Transparent Low Molecular Weight Poly(Ethylene Glycol) Diacrylate-Based Hydrogels as Film Media for Photoswitchable Drugs

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
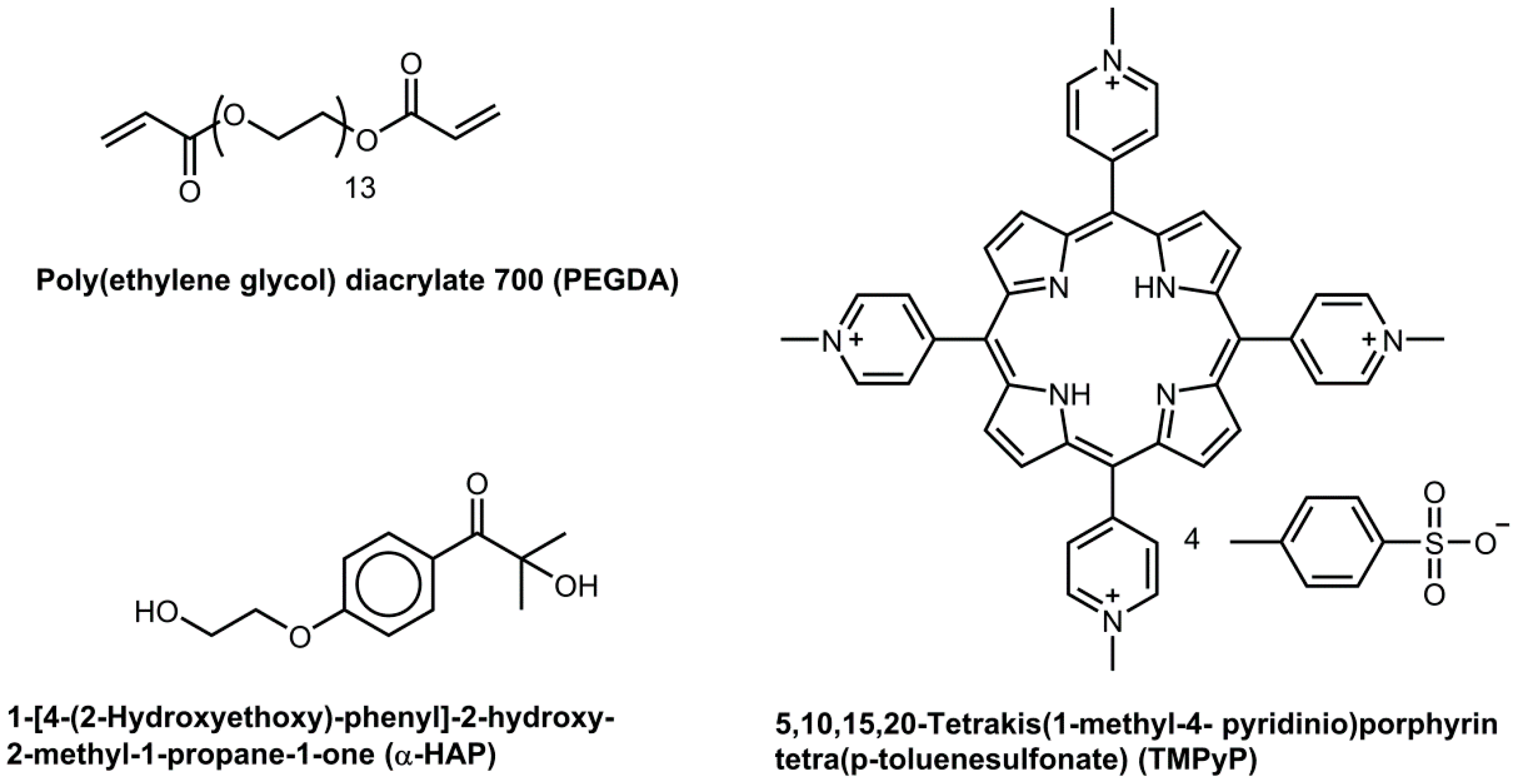
2.1. Materials
2.2. Formulations
2.3. Real-Time Infrared Spectroscopy
2.4. UV-Vis Spectroscopy
2.5. Scanning Electron Microscopy
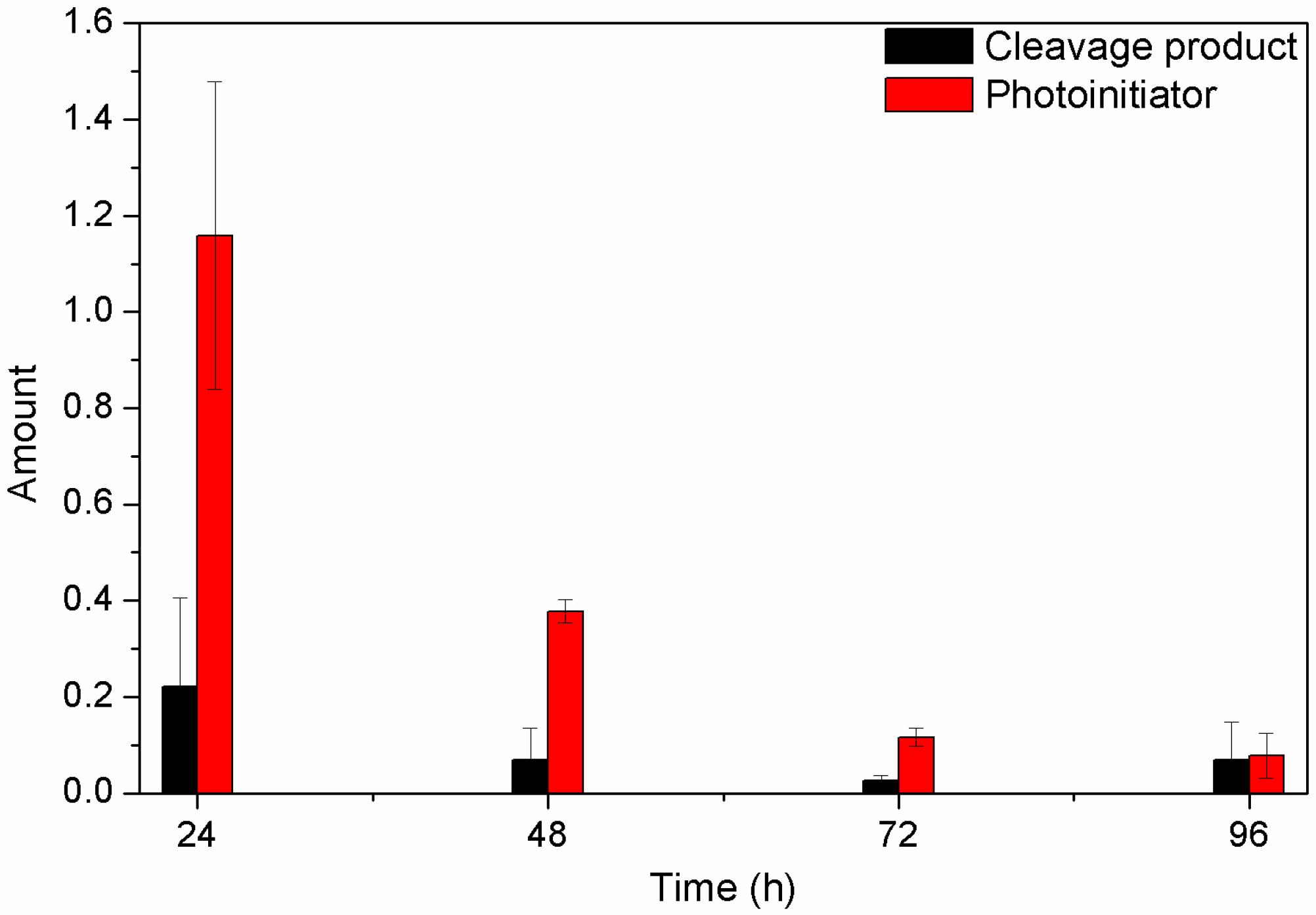
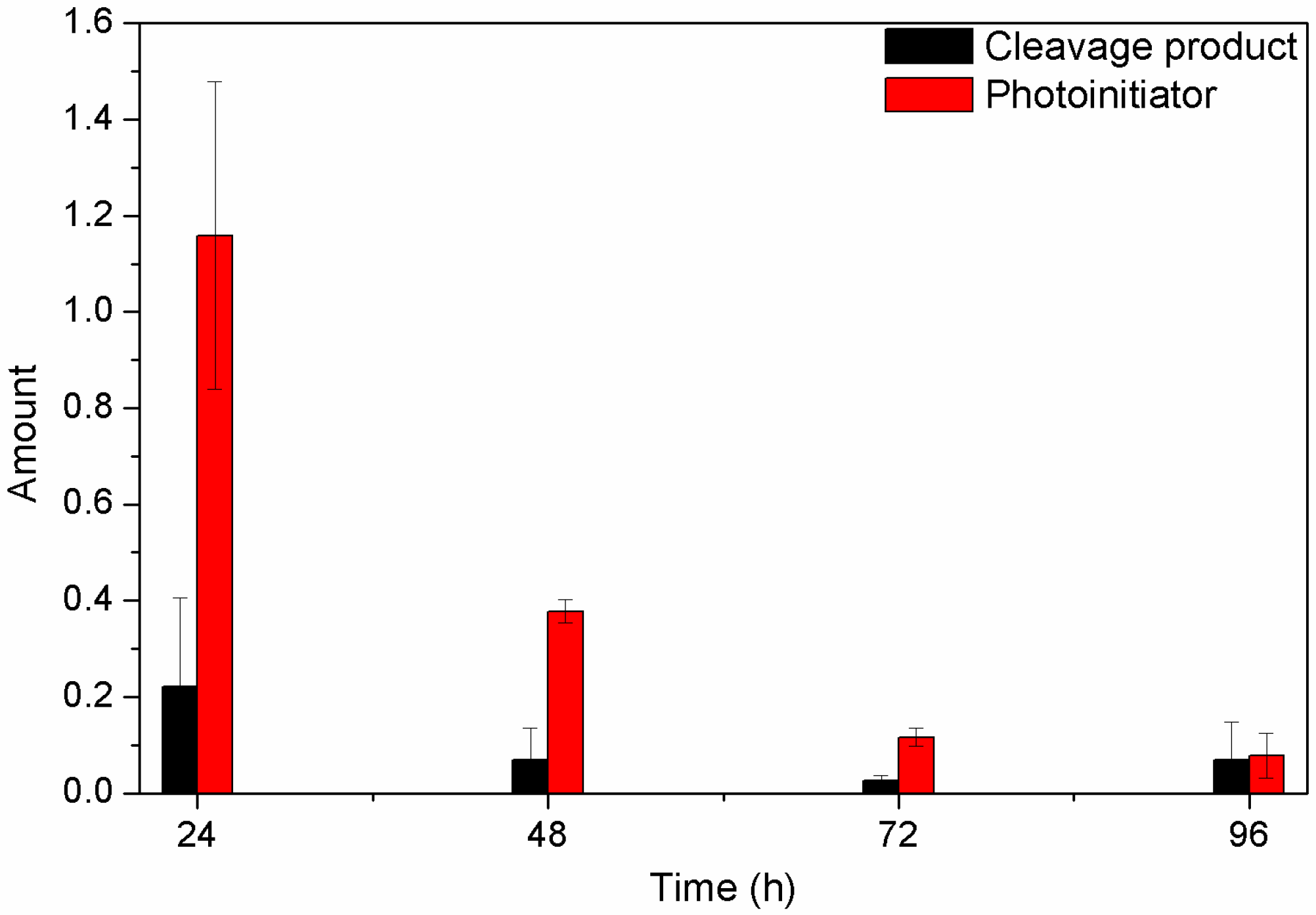
2.6. Detection of Unreacted Products
2.7. Swelling Ratio
3. Results & Discussion
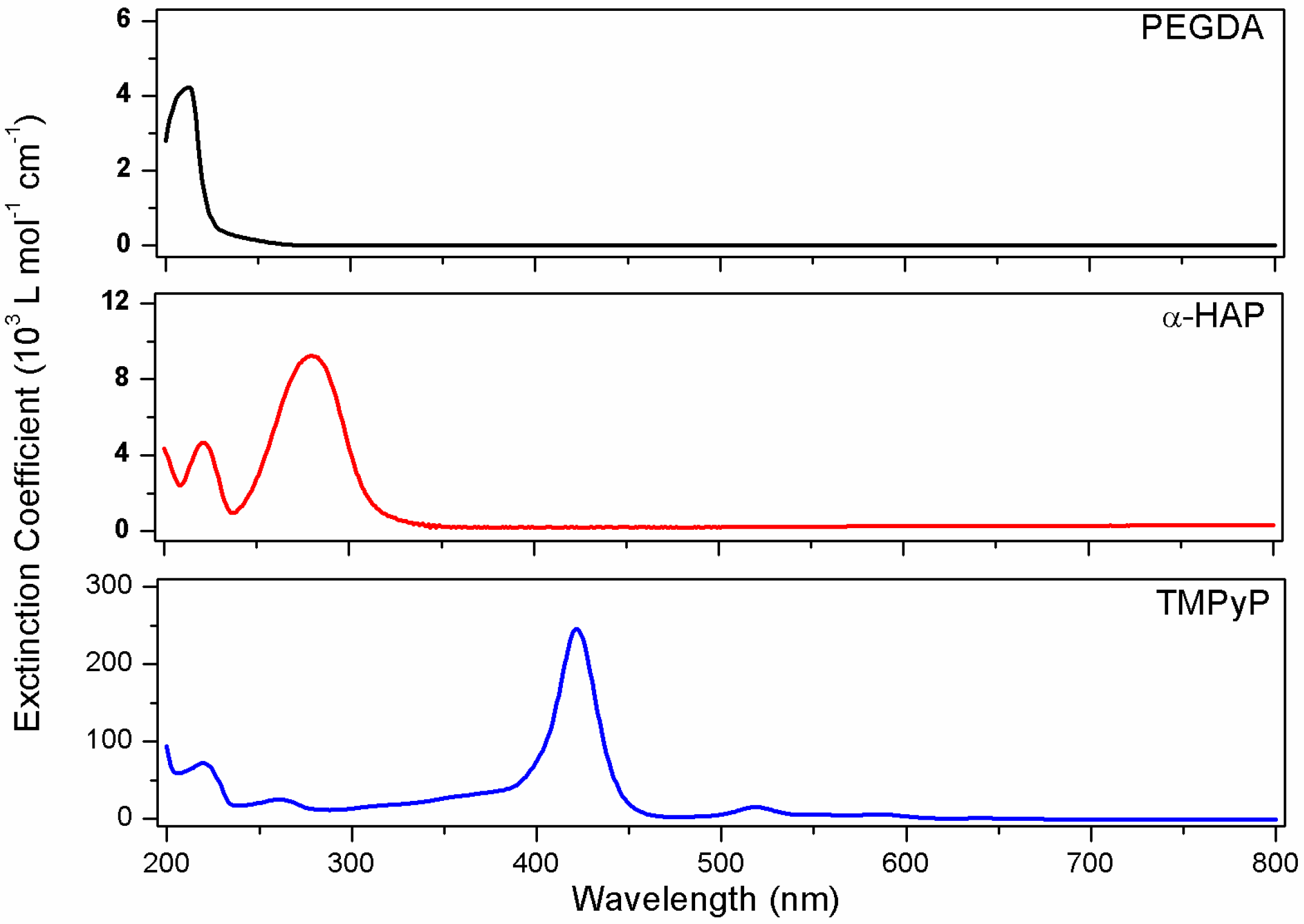
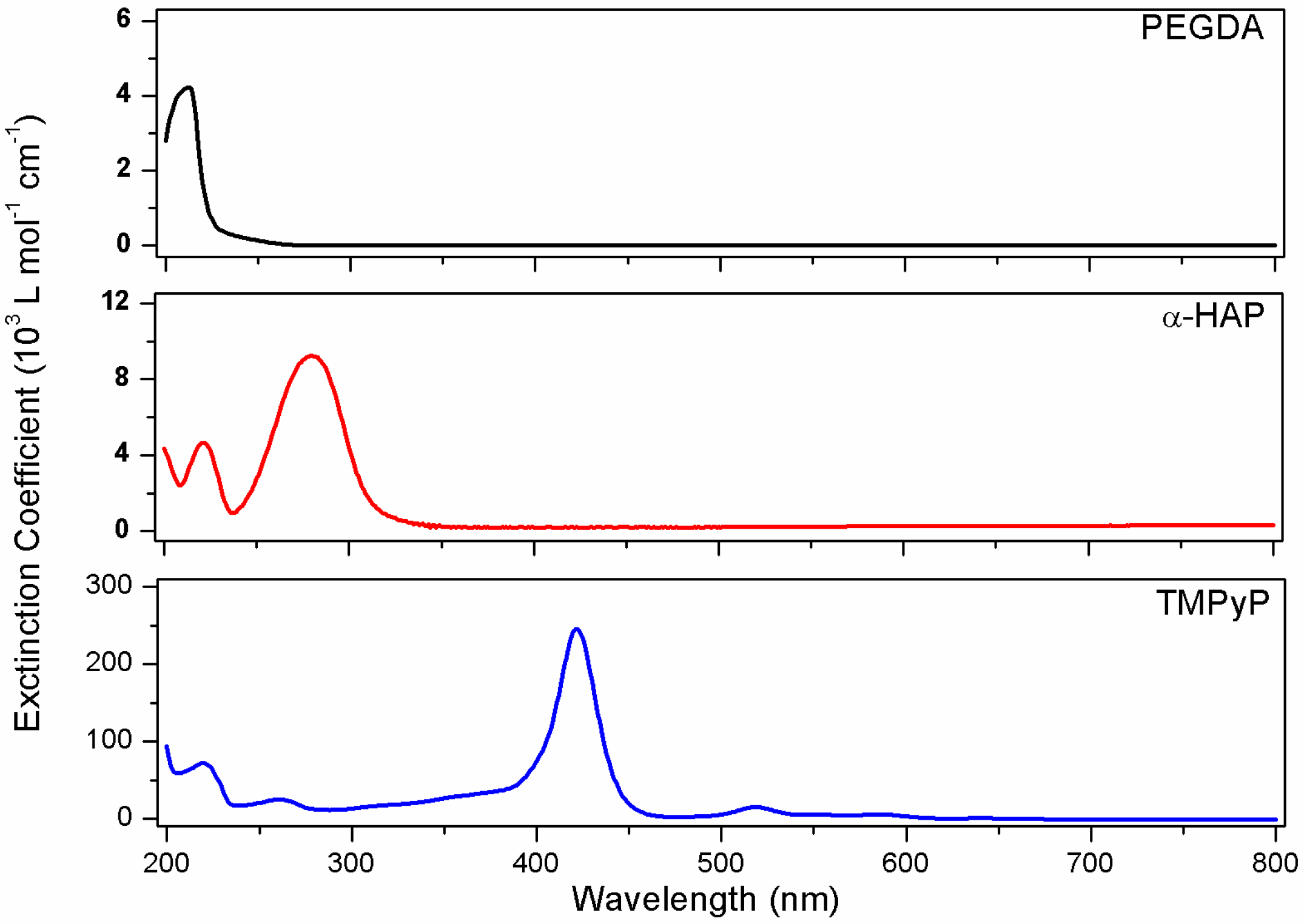
3.1. UV-Vis Spectroscopy
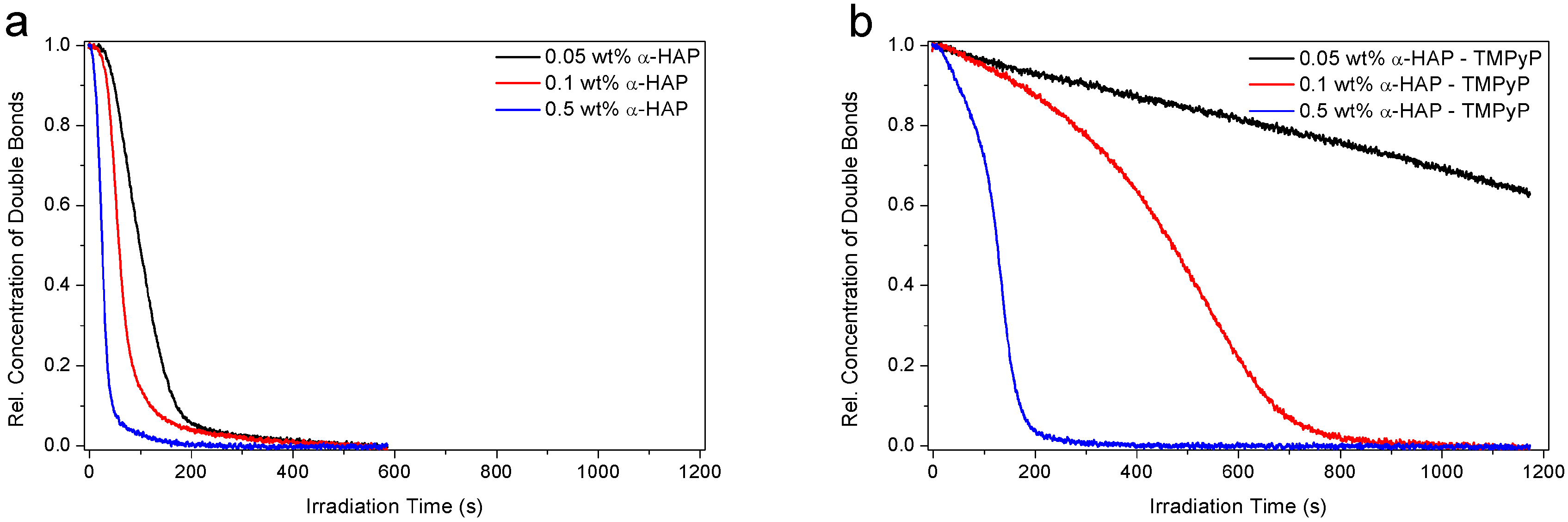
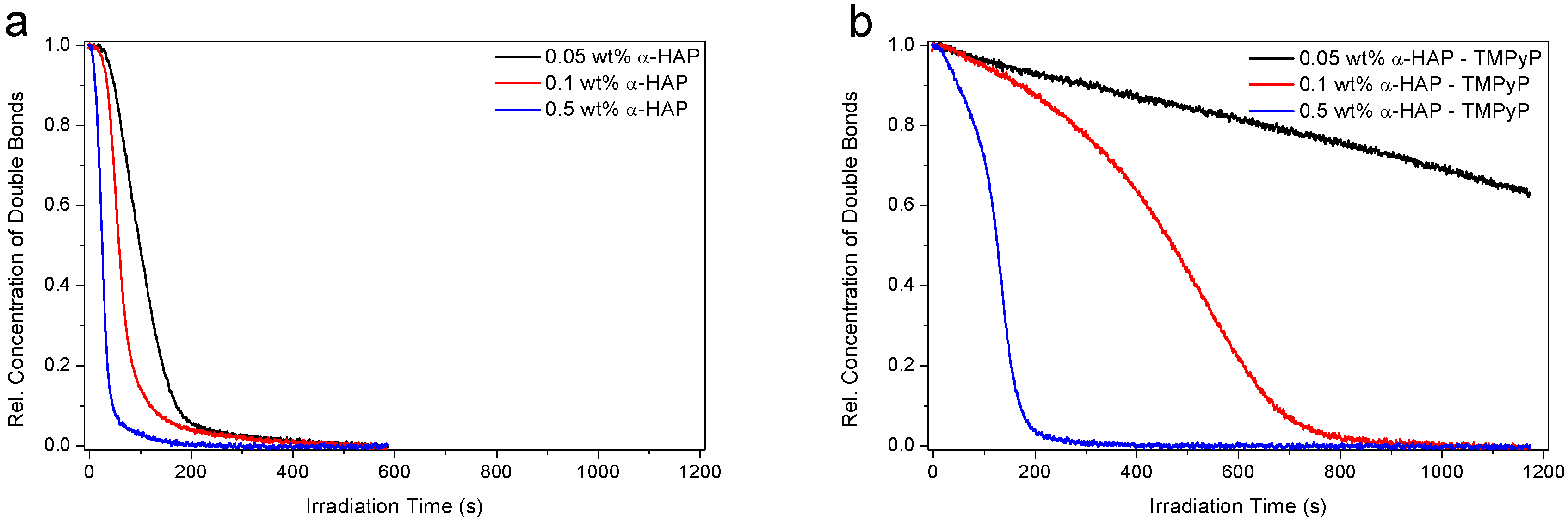
3.2. Investigation of the Hydrogel Photopolymerization
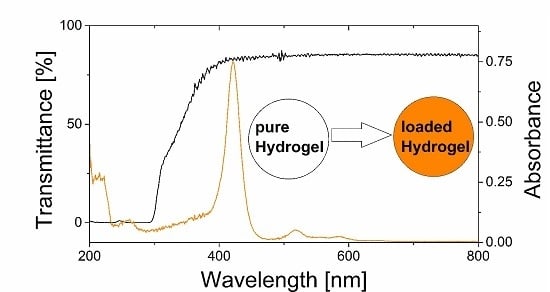
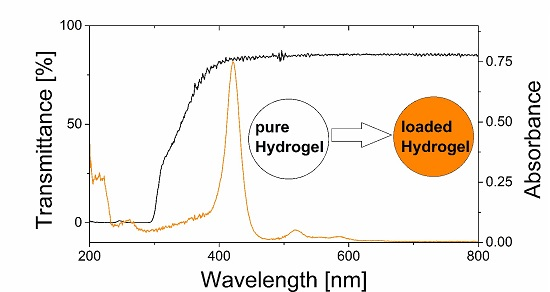
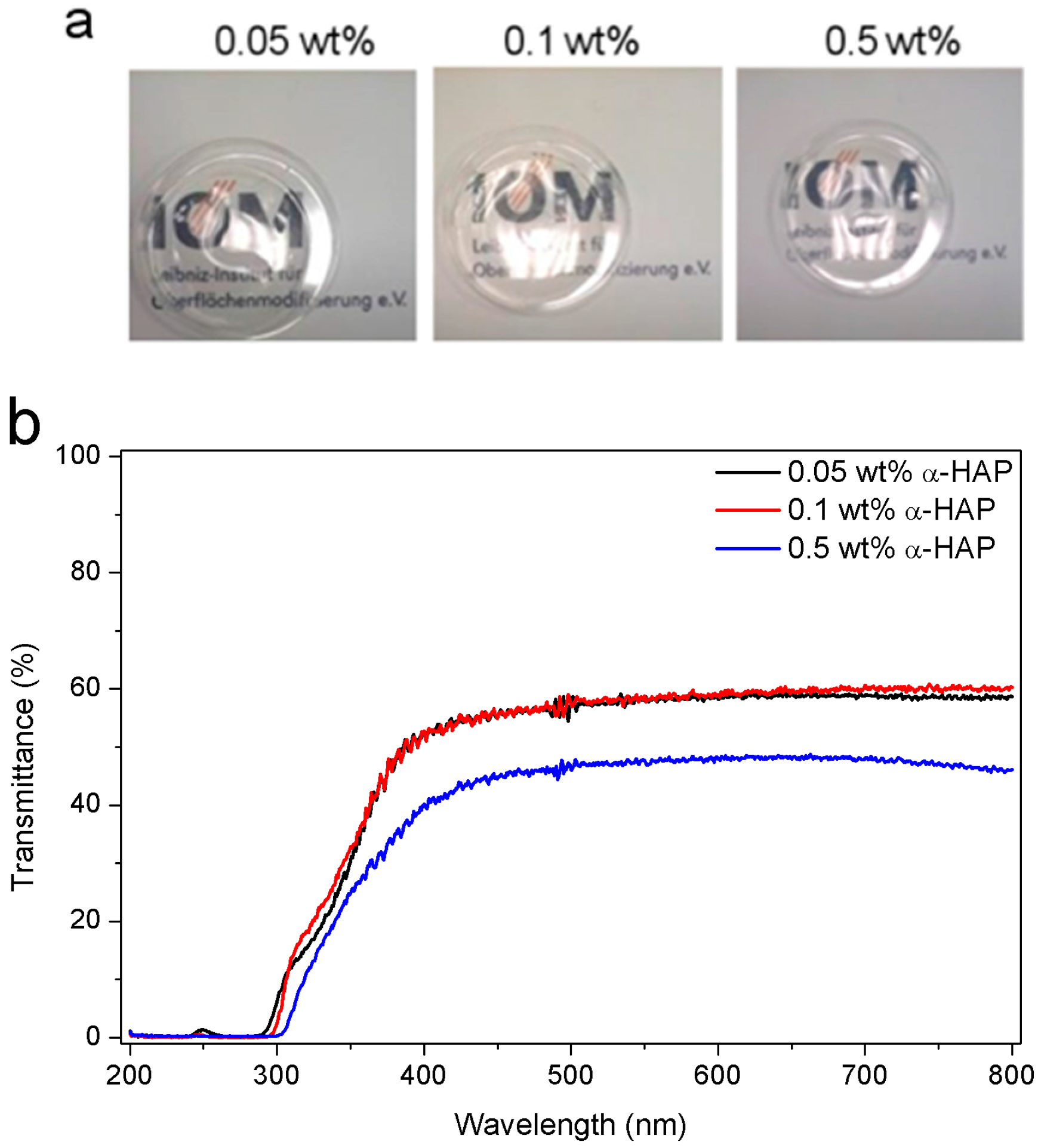
3.3. Transparency
3.4. Surface Morphology
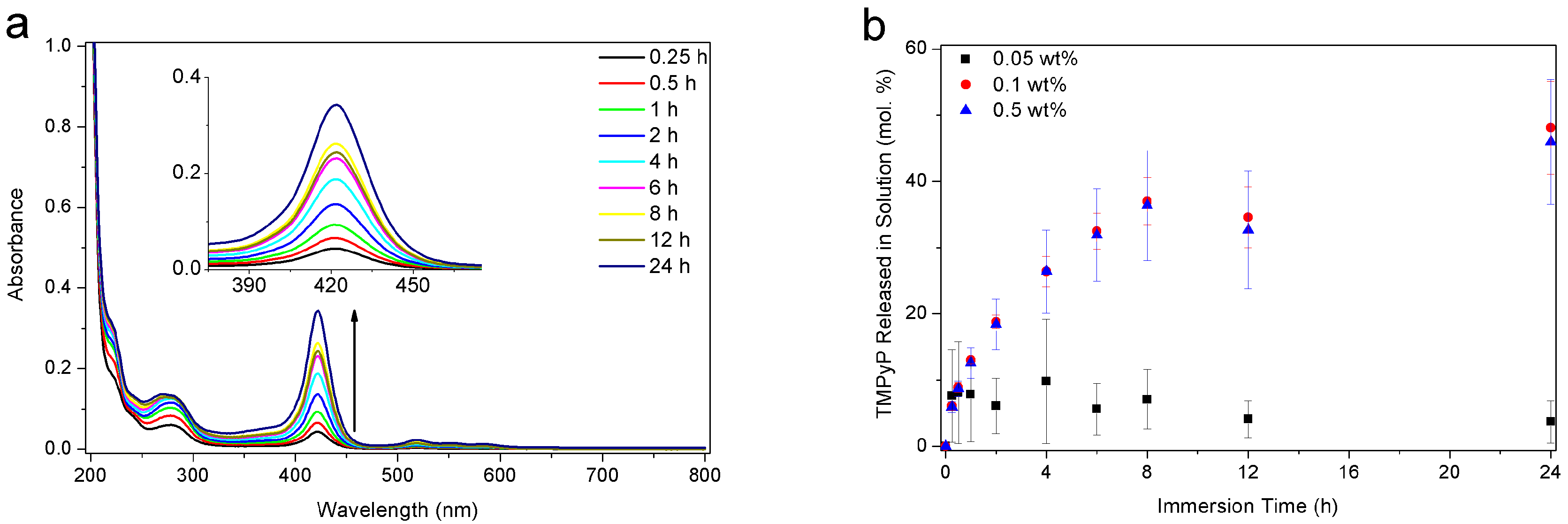
3.5. Release Behavior
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peppas, N.A.; Bures, P.; Khare, A.R. Preparation, structure and diffusional behavior of hydrogels in controlled release. Adv. Drug Deliv. Rev. 1993, 11, 1–35. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Calo, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Buwalda, S.J.; Boere, K.W.; Dijkstra, P.J.; Feijen, J.; Vermonden, T.; Hennink, W.E. Hydrogels in a historical perspective: From simple networks to smart materials. J. Control. Release 2014, 190, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Bioactive modification of poly(ethylene glycol)hydrogels for tissue engineering. Biomaterials 2010, 31, 4639–4656. [Google Scholar] [CrossRef] [PubMed]
- Wichterle, O.; Lim, D. Hydrophilic gels for biological use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Hwang, Y.; Chen, A.C.; Varghese, S.; Sah, R.L. Cartilage-like mechanical properties of poly(ethylene glycol)-diacrylate hydrogels. Biomaterials 2012, 33, 6682–6690. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1879. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.; Burban, J.H.; Cussler, E.L. Hydrogels as separation agents. In Responsive Gels: Volume Transitions II; Springer: Berlin, Germany, 1993; Volume 110. [Google Scholar]
- Jen, A.C.; Wake, M.C.; Mikos, A.G. Review: Hydrogels for cell immobilization. Biotechnol. Bioeng. 1996, 50, 357–364. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Sikareepaisan, P.; Ruktanonchai, U.; Supaphol, P. Preparation and characterization of asiaticoside-loaded alginate films and their potential for use as effectual wound dressings. Carbohydr. Polym. 2011, 83, 1457–1469. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Raab, O. Ueber die wirkung fluoreszierender stoffe auf infusorien. Z. Biol. 1900, 39, 524–546. [Google Scholar]
- Riyad, Y.M.; Naumov, S.; Schastak, S.; Griebel, J.; Kahnt, A.; Häupl, T.; Neuhaus, J.; Abel, B.; Hermann, R. Chemical modification of a tetrapyrrole-type photosensitizer: Tuning application and photochemical action beyond the singlet oxygen channel. J. Phys. Chem. B 2014, 118, 11646–11658. [Google Scholar] [CrossRef] [PubMed]
- Juzeniene, A.; Peng, Q.; Moan, J. Milestones in the development of photodynamic therapy and fluorescence diagnosis. Photochem. Photobiol. Sci. 2007, 6, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Hanakova, A.; Bogdanova, K.; Tomankova, K.; Pizova, K.; Malohlava, J.; Binder, S.; Bajgar, R.; Langova, K.; Kolar, M.; Mosinger, J.; et al. The application of antimicrobial photodynamic therapy on S. aureus and E. coli using porphyrin photosensitizers bound to cyclodextrin. Microbiol. Res. 2014, 169, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Konopka, K.; Goslinski, T. Photodynamic therapy in dentistry. J. Dent. Res. 2007, 86, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Divan, R.; Suthar, K.J.; Mancini, D.C.; Gemeinhart, R.A. Fabrication of poly(ethylene glycol) hydrogel structures for pharmaceutical applications using electron beam and optical lithography. J. Vac. Sci. Technol. B 2010, 28, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Brooks, H.J.; Moratti, S.C.; Hanton, L.R.; Cabral, J.D. Reducing the oxidation level of dextran aldehyde in a chitosan/dextran-based surgical hydrogel increases biocompatibility and decreases antimicrobial efficacy. Int. J. Mol. Sci. 2015, 16, 13798–13814. [Google Scholar] [CrossRef] [PubMed]
- Cabral, J.D.; Roxburgh, M.; Shi, Z.; Liu, L.; McConnell, M.; Williams, G.; Evans, N.; Hanton, L.R.; Simpson, J.; Moratti, S.C.; et al. Synthesis, physiochemical characterization, and biocompatibility of a chitosan/dextran-based hydrogel for postsurgical adhesion prevention. J. Mater. Sci. 2014, 25, 2743–2756. [Google Scholar] [CrossRef] [PubMed]
- Bode, F.; Alves da Silva, M.; Drake, A.F.; Ross-Murphy, S.B.; Dreiss, C.A. Enzymatically cross-linked tilapia gelatin hydrogels: Physical, chemical, and hybrid networks. Biomacromolecules 2011, 12, 3741–3752. [Google Scholar] [CrossRef] [PubMed]
- Wisotzki, E.I.; Hennes, M.; Schuldt, C.; Engert, F.; Knolle, W.; Decker, U.; Käs, J.A.; Zink, M.; Mayr, S.G. Tailoring the material properties of gelatin hydrogels by high energy electron irradiation. J. Mater. Chem. B 2014, 2, 4297–4309. [Google Scholar] [CrossRef]
- Wu, X.S.; Hoffman, A.S.; Yager, P. Synthesis and characterization of thermally reversible macroporous poly(N-isopropylacrylamide) hydrogels. J. Polym. Sci. Part A 1992, 30, 2121–2129. [Google Scholar] [CrossRef]
- Strauss, P.; Knolle, W.; Naumov, S. Radiation-induced radical formation and crosslinking in aqueous solutions of N-isopropylacrylamide. Macromol. Chem. Phys. 1998, 199, 2229–2235. [Google Scholar]
- Fujishige, S.; Kubota, K.; Ando, I. Phase transition of aqueous solutions of poly(N-isopropylacrylamide) and poly(N-isopropylmethacrylamide). J. Phys. Chem. 1989, 93, 3311–3313. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Rivera, C.; Wu, C.J.; Chan, B.K.; Schmidt, G. Photocrosslinked nanocomposite hydrogels from peg and silica nanospheres: Structural, mechanical and cell adhesion characteristics. Mater. Sci. Eng. C 2013, 33, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.C.L.; Betts, D.E.; Pandya, A.; Hillmyer, M.A.; DeSimone, J.M. Optically transparent, amphiphilic networks based on blends of perfluoropolyethers and poly(ehtylene glycol). J. Am. Chem. Soc. 2008, 130, 14244–14252. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, N.C.; Chatterji, P.R. Structural characteristics and swelling behavior of poly(ethylene glycol) diacrylate hydrogels. Macromolecules 1996, 29, 1976–1979. [Google Scholar] [CrossRef]
- Reichelt, S.; Abe, C.; Hainich, S.; Knolle, W.; Decker, U.; Prager, A.; Konieczny, R. Electron-beam derived polymeric cryogels. Soft Matter 2013, 9, 2484–2492. [Google Scholar] [CrossRef]
- Wang, L.; Ding, Y. Creating micro-structured hydrogel-forming polymer films by photopolymerization in an evaporating solvent: Compositional and morphological evolutions. Eur. Polym. J. 2015, 66, 99–107. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Ding, Y. Photocrosslinking-induced phase separation in evaporative solvents: Formation of skin layers and microspheres. Soft Matter 2013, 9, 4455–4463. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Dammu, S.A.; Canter, J.M.; Wu, C.J.; Schmidt, G. Highly extensible, tough, and elastomeric nanocomposite hydrogels from poly(ethylene glycol) and hydroxyapatite nanoparticles. Biomacromolecules 2011, 12, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Gaharwar, A.K.; Rivera, C.P.; Wu, C.J.; Schmidt, G. Transparent, elastomeric and tough hydrogels from poly(ethylene glycol) and silicate nanoparticles. Acta Biomater. 2011, 7, 4139–4148. [Google Scholar] [CrossRef] [PubMed]
- Beamish, J.A.; Zhu, J.; Kottke-Marchant, K.; Marchant, R.E. The effects of monoacrylated poly(ethylene glycol) on the properties of poly(ethylene glycol) diacrylate hydrogels used for tissue engineering. J. Biomed. Mater. Res. Part A 2009, 92A, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Brandl, F.; Kastner, F.; Gschwind, R.M.; Blunk, T.; Tessmar, J.; Goepferich, A. Hydrogel-based drug delivery systems: Comparison of drug diffusivity and release kinetics. J. Control. Release 2010, 142, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, B.D.; Schwartz, M.P.; Bowman, C.N.; Anseth, K.S. Photoinitiated polymerization of peg-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: Polymerization rate and cytocompatibility. Biomaterials 2009, 30, 6702–6707. [Google Scholar] [CrossRef] [PubMed]
- Abuteen, A.; Zanganeh, S.; Akhigbe, J.; Samankumara, L.P.; Aguirre, A.; Biswal, N.; Braune, M.; Vollertsen, A.; Roder, B.; Bruckner, C.; et al. The evaluation of NIR-absorbing porphyrin derivatives as contrast agents in photoacoustic imaging. Phys. Chem. Chem. Phys. 2013, 15, 18502–18509. [Google Scholar] [CrossRef] [PubMed]
- Ormond, A.B.; Freeman, H.S. Dye sensitizers for photodynamic therapy. Materials 2013, 6, 817–840. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.M.; dos Santos, F.V.; Lyon, J.P.; Maftoum-Costa, M.; Pacheco-Soares, C.; da Silva, N.S. Photodynamic therapy: Porphyrins and phthalocyanines as photosensitizers. Aust. J. Chem. 2008, 61, 741–754. [Google Scholar] [CrossRef]
- Scherzer, T.; Decker, U. Real-time FTIR–ATR spectroscopy to study the kinetics of ultrafast photopolymerization reactions induced by monochromatic uv light. Vib. Spectrosc. 1999, 19, 385–398. [Google Scholar] [CrossRef]
- Scherzer, T.; Knolle, W.; Naumov, S.; Elsner, C.; Buchmeiser, M.R. Self-initiation of the UV photopolymerization of brominated acrylates. J. Polym. Sci. Part A 2008, 46, 4905–4916. [Google Scholar] [CrossRef]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: San Diego, CA, USA, 1990. [Google Scholar]
- Ligon, S.C.; Husár, B.; Wutzel, H.; Holman, R.; Liska, R. Strategies to reduce oxygen inhibition in photoinduced polymerization. Chem. Rev. 2014, 114, 557–589. [Google Scholar] [CrossRef] [PubMed]
- Pynaert, R.; Buguet, J.; Croutxé-Barghorn, C.; Moireau, P.; Allonas, X. Effect of reactive oxygen species on the kinetics of free-radical photopolymerization. Polym. Chem. (UK) 2013, 4, 2475–2479. [Google Scholar] [CrossRef]
- Boyd, C.E.; Tucker, C.S. Pond Aquaculture Water Quality Management; Springer Science+Business Media, LLC: Berlin, Germany, 1998. [Google Scholar]
- Manahan, S.; Manahan, S.E. Environmental Chemistry, 9th ed.; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Ramsden, E.N. Chemistry of the Environment; Nelson Thornes: Cheltenham, UK, 1996. [Google Scholar]
- Courtecuisse, F.; Belbakra, A.; Croutxe-Barghorn, C.; Allonas, X.; Dietlin, C. Zirconium complexes to overcome oxygen inhibition in free-radical photopolymerization of acrylates: Kinetic, mechanism, and depth profiling. J. Polym. Sci. Part A 2011, 49, 5169–5175. [Google Scholar] [CrossRef]
- Belon, C.; Allonas, X.; Croutxé-Barghorn, C.; Lalevée, J. Overcoming the oxygen inhibition in the photopolymerization of acrylates: A study of the beneficial effect of triphenylphosphine. J. Polym. Sci. Part A 2010, 48, 2462–2469. [Google Scholar] [CrossRef]
- Kopecek, J. Hydrogels from soft contact lenses and implants to self-assembled nanomaterials. J. Polym. Sci. Part A 2009, 47, 5929–5946. [Google Scholar] [CrossRef] [PubMed]
- Yean, L.; Bunel, C.; Vairon, J.-P. Reversible immobilization of drugs on a hydrogel matrix, 2. Diffusion of free chloramphenicol from poly(2-hydroxyethyl methacrylate) hydrogels. Macromol. Chem. Phys. 1990, 191, 1119–1129. [Google Scholar] [CrossRef]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelras, T.; Glass, S.; Scherzer, T.; Elsner, C.; Schulze, A.; Abel, B. Transparent Low Molecular Weight Poly(Ethylene Glycol) Diacrylate-Based Hydrogels as Film Media for Photoswitchable Drugs. Polymers 2017, 9, 639. https://doi.org/10.3390/polym9120639
Pelras T, Glass S, Scherzer T, Elsner C, Schulze A, Abel B. Transparent Low Molecular Weight Poly(Ethylene Glycol) Diacrylate-Based Hydrogels as Film Media for Photoswitchable Drugs. Polymers. 2017; 9(12):639. https://doi.org/10.3390/polym9120639
Chicago/Turabian StylePelras, Théophile, Sarah Glass, Tom Scherzer, Christian Elsner, Agnes Schulze, and Bernd Abel. 2017. "Transparent Low Molecular Weight Poly(Ethylene Glycol) Diacrylate-Based Hydrogels as Film Media for Photoswitchable Drugs" Polymers 9, no. 12: 639. https://doi.org/10.3390/polym9120639