How Chain Intermixing Dictates the Polymorphism of PVDF in Poly(vinylidene fluoride)/Polymethylmethacrylate Binary System during Recrystallization: A Comparative Study on Core–Shell Particles and Latex Blend

 , , and
, , and
Abstract
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Sample Preparation
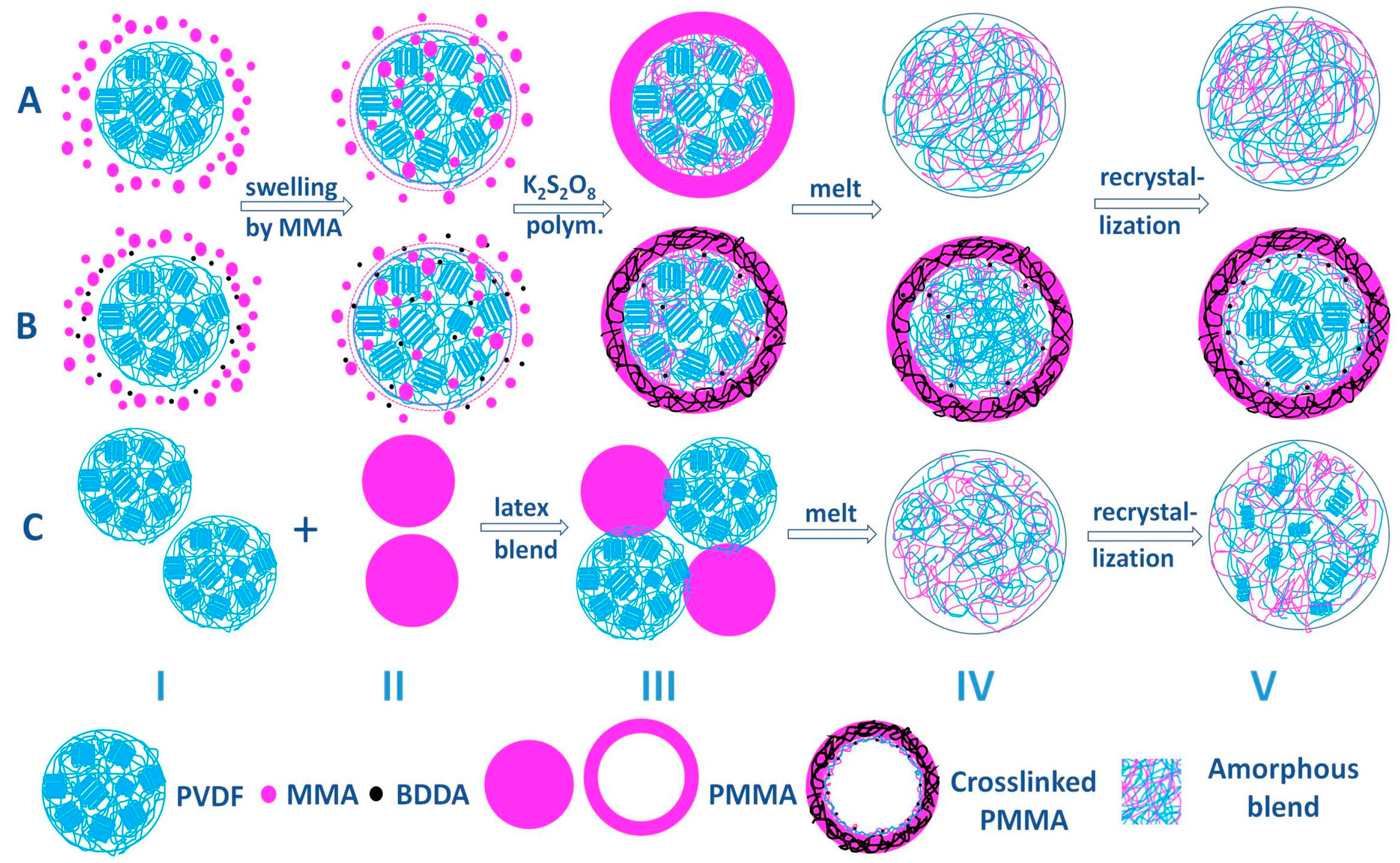
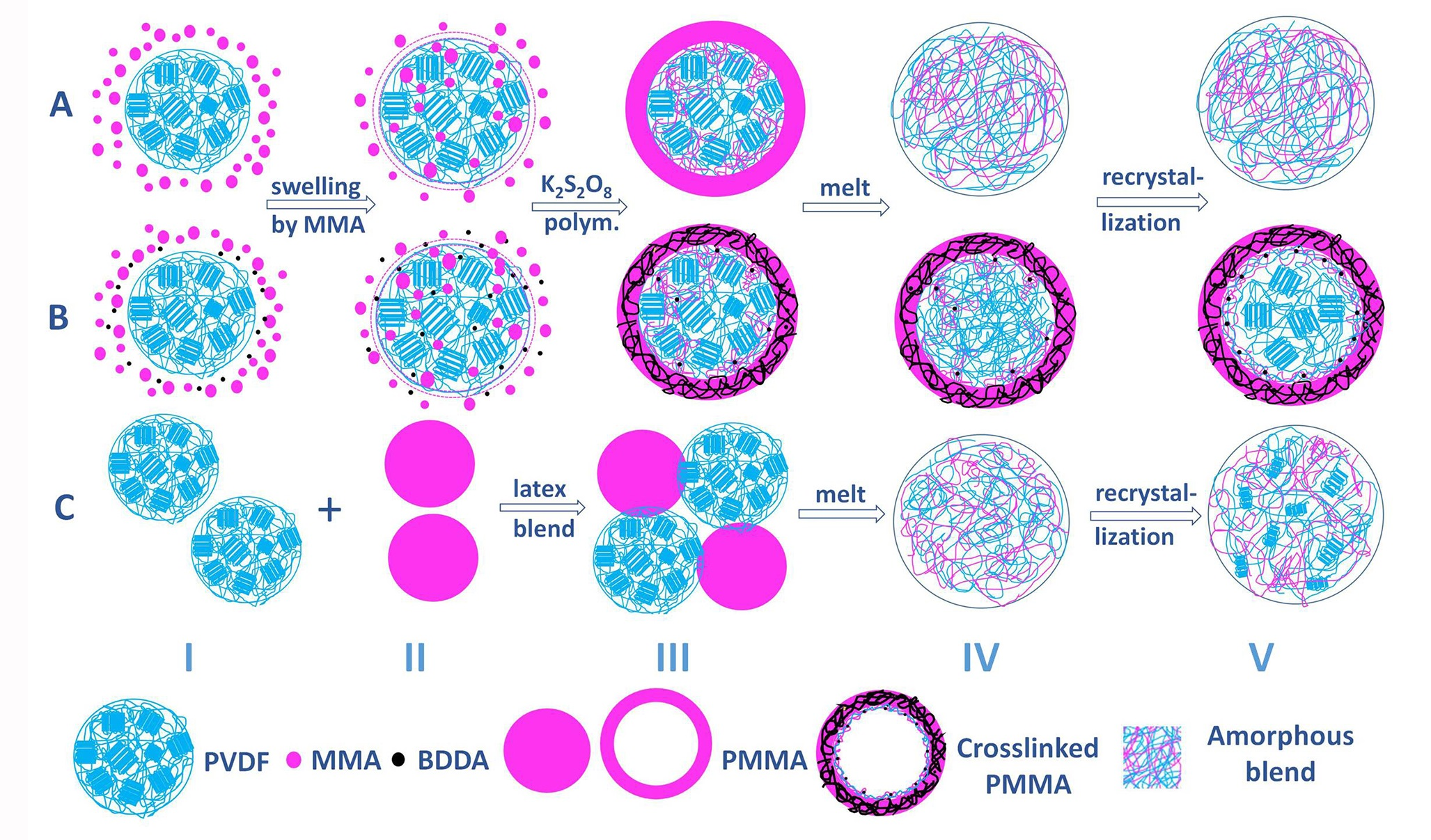
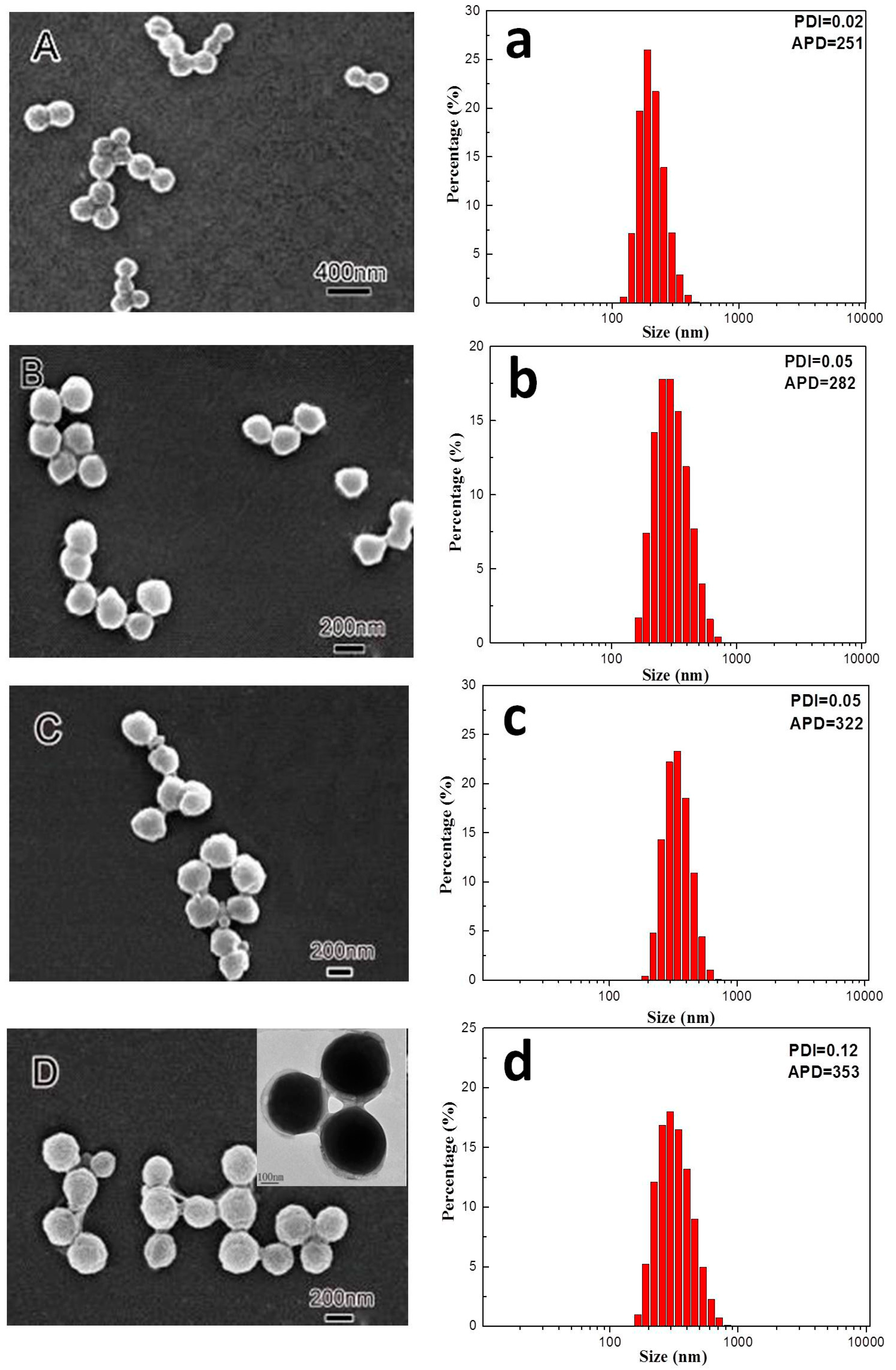
2.2.1. Preparation of Core–Shell PVDF@PMMA Nanoparticles
2.2.2. Preparation of PMMA Nanoparticles and PVDF/PMMA Blends
2.3. Characterization
3. Results and Discussion
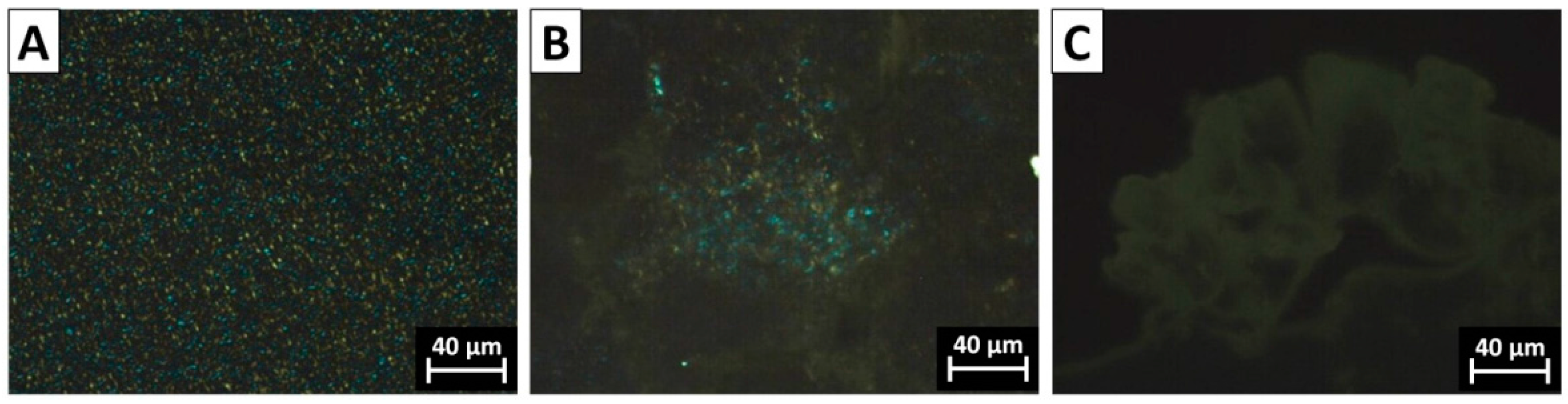
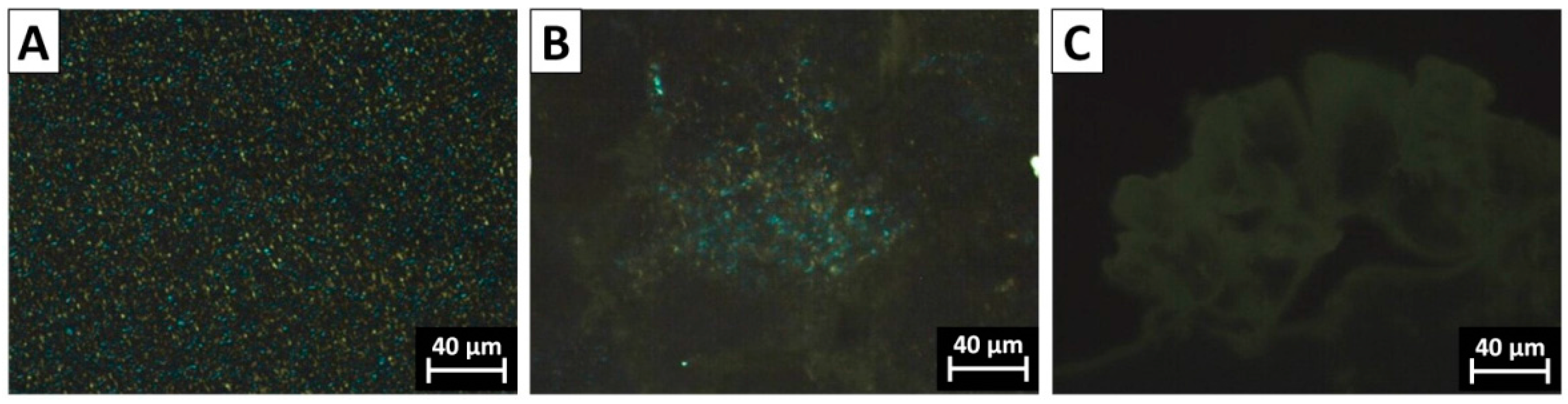
3.1. Morphology and Size Distribution of Core–Shell Latex Particles
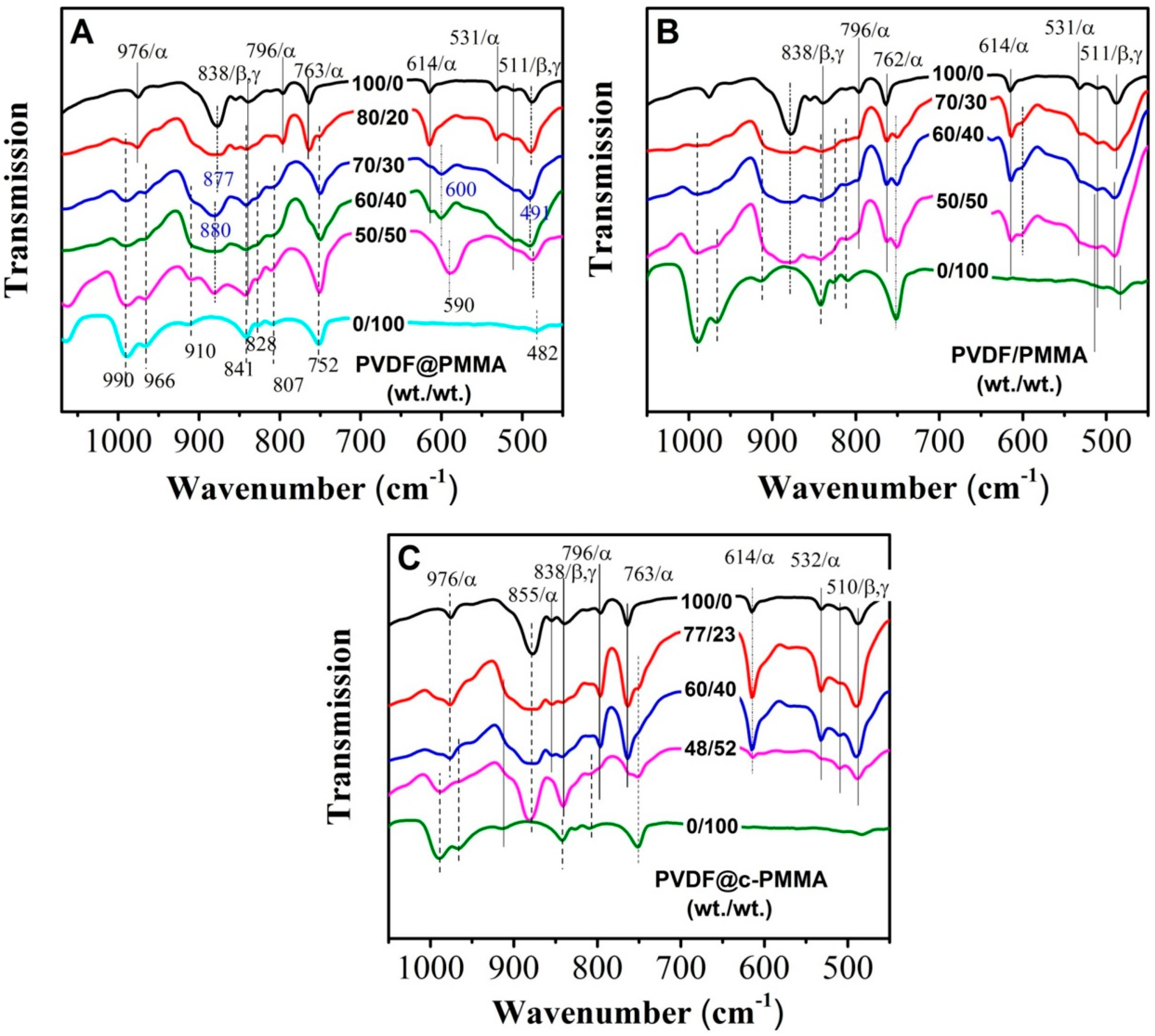
3.2. PVDF/PMMA Miscibility in Different Complexation Scenarios
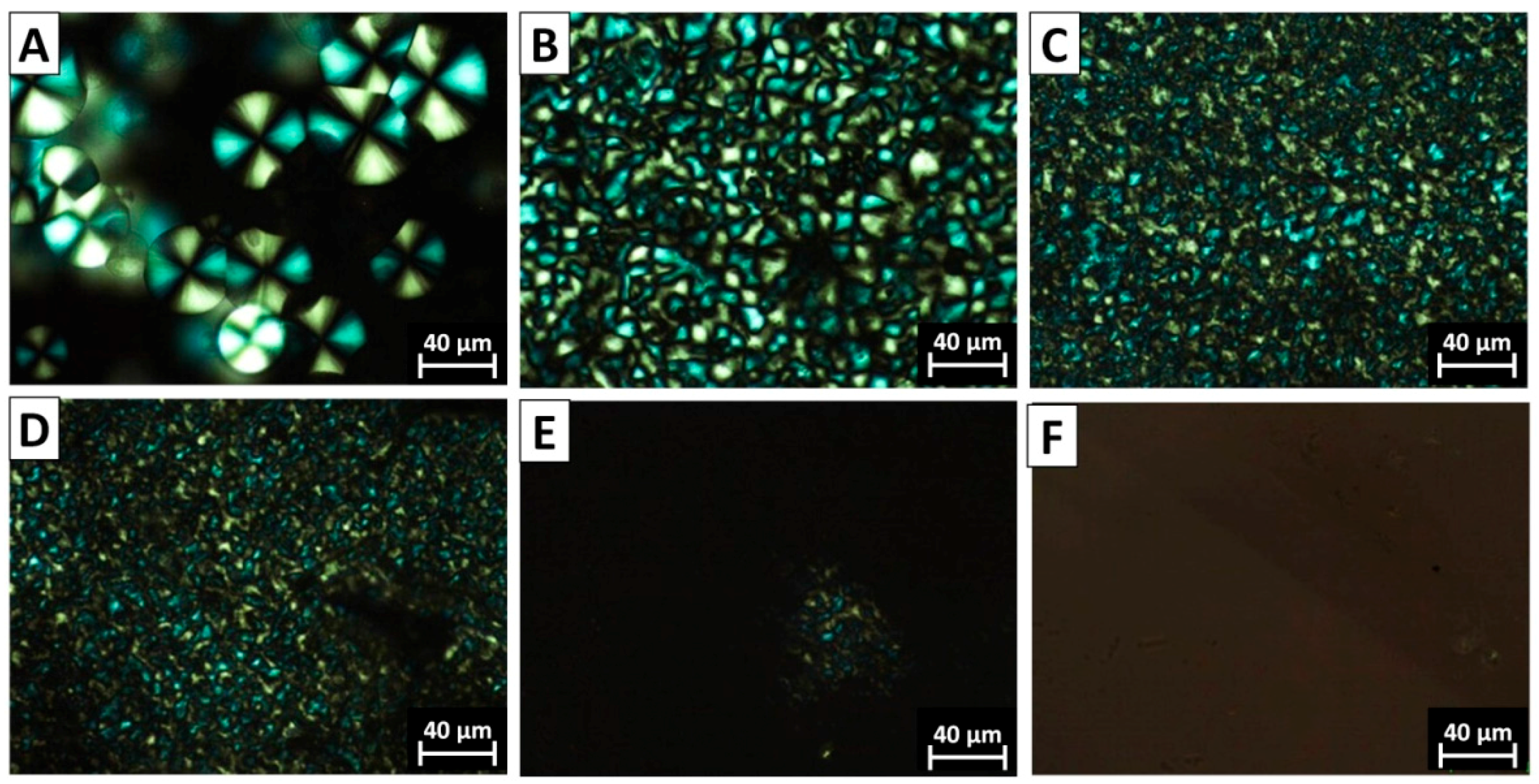
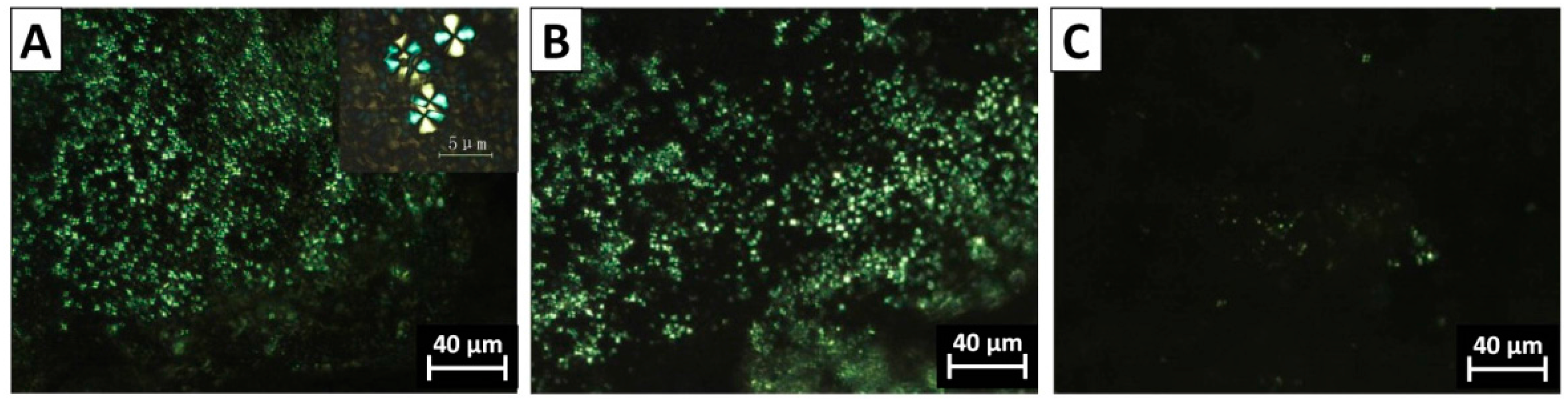
3.3. The Morphology of PVDF Crystal in Different Complexation Scenarios
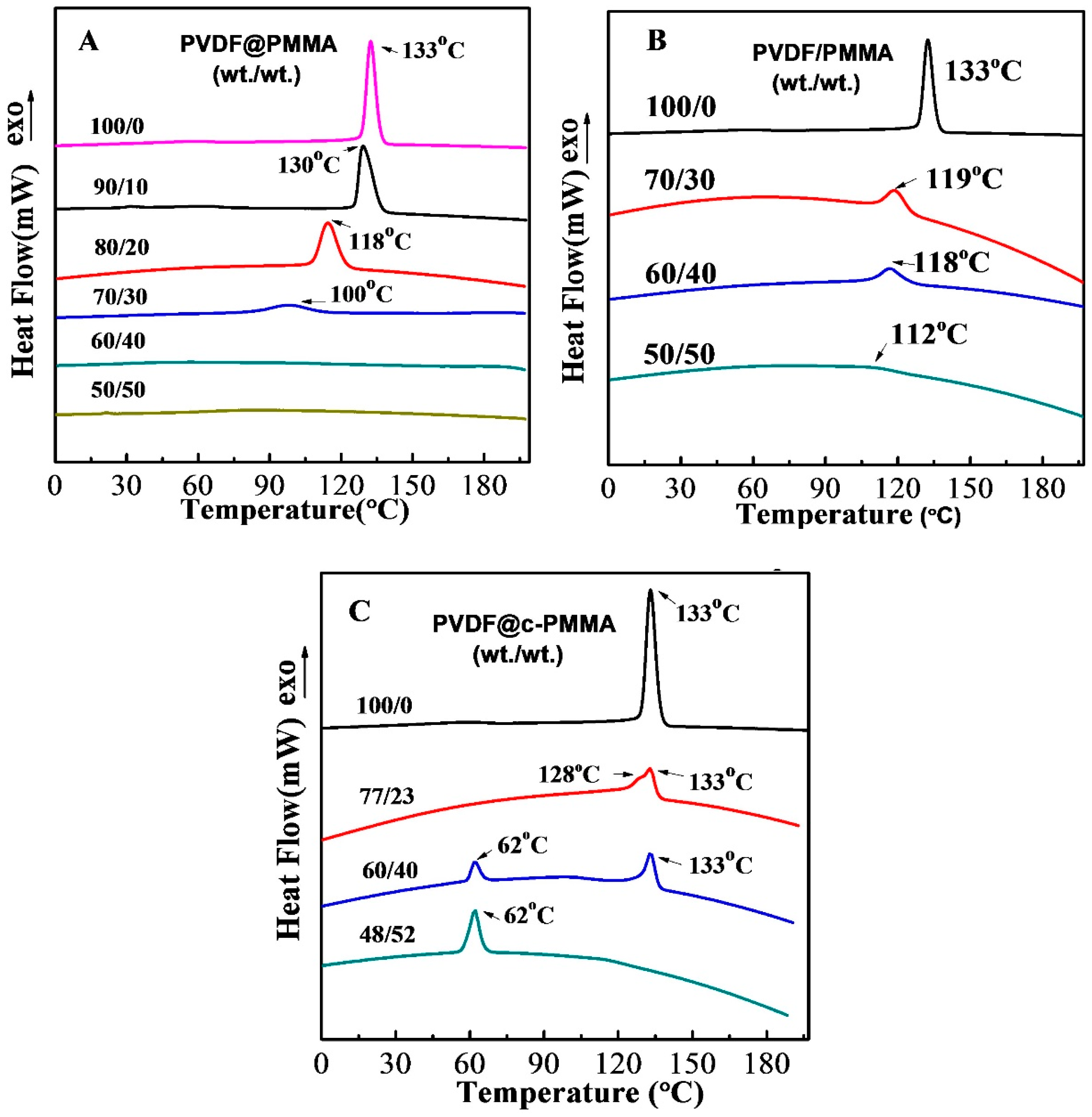
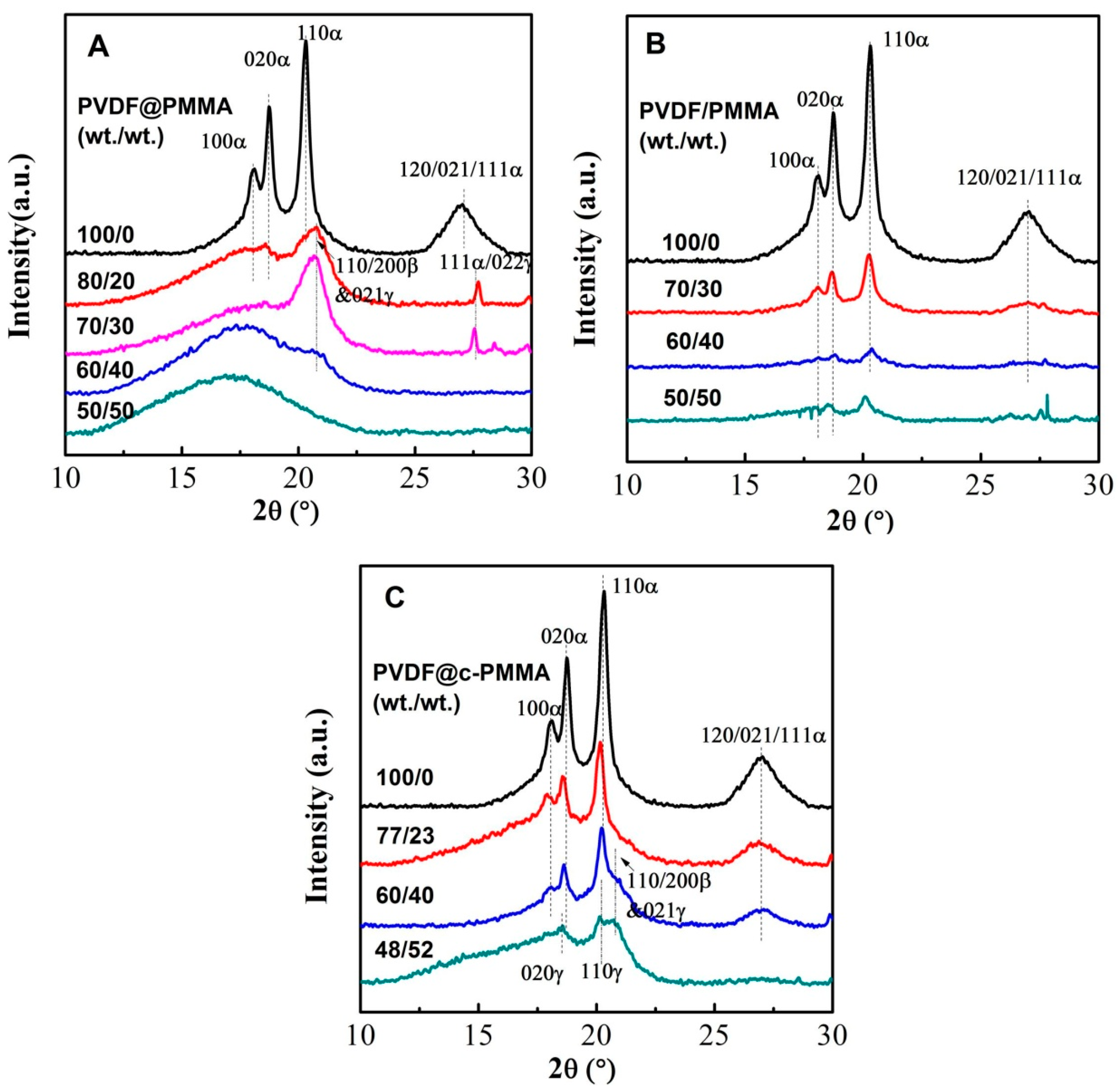
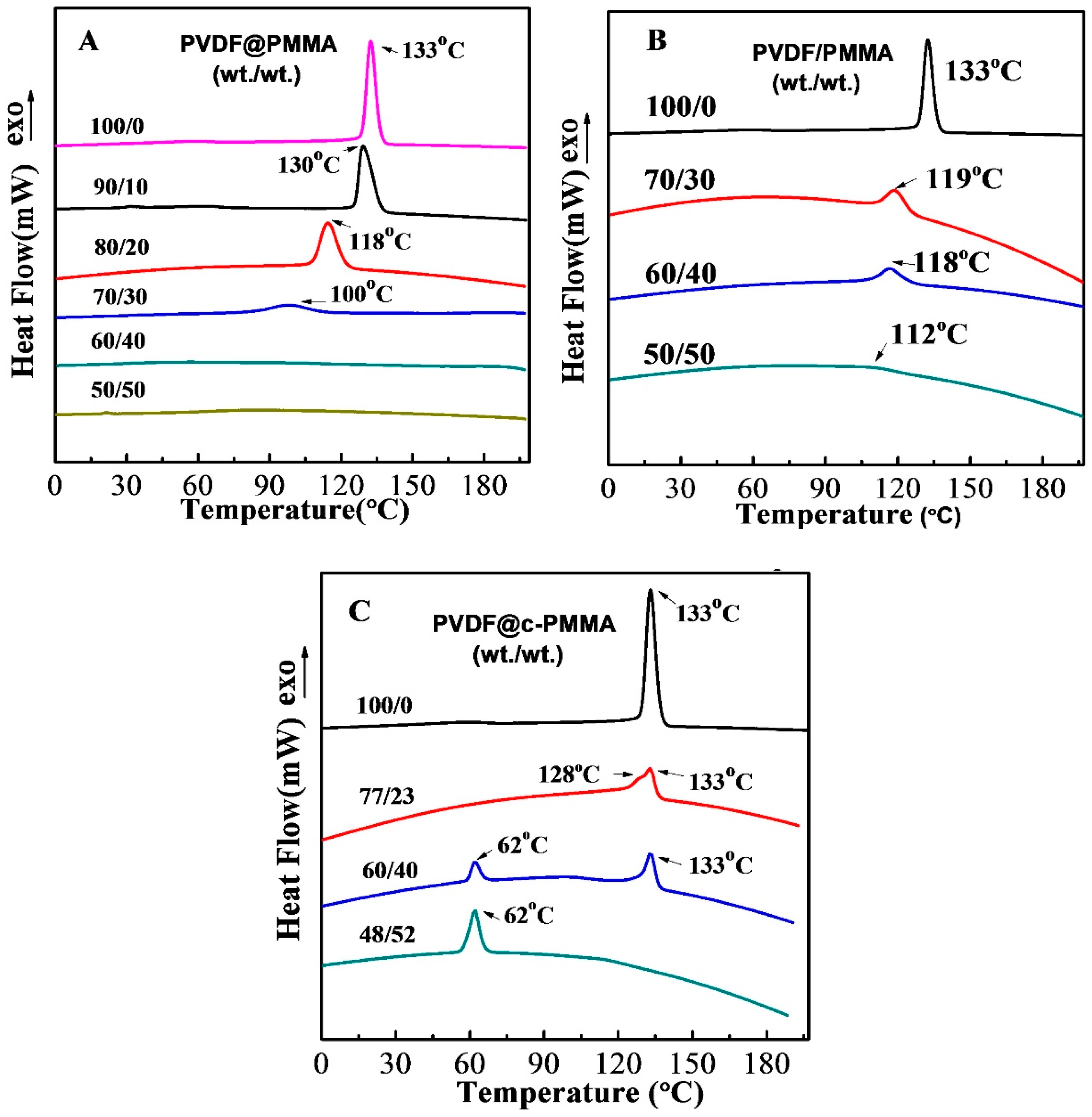
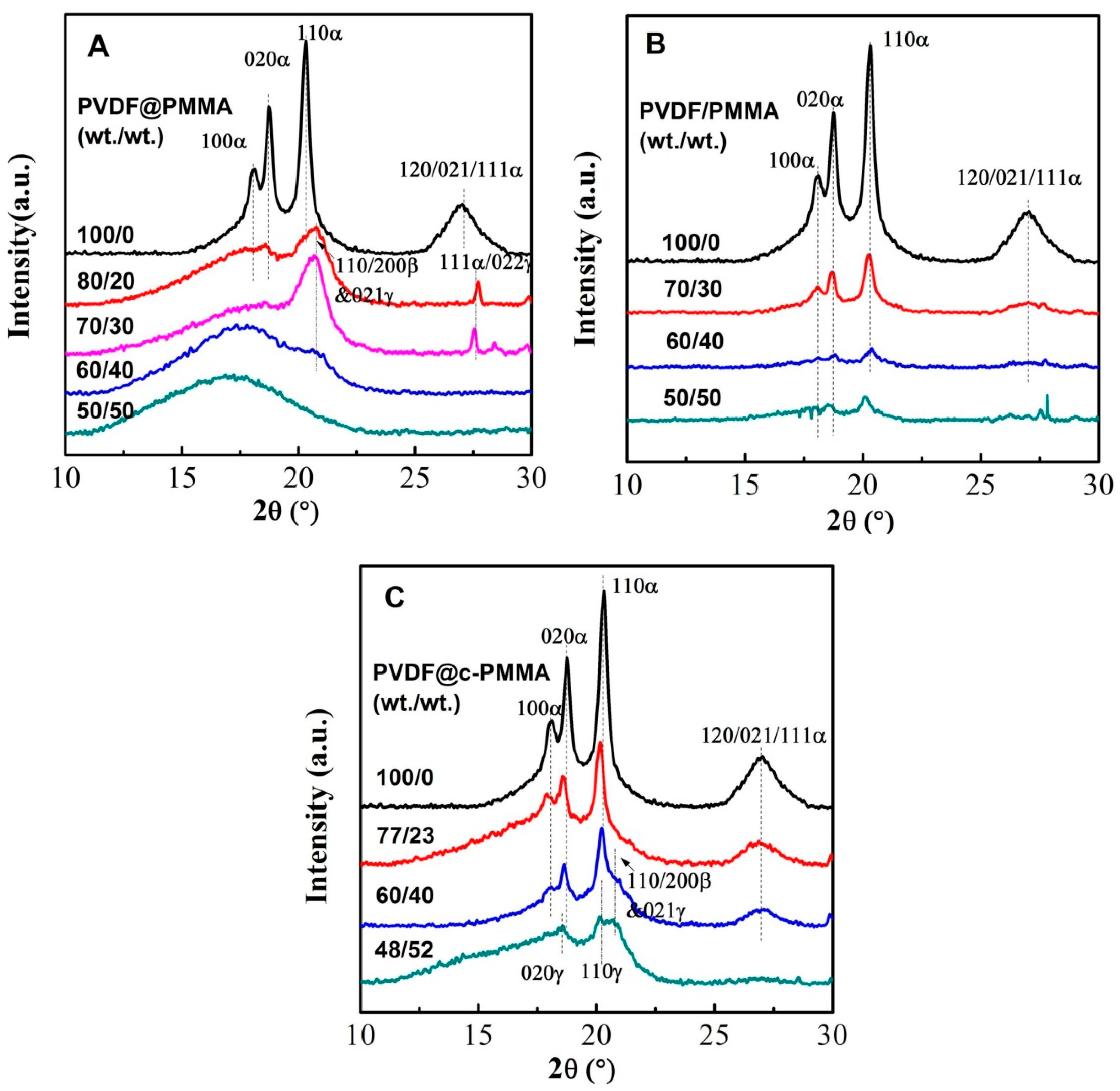
3.4. Polymorphic PVDF Crystallization Induced in Different Complexation Scenarios
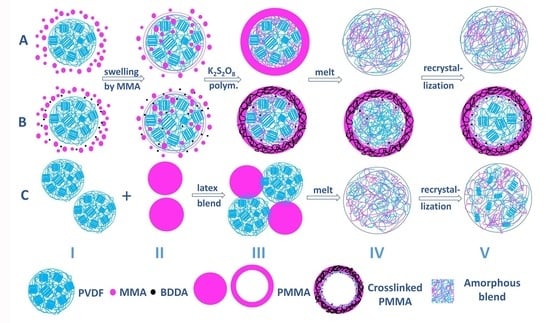
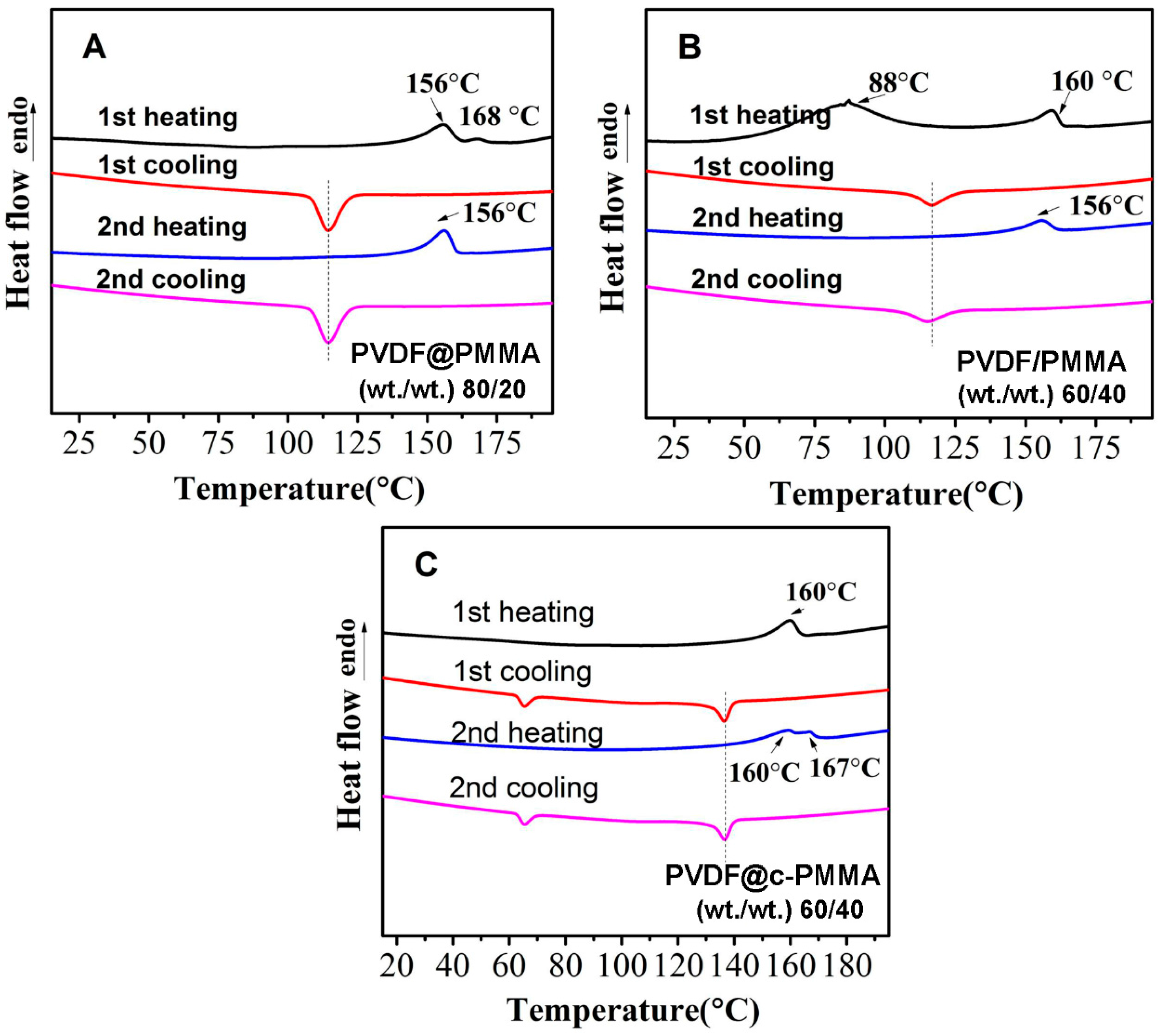
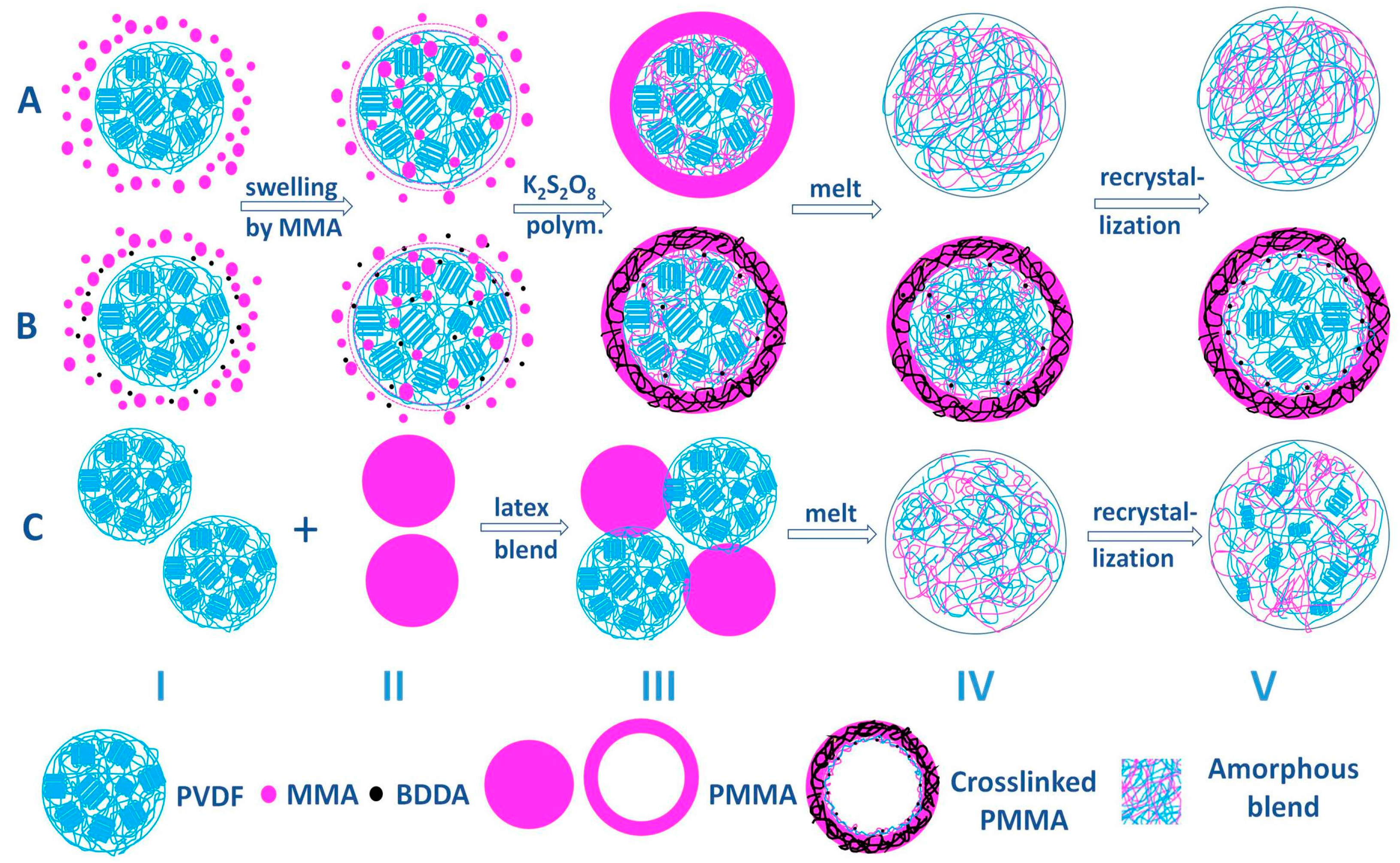
3.5. Structure Evolution in Different Complexation Scenarios
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lei, C.; Wang, X.; Tu, D.; Wang, H.; Du, Q. Charge distribution in PVDF/PMMA blends under DC field. Mater. Chem. Phys. 2009, 114, 272–278. [Google Scholar] [CrossRef]
- Ai, F.; Li, H.; Wang, Q.; Yuan, W.Z.; Chen, X.; Yang, L.; Zhao, J.; Zhang, Y. Surface characteristics and blood compatibility of PVDF/PMMA membranes. J. Mater. Sci. 2012, 47, 5030–5040. [Google Scholar] [CrossRef]
- Li, M.; Stingelin, N.; Michels, J.J.; Spijkman, M.-J.; Asadi, K.; Feldman, K.; Blom, P.W.M.; Leeuw, D.M. Ferroelectric phase diagram of PVDF: PMMA. Macromolecules 2012, 45, 7477–7485. [Google Scholar] [CrossRef]
- Fan, W.; Zheng, S. Miscibility and crystallization behavior in blends of poly (methyl methacrylate) and poly (vinylidene fluoride): Effect of star-like topology of poly (methyl methacrylate) chain. J. Polym. Sci. B 2007, 45, 2580–2593. [Google Scholar] [CrossRef]
- Sasaki, H.; Bala, P.K.; Yoshida, H.; Ito, E. Miscibility of PVDF/PMMA blends examined by crystallization dynamics. Polymer 1995, 36, 4805–4810. [Google Scholar] [CrossRef]
- Luciani, A.; Jarrin, J. Morphology development in immiscible polymer blends. Polym. Eng. Sci. 1996, 36, 1619–1626. [Google Scholar] [CrossRef]
- Okabe, Y.; Murakami, H.; Osaka, N.; Saito, H.; Inoue, T. Morphology development and exclusion of noncrystalline polymer during crystallization in PVDF/PMMA blends. Polymer 2010, 51, 1494–1500. [Google Scholar] [CrossRef]
- Tashiro, K. Crystal structure and phase transition of PVDF and related copolymers. Plast. Eng. N. Y. 1995, 28, 63–180. [Google Scholar]
- Lovinger, A.J. Annealing of poly (vinylidene fluoride) and formation of a fifth phase. Macromolecules 1982, 15, 40–44. [Google Scholar] [CrossRef]
- Takahashi, Y.; Matsubara, Y.; Tadokoro, H. Crystal structure of form II of poly (vinylidene fluoride). Macromolecules 1983, 16, 1588–1592. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tashiro, K.; Tadokoro, H. Molecular vibrations of three crystal forms of poly (vinylidene fluoride). Macromolecules 1975, 8, 158–171. [Google Scholar] [CrossRef]
- Takahashi, Y.; Tadokoro, H. Crystal structure of form III of poly (vinylidene fluoride). Macromolecules 1980, 13, 1317–1318. [Google Scholar] [CrossRef]
- Saito, H.; Okada, T.; Hamane, T.; Inoue, T. Crystallization kinetics in mixtures of poly (vinylidene fluoride) and poly (methyl methacrylate): Two-step diffusion mechanism. Macromolecules 1991, 24, 4446–4449. [Google Scholar] [CrossRef]
- Kim, K.J.; Cho, Y.J.; Kim, Y.H. Factors determining the formation of the β crystalline phase of poly (vinylidene fluoride) in poly (vinylidene fluoride)-poly (methyl methacrylate) blends. Vib. Spectrosc. 1995, 9, 147–159. [Google Scholar]
- Gradys, A.; Sajkiewicz, P.; Adamovsky, S.; Minakov, A.; Schick, C. Crystallization of poly (vinylidene fluoride) during ultra-fast cooling. Thermochim. Acta 2007, 461, 153–157. [Google Scholar] [CrossRef]
- Zhong, G.; Wang, K.; Zhang, L.; Li, Z.-M.; Fong, H.; Zhu, L. Nanodroplet formation and exclusive homogenously nucleated crystallization in confined electrospun immiscible polymer blend fibers of polystyrene and poly (ethylene oxide). Polymer 2011, 52, 5397–5402. [Google Scholar] [CrossRef]
- Pan, M.; Yang, L.; Wang, J.; Tang, S.; Zhong, G.; Su, R.; Sen, M.K.; Endoh, M.K.; Koga, T.; Zhu, L. Composite poly (vinylidene fluoride)/polystyrene latex particles for confined crystallization in 180 nm nanospheres via emulsifier-free batch seeded emulsion polymerization. Macromolecules 2014, 47, 2632–2644. [Google Scholar] [CrossRef]
- Horibe, H.; Hosokawa, Y.; Oshiro, H.; Sasaki, Y.; Takahashi, S.; Kono, A.; Nishiyama, T.; Danno, T. Effect of heat-treatment temperature after polymer melt and blending ratio on the crystalline structure of PVDF in a PVDF/PMMA blend. Polym. J. 2013, 45, 1195–1201. [Google Scholar] [CrossRef]
- Lee, M.; Koo, T.; Lee, S.; Min, B.H.; Kim, J.H. Morphology and physical properties of nanocomposites based on poly (methyl methacrylate)/poly (vinylidene fluoride) blends and multiwalled carbon nanotubes. Polym. Compos. 2015, 36, 1195–1204. [Google Scholar] [CrossRef]
- Freire, E.; Bianchi, O.; Martins, J.N.; Monteiro, E.E.; Forte, M.M.C. Non-isothermal crystallization of PVDF/PMMA blends processed in low and high shear mixers. J. Non-Cryst. Solids 2012, 358, 2674–2681. [Google Scholar] [CrossRef]
- Li, W.; Li, H.; Zhang, Y.-M. Preparation and investigation of PVDF/PMMA/TiO2 composite film. J. Mater. Sci. 2009, 44, 2977–2984. [Google Scholar] [CrossRef]
- Pan, M.; Yang, L.; Guan, B.; Lu, M.; Zhong, G.; Zhu, L. Surface nucleation-induced fluoropolymer Janus nanoparticles via emulsifier-free batch-seeded emulsion polymerization. Soft Matter 2011, 7, 11187–11193. [Google Scholar] [CrossRef]
- Lorec, G.; Baley, C.; Sire, O.; Grohens, Y. Characterization of interdiffusion between PVDF and stereoregular PMMA by using ATR–FTIR Spectroscopy. Macromol. Symp. 2005, 222, 265–271. [Google Scholar] [CrossRef]
- Duckworth, P.; Richardson, H.; Carelli, C.; Keddie, J. Infrared ellipsometry of interdiffusion in thin films of miscible polymers. Surf. Interface Anal. 2004, 36, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Zhang, J.; Wang, X.; Wang, S. Effect of PMMA on crystallization behavior and hydrophilicity of poly (vinylidene fluoride)/poly (methyl methacrylate) blend prepared in semi-dilute solutions. Appl. Surf. Sci. 2007, 253, 8377–8388. [Google Scholar] [CrossRef]
- Rios, L.; Hidalgo, M.; Cavaille, J.; Guillot, J.; Guyot, A.; Pichot, C. Polystyrene (1)/poly (butyl acrylate-methacrylic acid)(2) core–shell emulsion polymers. Part I. Synthesis and colloidal characterization. Colloid Polym. Sci. 1991, 269, 812–824. [Google Scholar] [CrossRef]
- Gilbert, R.G. Emulsion Polymerization: A Mechanistic Approach; Academic Press: London, UK, 1995. [Google Scholar]
- Suresh, K.I.; Pakula, T.; Bartsch, E. Synthesis Morphology and Rheological Behavior of Fluoropolymer-Polyacrylate Nanocomposites. Macromol. React. Eng. 2007, 1, 253–263. [Google Scholar] [CrossRef]
- Yoshida, H. Structure formation of PVDF/PMMA blends studied: Simultaneous DSC/FT–IR measurement. J. Thermal. Anal. 1997, 49, 101–105. [Google Scholar] [CrossRef]
- Nishi, T.; Wang, T. Melting point depression and kinetic effects of cooling on crystallization in poly (vinylidene fluoride)-poly (methyl methacrylate) mixtures. Macromolecules 1975, 8, 909–915. [Google Scholar] [CrossRef]
- Tashiro, K.; Kobayashi, M.; Tadokoro, H. Vibrational spectra and disorder-order transition of poly (vinylidene fluoride) form III. Macromolecules 1981, 14, 1757–1764. [Google Scholar] [CrossRef]
- Bachmann, M.; Gordon, W.; Koenig, J.; Lando, J. An infrared study of phase-III poly (vinylidene fluoride). J. Appl. Phys. 1979, 50, 6106–6112. [Google Scholar] [CrossRef]
- Tashiro, K.; Takano, K.; Kobayashi, M.; Chatani, Y.; Tadokoro, H. A preliminary X-ray study on ferroelectric phase transition of poly (vinylidene ruoride) crystal form I. Polym. Bull. 1983, 10, 464–469. [Google Scholar] [CrossRef]
- Li, Y.; Tang, S.; Pan, M.-W.; Zhu, L.; Zhong, G.-J.; Li, Z.-M. Polymorphic Extended-Chain and Folded-Chain Crystals in Poly (vinylidene fluoride) Achieved by Combination of High Pressure and Ion–Dipole Interaction. Macromolecules 2015, 48, 8565–8573. [Google Scholar] [CrossRef]
- Haris, M.R.; Kathiresan, S.; Mohan, S. FT–IR and FT–Raman spectra and normal coordinate analysis of poly methyl methacrylate. Der Pharma Chemica 2010, 2, 316–323. [Google Scholar]
- Peng, Y.; Wu, P. A two dimensional infrared correlation spectroscopic study on the structure changes of PVDF during the melting process. Polymer 2004, 45, 5295–5299. [Google Scholar] [CrossRef]
- Mohammadi, B.; Yousefi, A.A.; Bellah, S.M.; Yousefi, A.A.; Bellah, S.M. Effect of tensile strain rate and elongation on crystalline structure and piezoelectric properties of PVDF thin films. Polym. Test. 2007, 26, 42–50. [Google Scholar] [CrossRef]
- Salimi, A.; Yousefi, A. Analysis method: FTIR studies of β-phase crystal formation in stretched PVDF films. Polym. Test. 2003, 22, 699–704. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, X.; Zhou, D.; Shen, Q.D.; Hu, W. Low-temperature crystallization of P(VDF–TrFE–CFE) studied by Flash DSC. Polymer 2016, 84, 319–327. [Google Scholar] [CrossRef]
- Shukla, N.; Thakur, A.K. Nanocrystalline filler induced changes in electrical and stability properties of a polymer nanocomposite electrolyte based on amorphous matrix. J. Mater. Sci. 2010, 45, 4236–4250. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample series | PVDF/MMA ratio (wt/wt) | Polymerization temperature (°C) | Reaction time (h) | Composition ratio of PVDF to PMMA (wt/wt) |
|---|---|---|---|---|
| PVDF@PMMA | 1:4 | 75 | 0.5 | 70/30 |
| 1:4 | 75 | 1 | 60/40 | |
| 1:4 | 75 | 2 | 50/50 | |
| PVDF@c–PMMA | 1:5 | 65 | 4 | 77/23 |
| 1:5 | 65 | 5 | 60/40 | |
| 1:5 | 65 | 6 | 48/52 | |
| PVDF/PMMA 1 | 7:3 | 70/30 | ||
| 6:4 | 60/40 | |||
| 5:5 | 50/50 |
| Sample | Composition ratio (wt/wt) | Area ratio (110/200)β/110α | Amorphous peak position (2θ/°) | Average interchain separation (Å) 1 | Crystallinity 2 (%) |
|---|---|---|---|---|---|
| PVDF latex | 100/0 | 0 | 19.09 | 5.81 | 38.20 |
| PVDF@PMMA | 80/20 | 1.39 | 17.99 | 6.16 | 23.85 |
| 70/30 | 1.69 | 17.93 | 6.19 | 18.06 | |
| 60/40 | ---- | 17.56 | 6.32 | 3.57 | |
| 50/50 | ---- | 16.90 | 6.56 | 0 | |
| PVDF/PMMA | 70/30 | 0.17 | 18.99 | 5.84 | 45.00 |
| 60/40 | 0.34 | 18.67 | 5.94 | 32.42 | |
| 50/50 | 0.33 | 18.07 | 6.14 | 23.86 | |
| PVDF@c–PMMA | 77/23 | 0.16 | 16.4/19.09 | 5.81 | 27.52 |
| 60/40 3 | 1.16 | 19.09 | ---- | 28.34 | |
| 48/52 | ---- | 15.2/19.10 | 5.80 | 24.47 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, G.; Song, S.; Xu, H.; Pan, M.; Zhong, G.-J. How Chain Intermixing Dictates the Polymorphism of PVDF in Poly(vinylidene fluoride)/Polymethylmethacrylate Binary System during Recrystallization: A Comparative Study on Core–Shell Particles and Latex Blend. Polymers 2017, 9, 448. https://doi.org/10.3390/polym9090448
Li Y, Zhang G, Song S, Xu H, Pan M, Zhong G-J. How Chain Intermixing Dictates the Polymorphism of PVDF in Poly(vinylidene fluoride)/Polymethylmethacrylate Binary System during Recrystallization: A Comparative Study on Core–Shell Particles and Latex Blend. Polymers. 2017; 9(9):448. https://doi.org/10.3390/polym9090448
Chicago/Turabian StyleLi, Yue, Guoqiang Zhang, Shaofeng Song, Haijun Xu, Mingwang Pan, and Gan-Ji Zhong. 2017. "How Chain Intermixing Dictates the Polymorphism of PVDF in Poly(vinylidene fluoride)/Polymethylmethacrylate Binary System during Recrystallization: A Comparative Study on Core–Shell Particles and Latex Blend" Polymers 9, no. 9: 448. https://doi.org/10.3390/polym9090448