
Role of the Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis

Abstract
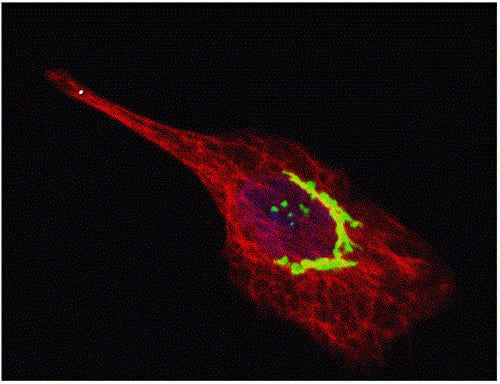
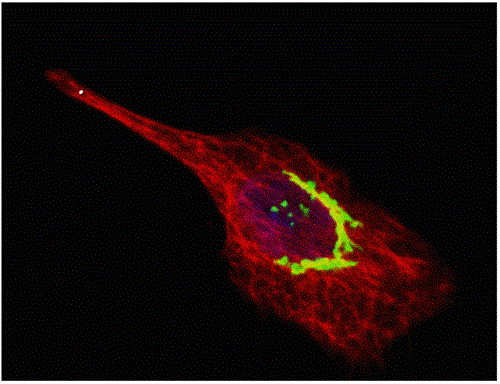
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
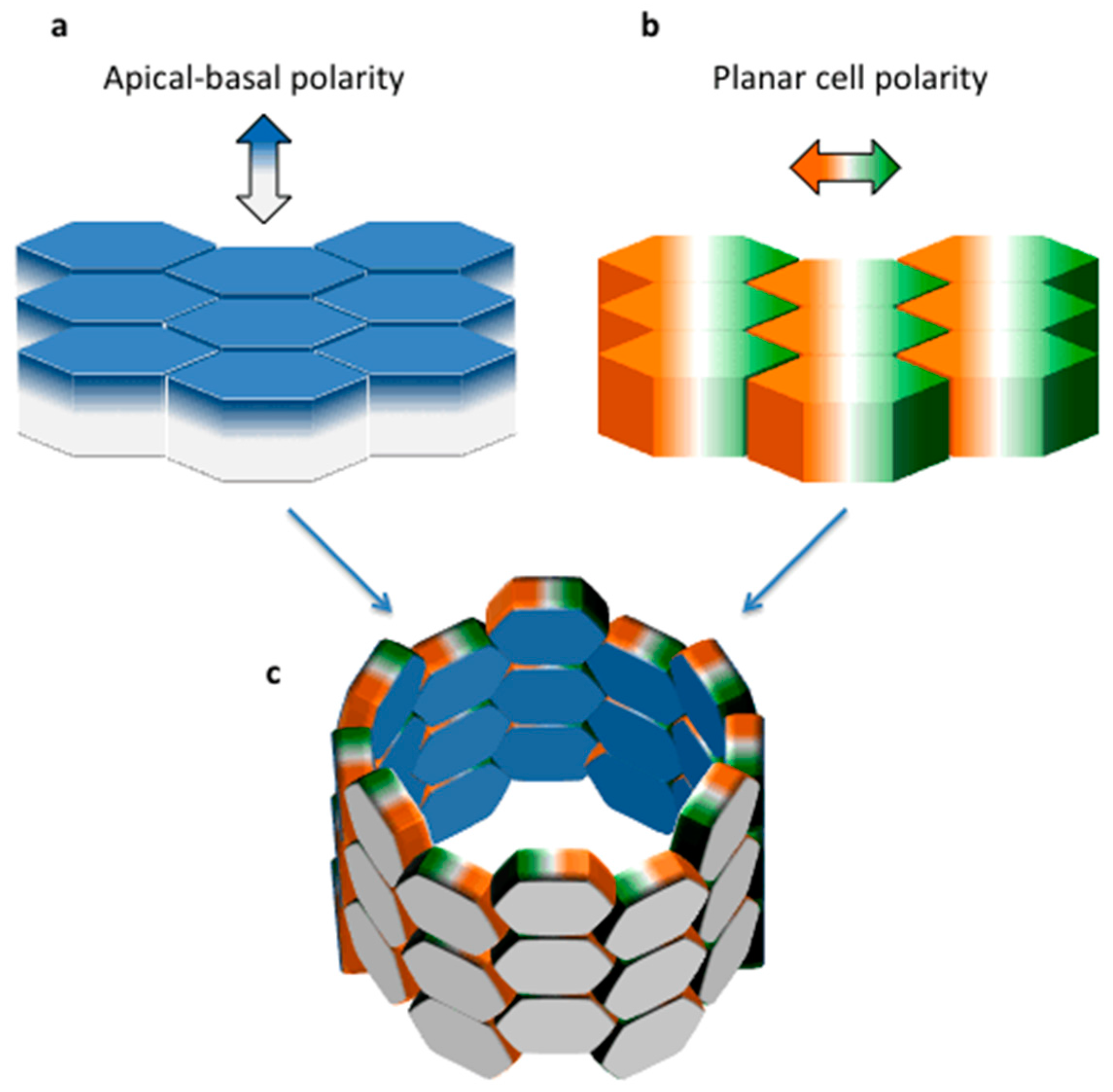
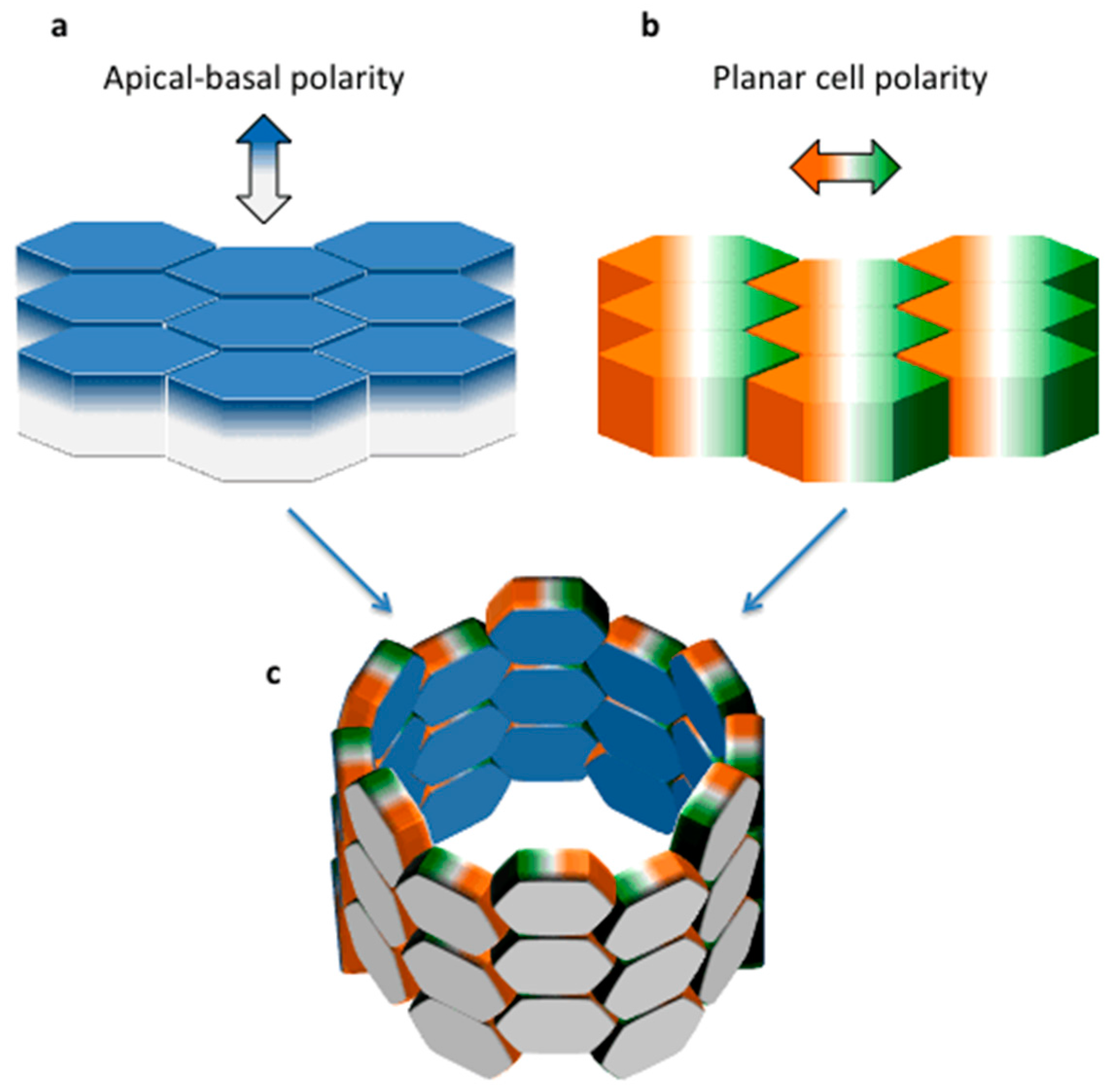
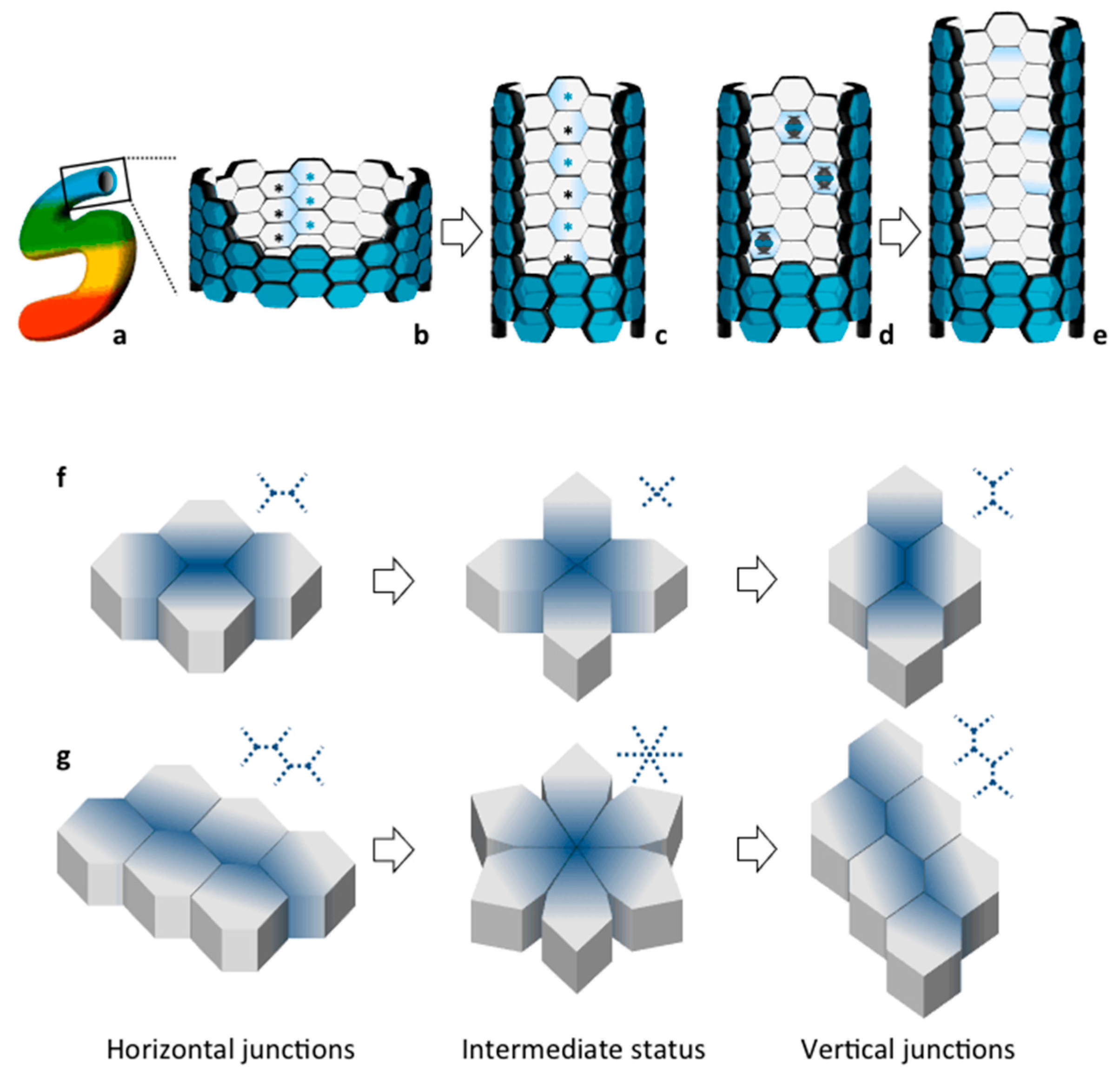
2. Planar Cell Polarity (PCP) in Tissue Morphogenesis: Lessons from Lower Organisms
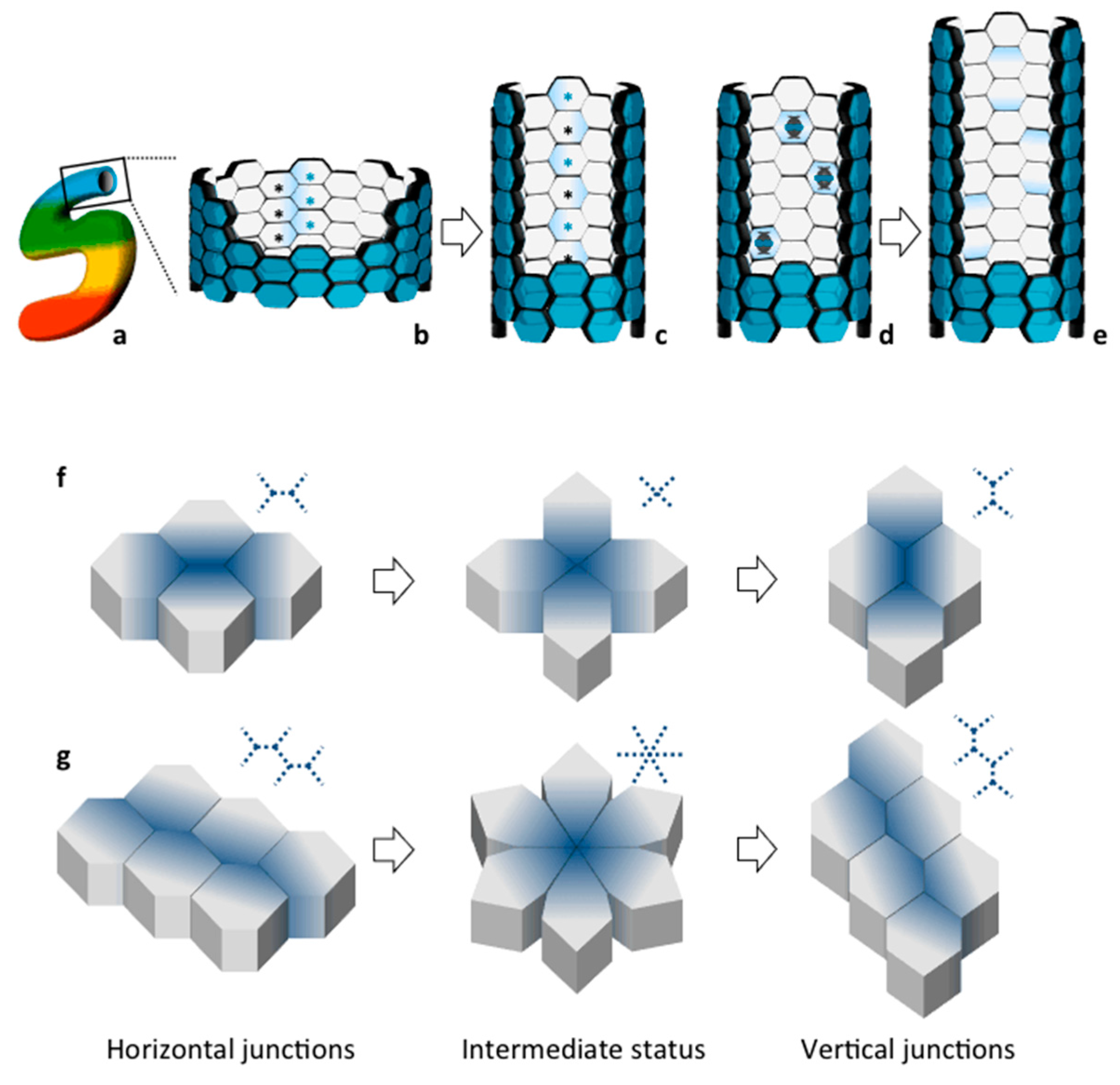
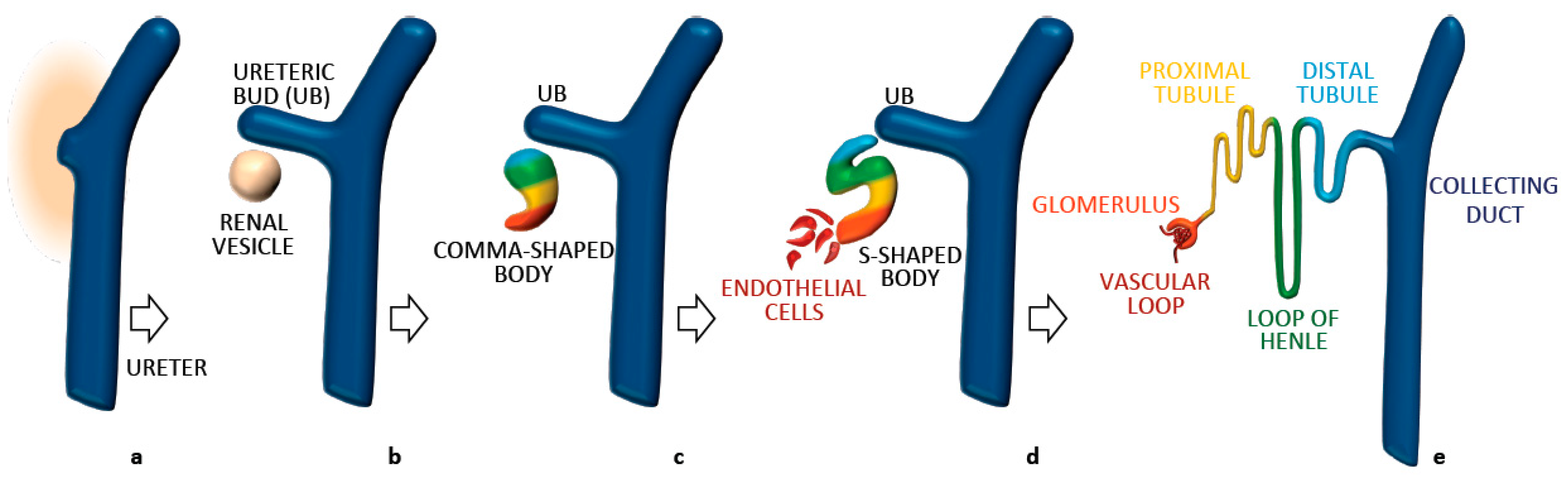
3. Establishment and Maintenance of Tubular Diameter in the Developing Kidney
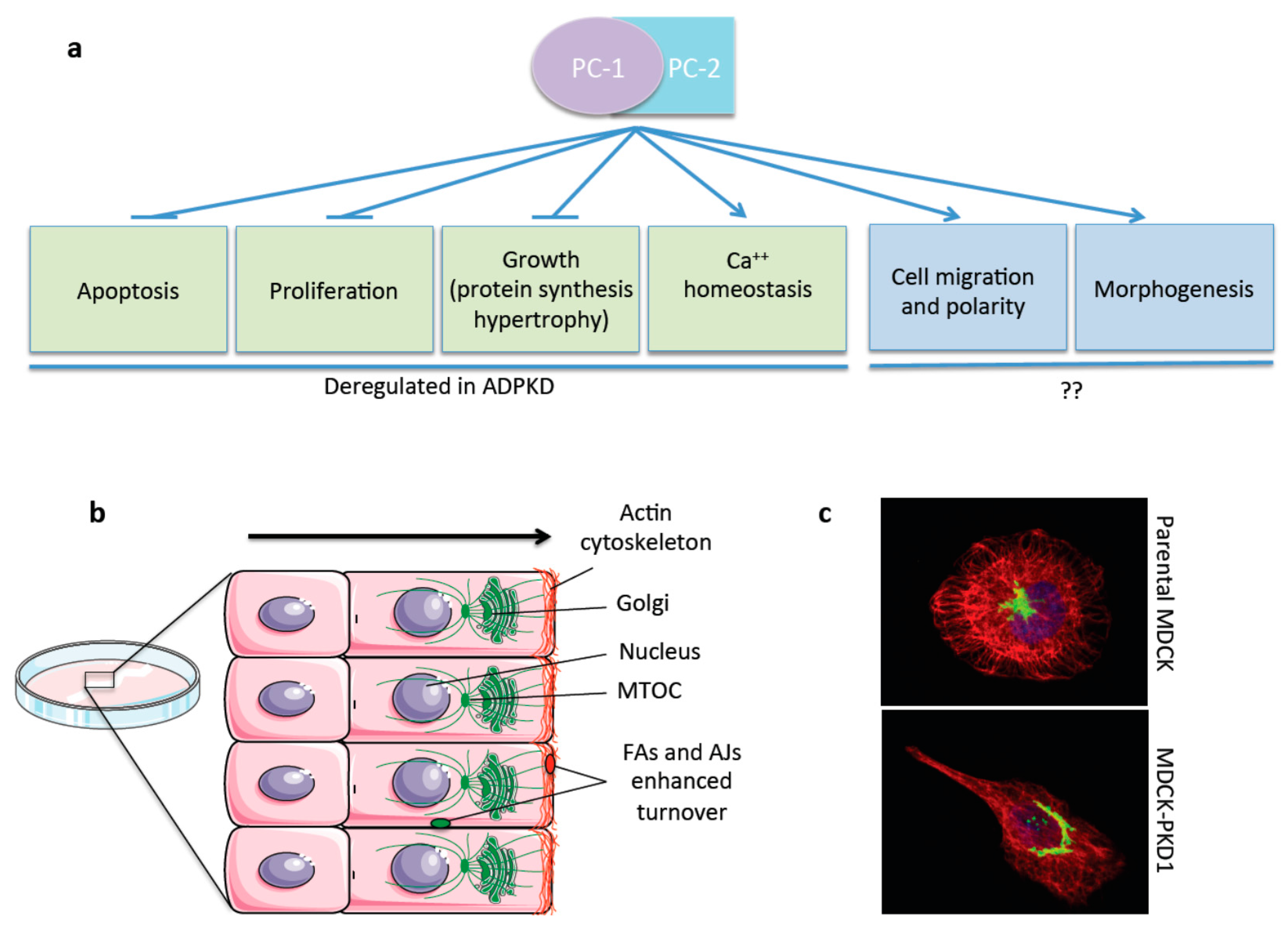
4. The Polycystins: A Multitasking Complex

5. The Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis
6. Defective Planar Cell Polarity as a Cause of Cystogenesis?
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grantham, J.J. Clinical practice. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2008, 359, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Attanasio, M.; Otto, E. Nephronophthisis: Disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009, 20, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [PubMed]
- Brown, J.M.; Witman, G.B. Cilia and diseases. Bioscience 2014, 64, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Boca, M.; Chiaravalli, M.; Ramalingam, H.; Rowe, I.; Distefano, G.; Carroll, T.; Boletta, A. Polycystin-1 binds par3/aPKC and controls convergent extension during renal tubular morphogenesis. Nat. Commun. 2013, 4, 2658. [Google Scholar] [CrossRef] [PubMed]
- Outeda, P.; Huso, D.L.; Fisher, S.A.; Halushka, M.K.; Kim, H.; Qian, F.; Germino, G.G.; Watnick, T. Polycystin signaling is required for directed endothelial cell migration and lymphatic development. Cell Rep. 2014, 7, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Su, X.; Nguyen, V.; Roberts, K.; Li, X.; Takakura, A.; Plomann, M.; Zhou, J. Polycystin-1 regulates actin cytoskeleton organization and directional cell migration through a novel PC1-Pacsin 2-N-Wasp complex. Hum. Mol. Genet. 2014, 23, 2769–2779. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; de Pascalis, C.; Distefano, G.; Ducano, N.; Oldani, A.; Lanzetti, L.; Boletta, A. Regulation of the microtubular cytoskeleton by polycystin-1 favors focal adhesions turnover to modulate cell adhesion and migration. BMC Cell Biol. 2015, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.C.; Harris, P.C. A polycystin-centric view of cyst formation and disease: The polycystins revisited. Kidney Int. 2015, 88, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Fanto, M.; McNeill, H. Planar polarity from flies to vertebrates. J. Cell Sci. 2004, 117, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Strutt, D. Principles of planar polarity in animal development. Development 2011, 138, 1877–1892. [Google Scholar] [CrossRef] [PubMed]
- Rida, P.C.; Chen, P. Line up and listen: Planar cell polarity regulation in the mammalian inner ear. Semin. Cell Dev. Biol. 2009, 20, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Ezan, J.; Montcouquiol, M. Revisiting planar cell polarity in the inner ear. Semin. Cell Dev. Biol. 2013, 24, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.; Davidson, L.; Edlund, A.; Elul, T.; Ezin, M.; Shook, D.; Skoglund, P. Mechanisms of convergence and extension by cell intercalation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 897–922. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Heisenberg, C.P. Convergent extension: Using collective cell migration and cell intercalation to shape embryos. Development 2012, 139, 3897–3904. [Google Scholar] [CrossRef] [PubMed]
- Zallen, J.A.; Wieschaus, E. Patterned gene expression directs bipolar planar polarity in drosophila. Dev. Cell 2004, 6, 343–355. [Google Scholar] [CrossRef]
- Ossipova, O.; Chu, C.W.; Fillatre, J.; Brott, B.K.; Itoh, K.; Sokol, S.Y. The involvement of PCP proteins in radial cell intercalations during xenopus embryonic development. Dev. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sokol, S.Y. Spatial and temporal aspects of Wnt signaling and planar cell polarity during vertebrate embryonic development. Semin. Cell Dev. Biol. 2015, 42, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, M.J.; Itoh, K.; Sokol, S.Y. A positive role for the PP2A catalytic subunit in Wnt signal transduction. J. Biol. Chem. 2000, 275, 35680–35683. [Google Scholar] [CrossRef] [PubMed]
- Wallingford, J.B.; Harland, R.M. Neural tube closure requires dishevelled-dependent convergent extension of the midline. Development 2002, 129, 5815–5825. [Google Scholar] [CrossRef] [PubMed]
- Wallingford, J.B.; Fraser, S.E.; Harland, R.M. Convergent extension: The molecular control of polarized cell movement during embryonic development. Dev. Cell 2002, 2, 695–706. [Google Scholar] [CrossRef]
- Keller, R. Shaping the vertebrate body plan by polarized embryonic cell movements. Science 2002, 298, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.; Keller, R. Cell motility driving mediolateral intercalation in explants of Xenopus laevis. Development 1992, 116, 901–914. [Google Scholar] [PubMed]
- Jessen, J.R.; Topczewski, J.; Bingham, S.; Sepich, D.S.; Marlow, F.; Chandrasekhar, A.; Solnica-Krezel, L. Zebrafish trilobite identifies new roles for strabismus in gastrulation and neuronal movements. Nat. Cell Biol. 2002, 4, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Wallingford, J.B.; Rowning, B.A.; Vogeli, K.M.; Rothbacher, U.; Fraser, S.E.; Harland, R.M. Dishevelled controls cell polarity during Xenopus gastrulation. Nature 2000, 405, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Shindo, A.; Wallingford, J.B. PCP and septins compartmentalize cortical actomyosin to direct collective cell movement. Science 2014, 343, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Carreira-Barbosa, F.; Concha, M.L.; Takeuchi, M.; Ueno, N.; Wilson, S.W.; Tada, M. Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development 2003, 130, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Formstone, C.J.; Mason, I. Combinatorial activity of flamingo proteins directs convergence and extension within the early zebrafish embryo via the planar cell polarity pathway. Dev. Biol. 2005, 282, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Heisenberg, C.P.; Tada, M.; Rauch, G.J.; Saude, L.; Concha, M.L.; Geisler, R.; Stemple, D.L.; Smith, J.C.; Wilson, S.W. Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature 2000, 405, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Smith, J.C. Xwnt11 is a target of xenopus brachyury: Regulation of gastrulation movements via dishevelled, but not through the canonical wnt pathway. Development 2000, 127, 2227–2238. [Google Scholar] [PubMed]
- Goto, T.; Keller, R. The planar cell polarity gene strabismus regulates convergence and extension and neural fold closure in xenopus. Dev. Biol. 2002, 247, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Song, H.; Kim, W.; Kispert, A.; Yang, Y. Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development 2015, 142, 1516–1527. [Google Scholar] [CrossRef] [PubMed]
- Gillies, T.E.; Cabernard, C. Cell division orientation in animals. Curr. Biol. 2011, 21, R599–R609. [Google Scholar] [CrossRef] [PubMed]
- Castanon, I.; Gonzalez-Gaitan, M. Oriented cell division in vertebrate embryogenesis. Curr. Opin. Cell Biol. 2011, 23, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Concha, M.L.; Adams, R.J. Oriented cell divisions and cellular morphogenesis in the zebrafish gastrula and neurula: A time-lapse analysis. Development 1998, 125, 983–994. [Google Scholar] [PubMed]
- Gong, Y.; Mo, C.; Fraser, S.E. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature 2004, 430, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Hernandez, E.; Caneparo, L.; Schneider, S.; Winkler, S.; Liebling, M.; Fraser, S.E.; Heisenberg, C.P. Stereotypical cell division orientation controls neural rod midline formation in zebrafish. Curr. Biol. 2010, 20, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.D.; Strauss, B.; Papalopulu, N. Oriented cell divisions asymmetrically segregate aPKC and generate cell fate diversity in the early xenopus embryo. Development 2003, 130, 2657–2668. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.M.; Vincent, J.P. Oriented cell divisions in the extending germband of drosophila. Development 2007, 134, 3049–3054. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F. Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Legue, E.; Doyen, A.; Nato, F.; Nicolas, J.F.; Torres, V.; Yaniv, M.; Pontoglio, M. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 2006, 38, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Karner, C.M.; Chirumamilla, R.; Aoki, S.; Igarashi, P.; Wallingford, J.B.; Carroll, T.J. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat. Genet. 2009, 41, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F.; Kopan, R. Patterning a complex organ: Branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 2010, 18, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Lienkamp, S.S.; Liu, K.; Karner, C.M.; Carroll, T.J.; Ronneberger, O.; Wallingford, J.B.; Walz, G. Vertebrate kidney tubules elongate using a planar cell polarity-dependent, rosette-based mechanism of convergent extension. Nat. Genet. 2012, 44, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Boletta, A.; Germino, G.G. Role of polycystins in renal tubulogenesis. Trends Cell Biol. 2003, 13, 484–492. [Google Scholar] [CrossRef]
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J Cell Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [PubMed]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hempson, S.J.; Reif, G.A.; Hedge, A.M.; Wallace, D.P. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 2006, 17, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- DeCaen, P.G.; Delling, M.; Vien, T.N.; Clapham, D.E. Direct recording and molecular identification of the calcium channel of primary cilia. Nature 2013, 504, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Chapin, H.C.; Rajendran, V.; Caplan, M.J. Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the g protein-coupled receptor proteolytic site. Mol. Biol. Cell 2010, 21, 4338–4348. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Okuhara, D.; Yu, Z.; Tian, X.; Cai, Y.; Shibazaki, S.; Somlo, S. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J. Cell Sci. 2006, 119, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.H.; Brown-Glaberman, U.; Wang, J.; Morita, Y.; Alper, S.L.; Bedrick, E.J.; Gattone, V.H., 2nd; Deretic, D.; Wandinger-Ness, A. A conserved signal and gtpase complex are required for the ciliary transport of polycystin-1. Mol. Biol. Cell 2011, 22, 3289–3305. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, H.; Babinger, K.; Gurster, S.; Cedzich, A.; Meese, C.; Schadendorf, K.; Osten, L.; de Vries, U.; Rascle, A.; Witzgall, R. Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol. 2011, 192, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; Yao, Q.; Li, W.; Huang, Q.; Outeda, P.; Cebotaru, V.; Chiaravalli, M.; Boletta, A.; Piontek, K.; et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat. Commun. 2014, 5, 5482. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Fedeles, S.V.; Dong, K.; Anyatonwu, G.; Onoe, T.; Mitobe, M.; Gao, J.D.; Okuhara, D.; Tian, X.; Gallagher, A.R.; et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J. Clin. Investig. 2014, 124, 5129–5144. [Google Scholar] [CrossRef] [PubMed]
- Gainullin, V.G.; Hopp, K.; Ward, C.J.; Hommerding, C.J.; Harris, P.C. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Investig. 2015, 125, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Follit, J.A.; San Agustin, J.T.; Xu, F.; Jonassen, J.A.; Samtani, R.; Lo, C.W.; Pazour, G.J. The Golgin GMAP210/TRIP11 anchors IFT20 to the Golgi complex. PLoS Genet. 2008, 4, e1000315. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Boletta, A.; Bhunia, A.K.; Xu, H.; Liu, L.; Ahrabi, A.K.; Watnick, T.J.; Zhou, F.; Germino, G.G. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. USA 2002, 99, 16981–16986. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Hackmann, K.; Gao, J.; He, X.; Piontek, K.; Garcia-Gonzalez, M.A.; Menezes, L.F.; Xu, H.; Germino, G.G.; Zuo, J.; et al. Essential role of cleavage of polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. USA 2007, 104, 18688–18693. [Google Scholar] [CrossRef] [PubMed]
- Boletta, A.; Qian, F.; Onuchic, L.F.; Bhunia, A.K.; Phakdeekitcharoen, B.; Hanaoka, K.; Guggino, W.; Monaco, L.; Germino, G.G. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol. Cell 2000, 6, 1267–1273. [Google Scholar] [CrossRef]
- Boca, M.; Distefano, G.; Qian, F.; Bhunia, A.K.; Germino, G.G.; Boletta, A. Polycystin-1 induces resistance to apoptosis through the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Am. Soc. Nephrol. 2006, 17, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.K.; Piontek, K.; Boletta, A.; Liu, L.; Qian, F.; Xu, P.N.; Germino, F.J.; Germino, G.G. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002, 109, 157–168. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mtor pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Olsan, E.E.; Mukherjee, S.; Wulkersdorfer, B.; Shillingford, J.M.; Giovannone, A.J.; Todorov, G.; Song, X.; Pei, Y.; Weimbs, T. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2011, 108, 18067–18072. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.; Boca, M.; Rowe, I.; Wodarczyk, C.; Ma, L.; Piontek, K.B.; Germino, G.G.; Pandolfi, P.P.; Boletta, A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell Biol. 2009, 29, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Yang, J.; Wang, Z.; Li, Q.; Tang, Y.; Chen, X.Z. Polycystin-2 down-regulates cell proliferation via promoting PERK-dependent phosphorylation of eIF2alpha. Hum. Mol. Genet. 2008, 17, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Lal, M.; Song, X.; Pluznick, J.L.; di Giovanni, V.; Merrick, D.M.; Rosenblum, N.D.; Chauvet, V.; Gottardi, C.J.; Pei, Y.; Caplan, M.J. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum. Mol. Genet. 2008, 17, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Arnould, T.; Sellin, L.K.; Benzing, T.; Fan, M.J.; Gruning, W.; Sokol, S.Y.; Drummond, I.; Walz, G. The polycystic kidney disease 1 gene product modulates wnt signaling. J. Biol. Chem. 1999, 274, 4947–4953. [Google Scholar] [CrossRef] [PubMed]
- Parnell, S.C.; Magenheimer, B.S.; Maser, R.L.; Zien, C.A.; Frischauf, A.M.; Calvet, J.P. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J. Biol. Chem. 2002, 277, 19566–19572. [Google Scholar] [CrossRef] [PubMed]
- Boca, M.; D’Amato, L.; Distefano, G.; Polishchuk, R.S.; Germino, G.G.; Boletta, A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol. Biol. Cell 2007, 18, 4050–4061. [Google Scholar] [CrossRef] [PubMed]
- Low, S.H.; Vasanth, S.; Larson, C.H.; Mukherjee, S.; Sharma, N.; Kinter, M.T.; Kane, M.E.; Obara, T.; Weimbs, T. Polycystin-1, STAT6, and p100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell 2006, 10, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Magenheimer, B.S.; Maser, R.L.; Ryan, E.M.; Zien, C.A.; Walker, D.D.; Wallace, D.P.; Hempson, S.J.; Calvet, J.P. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J. Biol. Chem. 2004, 279, 55455–55464. [Google Scholar] [CrossRef] [PubMed]
- Sharif-Naeini, R.; Folgering, J.H.; Bichet, D.; Duprat, F.; Lauritzen, I.; Arhatte, M.; Jodar, M.; Dedman, A.; Chatelain, F.C.; Schulte, U.; et al. Polycystin-1 and -2 dosage regulates pressure sensing. Cell 2009, 139, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Peyronnet, R.; Martins, J.R.; Duprat, F.; Demolombe, S.; Arhatte, M.; Jodar, M.; Tauc, M.; Duranton, C.; Paulais, M.; Teulon, J.; et al. Piezo1-dependent stretch-activated channels are inhibited by polycystin-2 in renal tubular epithelial cells. EMBO Rep. 2013, 14, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Pedrozo, Z.; Criollo, A.; Battiprolu, P.K.; Morales, C.R.; Contreras-Ferrat, A.; Fernandez, C.; Jiang, N.; Luo, X.; Caplan, M.J.; Somlo, S.; et al. Polycystin-1 is a cardiomyocyte mechanosensor that governs l-type Ca2+ channel protein stability. Circulation 2015, 131, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Nickel, C.; Benzing, T.; Sellin, L.; Gerke, P.; Karihaloo, A.; Liu, Z.X.; Cantley, L.G.; Walz, G. The polycystin-1 C-terminal fragment triggers branching morphogenesis and migration of tubular kidney epithelial cells. J. Clin. Investig. 2002, 109, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.H.; Karihaloo, A.; Cai, Y.; Somlo, S.; Cantley, L.G.; Caplan, M.J. Polycystin-2 regulates proliferation and branching morphogenesis in kidney epithelial cells. J. Biol. Chem. 2006, 281, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Merrick, D.; Chapin, H.; Baggs, J.E.; Yu, Z.; Somlo, S.; Sun, Z.; Hogenesch, J.B.; Caplan, M.J. The gamma-secretase cleavage product of polycystin-1 regulates TCF and CHOP-mediated transcriptional activation through a p300-dependent mechanism. Dev. Cell 2012, 22, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Piontek, K.B.; Germino, G.G.; Boletta, A. Impaired glomerulogenesis and endothelial cell migration in PKD1-deficient renal organ cultures. Biochem. Biophys. Res. Commun. 2014, 444, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Coxam, B.; Sabine, A.; Bower, N.I.; Smith, K.A.; Pichol-Thievend, C.; Skoczylas, R.; Astin, J.W.; Frampton, E.; Jaquet, M.; Crosier, P.S.; et al. PKD1 regulates lymphatic vascular morphogenesis during development. Cell Rep. 2014, 7, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirao, B.; Meunier, A.; Mortaud, S.; Aguilar, A.; Corsi, J.M.; Strehl, L.; Hirota, Y.; Desoeuvre, A.; Boutin, C.; Han, Y.G.; et al. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat. Cell Biol. 2010, 12, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Vladar, E.K.; Bayly, R.D.; Sangoram, A.M.; Scott, M.P.; Axelrod, J.D. Microtubules enable the planar cell polarity of airway cilia. Curr. Biol. 2012, 22, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; McManus, E.J.; Hall, A. Cdc42 and noncanonical wnt signal transduction pathways cooperate to promote cell polarity. J. Cell Biol. 2007, 178, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Peifer, M. Adherens junction-dependent and -independent steps in the establishment of epithelial cell polarity in drosophila. J. Cell Biol. 2004, 167, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Peifer, M. aPKC controls microtubule organization to balance adherens junction symmetry and planar polarity during development. Dev. Cell 2007, 12, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Steigelman, K.A.; Lelli, A.; Wu, X.; Gao, J.; Lin, S.; Piontek, K.; Wodarczyk, C.; Boletta, A.; Kim, H.; Qian, F.; et al. Polycystin-1 is required for stereocilia structure but not for mechanotransduction in inner ear hair cells. J. Neurosci. 2011, 31, 12241–12250. [Google Scholar] [CrossRef] [PubMed]
- Ohata, S.; Herranz-Pérez, V.; Nakatani, J.; Boletta, A.; García-Verdugo, J.; Álvarez-Buylla, A. Mechanosensory genes PKD1 and PKD2 contribute to the planar polarization of brain ventricular epithelium. J. Neurosci. 2015, 35, 11153–11168. [Google Scholar] [CrossRef] [PubMed]
- McNeill, H. Planar cell polarity and the kidney. J. Am. Soc. Nephrol. 2009, 20, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type ii, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Tian, X.; Gallagher, A.R.; Yu, Z.; Patel, V.; Igarashi, P.; Somlo, S. Loss of oriented cell division does not initiate cyst formation. J. Am. Soc. Nephrol. 2010, 21, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Saburi, S.; Hester, I.; Fischer, E.; Pontoglio, M.; Eremina, V.; Gessler, M.; Quaggin, S.E.; Harrison, R.; Mount, R.; McNeill, H. Loss of FAT4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 2008, 40, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Luyten, A.; Su, X.; Gondela, S.; Chen, Y.; Rompani, S.; Takakura, A.; Zhou, J. Aberrant regulation of planar cell polarity in polycystic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, J.A.; SanAgustin, J.; Baker, S.P.; Pazour, G.J. Disruption of ift complex a causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol. 2012, 23, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.L.; Papakrivopoulou, J.; Long, D.A.; Goggolidou, P.; Connolly, J.O.; Woolf, A.S.; Dean, C.H. The planar cell polarity gene vangl2 is required for mammalian kidney-branching morphogenesis and glomerular maturation. Hum. Mol. Genet. 2010, 19, 4663–4676. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J.; Cook, L.T.; Wetzel, L.H.; Cadnapaphornchai, M.A.; Bae, K.T. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin. J. Am. Soc. Nephrol. 2010, 5, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Takakura, A.; Contrino, L.; Zhou, X.; Bonventre, J.V.; Sun, Y.; Humphreys, B.D.; Zhou, J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum. Mol. Genet. 2009, 18, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Li, L.; Cobo-Stark, P.; Shao, X.; Somlo, S.; Lin, F.; Igarashi, P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 2008, 17, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.H.; Conklin, J.; Chan, W.; Roslin, N.M.; Liu, J.; He, N.; Wang, K.; Sundsbak, J.L.; Heyer, C.M.; Haider, M.; et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigro, E.A.; Castelli, M.; Boletta, A. Role of the Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis. Cells 2015, 4, 687-705. https://doi.org/10.3390/cells4040687
Nigro EA, Castelli M, Boletta A. Role of the Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis. Cells. 2015; 4(4):687-705. https://doi.org/10.3390/cells4040687
Chicago/Turabian StyleNigro, Elisa Agnese, Maddalena Castelli, and Alessandra Boletta. 2015. "Role of the Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis" Cells 4, no. 4: 687-705. https://doi.org/10.3390/cells4040687