HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic mTOR Suppression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs
2.3. Tumor Cell Binding to HUVECs
2.4. Attachment to Extracellular Matrix Components
2.5. Tumor Cell Motility (Chemotaxis), Migration, and Invasion
2.6. Integrin Surface Expression
2.7. Western Blot Analysis
2.8. Real-Time (RT)-qPCR
2.9. Blocking Studies
2.10. Statistics
3. Results
3.1. Adhesion Characteristics
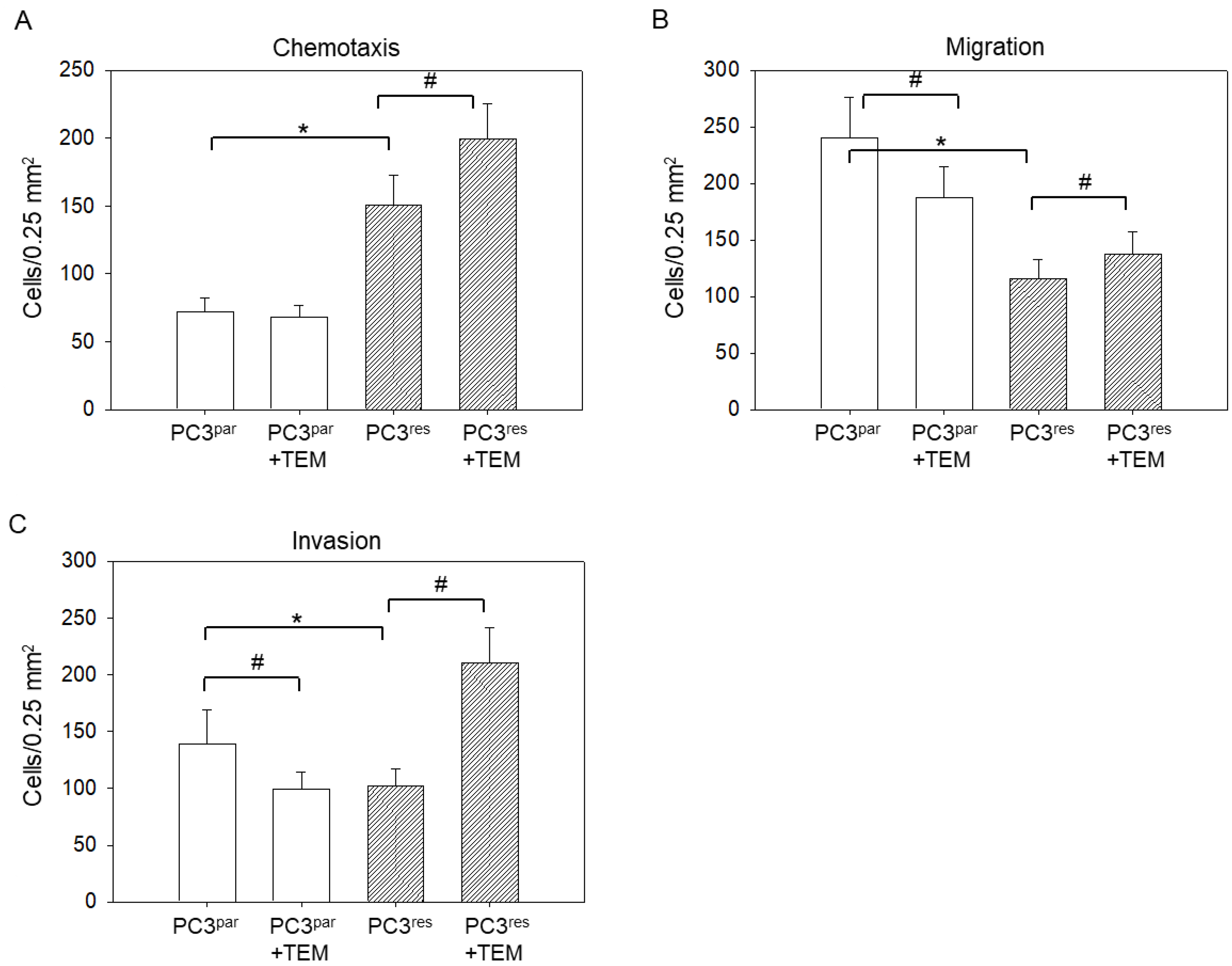
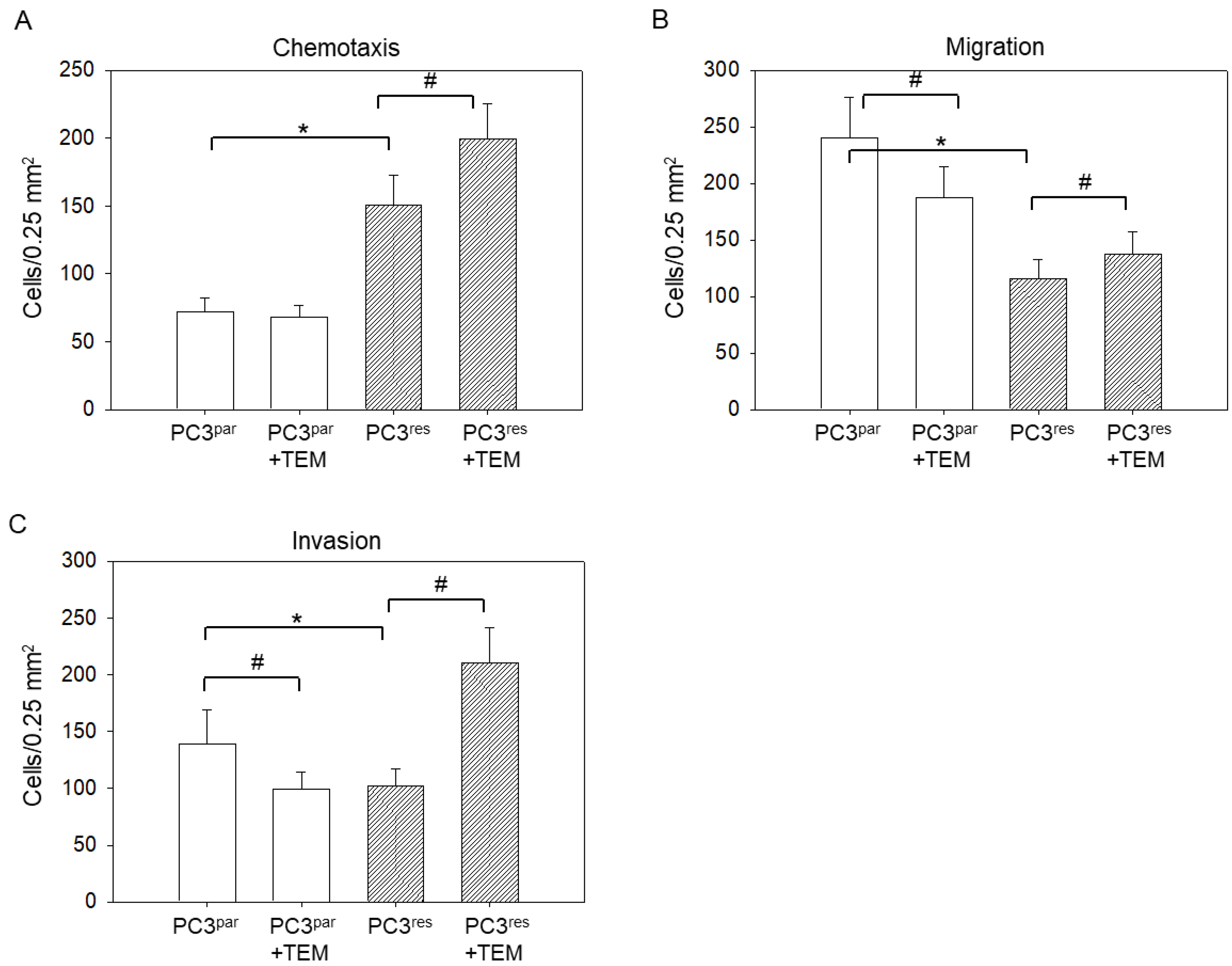
3.2. Tumor Motility, Migration, and Invasion
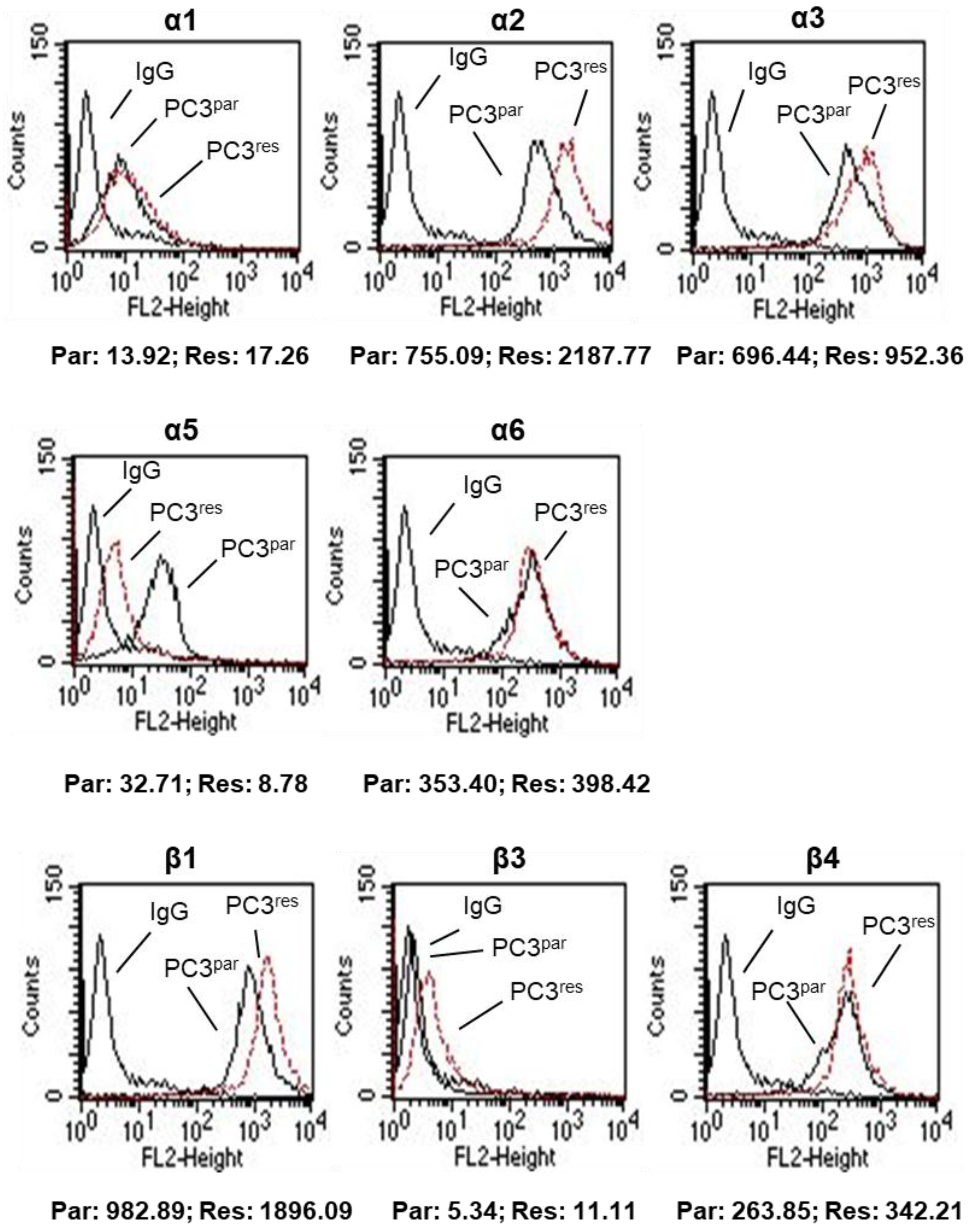
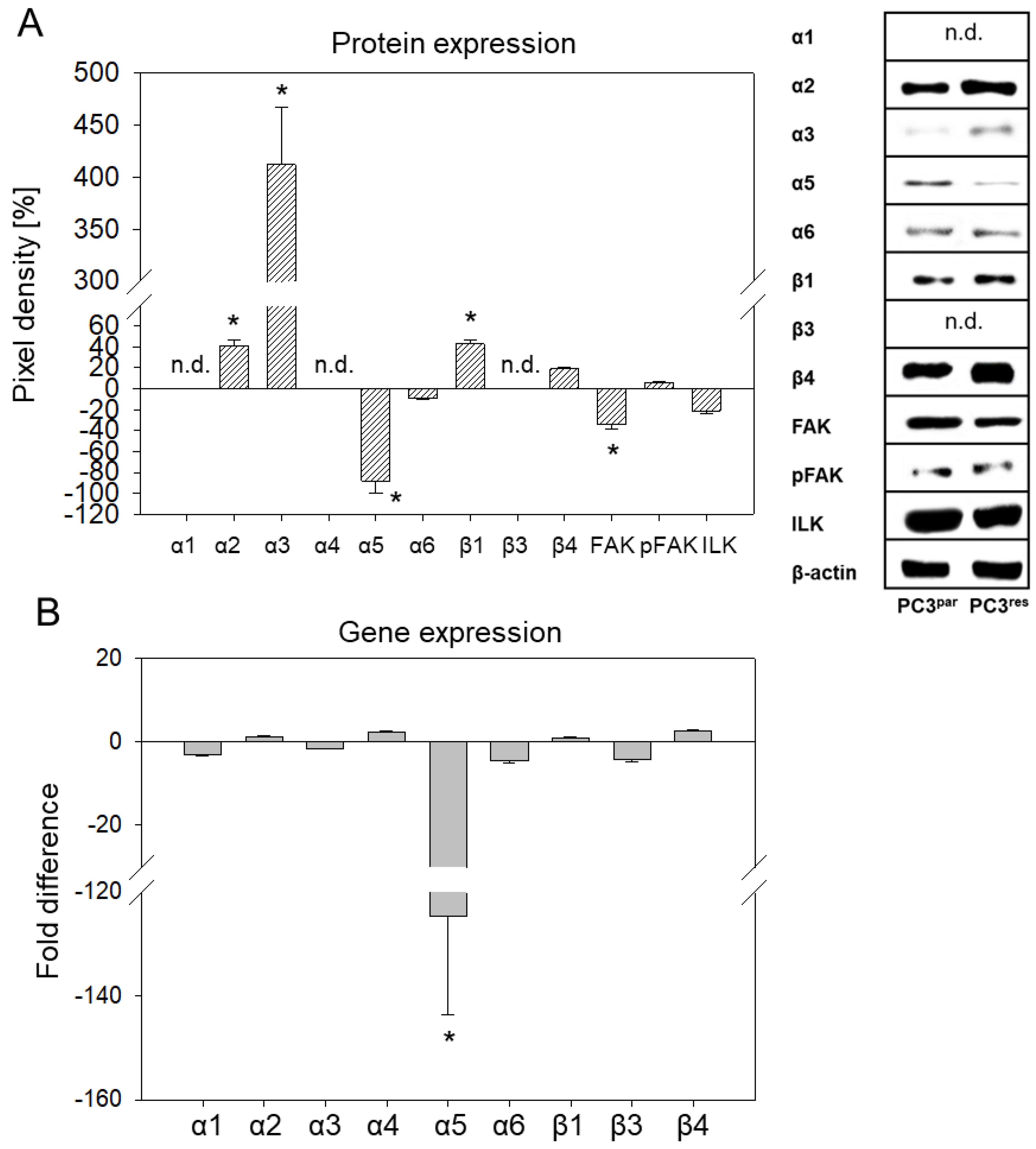
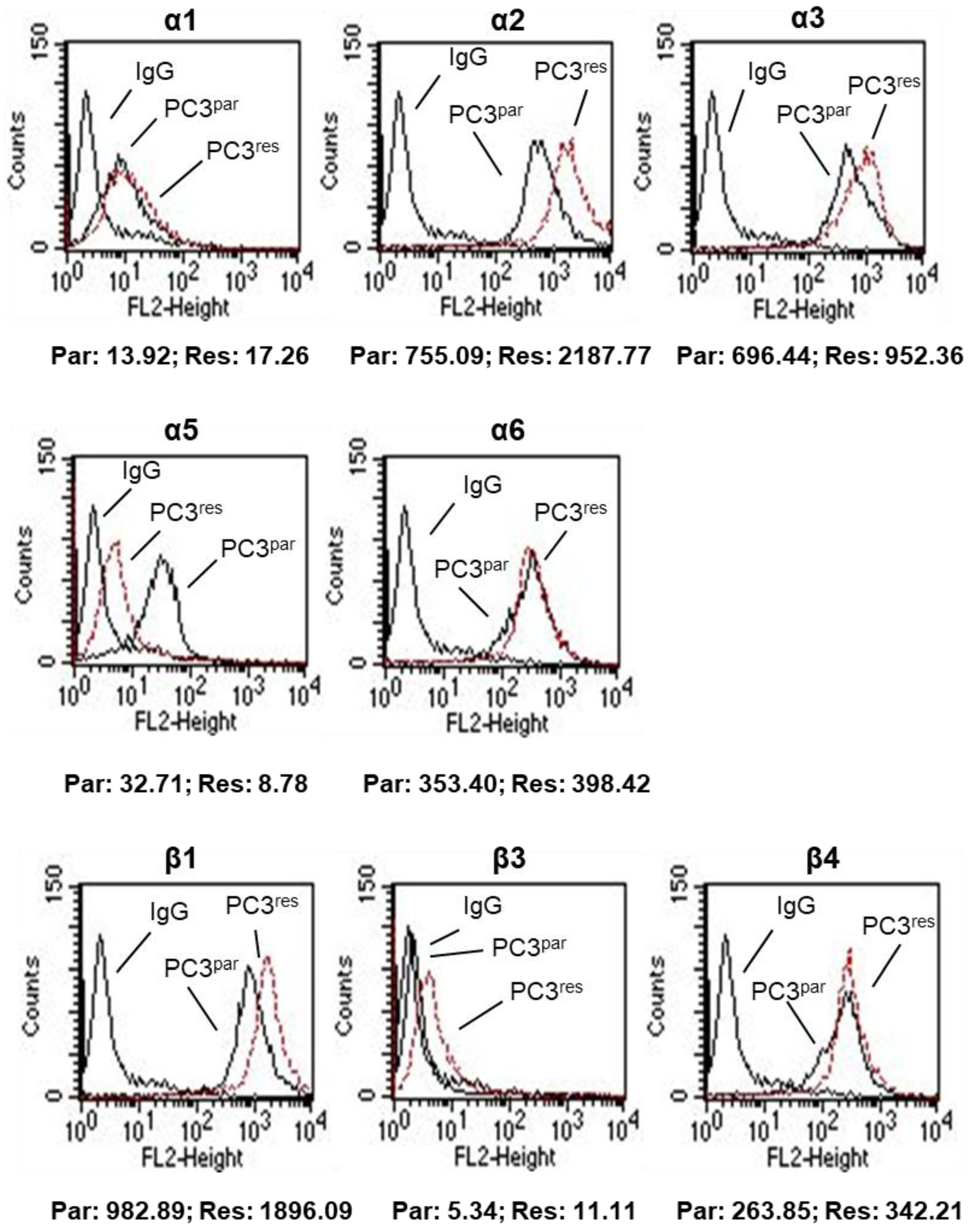
3.3. Integrin Expression Pattern in PC3par and PC3res Cells
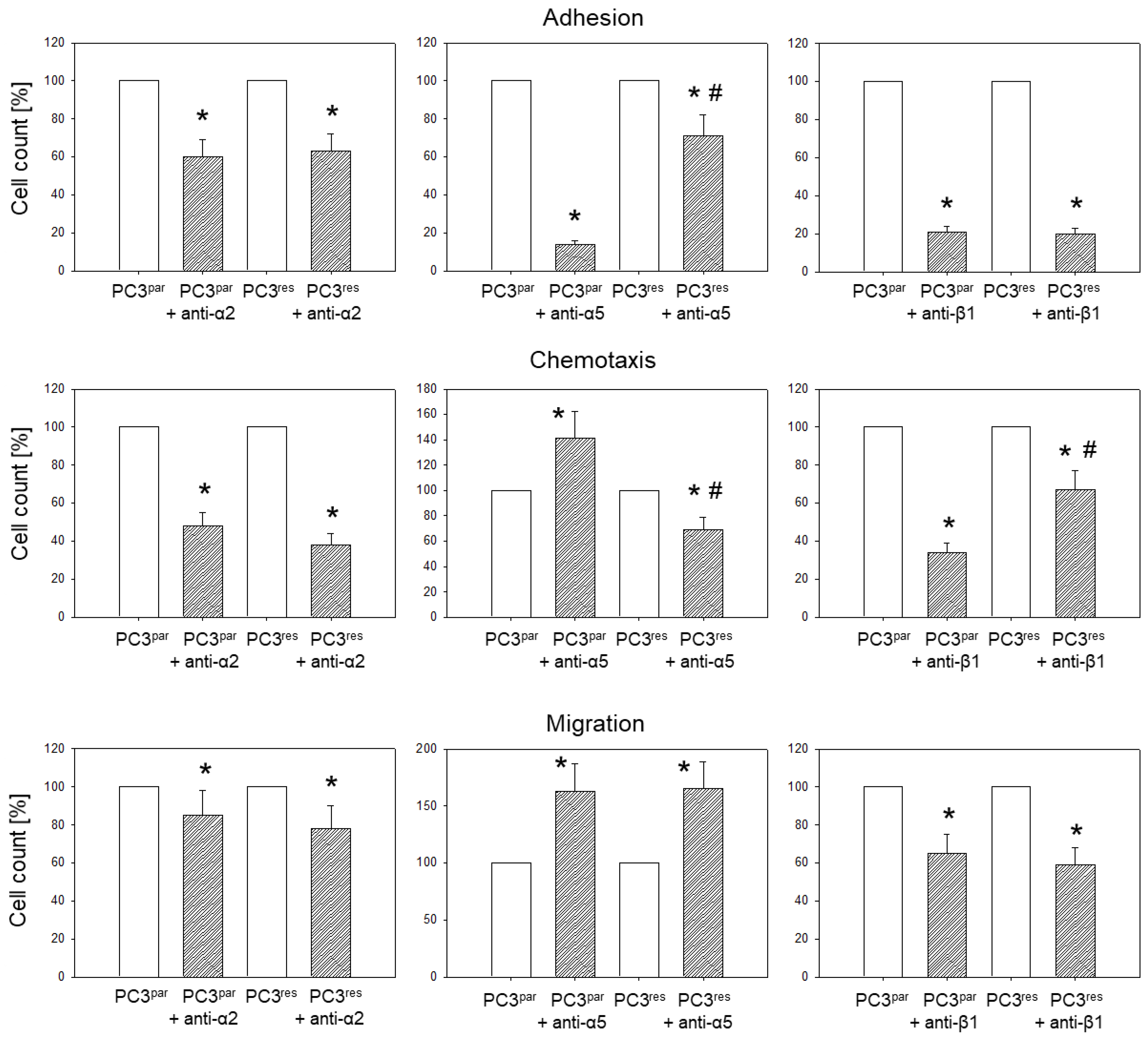
3.4. Blocking Studies
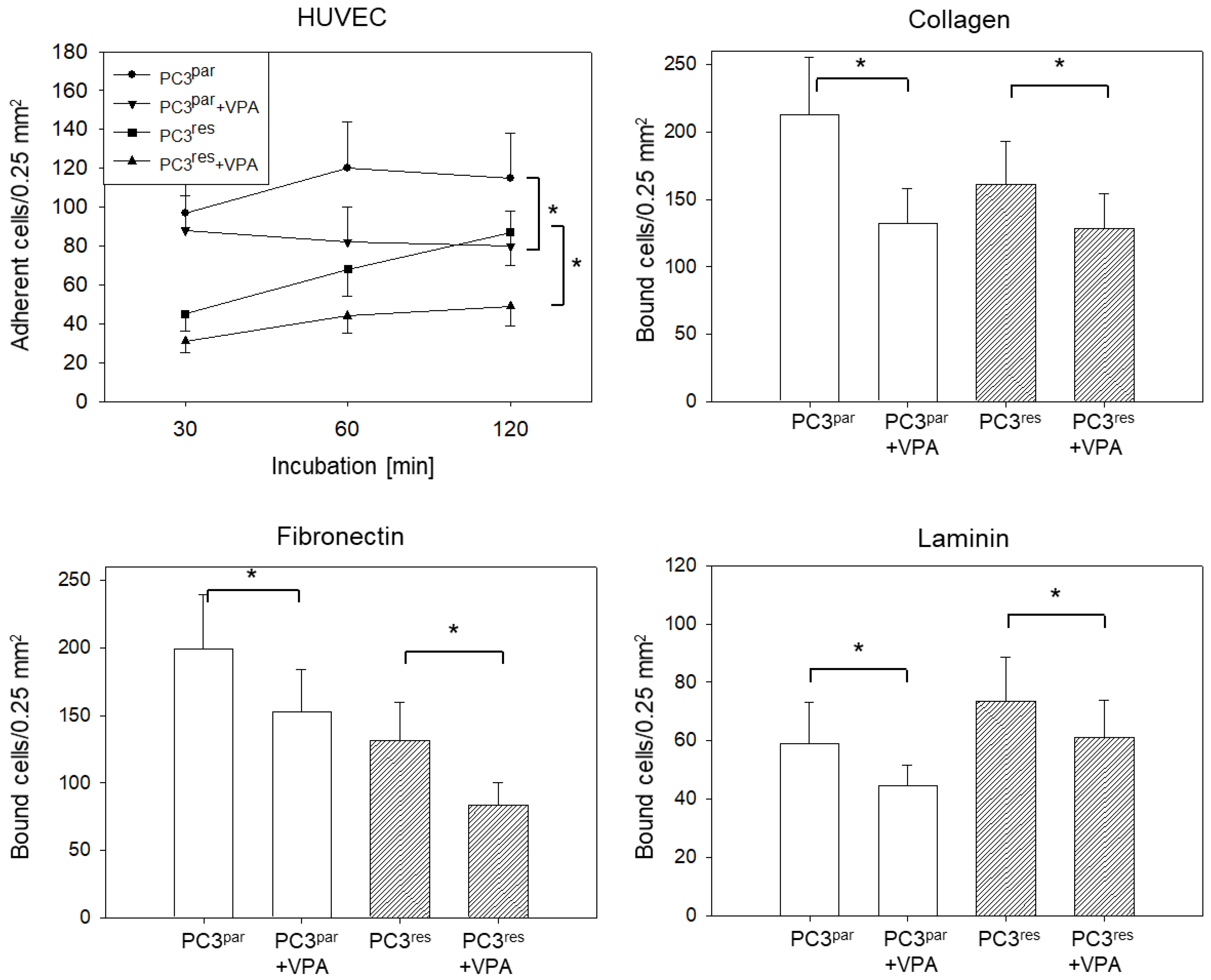
3.5. Influence of VPA on Adhesion, Chemotaxis, Migration, and Integrin Expression of PC3par and PC3res Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ritch, C.R.; Cookson, M.S. Advances in the management of castration resistant prostate cancer. BMJ 2016, 355, i4405. [Google Scholar] [CrossRef] [PubMed]
- Calimeri, T.; Ferreri, A.J.M. m-TOR inhibitors and their potential role in haematological malignancies. Br. J. Haematol. 2017, 177, 684–702. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Leite, R.; Arantes-Rodrigues, R.; Sousa, N.; Oliveira, P.A.; Santos, L. mTOR inhibitors in urinary bladder cancer. Tumour Biol. 2016, 37, 11541–11551. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Ru, Y.; Wu, K.; Yan, F.; Wang, Q.; Wang, J.; Pan, T.; Zhang, M.; Han, H.; Li, X.; et al. GOLM1 promotes prostate cancer progression through activating PI3K-AKT-mTOR signaling. Prostate 2018, 78, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Statz, C.M.; Patterson, S.E.; Mockus, S.M. mTOR Inhibitors in Castration-Resistant Prostate Cancer: A Systematic Review. Target Oncol. 2017, 12, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Makarević, J.; Hudak, L.; Juengel, E.; Kurosch, M.; Wiesner, C.; Bartsch, G.; Harder, S.; Haferkamp, A.; Blaheta, R.A. The cdk1-cyclin B complex is involved in everolimus triggered resistance in the PC3 prostate cancer cell line. Cancer Lett. 2011, 313, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Nowaz, S.; Makarevi, J.; Natsheh, I.; Werner, I.; Nelson, K.; Reiter, M.; Tsaur, I.; Mani, J.; Harder, S.; et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol. Cancer 2014, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Shen, T.; Halabi, S.; Kemeny, G.; Bitting, R.L.; Kartcheske, P.; Embree, E.; Morris, K.; Winters, C.; Jaffe, T.; et al. A phase II trial of temsirolimus in men with castration-resistant metastatic prostate cancer. Clin. Genitourin. Cancer 2013, 11, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Lai, C.J.; Bao, R.; Wang, D.G.; Wang, J.; Xu, G.X.; Atoyan, R.; Qu, H.; Yin, L.; Samson, M.; et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin. Cancer Res. 2012, 18, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Beagle, B.R.; Nguyen, D.M.; Mallya, S.; Tang, S.S.; Lu, M.; Zeng, Z.; Konopleva, M.; Vo, T.T.; Fruman, D.A. mTOR kinase inhibitors synergize with histone deacetylase inhibitors to kill B-cell acute lymphoblastic leukemia cells. Oncotarget 2015, 6, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Thelen, P.; Krahn, L.; Bremmer, F.; Strauss, A.; Brehm, R.; Loertzer, H. Synergistic effects of histone deacetylase inhibitor in combination with mTOR inhibitor in the treatment of prostate carcinoma. Int. J. Mol. Med. 2013, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zibelman, M.; Wong, Y.N.; Devarajan, K.; Malizzia, L.; Corrigan, A.; Olszanski, A.J.; Denlinger, C.S.; Roethke, S.K.; Tetzlaff, C.H.; Plimack, E.R. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Investig. New Drugs 2015, 33, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembla, V.; Groisberg, R.; Hess, K.; Fu, S.; Wheler, J.; Hong, D.S.; Janku, F.; Zinner, R.; Piha-Paul, S.A.; Ravi, V.; et al. Outcomes of patients with sarcoma enrolled in clinical trials of pazopanib combined with histone deacetylase, mTOR, Her2, or MEK inhibitors. Sci. Rep. 2017, 7, 15963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, L.; Roberts, J.L.; Sander, C.; Lee, J.; Kirkwood, J.M.; Poklepovic, A.; Dent, P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 2017, 8, 16367–16386. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Najafi, R.; Rutz, J.; Maxeiner, S.; Makarevic, J.; Roos, F.; Tsaur, I.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition as a treatment concept to combat temsirolimus-resistant bladder cancer cells. Oncotarget 2017, 8, 110016–110028. [Google Scholar] [CrossRef] [PubMed]
- Liberio, M.S.; Sadowski, M.C.; Soekmadji, C.; Davis, R.A.; Nelson, C.C. Differential effects of tissue culture coating substrates on prostate cancer cell adherence, morphology and behavior. PLoS ONE 2014, 9, e112122. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Entersz, I.; Butler, C.; Foty, R.A. Fibronectin matrix-mediated cohesion suppresses invasion of prostate cancer cells. BMC Cancer 2012, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Makarević, J.; Juengel, E.; Gasser, M.; Waaga-Gasser, A.M.; Kurosch, M.; Reiter, M.; Wedel, S.; Bartsch, G.; Haferkamp, A.; et al. Resistance to the mTOR-inhibitor RAD001 elevates integrin α2- and β1-triggered motility, migration and invasion of prostate cancer cells. Br. J. Cancer 2012, 107, 847–855. [Google Scholar] [PubMed] [Green Version]
- Juengel, E.; Makarević, J.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Resistance to the mTOR inhibitor temsirolimus alters adhesion and migration behavior of renal cell carcinoma cells through an integrin α5- and integrin β3-dependent mechanism. Neoplasia 2014, 16, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Altieri, D.C. Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle 2015, 14, 3242–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, M.M.; Williams, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Young, C.D.; Sarbassov, D.D.; Muller, W.J.; Brantley-Sieders, D.; Cook, R.S. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 2017, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Gu, X.; Dong, Y.; Tan, F.; Liu, Z.; Thiele, C.J.; Li, Z. PI3K and MAPK pathways mediate the BDNF/TrkB-increased metastasis in neuroblastoma. Tumour Biol. 2016, 37, 16227–16236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Slambrouck, S.; Jenkins, A.R.; Romero, A.E.; Steelant, W.F. Reorganization of the integrin α2 subunit controls cell adhesion and cancer cell invasion in prostate cancer. Int. J. Oncol. 2009, 34, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, N.E.; Zhang, Z.; Madamanchi, A.; Boyd, K.L.; O’Rear, L.D.; Nashabi, A.; Li, Z.; Dupont, W.D.; Zijlstra, A.; Zutter, M.M. The α2β1 integrin is a metastasis suppressor in mouse models and human cancer. J. Clin. Investig. 2011, 121, 226–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Slambrouck, S.; Hilkens, J.; Bisoffi, M.; Steelant, W.F. AsialoGM1 and integrin α2β1 mediate prostate cancer progression. Int. J. Oncol. 2009, 35, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Daignault-Newton, S.; Zhang, X.; Morrissey, C.; Hussain, M.H.; Keller, E.T.; Hall, C.L. Integrin α2β1 (α2β1) promotes prostate cancer skeletal metastasis. Clin. Exp. Metastasis 2013, 30, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hatakeyama, S.; Yu, S.Y.; Bao, X.; Ohyama, C.; Khoo, K.H.; Fukuda, M.N.; Fukuda, M. Core3 O-glycan synthase suppresses tumor formation and metastasis of prostate carcinoma PC3 and LNCaP cells through down-regulation of α2β1 integrin complex. J. Biol. Chem. 2009, 284, 17157–17169. [Google Scholar] [CrossRef] [PubMed]
- Pollan, S.G.; Huang, F.; Sperger, J.M.; Lang, J.M.; Morrissey, C.; Cress, A.E.; Chu, C.Y.; Bhowmick, N.A.; You, S.; Freeman, M.R.; et al. Regulation of inside-out β1-integrin activation by CDCP1. Oncogene 2018, 37, 2817–2836. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, A.; Goto, Y.; Matsushita, R.; Fukumoto, I.; Kato, M.; Nishikawa, R.; Sakamoto, S.; Enokida, H.; Nakagawa, M.; Ichikawa, T.; et al. Tumor-suppressive microRNA-223 inhibits cancer cell migration and invasion by targeting ITGA3/ITGB1 signaling in prostate cancer. Cancer Sci. 2016, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.K.; Tien, P.C.; Cheng, C.J.; Song, J.H.; Huang, C.; Lin, S.H.; Gallick, G.E. Talin1 phosphorylation activates β1 integrins: A novel mechanism to promote prostate cancer bone metastasis. Oncogene 2015, 34, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, J.; Muller, D.J.; Helenius, J. In PC3 prostate cancer cells ephrin receptors crosstalk to β1-integrins to strengthen adhesion to collagen type I. Sci. Rep. 2015, 5, 8206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtakoivu, R.; Pellinen, T.; Rantala, J.K.; Perälä, M.; Ivaska, J. Distinct roles of AKT isoforms in regulating β1-integrin activity, migration, and invasion in prostate cancer. Mol. Biol. Cell 2012, 23, 3357–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfay, L.; Schulz, V.V.; Frank, S.B.; Lamb, L.E.; Miranti, C.K. Receptor tyrosine kinase Met promotes cell survival via kinase-independent maintenance of integrin α3β1. Mol. Biol. Cell 2016, 27, 2493–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 4, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, A.; Goto, Y.; Matsushita, R.; Fukumoto, I.; Kato, M.; Nishikawa, R.; Sakamoto, S.; Enokida, H.; Nakagawa, M.; Ichikawa, T.; et al. Tumor-suppressive microRNA-223 inhibits cancer cell migration and invasion by targeting ITGA3/ITGB1 signaling in prostate cancer. Cancer Sci. 2016, 10, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Anderson, T.A.; Gard, J.M.; Sroka, I.C.; Strautman, S.R.; Nagle, R.B.; Morrissey, C.; Knudsen, B.S.; Cress, A.E. Characterization of Laminin Binding Integrin Internalization in Prostate Cancer Cells. J. Cell. Biochem. 2017, 118, 1038–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drivalos, A.; Chrisofos, M.; Efstathiou, E.; Kapranou, A.; Kollaitis, G.; Koutlis, G.; Antoniou, N.; Karanastasis, D.; Dimopoulos, M.A.; Bamias, A. Expression of α5-integrin, α7-integrin, Ε-cadherin, and N-cadherin in localized prostate cancer. Urol. Oncol. 2016, 34, 165.e11–165.e18. [Google Scholar] [CrossRef] [PubMed]
- Varzavand, A.; Drake, J.M.; Svensson, R.U.; Herndon, M.E.; Zhou, B.; Henry, M.D.; Stipp, C.S. Integrin α3β1 regulates tumor cell responses to stromal cells and can function to suppress prostate cancer metastatic colonization. Clin. Exp. Metastasis 2013, 30, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Dauselt, A.; Makarević, J.; Wiesner, C.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Lett. 2012, 324, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Guo, J.; Liao, Q.; Zhao, Y. β1 and β3 integrins in breast, prostate and pancreatic cancer: A novel implication. Oncol. Lett. 2018, 15, 5412–5416. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Yasui, H.; Outani, H.; Wakamatsu, T.; Hamada, K.; Nakai, T.; Yamada, S.; Myoui, A.; Araki, N.; Ueda, T.; et al. Combined targeting of mTOR and c-MET signaling pathways for effective management of epithelioid sarcoma. Mol. Cancer 2014, 13, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etnyre, D.; Stone, A.L.; Fong, J.T.; Jacobs, R.J.; Uppada, S.B.; Botting, G.M.; Rajanna, S.; Moravec, D.N.; Shambannagari, M.R.; Crees, Z.; et al. Targeting c-Met in melanoma: Mechanism of resistance and efficacy of novel combinatorial inhibitor therapy. Cancer Biol. Ther. 2014, 15, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; Nguyen, A.; Chandra, A.; Sidorov, M.K.; Yagnik, G.; Rick, J.; Han, S.W.; Chen, W.; Flanigan, P.M.; Schneidman-Duhovny, D.; et al. Cross-activating c-Met/β1 integrin complex drives metastasis and invasive resistance in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E8685–E8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarević, J.; Tawanaie, N.; Juengel, E.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Cross-communication between histone H3 and H4 acetylation and Akt-mTOR signalling in prostate cancer cells. J. Cell. Mol. Med. 2014, 18, 1460–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedel, S.; Hudak, L.; Seibel, J.M.; Makarević, J.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. Impact of combined HDAC and mTOR inhibition on adhesion, migration and invasion of prostate cancer cells. Clin. Exp. Metastasis 2011, 28, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Siva, A.C.; Kirkland, R.E.; Lin, B.; Maruyama, T.; McWhirter, J.; Yantiri-Wernimont, F.; Bowdish, K.S.; Xin, H. Selection of anti-cancer antibodies from combinatorial libraries by whole-cell panning and stringent subtraction with human blood cells. J. Immunol. Methods 2008, 330, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Veine, D.M.; Yao, H.; Stafford, D.R.; Fay, K.S.; Livant, D.L. A D-amino acid containing peptide as a potent, noncovalent inhibitor of α5β1 integrin in human prostate cancer invasion and lung colonization. Clin. Exp. Metastasis 2014, 31, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Salminen, J.K.; Tammela, T.L.; Auvinen, A.; Murtola, T.J. Antiepileptic drugs with histone deacetylase inhibition activity and prostate cancer risk: A population-based case-control study. Cancer Causes Control 2016, 27, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Gillespie, T.W.; Goodman, M.; Brodie, S.A.; Brandes, M.; Ribeiro, M.; Ramalingam, S.S.; Shin, D.M.; Khuri, F.R.; Brandes, J.C. Long-term use of valproic acid in US veterans is associated with a reduced risk of smoking-related cases of head and neck cancer. Cancer 2014, 120, 1394–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motawi, T.K.; Darwish, H.A.; Diab, I.; Helmy, M.W.; Noureldin, M.H. Combinatorial strategy of epigenetic and hormonal therapies: A novel promising approach for treating advanced prostate cancer. Life Sci. 2018, 198, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, X.; Zhu, S.; Hu, X.; Niu, H.; Zhang, X.; Zhu, D.; Nesa, E.U.; Tian, K.; Yuan, H. Hyper-acetylation contributes to the sensitivity of chemo-resistant prostate cancer cells to histone deacetylase inhibitor Trichostatin A. J. Cell. Mol. Med. 2018, 22, 1909–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Hudak, L.; Makarević, J.; Juengel, E.; Mani, J.; Borgmann, H.; Gust, K.M.; Schilling, D.; Bartsch, G.; Nelson, K.; et al. Intensified antineoplastic effect by combining an HDAC-inhibitor, an mTOR-inhibitor and low dosed interferon alpha in prostate cancer cells. J. Cell. Mol. Med. 2015, 19, 1795–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.Y.; Ding, W.; Li, T.Q.; Zhang, Y.X.; Zhao, S.C. Histone Deacetylase (HDAC) Inhibitor, Suberoylanilide Hydroxamic Acid (SAHA), Induces Apoptosis in Prostate Cancer Cell Lines via the Akt/FOXO3a Signaling Pathway. Med. Sci. Monit. 2017, 23, 5793–5802. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Wenig, S.C.; Tseng, P.H.; Lin, H.P.; Chen, C.S. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 2005, 280, 38879–38887. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Salumbides, B.; Van Erp, K.; Hammers, H.; Qian, D.Z.; Sanni, T.; Atadja, P.; Pili, R. Combination strategy targeting the hypoxia inducible factor-1α with mammalian target of rapamycin and histone deacetylase inhibitors. Clin. Cancer Res. 2008, 14, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Ku, S.Y.; Ramakrishnan, S.; Lasorsa, E.; Azabdaftari, G.; Godoy, A.; Pili, R. Combinatorial antitumor effect of HDAC and the PI3K-Akt-mTOR pathway inhibition in a Pten defecient model of prostate cancer. Oncotarget 2013, 4, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; An, J.; Yang, Y.; Wu, D.; Bai, Y.; Cao, W.; Ma, L.; Chen, J.; Yu, Z.; He, Y.; et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol. Med. 2018, 10, e8478. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Mani, J.; Vallo, S.; Tsaur, I.; Roos, F.; Chun, F.K.-H.; Blaheta, R.A. HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic mTOR Suppression. Cells 2018, 7, 129. https://doi.org/10.3390/cells7090129
Makarević J, Rutz J, Juengel E, Maxeiner S, Mani J, Vallo S, Tsaur I, Roos F, Chun FK-H, Blaheta RA. HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic mTOR Suppression. Cells. 2018; 7(9):129. https://doi.org/10.3390/cells7090129
Chicago/Turabian StyleMakarević, Jasmina, Jochen Rutz, Eva Juengel, Sebastian Maxeiner, Jens Mani, Stefan Vallo, Igor Tsaur, Frederik Roos, Felix K.-H. Chun, and Roman A. Blaheta. 2018. "HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic mTOR Suppression" Cells 7, no. 9: 129. https://doi.org/10.3390/cells7090129