang, J. MICA Gene Deletion in 3411 DNA Samples from Five Distinct Populations in Mainland China and Lack of Association with Nasopharyngeal Carcinoma (NPC) in a Southern Chinese Han population. Ann. Hum. Genet. 2016, 80, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Caillat-Zucman, S. New insights into the understanding of MHC associations with immune-mediated disorders: MHC-associated immune diseases. HLA 2017, 89, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Medici, M.; Porcu, E.; Pistis, G.; Teumer, A.; Brown, S.J.; Jensen, R.A.; Rawal, R.; Roef, G.L.; Plantinga, T.S.; Vermeulen, S.H.; et al. Identification of Novel Genetic Loci Associated with Thyroid Peroxidase Antibodies and Clinical Thyroid Disease. PLoS Genet. 2014, 10, e1004123. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A Novel Genetic Variant in Long Non-coding RNA Gene NEXN-AS1 is Associated with Risk of Lung Cancer. Sci. Rep. 2016, 6, 34234. [Google Scholar] [CrossRef] [Green Version]
- The International HIV Controllers Study. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science 2010, 330, 1551–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Limou, S.; Le Clerc, S.; Coulonges, C.; Carpentier, W.; Dina, C.; Delaneau, O.; Labib, T.; Taing, L.; Sladek, R.; ANRS Genomic Group; et al. Genomewide Association Study of an AIDS-Nonprogression Cohort Emphasizes the Role Played by HLA Genes (ANRS Genomewide Association Study 02). J. Infect. Dis. 2009, 199, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Helms, C.; Liao, W.; Zaba, L.C.; Duan, S.; Gardner, J.; Wise, C.; Miner, A.; Malloy, M.J.; Pullinger, C.R.; et al. A Genome-Wide Association Study of Psoriasis and Psoriatic Arthritis Identifies New Disease Loci. PLoS Genet. 2008, 4, e1000041. [Google Scholar] [CrossRef]
- Rodriguez-Novoa, S.; Cuenca, L.; Morello, J.; Cordoba, M.; Blanco, F.; Jimenez-Nacher, I.; Soriano, V. Use of the HCP5 single nucleotide polymorphism to predict hypersensitivity reactions to abacavir: Correlation with HLA-B*5701. J. Antimicrob. Chemother. 2010, 65, 1567–1569. [Google Scholar] [CrossRef]
- Suhre, K.; Arnold, M.; Bhagwat, A.M.; Cotton, R.J.; Engelke, R.; Raffler, J.; Sarwath, H.; Thareja, G.; Wahl, A.; DeLisle, R.K.; et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 2017, 8, 14357. [Google Scholar] [CrossRef] [Green Version]
- Crosslin, D.R.; Carrell, D.S.; Burt, A.; Kim, D.S.; Underwood, J.G.; Hanna, D.S.; Comstock, B.A.; Baldwin, E.; de Andrade, M.; Kullo, I.J.; et al. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Feenstra, B.; Pasternak, B.; Geller, F.; Carstensen, L.; Wang, T.; Huang, F.; Eitson, J.L.; Hollegaard, M.V.; Svanström, H.; Vestergaard, M.; et al. Common variants associated with general and MMR vaccine–related febrile seizures. Nat. Genet. 2014, 46, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, F.W.; Chen, W.; O’Hanlon, T.P.; Cooper, R.G.; Vencovsky, J.; Rider, L.G.; Danko, K.; Wedderburn, L.R.; Lundberg, I.E.; Pachman, L.M.; et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. 2015, 16, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, S.; Cooper, R.G.; Lundberg, I.E.; Miller, F.W.; Gregersen, P.K.; Bowes, J.; Vencovsky, J.; Danko, K.; Limaye, V.; Selva-O’Callaghan, A.; et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann. Rheum. Dis. 2016, 75, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-P.; Chang, S.-W.; Jaing, T.-H.; Wang, W.-T.; Hus, F.-P.; Tseng, C.-P. Single nucleotide polymorphisms within HLA region are associated with disease relapse for patients with unrelated cord blood transplantation. PeerJ 2018, 6, e5228. [Google Scholar] [CrossRef]
- van Manen, D.; Kootstra, N.A.; Boeser-Nunnink, B.; Handulle, M.A.; van’t Wout, A.B.; Schuitemaker, H. Association of HLA-C and HCP5 gene regions with the clinical course of HIV-1 infection. AIDS 2009, 23, 19–28. [Google Scholar] [CrossRef] [Green Version]
- van Manen, D.; van ‘t Wout, A.B.; Schuitemaker, H. Genome-wide association studies on HIV susceptibility, pathogenesis and pharmacogenomics. Retrovirology 2012, 9, 70. [Google Scholar] [CrossRef]
- Shrestha, S.; Aissani, B.; Song, W.; Wilson, C.M.; Kaslow, R.A.; Tang, J. Host genetics and HIV-1 viral load set-point in African–Americans. AIDS 2009, 23, 673–677. [Google Scholar] [CrossRef]
- Thørner, L.W.; Erikstrup, C.; Harritshøj, L.H.; Larsen, M.H.; Kronborg, G.; Pedersen, C.; Larsen, C.S.; Pedersen, G.; Gerstoft, J.; Obel, N.; et al. Impact of polymorphisms in the HCP5 and HLA-C, and ZNRD1 genes on HIV viral load. Infect. Genet. Evol. 2016, 41, 185–190. [Google Scholar] [CrossRef]
- Catano, G.; Kulkarni, H.; He, W.; Marconi, V.C.; Agan, B.K.; Landrum, M.; Anderson, S.; Delmar, J.; Telles, V.; Song, L.; et al. HIV-1 Disease-Influencing Effects Associated with ZNRD1, HCP5 and HLA-C Alleles Are Attributable Mainly to Either HLA-A10 or HLA-B*57 Alleles. PLoS ONE 2008, 3, e3636. [Google Scholar] [CrossRef]
- Colombo, S.; Rauch, A.; Rotger, M.; Fellay, J.; Martinez, R.; Fux, C.; Thurnheer, C.; Günthard, H.F.; Goldstein, D.B.; Furrer, H.; et al. The HCP5 Single-Nucleotide Polymorphism: A Simple Screening Tool for Prediction of Hypersensitivity Reaction to Abacavir. J. Infect. Dis. 2008, 198, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Pinto, B.; Correia, C.; Gomes, L.; Gil-Mata, S.; Araújo, L.; Correia, O.; Delgado, L. HLA and Delayed Drug-Induced Hypersensitivity. Int. Arch. Allergy Immunol. 2016, 170, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.F.; et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. 2012, 109, 9959–9964. [Google Scholar] [CrossRef] [Green Version]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Kisiel, B.; Kisiel, K.; Szymański, K.; Mackiewicz, W.; Biało-Wójcicka, E.; Uczniak, S.; Fogtman, A.; Iwanicka-Nowicka, R.; Koblowska, M.; Kossowska, H.; et al. The association between 38 previously reported polymorphisms and psoriasis in a Polish population: High predicative accuracy of a genetic risk score combining 16 loci. PLoS ONE 2017, 12, e0179348. [Google Scholar] [CrossRef] [PubMed]
- Borgiani, P.; Di Fusco, D.; Erba, F.; Marazzi, M.C.; Mancinelli, S.; Novelli, G.; Palombi, L.; Ciccacci, C. HCP5 genetic variant (RS3099844) contributes to Nevirapine-induced Stevens Johnsons Syndrome/Toxic Epidermal Necrolysis susceptibility in a population from Mozambique. Eur. J. Clin. Pharmacol. 2014, 70, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, F.; Perricone, C.; Borgiani, P.; Ciccacci, C.; Rufini, S.; Cipriano, E.; Alessandri, C.; Spinelli, F.R.; Sili Scavalli, A.; Novelli, G.; et al. Genetic Factors in Systemic Lupus Erythematosus: Contribution to Disease Phenotype. J. Immunol. Res. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Ciccacci, C.; Priori, R.; Latini, A.; Picarelli, G.; Arienzo, F.; Novelli, G.; Valesini, G.; Perricone, C.; Borgiani, P. STAT4, TRAF3IP2, IL10, and HCP5 Polymorphisms in Sjögren’s Syndrome: Association with Disease Susceptibility and Clinical Aspects. J. Immunol. Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Taylor, J.R.; Ding, J.; Lohman, K.; Johnson, C.; Siscovick, D.; Burke, G.; Post, W.; Shea, S.; Jacobs, D.R.; et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 2014, 5, 5366. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Nititham, J.; Elboudwarej, E.; Quach, H.L.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Genome-Wide Assessment of Differential DNA Methylation Associated with Autoantibody Production in Systemic Lupus Erythematosus. PLoS ONE 2015, 10, e0129813. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef]
- Zimmermann, M.T.; Oberg, A.L.; Grill, D.E.; Ovsyannikova, I.G.; Haralambieva, I.H.; Kennedy, R.B.; Poland, G.A. System-Wide Associations between DNA-Methylation, Gene Expression, and Humoral Immune Response to Influenza Vaccination. PLoS ONE 2016, 11, e0152034. [Google Scholar] [CrossRef] [PubMed]
- Kukushkina, V.; Modhukur, V.; Suhorutšenko, M.; Peters, M.; Mägi, R.; Rahmioglu, N.; Velthut-Meikas, A.; Altmäe, S.; Esteban, F.J.; Vilo, J.; et al. DNA methylation changes in endometrium and correlation with gene expression during the transition from pre-receptive to receptive phase. Sci. Rep. 2017, 7, 3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Sui, W.; Tan, Q.; Chen, J.; Zhang, Y.; Ou, M.; Xue, W.; Li, F.; Cao, C.; Sun, Y.; et al. Integrated analyses of a major histocompatibility complex, methylation and transcribed ultra-conserved regions in systemic lupus erythematosus. Int. J. Mol. Med. 2016, 37, 139–148. [Google Scholar] [CrossRef]
- Cheng, L.; Sun, B.; Xiong, Y.; Hu, L.; Gao, L.; Lv, Q.; Zhou, M.; Li, J.; Chen, X.; Zhang, W.; et al. The minor alleles HCP5 rs3099844 A and PSORS1C1 rs3131003 G are associated with allopurinol-induced severe cutaneous adverse reactions in Han Chinese: A multicentre retrospective case-control clinical study. Br. J. Dermatol. 2018, 178, e191–e193. [Google Scholar] [CrossRef]
- Cvitic, S.; Longtine, M.S.; Hackl, H.; Wagner, K.; Nelson, M.D.; Desoye, G.; Hiden, U. The Human Placental Sexome Differs between Trophoblast Epithelium and Villous Vessel Endothelium. PLoS ONE 2013, 8, 12. [Google Scholar] [CrossRef]
- Rosenfeld, C.S. Sex-Specific Placental Responses in Fetal Development. Endocrinology 2015, 156, 3422–3434. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Yan, Z.; Wu, C.; Zhang, Q.; Zhu, Y.; Li, K.; Xu, Y. Integrated analysis of dosage effect lncRNAs in lung adenocarcinoma based on comprehensive network. Oncotarget 2017, 8, 71430–71446. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; Kaushik, P.; Kerr, G.; Walsh, J.; Dubreuil, M.; Reimold, A.; Sawalha, A. Genome-Wide DNA Methylation Analysis in Ankylosing Spondylitis [abstract]. Arthritis Rheumatol. 2018, 70. abstract number 2061. [Google Scholar]
- Koga, M.; Kawasaki, A.; Ito, I.; Furuya, T.; Ohashi, J.; Kyogoku, C.; Ito, S.; Hayashi, T.; Matsumoto, I.; Kusaoi, M.; et al. Cumulative association of eight susceptibility genes with systemic lupus erythematosus in a Japanese female population. J. Hum. Genet. 2011, 56, 503–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houshdaran, S.; Zelenko, Z.; Irwin, J.C.; Giudice, L.C. Human endometrial DNA methylome is cycle-dependent and is associated with gene expression regulation. Mol. Endocrinol. 2014, 28, 1118–1135. [Google Scholar] [CrossRef] [PubMed]
- Saare, M.; Modhukur, V.; Suhorutshenko, M.; Rajashekar, B.; Rekker, K.; Sõritsa, D.; Karro, H.; Soplepmann, P.; Sõritsa, A.; Lindgren, C.M.; et al. The influence of menstrual cycle and endometriosis on endometrial methylome. Clin. Epigenetics 2016, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Orvis, T.; Hepperla, A.; Walter, V.; Song, S.; Simon, J.; Parker, J.; Wilkerson, M.D.; Desai, N.; Major, M.B.; Hayes, D.N.; et al. BRG1/SMARCA4 Inactivation Promotes Non-Small Cell Lung Cancer Aggressiveness by Altering Chromatin Organization. Cancer Res. 2014, 74, 6486–6498. [Google Scholar] [CrossRef]
- Warner, N.; Burberry, A.; Pliakas, M.; McDonald, C.; Núñez, G. A Genome-wide Small Interfering RNA (siRNA) Screen Reveals Nuclear Factor-κB (NF-κB)-independent Regulators of NOD2-induced Interleukin-8 (IL-8) Secretion. J. Biol. Chem. 2014, 289, 28213–28224. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Krishnan, A.; Zhu, Q.; Wong, A.K.; Lee, Y.-S.; Troyanskaya, O.G. Tissue-aware data integration approach for the inference of pathway interactions in metazoan organisms. Bioinformatics 2015, 31, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Rieswijk, L.; Claessen, S.M.H.; Bekers, O.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.M.C.M.; Kleinjans, J.C.S.; van Breda, S.G.J. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 2016, 350–352, 31–39. [Google Scholar] [CrossRef]
- Lerner, P.; Guadalupe, M.; Donovan, R.; Hung, J.; Flamm, J.; Prindiville, T.; Sankaran-Walters, S.; Syvanen, M.; Wong, J.K.; George, M.D.; et al. The Gut Mucosal Viral Reservoir in HIV-Infected Patients Is Not the Major Source of Rebound Plasma Viremia following Interruption of Highly Active Antiretroviral Therapy. J. Virol. 2011, 85, 4772–4782. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef]
- Borjabad, A.; Morgello, S.; Chao, W.; Kim, S.-Y.; Brooks, A.I.; Murray, J.; Potash, M.J.; Volsky, D.J. Significant Effects of Antiretroviral Therapy on Global Gene Expression in Brain Tissues of Patients with HIV-1-Associated Neurocognitive Disorders. Plos Pathog. 2011, 7, e1002213. [Google Scholar] [CrossRef]
- Ockenhouse, C.F.; Bernstein, W.B.; Wang, Z.; Vahey, M.T. Functional Genomic Relationships in HIV-1 Disease Revealed by Gene-Expression Profiling of Primary Human Peripheral Blood Mononuclear Cells. J. Infect. Dis. 2005, 191, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.N.; Kohler, J.J.; Coberley, C.R.; Sleasman, J.W.; Goodenow, M.M. HIV-1 Activates Macrophages Independent of Toll-Like Receptors. PLoS ONE 2008, 3, e3664. [Google Scholar] [CrossRef]
- Hyrcza, M.D.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Heisler, L.; Yang, S.; Wilkins, O.; Ostrowski, M.; Der, S.D. Distinct Transcriptional Profiles in Ex Vivo CD4+ and CD8+ T Cells Are Established Early in Human Immunodeficiency Virus Type 1 Infection and Are Characterized by a Chronic Interferon Response as Well as Extensive Transcriptional Changes in CD8+ T Cells. J. Virol. 2007, 81, 3477–3486. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-Wide Analysis of Chromosomal Features Repressing Human Immunodeficiency Virus Transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, R.; Zhao, X.; Su, J.; Bian, X.; Ni, J.; Yue, Y.; Cai, Y.; Jin, J. A potential diagnostic marker for ovarian cancer: Involvement of the histone acetyltransferase, human males absent on the first. Oncol. Lett. 2013, 6, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Shenoy, A.K.; Li, X.; Jin, Y.; Jin, L.; Cai, Q.; Tang, M.; Liu, Y.; Chen, H.; Reisman, D.; et al. MOF Acetylates the Histone Demethylase LSD1 to Suppress Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, H.; Wang, P.; Xue, Y.; Liu, X.; Ma, J.; Cai, H.; Xi, Z.; Li, Z.; Liu, Y. Role of HCP5-miR-139-RUNX1 Feedback Loop in Regulating Malignant Behavior of Glioma Cells. Mol. Ther. 2016, 24, 1806–1822. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-J.; Kim, M.; Kim, H.-P. Epigenetic regulation of long noncoding RNA UCA1 by SATB1 in breast cancer. Bmb Rep. 2016, 49, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Lu, B.; Sun, L.; Yan, X.; Xu, J. Identification of candidate genes or microRNAs associated with the lymph node metastasis of SCLC. Cancer Cell Int. 2018, 18, 161–171. [Google Scholar] [CrossRef]
- Yang, C.; Sun, J.; Liu, W.; Yang, Y.; Chu, Z.; Yang, T.; Gui, Y.; Wang, D. Long noncoding RNA HCP5 contributes to epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation and interacting with miR-139-5p. 11. Am. J. Transl. Res. 2019, 11, 953–963. [Google Scholar]
- Yun, W.-K.; Hu, Y.-M.; Zhao, C.-B.; Yu, D.-Y.; Tang, J.-B. HCP5 promotes colon cancer development by activating AP1G1 via PI3K/AKT pathway. Eur. Rev. Med Pharmacol. Sci. 2019, 23, 2786–2793. [Google Scholar]
- Liang, L.; Xu, J.; Wang, M.; Xu, G.; Zhang, N.; Wang, G.; Zhao, Y. LncRNA HCP5 promotes follicular thyroid carcinoma progression via miRNAs sponge. Cell Death Dis. 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Shen, H.; Fang, D.; Meng, Q.; Xin, Y. LncRNA HCP5 promotes the development of cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4812–4819. [Google Scholar] [PubMed]
- Olgun, G.; Sahin, O.; Tastan, O. Discovering lncRNA mediated sponge interactions in breast cancer molecular subtypes. BMC Genomics 2018, 19, 650. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Ma, R.; Wang, J.; Wu, X.; Jin, S.; Peng, J.; Tan, R.; Zhang, T.; Li, Y.; Wang, Y. LncRNA2Function: A comprehensive resource for functional investigation of human lncRNAs based on RNA-seq data. BMC Genom. 2015, 16, S2. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef]
- Lanzós, A.; Carlevaro-Fita, J.; Mularoni, L.; Reverter, F.; Palumbo, E.; Guigó, R.; Johnson, R. Discovery of Cancer Driver Long Noncoding RNAs across 1112 Tumour Genomes: New Candidates and Distinguishing Features. Sci. Rep. 2017, 7, 41544. [Google Scholar] [CrossRef] [Green Version]
- De Giorgio, A.; Krell, J.; Harding, V.; Stebbing, J.; Castellano, L. Emerging Roles of Competing Endogenous RNAs in Cancer: Insights from the Regulation of PTEN. Mol. Cell. Biol. 2013, 33, 3976–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Mo, X.; Li, T.; Xie, Y.; Zhu, L.; Xiao, B.; Liao, Q.; Guo, J. Identification and functional annotation of metabolism-associated lncRNAs and their related protein-coding genes in gastric cancer. Mol. Genet. Genom. Med. 2018, 6, 728–738. [Google Scholar] [CrossRef]
- Tian, L.; He, Y.; Zhang, H.; Wu, Z.; Li, D.; Zheng, C. Comprehensive analysis of differentially expressed profiles of lncRNAs and mRNAs reveals ceRNA networks in the transformation of diffuse large B-cell lymphoma. Oncol. Lett. 2018, 16, 882–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, Y.; Wang, H.; Ji, X. Targeting Long Noncoding RNA in Glioma: A Pathway Perspective. Mol. Ther. Nucleic Acids 2018, 13, 431–441. [Google Scholar] [CrossRef]
- Klase, Z.; Yedavalli, V.S.R.K.; Houzet, L.; Perkins, M.; Maldarelli, F.; Brenchley, J.; Strebel, K.; Liu, P.; Jeang, K.-T. Activation of HIV-1 from Latent Infection via Synergy of RUNX1 Inhibitor Ro5-3335 and SAHA. PLoS Pathog. 2014, 10, e1003997. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; He, Z.; Li, L.; Yang, D.; Liu, G. Expression profiles analysis of long non-coding RNAs identified novel lncRNA biomarkers with predictive value in outcome of cutaneous melanoma. Oncotarget 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Nohata, N.; Abba, M.C.; Gutkind, J.S. Unraveling the oral cancer lncRNAome: Identification of novel lncRNAs associated with malignant progression and HPV infection. Oral Oncol. 2016, 59, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Gallant-Behm, C.; Espinosa, J. ΔNp63α utilizes multiple mechanisms to repress transcription in squamous cell carcinoma cells. Cell Cycle 2013, 12, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Goossens, N.; Hoshida, Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 105. [Google Scholar] [CrossRef]
- Sanford, T.; Porten, S.; Meng, M.V. Molecular Analysis of Upper Tract and Bladder Urothelial Carcinoma: Results from a Microarray Comparison. PLoS ONE 2015, 10, e0137141. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Wong, W.; Jeng, M.; Zehnder, J.L. Gene expression profile of idiopathic thrombocytopenic purpura (ITP). Pediatr. Blood Cancer 2006, 47, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Gaudieri, S.; Kulski, J.K.; Dawkins, R.L. MHC and Disease Associations in Nonhuman Primates. In Molecular Biology and Evolution of Blood Group and MHC Antigens in Primates; Blancher, A., Klein, J., Socha, W.W., Eds.; Springer: Berlin, Germany, 1997; pp. 464–490. [Google Scholar]
Figure 1.
Location of the HCP5 gene (d) within the ERV16 element (c) and the human leukocyte antigen (HLA) class I region of the beta-block (b) on chromosome 6 at 6p21.33 (a). The HLA class I promoter region (the green rectangle labeled as Pr) for the 2630 bp HCP5 gene (d) initiates transcription of the 2547 bp lnc HCP5 RNA (e). The 91 bp intron in the HCP5 gene is represented by the thin grey line between the violet rectangular lines (d). The AluSp, THE1B, and LTR162B insertions within the 6173 bp ERV16 sequence [30] (c) are indicated by the labeled circle, triangle, and rectangles, respectively (see Table 1 for more details). The H3K27Ac binding (orange curve) and Dnase I hypersensitivity region (grey horizontal rectangle) associated with the HCP5 gene sequence (d) and sourced from the University of California, Santa Cruz (UCSC) genomic browser (Table S1) are shown on line (f).
Figure 1.
Location of the HCP5 gene (d) within the ERV16 element (c) and the human leukocyte antigen (HLA) class I region of the beta-block (b) on chromosome 6 at 6p21.33 (a). The HLA class I promoter region (the green rectangle labeled as Pr) for the 2630 bp HCP5 gene (d) initiates transcription of the 2547 bp lnc HCP5 RNA (e). The 91 bp intron in the HCP5 gene is represented by the thin grey line between the violet rectangular lines (d). The AluSp, THE1B, and LTR162B insertions within the 6173 bp ERV16 sequence [30] (c) are indicated by the labeled circle, triangle, and rectangles, respectively (see Table 1 for more details). The H3K27Ac binding (orange curve) and Dnase I hypersensitivity region (grey horizontal rectangle) associated with the HCP5 gene sequence (d) and sourced from the University of California, Santa Cruz (UCSC) genomic browser (Table S1) are shown on line (f).

Figure 2.
DNA nucleotide alignment between the papillomavirus minor structural protein (PMSP) (AJ437509.2) and HCP5 RNA sequence (NR_040662.1).
Figure 2.
DNA nucleotide alignment between the papillomavirus minor structural protein (PMSP) (AJ437509.2) and HCP5 RNA sequence (NR_040662.1).

Figure 3.
HCP5 (a) and HLA-B (b) RNA sequences in 27 different normal tissues from 95 human individuals. NCBI BioProject PREJEB4337. RPKM (reads per kilobase of transcript per million mapped reads) on the y-axis is a normalized unit of transcript expression [60].
Figure 3.
HCP5 (a) and HLA-B (b) RNA sequences in 27 different normal tissues from 95 human individuals. NCBI BioProject PREJEB4337. RPKM (reads per kilobase of transcript per million mapped reads) on the y-axis is a normalized unit of transcript expression [60].

Figure 4.
CTCF-binding sites proximal to and within the HCP5 gene. (A) ENCODE accession number (EH37E1260344) and statistical scores for presence of binding sites for CTCF, H3K4me3, and H3K27ac and DNaseI hypersensitivity clusters. (B) The ENCODE regulation tracking details from the UCSC genome browser (Table S1) show the CTCF locations (blue horizontal blocks on line 3) relative to (C) the HCP5 and HLA-X gene positions below the horizontal red line. In (B) the cREs with a high probability for binding H3K4me3 are the red blocks on line 1, and those with high probability for binding H3K27ac are the yellow blocks on line 2. (D) The location of the DNaseI hypersensitivity clusters and 161 transcription factor binding sites from ChIP-seq data analysis archived in ENCODE are shown below the HLA-X and HCP5 genes.
Figure 4.
CTCF-binding sites proximal to and within the HCP5 gene. (A) ENCODE accession number (EH37E1260344) and statistical scores for presence of binding sites for CTCF, H3K4me3, and H3K27ac and DNaseI hypersensitivity clusters. (B) The ENCODE regulation tracking details from the UCSC genome browser (Table S1) show the CTCF locations (blue horizontal blocks on line 3) relative to (C) the HCP5 and HLA-X gene positions below the horizontal red line. In (B) the cREs with a high probability for binding H3K4me3 are the red blocks on line 1, and those with high probability for binding H3K27ac are the yellow blocks on line 2. (D) The location of the DNaseI hypersensitivity clusters and 161 transcription factor binding sites from ChIP-seq data analysis archived in ENCODE are shown below the HLA-X and HCP5 genes.



{kind=link}
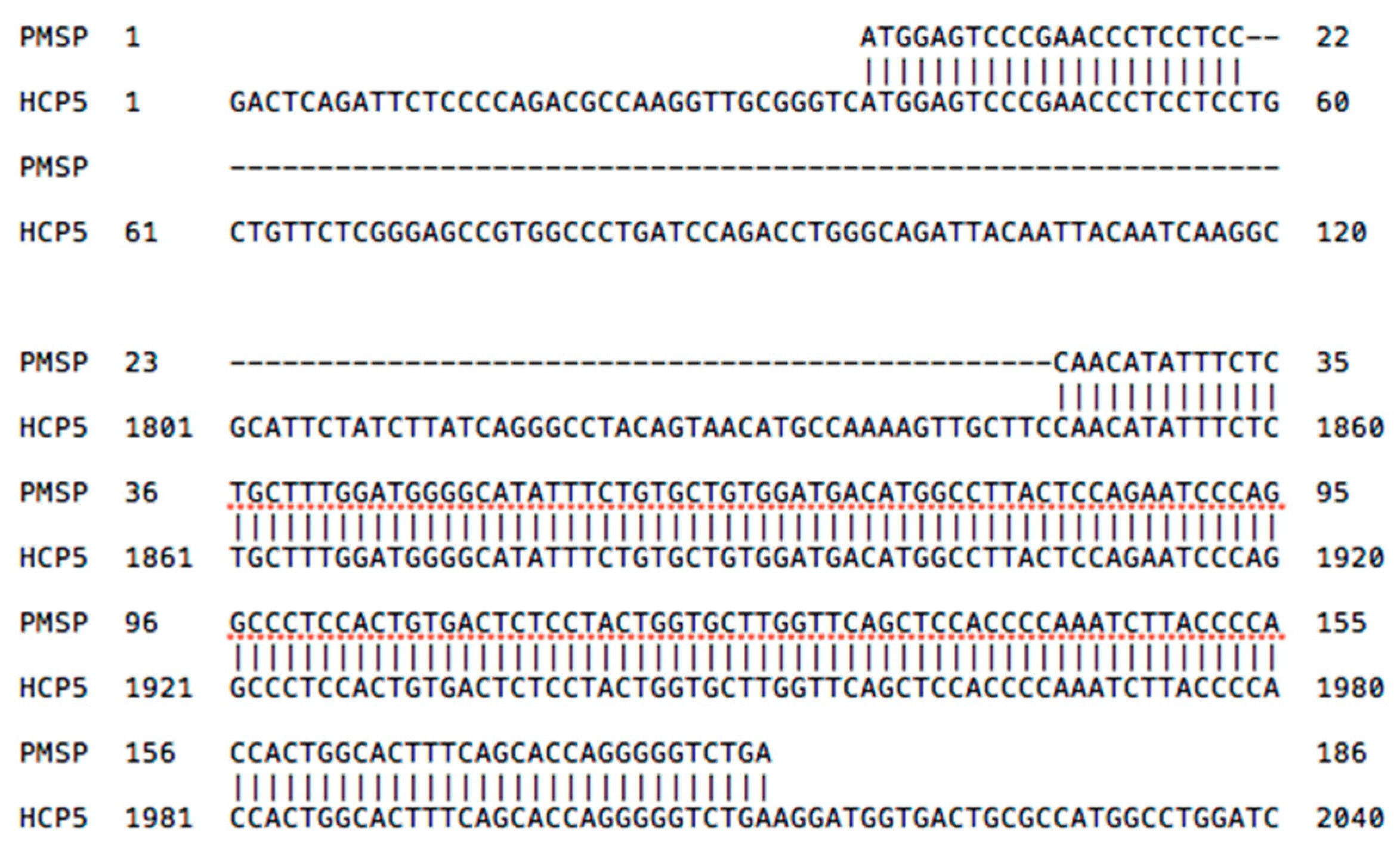{kind=link}
{kind=link}
{kind=link}
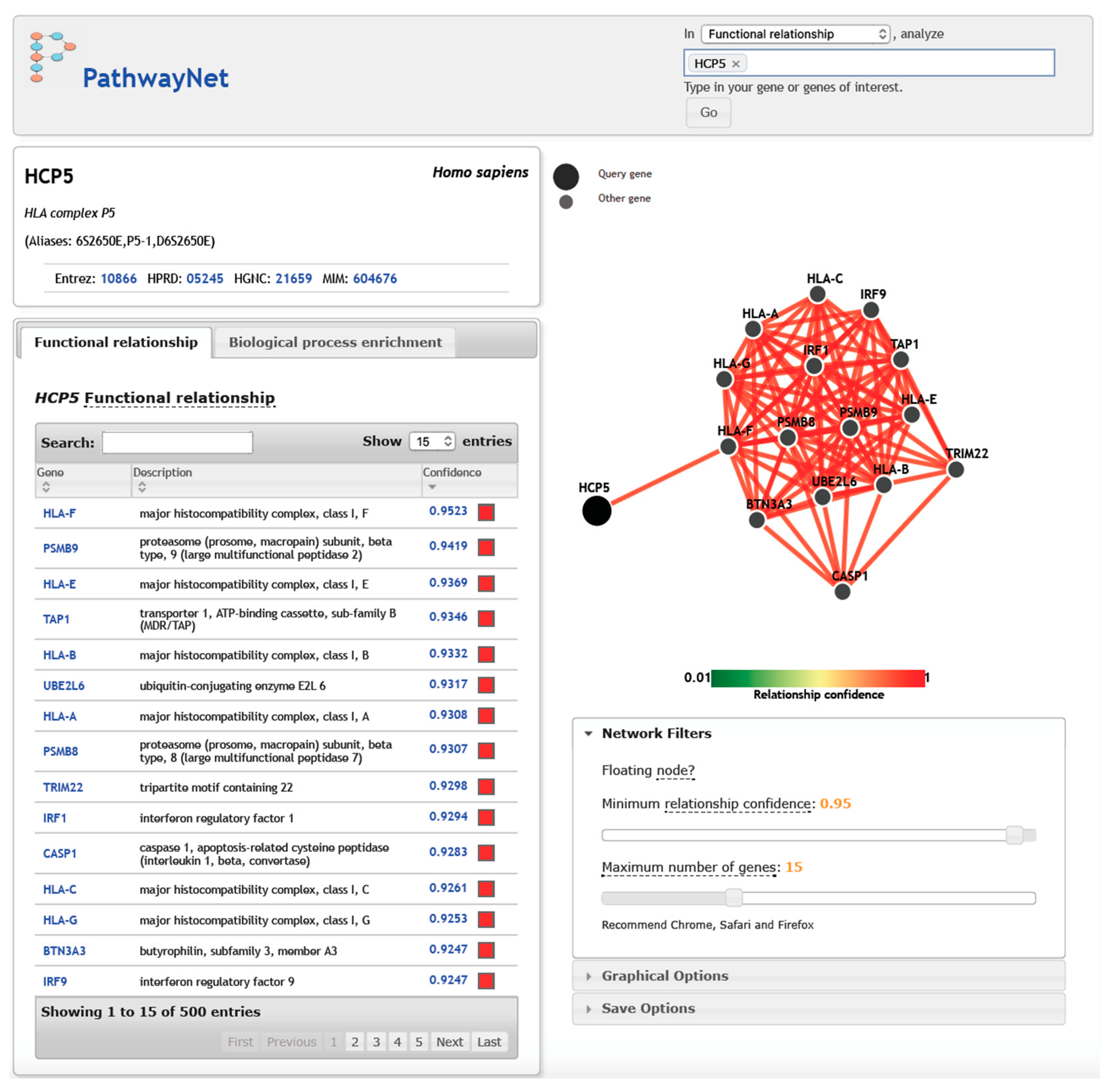{kind=link}
Table 1.
The chromosomal location of HCP5 and the ERV16 (LTR16B and ERV3-16A3) retroelement between the pseudogene HLA-X and the HCG26 lncRNA gene within the HLA class I region [30].
Table 1.
The chromosomal location of HCP5 and the ERV16 (LTR16B and ERV3-16A3) retroelement between the pseudogene HLA-X and the HCG26 lncRNA gene within the HLA class I region [30].
| Sequence Name | Location on Chr6 * | Length bp | Orient. | Feature |
|---|---|---|---|---|
| MERC21 | 31,461,669–31,462,057 | 389 | + | LTR fragment |
| HLA-X | 31,461,846–31,462,490 | 645 | + | Silent pseudogene |
| MERC21 | 31,462,544–31,462,625 | 82 | + | LTR fragment |
| HLA-UTR | 31,462,625–31,463,419 | 794 | + | HLA-5′UTR + exon 1 |
| HCP5-5′ncr | 31,462,464–31,463,413 | 950 | + | HLA class I promoter homologs |
| DNase region | 31,463,001–31,463,190 | 190 | + | 100% reliability score |
| HCP5 | 31,463,180–31,465,809 | 2630 | + | lncRNA gene |
| 3′LTR16B | 31,463,420–31,463,715 | 296 | − | 3′LTR (nt 150–446 in HCP5 RNA) |
| ERV3-16A3 | 31,463,716–31,464,933 | 1218 | − | Internal (nt 447–2547 in HCP5 RNA) |
| AluSp | 31,465,920–31,466,225 | 306 | − | Insertion within ERV3-16A3 |
| ERV3-16A3 | 31,466,237–31,467,707 | 1471 | − | Internal |
| THE1B | 31,467,708–31,468,050 | 343 | + | Fragmented insertion within ERV3-16A3 |
| ERV3-16A3 | 31,468,051–31,468,201 | 151 | − | Internal |
| ERV3-16A3 | 31,468,362–31,468,644 | 283 | − | Internal |
| 5′LTR16B2 | 31,468,881–31,469,043 | 163 | − | 5′LTR |
| AluSx | 31,469,044–31,469,282 | 239 | − | Insertion within LTR16B |
| 5′LTR16B2 | 31,469,283–31,469,592 | 310 | − | 5′LTR |
| L2 | 31,469,593–31,470,361 | 769 | − | L2 LINE |
| MLT1E2 | 31,470,734–31,471,317 | 584 | − | ERL-MaLR, LTR fragment |
| L2 | 31,471,325–31,472,047 | 723 | − | L2 LINE |
| HCG26 | 31,471,229–31,472,408 | 1180 | + | LncRNA |
* Chr6:31461669-31472408, GRCh38.p12 assembly (annotation release 109 in March 2018). 5′ncr is 5′ noncoding region. Orientation of the sequence is on the +ve DNA strand or the −ve (complementary) DNA strand.
Table 2.
Effect of interferons and cytokines on HCP5 RNA expression (nd, no difference).
| Cytokine | Cell Type | No: Controls/Tests | Up or Down | Geoprofile (206082_at) | Reference PMID |
|---|---|---|---|---|---|
| IL10 | Peripheral blood mononuclear cells | 4/4 | down | GDS4551 | 23449998 |
| KSHV-IRF4 | lymphoma | 2/2 | up | GDS4956 | 24335298 |
| IFN-alpha | natural killer cells | 1/3 | up | GDS4163 | 20334827 |
| IFN-gamma | keratinocytes | 5/4 | up | GDS1846 | 14760888 |
| IFN-alpha | hepatocytes (24 h) | 3/3 | up | GDS4390 | 22248663 |
| IL28B | hepatocytes (24 h) | 3/3 | up | GDS4390 | 22248663 |
| IFN-II | lung epithelium (24 h | 4/4 | up | GDS2341 | 16800785 |
| IFN-gamma | bronchial epithelium (24 h) | 5/5 | up | GDS1256 | 15985639 |
| IFNg + DXM | bronchial epithelium (24 h) | 5/5 | up | GDS1256 | 15985639 |
| IFN-gamma | macrophages | 6/6 | up | GDS4232 | 22140520 |
| IFN-gamma | microglia (24 h) | 4/4 | up | GDS1036 | 16163375 |
| TNF | endothelial | 4/4 | nd | GDS1542 | 16617158 |
| TNF | keratinocytes | 4/4 | up | GDS1289 | 15722350 |
| IL1 | macrophages | 5/5 | nd | GDS3005 | 18498781 |
| IL6 | macrophages | 5/5 | nd | GDS3005 | 18498781 |
| IL6 | macrophages | 3/3 | nd | GDS3290 | 18511485 |
| Cigarette smoke | small airway cells | 12/12 | down | GDS2486 | 19106307 |
DXM is dexamethasone.
Table 3.
Diseases and phenotypes associated with HCP5 SNV and retroelement (RE).
| SNV ID | Chr Position | RE or Pseudogene | HCP5 Position | Ref | Disease or Phenotype |
|---|---|---|---|---|---|
| rs3094228 | 6:31462150 | HLA-X | - | [91] | Thyroid peroxidase antibody positivity |
| rs115846244 | 6:31462283 | HLA-X | - | [43] | Tonsillectomy |
| rs4360170 | 6:31462582 | MERC21 | - | [43] | Cold sores |
| rs3094605 | 6:31462917 | HLA promoter | 5’UTR | [92] | Lung cancer |
| rs3099840 | 6:31462944 | HLA promoter | 5’UTR | [92] | Lung cancer |
| rs3132090 | 6:31462975 | HLA promoter | 5’UTR | [92] | Lung cancer |
| rs2255221 | 6:31463914 | ERV3-16A3 | internal | [93] | HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [94] | Flucloxacillin drug liver injury |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [33,34] | HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [95] | AIDS and HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [96] | Psoriasis and psoriatic arthritis |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [93] | HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [97] | Abacavir-induced hypersensitivity |
| rs3130907 | 6:31464036 | ERV3-16A3 | internal | [92] | Lung cancer |
| rs2284178 | 6:31464348 | ERV3-16A3 | internal | [98] | MICB measurement |
| rs3094014 | 6:31465781 | ERV3-16A3 | 3′UTR | [92] | Lung cancer |
| rs75640364 | 6:31465789 | ERV3-16A3 | 3′UTR | [99] | Herpes zoster |
| rs3128986 | 6:31465916 | ERV3-16A3 | 3′UTR | [92] | Lung cancer |
| rs79022003 | 6:31465935 | AluSp | 3’UTR | [100] | Eosinophil counts |
| rs3131620 | 6:31466054 | AluSp | 3′UTR | [92] | Lung cancer |
| rs3094604 | 6:31466334 | ERV3-16A3 | 3’UTR | [101] | Squamous cell lung carcinoma |
| rs3094604 | 6:31466334 | ERV3-16A3 | 3′UTR | [92] | Lung cancer |
| rs3094604 | 6:31466334 | ERV3-16A3 | 3’UTR | [102] | MMR vaccine-related febrile seizures |
| rs3131619 | 6:31466554 | ERV3-16A3 | 3’UTR | [103] | Myositis |
| rs3094013 | 6:31466589 | ERV3-16A3 | 3’UTR | [104] | Idiopathic inflammatory myopathies |
| rs3131618 | 6:31466589 | ERV3-16A3 | 3’UTR | [103] | Myositis |
| rs3094013 | 6:31466844 | ERV3-16A3 | 3’UTR | [43] | Tonsillectomy |
| rs2523675 | 6:31468255 | ERV3-16A3 | 3’UTR | [105] | Relapse after cord blood transplantation |
| rs2518028 | 6:31468270 | ERV3-16A3 | 3’UTR | [105] | Relapse after cord blood transplantation |
| rs2523673 | 6:31469218 | LTR16B/AluSx | 3’UTR | [100] | Platelet count |
Table 4.
Differentially methylated positions of HCP5′ in six recent studies.
| Disease/Phenotype | CpG Site | Position Genecode | Feature | Delta | p-Value | Reference |
|---|---|---|---|---|---|---|
| Age-related expression | cg25843003 | 31431312 | LTR16B2 | −0.23 | 2.3 × 10−13 | [121] |
| in monocytes and T cells | cg01082299 | 31431969 | ERV3-16 | −0.001 | 6.86 × 10−6 | |
| SLE: Anti-dsDNA’ | cg25843003 | 31431312 | LTR16B2 | −0.044 | 5.2 × 10−10 | [122] |
| cg00218406 | 31431407 | LTR16B2 | −0.067 | 8.7 × 10−9 | ||
| cg01082299 | 31431969 | ERV3-16 | −0.04 | 1.2 × 10−7 | ||
| cg18808777 | 31431503 | ERV3-16 | −0.051 | 3.0 × 10−10 | ||
| HIV associated marker | cg00218406 | 31431407 | LTR16B2 | [123] | ||
| Influenza vaccination | CpGsite | 31428956 | MER21B | −0.301 | 1.31 × 10−4 | [124] |
| 31431456 | LTR16B2 | |||||
| 31431457 | LTR16B2 | −0.401 | 2.0 × 10−7 | |||
| 31433586 | 3′UTR | |||||
| Endometrium | cg25843003 | 31431312 | LTR16B2 | −0.064 | [125] | |
| Pre to receptive | cg00218406 | 31431407 | LTR16B2 | −0.058 | ||
| cg21684411 | 31431573 | ERV3-16 | −0.071 | |||
| BMI | cg00218406 | 31431407 | LTR16B2 | 0.005 | 8.03 × 10−7 | [64] |
| BMI | cg25843003 | 31431312 | LTR16B2 | 0.002 | 1.51 × 10−6 | |
| Obesity | cg00218406 | 31431407 | LTR16B2 | 0.048 | 4.34 × 10−5 | |
| Obesity | cg25843003 | 31431312 | LTR16B2 | 0.020 | 5.98 × 10−5 |
The negative delta values reflect hypomethylation and gene upregulation and the positive delta values reflect hypermethylation and gene downregulation. SLE is Systematic Lupus Erythematosus. The p-values were mostly False Discovery Rate (FDR) adjusted.
Table 5.
Top 52 HCP5 gene interactions associated with Comparative Toxicogenomics Database (CTD) studies.
Table 5.
Top 52 HCP5 gene interactions associated with Comparative Toxicogenomics Database (CTD) studies.
| Gene Symbol | Gene ID | Interaction Count | Gene Symbol | Gene ID | Interaction Count |
|---|---|---|---|---|---|
| PTGS2 | 5743 | 71 | ICAM1 | 3383 | 9 |
| TNF | 7124 | 55 | NOS3 | 4846 | 9 |
| IL1B | 3553 | 52 | PON1 | 5444 | 9 |
| PTGS1 | 5742 | 42 | SCARB1 | 949 | 9 |
| CASP3 | 836 | 28 | AHR | 196 | 8 |
| BCL2 | 596 | 21 | APOA1 | 335 | 8 |
| AGT | 183 | 20 | TXN1 | 22166 | 8 |
| CTNNB1 | 1499 | 20 | VEGFA | 7422 | 8 |
| CCND1 | 595 | 18 | ABCA1 | 19 | 7 |
| NFKB1 | 4790 | 18 | ALB | 213 | 7 |
| RELA | 5970 | 17 | CCL2 | 6347 | 7 |
| TNFSF10 | 8743 | 17 | IKBKB | 3551 | 7 |
| NOS2 | 4843 | 14 | ITGA2B | 3674 | 7 |
| PARP1 | 142 | 14 | MAPK1 | 5594 | 7 |
| NFKBIA | 4792 | 13 | MAPK3 | 5595 | 7 |
| BAX | 581 | 12 | MYC | 4609 | 7 |
| CDKN1A | 1026 | 12 | PPARG | 5468 | 7 |
| IL4 | 3565 | 12 | SELP | 6403 | 7 |
| IL6 | 3569 | 11 | TCF4 | 6925 | 7 |
| ITGB3 | 3690 | 11 | ABCB1 | 5243 | 6 |
| MMP9 | 4318 | 11 | AKT1 | 207 | 6 |
| CASP9 | 842 | 10 | CYSLTR1 | 10800 | 6 |
| IL1A | 3552 | 10 | JUN | 3725 | 6 |
| PPARA | 5465 | 10 | MMP2 | 4313 | 6 |
| BIRC5 | 332 | 9 | PLAUR | 5329 | 6 |
| CASP8 | 841 | 9 | RB1 | 5925 | 6 |
Table 6.
HCP5–miRNA–protein coding gene regulator interactions.
| Antioncogenic miRNA | HCP5 Action | Gene Symbol and Up- or Downregulated | Cancer Type | Reference |
|---|---|---|---|---|
| miR-203 | sponge | TGFb/SMAD3 | Lung | [56] |
| miR-106b-5p | network | CTSS/FGL2 | Lung | [130] |
| miR-17-5b | network | PDCD1LG2/PDL2 | Lung | [130] |
| miR-126 | GSR/ASCL1 ↑ | Lung metastasis | [152] | |
| MET/GRM8/DACH1 ↓ | Lung metastasis | [152] | ||
| miR-139 | absorption | RUNX1 ↑ | Glioma | [150] |
| miR-139-5p | ZEB1 | Colorectal | [153] | |
| APIG1 ↓ | Colon | [154] | ||
| miR-22-3p | sponge | ST6GAL2 ↑ | Thyroid | [155] |
| mi-186-5p | sponge | ST6GAL2 ↑ | Thyroid | [155] |
| miR-216a-5p | sponge | ST6GAL2 ↑ | Thyroid | [155] |
| miR-15a | adsorption | MACC1 ↑ | Cervical | [156] |
| miR-155 | sponge | Complement genes | Breast | [157] |
| miR-128 | sponge | MACC1 ↑ | Gastric | [163] |
| miR-101 | sponge | NDUFB6 ↓ | Gastric | [163] |
| miR-103a | sponge | NDUFB6 ↓ | Gastric | [163] |
| 24 different mIR | ceRNA | TAP1 ↑ | Lymphoma | [164] |
| ceRNA | PSMB9 ↑ | Lymphoma | [164] | |
| ceRNA | KLF2 ↑ | Lymphoma | [164] | |
| ceRNA | GIPC1 ↑ | Lymphoma | [164] | |
| ceRNA | ETN3A3 ↑ | Lymphoma | [164] | |
| ceRNA | ETN3A1 ↑ | Lymphoma | [164] | |
| ceRNA | CD47 ↑ | Lymphoma | [164] | |
| ceRNA | CCDC50 ↑ | Lymphoma | [164] | |
| ceRNA | LMBR1L ↑ | Lymphoma | [164] | |
| ceRNA | HERC6 ↑ | Lymphoma | [164] |
↑ is upregulation and ↓ is downregulation.
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kulski, J.K. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells 2019, 8, 480. https://doi.org/10.3390/cells8050480
AMA Style
Kulski JK. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells. 2019; 8(5):480. https://doi.org/10.3390/cells8050480
Chicago/Turabian StyleKulski, Jerzy K. 2019. "Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations" Cells 8, no. 5: 480. https://doi.org/10.3390/cells8050480
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.