NFKB1 and Cancer: Friend or Foe?


Newcastle Fibrosis Research Group, Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, Tyne and Wear NE2 4HH, UK
*
Author to whom correspondence should be addressed.
Cells 2018, 7(9), 133; https://doi.org/10.3390/cells7090133
Submission received: 15 August 2018
/
Revised: 30 August 2018
/
Accepted: 4 September 2018
/
Published: 7 September 2018
(This article belongs to the Special Issue NF-κB in Cancer)
Abstract
:Current evidence strongly suggests that aberrant activation of the NF-κB signalling pathway is associated with carcinogenesis. A number of key cellular processes are governed by the effectors of this pathway, including immune responses and apoptosis, both crucial in the development of cancer. Therefore, it is not surprising that dysregulated and chronic NF-κB signalling can have a profound impact on cellular homeostasis. Here we discuss NFKB1 (p105/p50), one of the five subunits of NF-κB, widely implicated in carcinogenesis, in some cases driving cancer progression and in others acting as a tumour-suppressor. The complexity of the role of this subunit lies in the multiple dimeric combination possibilities as well as the different interacting co-factors, which dictate whether gene transcription is activated or repressed, in a cell and organ-specific manner. This review highlights the multiple roles of NFKB1 in the development and progression of different cancers, and the considerations to make when attempting to manipulate NF-κB as a potential cancer therapy.
1. Introduction
One of the emerging questions in cancer biology is: “How are inflammation and dysregulated immune responses linked to cancer?” It is now widely accepted that chronic inflammation and infection represent major risk factors for certain cancers. Increasing evidence suggests that the cellular effectors and signalling pathways involved in these pathological contexts play a substantial role in defining both the development and progression of cancer. Among those, NF-κB transcription factors play a fundamental role in the regulation of immune responses, apoptosis, and cell survival, and hence, are implicated as central mediators of cancer initiation and progression. Examples include strong links to inflammation-driven cancers such as colitis-associated cancer, MALT (mucosal-associated lymphoid tissue) lymphoma, and HBV (hepatitis B virus) or HCV (Hepatitis C virus) driven HCC (hepatocellular carcinoma) [1]. Whilst mutations in the upstream activators and individual subunits of NF-κB are rare, it is far more likely that increased and even consistent NF-κB activation is promoted by inflammatory stimuli, the source of which is likely to vary widely from underlying inflammatory conditions (i.e., viral hepatitis and EBV (Epstein-Barr virus), cellular senescence and senescence associated secretory proteins and immune activation by malignant cells) [2,3]. Indeed, dynamic cross-talk between malignant and immune cells further promote this activation to support tumour growth and survival [4]. NF-κB activation leads to the NF-κB-dependent transcription of genes coding for inflammatory cytokines, cell-cycle modulators, survival signals, and growth and angiogenic factors, all key drivers in a tumour-promoting environment. Due to the vast array of proteins under the control of NF-κB, attempts to inhibit this process with blanket inhibitors of upstream effectors such as IκB Kinase (IKK) inhibitors have had limited success, compounded by the off-target effects [5]. Balance between NF-κB activation and control is lost during pathological conditions such as chronic inflammation and cancer. To understand and more importantly regain control over this aberrant NF-κB activation, there is a need to understand more about de-activating NF-κB at both a cell-specific and molecular level (i.e., the NF-κB subunits).
2. NF-κB Subunits
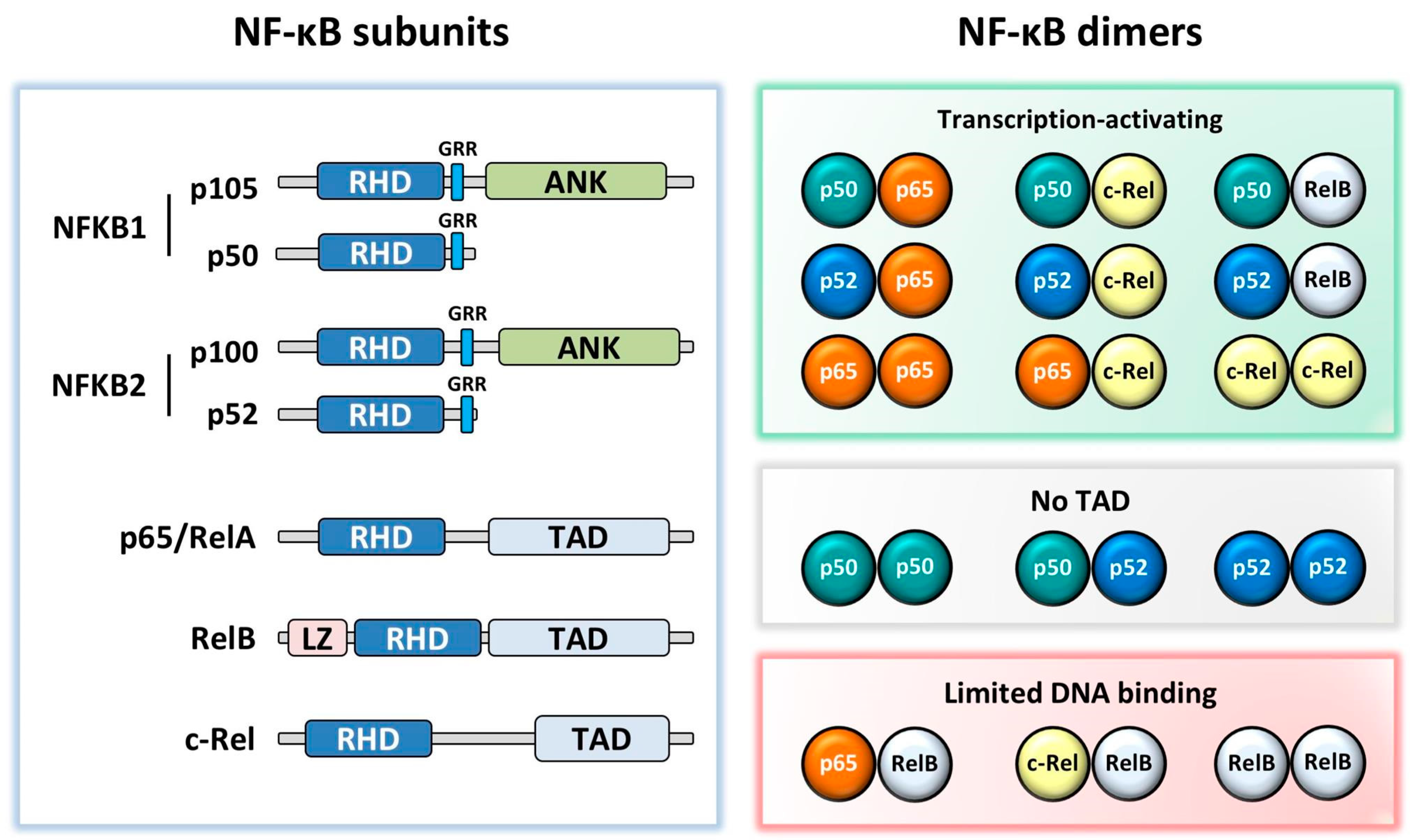
NF-κB consists of five subunits: p65 (RelA), RelB, c-Rel, p50 (NFKB1), and p52 (NFKB2). Unlike the other subunits, NFKB1 and NFKB2 are synthesized as precursors (p105 and p100) which are proteolytically cleaved to p50 and p52, respectively. The NF-κB subunits, which form homo- and hetero-dimers, are kept inactive in the cytoplasm by inhibitor of κB proteins (IκB); phosphorylation and degradation of IκB through activation of the NF-κB signalling pathway leads to the translocation of NF-κB dimers into the nucleus. These bind to target gene κB promoter sites via their Rel homology domain. Whilst the most well-known and commonly studied NF-κB dimer is p50:p65, NF-κB exists in several other dimer combinations with different and important functional outputs [6]. p65, RelB, and c-Rel all contain a transcriptional activation domain (TAD) enabling them to drive gene transcription in their homodimer form. However, this TAD is absent in p50 and p52, therefore they must either form a heterodimer with p65, c-Rel or RelB, or recruit co-activators to drive gene transcription, and when bound to DNA as homodimers they are generally thought to be transcriptional repressors (Figure 1) [7].
3. NFKB1 Processing
The NFKB1 gene, which comprises 24 exons, is located on chromosome 4q24 [11]. Upon stimuli, p105 is phosphorylated in its C-terminal domain containing an ankyrin repeat (similar to that of the IκB inhibitory proteins), followed by partial degradation in an ubiquitin-proteasome dependent manner. The degradation of the inhibitory C-terminal region subsequently leads to p50 activation [12]. Distinct from the role of p50, p105 also plays an important role in the stabilisation of pre-formed NF-κB dimers. In mice where p105 serine residues 927 and 932 are mutated to alanine, there is impaired p105 processing and as a result p50:p65 release and activity is inhibited [13,14]. Aside from its direct role in the control of NF-κB activation, p105 also plays an additional role in stabilising Tpl2 kinase (tumour progression locus 2). p105 inhibits Tpl2-mediated activation of the MEK/ERK (mitogen-activated protein kinase kinase/extracellular regulated kinase) pathway and this inhibition is abrogated following IKK-induced proteolysis of p105 [15]. The role of p105 in the regulation of this pathway has been extensively reviewed [16].
4. Co-Factor Recruitment
p50 homodimers repress inflammation by competing with activating NF-κB dimers and preventing them from binding to κB sites on the promoters of target genes, further strengthened by the recruitment of co-repressors. Histone deacetylases (HDAC) repress gene transcription by removing acetyl groups from histones, often leading to chromatin condensation and transcriptional repression. The p50:p50:HDAC1 complex is able to repress multiple inflammatory genes, including GM-CSF, CCL2, CXCL-10, and MMP-13 [17]. We have recently identified the site of p50:HDAC1 interaction and shown that mutagenesis of this binding site on p50 prevents HDAC1 binding with p50 homodimers. As a result of this loss of P50:HDAC1 interaction, we observed increased expression of the inflammatory genes CXCL1, CXCL2, and IL-6 both basally and in response to stimulus (in press).
The IκB family protein B cell lymphoma 3 (Bcl-3) plays a dual role in p50 transcriptional regulation. As a co-activator it can bind p50 via ankyrin repeats and help drive gene transcription when complexed with p50 homodimers, and is also able to recruit other transcriptional regulators including STAT1, AP-1, c-Jun, and MMP-13 c-fos [18]. However, Bcl-3 can also stabilize the p50 homodimer by masking ubiquitination sites and preventing removal from gene promoters [19]. Therefore, Bcl-3 can also function as a co-repressor of gene transcription by preventing transcriptionally active NF-κB dimers from binding to these gene promoters and driving transcription [19]. Indeed, following LPS (lipopolysaccharide) stimulation of macrophages, p50:p50:Bcl-3 complexes negatively regulate TNFα (tumour necrosis factor α), IL-1α, and IL-1β expression, while activating anti-inflammatory IL-10 gene transcription. In this context Bcl-3 enhances p50 homodimer-dependent TNFα repression, as p50 is also able to repress TNFα in the absence of Bcl-3, though to a lesser extent [20]. However, it is highly unlikely that these complexes are as simple as this; it is far more realistic to view these known protein interactions as parts of the jigsaw when in reality they are likely to form large multimeric complexes consisting of numerous co-factors. In support of this, both HDAC1 and Bcl-3 have also been found in nuclear complexes together in LPS stimulated cells [20].
Other factors known to interact with and modulate p50 homodimer activity include C/EBP (CCAAT enhancer binding protein) which has been shown to displace HDAC1 and HDAC3 bound to p50 homodimers, abrogating anti-apoptotic gene repression and contributing to aberrant p50 activity in acute myeloid leukemia [21]. Also, the co-activator p300 regulated by PARP (Poly (ADP-ribose) polymerase) is necessary for p50 transcriptional activation in primary lung fibroblasts following TNFα and LPS stimulation [22].
The dual ability of p50 to both activate and repress gene transcription instigates a complex relationship between this NF-κB subunit and cancer, with much controversy as to its role played in carcinogenesis. Given the complex and diverse nature of cancer as a disease, it is not surprising that the function of NFKB1 can differ substantially in a cell, organ, and cancer specific manner. In this review, we will discuss how NFKB1 is implicated in carcinogenesis, acting both as a pro- and anti-tumorigenic transcription factor.
5. NFKB1 Tumour Suppressor
p50 homodimers are primarily considered to repress gene transcription, dampen inflammatory responses, and abrogate anti-apoptotic signalling [17,23,24]. Chronic inflammation and the evasion of apoptosis are hallmarks of cancer [25], hence it is no surprise that there is an abundance of emerging evidence supporting a role for p50 homodimers as tumour-suppressors. KPC1 (KIP1 ubiquitination-promoting complex 1) was recently identified as the ubiquitin ligase that mediates the processing of p105 to p50. Kravtsova-Ivantsiv and colleagues [26] showed that KPC1 overexpression in a mouse xenograft tumour model derived from either the glioblastoma cell line (U87-MG cells) or the human breast cancer cell line (MDA-MB 231 cells) inhibits tumour growth via increased p50 generation. This was further supported by p50 overexpression that also reduced the growth of both tumours. Interestingly, p65 expression was significantly decreased in KPC1 and p50 overexpressing tumours, and an increase in the formation of p50 homodimers over p50-p65 heterodimers was confirmed by EMSA. The increase in p50 homodimer formation resulted in downregulated genes including HMGI-C, lin-28 homolog A, IL-6, IL-6R and VEGFA and an upregulation of tumour suppressor genes including PTEN (Phosphatase and tensin homolog). Moreover, in support of this, in a human context both p50 and KPC1 expression was significantly decreased in human head, neck, and glioblastoma tumours compared to normal tissue, and p50 expression decreased in breast cancer tumours [26]. Together their findings strongly suggest that KPC1, and hence p50, inhibit the growth of various tumours, likely via inhibition of p50:p65-mediated pro-tumorigenic gene transcription.
5.1. Liver Cancer
The role of NF-κB in liver cancer has been extensively studied and its first role in liver homeostasis highlighted in p65 deficient mice which suffer embryonic lethality due to TNFα-induced hepatocyte death during development [27]. Contrasting and cell-specific effects of IKK deletion in liver epithelial versus myeloid cells highlight the complexity of its role at different stages of HCC development. Knockdown of IKKβ in hepatocytes leads to increased liver cancer, whereas knockdown in myeloid cells results in significantly decreased hepatocarcinogenesis [28]. These studies of course merely highlight the implications of inhibiting the entire canonical NF-κB pathway which researchers are beginning to appreciate is far more complex and requires further refinement if we are to manipulate it in a more specific and productive way, including a more in-depth understanding of the epigenetic regulation and post-translational modifications of the individual subunits.
We have previously provided evidence that NFKB1, acting through p50 homodimers, is a suppressor of neutrophil-driven hepatocellular carcinoma (HCC). We previously demonstrated using a model of diethylnitrosamine (DEN)-induced HCC that Nfkb1−/− mice exhibited accelerated HCC and significantly increased tumour numbers compared to wild type mice. This was mediated in part by p50 homodimers complexed with HDAC1 repressing the hepatic expression of the neutrophil chemokines S100A9, CXCL1, and CXCL2. Treatment with a neutrophil-depleting antibody (anti-Ly6G) reduced tumour growth considerably in Nfkb1−/− mice. Furthermore, we identified that the phosphorylation of Ser342 is critical for p50 homodimer assembly, and that Nfkb1−/− mice with a serine to alanine mutation at position 342(S342A) display increased neutrophil numbers, neutrophil chemokine expression, and increased tumour burden in the DEN HCC mouse model. Combined this data confirmed that p50 homodimers in complex with HDAC1 play an important role in preventing liver inflammation and tumorigenesis by repressing neutrophil recruitment and as a result, neutrophil tumour-promoting effects. Interestingly, in a non-experimentally-induced context, aged 20 month old Nfkb1−/− mice also develop spontaneous chronic liver disease and liver cancer characterised by dysplastic nodules, increased tumour incidence, features of steatohepatitis and fibrosis [23].
In HCC patients where ~90% of cancers develop on a background of aberrant liver inflammation, we have also confirmed that an increase in neutrophil to lymphocyte ratio is associated with a poorer prognosis and worsened survival outcome [29]. Furthermore, mRNA, miRNA, and methylation profiling of 337 HCC patients identified NFKB1 and the MAPK (mitogen-activated protein kinase) pathway as important players in HCC progression. Here, NFKB1 inhibition was mediated via the miRNA let-7a at all stages of HCC, resulting in aberrant target gene regulation including Gadd45β and dysregulated cell proliferation and apoptosis. In addition, NFKB1 was differentially methylated in stage II and III HCC, but not stage IV [30]. The miRNA silencing of NFKB1 and its differential methylation could therefore be a potential mediator of HCC development; however, further research is needed in order to elucidate the specific role of p50 in different stages of HCC including liver inflammation and disease, and cancer initiation and progression. Whilst contrasting studies exist including correlating p50 expression with early recurrence of HCC and Akt phosphorylation, p65 expression was not assessed here [31]. Increased p50 and Bcl-3 co-expression in tumours compared to adjacent tissue has also been described [32]. However, these studies simply describe p50 expression levels and fail to address functionality and mechanism.
5.2. Gastric Cancer
Drivers of gastric cancer (GC) include poor diet, obesity, and chronic infection. One of the main infection-driven gastric cancers is caused by Helicobacter pylori. Helicobacter Pylori produces a number of virulence factors able to activate the NF-κB pathway, which in turn lead to the recruitment of inflammatory cells and the production of inflammatory mediators that help drive tumorigenesis [33]. In conjunction with liver cancer studies the complexity of NF-κB activation in gastric tumorigenesis has also been highlighted using IKK cell-specific knockouts. IKKβ−/− in the gastric epithelial cell compartment leads to accelerated development of dysplasia, whereas knockout in only myeloid cells inhibited progression to gastric atrophy and dysplasia [34]. However, also in correlation with liver cancer development, mice lacking Nfkb1 develop spontaneous invasive GC. Here the Nfkb1−/− mice display characteristics of gastric cancer in humans including T cell and NK cell infiltration, increased pro-inflammatory gene expression including IL-1β, IL-6, TNFα and osteopontin, and the metalloproteinases MMP-7, MMP-9 and MMP-13. Mechanistically, this is explained by aberrant JAK-STAT (Janus kinase-Signal transducer and activator of transcription) signalling via increased pro-inflammatory mediators, STAT1 expression and increased expression of the immune checkpoint inhibitor PD-L1 [35]. Importantly, in this model Nfkb1 expression was required in both the epithelial and immune cell compartments in order to prevent GC. The findings of this study are consistent with human GC patient data linking NFKB1 gene polymorphisms, including rs28362491, with reduced p105/p50 expression and GC development [36,37]. In contrast, both NFKB1 and p65 have been shown at higher levels in GC cell lines and primary human GC tumours when compared to normal gastric epithelial cells which correlated with poor survival in patients [38]. Mechanistically, it was revealed that the NFKB1 targeting miRNA miR-508-3p was downregulated in GC cells, suggesting a tumour-suppressive function for this miRNA [38]. This reiterates the potentiality that p50:p65 dimers are pro-tumorigenic drivers as opposed to p50 homodimers in this context.
5.3. Lung Cancer
Aside from the suppressive effects of p50 homodimers, p105 itself can also exert a tumour-suppressive function. A recent study highlighted a role for p105 as a tumour suppressor via stabilisation of Tpl2. In a urethane-induced lung cancer mouse model which re-enacts human lung cancers associated with tobacco smoking, both Nfkb1−/− and Tpl2−/− mice showed a significant increase in lung tumour size and number when compared to wild type mice. Following the observation that Tpl2 expression was also decreased in a NFKB1−/− human lung cancer cell line, Fan Sun and colleagues [39] demonstrated that re-expressing p105, and not p50, restored Tpl2 levels in these cells, resulting in reduced tumour growth. Contrastingly, Tpl2 knockdown increased lung tumour cell growth. This study highlights the importance of p105 stabilisation of Tpl2 in suppressing lung carcinogenesis [39]. Analysis of human non-small-cell lung carcinoma (NSCLC) tumours graded from stage 1 to 3A revealed that p105 stromal and epithelial tumour cell expression was a positive prognostic indicator of disease-specific survival [40]. However, it has also recently been shown that co-expression of p65 and phosphorylated p105 is associated with poor prognosis in NSCLC, whereas expression of p65 or phosphorylated p105 alone was not associated with this [41]. Again, this strongly suggests that it is p50:p65 dimer activity and not p50 homodimer activity that is detrimental in NSCLC.
5.4. Haematological Malignancies
mRNA expression analysis has revealed that NFKB1 is downregulated in multiple human haematological malignancies, including T and B-cell lymphoma and acute myeloid leukemia, while p65 expression is upregulated. DNA alkylation damage leads to increased lymphomas in Nfkb1−/− mice when compared with wild type mice. Mice heterozygous for Nfkb1 exhibit an intermediary phenotype whilst maintaining p50 function, indicating the haploinsufficient nature of Nfkb1 as a tumour suppressor [42]. Interestingly, p50 phosphorylation by ATR (ataxia telangiectasia mutated and Rad3-related kinase) at serine 329 ensures the elimination of replication-associated DNA-damaged cells and is necessary for genome maintenance. Phosphorylation of p50 at this site during the S phase of the cell cycle inhibits its activity and DNA binding, downregulating the expression of the anti-apoptotic protein Bcl-xL sensitizing cells to DNA strand breaks. Transfection of Nfkb1−/− MEFs (mouse embryonic fibroblasts) with S329A mutant p50 prevents p50 phosphorylation and inhibition, and blocks the observed decrease in Bcl-xL expression during S phase [43]. Similarly, it has also been shown that S329 p50 phosphorylation is involved in promoting apoptosis in cells that have undergone O6-methylguanine (O6-MeG) DNA lesions, by preventing the expression of anti-apoptotic genes [24]. Therefore, p50 may play a protective role in preventing the survival of DNA-damaged cells that have the potential to become cancerous. In support of this, it was shown by EMSA that in adult T-cell leukemia (ATL) patients, NF-κB-DNA complexes consist of both p50:p65 heterodimers and p50 homodimers, whereas in healthy patients they are predominantly p50 homodimers. Moreover, only p50:p65 dimers, and not p50 homodimers, were able to activate the transcription of IL-2Rα in ATL patients, demonstrating that constitutive activation of p50:p65 is a major driver of ATL [44]. These studies highlight that aberrant p50:p65, rather than p50:p50 signalling could be implicated in haematological malignancies where DNA-damaged cells have evaded apoptosis.
6. NFKB1 Tumour Promoter
In contrast to p50:p50 homodimers, an increase in DNA bound p50:p65 heterodimers leads to increased expression of NF-κB regulated genes including pro-inflammatory (IL-1β, TNFα, CXCL1), anti-apoptotic (Bcl-xL, Bcl-2, Gadd45β) and proliferative (IL-6, GM-CSF) to name a few. Therefore, it is not surprising that NFKB1 acts as a tumour promoter in this context. In addition, there are also a number of cancers including lymphomas and colorectal cancer where recruitment of co-activators will actually promote the expression of a similar array of tumour-promoting genes.
6.1. Bcl-3-Associated Cancers
The aforementioned Bcl-3 complexed with p50 homodimers is associated with tumorigenesis in several malignancies [45]. Notably, classical Hodgkin/Reed-Sternberg (HRS), anaplastic large cell lymphoma (ALCL) and T cell lymphomas, exhibit constitutive p50 homodimer activity associated with Bcl-3, verified by EMSA and IP shift [45]. Additionally, increased p50 and Bcl-3 expression in HRS and ALCL cell lines are mainly in the nuclear fractions verified by co-immunoprecipitation and is associated with increased expression of the anti-apoptotic genes Bcl-xL, c-IAP2, and TRAF-1 in the HRS cell lines [45].
In accordance with this, several other studies confirm that increased tumour expression of p50 (and not p65) correlates with an increased expression of Bcl-3. For instance, mouse skin papillomas and squamous cell carcinomas (SCC) display increased p50 and p52 expression compared to normal epithelial tissue from the middle stages of cancer development, while p65 levels remained unchanged. This correlates with increased Bcl-3 expression in late stage skin papillomas and SCC, suggesting a role for the p50:p50:Bcl-3 complex in tumour promotion [46]. Other examples of co-expression in malignant tissue include HPV16 (human papilloma virus 16) positive oral cancers, nasopharyngeal carcinoma and breast cancer [47,48,49]. Together these findings suggest that p50 homodimers in complex with Bcl-3 may play an important role in carcinogenesis, likely via the upregulation of NF-κB target genes. Importantly, to verify where p50:p50:Bcl3 complexes truly function as tumour promoters in-depth molecular analysis such as EMSA, nuclear localisation, IP and analysis of other NF-κB subunits should be included.
6.2. Breast and Gynaecological Cancers
Studies involving breast and gynaecological cancers suggest an association with increased NFKB1 expression and nuclear localisation. Increased p50 DNA binding and to a lesser extent p65 binding has been observed in a subset of high-risk estrogen receptor positive breast cancers [50]. Also, knock-down of NFKB1 in the inflammatory breast cancer cell line (SUM-149) suggests that NFKB1 expression positively regulates Rho C expression and cell motility, which may contribute to the metastatic phenotype of inflammatory breast cancer [51]. Overexpression of p105 along with p100 in cervical carcinoma-derived keratinocytes expressing the HPV16 oncoproteins E7 or E6, show localisation is predominantly cytoplasmic when E7 is expressed, and nuclear in the case of E6; therefore, these viral oncoproteins may actually dictate the subcellular localisation of p105 and p100 and hence activity of p105 and p100 [52]. Contrasting results exist in cervical cancer describing both increased nuclear p50 and p65 correlating with poor tumour grade and larger tumour size, and increased expression of predominantly p50 homodimers with no observed increase in p65 [53,54]. Whereas in ovarian cancer it is both p50 and p65 that are increased when compared to borderline and benign tumours, indicating the involvement of p50:p65 heterodimers rather than p50 homodimers [55].
However, caution must be applied when thinking about mechanism and the cell-specific implications of NF-κB. Whilst there are many studies focused on subunit expression in tumours, specifically that of p50 and p65, as suggested earlier, epithelial versus immune cell NF-κB activation could have very different outputs. For instance, p50 and not p65 is overexpressed in tumour-associated macrophages from human ovarian cancer. As a result, p50:p50 homodimers repress the activation signals required for M1 polarisation leading to defective IL-12 production. When Nfkb1−/− mice are implanted with murine fibrosarcoma there is no observed defect in M1 cytokine production and tumour growth is significantly reduced. The Nfkb1−/− mice also show enhanced CXCL-10 chemokine expression and hence CD4+ and CD8+ T cell infiltration that help combat tumour growth in this model [56]. Thus, highlighting the importance of dissecting cell-specific roles of NFKB1 if we are to attempt to manipulate its function as a potential future therapy for inflammation and cancer.
6.3. Colorectal and Pancreatic Cancers
Similar to the previously mentioned study [56], it has also been recently described that p50 homodimers impair macrophage M1 polarisation, driving the development of colorectal cancer [57]. In this study, Nfkb1−/− mice displayed fewer colorectal tumours and increased expression of IL-12 and CXCL-10, supporting the idea that p50 homodimers repress these genes in macrophages, orchestrating their pro-tumorigenic phenotype [57]. BAG-1 (Bcl-2 associated athanogene) is an anti-apoptotic protein highly expressed in pre-malignant and colon cancer tissue. The interaction between BAG-1 and p50 homodimers has been demonstrated in both colorectal epithelial cells and in the colorectal carcinoma cell line HCT116 with knockdown of either protein leading to cell death. This complex is present at both the EGFR and the COX2 gene (PTGS2) promoters. Furthermore, the complex is able to differentially regulate gene expression by suppressing transcription of EGFR and promoting COX-2 transcription, both known to promote colon cancer [58].
Increased p50 signalling is also implicated in pancreatic carcinogenesis. Annexin A2 (ANXA2), is a calcium-dependent phospholipid binding protein involved in the progression and metastasis of a number of tumours and is shown to interact and translocate to the nucleus in a complex with p50 [59]. Increased NF-κB activity is observed in both resting and TNFα stimulated pancreatic cancer cells (Mia-Paca2). Furthermore, TNFα treatment of HeLa cells expressing ANXA2 leads to the induction of several NF-κB target genes linked to anti-apoptotic signalling and drug resistance in cancer, including GM-CSF, IL-1β, IL-6 and Gadd45β. This suggests a link between increased ANXA2 expression and p50 interaction leading to increased anti-apoptotic gene expression and drug resistance in pancreatic cancer cells [59]. However, in a mouse pancreatic tumour model, increased tumour growth was observed when pancreatic cancer cells were co-injected with p50−/− compared to wild type PSC (pancreatic stellate cells), suggesting a potential role for PSC p50 in pancreatic prevention [60]. Thus, NFKB1 plays very contrasting roles in the development of colon and pancreatic cancers, and further research is needed to elucidate its cellular and context-dependent function.
6.4. Haematological Malignancies
In an Eμ-TCL1 mouse model of chronic lymphocytic leukemia, the incidence of leukemia is significantly lower in mice lacking Nfkb1, with CD19/CD5+ B cell numbers decreased despite no difference in overall survival. Nfkb1+/− TCL1 mice still show a significant reduction in leukemia incidence, demonstrating that even a partial reduction in Nfkb1 can impede disease development [61]. It is known that intrinsic B cell defects are characteristic of mice deficient in Nfkb1, including proliferative defects and impairment in antibody class-switching [62]. Therefore, NFKB1 could be directly linked to B cell proliferation in chronic lymphocytic leukemia, characterised by an abnormally increased level of B cells in the lymph nodes, bone marrow, and blood [63].
Diffuse large B-cell lymphoma (DLBCL) has two distinct molecular subtypes: germinal centre B cell like (GCB) and activated B cell like (ABC). ShRNA silencing of either p105 or p100 in two lymphoma cell lines identified a set of distinctly regulated genes that were later confirmed in primary DLBCL samples. Pathway analysis identified patterns of predominantly p105 signalling associated with the GCB lymphoma subtype, whereas p100 was associated with ABC potentially as a consequence of mutations in upstream activators of either the canonical or non-canonical pathway respectively [64]. Moreover, in a study conducted on 465 patients, p50 activation in ABC DLBCL was associated with poorer survival, despite significant correlations between p50 nuclear expression and downregulation of Bcl-2, p53, phospho-AKT, CXCR4 and Myc. The poor outcome in p50 positive patients was attributed to immune dysregulation including immune suppression by TIM-3 and TNFα upregulation. A similar correlation was found in GCB DLBCL patients with wild-type TP53, however p50 expression in GBC DLBCL patients with TP53 mutants was associated with a significantly improved outcome, as well as reduced expression of Bcl-2, Myc and p53 [65].
The role of p50 therefore differs substantially depending on its dimerization partner and the co-factors involved, as well as the cell type and cancer type, whereby gene expression can either be repressed or activated, hindering or driving tumorigenesis (summarised in Figure 2).
7. NFKB1 Polymorphisms in Cancer
NFKB1 polymorphisms have been largely associated with immune defects including autoimmunity [66,67]. However, there are also links to either increased or decreased cancer risk in several different populations and cancer types (Table 1). Polymorphisms in the NFKB1 gene have been suggested as risk factors for the development of several cancers including liver, gastric and ovarian cancers. These small changes in the NFKB1 nucleotide sequence may impact protein function and expression through transcriptional regulation, since the polymorphisms are often located in potential regulatory regions such as introns and promoter sites. These polymorphisms have been predominantly studied in Asian populations, where it may confer a selective advantage for other cellular processes or diseases. However, fewer studies regarding NFKB1 polymorphisms and cancer risk have been conducted in Western populations. Hence, further studies are required in order to extrapolate the cancer risk association to a wider population. In addition, there is a need to understand the functional effects of the different polymorphisms not only to give us more information about the physiological processes governed by this gene, but also to provide us with more information on how to harness its function in a variety of inflammatory scenarios and disease settings. Indeed, restoring the advantageous NFKB1 nucleotide sequence through gene editing may also represent an effective therapeutic intervention in future years.
8. Potential Interventions
Drugs that stabilize or promote p50 homodimers have the potential to act as powerful anti-inflammatory and anti-cancer therapies. However, we still know very little about what dictates dimer partner choice and co-factor recruitment. Ongoing research focused on uncovering the specific amino acids responsible for the regulation and stabilization of the NF-κB subunits will prove vital in understanding their regulatory roles and post-translational modifications for future drug development. These include in vivo work exploring the use of peptide mimetics to strengthen p50 homodimer:DNA complexes to prevent inflammation which have huge potential as anti-cancer therapeutics [85].
9. Conclusions and Future Perspectives
The overarching role played by NFKB1 in carcinogenesis still remains incompletely understood. Contrasting mechanisms exist depending on the cancer and cell type, and uncertainty as to which dimerization partners and co-factors are involved form the basis of this conundrum. For these reasons more research is required to better delineate the specific role of NFKB1 in each cancer type. The emergence of data from genetic mouse models such as the S340A defective in p50 homodimers and the cell-specific knockouts Nfkb1−/− floxed mice will provide more functional insights into its role in a cancer-specific context. Overall, strong evidence suggests a protective and anti-tumorigenic role for p50 homodimers, particularly in HCC for example, whereas p50:p65 heterodimers are generally considered to be pro-inflammatory and pro-tumorigenic particularly where there is aberrant NF-κB activation. Therapeutic interventions targeting upstream activators of NF-κB directly may prove unsafe and non-specific causing toxicity to normal cells and tissues. However, manipulating NF-κB subunit dimerization, degradation or co-factor interaction may represent a safer, more targeted therapy in the treatment of cancer. For example, the emerging potential therapies aimed at stabilising p50 homodimer co-repressor complexes and target gene promoter binding are a promising approach. A number of NFKB1 polymorphisms are now associated with cancer progression; therefore, further elucidating the functional effect of these polymorphisms may also bring us one step closer to the development of powerful anti-inflammatory and anti-cancer therapies in the future.
Author Contributions
J.C. and C.L.W. conceived the ideas and wrote the manuscript. C.L.W. edited the manuscript.
Funding
This research was funded by Cancer Research UK programme Grant No. C18342/A23390.
Acknowledgments
We apologize to our colleagues whose work we have not been able to cite in this review. We would like to thank Tyrell Cartwright and Edith M. Hessel for their assistance whilst writing this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ALCL | anaplastic large-cell lymphoma |
| BAG-1 | BCL-2 associated athanogene |
| Bcl-3 | B-cell lymphoma 3 |
| C/EBP | CCAAT enhancer-binding protein |
| EBV | Epstein–Barr virus |
| EGFR | epidermal growth factor receptor |
| EMSA | electrophoresis mobility shift assay |
| ERK | extracellular regulated kinase |
| HCC | hepatocellular carcinoma |
| HDAC1 | histone deacetylase 1 |
| HPV16 | human papilloma virus 16 |
| IFN | interferon |
| IKK | IκB kinase |
| IL | interleukin |
| IκB | inhibitor of kappa light polypeptide gene enhancer in B-cells |
| JAK-STAT | Janus kinase-Signal transducer and activator of transcription |
| KPC1 | Kip1 ubiquitination-promoting complex 1 |
| LPS | lipopolysaccharide |
| MEK | mitogen-activated protein kinase kinase |
| NFKB1 | nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 |
| NF-κB | nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| NSCLC | non-small-cell lung carcinoma |
| PD-L1 | programmed death-ligand 1 |
| PTEN | phosphatase and tensin homolog |
| SCCHN | squamous cell carcinoma of the head and neck |
| STAT | signal transducer and activator of transcription |
| TAD | transcriptional activation domain |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TNFα | tumour necrosis factor α |
| TPL-2 | tumour progression locus 2 |
References
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yokosuka, O.; Nagao, K.; Saisho, H. State of hepatitis C viral replication enhances activation of NF-kB- and AP-1-signaling induced by hepatitis B virus X. Cancer Lett. 2006, 234, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chung, G.T.-Y.; Lou, W.P.-K.; Chow, C.; To, K.-F.; Choy, K.-W.; Leung, A.W.-C.; Tong, C.Y.-K.; Yuen, J.W.-F.; Ko, C.-W.; Yip, T.T.-C.; et al. Constitutive activation of distinct NF-κB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa B kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Tchenio, T.; Piton, G.; Romeo, P.H.; Baud, V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 14635–14640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marienfeld, R.; May, M.J.; Berberich, I.; Serfling, E.; Ghosh, S.; Neumann, M. RelB forms transcriptionally inactive complexes with RelA/p65. J. Biol. Chem. 2003, 278, 19852–19860. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Murty, V.V.; Dalla-Favera, R.; Chaganti, R.S. Chromosomal localization of genes encoding the transcription factors, c-rel, NF-kappa Bp50, NF-kappa Bp65, and lyt-10 by fluorescence in situ hybridization. Oncogene 1993, 8, 191–193. [Google Scholar] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Sriskantharajah, S.; Belich, M.P.; Papoutsopoulou, S.; Janzen, J.; Tybulewicz, V.; Seddon, B.; Ley, S.C. Proteolysis of NF-kappaB1 p105 is essential for T cell antigen receptor-induced proliferation. Nat. Immunol. 2009, 10, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Schweighoffer, E.; Visekruna, A.; Papoutsopoulou, S.; Janzen, J.; Zillwood, R.; Tarlinton, D.M.; Tybulewicz, V.L.J.; Ley, S.C. IKK-induced NF-κB1 p105 proteolysis is critical for B cell antibody responses to T cell–dependent antigen. J. Exp. Med. 2014, 211, 2085–2101. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Robinson, M.J.; Hugunin, M.; Ley, S.C. Lipopolysaccharide Activation of the TPL-2/MEK/Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase Cascade Is Regulated by IκB Kinase-Induced Proteolysis of NF-κB1 p105. Mol. Cell. Biol. 2004, 24, 9658–9667. [Google Scholar] [CrossRef] [PubMed]
- Gantke, T.; Sriskantharajah, S.; Sadowski, M.; Ley, S.C. IκB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol. Rev. 2012, 246, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Oakley, F.; Lin, F.; Packham, G.; Mann, D.A.; Mann, J. The NF-κB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010, 53, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-P.; Vancurova, I. Bcl3 regulates pro-survival and pro-inflammatory gene expression in cutaneous T-cell lymphoma. Biochim. Biophys. Acta 2014, 1843, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Kiely, P.A.; Carmody, R.J. Inhibition of transcription by B cell Leukemia 3 (Bcl-3) protein requires interaction with nuclear factor kappaB (NF-kappaB) p50. J. Biol. Chem. 2014, 289, 7059–7067. [Google Scholar] [CrossRef] [PubMed]
- Wessells, J.; Baer, M.; Young, H.A.; Claudio, E.; Brown, K.; Siebenlist, U.; Johnson, P.F. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J. Biol. Chem. 2004, 279, 49995–50003. [Google Scholar] [CrossRef] [PubMed]
- Paz-Priel, I.; Houng, S.; Dooher, J.; Friedman, A.D. C/EBPα and C/EBPα oncoproteins regulate nfkb1 and displace histone deacetylases from NF-κB p50 homodimers to induce NF-κB target genes. Blood 2011, 117, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Buerki, C.; Lombardi, C.; Imhof, R.; Hottiger, M.O. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J. Biol. Chem. 2003, 278, 45145–45153. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.M.; Gieling, R.G.; Chakraborty, J.B.; Fox, C.; et al. NFkappaB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Crawley, C.D.; Kang, S.; Raleigh, D.R.; Yu, X.; Wahlstrom, J.S.; Voce, D.J.; Darga, T.E.; Weichselbaum, R.R.; Yamini, B. p50 (NF-κB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol. Cell 2011, 44, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. KPC1-mediated ubiquitination and proteasomal processing of NF-kappaB1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.S.; Marino, M.W.; Takahashi, T.; Yoshida, T.; Sakakura, T.; Old, L.J.; Obata, Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl. Acad. Sci. USA 1999, 96, 2994–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKβ Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation that Promotes Chemical Hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margetts, J.; Ogle, L.F.; Chan, S.L.; Chan, A.W.H.; Chan, K.C.A.; Jamieson, D.; Willoughby, C.E.; Mann, D.A.; Wilson, C.L.; Manas, D.M.; et al. Neutrophils: Driving progression and poor prognosis in hepatocellular carcinoma? Br. J. Cancer 2017, 118, 248. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Chang, P.Y.; Chen, B.S. Investigating the mechanism of hepatocellular carcinoma progression by constructing genetic and epigenetic networks using NGS data identification and big database mining method. Oncotarget 2016, 7, 79453–79473. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, H.; Yasuda, J.; Nakanishi, K.; Chuma, M.; Kamiyama, T.; Todo, S.; Hirohashi, S.; Sakamoto, M. Clinicopathological significance of nuclear factor-kappaB activation in hepatocellular carcinoma. Hepatol. Res. 2011, 41, 240–249. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Farrah, H.; Kelly, H.; Baldwin, A.S.; Funkhouser, W.K. Analysis of NF-kappa B in hepatocellular carcinoma (HCC) reveals frequent activation of p50 and bcl-3. J. Clin. Oncol. 2005, 23 (Suppl. 16), 9621. [Google Scholar] [CrossRef]
- Sokolova, O.; Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IkappaB-kinase beta (IKKβ) accelerates Helicobacter-dependent gastric apoptosis, proliferation and preneoplasia. Gastroenterology 2010, 138, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.A.; Putoczki, T.L.; Mielke, L.A.; Low, J.T.; Lin, A.; Preaudet, A.; Herold, M.J.; Yaprianto, K.; Tai, L.; Kueh, A.; et al. Loss of NF-κB1 Causes Gastric Cancer with Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a STAT-1-Dependent Manner. Immunity 2018, 48, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, T.; Tahara, T.; Shiroeda, H.; Yamada, K.; Nomura, T.; Yamada, H.; Hayashi, R.; Matsunaga, K.; Otsuka, T.; Nakamura, M.; et al. Functional promoter polymorphisms of NFKB1 influence susceptibility to the diffuse type of gastric cancer. Oncol. Rep. 2013, 30, 3013–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, T.; Qinsheng, W.; Xuxia, W.; Shuguang, Z.; Ming, Q.; Zhenxiong, L.; Jingjie, W. Nuclear Factor-Kappa B1 is Associated With Gastric Cancer in a Chinese Population. Medicine 2014, 93, e279. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kang, W.; Zhang, B.; Wu, F.; Dong, Y.; Tong, J.H.M.; Yang, W.; Zhou, Y.; Zhang, L.; Cheng, A.S.L.; et al. miR-508-3p concordantly silences NFKB1 and RELA to inactivate canonical NF-κB signaling in gastric carcinogenesis. Mol. Cancer 2016, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Qu, Z.; Xiao, Y.; Zhou, J.; Burns, T.F.; Stabile, L.P.; Siegfried, J.M.; Xiao, G. NF-kappaB1 p105 suppresses lung tumorigenesis through the Tpl2 kinase but independently of its NF-kappaB function. Oncogene 2016, 35, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Al-Saad, S.; Al-Shibli, K.; Donnem, T.; Persson, M.; Bremnes, R.M.; Busund, L.T. The prognostic impact of NF-κB p105, vimentin, E-cadherin and Par6 expression in epithelial and stromal compartment in non-small-cell lung cancer. Br. J. Cancer 2008, 99, 1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Li, C.; Huang, C.; Zhuang, W.; Huang, Y.; Xu, H.; Miao, Q.; Hu, D. Co-expression of NF-kappaB-p65 and phosphorylated NF-kappaB-p105 is associated with poor prognosis in surgically resectable non-small cell lung cancer. J. Cell. Mol. Med. 2018, 22, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Voce, D.J.; Schmitt, A.M.; Uppal, A.; McNerney, M.E.; Bernal, G.M.; Cahill, K.E.; Wahlstrom, J.S.; Nassiri, A.; Yu, X.; Crawley, C.D.; et al. Nfkb1 is a haploinsufficient DNA damage-specific tumor suppressor. Oncogene 2015, 34, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Crawley, C.D.; Kang, S.; Bernal, G.M.; Wahlstrom, J.S.; Voce, D.J.; Cahill, K.E.; Garofalo, A.; Raleigh, D.R.; Weichselbaum, R.R.; Yamini, B. S-phase-dependent p50/NF-кB1 phosphorylation in response to ATR and replication stress acts to maintain genomic stability. Cell Cycle 2015, 14, 566–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Fujii, M.; Ikeda, S.; Yamada, Y.; Tomonaga, M.; Ballard, D.W.; Yamamoto, N. Constitutive Activation of NF-κB in Primary Adult T-Cell Leukemia Cells. Blood 1999, 93, 2360–2368. [Google Scholar] [PubMed]
- Mathas, S.; Johrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P.; et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Budunova, I.V.; Perez, P.; Vaden, V.R.; Spiegelman, V.S.; Slaga, T.J.; Jorcano, J.L. Increased expression of p50-NF-kappaB and constitutive activation of NF-kappaB transcription factors during mouse skin carcinogenesis. Oncogene 1999, 18, 7423–7431. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bharti, A.C.; Varghese, P.; Saluja, D.; Das, B.C. Differential expression and activation of NF-kappaB family proteins during oral carcinogenesis: Role of high risk human papillomavirus infection. Int. J. Cancer 2006, 119, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, N.J.; Pathmanathan, R.; Raab-Traub, N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res. 2003, 63, 8293–8301. [Google Scholar] [PubMed]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.S., Jr. Selective activation of NF-kappa B subunits in human breast cancer: Potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Eppenberger-Castori, S.; Marx, C.; Yau, C.; Scott, G.K.; Eppenberger, U.; Benz, C.C. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int. J. Biochem. Cell. Biol. 2005, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Rosenthal, D.; Perec, L.; Merajver, S. Down regulation of NfkB1 expression blocks cell motility in a cell line model of inflammatory breast cancer. Cancer Res. 2008, 68, 1991. [Google Scholar]
- Havard, L.; Rahmouni, S.; Boniver, J.; Delvenne, P. High levels of p105 (NFKB1) and p100 (NFKB2) proteins in HPV16-transformed keratinocytes: Role of E6 and E7 oncoproteins. Virology 2005, 331, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Xie, L.; Wang, X.; He, H.; Lin, Y.; Hu, L. Association of constitutive nuclear factor-kappaB activation with aggressive aspects and poor prognosis in cervical cancer. Int. J. Gynecol. Cancer 2009, 19, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Prusty, B.K.; Husain, S.A.; Das, B.C. Constitutive activation of nuclear factor -kB: Preferntial homodimerization of p50 subunits in cervical carcinoma. Front. Biosci. 2005, 10, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Giopanou, I.; Bravou, V.; Papanastasopoulos, P.; Lilis, I.; Aroukatos, P.; Papachristou, D.; Kounelis, S.; Papadaki, H. Metadherin, p50, and p65 expression in epithelial ovarian neoplasms: An immunohistochemical study. Biomed. Res. Int. 2014, 2014, 178410. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Ippolito, A.; Consonni, F.M.; Carraro, L.; Celesti, G.; Correale, C.; Grizzi, F.; Pasqualini, F.; Tartari, S.; Rinaldi, M.; et al. Protumor Steering of Cancer Inflammation by p50 NF-κB Enhances Colorectal Cancer Progression. Cancer Immunol. Res. 2018, 6, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Southern, S.L.; Collard, T.J.; Urban, B.C.; Skeen, V.R.; Smartt, H.J.; Hague, A.; Oakley, F.; Townsend, P.A.; Perkins, N.D.; Paraskeva, C.; et al. BAG-1 interacts with the p50-p50 homodimeric NF-kappaB complex: Implications for colorectal carcinogenesis. Oncogene 2012, 31, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, J.S.; Kim, W.K.; Oh, K.J.; Kim, J.M.; Lee, H.J.; Han, B.S.; Kim, D.S.; Seo, Y.S.; Lee, S.C.; et al. Intracellular annexin A2 regulates NF-kappaB signaling by binding to the p50 subunit: Implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis. 2015, 6, e1606. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Garg, B.; Modi, S.; George, J.; Sethi, V.; Banerjee, S.; Saluja, A.; Dudeja, V. NF-κB P50 Subunit in Stellate-Cells Actively Modulates Cancer Growth in Mouse Models of Pancreatic Cancer. J. Am. Coll. Surg. 2016, 223, S142. [Google Scholar] [CrossRef]
- Chen, T.L.; Tran, M.; Lakshmanan, A.; Harrington, B.K.; Goettl, V.M.; Lehman, A.M.; Trudeau, S.; Lucas, D.M.; Johnson, A.J.; Byrd, J.C.; et al. NF-κB p50 (nfkb1) contributes to pathogenesis in the Eμ-TCL1 mouse model of chronic lymphocytic leukemia. Blood 2017, 130, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.C.; Liou, H.-C.; Tuomanen, E.I.; Baltimore, D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell 1995, 80, 321–330. [Google Scholar] [CrossRef]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Koff, J.L.; Moffitt, A.B.; Cinar, M.; Ramachandiran, S.; Chen, Z.; Switchenko, J.M.; Mosunjac, M.; Neill, S.G.; Mann, K.P.; et al. Molecular impact of selective NFKB1 and NFKB2 signaling on DLBCL phenotype. Oncogene 2017, 36, 4224–4232. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Tu, M.; Xu-Monette, Z.Y.; Sun, R.; Manyam, G.C.; Xu, X.; Tzankov, A.; His, E.D.; Møller, M.B.; Medeiros, L.J.; et al. NF-κB p50 activation associated with immune dysregulation confers poorer survival for diffuse large B-cell lymphoma patients with wild-type p53. Mod. Pathol. 2017, 30, 854. [Google Scholar] [CrossRef] [PubMed]
- Yenmis, G.; Oner, T.; Cam, C.; Koc, A.; Kucuk, O.S.; Yakicier, M.C.; Dizman, D.; Sultuybek, G.K. Association of NFKB1 and NFKBIA polymorphisms in relation to susceptibility of Behcet’s disease. Scand. J. Immunol. 2015, 81, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Koc, A.; Batar, B.; Celik, O.; Onaran, I.; Tasan, E.; Sultuybek, G.K. Polymorphism of the NFKB1 affects the serum inflammatory levels of IL-6 in Hashimoto thyroiditis in a Turkish population. Immunobiology 2014, 219, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Lewander, A.; Butchi, A.K.; Gao, J.; He, L.J.; Lindblom, A.; Arbman, G.; Carstensen, J.; Zhang, Z.Y.; Sun, X.F. Polymorphism in the promoter region of the NFKB1 gene increases the risk of sporadic colorectal cancer in Swedish but not in Chinese populations. Scand. J. Gastroenterol. 2007, 42, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Mohd Suzairi, M.S.; Tan, S.C.; Ahmad Aizat, A.A.; Mohd Aminudin, M.; Siti Nurfatimah, M.S.; Andee, Z.D.; Ankathil, R. The functional -94 insertion/deletion ATTG polymorphism in the promoter region of NFKB1 gene increases the risk of sporadic colorectal cancer. Cancer Epidemiol. 2013, 37, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Su, J.-L.; Lin, C.-W.; Su, C.-W.; Shih, C.-H.; Yang, S.-F.; Chien, M.-H. Effects of NFKB1 and NFKBIA Gene Polymorphisms on Hepatocellular Carcinoma Susceptibility and Clinicopathological Features. PLoS ONE 2013, 8, e56130. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gu, J.; Yang, X.; Cai, H.; Tao, J.; Yang, X.; Lu, Q.; Wang, Z.; Yin, C.; Gu, M. Functional promoter -94 ins/del ATTG polymorphism in NFKB1 gene is associated with bladder cancer risk in a Chinese population. PLoS ONE 2013, 8, e71604. [Google Scholar] [CrossRef] [PubMed]
- Umar, M.; Upadhyay, R.; Kumar, S.; Ghoshal, U.C.; Mittal, B. Association of Common Polymorphisms in TNFA, NFkB1 and NFKBIA with Risk and Prognosis of Esophageal Squamous Cell Carcinoma. PLoS ONE 2013, 8, e81999. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, H.; Liang, Y.; Sun, R.; Wei, T.; Li, Z.; Gong, Y.; Gong, R.; Liu, F.; Zhang, L.; et al. A Functional Insertion/Deletion Polymorphism in the Promoter Region of the NFKB1 Gene Increases the Risk of Papillary Thyroid Carcinoma. Genet. Test. Mol. Biomark. 2015, 19, 167–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, G.F.; Arraes, J.A.; Bakos, L.; Ashton-Prolla, P.; Giugliani, R.; Callegari-Jacques, S.M.; Santos, S.; Bakos, R.M. Polymorphisms in CYP19A1 and NFKB1 genes are associated with cutaneous melanoma risk in southern Brazilian patients. Melanoma Res. 2016, 26, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.; Rosdahl, I.; Sun, X.F.; Zhang, H. Importance of polymorphisms in NF-kappaB1 and NF-kappaBIalpha genes for melanoma risk, clinicopathological features and tumor progression in Swedish melanoma patients. J. Cancer Res. Clin. Oncol. 2007, 133, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Rao, L.; Li, Y.; Gao, L.; Wang, Y.; Chen, Y.; Xue, H.; Song, Y.; Peng, Y.; Liao, M.; et al. A functional insertion/deletion polymorphism in the promoter region of NFKB1 gene increases susceptibility for nasopharyngeal carcinoma. Cancer Lett. 2009, 275, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Hsieh, Y.S.; Hsin, C.H.; Su, C.W.; Lin, C.H.; Wei, L.H.; Yang, S.F.; Chien, M.H. Effects of NFKB1 and NFKBIA gene polymorphisms on susceptibility to environmental factors and the clinicopathologic development of oral cancer. PLoS ONE 2012, 7, e35078. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Begin, L.R.; Gleave, M.E.; Mes-Masson, A.M.; Saad, F. Nuclear localisation of nuclear factor-kappaB transcription factors in prostate cancer: An immunohistochemical study. Br. J. Cancer 2005, 93, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Qie, M.; Wang, Y.; Yan, L.; Zhang, Z.; Liang, A.; Wang, T.; Wang, X.; Song, Y.; Zhang, L. Relationship between NFKB1 -94 insertion/deletion ATTG polymorphism and susceptibility of cervical squamous cell carcinoma risk. Ann. Oncol. 2010, 21, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, S.; Anoop, K.; Showket, H.; Alo, N.; Mausumi, B. NFKB1/NFKBIa polymorphisms are associated with the progression of cervical carcinoma in HPV-infected postmenopausal women from rural area. Tumor Biol. 2015, 36, 6265–6276. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, H.-L.; Gao, S.; Zhang, W.; Tan, Y.-T.; Rothman, N.; Purdue, M.; Gao, Y.-T.; Zheng, W.; Shu, X.-O.; et al. Genetic polymorphism of NFKB1 and NFKBIA genes and liver cancer risk: A nested case–control study in Shanghai, China. BMJ Open 2014, 4, e004427. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, R.; Zheng, H.; Xiao, R.; Feng, J.; Wang, H.; Gao, X.; Guo, L. The NFKB1 polymorphism (rs4648068) is associated with the cell proliferation and motility in gastric cancer. BMC Gastroenterol. 2015, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Sunakawa, Y.; Stremitzer, S.; Cao, S.; Zhang, W.; Yang, D.; Wakatsuki, T.; Ning, Y.; Yamauchi, S.; Stintzing, S.; Sebio, A.; et al. Association of variants in genes encoding for macrophage-related functions with clinical outcome in patients with locoregional gastric cancer. Ann. Oncol. 2015, 26, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.P.; Cai, P.S.; Liang, H.B. Association of the genetic polymorphisms of NFKB1 with susceptibility to ovarian cancer. Genet. Mol. Res. 2015, 14, 8273–8282. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Grassia, G.; Colleran, A.; Kiely, P.A.; Ialenti, A.; Maffia, P.; Carmody, R.J. Mapping the Interaction of B Cell Leukemia 3 (BCL-3) and Nuclear Factor kappaB (NF-kappaB) p50 Identifies a BCL-3-mimetic Anti-inflammatory Peptide. J. Biol. Chem. 2015, 290, 15687–15696. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
NF-κB subunit structures and dimeric combinations. All subunits contain a Rel homology domain (RHD). NFKB1 and NFKB2 have a glycine rich region (GRR), followed by an Ankyrin repeat domain (ANK) in the precursors p105 and p100. p65, RelB, and c-Rel contain a transactivation domain (TAD), with RelB additionally containing a leucine zipper (LZ) motif. While most dimers activate transcription, p50:p50, p50:p52, and p52:p52 dimers lack a TAD, and therefore repress transcription in the absence of co-activating factors and p65:RelB, c-Rel:RelB, and RelB:RelB dimers are thought to have limited DNA binding [8,9,10].
Figure 1.
NF-κB subunit structures and dimeric combinations. All subunits contain a Rel homology domain (RHD). NFKB1 and NFKB2 have a glycine rich region (GRR), followed by an Ankyrin repeat domain (ANK) in the precursors p105 and p100. p65, RelB, and c-Rel contain a transactivation domain (TAD), with RelB additionally containing a leucine zipper (LZ) motif. While most dimers activate transcription, p50:p50, p50:p52, and p52:p52 dimers lack a TAD, and therefore repress transcription in the absence of co-activating factors and p65:RelB, c-Rel:RelB, and RelB:RelB dimers are thought to have limited DNA binding [8,9,10].
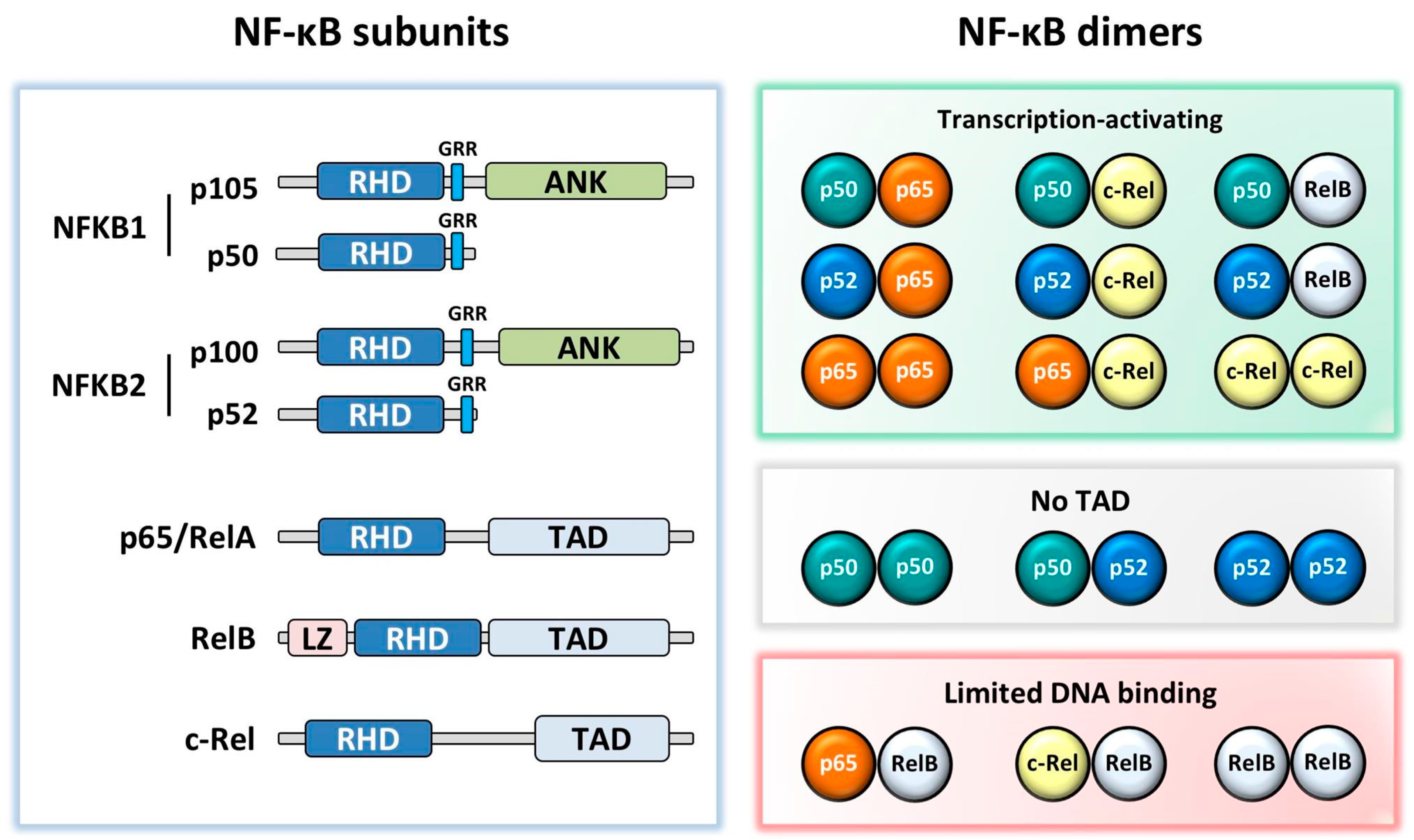
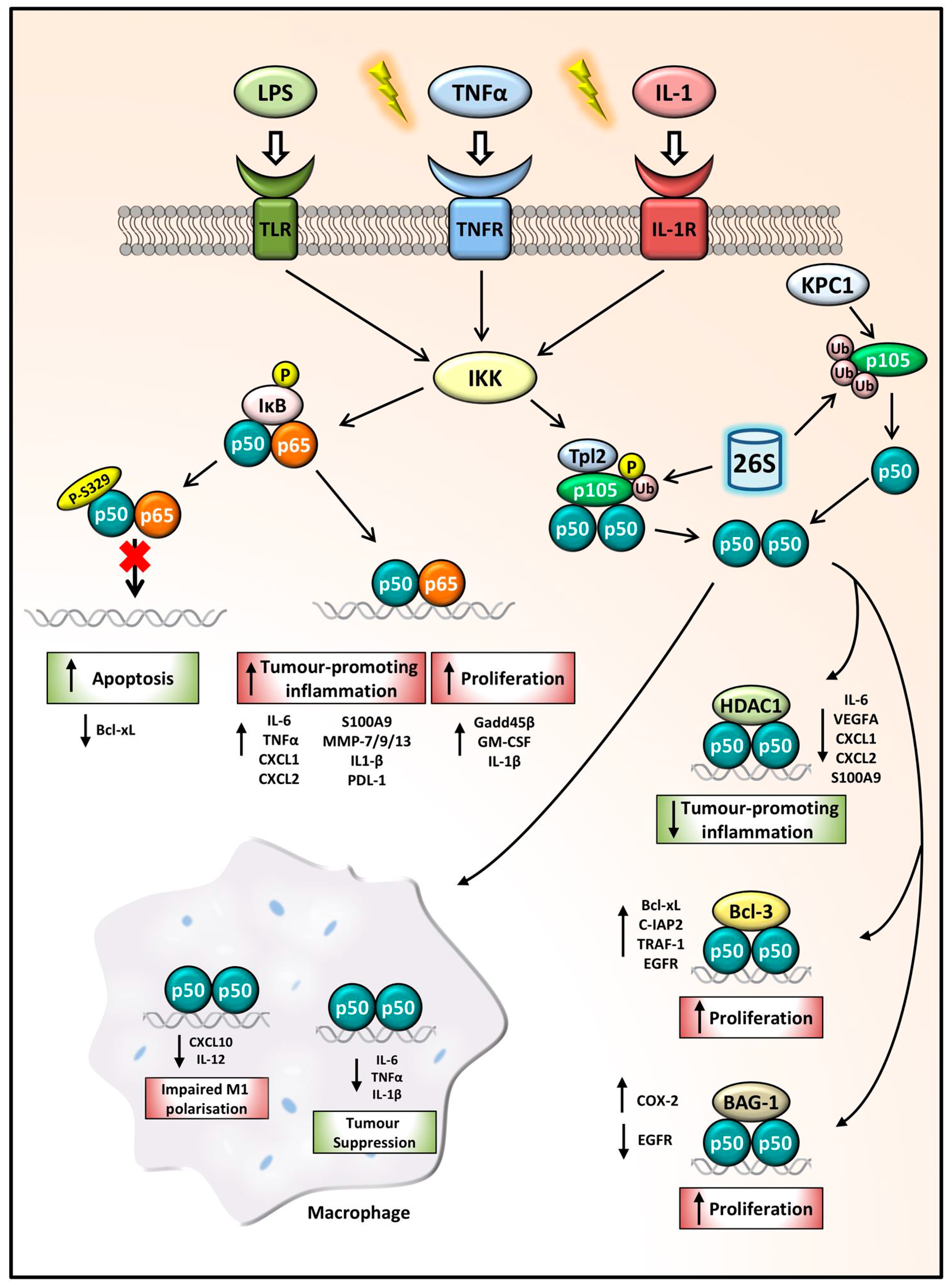
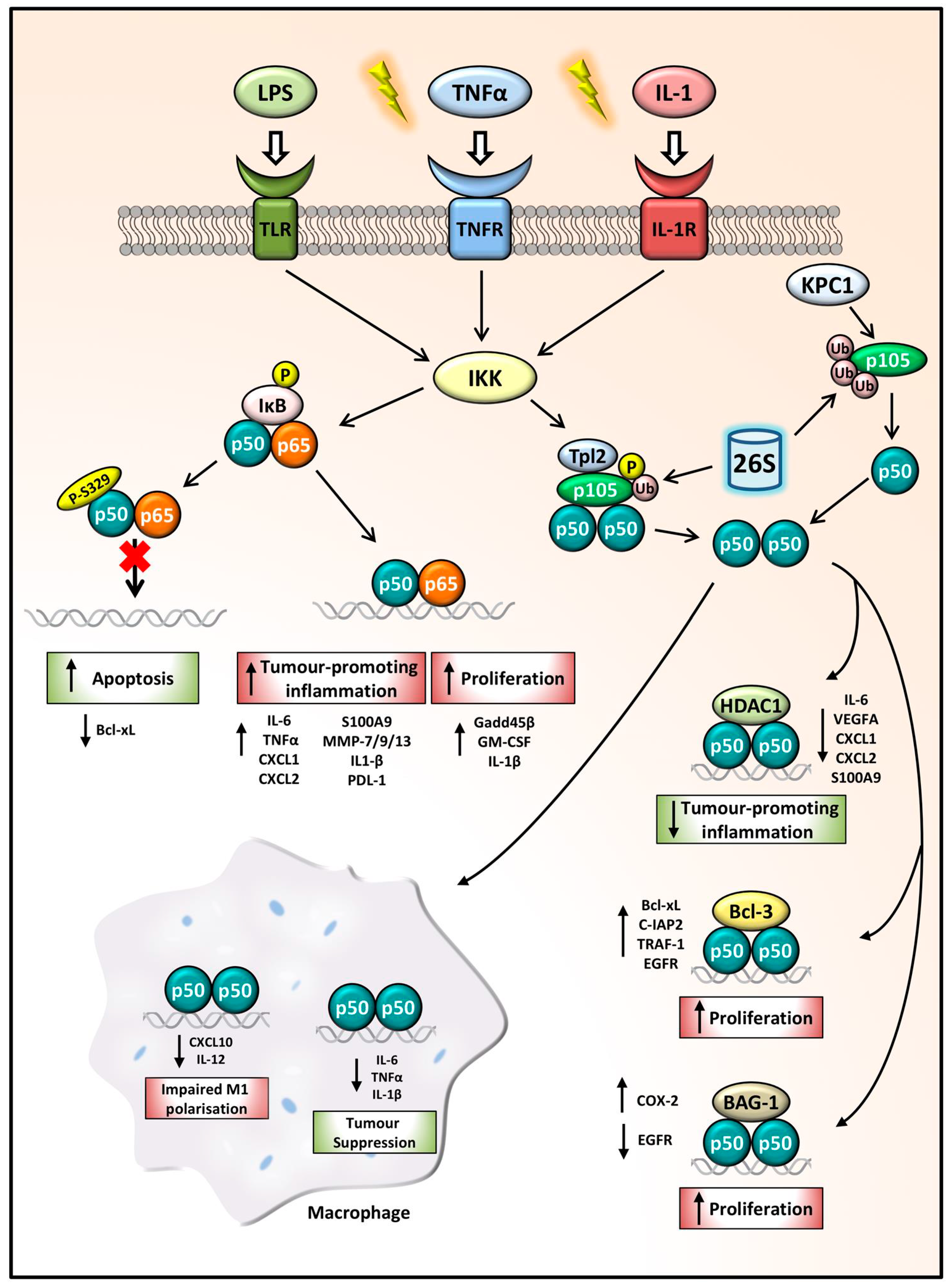
Figure 2.
NFKB1 p105/p50 regulation of gene expression in cancer. Following inflammatory stimulation, IKK functions as the key activator of the NFKB1 signalling pathway. Phosphorylation and ubiquitination of IκB releases p50:p65 dimers which can drive the transcription of tumour-promoting inflammatory genes and anti-apoptotic and proliferative genes. p50 phosphorylation at the S329 site impairs p50:p65 dimer binding to DNA, decreasing Bcl-xL expression leading to increased apoptosis, hindering cancer progression. p105 phosphorylation and ubiquitination leads to its processing by the 26S proteasome, releasing p50 homodimers. The p50:p50:HDAC1 complex represses tumour-promoting inflammatory gene expression thus acting as a tumour-suppressor complex, while p50 homodimers in complex with Bcl-3 or BAG-1 can drive proliferation. In macrophages, aberrant p50 homodimer repression of CXCL10 and IL-12 leads to impaired M1 polarisation. Whilst p50:p50 repression of pro-inflammatory and proliferative mediators can also promote tumour suppression.
Figure 2.
NFKB1 p105/p50 regulation of gene expression in cancer. Following inflammatory stimulation, IKK functions as the key activator of the NFKB1 signalling pathway. Phosphorylation and ubiquitination of IκB releases p50:p65 dimers which can drive the transcription of tumour-promoting inflammatory genes and anti-apoptotic and proliferative genes. p50 phosphorylation at the S329 site impairs p50:p65 dimer binding to DNA, decreasing Bcl-xL expression leading to increased apoptosis, hindering cancer progression. p105 phosphorylation and ubiquitination leads to its processing by the 26S proteasome, releasing p50 homodimers. The p50:p50:HDAC1 complex represses tumour-promoting inflammatory gene expression thus acting as a tumour-suppressor complex, while p50 homodimers in complex with Bcl-3 or BAG-1 can drive proliferation. In macrophages, aberrant p50 homodimer repression of CXCL10 and IL-12 leads to impaired M1 polarisation. Whilst p50:p50 repression of pro-inflammatory and proliferative mediators can also promote tumour suppression.

{kind=link}
{kind=link}
Table 1.
NFKB1 SNPs (Single Nucleotide Polymorphisms) associated with cancer. NFKB1 polymorphisms in different c-94ancer types are associated with either an advantageous or disadvantageous prognosis in different population cohorts.
Table 1.
NFKB1 SNPs (Single Nucleotide Polymorphisms) associated with cancer. NFKB1 polymorphisms in different c-94ancer types are associated with either an advantageous or disadvantageous prognosis in different population cohorts.
| SNP | Cancer Type | Sequence; Location; Consequence (If Known) | Prognostic | Cohort | Reference |
|---|---|---|---|---|---|
| rs28362491 | Colorectal | ATTG del; -94 promoter region; reduces promoter activity and nuclear protein binding ability, decreasing NFKB1 expression | Disadvantageous | Swedish; Malaysian | [68,69] |
| Gastric | Disadvantageous | Japanese; Chinese | [36,37] | ||
| Liver | Disadvantageous | Taiwanese | [70] | ||
| Bladder | Disadvantageous | Chinese | [71] | ||
| Esophageal | Disadvantageous | Northern Indian | [72] | ||
| Thyroid | Disadvantageous | Chinese | [73] | ||
| Melanoma | Disadvantageous | Brazilian, Swedish | [74,75] | ||
| Nasopharyngeal | Disadvantageous | Chinese | [76] | ||
| Oral | Disadvantageous | Taiwanese | [77] | ||
| Prostate | Advantageous | Canadian | [78] | ||
| Ovarian | Advantageous | Greek | [55] | ||
| Cervical | Advantageous | Chinese, Indian | [79,80] | ||
| rs230496 | Liver | AG and GG genotypes | Disadvantageous | Chinese | [81] |
| rs230525 | Liver | GA genotypes | Disadvantageous | Chinese | [81] |
| rs230530 | Liver | AA genotypes | Disadvantageous | Chinese | [81] |
| rs78696119 | Gastric | GG genotypes; increased inflammatory cell infiltration, diffuse type gastric cancer and cancer progression | Disadvantageous | Japanese | [36] |
| rs72696119 | Gastric | GG and del/del genotypes; -449 in 5′ UTR region; muscle layer tumour invasion and lymph node metastasis | Disadvantageous | Japanese | [36] |
| rs4648068 | Gastric | GG genotype; intron 12; increased incorporation of the H3K9me1 and H3K4me1 histones, increased chemomodification, enhanced transcriptional activity, cell proliferation and motility | Disadvantageous | Chinese | [37,82] |
| rs230510 | Gastric | A genotype; intronic region | Advantageous | Japanese, US | [83] |
| Ovarian | T genotype; intron 12 | Advantageous | Chinese | [84] | |
| rs230521 | Ovarian | G genotype; intron 4 | Advantageous | Chinese | [84] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Concetti, J.; Wilson, C.L. NFKB1 and Cancer: Friend or Foe? Cells 2018, 7, 133. https://doi.org/10.3390/cells7090133
AMA Style
Concetti J, Wilson CL. NFKB1 and Cancer: Friend or Foe? Cells. 2018; 7(9):133. https://doi.org/10.3390/cells7090133
Chicago/Turabian StyleConcetti, Julia, and Caroline L. Wilson. 2018. "NFKB1 and Cancer: Friend or Foe?" Cells 7, no. 9: 133. https://doi.org/10.3390/cells7090133
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.