Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling

 ,
,
Abstract
: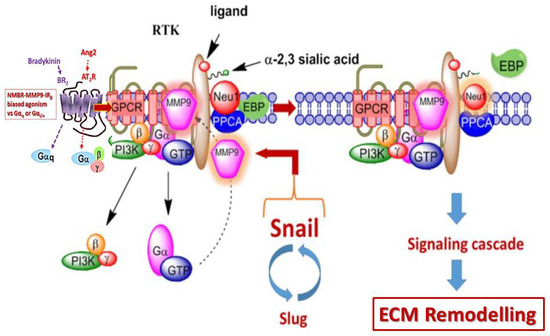
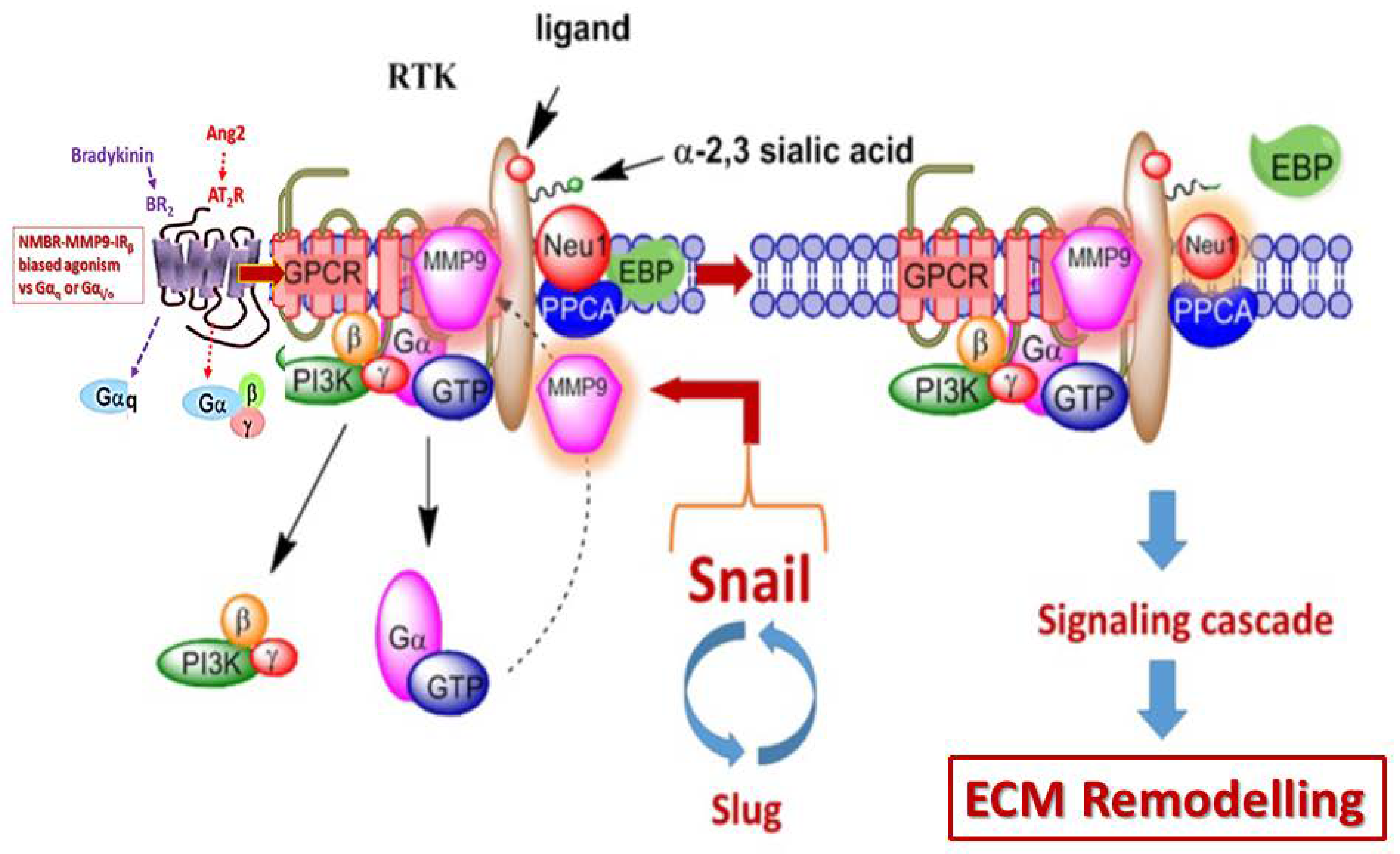{kind=link}
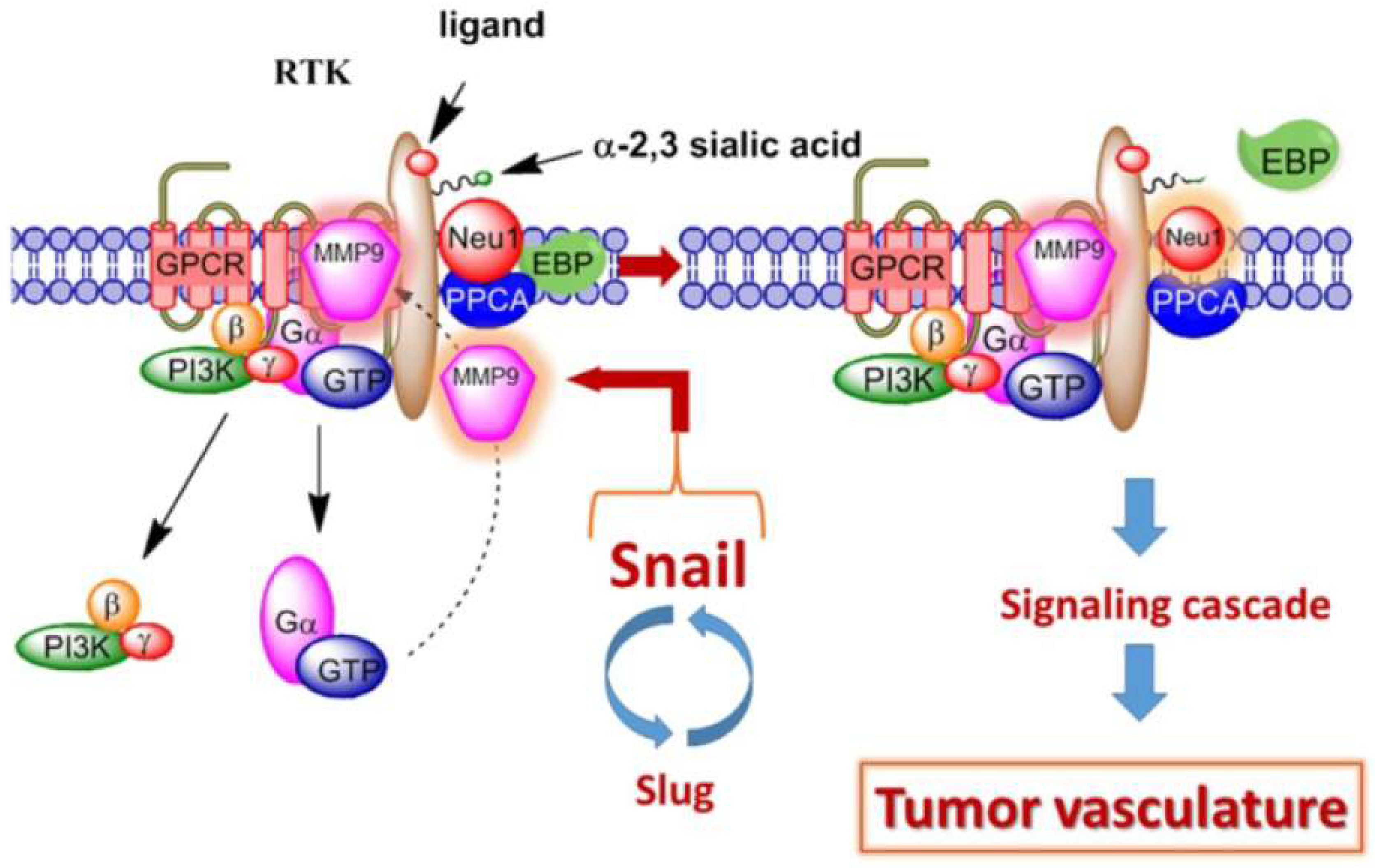{kind=link}
{kind=link}
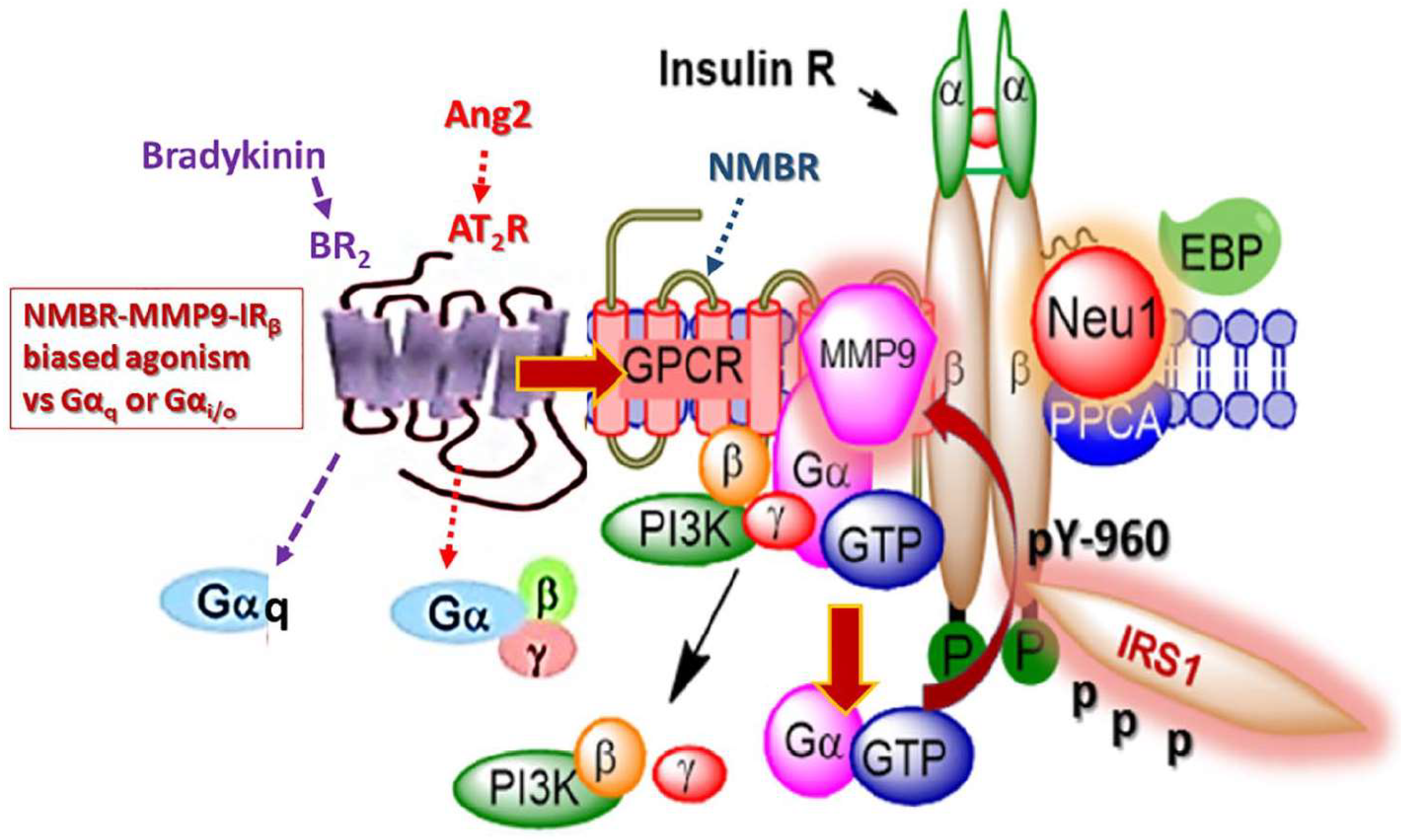{kind=link}
{kind=link}
1. Introduction
2. MMPs
2.1. Matrix Metalloproteinase-9 Crosstalk with Neuraminidase-1 on Cell-Surface Receptors
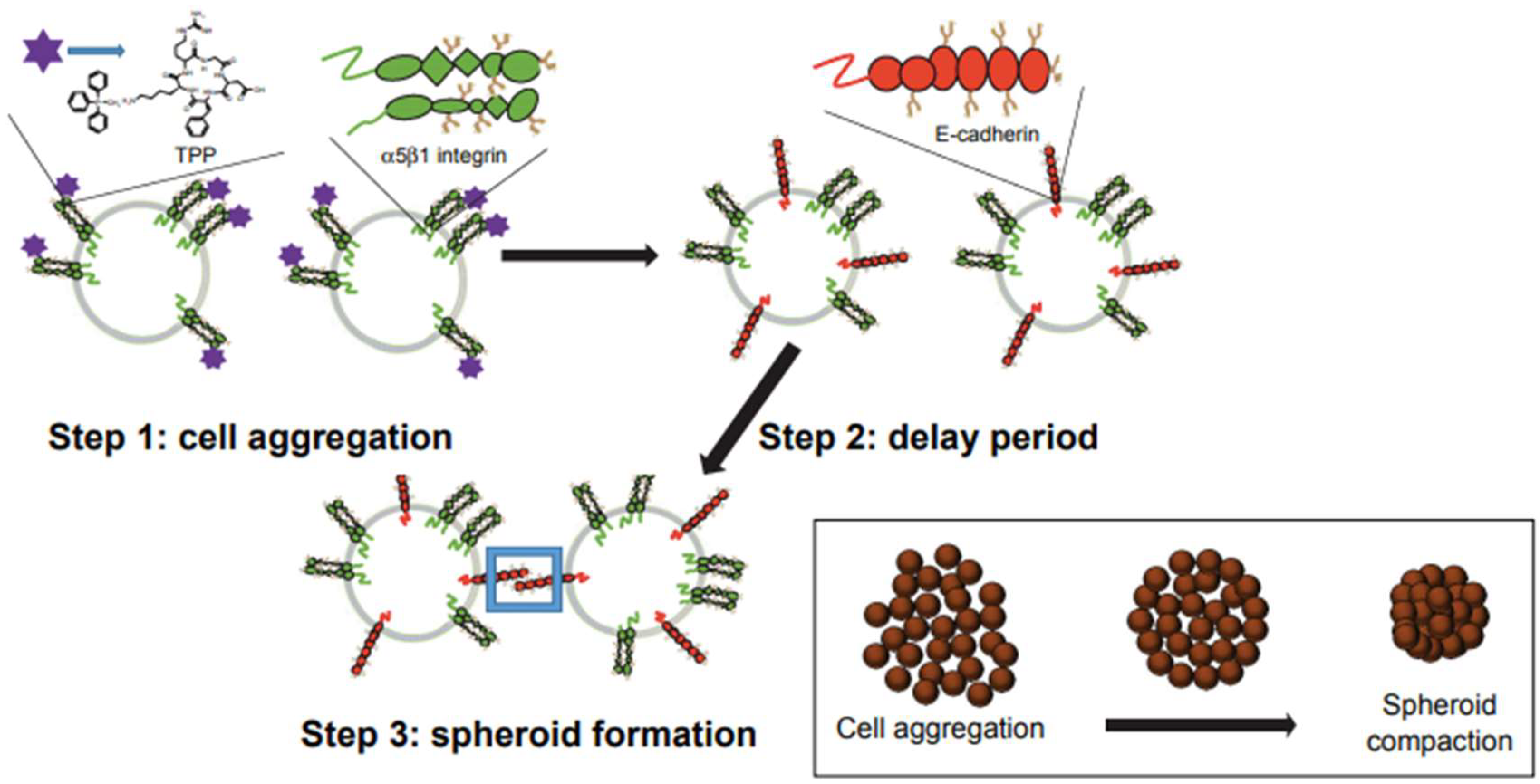
2.2. Integrins and Matrix Metalloproteinase-9
2.3. Matrix Metalloproteinase-9 and the TrkA Receptor
2.4. G protein-Coupled Receptors Biased Agonism to Activate TrkA
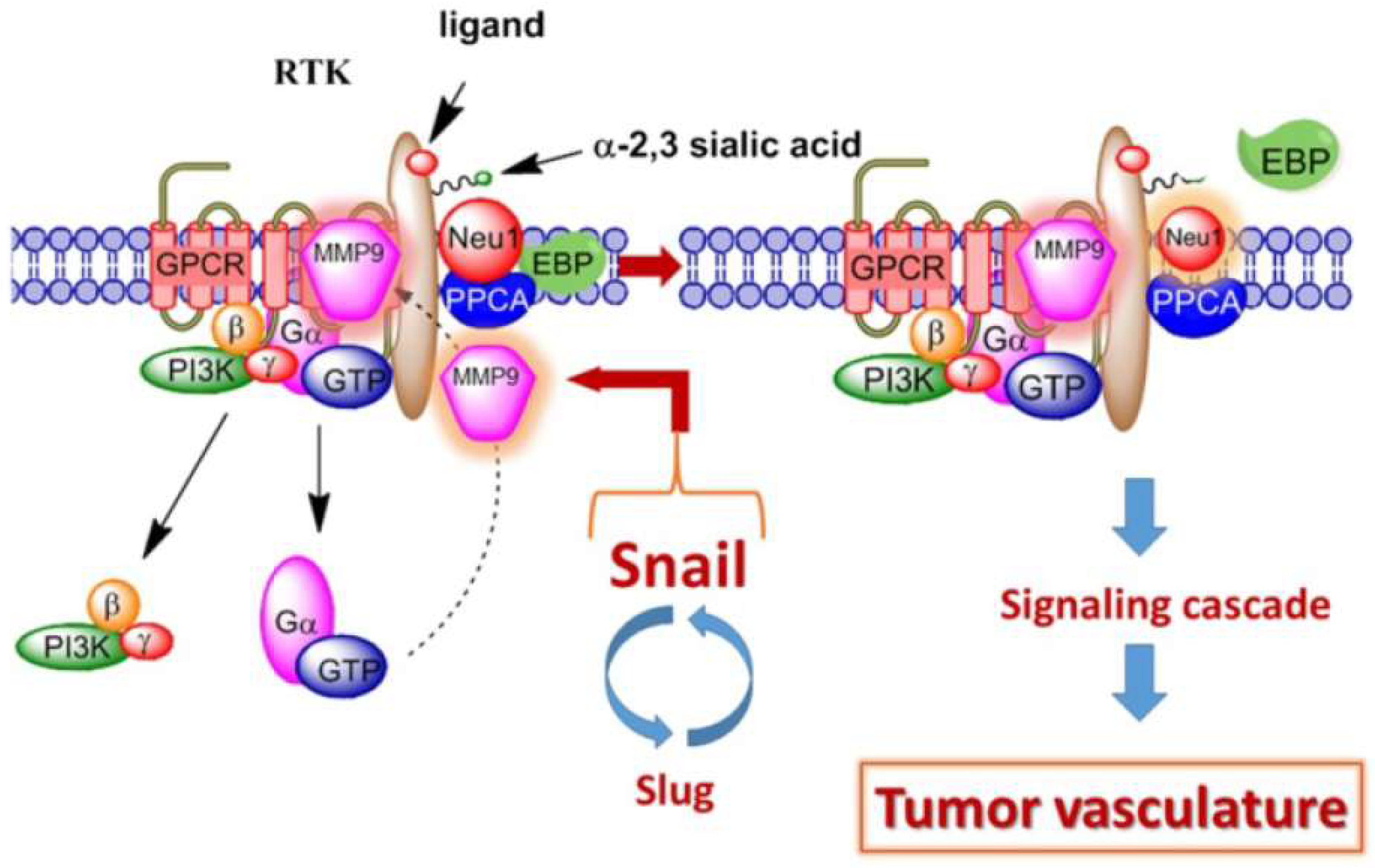
2.5. TrkA, MMP-9, and Angiogenesis
2.6. MMP-9 and EGFR Signaling
2.7. Biased Agonism of G-Protein Coupled Receptors to Activate EGFRs
2.8. MMP-9 and IR Signaling
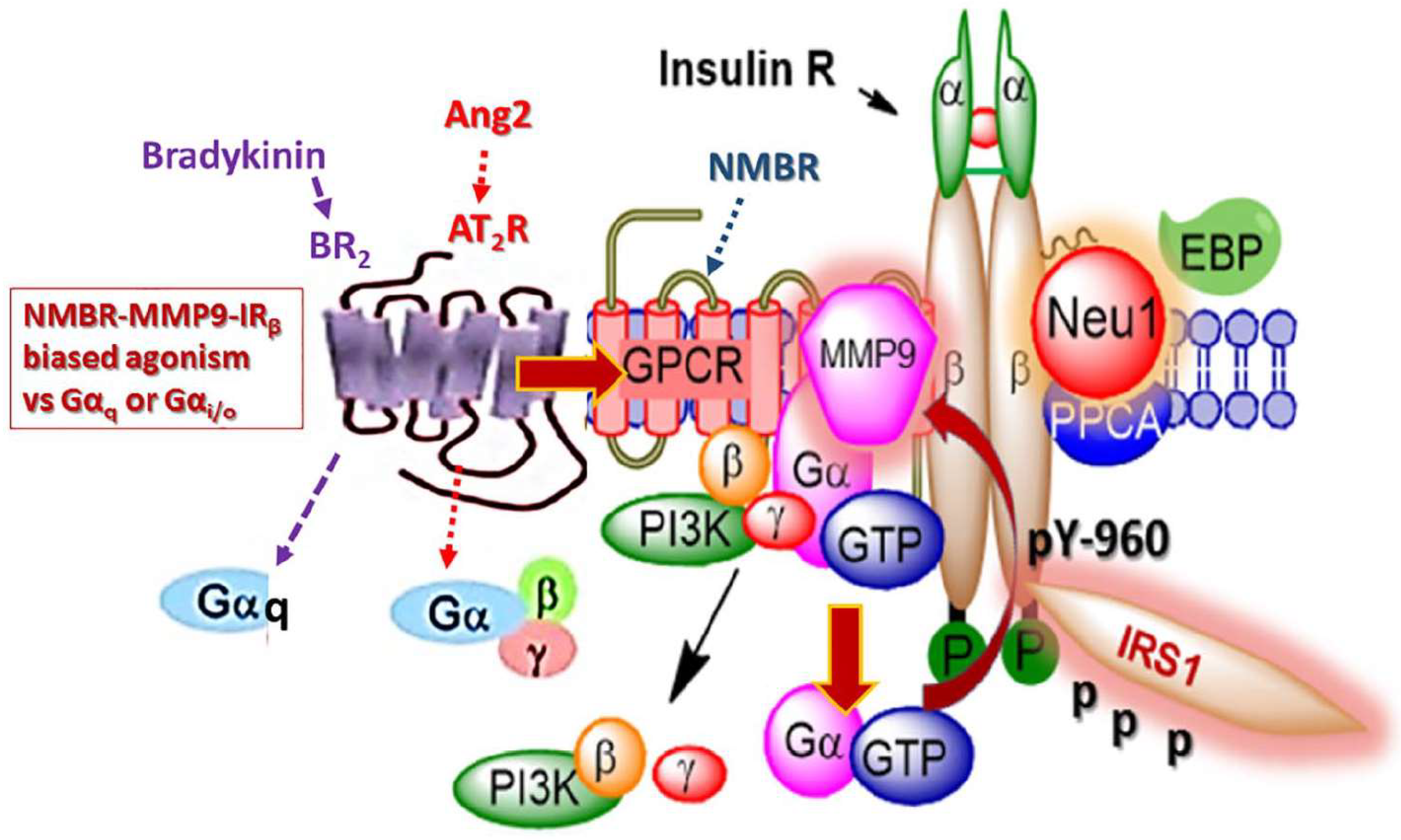
2.9. Biased Agonism of G-Protein-Coupled Receptors to Activate the Insulin Receptor
2.10. IR and Cardiac Extracellular Matrix Remodeling
2.11. Matrix Metalloproteinase-9 and TLRs
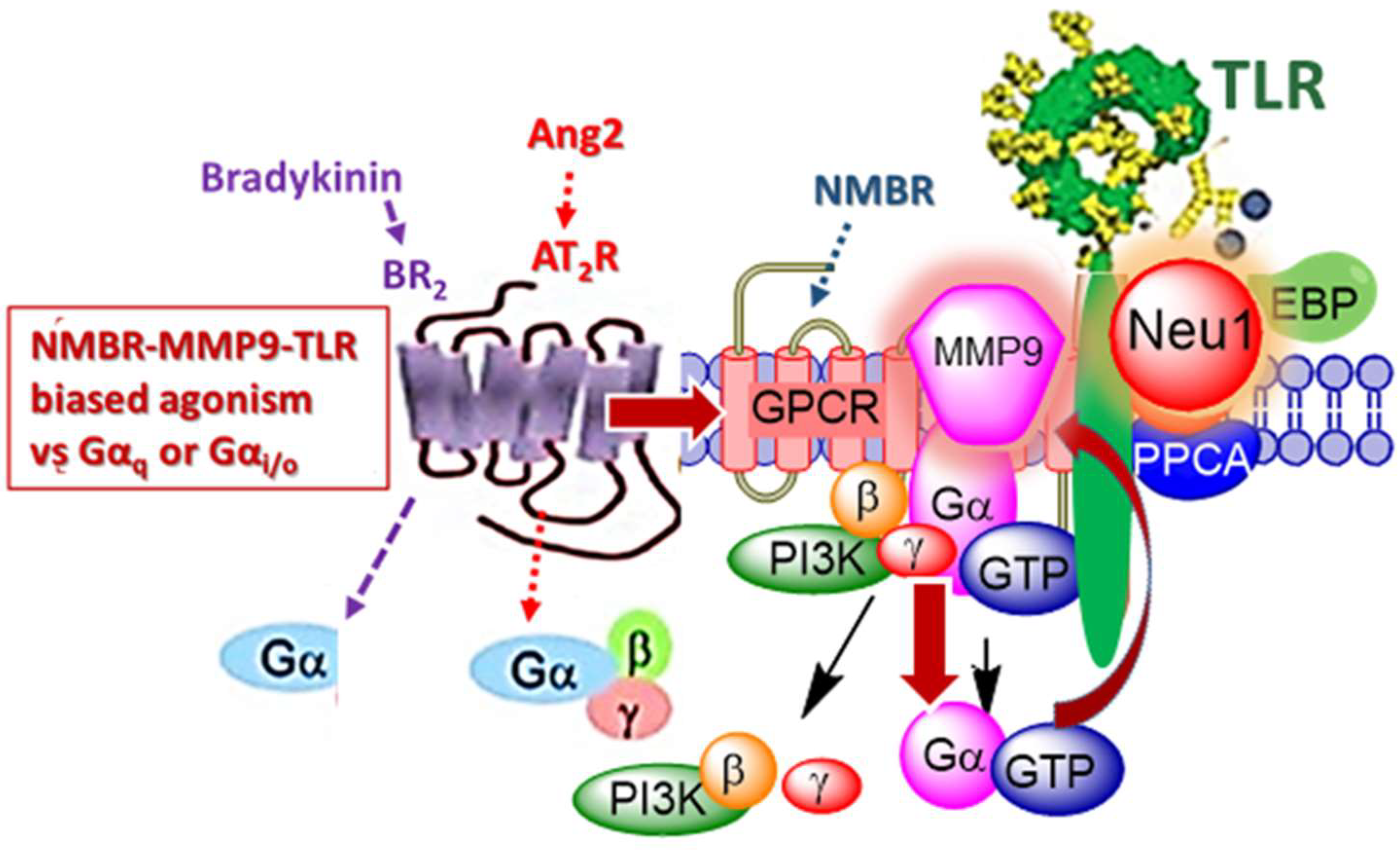
2.12. Biased G-Protein-Coupled Receptors Agonism to Activate TLRs
2.13. TLR Induced MMP-9 Activity in Chronic Inflammation
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloproteinase |
| ADAMTS | ADAMs with thrombospondin motifs |
| Akt | protein kinase B |
| Ang II | angiotensin II |
| ASM | airway smooth muscle |
| AT2R | angiotensin II receptor type I |
| BR2 | bradykinin |
| cycloRGDfK | cyclic Arg-Gly-Asp-D-Phe-Lys |
| DAMP | damage-associated molecular pattern |
| DR | diabetic retinopathy |
| EBP | elastin binding protein |
| ECM | extracellular matrix |
| EDA | extra domain A |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| FGF | fibroblast growth factor |
| GnRH | gonadotropin-releasing hormone |
| GPCR | G protein-coupled receptor |
| GPI | glycosylphosphatidylinositol |
| Grb-2 | growth factor receptor-bound protein 2 |
| H-RAS | RasV12 |
| ICAM | intercellular adhesion molecule-1 |
| IFN | type I interferon |
| Ig | immunoglobulin |
| IGF-R1 | insulin growth factor receptor-1 |
| IL-1 | interleukin-1 |
| IR | insulin receptor |
| IRS1 | insulin receptor substrate 1 |
| IRβ | insulin receptor β subunit |
| LH | leuteinizing hormone |
| LPS | lipopolysaccharide |
| LRP | low density lipoprotein receptor-related protein |
| LRR | leucine-rich region |
| LTP | long-term potentiation |
| Mal | MyD88 adaptor-like |
| MI | myocardia infarction |
| MMP | matrix metalloproteinase |
| MMP-9 | matrix metalloproteinase-9 |
| MT-MMP | membrane-type MMP |
| MT1-MMP | membrane-type 1 MMP |
| MUC1 | mucin-1 |
| MyD88 | myeloid differentiating factor-88 |
| Neu-1 | neuraminidase-1 |
| NGF | nerve growth factor |
| NMBR | neuromedin B receptor |
| NSCLC | non-small cell lung cancer |
| ODN | oligodeoxynucleotide |
| PACAP | pituitary adenylate cyclase-activating polypeptide |
| PAMP | pathogen-associated molecular pattern |
| PEX | hemopexin-like |
| PI3K | phophatidylinositol 3-kinase |
| PI3K-AKT | phosphoinositide 3-kinase-protein kinase B |
| PPCA | protective protein cathepsin A |
| PRR | pattern-recognition receptor |
| RAAS | renin-angiotensin-aldosterone system |
| RECK | reversion inducing cysteine-rich protein with Kazal motifs |
| RGD | arginine-glycine-aspartic acid |
| RSD | renal sympathetic denervation |
| RTK | receptor tyrosine kinase |
| T2DM | type 2 diabetes mellitus |
| TGF-β1 | tumor growth factor β1 |
| TIMP | tissue inhibitors of metalloproteinase |
| TIR | Toll/interleukin-1 receptor |
| TLR | Toll-like receptor |
| TLR-2 | Toll-like receptor-2 |
| TLR-4 | Toll-like receptor-4 |
| TNF-α | tumor necrosis factor-α |
| TPP | triphenylphosphonium cation |
| TRAM | TRIF-related adaptor molecule |
| TRIF | TIR domain-containing adaptor-inducing IFN-β |
| TrkA | tropomyosin receptor kinase A |
| VEGF | vascular endothelial growth factor |
References
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Friedl, P. Interstitial cell migration: Integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 2009, 339, 83. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Ivan, S. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and timps. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, B.; Franz-Xaver, G.-R.; Walter, S. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 1993, 331, 134–140. [Google Scholar] [Green Version]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Rodriguez, D.; Petitclerc, E.; Kim, J.J.; Hangai, M.; Yuen, S.M.; Davis, G.E.; Brooks, P.C. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J. Cell Biol. 2001, 154, 1069–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galasso, O.; Familiari, F.; De Gori, M.; Gasparini, G. Recent findings on the role of gelatinases (matrix metalloproteinase-2 and-9) in osteoarthritis. Adv. Orthop. 2012, 2012, 834208. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. BioMed Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur. J. Cell Biol. 1997, 74, 111–122. [Google Scholar] [PubMed]
- Fridman, R.; Toth, M.; Chvyrkova, I.; Meroueh, S.O.; Mobashery, S. Cell surface association of matrix metalloproteinase-9 (gelatinase B). Cancer Metastasis Rev. 2003, 22, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, R.; Zucker, S.; Toole, B.P. EMMPRIN (CD147), an inducer of matrix metalloproteinase synthesis, also binds interstitial collagenase to the tumor cell surface. Cancer Res. 2000, 60, 888–891. [Google Scholar] [PubMed]
- Toth, M.; Sado, Y.; Ninomiya, Y.; Fridman, R. Biosynthesis of α2(IV) and α(IV) chains of collagen IV and interactions with matrix metalloproteinase-9. J. Cell. Physiol. 1999, 180, 131–139. [Google Scholar] [CrossRef]
- Fiore, E.; Fusco, C.; Romero, P.; Stamenkovic, I. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene 2002, 21, 5213–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, C.A.; Phillips, P.G.; Niedbala, M.J.; Jeffrey, J.J. Localization and activation of type IV collagenase/gelatinase at endothelial focal contacts. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997, 272, L813–L822. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn-Dantona, E.; Ruiz, J.F.; Bornstein, P.; Strickland, D.K. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J. Biol. Chem. 2001, 276, 15498–15503. [Google Scholar] [CrossRef] [PubMed]
- Ramos-DeSimone, N.; Hahn-Dantona, E.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 1999, 274, 13066–13076. [Google Scholar] [CrossRef] [PubMed]
- Puyraimond, A.; Fridman, R.; Lemesle, M.; Arbeille, B.; Menashi, S. MMP-2 colocalizes with caveolae on the surface of endothelial cells. Exp. Cell Res. 2001, 262, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Tomari, T.; Koshikawa, N.; Kajita, M.; Itoh, Y.; Sato, H.; Tojo, H.; Yana, I.; Seiki, M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002, 21, 3949–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulkhalek, S.; Szewczuk, M.R. NEU1 sialidase and matrix metalloproteinase-9 cross-talk regulates nucleic acid-induced endosomal toll-like receptor-7 and -9 activation, cellular signaling and pro-inflammatory responses. Cell. Signal. 2013, 25, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Jayanth, P.; Amith, S.R.; Gee, K.; Szewczuk, M.R. NEU1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell. Signal. 2010, 22, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, A.M.; Abdulkhalek, S.; Cheng, T.S.W.; Alghamdi, F.; Jayanth, P.; O’Shea, L.K.; Geen, O.; Arvizu, L.A.; Szewczuk, M.R. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell. Signal. 2013, 25, 2587–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghamdi, F.; Guo, M.; Abdulkhalek, S.; Crawford, N.; Amith, S.R.; Szewczuk, M.R. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell. Signal. 2014, 26, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Amith, S.R.; Franchuk, S.L.; Jayanth, P.; Guo, M.; Finlay, T.; Gilmour, A.; Guzzo, C.; Gee, K.; Beyaert, R.; et al. NEU1 sialidase and matrix metalloproteinase-9 cross-talk is essential for toll-like receptor activation and cellular signaling. J. Biol. Chem. 2011, 286, 36532–36549. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Seyrantepe, V.; Landry, K.; Ahmad, R.; Ahmad, A.; Stamatos, N.M.; Pshezhetsky, A.V. Monocyte differentiation up-regulates the expression of the lysosomal sialidase, NEU1, and triggers its targeting to the plasma membrane via major histocompatibility complex class II-positive compartments. J. Biol. Chem. 2006, 281, 27526–27538. [Google Scholar] [CrossRef] [PubMed]
- Hinek, A.; Pshezhetsky, A.V.; von Itzstein, M.; Starcher, B. Lysosomal sialidase (neuraminidase-1) is targeted to the cell surface in a multiprotein complex that facilitates elastic fiber assembly. J. Biol. Chem. 2006, 281, 3698–3710. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Elsliger, M.-A.; Chang, Y.; Richard, C.; Thomas, G.; Carey, W.; Tylki-Szymanska, A.; Czartoryska, B.; Buchholz, T.; Criado, G.R. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum. Mol. Genet. 2000, 9, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Carubelli, I.; Stamatos, N.M. Sialidase expression in activated human T lymphocytes influences production of IFN-γ. J. Leukoc. Biol. 2007, 81, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Hrynyk, M.; Szewczuk, M.R. A novel G-protein-coupled receptor-signaling platform and its targeted translation in human disease. Res. Rep. Biochem. 2013, 2013, 17–30. [Google Scholar]
- Abdulkhalek, S.; Geen, O.D.; Brodhagen, L.; Haxho, F.; Alghamdi, F.; Allison, S.; Simmons, D.J.; O’Shea, L.K.; Neufeld, R.J.; Szewczuk, M.R. Transcriptional factor snail controls tumor neovascularization, growth and metastasis in mouse model of human ovarian carcinoma. Clin. Transl. Med. 2014, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002, 21, 3241–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorda, M.; Olmeda, D.; Vinyals, A.; Valero, E.; Cubillo, E.; Llorens, A.; Cano, A.; Fabra, A. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J. Cell Sci. 2005, 118, 3371–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, J.; Zhang, K.; Chen, J. Role of integrins in regulating proteases to mediate extracellular matrix remodeling. Cancer Microenviron. 2012, 5, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Heino, J. Adhesion receptors and cell invasion: Mechanisms of integrin-guided degradation of extracellular matrix. Cell. Mol. Life Sci. 2000, 57, 16–24. [Google Scholar] [CrossRef] [PubMed]
- DiPersio, C.M.; Shao, M.; Di Costanzo, L.; Kreidberg, J.A.; Hynes, R.O. Mouse keratinocytes immortalized with large T antigen acquire alpha3beta1 integrin-dependent secretion of MMP-9/gelatinase B. J. Cell Sci. 2000, 113, 2909–2921. [Google Scholar] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Sambi, M.; Qorri, B.; Frank, S.S.; Mouhamed, Y.; Kalaydina, R.-V.; Mendonza, N.; Szewczuk, M.R. Novel use of peptides to facilitate the formation of 3D multicellular tumor spheroids. Curr. Top. Pept. Protein Res. 2017, 18, 25–34. [Google Scholar]
- Haq, S.; Samuel, V.; Haxho, F.; Akasov, R.; Leko, M.; Burov, S.V.; Markvicheva, E.; Szewczuk, M.R. Sialylation facilitates self-assembly of 3D multicellular prostaspheres by using cyclo-RGDFK(TPP) peptide. OncoTargets Ther. 2017, 10, 2427–2447. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Stefanidakis, M.; Koivunen, E. Cell-surface association between matrix metalloproteinases and integrins: Role of the complexes in leukocyte migration and cancer progression. Blood 2006, 108, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Stawarski, M.; Stefaniuk, M.; Wlodarczyk, J. Matrix metalloproteinase-9 involvement in the structural plasticity of dendritic spines. Front. Neuroanat. 2014, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Bijata, M.; Labus, J.; Guseva, D.; Stawarski, M.; Butzlaff, M.; Dzwonek, J.; Schneeberg, J.; Bohm, K.; Michaluk, P.; Rusakov, D.A.; et al. Synaptic remodeling depends on signaling between serotonin receptors and the extracellular matrix. Cell Rep. 2017, 19, 1767–1782. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, A.; Lapinska, J.; Rylski, M.; McKay, R.D.; Kaczmarek, L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J. Neurosci. 2002, 22, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-b.; Bozdagi, O.; Nikitczuk, J.S.; Zhai, Z.W.; Zhou, Q.; Huntley, G.W. Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc. Natl. Acad. Sci. USA 2008, 105, 19520–19525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaluk, P.; Mikasova, L.; Groc, L.; Frischknecht, R.; Choquet, D.; Kaczmarek, L. Matrix metalloproteinase-9 controls nmda receptor surface diffusion through integrin β1 signaling. J. Neurosci. 2009, 29, 6007–6012. [Google Scholar] [CrossRef] [PubMed]
- Bellone, C.; Nicoll, R.A. Rapid bidirectional switching of synaptic nmda receptors. Neuron 2007, 55, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, I.; Sariyer, I.K.; Lecht, S.; Brown, M.C.; Walsh, E.M.; Tuszynski, G.P.; Safak, M.; Lazarovici, P.; Marcinkiewicz, C. Integrin α9β1 is a receptor for nerve growth factor and other neurotrophins. J. Cell Sci. 2008, 121, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bothwell, M. Functional interactions of neurotrophins and neurotrophin receptors. Ann. Rev. Neurosci. 1995, 18, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Snider, W.D. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell 1994, 77, 627–638. [Google Scholar] [CrossRef]
- Jing, S.; Tapley, P.; Barbacid, M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron 1992, 9, 1067–1079. [Google Scholar] [CrossRef]
- Johnson, D.; Lanahan, A.; Buck, C.R.; Sehgal, A.; Morgan, C.; Mercer, E.; Bothwell, M.; Chao, M. Expression and structure of the human NGF receptor. Cell 1986, 47, 545–554. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Hempstead, B.L.; Martin-Zanca, D.; Chao, M.V.; Parada, L.F. The trk proto-oncogene product: A signal transducing receptor for nerve growth factor. Science 1991, 252, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Jing, S.Q.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Schneider, R.; Schweiger, M. A novel modular mosaic of cell adhesion motifs in the extracellular domains of the neurogenic trk and trkB tyrosine kinase receptors. Oncogene 1991, 6, 1807–1811. [Google Scholar] [PubMed]
- Woronowicz, A.; Amith, S.R.; De Vusser, K.; Laroy, W.; Contreras, R.; Basta, S.; Szewczuk, M.R. Dependence of neurotrophic factor activation of trk tyrosine kinase receptors on cellular sialidase. Glycobiology 2007, 17, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Woronowicz, A.; Amith, S.R.; Davis, V.W.; Jayanth, P.; De Vusser, K.; Laroy, W.; Contreras, R.; Meakin, S.O.; Szewczuk, M.R. Trypanosome trans-sialidase mediates neuroprotection against oxidative stress, serum/glucose deprivation, and hypoxia-induced neurite retraction in Trk-expressing PC12 cells. Glycobiology 2007, 17, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dridi, L.; Seyrantepe, V.; Fougerat, A.; Pan, X.; Bonneil, É.; Thibault, P.; Moreau, A.; Mitchell, G.A.; Heveker, N.; Cairo, C.W.; et al. Positive regulation of insulin signaling by neuraminidase 1. Diabetes 2013, 62, 2338–2346. [Google Scholar] [CrossRef] [PubMed]
- Arabkhari, M.; Bunda, S.; Wang, Y.; Wang, A.; Pshezhetsky, A.V.; Hinek, A. Desialylation of insulin receptors and IGF-1 receptors by neuraminidase-1 controls the net proliferative response of l6 myoblasts to insulin. Glycobiology 2010, 20, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Guo, M.; Amith, S.R.; Jayanth, P.; Szewczuk, M.R. G-protein coupled receptor agonists mediate neu1 sialidase and matrix metalloproteinase-9 cross-talk to induce transactivation of toll-like receptors and cellular signaling. Cell. Signal. 2012, 24, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Amith, S.R.; Jayanth, P.; Finlay, T.; Franchuk, S.; Gilmour, A.; Abdulkhalek, S.; Szewczuk, M.R. Detection of Neu1 sialidase activity in regulating toll-like receptor activation. J. Vis. Exp. 2010, 43, 2142. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V.; Ashmarina, L.I. Desialylation of surface receptors as a new dimension in cell signaling. Biochemistry 2013, 78, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V.; Hinek, A. Where catabolism meets signalling: Neuraminidase 1 as a modulator of cell receptors. Glycoconj. J. 2011, 28, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase–G-protein-coupled receptor signalling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Waters, C.; Moughal, N.A.; Sambi, B.S.; Pyne, S. Receptor tyrosine kinase–GPCR signal complexes. Biochem. Soc. Trans. 2003, 31, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Waters, C.M.; Long, J.S.; Moughal, N.A.; Tigyi, G.; Pyne, S. Receptor tyrosine kinase-G-protein coupled receptor complex signaling in mammalian cells. Adv. Enzym. Regul. 2007, 47, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haxho, F.; Alghamdi, F.; Neufeld, R.J.; Szewczuk, M.R. Novel insulin receptor signaling platform. Int. J. Diabetes Clin. Res. 2014, 1, 005. [Google Scholar] [CrossRef]
- Onfroy, L.; Galandrin, S.; Pontier, S.M.; Seguelas, M.H.; N’Guyen, D.; Senard, J.M.; Gales, C. G protein stoichiometry dictates biased agonism through distinct receptor-G protein partitioning. Sci. Rep. 2017, 7, 7885. [Google Scholar] [CrossRef] [PubMed]
- Khoury, E.; Clément, S.; Laporte, S.A. Allosteric and biased G protein-coupled receptor signaling regulation: Potentials for new therapeutics. Front. Endocrinol. 2014, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the promise of “biased” G protein–coupled receptor agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; May, L.T.; Parton, R.G.; Sexton, P.M.; Christopoulos, A. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 2017, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, S.J.; Changeux, J.-P. Biased allostery. Biophys. J. 2016, 111, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Sengmany, K.; Singh, J.; Stewart, G.D.; Conn, P.J.; Christopoulos, A.; Gregory, K.J. Biased allosteric agonism and modulation of metabotropic glutamate receptor 5: Implications for optimizing preclinical neuroscience drug discovery. Neuropharmacology 2017, 115, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, F.S.; Chao, M.V. Activation of trk neurotrophin receptors in the absence of neurotrophins. Proc. Natl. Acad. Sci. USA 2001, 98, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, R.; Chen, Z.-Y.; Lee, F.S.; Chao, M.V. Transactivation of trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J. Neurosci. 2004, 24, 6650–6658. [Google Scholar] [CrossRef] [PubMed]
- Stephens, R.M.; Loeb, D.M.; Copeland, T.D.; Pawson, T.; Greene, L.A.; Kaplan, D.R. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron 1994, 12, 691–705. [Google Scholar] [CrossRef]
- Nickols, H.H.; Conn, P.J. Development of allosteric modulators of gpcrs for treatment of cns disorders. Neurobiol. Dis. 2014, 61, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Conn, P.J. Allosteric modulation of gpcrs: New insights and potential utility for treatment of schizophrenia and other cns disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D.; Klagsbrun, M.; Folkman, J. Vasculogenesis, angiogenesis, and growth factors: Ephrins enter the fray at the border. Cell 1998, 93, 661–664. [Google Scholar] [CrossRef]
- Calza, L.; Giardino, L.; Giuliani, A.; Aloe, L.; Levi-Montalcini, R. Nerve growth factor control of neuronal expression of angiogenetic and vasoactive factors. Proc. Natl. Acad. Sci. USA 2001, 98, 4160–4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verslegers, M.; Lemmens, K.; Van Hove, I.; Moons, L. Matrix metalloproteinase-2 and -9 as promising benefactors in development, plasticity and repair of the nervous system. Prog. Neurobiol. 2013, 105, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Mangieri, D.; Benagiano, V.; Crivellato, E.; Ribatti, D. Nerve growth factor as an angiogenic factor. Microvasc. Res. 2008, 75, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Salis, M.B.; Pinna, A.; Graiani, G.; Manni, L.; Madeddu, P. Nerve growth factor promotes angiogenesis and arteriogenesis in ischemic hindlimbs. Circulation 2002, 106, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Lempereur, L.; Presta, M.; Ribatti, D.; Lombardo, G.; Lazarovici, P.; Zappalà, G.; Pafumi, C.; Bernardini, R. Nerve growth factor–endothelial cell interaction leads to angiogenesis in vitro and in vivo. FASEB J. 2002, 16, 1307–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bray, P.; McCaffrey, T.; March, K.; Hempstead, B.L.; Kraemer, R. P75ntr mediates neurotrophin-induced apoptosis of vascular smooth muscle cells. Am. J. Pathol. 2000, 157, 1247–1258. [Google Scholar] [CrossRef]
- Graiani, G.; Emanueli, C.; Desortes, E.; Van Linthout, S.; Pinna, A.; Figueroa, C.; Manni, L.; Madeddu, P. Nerve growth factor promotes reparative angiogenesis and inhibits endothelial apoptosis in cutaneous wounds of type 1 diabetic mice. Diabetologia 2004, 47, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-J.; Kwak, H.-J.; Lee, H.-C.; Yoo, D.-H.; Park, I.-C.; Kim, M.-S.; Lee, S.-H.; Rhee, C.H.; Hong, S.-I. Nerve growth factor induces endothelial cell invasion and cord formation by promoting matrix metalloproteinase-2 expression through the phosphatidylinositol 3-kinase/akt signaling pathway and AP-2 transcription factor. J. Biol. Chem. 2007, 282, 30485–30496. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, R.; Hempstead, B.L. Neurotrophins: Novel mediators of angiogenesis. Front. Biosci. 2003, 8, s1181–s1186. [Google Scholar] [PubMed]
- Arroyo, A.G.; Iruela-Arispe, M.L. Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc. Res. 2010, 86, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.W.; Zhou, X.F.; Foster, B.K.; Grills, B.L.; Xu, J.; Xian, C.J. Roles of neurotrophins in skeletal tissue formation and healing. J. Cell. Physiol. 2018, 233, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.W.; Chung, R.; Ruan, C.S.; Chim, S.M.; Kuek, V.; Dwivedi, P.P.; Hassanshahi, M.; Chen, K.M.; Xie, Y.; Chen, L.; et al. Neurotrophin-3 induces BMP-2 and VEGF activities and promotes the bony repair of injured growth plate cartilage and bone in rats. J. Bone Miner. Res. 2016, 31, 1258–1274. [Google Scholar] [CrossRef] [PubMed]
- Saran, U.; Gemini Piperni, S.; Chatterjee, S. Role of angiogenesis in bone repair. Arch. Biochem. Biophys. 2014, 561, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The erbb signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, T.; Fujita, Y.; Naoki, H.; Aoki, K.; Kamioka, Y.; Matsuda, M. Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. eLife 2015, 4, e05178. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Prenzel, N.; Ullrich, A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002, 62, 6329–6336. [Google Scholar] [PubMed]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884. [Google Scholar] [CrossRef] [PubMed]
- Paye, A.; Truong, A.; Yip, C.; Cimino, J.; Blacher, S.; Munaut, C.; Cataldo, D.; Foidart, J.M.; Maquoi, E.; Collignon, J. EGFR activation and signaling in cancer cells are enhanced by the membrane-bound metalloprotease MT4-MMP. Cancer Res. 2014, 74, 6758–6770. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Dai, J.; Park, Y.-h.; Fai, L.Y.; Wang, L.; Pratheeshkumar, P.; Son, Y.-O.; Kondo, K.; Xu, M.; Luo, J. Activation of epidermal growth factor receptor/p38/hypoxia-inducible factor-1α is pivotal for angiogenesis and tumorigenesis of malignantly transformed cells induced by hexavalent chromium. J. Biol. Chem. 2016, 291, 16271–16281. [Google Scholar] [CrossRef] [PubMed]
- Overland, A.C.; Insel, P.A. Heterotrimeric G proteins directly regulate MMP14/membrane type-1 matrix metalloprotease: A novel mechanism for GPCR-EGFR transactivation. J. Biol. Chem. 2015, 290, 9941–9947. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.D.C.; Symowicz, J.; Ning, Y.; Gutierrez, E.; Fishman, D.A.; Adley, B.P.; Stack, M.S.; Hudson, L.G. Matrix metalloproteinase 9 is a mediator of epidermal growth factor–dependent E-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008, 68, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Berna, M.J.; Mantey, S.; Sancho, V.; Ridnour, L.; Wink, D.A.; Chan, D.; Giaccone, G.; Jensen, R.T. Neuromedin B receptors regulate egf receptor tyrosine phosphorylation in lung cancer cells. Eur. J. Pharmacol. 2010, 637, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Lillehoj, E.P.; Hyun, S.W.; Feng, C.; Zhang, L.; Liu, A.; Guang, W.; Nguyen, C.; Luzina, I.G.; Atamas, S.P.; Passaniti, A.; et al. NEU1 sialidase expressed in human airway epithelia regulates epidermal growth factor receptor (EGFR) and MUC1 protein signaling. J. Biol. Chem. 2012, 287, 8214–8231. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Moss, N.M.; Stack, M.S. EGF-receptor regulation of matrix metalloproteinases in epithelial ovarian carcinoma. Future Oncol. 2009, 5, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by G protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.W.; Lawrence, M.C. Ligand-induced activation of the insulin receptor: A multi-step process involving structural changes in both the ligand and the receptor. Bioessays 2009, 31, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Molecular basis of signaling specificity of insulin and igf receptors: Neglected corners and recent advances. Front. Endocrinol. 2012, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. The insulin receptor and its signal transduction network. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hirabara, S.M.; Gorjao, R.; Vinolo, M.A.; Rodrigues, A.C.; Nachbar, R.T.; Curi, R. Molecular targets related to inflammation and insulin resistance and potential interventions. J. Biomed. Biotechnol. 2012, 2012, 379024. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.R.; Nachbar, R.T.; Gorjao, R.; Vinolo, M.A.; Festuccia, W.T.; Lambertucci, R.H.; Cury-Boaventura, M.F.; Silveira, L.R.; Curi, R.; Hirabara, S.M. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: Importance of the mitochondrial function. Lipids Health Dis. 2012, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaise, S.; Romier, B.; Kawecki, C.; Ghirardi, M.; Rabenoelina, F.; Baud, S.; Duca, L.; Maurice, P.; Heinz, A.; Schmelzer, C.E.; et al. Elastin-derived peptides are new regulators of insulin resistance development in mice. Diabetes 2013, 62, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Fischoeder, A.; Meyborg, H.; Stibenz, D.; Fleck, E.; Graf, K.; Stawowy, P. Insulin augments matrix metalloproteinase-9 expression in monocytes. Cardiovasc. Res. 2007, 73, 841–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karoor, V.; Wang, L.; Wang, H.Y.; Malbon, C.C. Insulin stimulates sequestration of beta-adrenergic receptors and enhanced association of beta-adrenergic receptors with GRB2 via tyrosine 350. J. Biol. Chem. 1998, 273, 33035–33041. [Google Scholar] [CrossRef] [PubMed]
- Karoor, V.; Malbon, C.C. Insulin-like growth factor receptor-1 stimulates phosphorylation of the beta2-adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J. Biol. Chem. 1996, 271, 29347–29352. [Google Scholar] [CrossRef] [PubMed]
- Baltensperger, K.; Karoor, V.; Paul, H.; Ruoho, A.; Czech, M.P.; Malbon, C.C. The beta-adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J. Biol. Chem. 1996, 271, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Karoor, V.; Baltensperger, K.; Paul, H.; Czech, M.P.; Malbon, C.C. Phosphorylation of tyrosyl residues 350/354 of the beta-adrenergic receptor is obligatory for counterregulatory effects of insulin. J. Biol. Chem. 1995, 270, 25305–25308. [Google Scholar] [CrossRef] [PubMed]
- Haxho, F.; Haq, S.; Szewczuk, M.R. Biased G protein-coupled receptor agonism mediates NEU1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell. Signal. 2018, 43, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Kroeze, W.K.; Sheffler, D.J.; Roth, B.L. G-protein-coupled receptors at a glance. J. Cell Sci. 2003, 116, 4867–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayashree, K.; Yasir, M.; Senthilkumar, G.P.; Ramesh Babu, K.; Mehalingam, V.; Mohanraj, P.S. Circulating matrix modulators (MMP-9 and TIMP-1) and their association with severity of diabetic retinopathy. Diabetes Metab. Syndr. Clin. Res. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gentry, P.R.; Sexton, P.M.; Christopoulo, A. Novel allosteric modulators of G protein-coupled receptors. J. Biol. Chem. 2015, 290, 19478–19488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, A.S.; Duarte, D.A.; Lima, V.; Teixeira, L.B.; Parreiras-e-Silva, L.T.; Costa-Neto, C.M. Recent updates on GPCR biased agonism. Pharmacol. Res. 2016, 112, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Chawla, R.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J. Endocrinol. Metabol. 2016, 20, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Sayin, N.; Kara, N.; Pekel, G. Ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Navaratna, D.; McGuire, P.G.; Menicucci, G.; Das, A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes 2007, 56, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.J.; Menicucci, G.; McGuire, P.G.; Das, A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab. Investig. 2005, 85, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by H-Ras. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4320–4326. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Chakrabarti, S.; Khan, Z. Potential contributory role of H-Ras, a small G-protein, in the development of retinopathy in diabetic rats. Diabetes 2004, 53, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G.; dos Santos, J.M.; Zhong, Q. Abrogation of mmp-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes 2011, 60, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; D’Angelo, A.; Tinelli, C.; Devangelio, E.; Consoli, A.; Miccoli, R.; Penno, G.; Del Prato, S.; Paniga, S.; Cicero, A.F. Evaluation of metalloproteinase 2 and 9 levels and their inhibitors in diabetic and healthy subjects. Diabetes Metab. 2007, 33, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Jacqueminet, S.; Ben Abdesselam, O.; Chapman, M.J.; Nicolay, N.; Foglietti, M.J.; Grimaldi, A.; Beaudeux, J.L. Elevated circulating levels of matrix metalloproteinase-9 in type 1 diabetic patients with and without retinopathy. Clin. Chim. Acta 2006, 367, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Gallinat, S.; Bohle, R.M.; Reinecke, A.; Seebeck, J.; Franke, F.; Fink, L.; Zhu, M.; Sumners, C.; Unger, T. Expression of angiotensin AT1 and AT2 receptors in adult rat cardiomyocytes after myocardial infarction: A single-cell reverse transcriptase-polymerase chain reaction study. Am. J. Pathol. 2000, 157, 605–611. [Google Scholar] [CrossRef]
- Stauss, H.M.; Zhu, Y.C.; Redlich, T.; Adamiak, D.; Mott, A.; Kregel, K.C.; Unger, T. Angiotensin-converting enzyme inhibition in infarct-induced heart failure in rats: Bradykinin versus angiotensin II. J. Cardiovasc. Risk 1994, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Cockerill, G.; Ng, L.L.; Thompson, M.; Khan, S.; Samani, N.J.; Squire, I.B. Plasma matrix metalloproteinase-9 and left ventricular remodelling after acute myocardial infarction in man: A prospective cohort study. Eur. Heart J. 2007, 28, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Li, J. The role of the renin-angiotensin-aldosterone system in heart failure. J. Renin Angiotensin Aldosterone Syst. 2004, 5 (Suppl. 1), S7–S10. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.-X.; Li, X.-Y.; Lyu, Y.-N.; He, Y.-Y.; Wan, W.-G.; Zhu, H.-L.; Jiang, X.-J. Possible mechanism by which renal sympathetic denervation improves left ventricular remodelling after myocardial infarction. Exp. Physiol. 2015, 101, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Liauchonak, I.; Dawoud, F.; Riat, Y.; Qorri, B.; Sambi, M.; Jain, J.; Kalaydina, R.V.; Mendonza, N.; Bajwa, K.; Szewczuk, M.R. The biased G-protein-coupled receptor agonism bridges the gap between the insulin receptor and the metabolic syndrome. Int. J. Mol. Sci. 2018, 19, 575. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Dalmaroni, M.J.; Gerswhin, M.E.; Adamopoulos, I.E. The critical role of toll-like receptors—From microbial recognition to autoimmunity: A comprehensive review. Autoimmun. Rev. 2016, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, E.C.; Murphy, C.; O’Neill, L.A.J.; Creagh, E.M. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 2009, 3, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicotra, L.; Loram, L.C.; Watkins, L.R.; Hutchinson, M.R. Toll-like receptors in chronic pain. Exp. Neurol. 2012, 234, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eun-Jung, L.; Sun-Hye, L.; Jin-Gu, L.; Byung-Ro, C.; Yoe-Sik, B.; Jae-Ryong, K.; Chu-Hee, L.; Suk-Hwan, B. Activation of toll-like receptor-9 induces matrix metalloproteinase-9 expression through AKT and tumor necrosis factor-α signaling. FEBS Lett. 2006, 580, 4533–4538. [Google Scholar]
- Lim, E.-J.; Lee, S.-H.; Lee, J.-G.; Kim, J.-R.; Yun, S.-S.; Baek, S.-H.; Lee, C. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp. Mol. Med. 2007, 39, 239–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodavance, S.Y.; Gareri, C.; Torok, R.D.; Rockman, H.A. G protein-coupled receptor biased agonism. J. Cardiovasc. Pharmacol. 2016, 67, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef]
- Li, H.; Xu, H.; Liu, S. Toll-like receptors 4 induces expression of matrix metalloproteinase-9 in human aortic smooth muscle cells. Mol. Biol. Rep. 2011, 38, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Liu, J.; Wang, Z.; Liu, N. Angiotensin ii induces inflammatory response partly via toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell. Physiol. Biochem. 2009, 23, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Bohlender, J.; Bondeva, T.; Roger, T.; Thaiss, F.; Wenzel, U.O. Angiotensin II upregulates toll-like receptor 4 on mesangial cells. J. Am. Soc. Nephrol. 2006, 17, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Venegas, G.; Arreguín-Cano, J.A.; Hernández-Bermúdez, C. Bradykinin promotes toll like receptor-4 expression in human gingival fibroblasts. Int. Immunopharmacol. 2012, 14, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.F. The extra domain a of fibronectin activates toll-like receptor 4. J. Biol. Chem. 2001, 276, 10229–10233. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S. Airway smooth muscle in airway reactivity and remodeling: What have we learned? Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L912–L933. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.R.A.; Burgess, J.K.; Underwood, P.A.; Au, W.; Poniris, M.H.; Tamm, M.; Ge, Q.; Roth, M.; Black, J.L. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J. Allergy Clin. Immunol. 2004, 113, 690–696. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qorri, B.; Kalaydina, R.-V.; Velickovic, A.; Kaplya, Y.; Decarlo, A.; Szewczuk, M.R. Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling. Cells 2018, 7, 117. https://doi.org/10.3390/cells7090117
Qorri B, Kalaydina R-V, Velickovic A, Kaplya Y, Decarlo A, Szewczuk MR. Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling. Cells. 2018; 7(9):117. https://doi.org/10.3390/cells7090117
Chicago/Turabian StyleQorri, Bessi, Regina-Veronicka Kalaydina, Aleksandra Velickovic, Yekaterina Kaplya, Alexandria Decarlo, and Myron R. Szewczuk. 2018. "Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling" Cells 7, no. 9: 117. https://doi.org/10.3390/cells7090117