
Correction of MHS Viscosimetric Constants upon Numerical Simulation of Temperature Induced Degradation Kinetic of Chitosan Solutions


Abstract
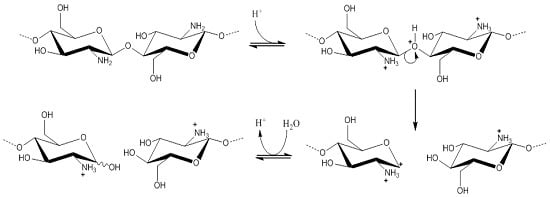
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Rheological Characterization
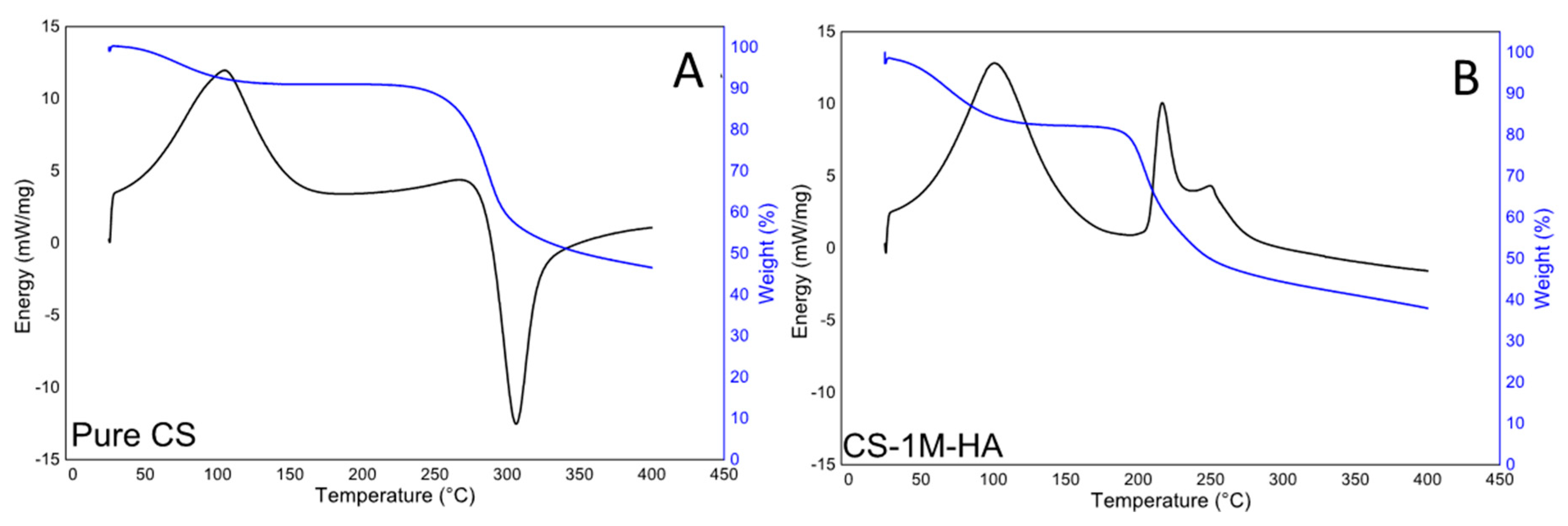
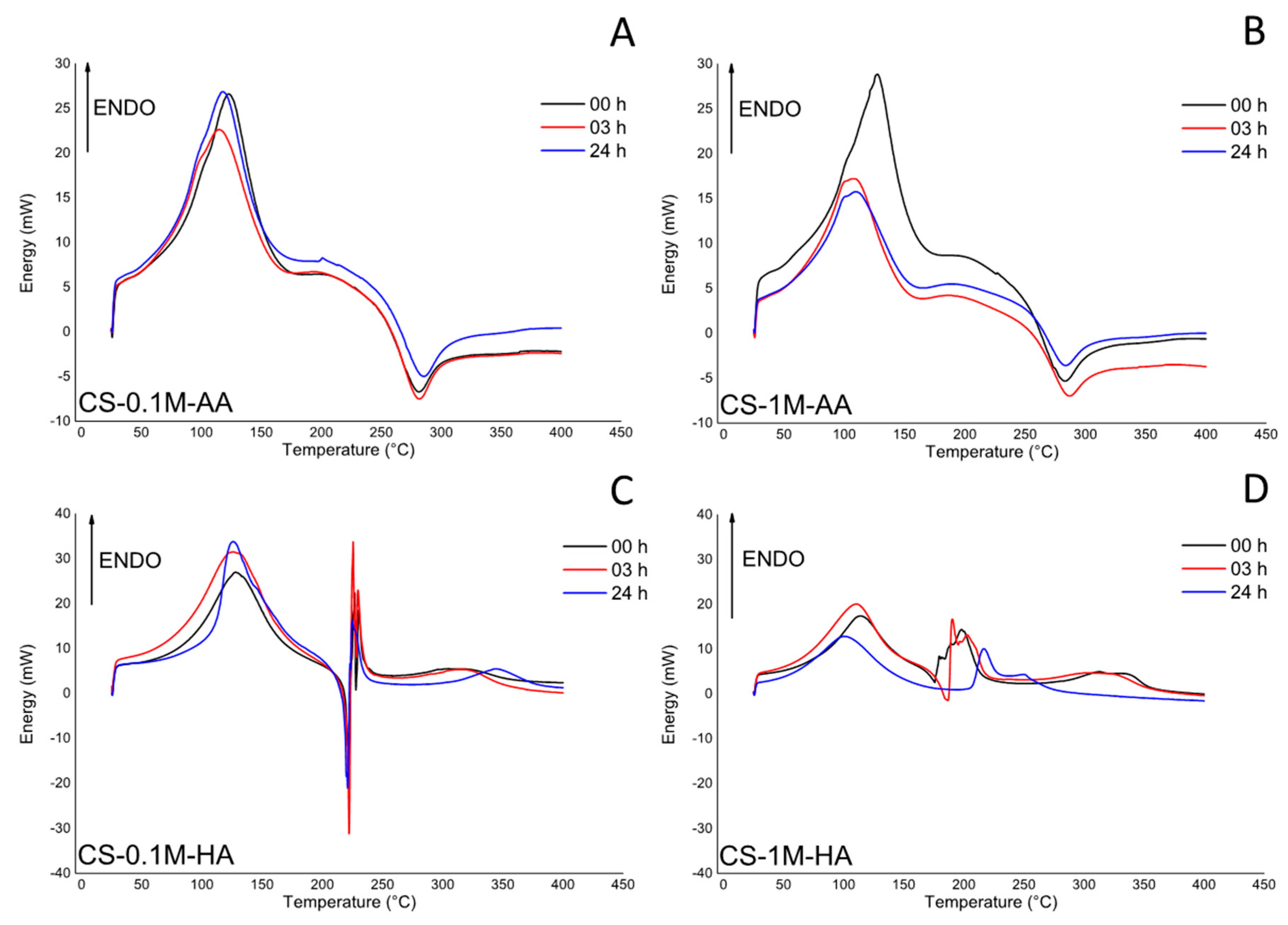
2.3. Thermo Gravimetrical (TG) and Differential Scanning Calorimeter (DSC)
2.4. Fourier Transformed Infrared Spectroscopy (FT-IR)
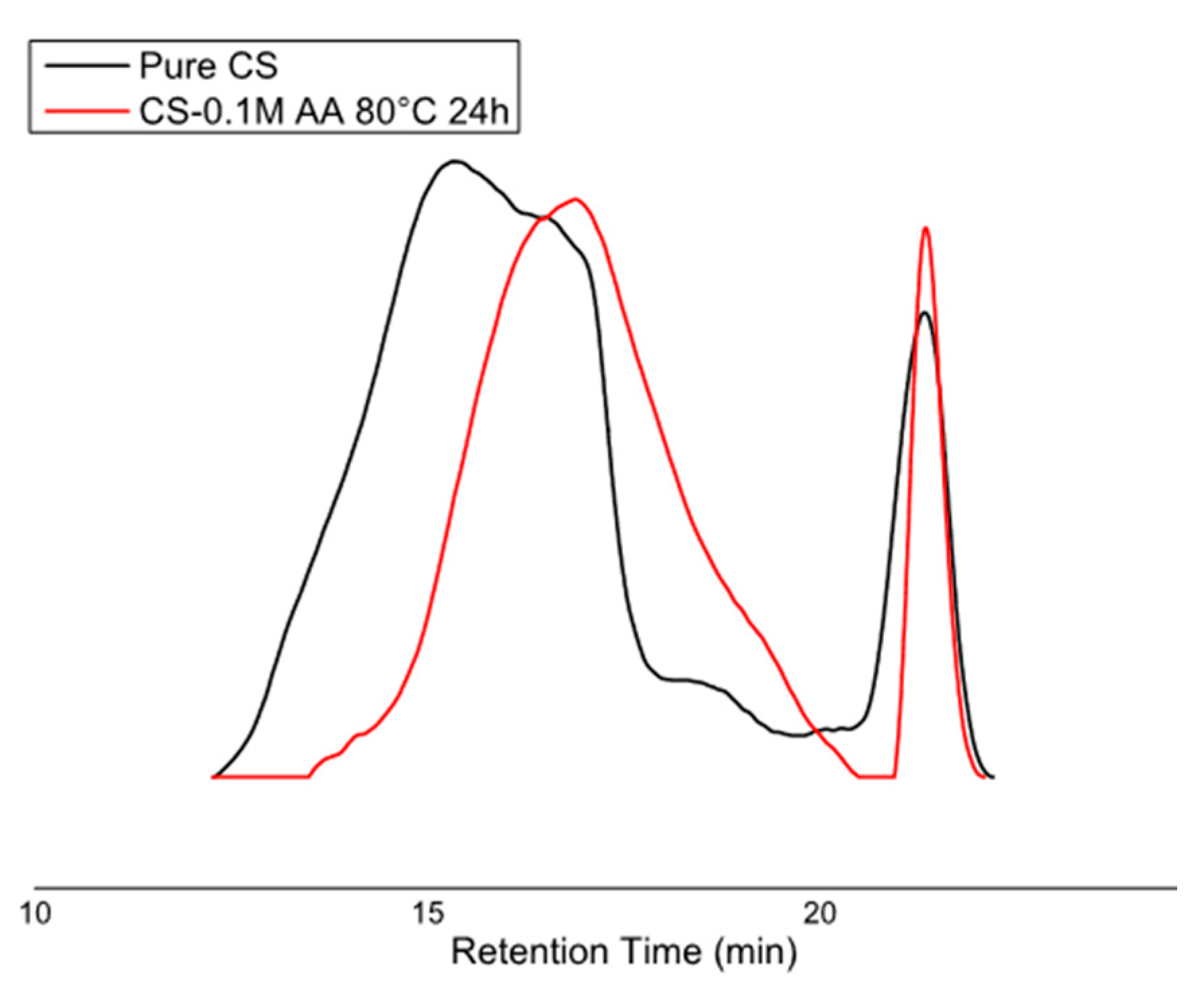
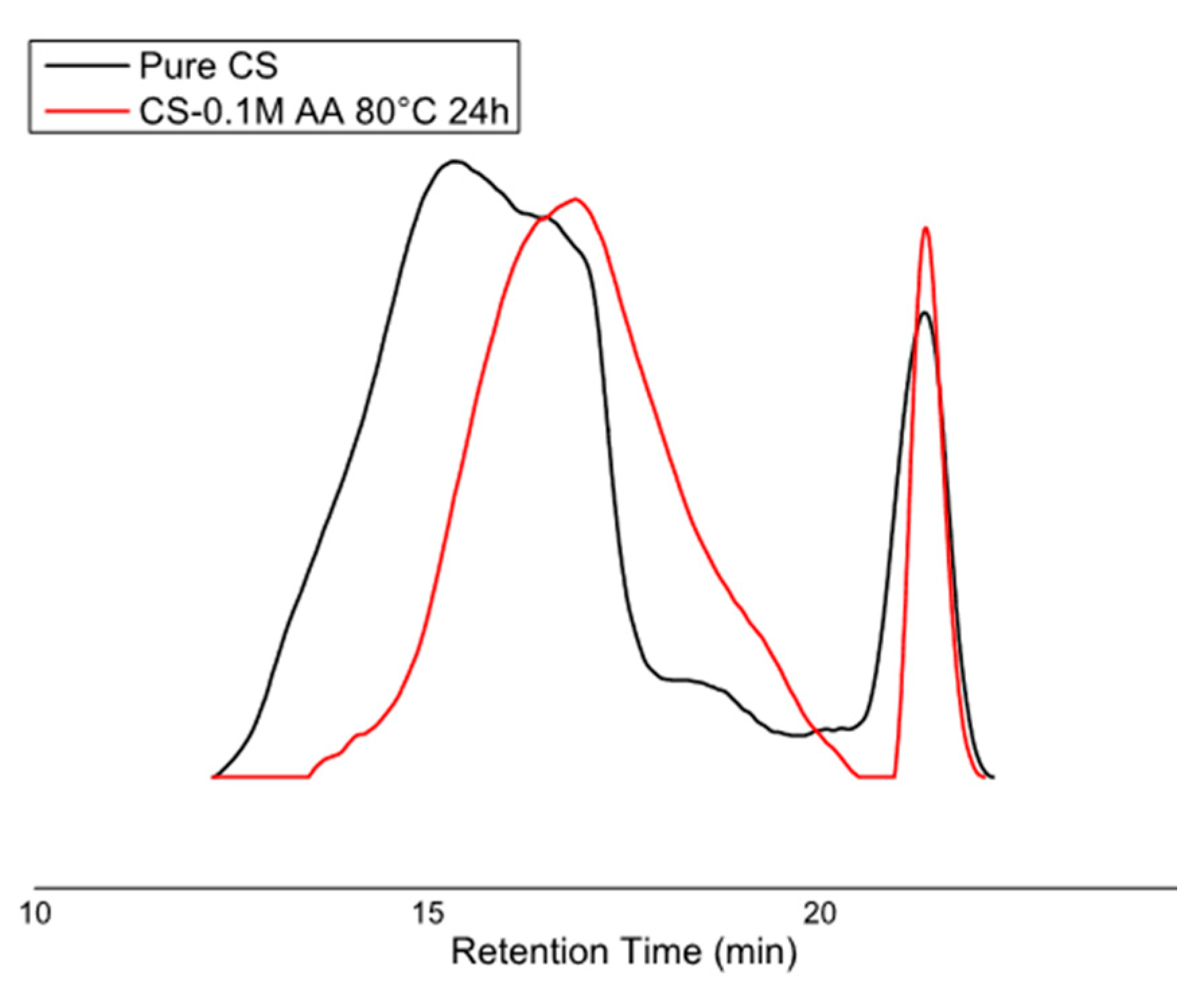
2.5. Gel Permeation Chromatography (GPC)
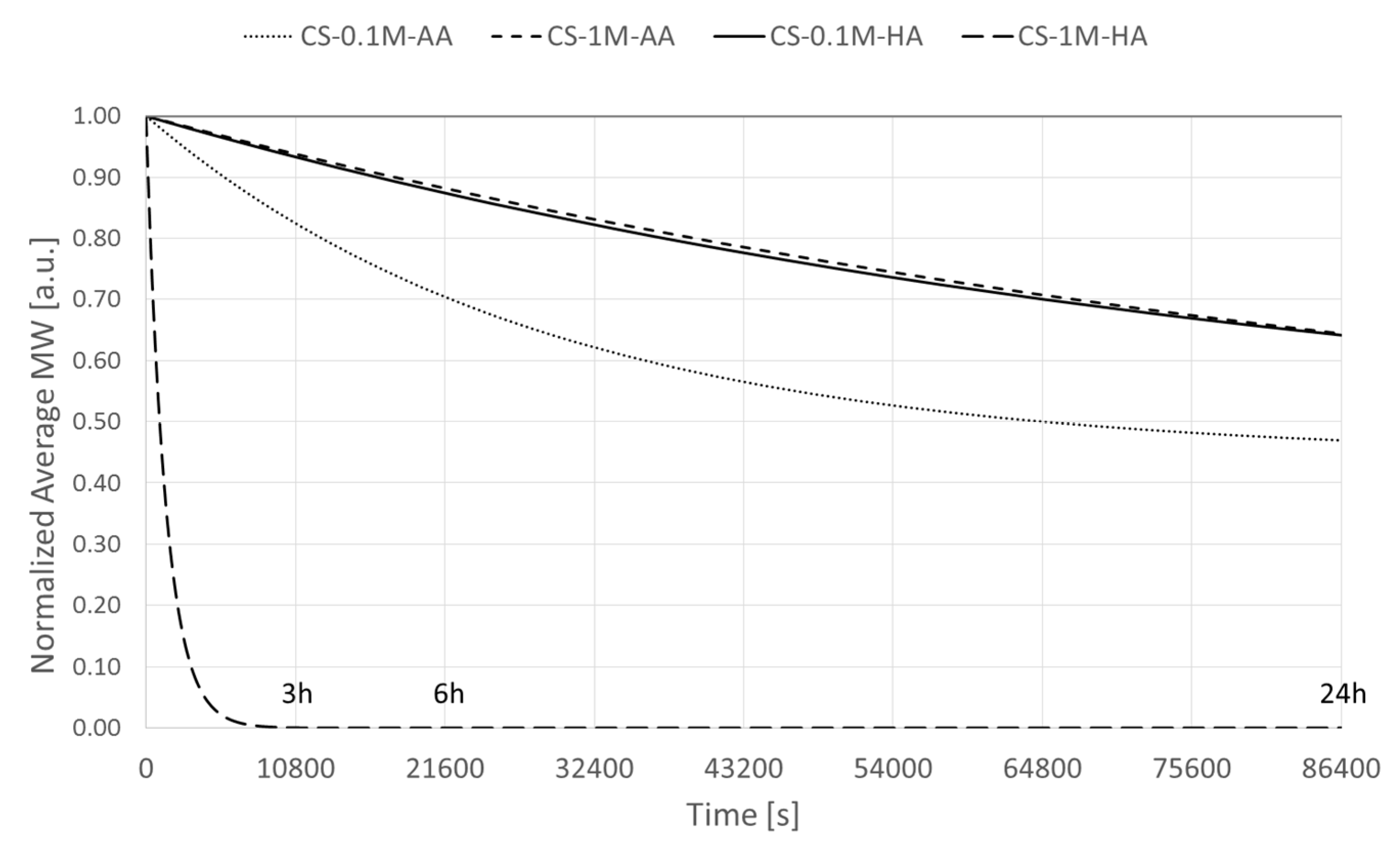
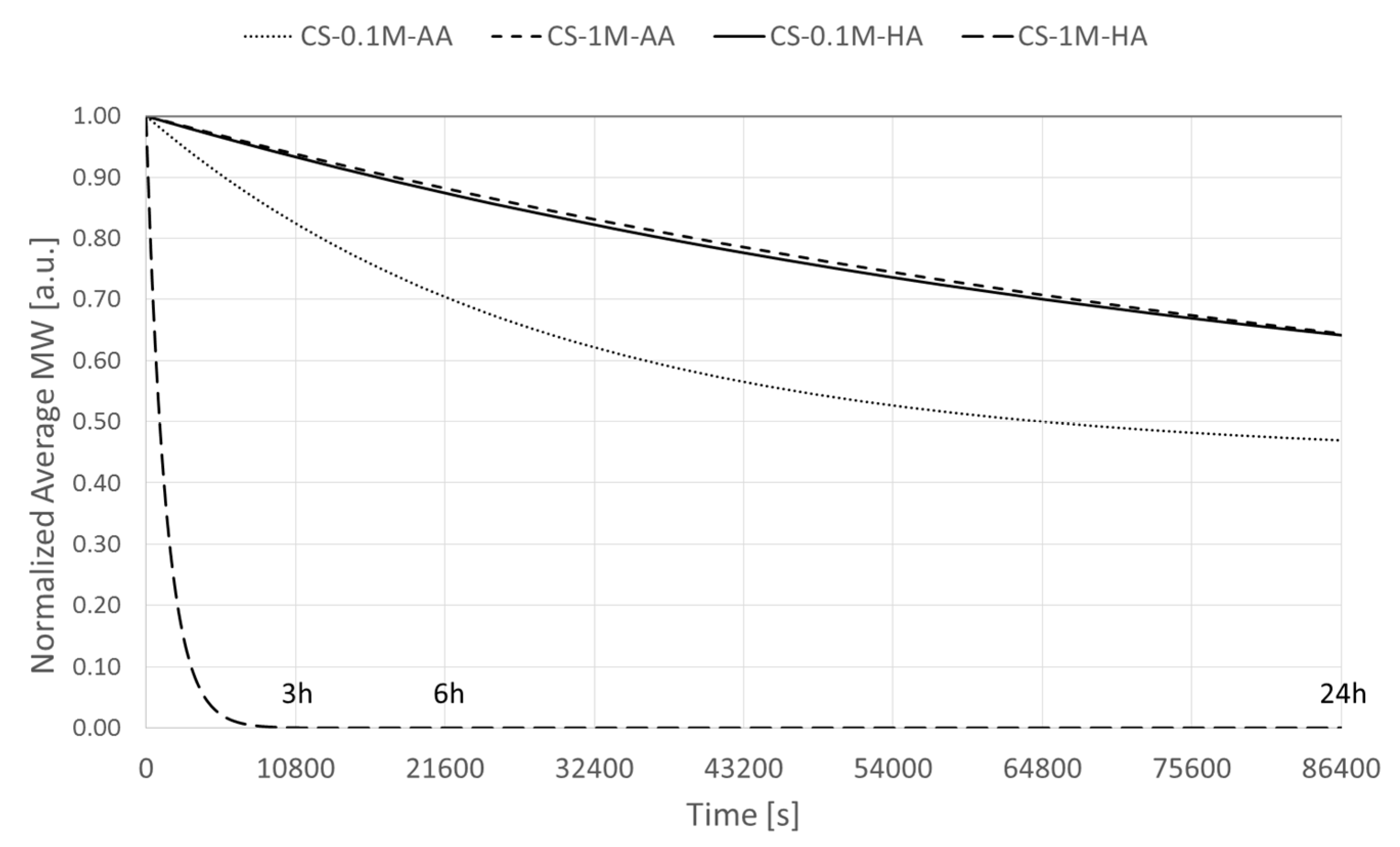
2.6. Determination of the Degradation Rate and Activation Energy
2.7. Numerical Simulations
2.8. Statistical Analysis
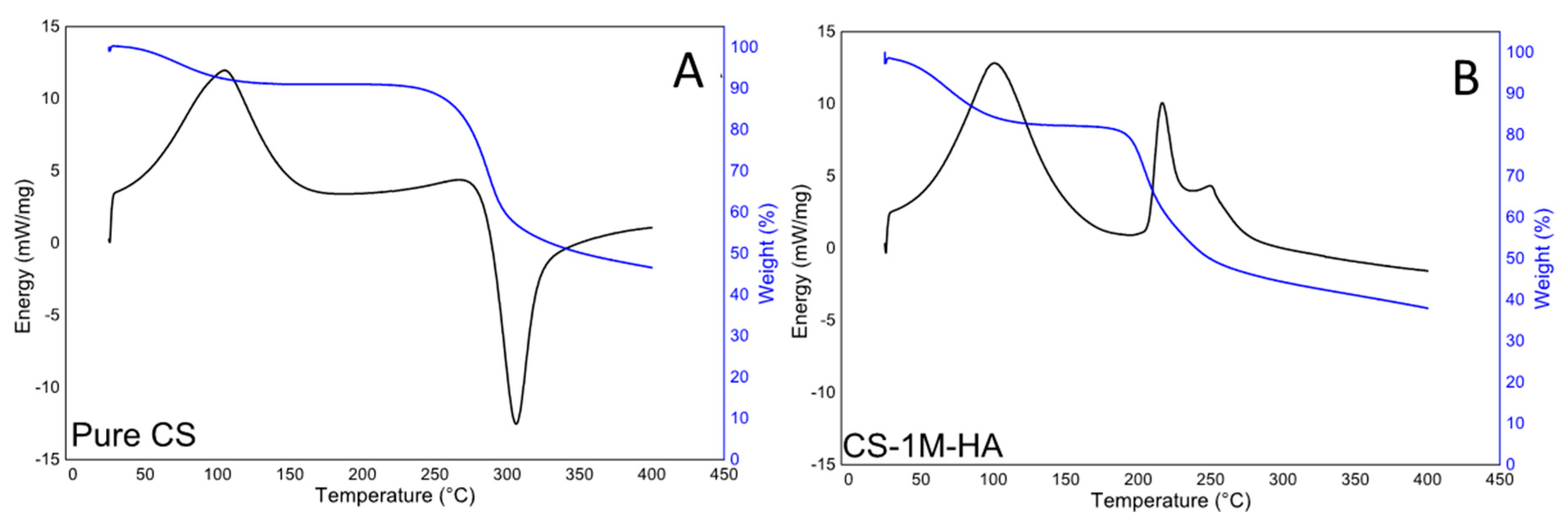
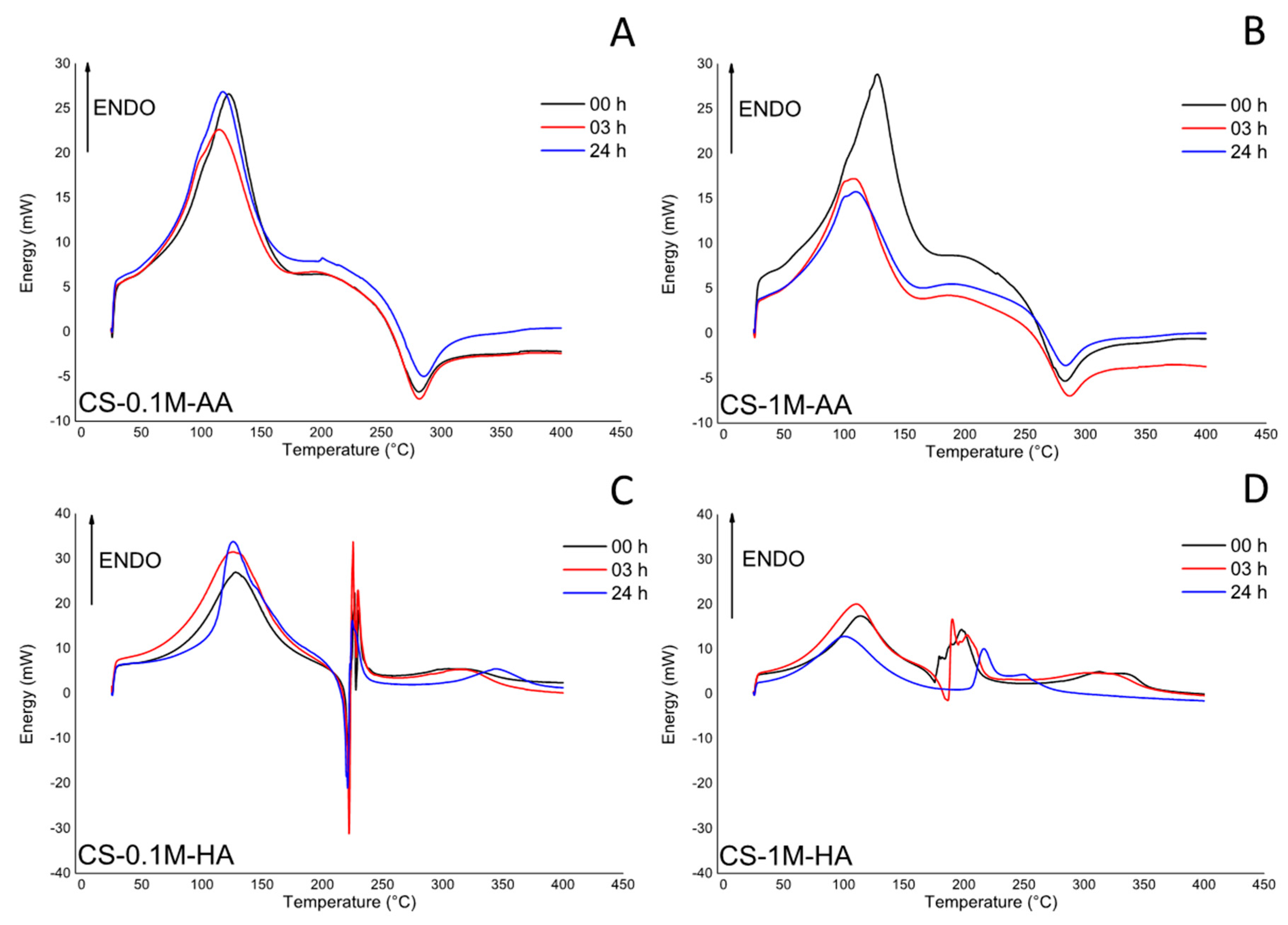
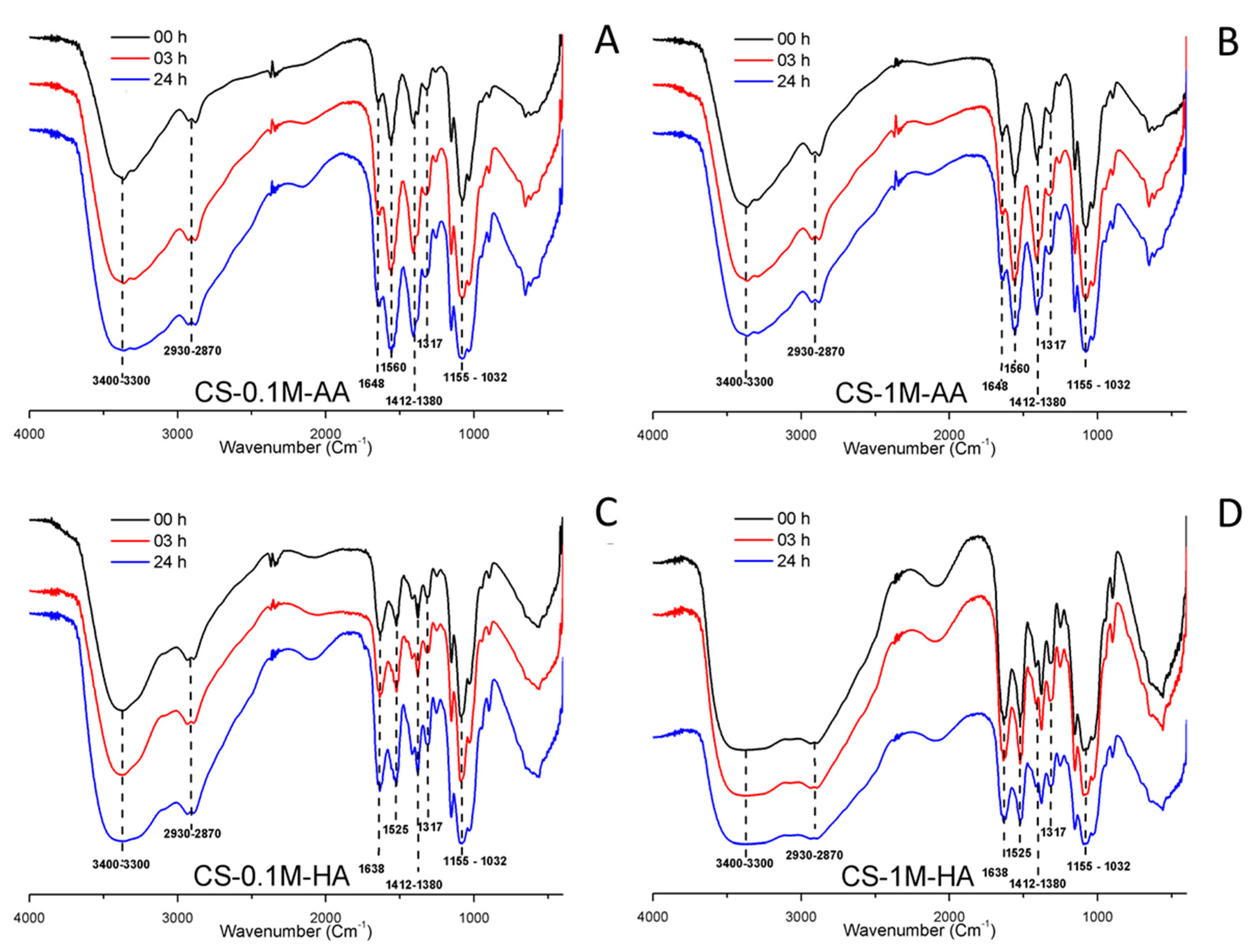
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carraher, C.E. Seymour/Carraher’s Polymer Chemistry: Sixth Edition; CRC Press: Boca Rtaon, FL, USA, 2003. [Google Scholar]
- Ferdous, J.; Kolachalama, V.B.; Shazly, T. Impact of polymer structure and composition on fully resorbable endovascular scaffold performance. Acta Biomater. 2013, 9, 6052–6061. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.A.F. Chitin Chemistry; Macmillan: New York, NY, USA, 1992. [Google Scholar]
- Di Mario, F.; Rapanà, P.; Tomati, U.; Galli, E. Chitin and chitosan from basidiomycetes. Int. J. Biol. Macromol. 2008, 43, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Kannan, M.; Nesakumari, M.; Rajarathinam, K.; Singh, A. Production and characterization of mushroom chitosan under solid-state fermentation conditions. Adv. Biol. Res. 2010, 4, 10–13. [Google Scholar]
- Vandamme, E.; de Baets, S.; Steinbüchel, A. Polysaccharides II: Polysaccharides from Eukaryotes; Wiley-Vch: Weinheim, Germany, 2002. [Google Scholar]
- Madaghiele, M.; Marotta, F.; Demitri, C.; Montagna, F.; Maffezzoli, A.; Sannino, A. Development of semi-and grafted interpenetrating polymer networks based on poly(ethylene glycol) diacrylate and collagen. J. Appl. Biomater. Funct. Mater. 2014, 12, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Cannazza, G.; Cataldo, A.; De Benedetto, E.; Demitri, C.; Madaghiele, M.; Sannino, A. Experimental assessment of the use of a novel superabsorbent polymer (SAP) for the optimization ofwater consumption in agricultural irrigation process. Water 2014, 6, 2056–2069. [Google Scholar] [CrossRef]
- Demitri, C.; Giuri, A.; Raucci, M.G.; Giugliano, D.; Madaghiele, M.; Sannino, A.; Ambrosio, L. Preparation and characterization of cellulose-based foams via microwave curing. Interface Focus 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Demitri, C.; Scalera, F.; Madaghiele, M.; Sannino, A.; Maffezzoli, A. Potential of cellulose-based superabsorbent hydrogels as water reservoir in agriculture. Int. J. Polym. Sci. 2013, 2013, 6. [Google Scholar] [CrossRef]
- Raucci, M.G.; Alvarez-Perez, M.A.; Demitri, C.; Sannino, A.; Ambrosio, L. Proliferation and osteoblastic differentiation of hmscs on cellulose-based hydrogels. J. Appl. Biomater. Funct. Mater. 2012, 10, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Sannino, A.; Madaghiele, M.; Demitri, C.; Scalera, F.; Esposito, A.; Esposito, V.; Maffezzoli, A. Development and characterization of cellulose-based hydrogels for use as dietary bulking agents. J. Appl. Polym. Sci. 2010, 115, 1438–1444. [Google Scholar] [CrossRef]
- Depan, D.; Shah, J.S.; Misra, R.D.K. Degradation mechanism and increased stability of chitosan-based hybrid scaffolds cross-linked with nanostructured carbon: Process–structure–functional property relationship. Polym. Degrad. Stab. 2013, 98, 2331–2339. [Google Scholar] [CrossRef]
- Depan, D.; Pratheep Kumar, A.; Singh, R.P.; Misra, R.D.K. Stability of chitosan/montmorillonite nanohybrid towards enzymatic degradation on grafting with poly(lactic acid). Mater. Sci. Technol. 2014, 30, 587–592. [Google Scholar] [CrossRef]
- Dudhani, A.R.; Kosaraju, S.L. Bioadhesive chitosan nanoparticles: Preparation and characterization. Carbohydr. Polym. 2010, 81, 243–251. [Google Scholar] [CrossRef]
- Depan, D.; Pesacreta, T.C.; Misra, R.D.K. The synergistic effect of a hybrid graphene oxide-chitosan system and biomimetic mineralization on osteoblast functions. Biomater. Sci. 2014, 2, 264–274. [Google Scholar] [CrossRef]
- Depan, D.; Misra, R.D.K. The interplay between nanostructured carbon-grafted chitosan scaffolds and protein adsorption on the cellular response of osteoblasts: Structure–function property relationship. Acta Biomater. 2013, 9, 6084–6094. [Google Scholar] [CrossRef] [PubMed]
- Depan, D.; Misra, R.D.K. Processing–structure–functional property relationship in organic–inorganic nanostructured scaffolds for bone-tissue engineering: The response of preosteoblasts. J. Biomed. Mater. Res. Part A 2012, 100A, 3080–3091. [Google Scholar] [CrossRef] [PubMed]
- Maganti, N.; Venkat Surya, P.K.C.; Thein-Han, W.W.; Pesacreta, T.C.; Misra, R.D.K. Structure–process–property relationship of biomimetic chitosan-based nanocomposite scaffolds for tissue engineering: Biological, physico-chemical, and mechanical functions. Adv. Eng. Mater. 2011, 13, B108–B122. [Google Scholar] [CrossRef]
- Depan, D.; Venkata Surya, P.K.C.; Girase, B.; Misra, R.D.K. Organic/inorganic hybrid network structure nanocomposite scaffolds based on grafted chitosan for tissue engineering. Acta Biomater. 2011, 7, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Depan, D.; Girase, B.; Shah, J.S.; Misra, R.D.K. Structure–process–property relationship of the polar graphene oxide-mediated cellular response and stimulated growth of osteoblasts on hybrid chitosan network structure nanocomposite scaffolds. Acta Biomater. 2011, 7, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Thein-Han, W.W.; Misra, R.D. Biomimetic chitosan-nanohydroxyapatite composite scaffolds for bone tissue engineering. Acta Biomater. 2009, 5, 1182–1197. [Google Scholar] [CrossRef] [PubMed]
- Thein-Han, W.W.; Saikhun, J.; Pholpramoo, C.; Misra, R.D.K.; Kitiyanant, Y. Chitosan–gelatin scaffolds for tissue engineering: Physico-chemical properties and biological response of buffalo embryonic stem cells and transfectant of GFP–buffalo embryonic stem cells. Acta Biomater. 2009, 5, 3453–3466. [Google Scholar] [CrossRef] [PubMed]
- Thein-Han, W.W.; Misra, R.D.K. Three-dimensional chitosan-nanohydroxyapatite composite scaffolds for bone tissue engineering. JOM 2009, 61, 41–44. [Google Scholar] [CrossRef]
- Thein-Han, W.W.; Kitiyanant, Y.; Misra, R.D.K. Chitosan as scaffold matrix for tissue engineering. Mater. Sci. Technol. 2008, 24, 1062–1075. [Google Scholar] [CrossRef]
- Demitri, C.; De Benedictis, V.M.; Madaghiele, M.; Corcione, C.E.; Maffezzoli, A. Nanostructured active chitosan-based films for food packaging applications: Effect of graphene stacks on mechanical properties. Measurement 2016, 90, 418–423. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Kurita, K. Chemistry and application of chitin and chitosan. Polym. Degrad. Stab. 1998, 59, 117–120. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, D.; Liu, F.; Yu, X.; Zhao, C.; Wan, C. Structure and properties of chitosan derivatives modified calcium polyphosphate scaffolds. Polym. Degrad. Stab. 2010, 95, 1205–1210. [Google Scholar] [CrossRef]
- Wong, Y.Y.; Yuan, S.; Choong, C. Degradation of PEG and non-PEG alginate–chitosan microcapsules in different ph environments. Polym. Degrad. Stab. 2011, 96, 2189–2197. [Google Scholar] [CrossRef]
- Yuan, Q.; Hein, S.; Misra, R.D.K. New generation of chitosan-encapsulated ZnO quantum dots loaded with drug: Synthesis, characterization and in vitro drug delivery response. Acta Biomater. 2010, 6, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Shah, J.; Hein, S.; Misra, R.D.K. Controlled and extended drug release behavior of chitosan-based nanoparticle carrier. Acta Biomater. 2010, 6, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Venkatasubramanian, R.; Hein, S.; Misra, R.D.K. A stimulus-responsive magnetic nanoparticle drug carrier: Magnetite encapsulated by chitosan-grafted-copolymer. Acta Biomater. 2008, 4, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Crini, G. Recent developments in polysaccharide-based materials used as adsorbents in wastewater treatment. Prog. Polym. Sci. 2005, 30, 38–70. [Google Scholar] [CrossRef]
- Dutta, P.; Tripathi, S.; Mehrotra, G.; Dutta, J. Perspectives for chitosan based antimicrobial films in food applications. Food Chem. 2009, 114, 1173–1182. [Google Scholar] [CrossRef]
- Anchisi, C.; Meloni, M.; Maccioni, A. Chitosan beads loaded with essential oils in cosmetic formulations. Int. J. Cosmet. Sci. 2007, 29, 485. [Google Scholar] [CrossRef]
- El-Hefian, E.A.; Elgannoudi, E.S.; Mainal, A.; Yahaya, A.H. Characterization of chitosan in acetic acid: Rheological and thermal studies. Turk. J. Chem. 2010, 34, 47–56. [Google Scholar]
- Balau, L.; Lisa, G.; Popa, M.; Tura, V.; Melnig, V. Physico-chemical properties of chitosan films. Cent. Eur. J. Chem. 2004, 2, 638–647. [Google Scholar] [CrossRef]
- Hwang, J.K.; Shin, H.H. Rheological properties of chitosan solutions. Korea Aust. Rheol. J. 2000, 12, 175–179. [Google Scholar]
- Desbrieres, J. Viscosity of semiflexible chitosan solutions: Influence of concentration, temperature, and role of intermolecular interactions. Biomacromolecules 2002, 3, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M.; Pavlov, G.; Desbrieres, J. Influence of acetic acid concentration on the solubilization of chitosan. Polymer 1999, 40, 7029–7032. [Google Scholar] [CrossRef]
- Kienzle-Sterzer, C.; Rodriguez-Sanchez, D.; Rha, C. Flow behavior of a cationic biopolymer: Chitosan. Polym. Bull. 1985, 13, 1–6. [Google Scholar] [CrossRef]
- Wang, W.; Xu, D. Viscosity and flow properties of concentrated solutions of chitosan with different degrees of deacetylation. Int. J. Biol. Macromol. 1994, 16, 149–152. [Google Scholar] [CrossRef]
- Mironov, A.V.; Vikhoreva, G.A.; Kil’deeva, N.R.; Uspenskii, S.A. Reasons for unstable viscous properties of chitosan solutions in acetic acid. Polym. Sci. Ser. B 2007, 49, 15–17. [Google Scholar] [CrossRef]
- Vårum, K.; Ottøy, M.; Smidsrød, O. Acid hydrolysis of chitosans. Carbohydr. Polym. 2001, 46, 89–98. [Google Scholar] [CrossRef]
- Moriana, R.; Zhang, Y.; Mischnick, P.; Li, J.; Ek, M. Thermal degradation behavior and kinetic analysis of spruce glucomannan and its methylated derivatives. Carbohydr. Polym. 2014, 106, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Enomoto-Rogers, Y.; Ohmomo, Y.; Iwata, T. Syntheses and characterization of konjac glucomannan acetate and their thermal and mechanical properties. Carbohydr. Polym. 2013, 92, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Wasikiewicz, J.M.; Yeates, S.G. “Green” molecular weight degradation of chitosan using microwave irradiation. Polym. Degrad. Stab. 2013, 98, 863–867. [Google Scholar] [CrossRef]
- Chattopadhyay, D.; Inamdar, M.S. Aqueous behavior of chitosan. Int. J. Polym. Sci. 2011, 2010. [Google Scholar] [CrossRef]
- Yue, W.; Yao, P.; Wei, Y. Influence of ultraviolet-irradiated oxygen on depolymerization of chitosan. Polym. Degrad. Stab. 2009, 94, 851–858. [Google Scholar] [CrossRef]
- Morris, G.A.; Castile, J.; Smith, A.; Adams, G.G.; Harding, S.E. The kinetics of chitosan depolymerization at different temperatures. Polym. Degrad. Stab. 2009, 94, 1344–1348. [Google Scholar] [CrossRef]
- Sionkowska, A.; Wisniewski, M.; Skopinska, J.; Poggi, G.; Marsano, E.; Maxwell, C.; Wess, T. Thermal and mechanical properties of UV irradiated collagen/chitosan thin films. Polym. Degrad. Stab. 2006, 91, 3026–3032. [Google Scholar] [CrossRef]
- BeMiller, J. Acid-catalyzed hydrolysis of glycosides. Adv. Carbohydr. Chem. Biochem. 1966, 22, 25–108. [Google Scholar]
- Holme, H.K.; Davidsen, L.; Kristiansen, A.; Smidsrød, O. Kinetics and mechanisms of depolymerization of alginate and chitosan in aqueous solution. Carbohydr. Polym. 2008, 73, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Tømmeraas, K.; Melander, C. Kinetics of hyaluronan hydrolysis in acidic solution at various pH values. Biomacromolecules 2008, 9, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Kasaai, M.R. Calculation of mark–houwink–sakurada (MHS) equation viscometric constants for chitosan in any solvent–temperature system using experimental reported viscometric constants data. Carbohydr. Polym. 2007, 68, 477–488. [Google Scholar] [CrossRef]
- Asimba, B.O.; Shigoli, A.K.; Suthar, K.J.; Ghantasala, M.K.; Mancini, D.C. Numerical simulation of pH-sensitive hydrogel response in different conditions. In Proceedings of the 2012 COMSOL Conference, Boston, MA, USA, 3–5 October 2012; pp. 2–5.
- Masuelli, M.A. Mark-houwink parameters for aqueous-soluble polymers and biopolymers at various temperatures. J. Polym. Biopolym. Phys. Chem. 2014, 2, 37–43. [Google Scholar]
- Çatiker, E.; Güner, A. Unperturbed dimensions and the theta temperature of dextran in ethylene glycol solutions. Eur. Polym. J. 2000, 36, 2143–2146. [Google Scholar] [CrossRef]
- Tanford, C. Physical Chemistry of Macromolecules; Wiley: New York, NY, USA, 1961. [Google Scholar]
- Solomon, O.F.; Ciutǎ, I.Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]
- Anthonsen, M.W.; Vårum, K.M.; Smidsrød, O. Solution properties of chitosans: Conformation and chain stiffness of chitosans with different degrees of n-acetylation. Carbohydr. Polym. 1993, 22, 193–201. [Google Scholar] [CrossRef]
- Demitri, C.; Raucci, M.G.; Giuri, A.; De Benedictis, V.M.; Giugliano, D.; Calcagnile, P.; Sannino, A.; Ambrosio, L. Cellulose-based porous scaffold for bone tissue engineering applications: Assessment of hmsc proliferation and differentiation. J. Biomed. Mater. Res. Part A 2016, 104, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Raucci, M.G.; Alvarez-Perez, M.A.; Demitri, C.; Giugliano, D.; De Benedictis, V.; Sannino, A.; Ambrosio, L. Effect of citric acid crosslinking cellulose-based hydrogels on osteogenic differentiation. J. Biomed. Mater. Res. Part A 2015, 103, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Dimida, S.; Demitri, C.; De Benedictis, V.M.; Scalera, F.; Gervaso, F.; Sannino, A. Genipin-cross-linked chitosan-based hydrogels: Reaction kinetics and structure-related characteristics. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Demitri, C.; Moscatello, A.; Giuri, A.; Raucci, M.G.; Esposito Corcione, C. Preparation and characterization of eg-chitosan nanocomposites via direct exfoliation: A green methodology. Polymers 2015, 7, 2584–2594. [Google Scholar] [CrossRef]
- Holme, H.; Foros, H.; Pettersen, H.; Dornish, M.; Smidsrød, O. Thermal depolymerization of chitosan chloride. Carbohydr. Polym. 2001, 46, 287–294. [Google Scholar] [CrossRef]
- Moggridge, R.; Neuberger, A. 139. Methylglucosaminide: Its structure, and the kinetics of its hydrolysis by acids. J. Chem. Soc. 1938. [Google Scholar] [CrossRef]
- Fee, M. Evaluation of Chitosan Stability in Aqueous Systems. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2005. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample code | Chitosan | Acetic acid | Hydrochloric acid | pH |
|---|---|---|---|---|
| CS-0.1M-AA | 2% | 0.1 M | na | 5.10 |
| CS-1M-AA | 2% | 1.0 M | na | 3.10 |
| CS-0.1M-HA | 2% | na | 0.1 M | 1.20 |
| CS-1M-HA | 2% | na | 1.0 M | 0.20 |
| Sample code | Molecular weight [kDa] t = 0, T = 0 °C | Molecular weight [kDa] t = 24 h, T = 80 °C |
|---|---|---|
| CS-0.1M-AA | 995.15 ± 4.03 | 824.0 ± 23.1 |
| CS-1M-AA | 1,003.00 ± 9.90 | 453.50 ± 67.17 |
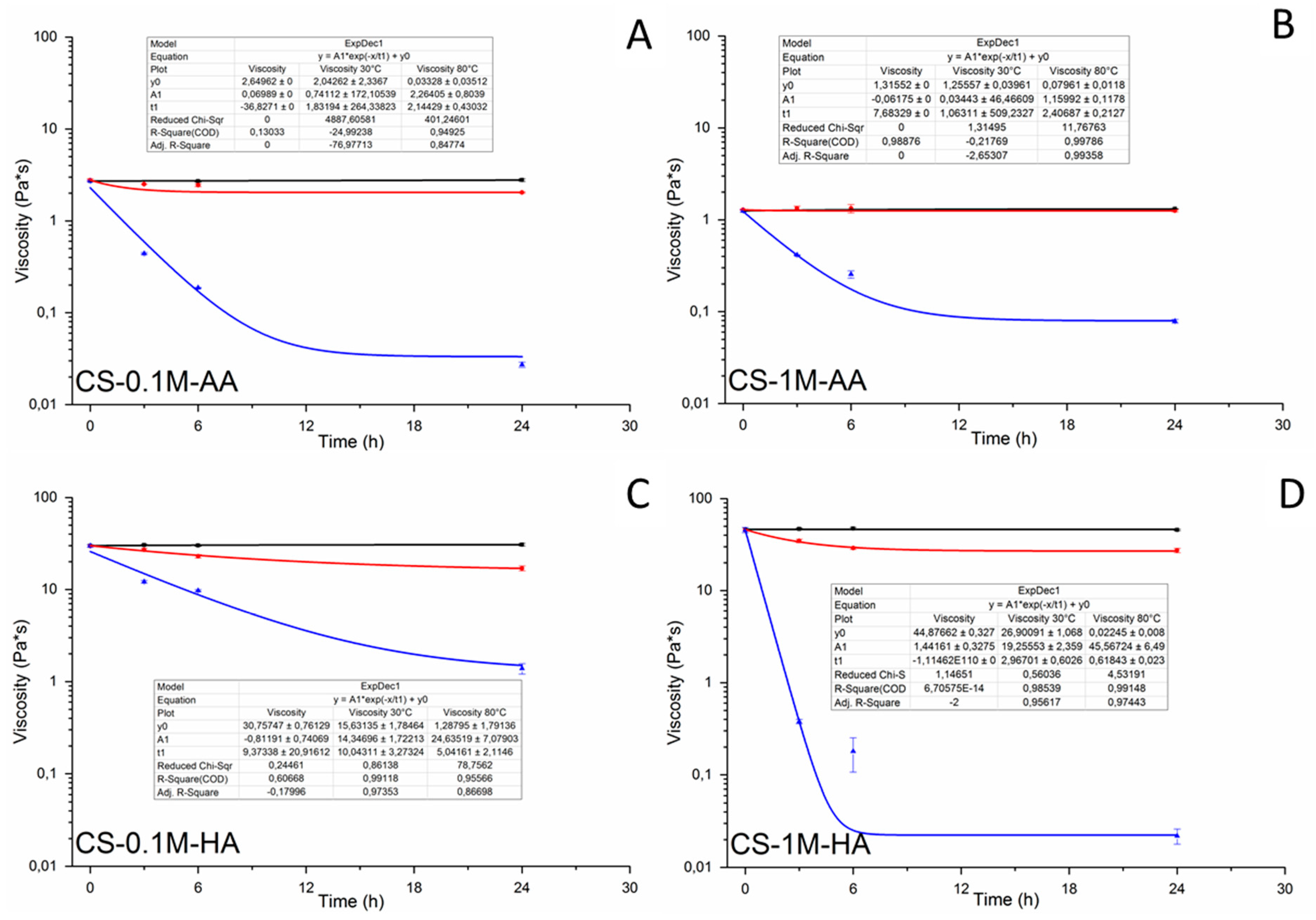
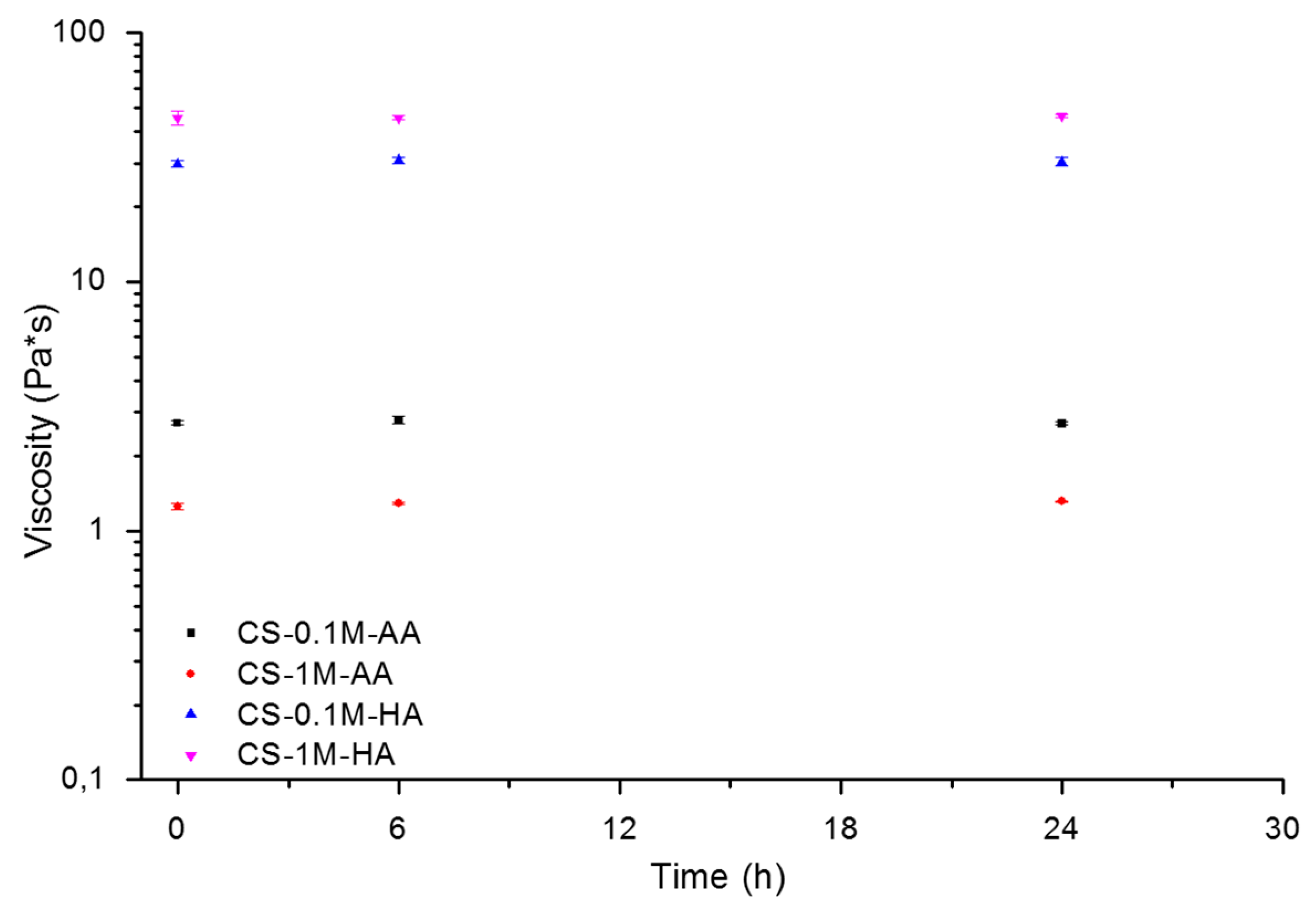
| Sample code | Degradation rate constant k30 °C (h−1) | Degradation rate constant k80 °C (h−1) | Rate constant ratio k80 °C/k30 °C | Activation energy Ea (kJ/mol) |
|---|---|---|---|---|
| CS-0.1M-AA | 0.0015 ± 0.0002 | 0.1261 ± 0.0008 | 84 | 72.325 ± 1.509 |
| CS-1M-AA | 0.00028 ± 0.00004 | 0.0348 ± 0.0025 | 124 | 55.860 ± 7.416 |
| CS-0.1M-HA | 0.0032 ± 0.0280 | 0.0423 ± 0.0018 | 13 | 59.478 ± 3.073 |
| CS-1M-HA | 0.0038 ± 00012 | 2.5330 ± 0.1084 | 672 | 75.704 ± 9.786 |
| Sample ID | Intrinsic viscosity difference (%) | |
|---|---|---|
| T = 0 h | T = 80; t = 24 h | |
| CS-0.1M-AA | −52.40 | 60.81 |
| CS-1.0M-AA | 13.92 | 51.65 |
| CS-0.1M-HA | 0.34 | 59.17 |
| CS-1.0M-HA | 1,802.28 | – |
| Sample ID | Exp | Calc | ||
|---|---|---|---|---|
| a | K | a | K | |
| CS-0.1M-AA | 1.26 | 3.04 × 10−5 | 2.72 | 3.01 × 10−5 |
| CS-1.0M-AA | 0.85 | 0.0138 | 3.68 | 3.63 × 10−8 |
| CS-0.1M-HA | 0.78 | 0.175 | 3.02 | 7.04 × 10−4 |
| CS-1.0M-HA | 0.66 | 0.0585 | 0.16 | 2,624.68 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Benedictis, V.M.; Soloperto, G.; Demitri, C. Correction of MHS Viscosimetric Constants upon Numerical Simulation of Temperature Induced Degradation Kinetic of Chitosan Solutions. Polymers 2016, 8, 210. https://doi.org/10.3390/polym8060210
De Benedictis VM, Soloperto G, Demitri C. Correction of MHS Viscosimetric Constants upon Numerical Simulation of Temperature Induced Degradation Kinetic of Chitosan Solutions. Polymers. 2016; 8(6):210. https://doi.org/10.3390/polym8060210
Chicago/Turabian StyleDe Benedictis, Vincenzo Maria, Giulia Soloperto, and Christian Demitri. 2016. "Correction of MHS Viscosimetric Constants upon Numerical Simulation of Temperature Induced Degradation Kinetic of Chitosan Solutions" Polymers 8, no. 6: 210. https://doi.org/10.3390/polym8060210