Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Generation of iPS Cell and Culture
2.2. Immunohistochemistry
2.3. Live Cell Oxygen Consumption
2.4. Western Blot Analysis
2.5. Detection of Autophagosomes, Autolysosomes and Mitochondria
2.6. Reactive Oxygen Species (ROS) Detection Using a Fluorescent Agent MitoSox
2.7. Measurement of Mitochondrial Membrane Potential
2.8. Detection of Intracellular Calcium Mobilization
2.9. Measurement of Intracellular ATP Content
2.10. Cell Viability Assay
2.11. Statistical Analysis
3. Results
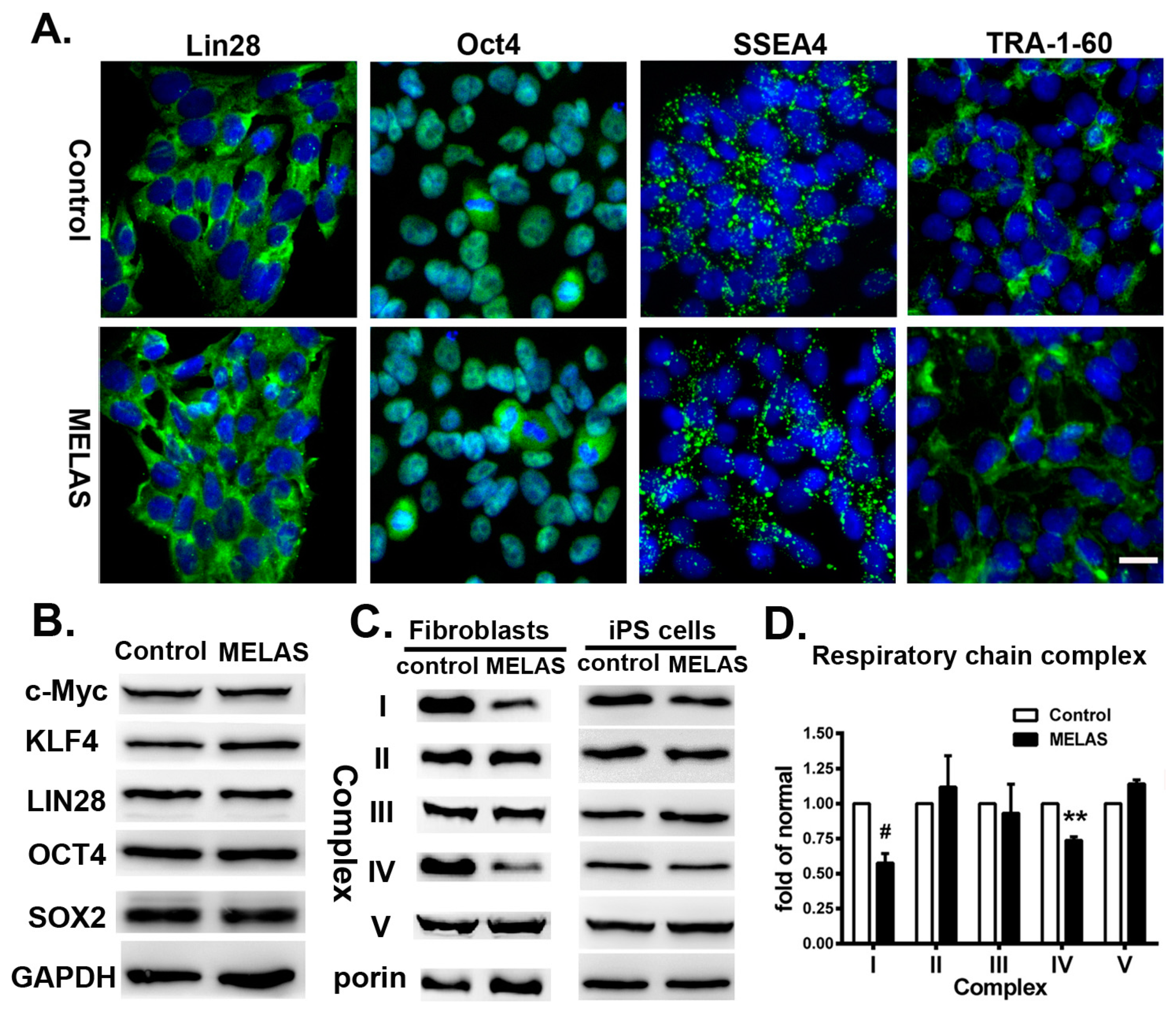
3.1. Identifying Cell Surface Markers and Respiratory Complexes for the iPS Cells
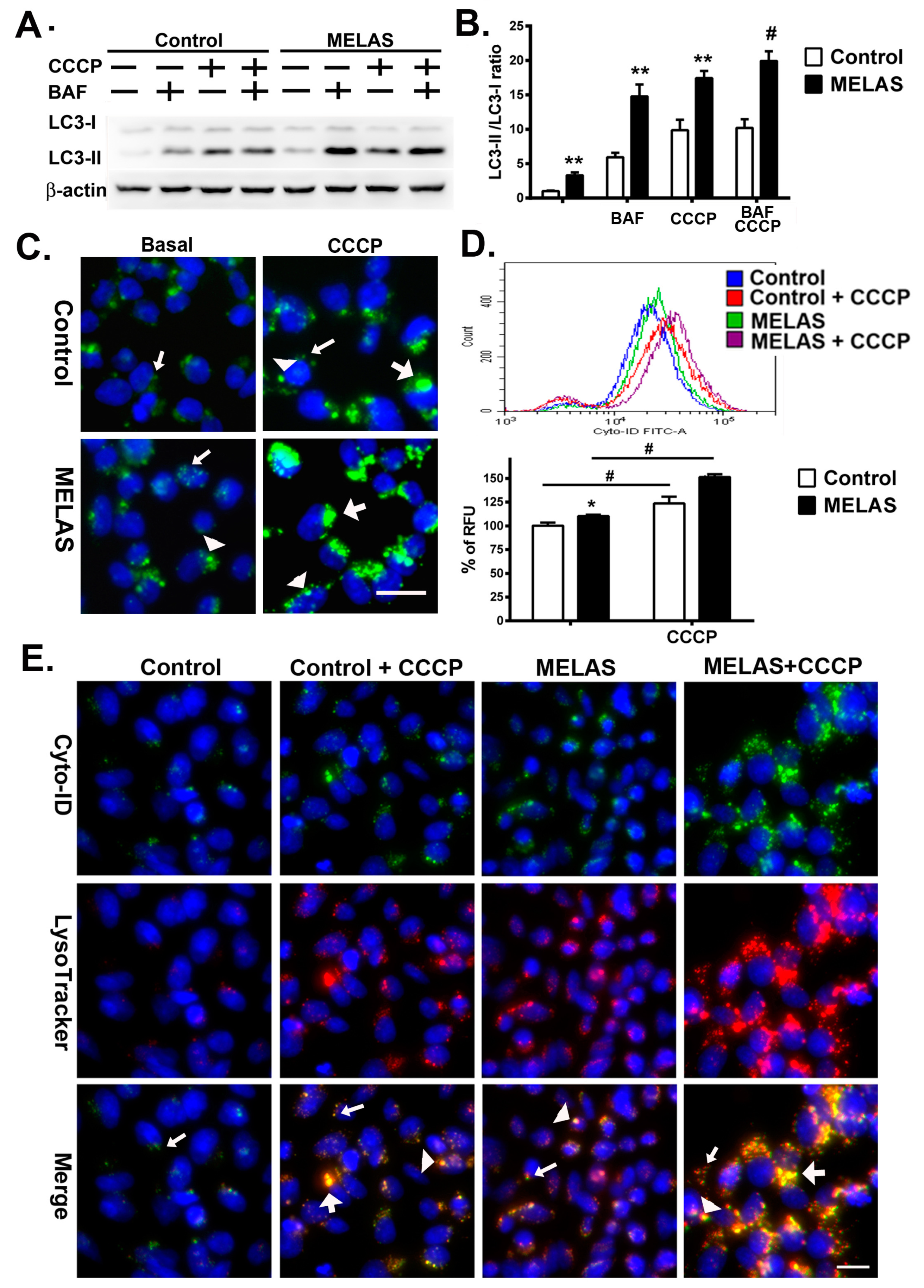
3.2. Enhanced Flux of Autophagy
3.3. Accumulation of Autophagosomes
3.4. Accumulation of Autolysosomes
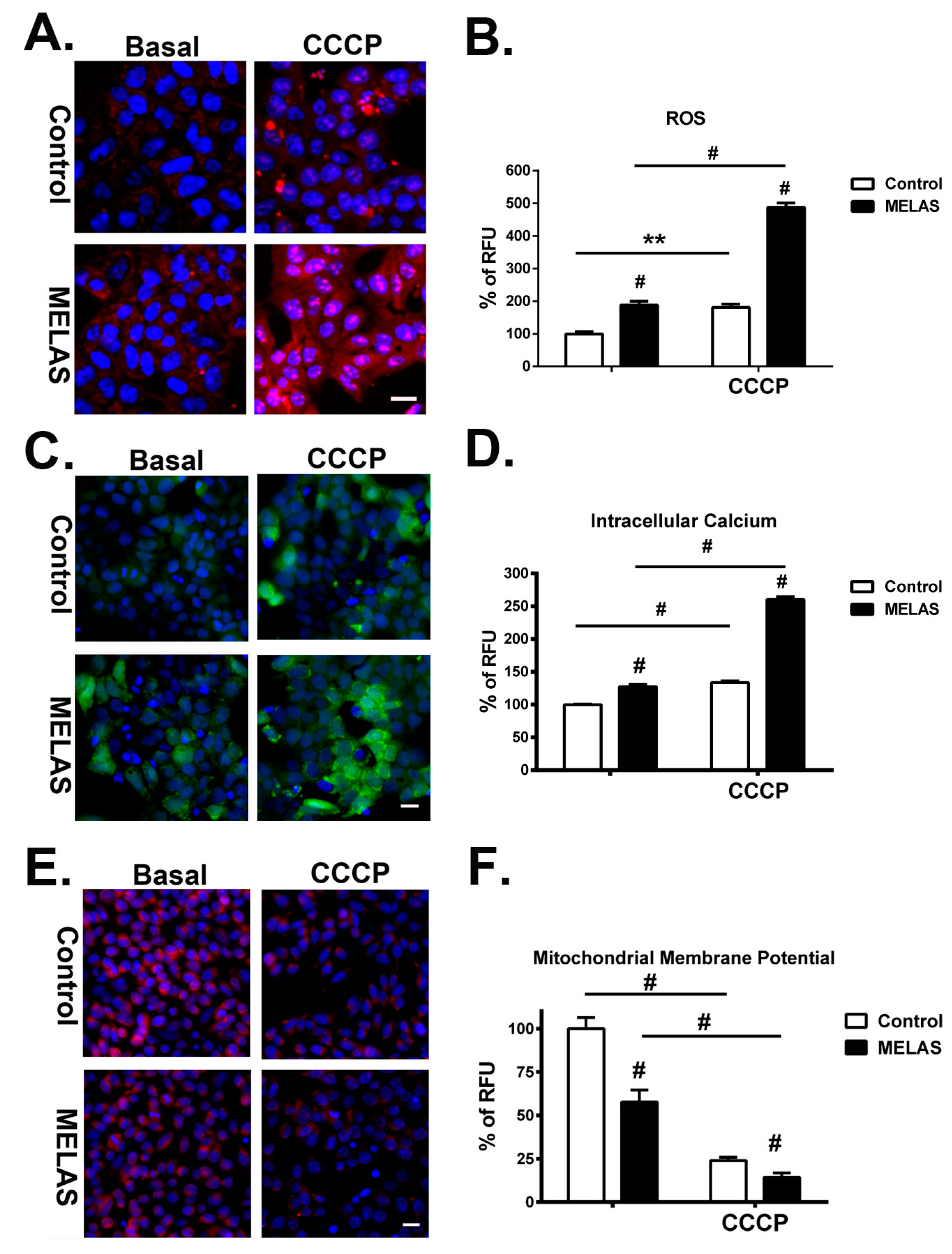
3.5. Remarked Increase of ROS
3.6. Overload of Cytoplasmic Calcium Flux
3.7. Robust Depolarization of Mitochondrial Membrane Potential
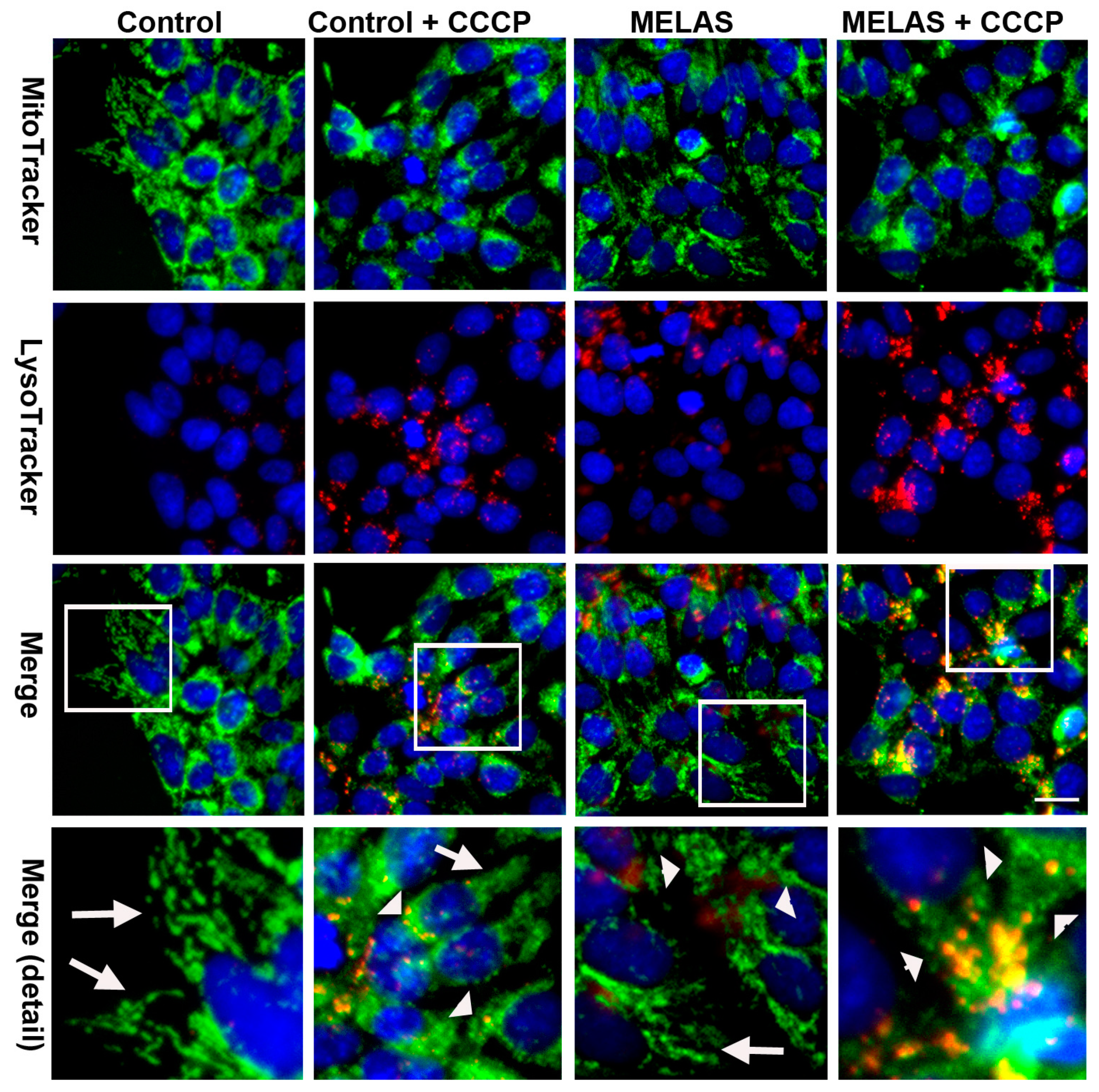
3.8. Activation of Mitophagy Upon Oxidative Insults
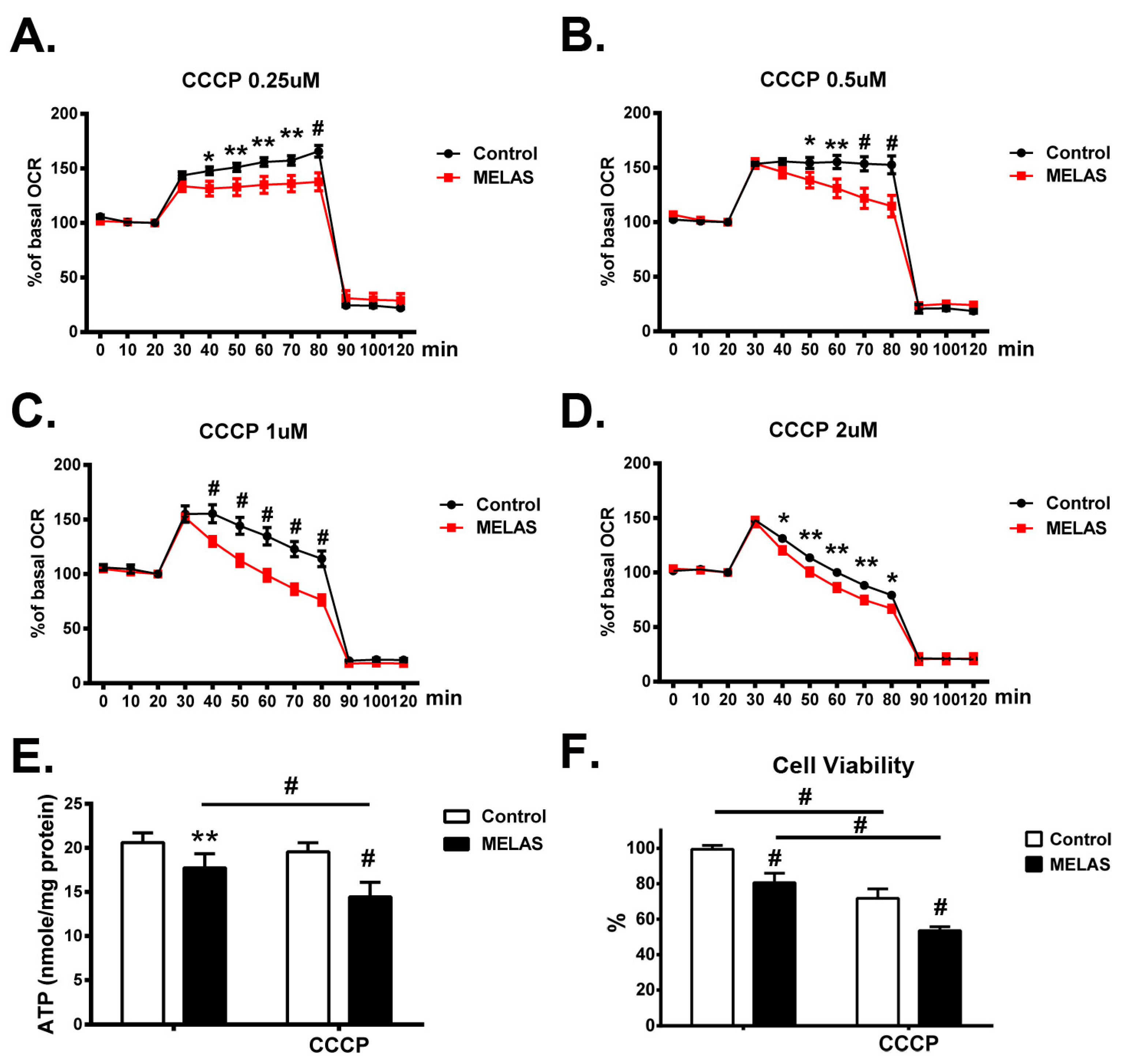
3.9. Decrease of Cellular Bioenergetics
3.10. Deficiency of Intracellular ATP Content
3.11. Decrease of Cell Viability
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S. Mitochondrial diseases. Biochim. Biophys. Acta 2004, 1658, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Kawai, H.; Sanada, M.; Terashima, T.; Ogawa, N.; Idehara, R.; Makiishi, T.; Yasuda, H.; Sato, S.; Hoshi, K.; et al. Clinical Phenotype and Segregation of Mitochondrial 3243A>G Mutation in 2 Pairs of Monozygotic Twins. JAMA Neurol. 2016, 73, 990–993. [Google Scholar] [CrossRef]
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, D.C. MITOMAP: A human mitochondrial genome database--2004 update. Nucleic Acids Res. 2005, 33, D611–D613. [Google Scholar] [CrossRef]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef]
- King, M.P.; Koga, Y.; Davidson, M.; Schon, E.A. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol. Cell. Biol. 1992, 12, 480–490. [Google Scholar] [CrossRef]
- Chomyn, A.; Enriquez, J.A.; Micol, V.; Fernandez-Silva, P.; Attardi, G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 2000, 275, 19198–19209. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Kao, S.H.; Ho, C.S.; Wei, Y.H.; Hung, P.L.; Hsu, M.H.; Wu, T.Y.; Wang, T.J.; Jian, Y.R.; Lee, T.H.; et al. Inflexibility of AMPK-mediated metabolic reprogramming in mitochondrial disease. Oncotarget 2017, 8, 73627–73639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotan, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Avila, M.; Rodriguez-Hernandez, A.; Gomez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011, 25, 2669–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Maraver, J.; Cordero, M.D.; Monino, I.D.; Pereira-Arenas, S.; Lechuga-Vieco, A.V.; Cotan, D.; De la Mata, M.; Oropesa-Avila, M.; De Miguel, M.; Bautista Lorite, J.; et al. Screening of effective pharmacological treatments for MELAS syndrome using yeasts, fibroblasts and cybrid models of the disease. Br. J. Pharmacol. 2012, 167, 1311–1328. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; De Vries, R.L.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef]
- Berezhnov, A.V.; Soutar, M.P.; Fedotova, E.I.; Frolova, M.S.; Plun-Favreau, H.; Zinchenko, V.P.; Abramov, A.Y. Intracellular pH Modulates Autophagy and Mitophagy. J. Biol. Chem. 2016, 291, 8701–8708. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Barteneva, N.S.; Ponomarev, E.D.; Tsytsykova, A.; Armant, M.; Vorobjev, I.A. Mitochondrial staining allows robust elimination of apoptotic and damaged cells during cell sorting. J. Histochem. Cytochem. 2014, 62, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.L.; Shen, D.; Wilkinson, A.R.; Patton, W.; Lai, N.; Chan, E.; Kuksin, D.; Lin, B.; Qiu, J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy 2012, 8, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Shlyahova, L. Cell nuclei generate DNA-nicking superoxide radicals. FEBS Lett. 1986, 194, 317–321. [Google Scholar] [CrossRef]
- Aboulmaouahib, S.; Madkour, A.; Kaarouch, I.; Sefrioui, O.; Saadani, B.; Copin, H.; Benkhalifa, M.; Louanjli, N.; Cadi, R. Impact of alcohol and cigarette smoking consumption in male fertility potential: Looks at lipid peroxidation, enzymatic antioxidant activities and sperm DNA damage. Andrologia 2018, 50, e12926. [Google Scholar] [CrossRef]
- Douiev, L.; Abu-Libdeh, B.; Saada, A. Cytochrome c oxidase deficiency, oxidative stress, possible antioxidant therapy and link to nuclear DNA damage. Eur. J. Hum. Genet. 2018, 26, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajnoczky, G.; Csordas, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006, 40, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Janssen, G.M.; Hensbergen, P.J.; van Bussel, F.J.; Balog, C.I.; Maassen, J.A.; Deelder, A.M.; Raap, A.K. The A3243G tRNALeu(UUR) mutation induces mitochondrial dysfunction and variable disease expression without dominant negative acting translational defects in complex IV subunits at UUR codons. Hum. Mol. Genet. 2007, 16, 2472–2481. [Google Scholar] [CrossRef] [Green Version]
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Folmes, C.D.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, M.; Hatakeyama, H.; Yuasa, S.; Seki, T.; Egashira, T.; Tohyama, S.; Kuroda, Y.; Tanaka, A.; Okata, S.; Hashimoto, H.; et al. Impaired respiratory function in MELAS-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio. 2015, 5, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Maraver, J.; Paz, M.V.; Cordero, M.D.; Bautista-Lorite, J.; Oropesa-Avila, M.; de la Mata, M.; Pavon, A.D.; de Lavera, I.; Alcocer-Gomez, E.; Galan, F.; et al. Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim. Biophys. Acta 2015, 1852, 2535–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender? J. Neurol. 2014, 261, 504–510. [Google Scholar] [CrossRef]
- Benit, P.; El-Khoury, R.; Schiff, M.; Sainsard-Chanet, A.; Rustin, P. Genetic background influences mitochondrial function: Modeling mitochondrial disease for therapeutic development. Trends Mol. Med. 2010, 16, 210–217. [Google Scholar] [CrossRef]
- De Andrade, P.B.; Rubi, B.; Frigerio, F.; van den Ouweland, J.M.; Maassen, J.A.; Maechler, P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia 2006, 49, 1816–1826. [Google Scholar] [CrossRef]
- Malena, A.; Pantic, B.; Borgia, D.; Sgarbi, G.; Solaini, G.; Holt, I.J.; Spinazzola, A.; Perissinotto, E.; Sandri, M.; Baracca, A.; et al. Mitochondrial quality control: Cell-type-dependent responses to pathological mutant mitochondrial DNA. Autophagy 2016, 12, 2098–2112. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.; Rivera, H.; Sanchez-Arago, M.; Blazquez, A.; Merinero, B.; Ugalde, C.; Arenas, J.; Cuezva, J.M.; Martin, M.A. Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts. Biochim. Biophys. Acta 2010, 1802, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.; Marin-Buera, L.; Gil-Borlado, M.C.; Rivera, H.; Blazquez, A.; Seneca, S.; Vazquez-Lopez, M.; Arenas, J.; Martin, M.A.; Ugalde, C. Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum. Mutat. 2010, 31, 930–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, M.; Delmiro, A.; Blazquez, A.; Ugalde, C.; Arenas, J.; Martin, M.A. Bulk autophagy, but not mitophagy, is increased in cellular model of mitochondrial disease. Biochim. Biophys. Acta 2014, 1842, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Shan, Y.; Lloyd, K.C.; Cortopassi, G.A. Mutant Twinkle increases dopaminergic neurodegeneration, mtDNA deletions and modulates Parkin expression. Hum. Mol. Genet. 2012, 21, 5147–5158. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Davies, V.J.; Hogan, V.E.; Piechota, M.J.; Nichols, P.P.; Turnbull, D.M.; Votruba, M. OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest. Ophthalmol. Vis. Sci. 2009, 50, 2567–2571. [Google Scholar] [CrossRef] [PubMed]
- Button, R.W.; Roberts, S.L.; Willis, T.L.; Hanemann, C.O.; Luo, S. Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef] [Green Version]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 334. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenerg. 2017, 1858, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, D.; Sun, Q.; Xu, L.; Lee, E.; Lewis, A.J.; Zuckerbraun, B.S.; Rosengart, M.R. Calcium/calmodulin-dependent protein kinase regulates the PINK1/Parkin and DJ-1 pathways of mitophagy during sepsis. FASEB J. 2017, 31, 4382–4395. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Narendra, D.P.; Tanaka, A.; Manfredi, G.; Youle, R.J. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11835–11840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laar, V.S.; Arnold, B.; Cassady, S.J.; Chu, C.T.; Burton, E.A.; Berman, S.B. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 2011, 20, 927–940. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; P, D.A.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604 e585. [Google Scholar] [CrossRef]
- Lim, G.G.; Lim, K.L. Parkin-independent mitophagy-FKBP8 takes the stage. EMBO Rep. 2017, 18, 864–865. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Hung, P.-L.; Hsu, M.-H.; Wang, T.-J.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Chang, P.-C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65. https://doi.org/10.3390/cells8010065
Lin D-S, Huang Y-W, Ho C-S, Hung P-L, Hsu M-H, Wang T-J, Wu T-Y, Lee T-H, Huang Z-D, Chang P-C, et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells. 2019; 8(1):65. https://doi.org/10.3390/cells8010065
Chicago/Turabian StyleLin, Dar-Shong, Yu-Wen Huang, Che-Sheng Ho, Pi-Lien Hung, Mei-Hsin Hsu, Tuan-Jen Wang, Tsu-Yen Wu, Tsung-Han Lee, Zo-Darr Huang, Po-Chun Chang, and et al. 2019. "Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease" Cells 8, no. 1: 65. https://doi.org/10.3390/cells8010065