Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Skin Fibroblasts and Culture Conditions
2.2. NSC34 Cells and Culture Conditions
2.3. Immunofluorescence and Live Microscopy
2.4. DQBSA (Self-Quenched BODIPY Dye Conjugates of Bovine Serum Albumin) Assay
2.5. Western Blotting
2.5.1. Maturation of Cathepsin D
2.5.2. Autophagic Flux
2.6. Nucleic Acid Extraction and Whole mtDNA Amplification, Sequencing and Mutation Screening
2.7. Senescence-Associated Beta-Galactosidase (SA-βgal) Activity Assay
2.8. Statistical Analysis
3. Results
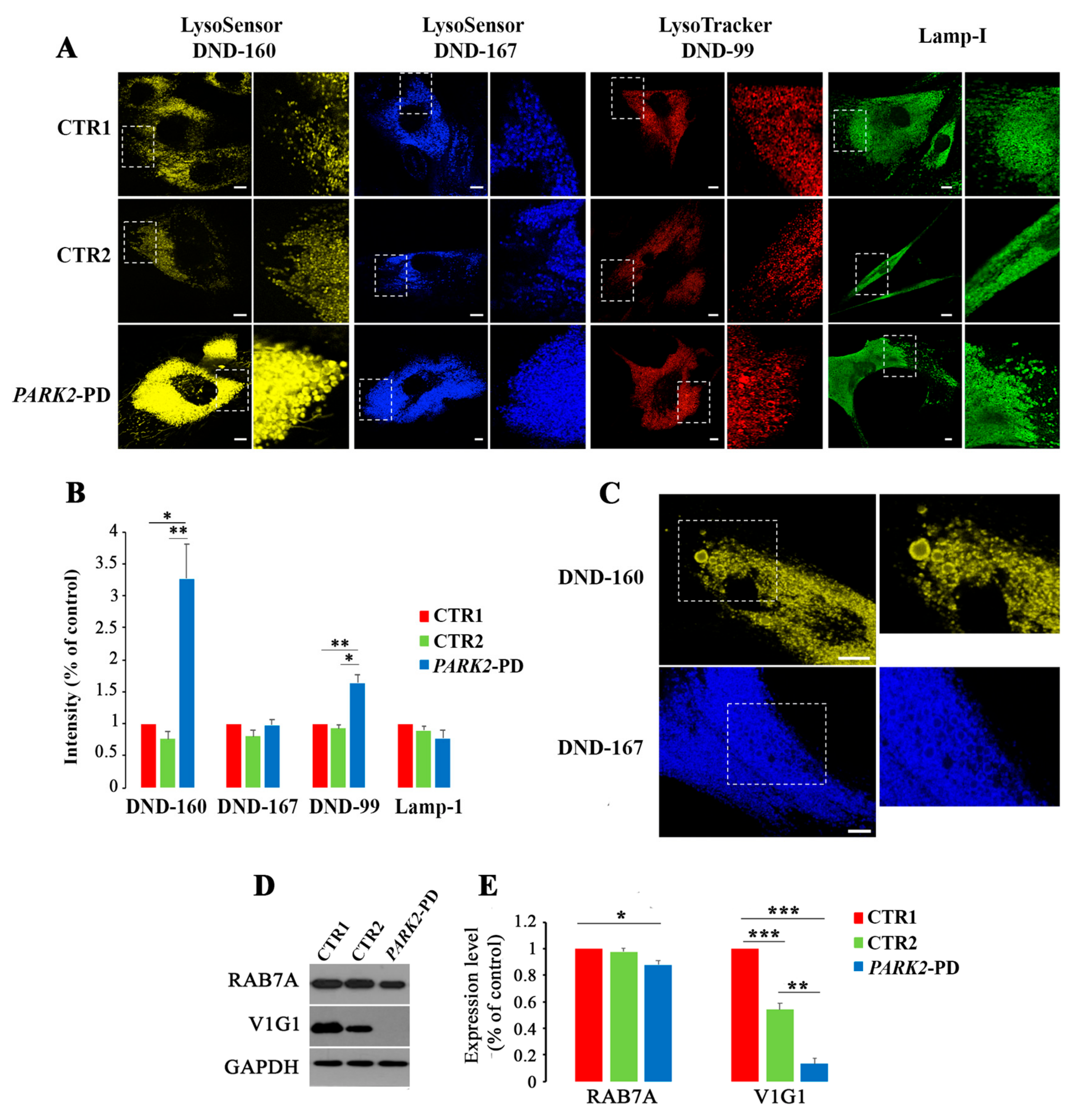
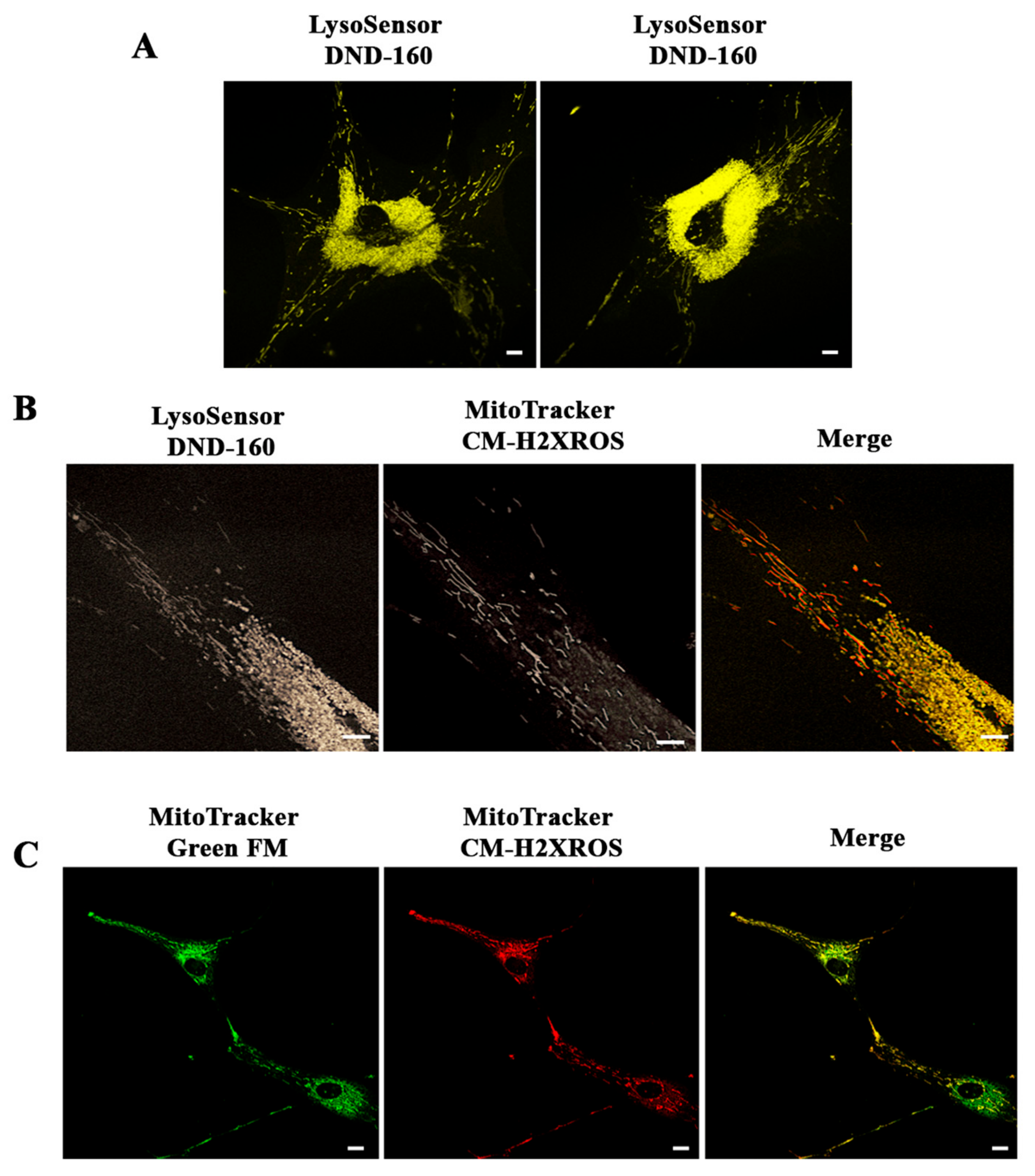
3.1. PARK2-PD Fibroblasts Display Abnormal Abundance, Acidification and Morphology of the Late Endocytic Compartment
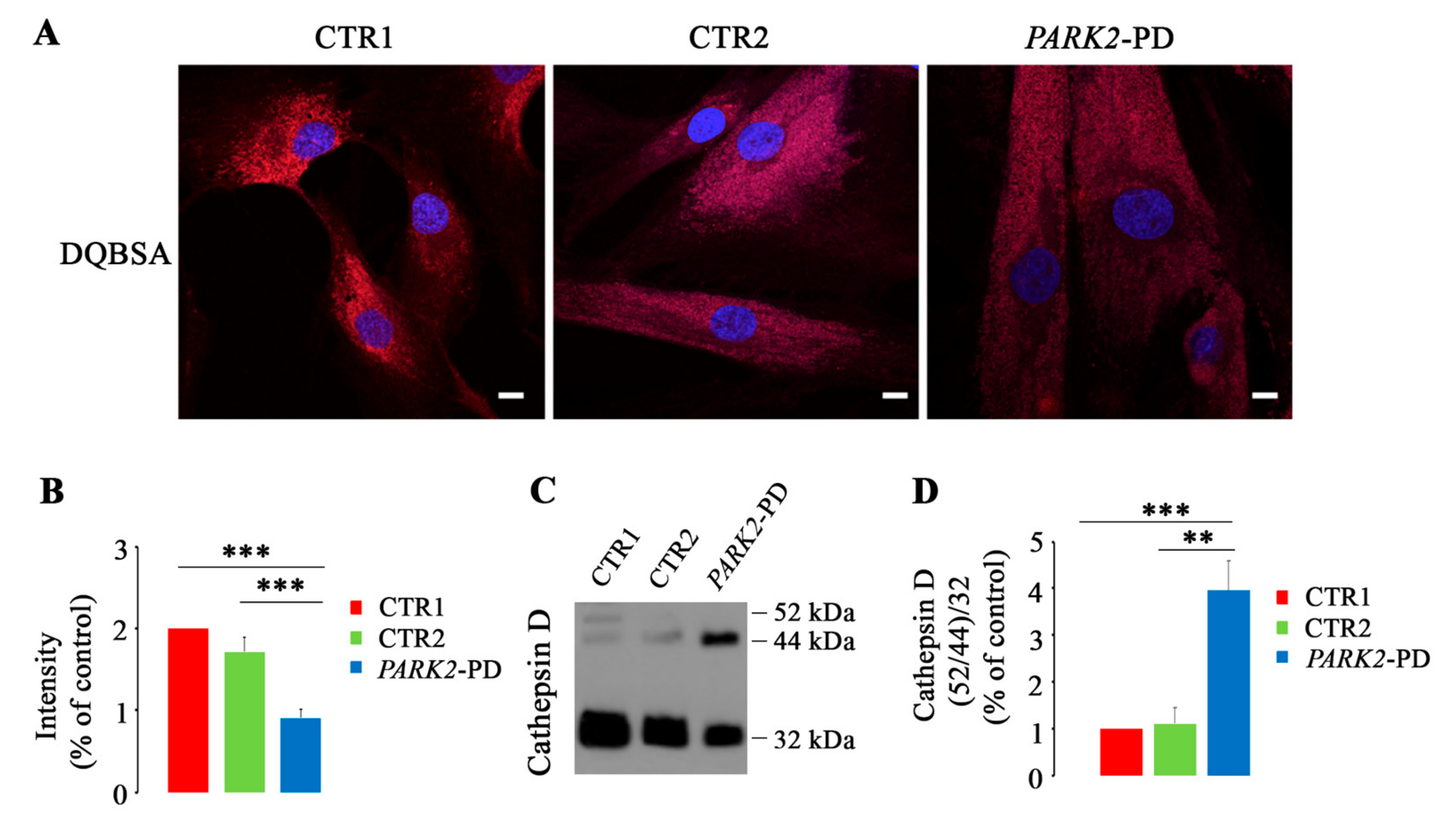
3.2. Lysosomal Function Is Impaired in PARK2-PD Fibroblasts
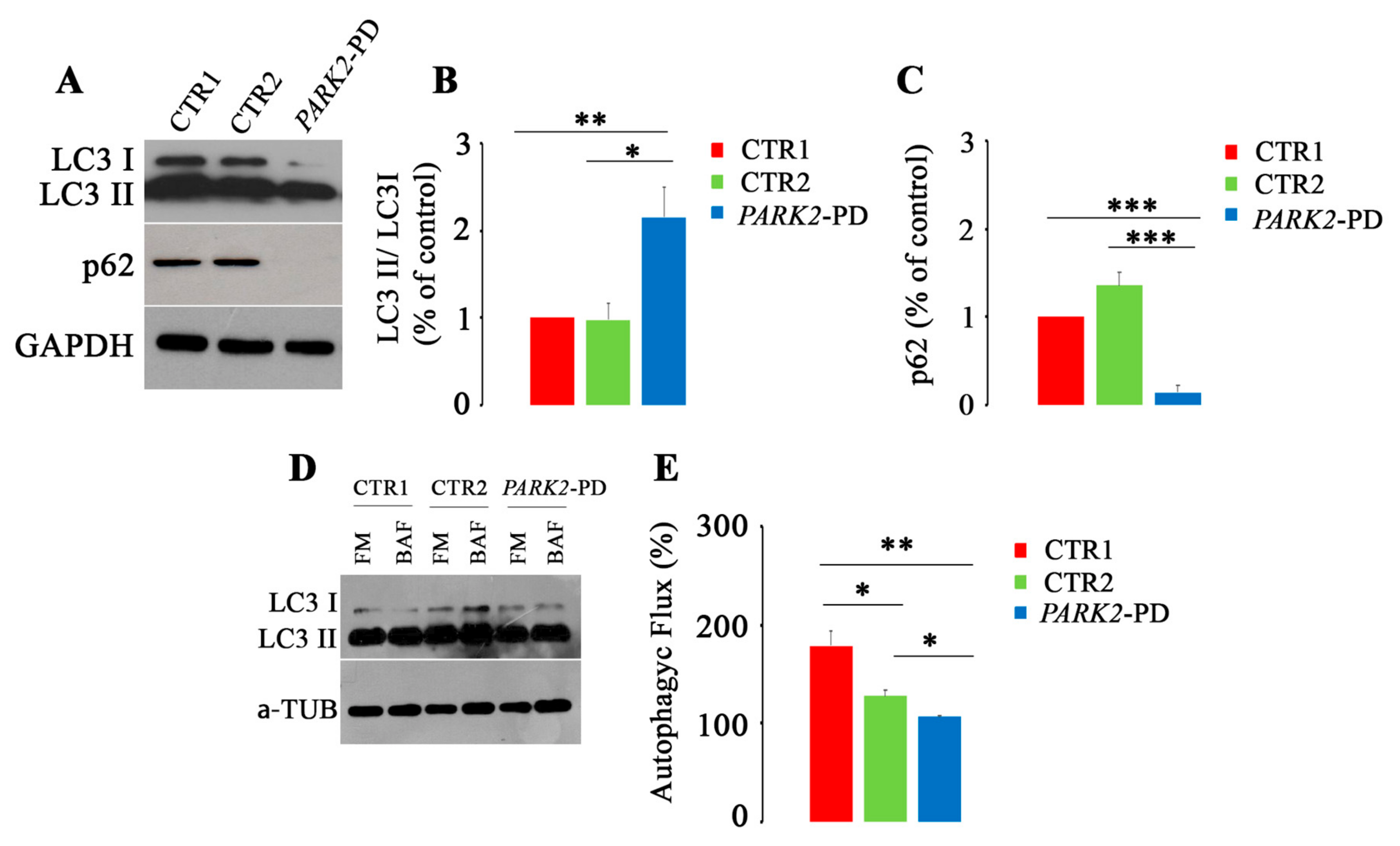
3.3. Autophagic Flux Is Impaired in PARK2-PD Fibroblasts
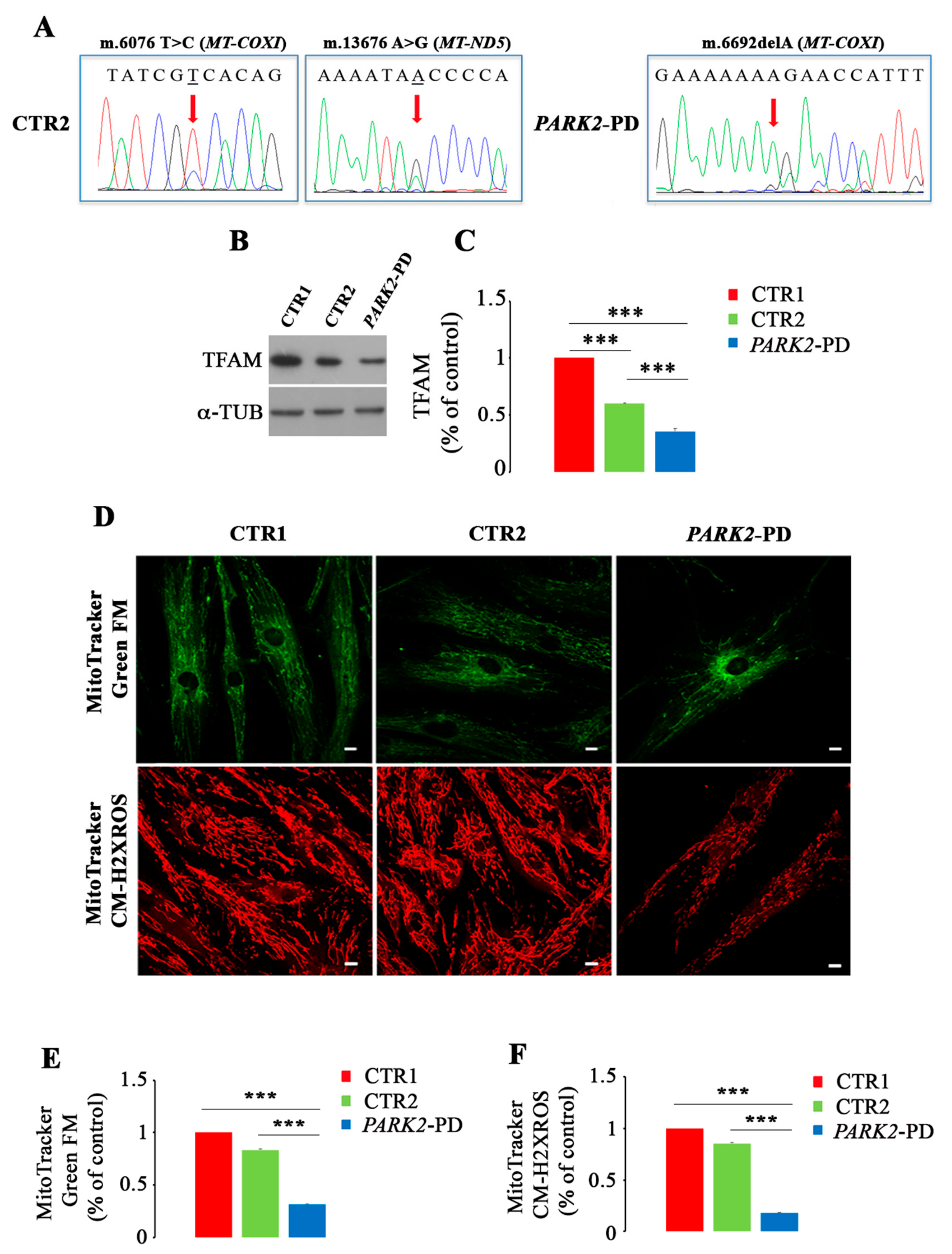
3.4. Mitochondrial DNA Mutations and Biogenesis Dysfunction in PARK2-PD Fibroblasts
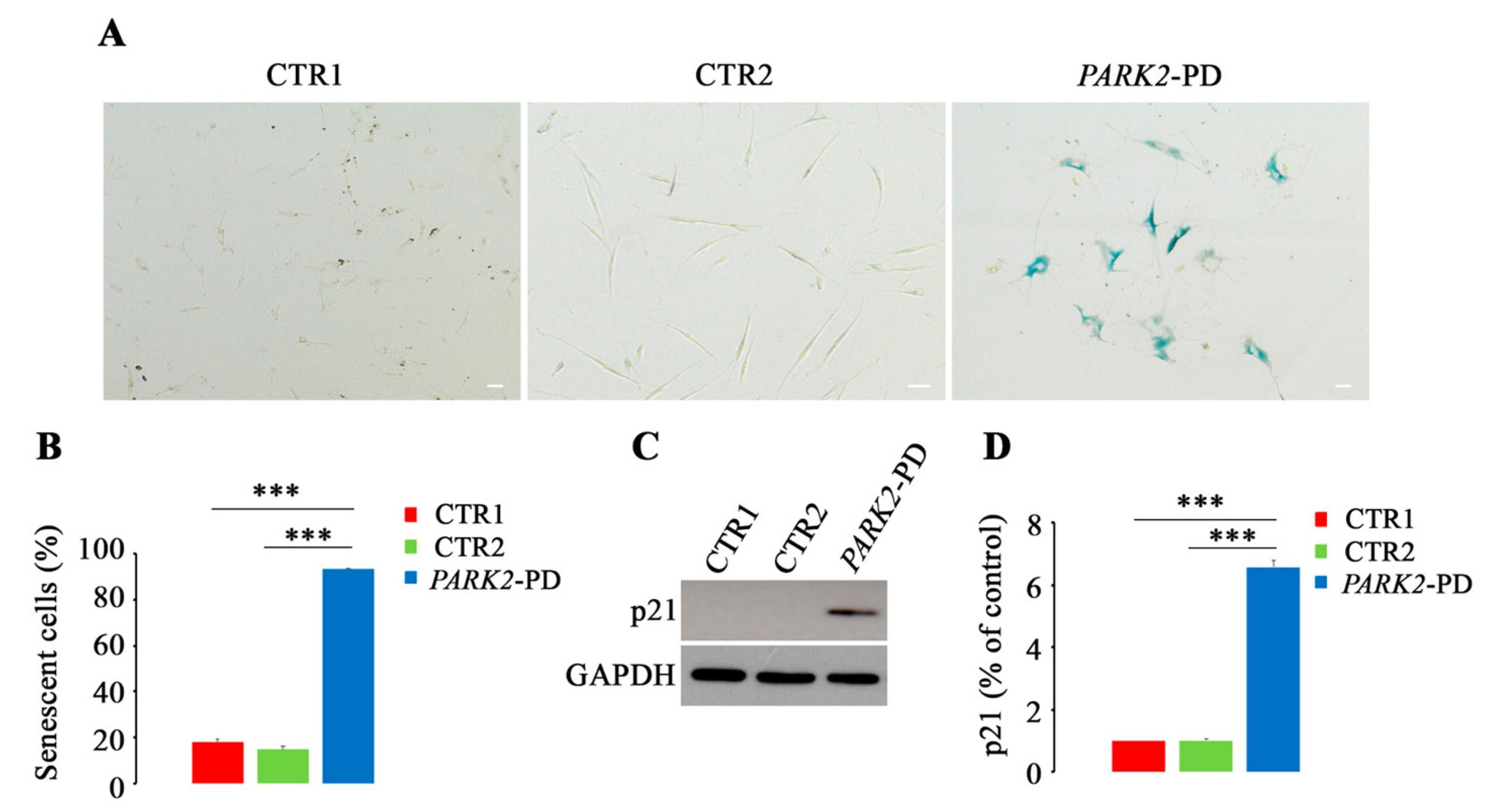
3.5. Premature Senescence in PARK2-PD Fibroblasts
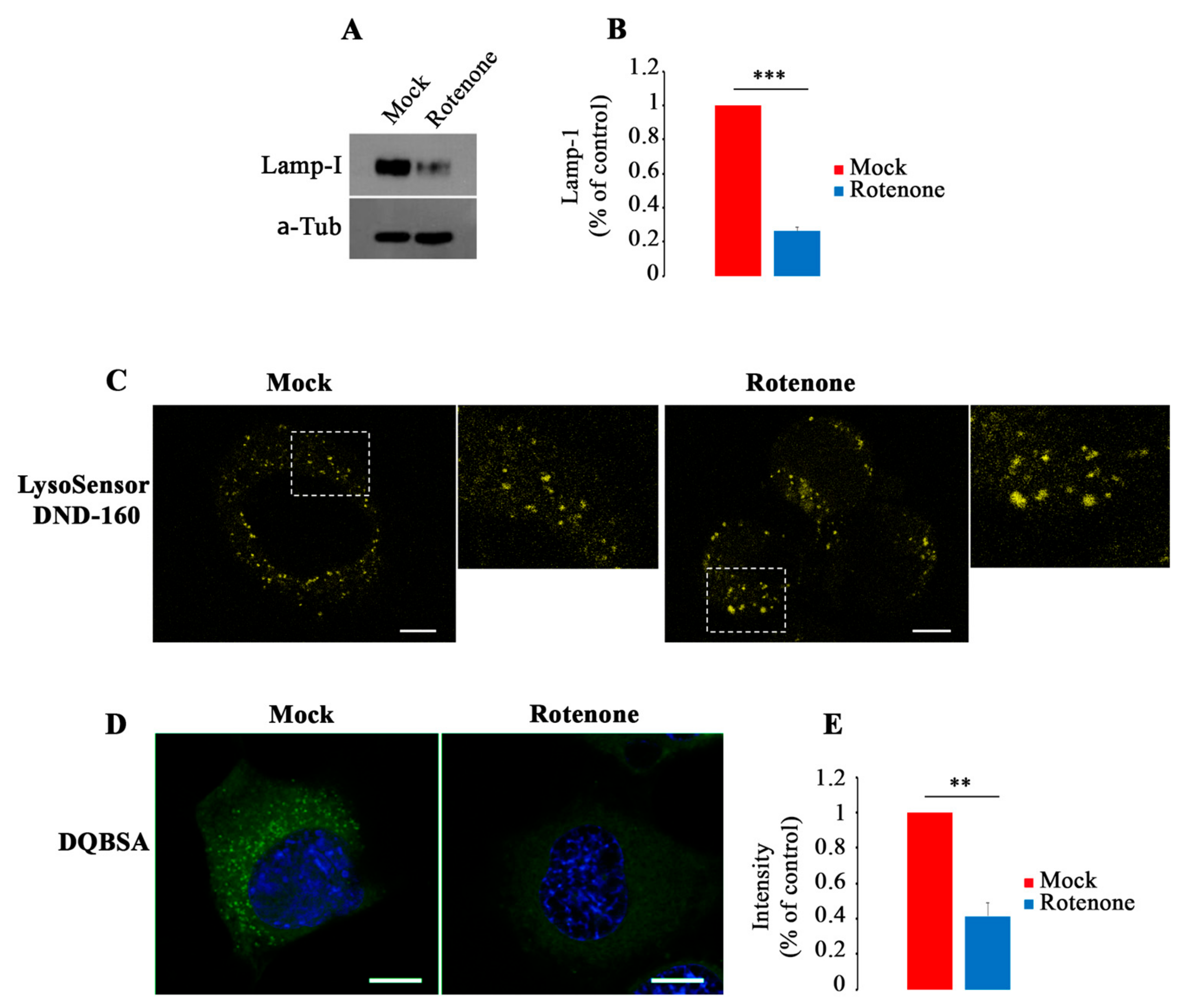
3.6. Inhibition of Complex I Activity Induce Lysosomal Dysfunction
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dauer, W.; Przedborski, S. parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and parkinson’s disease. Environ. Health Perspect 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause park8-linked parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset parkinson’s disease caused by mutations in pink1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. Dj-1( park7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in atp13a2, encoding a lysosomal type 5 p-type atpase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in unraveling the genetic etiology of parkinson disease in a genomic era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; De Rasmo, D. Complex i deficiencies in neurological disorders. Trends Mol. Med. 2013, 19, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex i deficiency in parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Rappold, P.M.; Cui, M.; Chesser, A.S.; Tibbett, J.; Grima, J.C.; Duan, L.; Sen, N.; Javitch, J.A.; Tieu, K. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc. Natl. Acad. Sci. USA 2011, 108, 20766–20771. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.D.; Song, L.K.; Li, J.; Chu, S.F.; Yuan, Y.H.; Chen, N.H. Environment-contact administration of rotenone: A new rodent model of parkinson’s disease. Behav. Brain Res. 2015, 294, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar alpha-synuclein oligomers promote complex i-dependent, ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Eitan, E.; Wu, T.Y.; Mattson, M.P. Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol. Aging 2018, 61, 52–65. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Hurley, J.H. Retromer. Curr. Opin. Cell Biol. 2008, 202, 427–436. [Google Scholar] [CrossRef]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. Vps35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine promotes α-synuclein pathology in mutant gba-associated parkinson’s disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, N.Y.; Lee, C.; Lee, H.J.; Kim, S.; Sardi, S.P.; Lee, S.J. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant vps35 causes mitochondrial dysfunction by recycling dlp1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. Atp13a2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. Park9-associated atp13a2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef]
- Niu, J.; Yu, M.; Wang, C.; Xu, Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via dynamin-like protein. J. Neurochem. 2012, 122, 650–658. [Google Scholar] [CrossRef]
- Papkovskaia, T.D.; Chau, K.Y.; Inesta-Vaquera, F.; Papkovsky, D.B.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.R.; Hardy, J.; Schapira, A.H.; et al. G2019s leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Hu, Y. Mitochondrial dysfunction driven by the lrrk2-mediated pathway is associated with loss of purkinje cells and motor coordination deficits in diabetic rat model. Cell Death Dis. 2014, 5, e1217. [Google Scholar] [CrossRef]
- Fernandez-Mosquera, L.; Diogo, C.V.; Yambire, K.F.; Santos, G.L.; Luna Sanchez, M.; Benit, P.; Rustin, P.; Lopez, L.C.; Milosevic, I.; Raimundo, N. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep. 2017, 7, 45076. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Muqit, M.M. Pink1 and parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial respiration controls lysosomal function during inflammatory t cell responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef]
- Girotto, S.; Cendron, L.; Bisaglia, M.; Tessari, I.; Mammi, S.; Zanotti, G.; Bubacco, L. Dj-1 is a copper chaperone acting on sod1 activation. J. Biol. Chem. 2014, 289, 10887–10899. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of dj-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. Dj-1 acts in parallel to the pink1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and pgc-1alpha dysfunction in parkin-associated familial parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef]
- Colecchia, D.; Stasi, M.; Leonardi, M.; Manganelli, F.; Nolano, M.; Veneziani, B.M.; Santoro, L.; Eskelinen, E.-L.; Chiariello, M.; Bucci, C. Alterations of autophagy in charcot-marie-tooth type 2b. Autophagy 2018, 14, 930–941. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (nsc) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Haugland, R.P. The Handbook: A Guide to Fluorescent Probes and Labeling Technologies, 10th ed.; Molecular Probes; Invitrogen Corp.: Carlsbad, CA, USA, 2005; p. 390. [Google Scholar]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. Beta-catenin knockdown affects mitochondrial biogenesis and lipid metabolism in breast cancer cells. Front. Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Western blotting: An introduction. Methods Mol. Biol. 2015, 1312, 17–30. [Google Scholar] [PubMed]
- Richo, G.; Conner, G.E. Proteolytic activation of human procathepsin d. Adv. Exp. Med. Biol. 1991, 306, 289–296. [Google Scholar]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin a1 disrupts autophagic flux by inhibiting both v-atpase-dependent acidification and ca-p60a/serca-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Mauvezin, C.; Nagy, P.; Juhasz, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of v-atpase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Girolimetti, G.; Guerra, F.; Iommarini, L.; Kurelac, I.; Vergara, D.; Maffia, M.; Vidone, M.; Amato, L.B.; Leone, G.; Dusi, S.; et al. Platinum-induced mitochondrial DNA mutations confer lower sensitivity to paclitaxel by impairing tubulin cytoskeletal organization. Hum. Mol. Genet. 2017, 26, 2961–2974. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using polyphen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Gutta, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. Mtoolbox: A highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef]
- Rubino, F.; Piredda, R.; Calabrese, F.M.; Simone, D.; Lang, M.; Calabrese, C.; Petruzzella, V.; Tommaseo-Ponzetta, M.; Gasparre, G.; Attimonelli, M. Hmtdb, a genomic re- source for mitochondrion-based human variability studies. Nucleic Acid Res. 2012, 40, D1150–D1159. [Google Scholar] [CrossRef] [PubMed]
- Preste, R.; Vitale, O.; Clima, R.; Gasparre, G.; Attimonelli, M. Hmtvar: A new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res. 2019, 47, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (sa-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nature 2009, 4, 1798–1806. [Google Scholar]
- Vergara, D.; Ferraro, M.M.; Cascione, M.; del Mercato, L.L.; Leporatti, S.; Ferretta, A.; Tanzarella, P.; Pacelli, C.; Santino, A.; Maffia, M.; et al. Cytoskeletal alterations and biomechanical properties of parkin-mutant human primary fibroblasts. Cell Biochem. Biophys. 2015, 71, 1395–1404. [Google Scholar] [CrossRef]
- Vergara, D.; Gaballo, A.; Signorile, A.; Ferretta, A.; Tanzarella, P.; Pacelli, C.; Di Paola, M.; Cocco, T.; Maffia, M. Resveratrol modulation of protein expression in parkin-mutant human skin fibroblasts: A proteomic approach. Oxid. Med. Cell Longev. 2017, 2017, 2198243. [Google Scholar] [CrossRef]
- Lippolis, R.; Siciliano, R.A.; Pacelli, C.; Ferretta, A.; Mazzeo, M.F.; Scacco, S.; Papa, F.; Gaballo, A.; Dell’Aquila, C.; De Mari, M.; et al. Altered protein expression pattern in skin fibroblasts from parkin-mutant early-onset parkinson’s disease patients. Biochim. Biophys. Acta 2015, 1852, 1960–1970. [Google Scholar] [CrossRef]
- Diogo, C.V.; Yambire, K.F.; Fernandez Mosquera, L.; Branco, F.T.; Raimundo, N. Mitochondrial adventures at the organelle society. Biochem. Biophys. Res. Commun. 2018, 500, 87–93. [Google Scholar] [CrossRef]
- Wang, T.; Ming, Z.; Xiaochun, W.; Hong, W. Rab7: Role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal. 2011, 23, 516–521. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple roles of the small gtpase rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Vitelli, R.; Santillo, M.; Lattero, D.; Chiariello, M.; Bifulco, M.; Bruni, C.; Bucci, C. Role of the small gtpase rab7 in the late endocytic pathway. J. Biol. Chem. 1997, 272, 4391–4397. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.; Cogli, L.; Progida, C.; Nisi, V.; Pascolutti, R.; Sigismund, S.; Di Fiore, P.P.; Bucci, C. Rilp regulates vacuolar atpase through interaction with the v1g1 subunit. J. Cell Sci. 2014, 127, 2697–2708. [Google Scholar] [CrossRef]
- De Luca, M.; Bucci, C. A new v-atpase regulatory mechanism mediated by the rab interacting lysosomal protein (rilp). Commun. Integr. Biol. 2014, 7, 1–4. [Google Scholar] [CrossRef]
- Voss, E.W., Jr.; Workman, C.J.; Mummert, M.E. Detection of protease activity using a fluorescence-enhancement globular substrate. Biotechniques 1996, 20, 286–291. [Google Scholar] [CrossRef]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The role of cathepsin d in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. Lc3, a mammalian homologue of yeast apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/sqstm1 binds directly to atg8/lc3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret lc3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous lc3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef]
- Manley, S.; Williams, J.A.; Ding, W.X. Role of p62/sqstm1 in liver physiology and pathogenesis. Exp. Biol. Med. (Maywood) 2013, 238, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type i endometrial carcinoma. Hum. Mol. Genet. 2011, 20, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Romeo, G.; Rugolo, M.; Porcelli, A.M. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim. Biophys. Acta 2011, 1807, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M. Regulation of mitochondrial biogenesis through tfam-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging (Albany NY) 2013, 5, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T. Mtor as regulator of lifespan, aging, and cellular senescence: A mini-review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef]
- Enriquez, J.A. Supramolecular organization of respiratory complexes. Annu. Rev. Physiol. 2016, 78, 533–561. [Google Scholar] [CrossRef]
- Blakely, E.L.; Mitchell, A.L.; Fisher, N.; Meunier, B.; Nijtmans, L.G.; Schaefer, A.M.; Jackson, M.J.; Turnbull, D.M.; Taylor, R.W. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005, 272, 3583–3592. [Google Scholar] [CrossRef]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (q352x) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes i and iii. Neuromuscul. Disord. 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex i in mouse fibroblasts. Mol. Cell Biol. 2006, 26, 4872–4881. [Google Scholar] [CrossRef]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex i plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 5, ra47. [Google Scholar] [CrossRef]
- Cieri, D.; Brini, M.; Cali, T. Emerging (and converging) pathways in parkinson’s disease: Keeping mitochondrial wellness. Biochem. Biophys. Res. Commun. 2017, 483, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.E. No parkin zone: Mitophagy without parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via rab7 gtp hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef]
- Sinha, P.; Islam, M.N.; Bhattacharya, S.; Bhattacharya, J. Intercellular mitochondrial transfer: Bioenergetic crosstalk between cells. Curr. Opin. Genet. Dev. 2016, 38, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtdna mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [PubMed]
- Desdin-Mico, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adh. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Kruger, R. The use of primary human fibroblasts for monitoring mitochondrial phenotypes in the field of parkinson’s disease. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [PubMed]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary skin fibroblasts as a model of parkinson’s disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef]
- Ambrosi, G.; Ghezzi, C.; Sepe, S.; Milanese, C.; Payan-Gomez, C.; Bombardieri, C.R.; Armentero, M.T.; Zangaglia, R.; Pacchetti, C.; Mastroberardino, P.G.; et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 1385–1394. [Google Scholar] [CrossRef]
- Hoepken, H.H.; Gispert, S.; Azizov, M.; Klinkenberg, M.; Ricciardi, F.; Kurz, A.; Morales-Gordo, B.; Bonin, M.; Riess, O.; Gasser, T.; et al. Parkinson patient fibroblasts show increased alpha-synuclein expression. Exp. Neurol. 2008, 212, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Romani-Aumedes, J.; Canal, M.; Martin-Flores, N.; Sun, X.; Perez-Fernandez, V.; Wewering, S.; Fernandez-Santiago, R.; Ezquerra, M.; Pont-Sunyer, C.; Lafuente, A.; et al. Parkin loss of function contributes to rtp801 elevation and neurodegeneration in parkinson’s disease. Cell Death Dis. 2014, 5, e1364. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.; Bravo-San Pedro, J.M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Rodriguez-Arribas, M.; Climent, V.; Aiastui, A.; Lopez de Munain, A.; Fuentes, J.M.; Gonzalez-Polo, R.A. G2019s lrrk2 mutant fibroblasts from parkinson’s disease patients show increased sensitivity to neurotoxin 1-methyl-4-phenylpyridinium dependent of autophagy. Toxicology 2014, 324, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Dodson, M.; Ravi, S.; Redmann, M.; Ouyang, X.; Darley Usmar, V.M.; Zhang, J. Bioenergetic adaptation in response to autophagy regulators during rotenone exposure. J. Neurochem. 2014, 131, 625–633. [Google Scholar] [CrossRef]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Gene | Amino Acid Change | Polyphen2 Score | Nucleotide Variability |
|---|---|---|---|---|
| m.6076T>C | MT-COXI | V58A | 0.783 | 0.000277 |
| m.13676A>G | MT-ND5 | D447S | 0.51 | 0.0005 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells 2019, 8, 452. https://doi.org/10.3390/cells8050452
Guerra F, Girolimetti G, Beli R, Mitruccio M, Pacelli C, Ferretta A, Gasparre G, Cocco T, Bucci C. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells. 2019; 8(5):452. https://doi.org/10.3390/cells8050452
Chicago/Turabian StyleGuerra, Flora, Giulia Girolimetti, Raffaella Beli, Marco Mitruccio, Consiglia Pacelli, Anna Ferretta, Giuseppe Gasparre, Tiziana Cocco, and Cecilia Bucci. 2019. "Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease" Cells 8, no. 5: 452. https://doi.org/10.3390/cells8050452