ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Tumor Samples
2.3. Antibodies
2.4. Cell Culture
2.5. Stable Cell Lines
2.6. Western Blotting
2.7. RNA Extraction and RT-qPCR
2.8. Cell Proliferation Assays
2.9. Cell Migration and Invasion Assays
2.10. Immunohistochemistry and Immunostaining Assessment
2.11. Statistical Analysis
3. Results
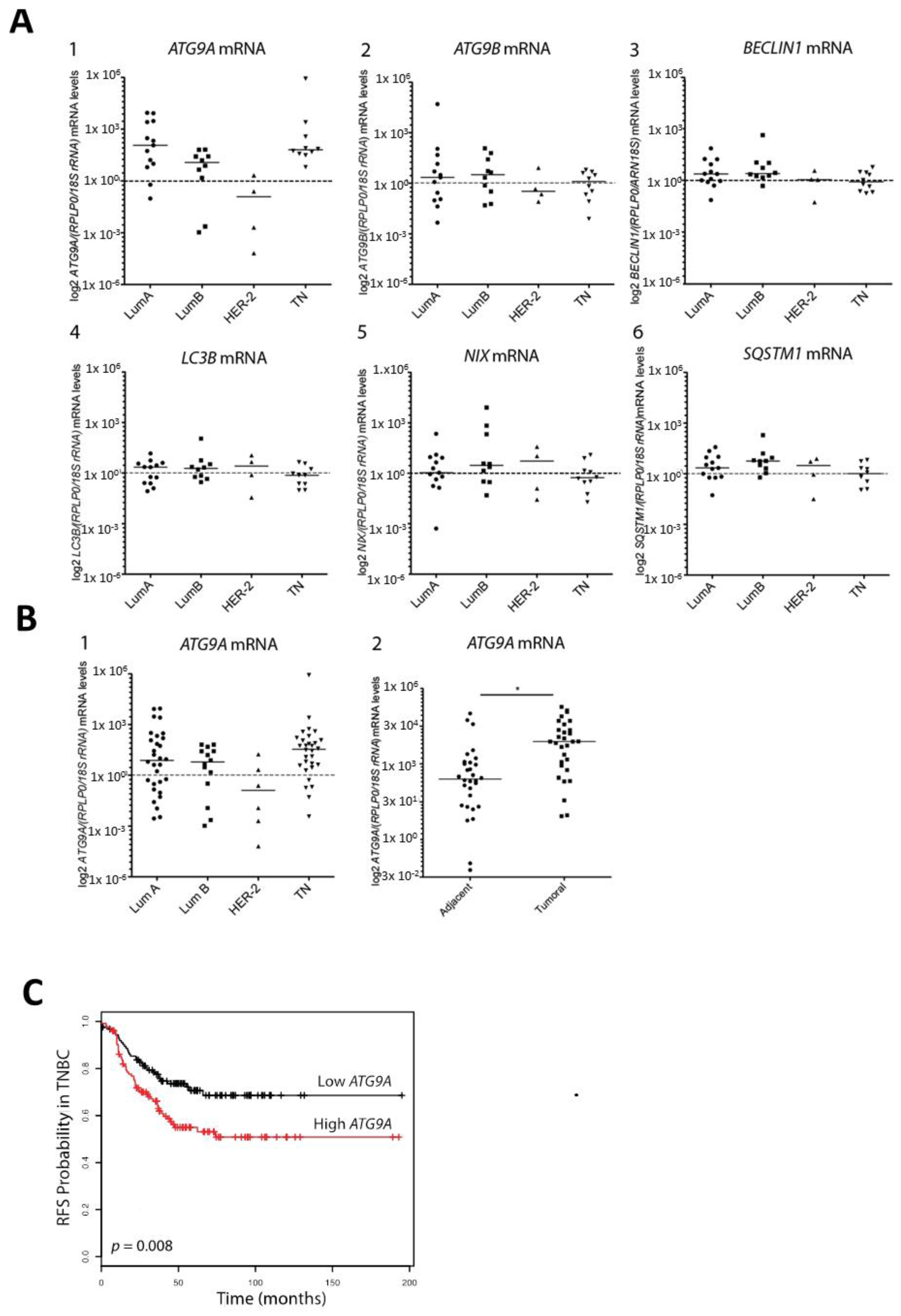
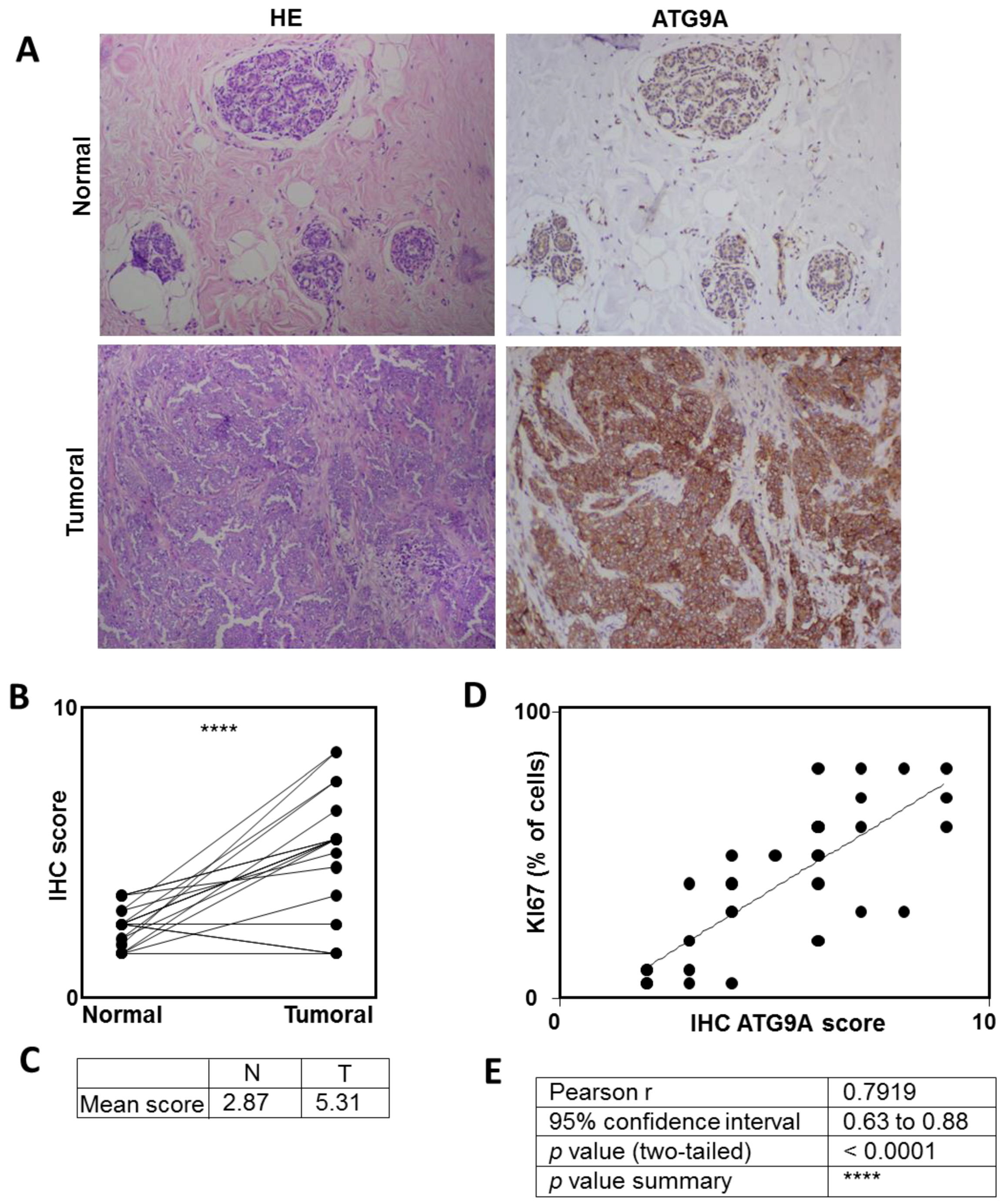
3.1. ATG9A Expression Was Increased in Breast Cancers
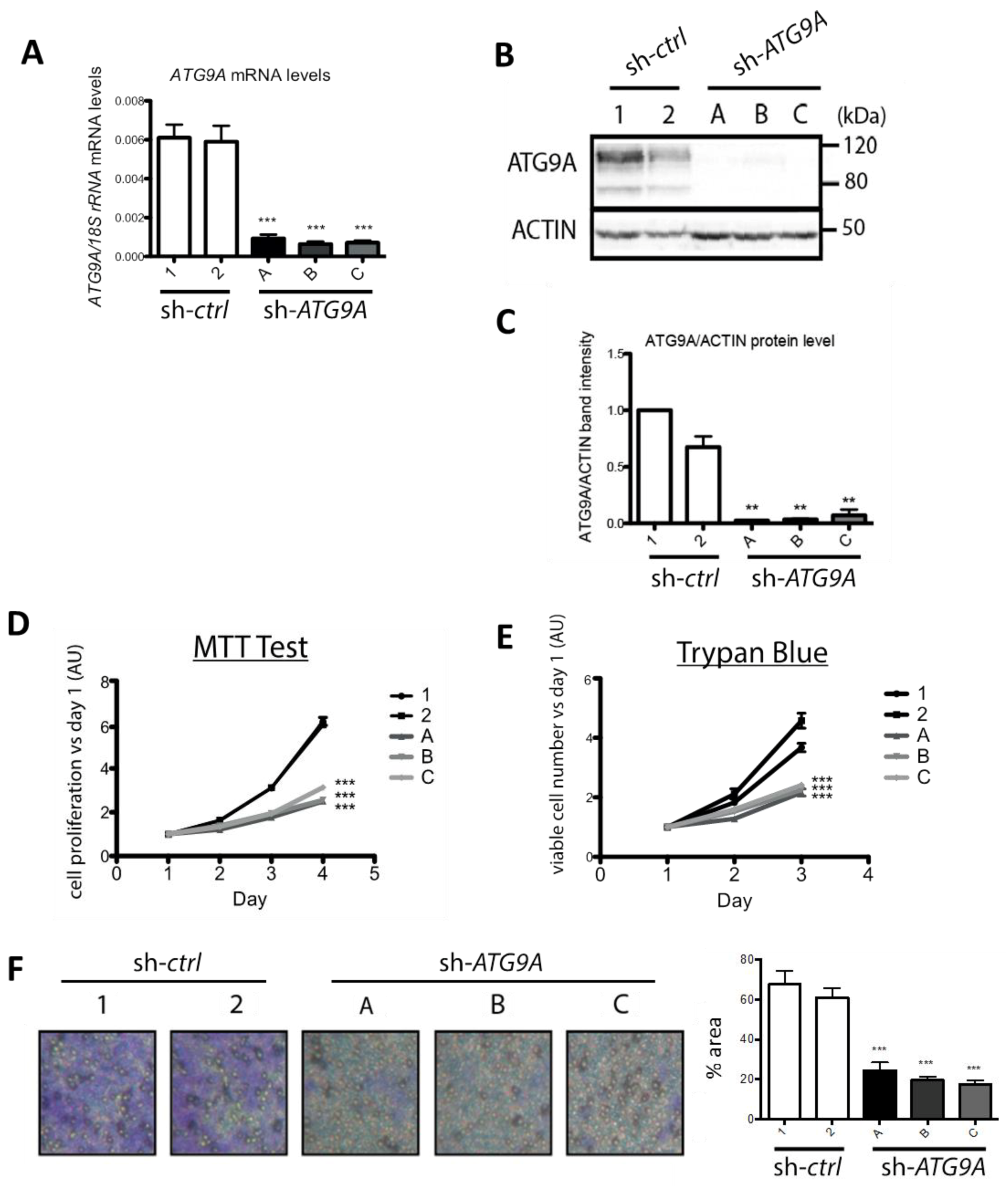
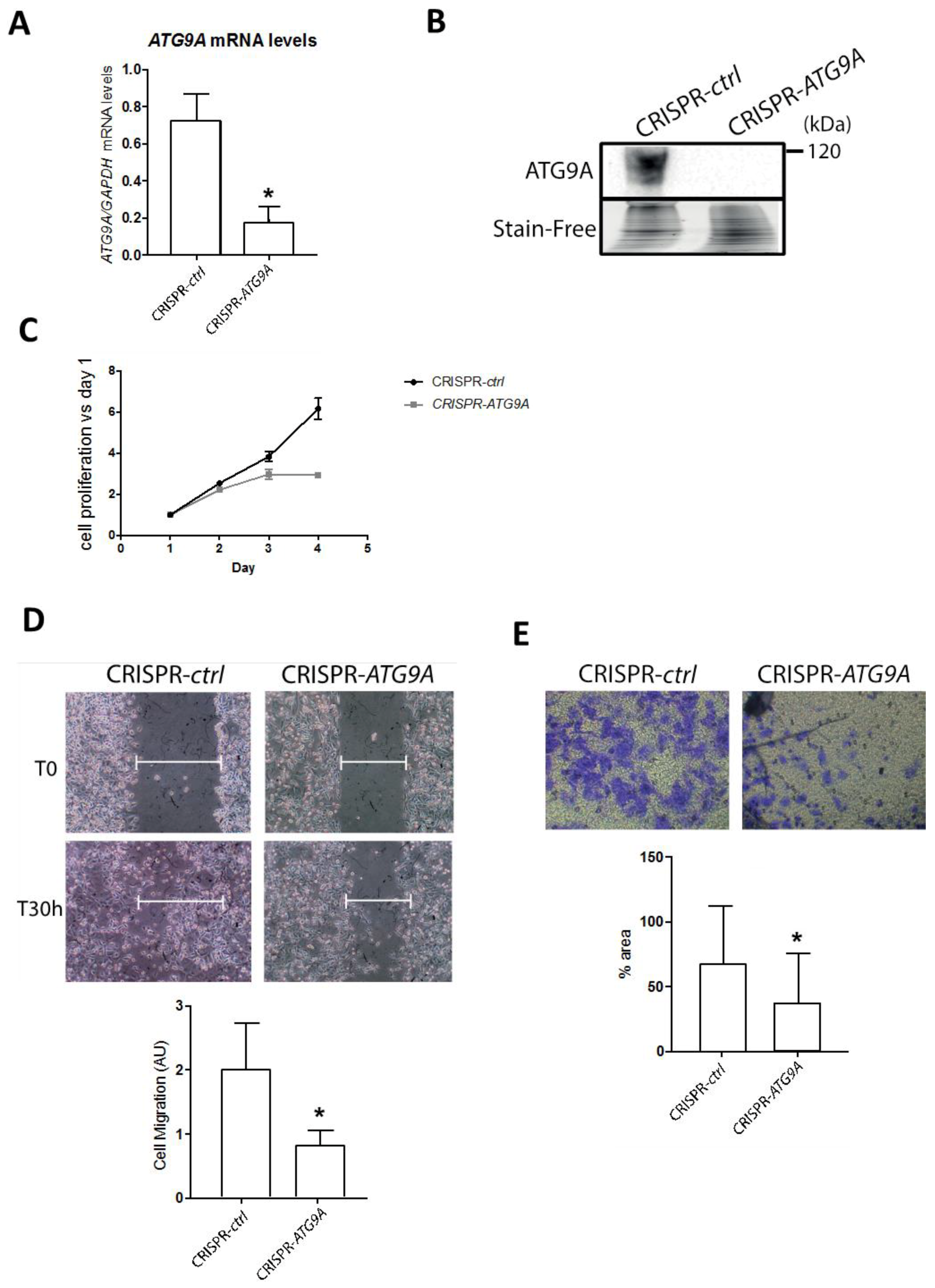
3.2. Inhibition of ATG9A Expression Using shRNA Inhibits In Vitro Cell Proliferation and Invasion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATG | autophagy-related |
| BC | breast cancer |
| CSCs | cancer stem cells |
| DMEM | dulbecco’s minimum essential medium |
| EBSS | earle’s balanced salt solution |
| ECM | extra cellular matrix |
| ER | estrogen receptor |
| FBS | fetal bovine serum |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HRP | horseradish peroxidase |
| IHC | immunohistochemistry |
| LC3B | microtubule-associated protein 1 light chain 3B |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | phosphate buffer saline |
| PBS-T | PBS triton-X-100 |
| pN | lymph node status |
| PR | Progesterone Receptor |
| pT | tumor size |
| PVDF | polyvinylidene difluoride |
| RT-qPCR | reverse transcription-quantitative polymerase chain reaction |
| SDS | sodium dodecylsulfate |
| sgRNA | single guide RNA |
| SISH | silver in situ hybridization |
| TN | triple negative |
| TNBC | triple negative breast cancer |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Cronin, K.A.; Plevritis, S.K.; Fryback, D.G.; Clarke, L.; Zelen, M.; Mandelblatt, J.S.; Yakovlev, A.Y.; Habbema, J.D.; Feuer, E.J. Effect of screening and adjuvant therapy on mortality from breast cancer. N. Engl. J. Med. 2005, 353, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.S.; Sharma, S.C.; Chanana, P.; Jhamb, S. Systemic treatment strategies for triple-negative breast cancer. World J. Clin. Oncol. 2014, 5, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.M.; Bourachot, B.; Zago, G.; Bieche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy 2014, 10, 2122–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Yeo, S.; Wang, C.; Chen, S.; Sun, S.; Haas, M.A.; Tu, W.; Jin, F.; Guan, J.L. Autophagy inhibition re-sensitizes pulse stimulation-selected paclitaxel-resistant triple negative breast cancer cells to chemotherapy-induced apoptosis. Breast Cancer Res. Treat. 2015, 149, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Yamada, T.; Carson, A.R.; Caniggia, I.; Umebayashi, K.; Yoshimori, T.; Nakabayashi, K.; Scherer, S.W. Endothelial nitric-oxide synthase antisense (NOS3AS) gene encodes an autophagy-related protein (APG9-like2) highly expressed in trophoblast. J. Biol. Chem. 2005, 280, 18283–18290. [Google Scholar] [CrossRef]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Kochl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [Green Version]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010, 29, 27–40. [Google Scholar] [CrossRef]
- Longatti, A.; Lamb, C.A.; Razi, M.; Yoshimura, S.; Barr, F.A.; Tooze, S.A. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. ATG16L1 meets ATG9 in recycling endosomes: Additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy 2014, 10, 182–184. [Google Scholar] [CrossRef]
- Imai, K.; Hao, F.; Fujita, N.; Tsuji, Y.; Oe, Y.; Araki, Y.; Hamasaki, M.; Noda, T.; Yoshimori, T. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell Sci. 2016, 129, 3781–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, D.; Dikic, I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014, 15, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Chen, Y.K.; Chu, P.Y.; Chai, C.Y. ATG9A overexpression is associated with disease recurrence and poor survival in patients with oral squamous cell carcinoma. Virchows Arch. 2013, 463, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.; Zhang, H.; Angelopoulos, N.; Chhetri, J.; Osipo, C.; Grothey, A.; Stebbing, J.; Giamas, G. ATG9A loss confers resistance to trastuzumab via c-Cbl mediated Her2 degradation. Oncotarget 2016, 7, 27599–27612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, C.; Wang, D.; Chen, Q.; Li, C.L.; Li, H.J. Aberrant methylation of ATG2B, ATG4D, ATG9A and ATG9B CpG island promoter is associated with decreased mRNA expression in sporadic breast carcinoma. Gene 2016, 590, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Jacquet, M.; Hervouet, E.; Gauthier, T.; Fraichard, A.; Borg, C.; Pallandre, J.R.; Gonzalez, B.J.; Ramdani, Y.; Boyer-Guittaut, M.; et al. GABARAPL1 tumor suppressive function is independent of its conjugation to autophagosomes in MCF-7 breast cancer cells. Oncotarget 2017, 8, 55998–56020. [Google Scholar] [CrossRef] [Green Version]
- Herfs, M.; Longuespee, R.; Quick, C.M.; Roncarati, P.; Suarez-Carmona, M.; Hubert, P.; Lebeau, A.; Bruyere, D.; Mazzucchelli, G.; Smargiasso, N.; et al. Proteomic signatures reveal a dualistic and clinically relevant classification of anal canal carcinoma. J. Pathol. 2017, 241, 522–533. [Google Scholar] [CrossRef]
- Hubert, P.; Herman, L.; Roncarati, P.; Maillard, C.; Renoux, V.; Demoulin, S.; Erpicum, C.; Foidart, J.M.; Boniver, J.; Noel, A.; et al. Altered alpha-defensin 5 expression in cervical squamocolumnar junction: Implication in the formation of a viral/tumour-permissive microenvironment. J. Pathol. 2014, 234, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Szabo, A.; Santarpia, L.; Gyorffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.J.; Willy, J.A.; Quirin, K.A.; Wek, R.C.; Korc, M.; Yin, X.M.; Kota, J. Novel role of miR-29a in pancreatic cancer autophagy and its therapeutic potential. Oncotarget 2016, 7, 71635–71650. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef]
- Berthier, A.; Seguin, S.; Sasco, A.J.; Bobin, J.Y.; De Laroche, G.; Datchary, J.; Saez, S.; Rodriguez-Lafrasse, C.; Tolle, F.; Fraichard, A.; et al. High expression of gabarapl1 is associated with a better outcome for patients with lymph node-positive breast cancer. Br. J. Cancer 2010, 102, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Nemos, C.; Mansuy, V.; Vernier-Magnin, S.; Fraichard, A.; Jouvenot, M.; Delage-Mourroux, R. Expression of gec1/GABARAPL1 versus GABARAP mRNAs in human: Predominance of gec1/GABARAPL1 in the central nervous system. Brain Res. Mol. Brain Res. 2003, 119, 216–219. [Google Scholar] [CrossRef]
- Ran, L.; Hong, T.; Xiao, X.; Xie, L.; Zhou, J.; Wen, G. GABARAPL1 acts as a potential marker and promotes tumor proliferation and metastasis in triple negative breast cancer. Oncotarget 2017, 8, 74519–74526. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, Y.; Zhang, J.; Cao, L.; He, M.; Liu, X.; Zhao, N.; Yin, A.; Huang, H.; Wang, L. TMEM74 promotes tumor cell survival by inducing autophagy via interactions with ATG16L1 and ATG9A. Cell Death Dis. 2017, 8, e3031. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rahim, S.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Wu, P.; Xia, R.; Yang, J.; Huo, X.Y.; Gu, D.Y.; Tang, C.J.; De, W.; Yang, F. STAT3-induced lncRNA HAGLROS overexpression contributes to the malignant progression of gastric cancer cells via mTOR signal-mediated inhibition of autophagy. Mol. Cancer 2018, 17, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef]
- Mukthavaram, R.; Ouyang, X.; Saklecha, R.; Jiang, P.; Nomura, N.; Pingle, S.C.; Guo, F.; Makale, M.; Kesari, S. Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J. Transl. Med. 2015, 13, 269. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort 1 (37) | Cohort 2 (80) | ||
|---|---|---|---|
| Characteristics | N | Characteristics | N |
| Age (year) (37) | Age (year) (80) | ||
| >57 | 18 | >58 | 40 |
| ≤57 | 19 | ≤58 | 40 |
| BC subtype (37) | BC subtype (80) | ||
| LumA | 13 | LumA | 30 |
| LumB | 10 | LumB | 14 |
| HER-2 | 4 | HER-2 | 6 |
| TN | 10 | TN | 30 |
| Grade (36) | Grade (79) | ||
| I | 5 | I | 9 |
| II | 21 | II | 37 |
| III | 10 | III | 33 |
| pT (mm) (35) | pT (mm) (78) | ||
| ≤20 | 18 | ≤20 | 35 |
| 20–50 | 16 | 20–50 | 41 |
| ≥50 | 1 | ≥50 | 2 |
| pN (36) | pN (80) | ||
| negative | 17 | negative | 41 |
| positive | 19 | positive | 39 |
| ER status (37) | ER status (80) | ||
| negative | 14 | negative | 36 |
| positive | 23 | positive | 44 |
| PR status (37) | PR status (80) | ||
| negative | 15 | negative | 39 |
| positive | 21 | positive | 41 |
| HER-2 status (37) | HER-2 status (80) | ||
| negative | 23 | negative | 60 |
| positive | 14 | positive | 20 |
| Gene | Forward | Reverse |
|---|---|---|
| 18S rRNA | 5’-GTAACCCGTTGAACCCCATT-3’ | 5’-CCATCCAATCGGTAGTAGCG-3’ |
| ATG9A | 5’-TTTGCGTTAGGGTGAAGACC-3’ | 5’-AGGGCAGCAAAGTATTTCCA-3’ |
| ATG9B | 5’-CCTTGGGCAGTTCTTCTTTG-3’ | 5’-CTTCCTGGTGCCTGGTACAT-3’ |
| BECLIN1 | 5’-TCACCATCCAGGAACTCACA-3’ | 5’-CCTGGCGAGGAGTTTCAATA-3’ |
| LC3B | 5’-CGGAAAGCAGCAGTGTACCA-3’ | 5’-GGCAGAAGGGAGTGTGTCTGA-3’ |
| NIX | 5’-AAGGCAGGCTTCATTTTTCA-3’ | 5’-CCAATAATTTCCACAACGGG-3’ |
| RPLP0 | 5’-TCGACAATGGCAGCATCTAC-3’ | 5’-GCCTTGACCTTTTCAGCAAG-3’ |
| SQSTM1 | 5’-ATCGGAGGATCCGAGTGT-3’ | 5’-TGGCTGTGAGCTGCTCTT-3’ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claude-Taupin, A.; Fonderflick, L.; Gauthier, T.; Mansi, L.; Pallandre, J.-R.; Borg, C.; Perez, V.; Monnien, F.; Algros, M.-P.; Vigneron, M.; et al. ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes. Cells 2018, 7, 248. https://doi.org/10.3390/cells7120248
Claude-Taupin A, Fonderflick L, Gauthier T, Mansi L, Pallandre J-R, Borg C, Perez V, Monnien F, Algros M-P, Vigneron M, et al. ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes. Cells. 2018; 7(12):248. https://doi.org/10.3390/cells7120248
Chicago/Turabian StyleClaude-Taupin, Aurore, Leïla Fonderflick, Thierry Gauthier, Laura Mansi, Jean-René Pallandre, Christophe Borg, Valérie Perez, Franck Monnien, Marie-Paule Algros, Marc Vigneron, and et al. 2018. "ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes" Cells 7, no. 12: 248. https://doi.org/10.3390/cells7120248