Appendix
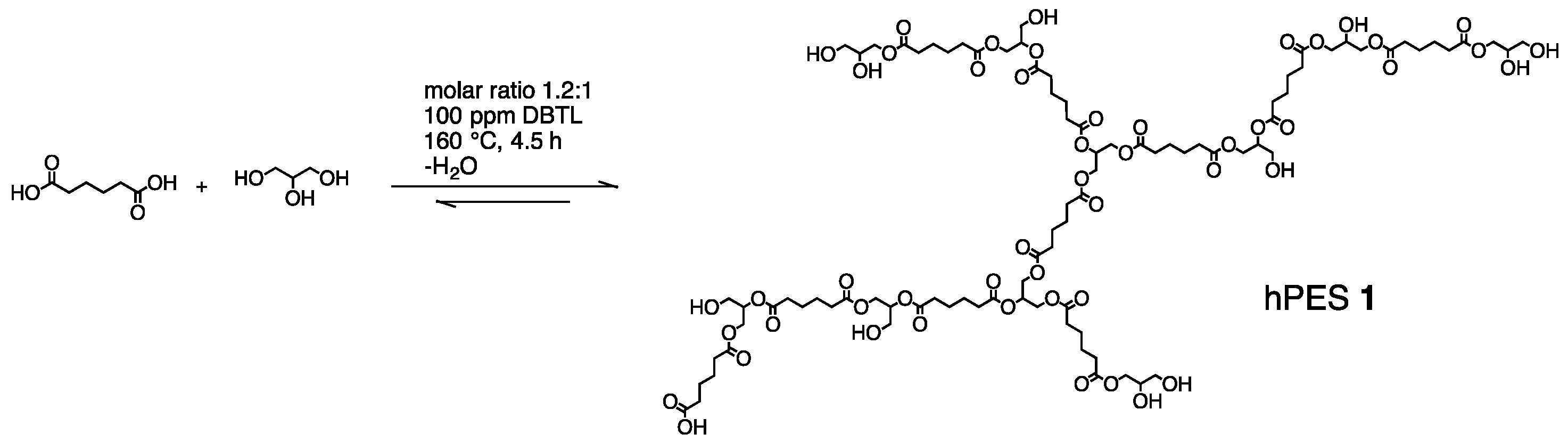
A.1. Synthesis of Hyperbranched Polyester (hPES 1)
At room temperature, adipic acid (39.9 g, 273 mmol, 1.2 eq) was charged into a three-neck glass vertical reactor, equipped with a mechanical stirrer and a Liebig condenser. After adding pre-dried glycerol (20.9 g, 228 mmol), the bulk monomer mixture was heated to 150 °C. Under stirring at 150 rpm, a 0.6 mL of a stock solution of DBTL in toluene (100 ppm) was added to the molten monomers using a syringe. The reaction temperature was increased to 160 °C. After 1 h at 160 °C, the formed volatiles were removed by cryo-distillation and collected in a round-bottom flask. The removal of volatiles was repeated every hour. With proceeding reaction time, the frequency of volatile removal was increased to once per every 10 min. Conversion of the reaction was controlled by determining the ratio of the unreacted acid groups to the total amount of acid groups, using 1H NMR spectroscopy. When the conversion almost reached the maximum conversion PA. as determined by Flory Equation, the reaction was stopped by completely removing the volatiles and cooling the reactor to RT. The viscous product hPES 1 was obtained without any further purification as a light yellow, viscous solid and dissolved in THF for easier handling.
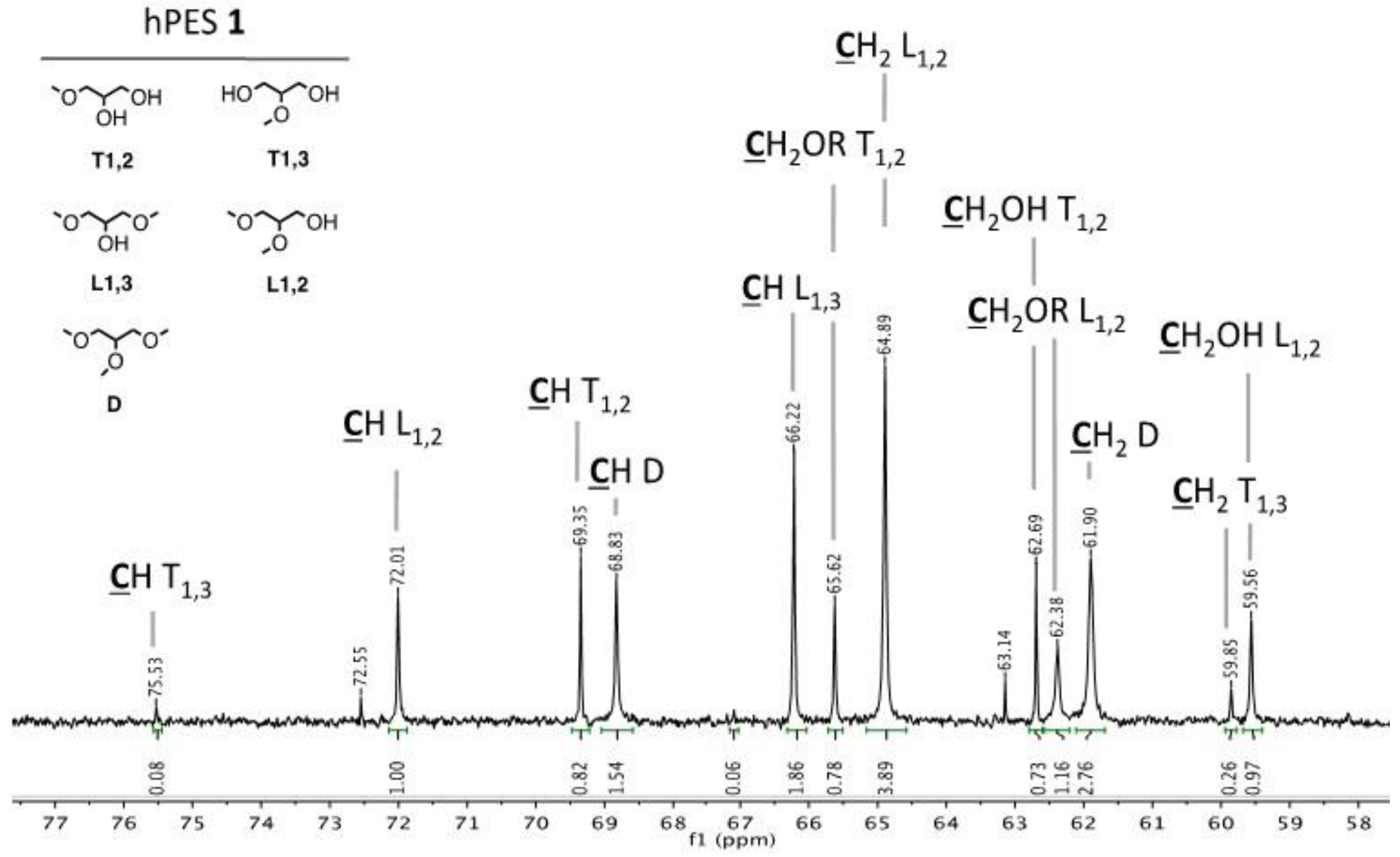
1H NMR (400 MHz, DMSO-d
6): δ = 12.00 (s, 1H, R–COO
H), 5.18 (m, 1H, C
HD), 4.94 (m, 1H, C
HL1,2), 4.72 (m, 1H, C
HT1,3), 4.26–4.23, 4.14–4.12 (2 m, 4H, C
H2D), 4.23–4.07 (m, 2H, C
H2L1,2), 4.03, 3.89 (2 m, 4H, C
H2T1,3), 3.98 (m, 4H, C
H2L1,3), 3.86 (m, 1H, C
HL1,3), 3.63 (q, 1H, C
HT1,2), 3.49–3.40 (m, 4H, C
H2T1,3), 3.49–3.48 (m, 2H, C
H2L1,2), 3.55–3.26 (m, 2H, C
H2T1,2), 2.30 (m, 2H, –C
H2–COOR), 2.20 (m, 2H, –C
H2–COOH), 1.52 (m, 4H, ROOC–CH
2–(C
H2)
2–CH
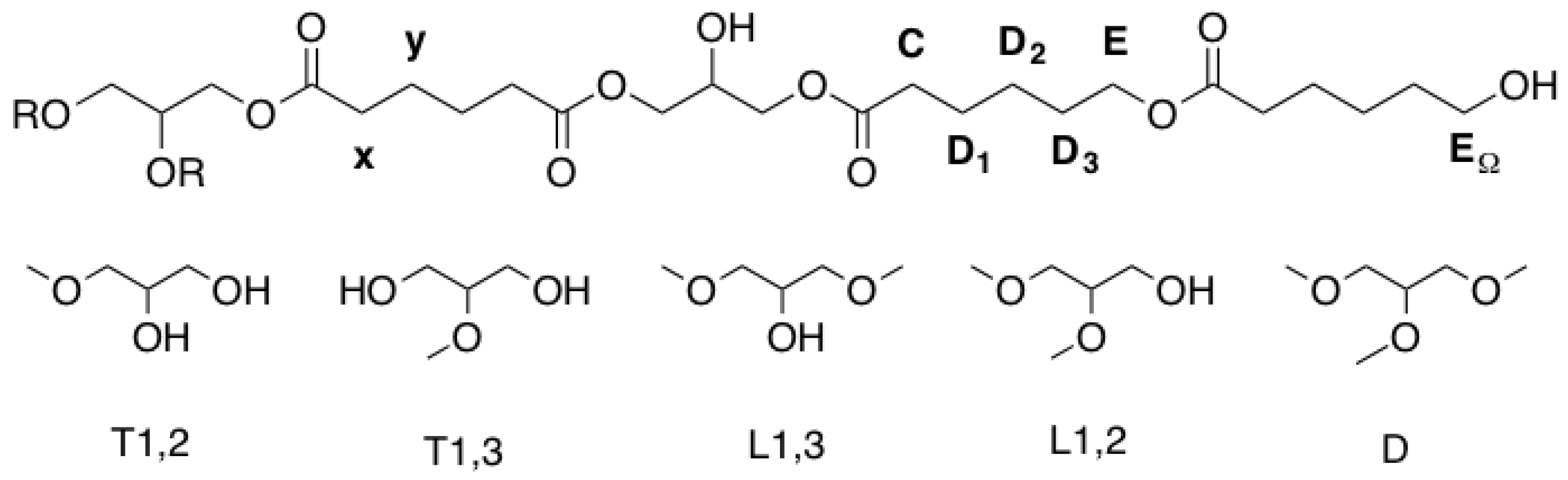
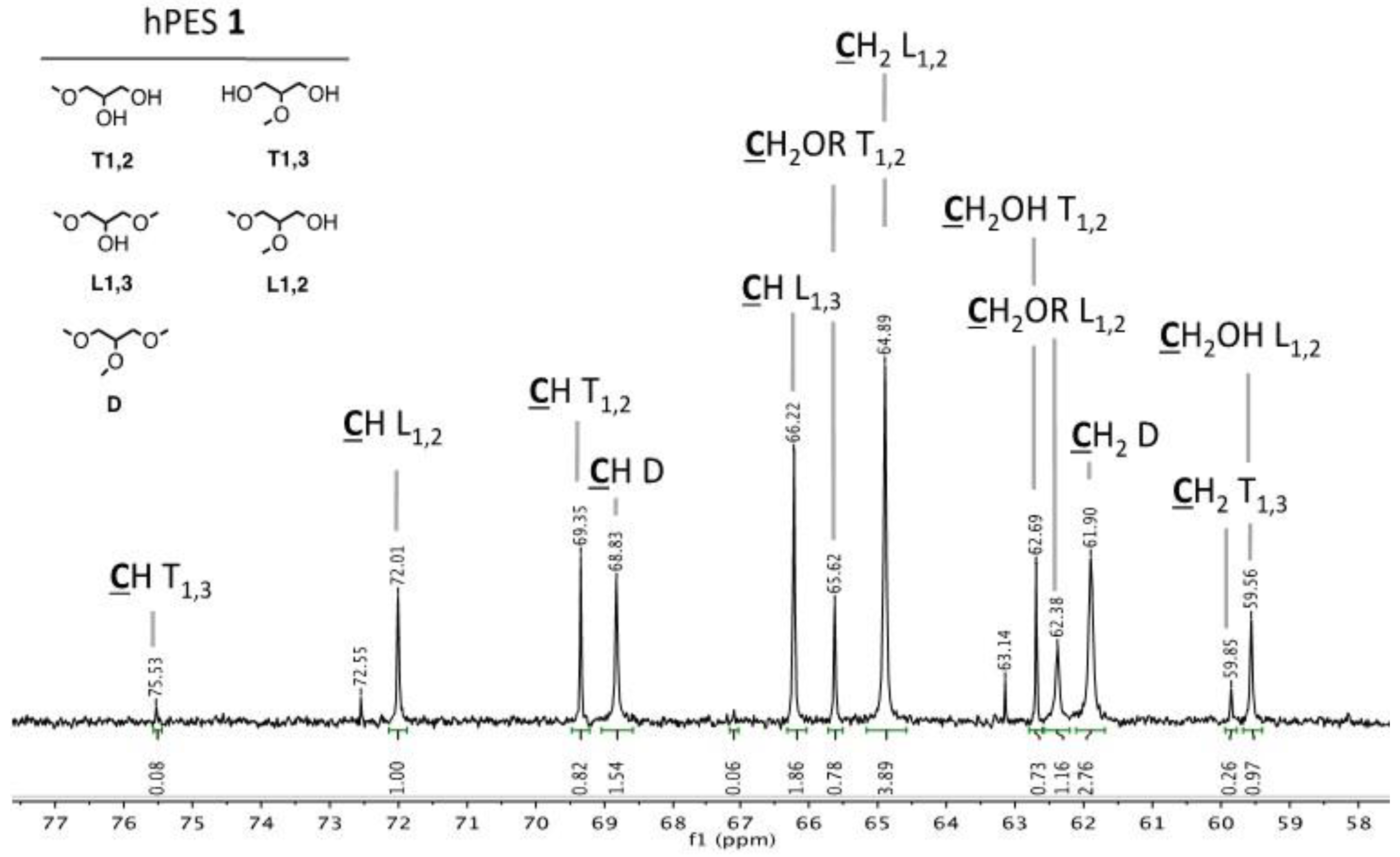
2–COOR) ppm. Abbreviations in accordance with
Figure A1.
IG 13C NMR (400 MHz, DMSO-d
6): δ = 174.42–174.34 (m, 1H, HOO
C–(CH
2)
4–COO–CH
2–(CHOR)–(CH
2OR)), 172.88–172.08 (m, HOO
C–(CH
2)
4–COO–CH
2–(CHOH)–(CH
2OR), HOOC–(CH
2)
4–
COO–CH
2–(CHOH)–(CH
2OR), HOOC–(CH
2)
4–
COO–CH
2–(CHOR)–(CH
2OR), (CH
2OR)–(CHOR)–CH
2–OO
C–(CH
2)
4–
COO–CH
2–(CHOR)–(CH
2OR)), 75.53 (s, 1 C,
CH
T1,3), 72.01 (s, 1 C,
CH
L1,2), 69.35 (s, 1 C,
CH
T1,2), 68.83 (s, 1 C,
CH
D), 66.22 (s, 1 C,
CH
L1,3), 65.62 (s, 1 C,
CH
2–OR
T1,2), 64.89 (s, 1 C,
CH
2L1,2), 62.69 (
CH
2–OH
T1,2), 62.38 (s, 1 C,
CH
2–OR
L1,2), 61.90 (s, 1 C,
CH
2D), 59.85 (s, 1 C,
CH
2T1,3), 59.56 (s, 1 C,
CH
2–OH
L1,2), 33.44–32.93 (m, 2 C, ROCO–
CH
2–(CH
2)
2 –
CH
2–COOR), 24.10–23.67 (m, 2 C, ROCO–CH
2–(
CH
2)
2–CH
2–COOR) ppm. Abbreviations in accordance with
Figure A1.
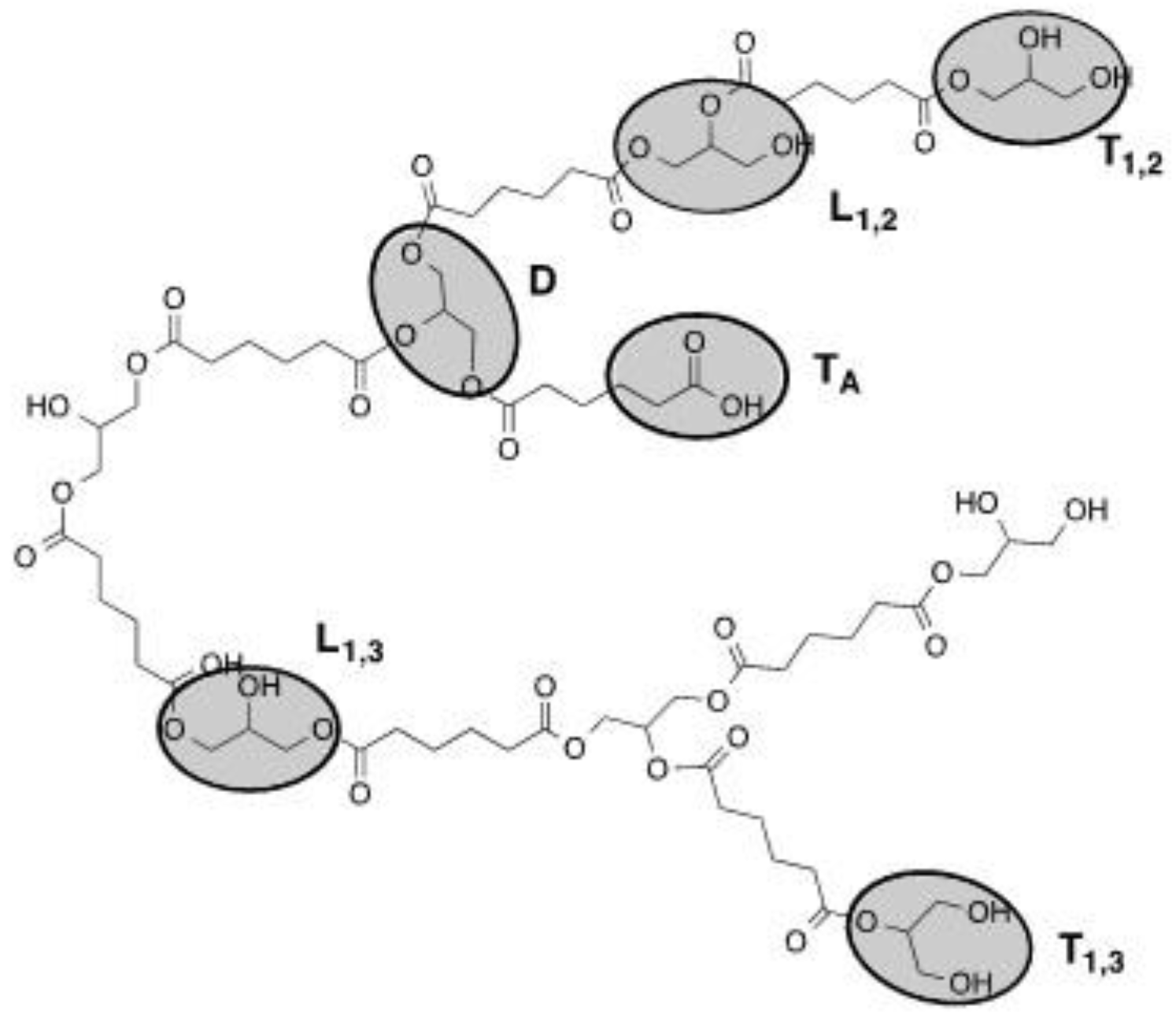
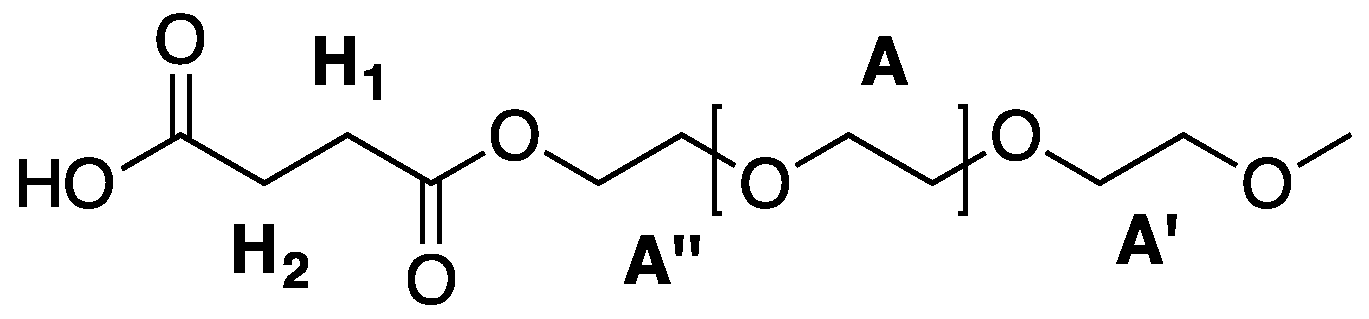
Figure A1.
Structural units of hPES 1.
Figure A1.
Structural units of hPES 1.
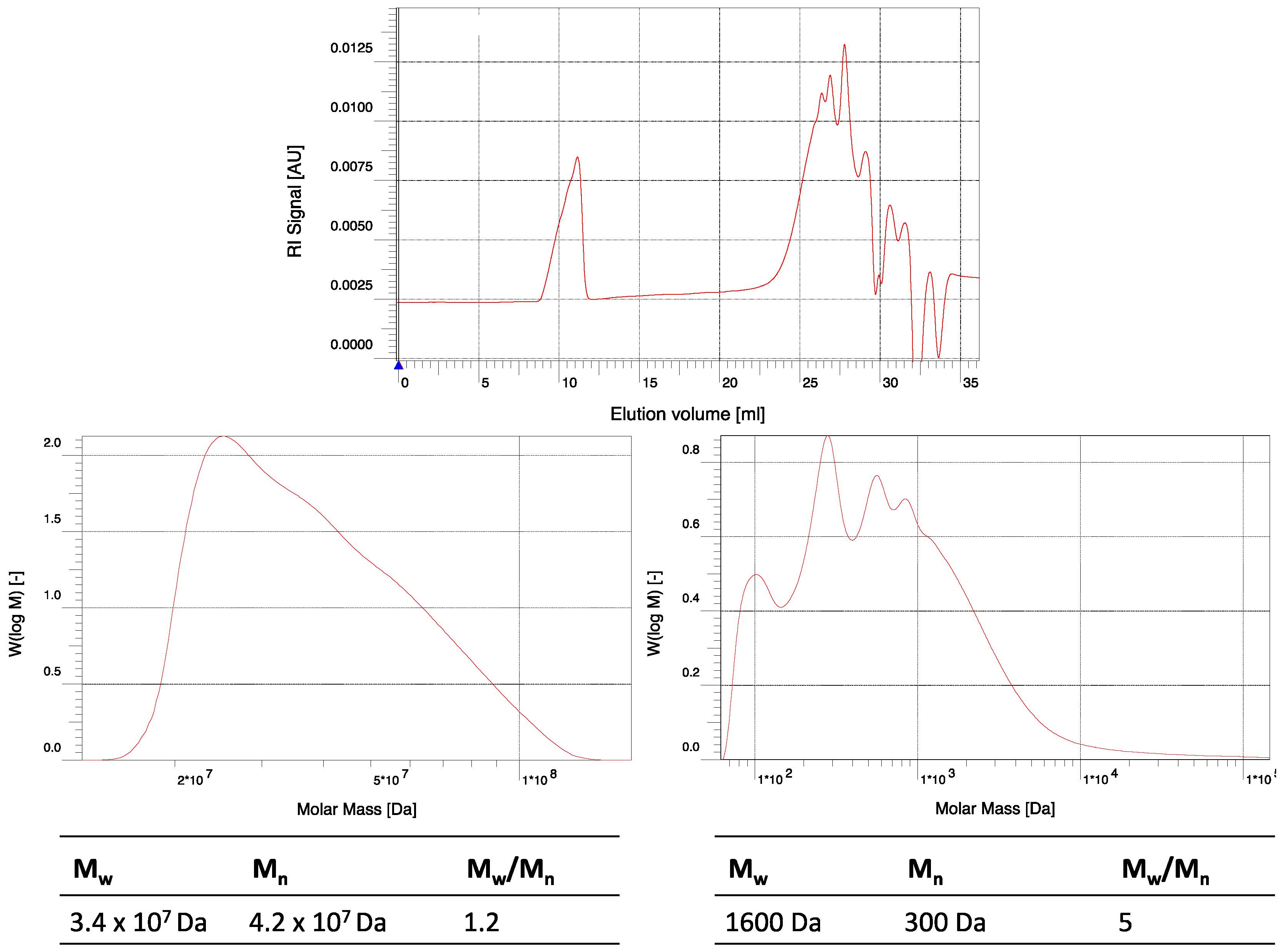
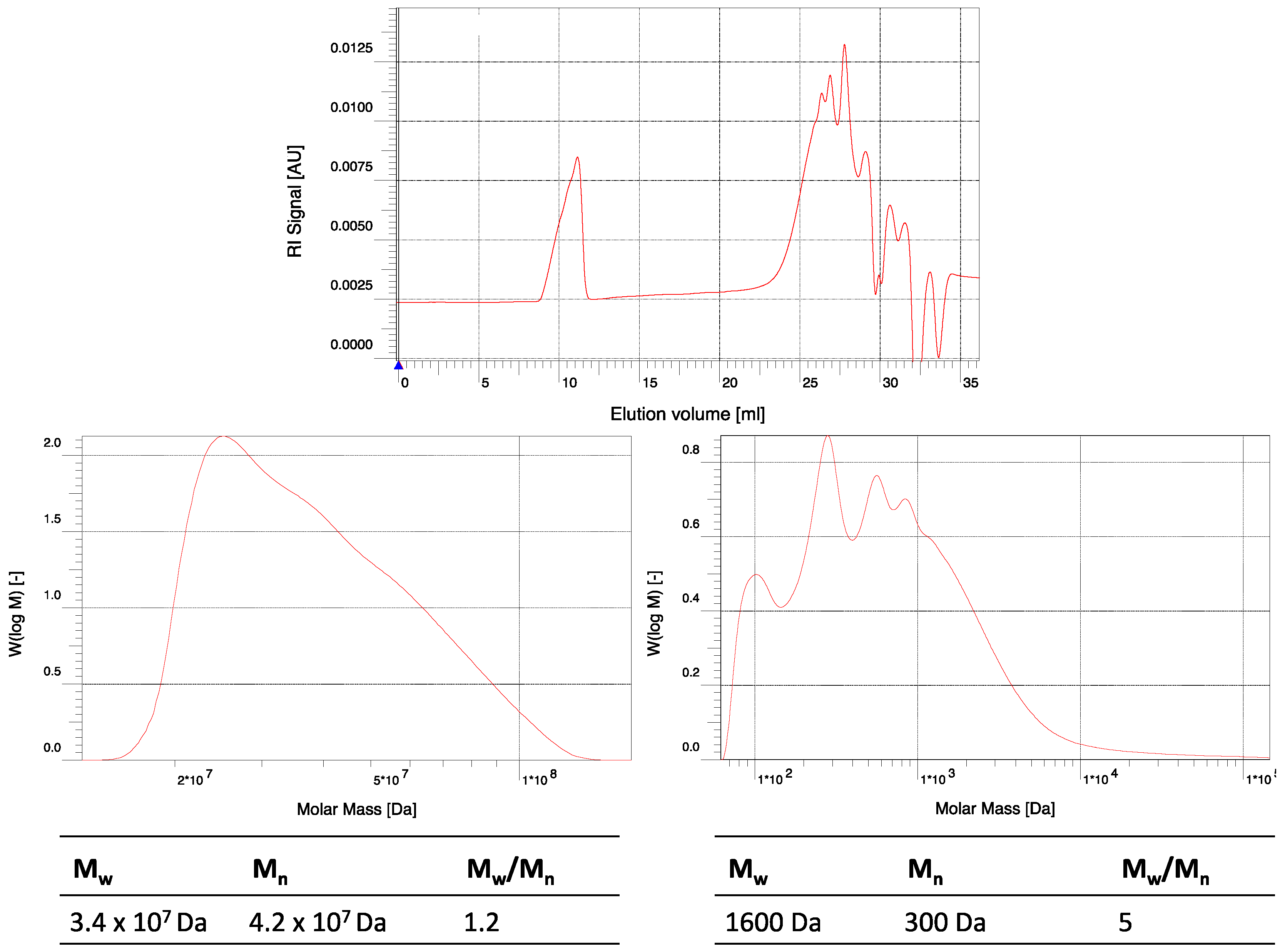
Table A1.
Molecular weight distribution of hPES 1.
Table A1.
Molecular weight distribution of hPES 1.
| Peak No. | Mn | Mw | Mw/Mn |
|---|
| 1 | 3.4 × 107 Da | 4.2 × 107 Da | 1.2 |
| 2 | 300 Da | 1,600 Da | 5 |
DB: 0.52
GPC (THF)
TAV: 1.80 mmol COOH/g polymer
THV: 2.25 mmol OH/g polymer
IR: ν = 3458.71, 2948.63, 1729.83, 1455.03, 1416.46, 1381.75, 1166.72, 1135.87, 1078.01, 1061.62, 943.02, 754.031 cm−1.
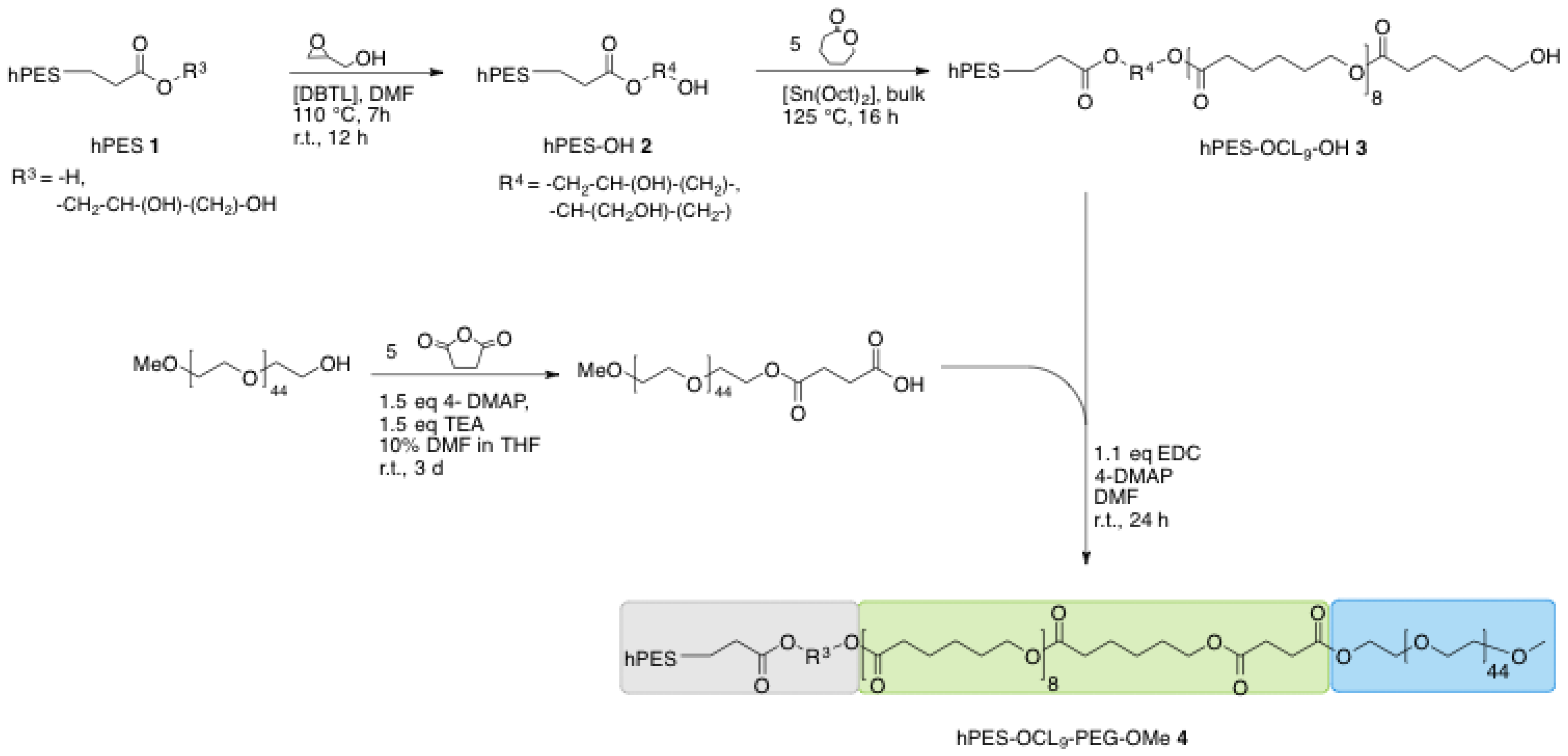
A.2. Synthesis of Polyesterol hPES-OH 2
30 mL of a solution of hyperbranched polyester hPES 1 in THF (c = 347 mg·mL−1, 10.41 g, 19 mmol COOH) was charged into a Schlenk flask. The residual catalyst DBTL (1.3 mg, 3.9 μmol) from the original product hPES 1 was used to catalyze the reaction; no further catalyst was added. After solubilization in 10 mL DMF under stirring at RT, the flask was heated to 85 °C. THF was removed from the mixture under controlled reduced pressure using cryo-distillation, and the completeness of the THF removal was controlled by 1H NMR. After THF was removed, the mixture was heated to 110 °C. Glycidol (1.25 mL, 1.388 g, 19 mmol, 1 eq) was added drop wise to the stirring yellow solution during a time period of 10 min using a syringe. The reaction mixture was stirred at 110 °C for 120 min, afterwards at RT overnight. Due to the incompleteness of reaction, the reaction was reheated to 110 °C and more glycidol (0.1 mL, 20 mmol in total) was added dropwise to the stirring reaction mixture. The reaction was stirred at 110 °C for 5.5 h and afterwards allowed to cool down to RT. The viscous product was obtained without further purification and stored in DMF.
1H NMR (500 MHz, DMSO-d
6): δ = 7.95 (s, DMF), 5.18 (s, 1 H, C
HD), 4.94 (s, 1 H, C
HL1,2), 4.72 (q, 1 H, C
HT1,3), 3.86–4.26 (m, 13 H, C
HL1,3, C
H2T1,2, C
H2L1,3, C
H2L1,2, C
H2D), 3.63 (q, 1 H, C
HT1,2), 3.60 (THF), 3.27–3.50 (m, 6 H, C
H2’T1,2, C
H2T1,3, C
H2’L1,2), 3.19, 3.02, 2.89, 2.73 (s, DMF), 2.59, 2.31 (m, 2H, –C
H2–CO
2–R), 2.10–2.12 (m, 2 H, –C
H2–CO
2H), 1.75 (THF), 1.54 (m, 4 H, –OCO–CH
2–(C
H2)
2–CH
2–COO–) ppm. Abbreviations in accordance with
Figure A1 and
Figure A2.
IG 13C NMR (500 MHz, DMSO-d
6): δ = 174.93–175.04 (CH
2–
COOH), 172.08–172.85 (CH
2–
COOR), 75.49 (
CH
T1,3), 72.98, 72.52, 71.97 (
CH
L1,2), 70.52, 69.33 (
CH
T1,2), 68.79 (
CH
D), 67.02 (THF), 66.19 (
CH
L1,3), 65.57 (
CH
2–OR
T1,2), 64.85 (s,
CH
2L1,3), 62.68 (s,
CH
2–OH
T1,2), 62.33 (s,
CH
2–OR
L1,2), 61.83 (s,
CH
2D), 59.79 (
CH
2T1,3), 59.52 (
CH
2–OH
L1,2), 35.75 (DMF), 32.85–33.36 (–OCO–
CH
2–(CH
2)
2–
CH
2–COO), 30.74 (DMF), 25.13 (THF), 23.62–23.91 (–OCO–CH
2–(
CH
2)
2–CH
2–COO–) ppm. Abbreviations in accordance with
Figure A1 and
Figure A2.
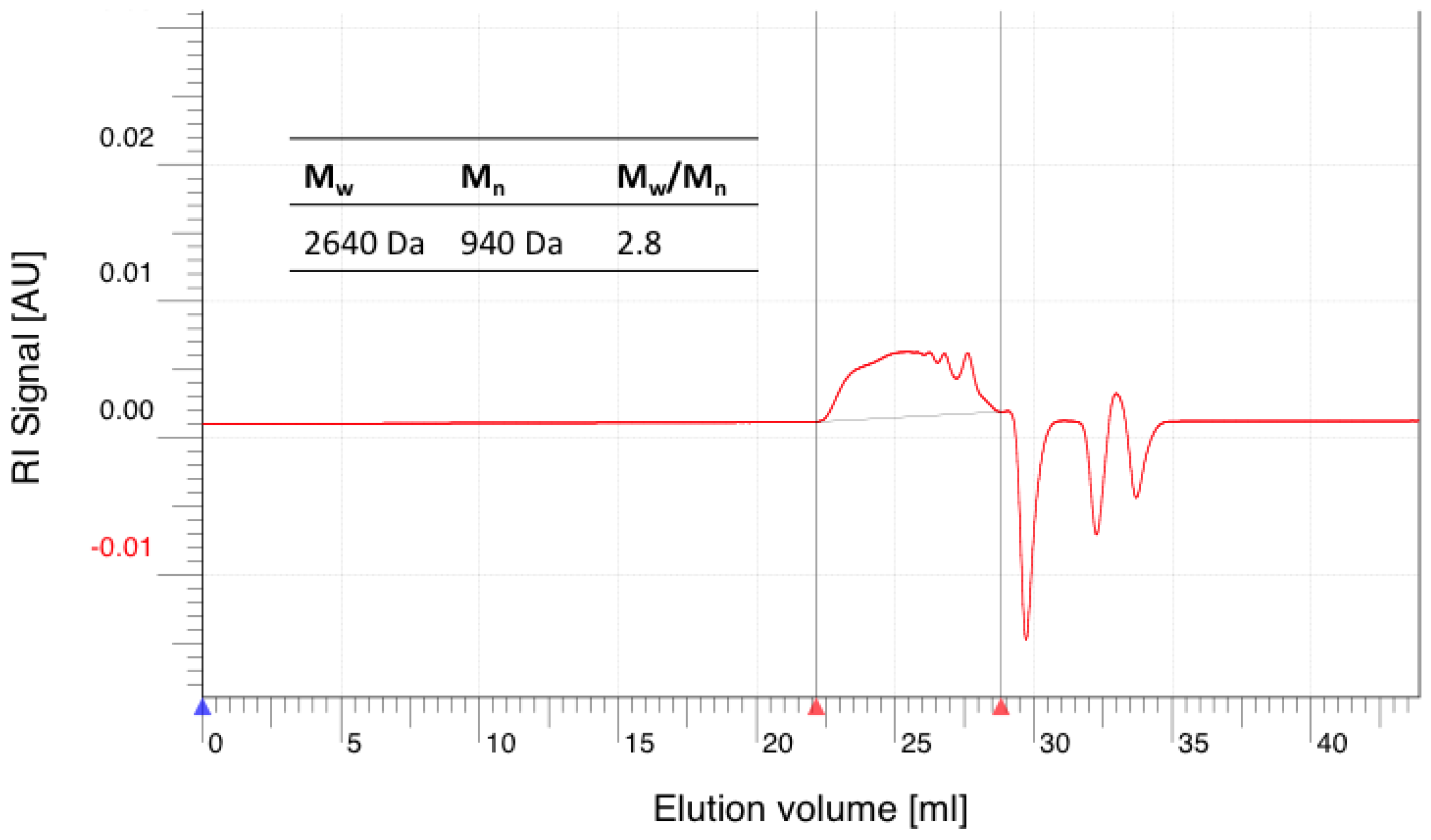
DB: 0.41
GPC: Mn = 900 Da; Mw = 2400 Da, Mw/Mn = 2.7
TAV: 0.045 mmol COOH/g polymer
THV: 5.3 mmol OH/g polymer
IR: ν = 3384.46, 2938.02, 2871.49, 2332.48, 1732.73, 1660.41, 1439.6, 1409.71, 1383.68, 1250.61, 1167.69, 1137.8, 1091.51, 1054.87, 937.24, 865.88 cm−1.
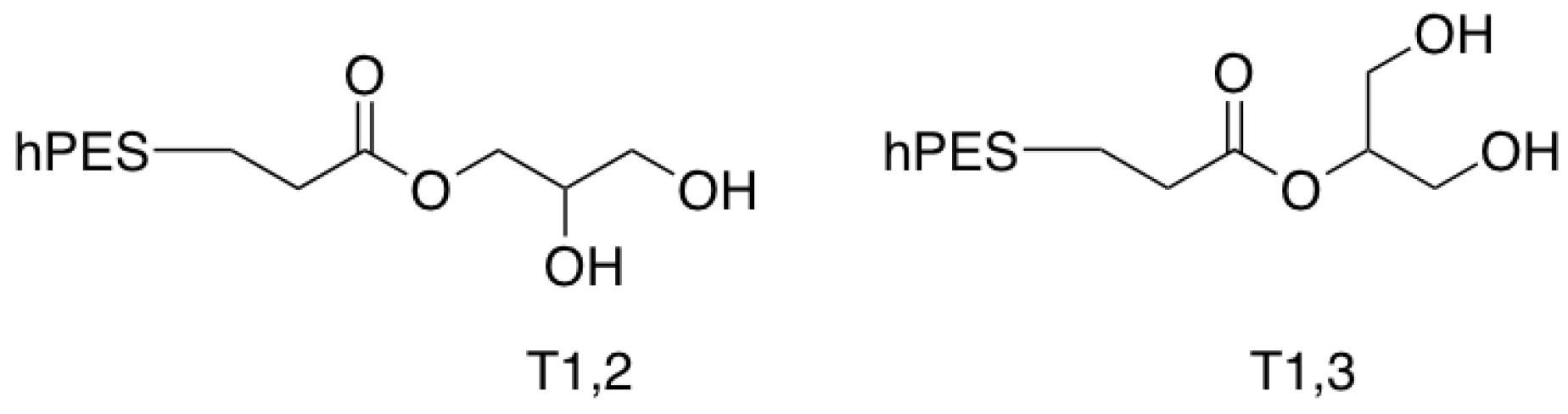
Figure A2.
Terminal glycerol units of hPES-OH 2.
Figure A2.
Terminal glycerol units of hPES-OH 2.
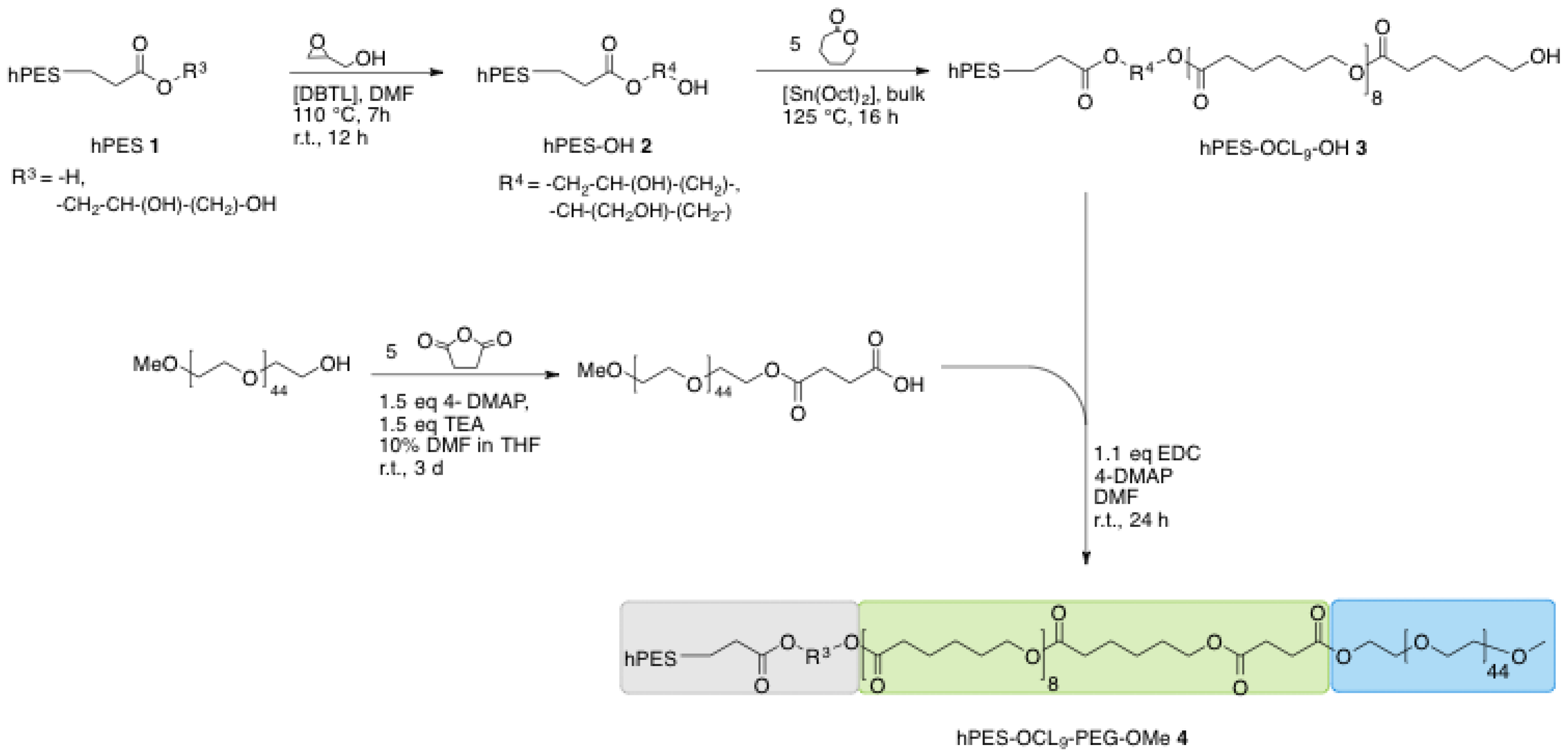
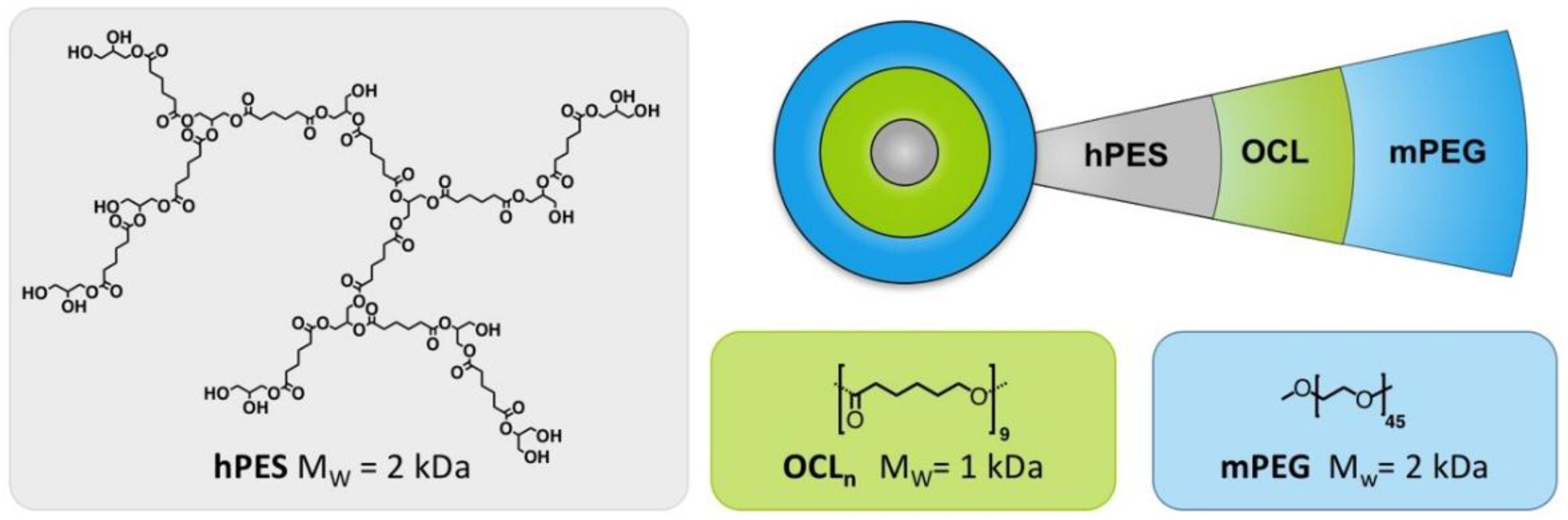
A.3. Synthesis of Linear Di-Block Copolymer hPES-OCL9-OH 3
In a Schlenk flask pre-dried macroinitiator hPES–OH 2 (2 g, 10.6 mmol OH) was dissolved in freshly distilled ε-caprolactone (6.18 g, 54 mmol) at 60 °C and two drops of Sn(Oct)2 were added to the stirring mixture, followed by an increase of the temperature to 125 °C. The bulk mixture was stirred at 125 °C for 18 h. Purification was performed by dissolving the crude reaction mixture in DCM in high dilution and precipitation in a high excess of ice-cold MeOH under vigorous stirring. The dispersion was separated from the formed gel, solvent was removed under reduced pressure, and the received solid was redissolved in DCM in high dilution. Another precipitation was performed by adding the DCM solution dropwise into vigorously stirred ice-cold diethyl ether. The mixture was separated by centrifugation at 4000 min−1 for 1 min, and the supernatant was collected. After drying the separated supernatant under reduced pressure, the received solid was once again purified using dialysis in DCM (benzoylated RC membrane, MWCO 1–2 kDa, 7 h) for removal of Sn(Oct)2 and traces of ε-caprolactone. A white, wax-like solid product was obtained after removing the solvent under reduced pressure (2.69 g, yield: 33%).
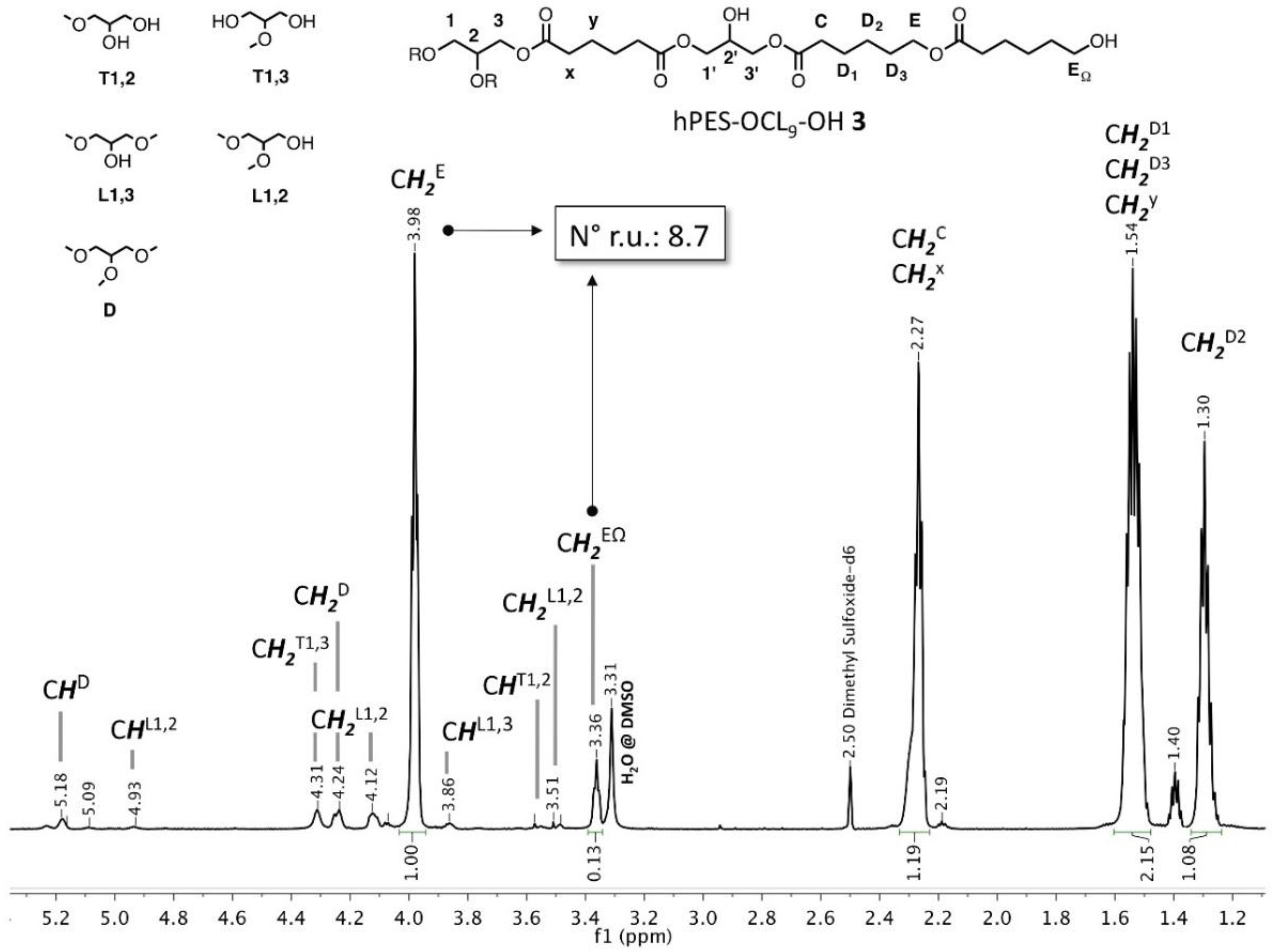
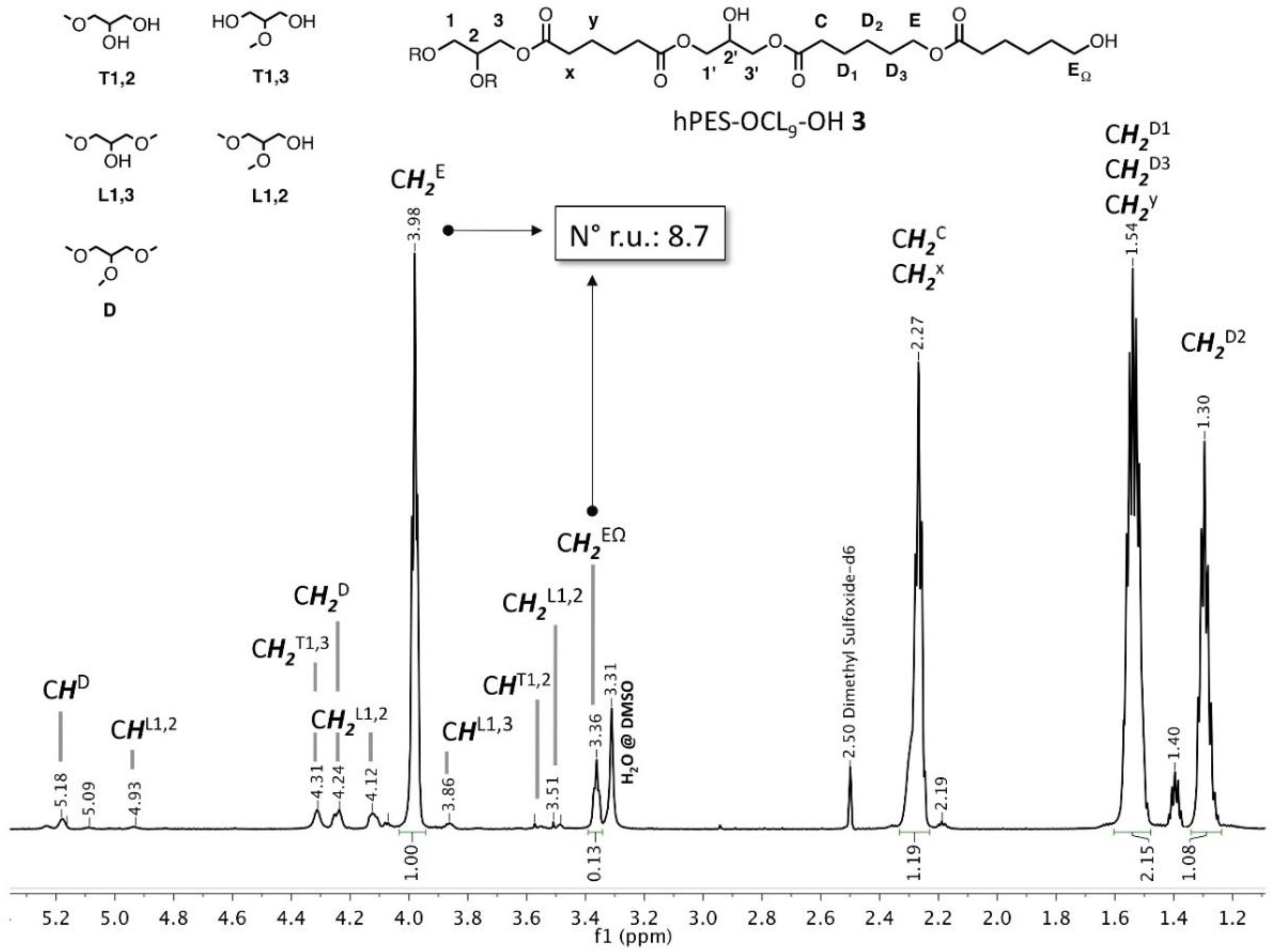
1H NMR (500 MHz, DMSO-d
6): δ = 5.18 (C
HD), 5.09 (new), 4.93 (C
HL1,2), 4.31 (C
H2T1,3), 4.24 (C
H2D), 4.12 (C
H2L1,2), 3.98 (C
H2E), 3.86 (C
HL1,3), 3.57 (C
HT1,2), 3.51 (C
H2L1,2), 3.36 (C
H2EΩ), 2.27 (m, C
H2C, C
H2X), 1.54 (C
H2D1, C
H2D3, C
H2y), 1.40, 1.30 (m, C
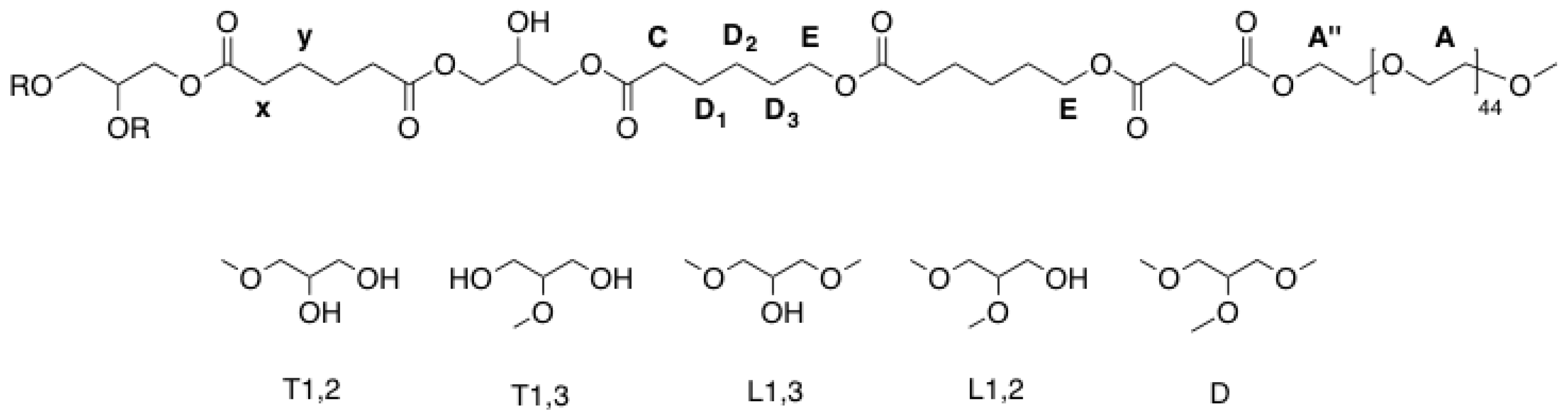
H2D2) ppm. Abbreviations in accordance with
Figure A3.
13C NMR (700 MHz, overnight, DMSO-d
6): δ = 172.8, 172.7, 172.4 (various CH
2-
COOR), 71.9 (
CH
L1,2), 69.8 (new), 69.3 (
CH
T1,2), 68.8 (
CH
D), 66.1 (
CH
L1,3), 64.7 (
CH
2L1,3), 63.5 (
CH
2E), 62.6 (
CH
2OH
T1,2), 62.3 (
CH
2OR
L1,2), 61.8 (
CH
2D), 60.5 (
CH
2EΩ), 59.5 (
CH
2OH
L1,2), 33.6 (
CH
2X), 33.3 (
CH
2C), 32.1, 27.8 (
CH
2D3), 25.0, 24.9 (
CH
2D2), 24.4, 24.1 (
CH
2D1) ppm. Abbreviations in accordance with
Figure A3.
DB: 0.74
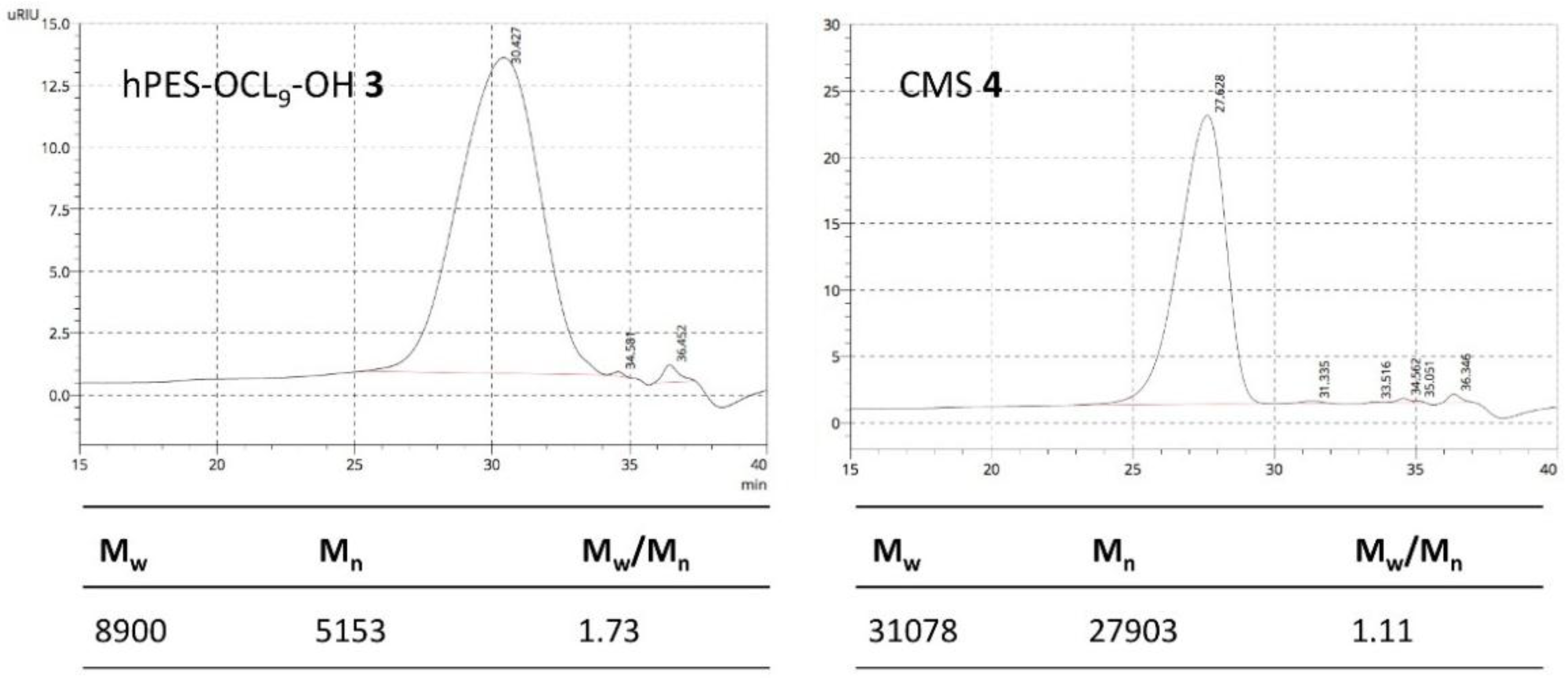
GPC: Mn = 5000 Da, Mw = 8900 Da, Mw/Mn = 1.72
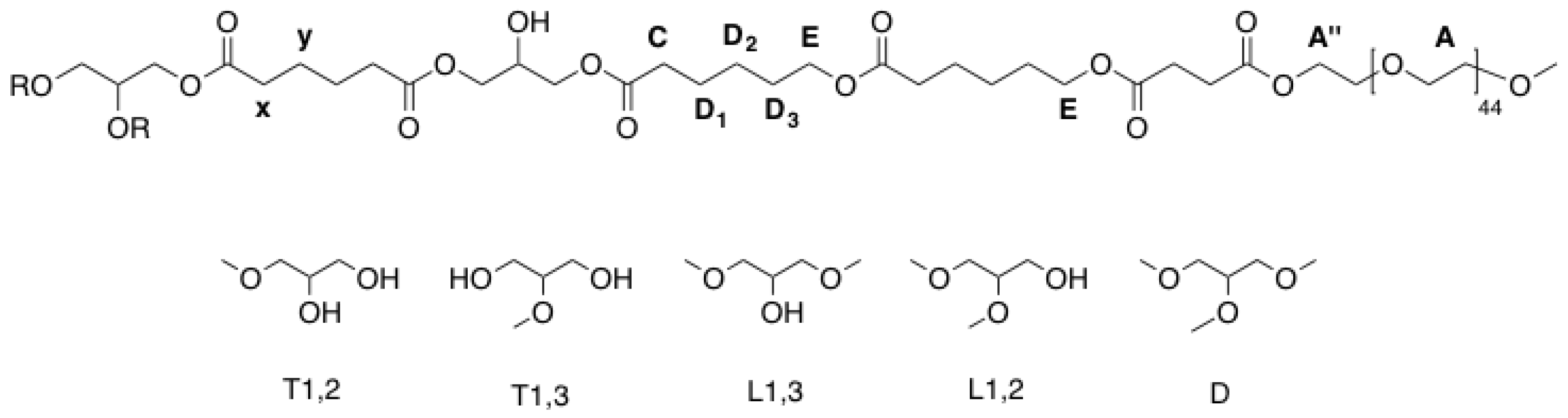
Figure A3.
Labelling of atoms and structural units of hPES-OCL9-OH 3 used for NMR description.
Figure A3.
Labelling of atoms and structural units of hPES-OCL9-OH 3 used for NMR description.
A.4. Functionalization of mPEG-OH (mPEG-COOH)
To a solution of pre-dried mPEG (4.786 g, 2.5 mmol) in a mixture of 10% anhydrous DMF in anhydrous THF (28 mL) in a 50 mL Schlenk flask, 4-DMAP (0.44 g, 3.8 mmol, 1.5 eq), TEA (0.5 mL, 3.8 mmol, 1.5 eq), and succinic anhydride (1.2 g, 12.5 mmol, 5 eq) were added under stirring at RT After stirring at RT for three days, the unreacted precipitated starting material was removed from the solution and the solution was dried under reduced pressure. The crude product was purified by precipitation from DCM solution into an ice-cold, 10-fold excess of Et2O. The formed precipitate was filtered off using a glass filter (P4), redissolved in DCM, and precipitated once more following the same procedure. The collected precipitate was dried under high vacuum and 3.5 g (1.67 mmol, yield: 70%) of pure product were obtained.
1H NMR (500 MHz, DMSO-d
6): δ = 4.21 (t, 2 H, C
H2A″), 3.73–3.45 (m, 185 H, various C
H2A), 3.33 (s, 3 H, PEG–O–C
H3), 2.64–2.58 (m, 4 H, C
H2H1, C
H2H2) ppm. Abbreviations in accordance with
Figure A4.
Figure A4.
Labelling of atoms of mPEG-COOH used for NMR description.
Figure A4.
Labelling of atoms of mPEG-COOH used for NMR description.
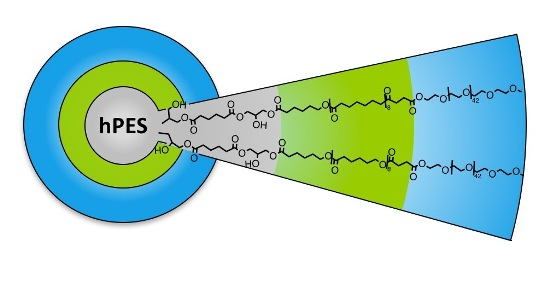
A.5. Synthesis of Core-Multishell Nanocarrier (hPES-OCL9-PEG-OMe 4)
In a dried 25 mL Schlenk flask, solid mPEG–COOH (1.110 g, 0.529 mmol, 1.1 eq) was added at RT to a stirring solution of hPES–OCL9–OH 3 (304 mg, 0.48 mmol OH) in 6 mL anhydrous DMF. After the addition of 4-DMAP (0.016 g, 0.106 mmol, 20 mol %), EDCl (0.110 g, 0.574 mmol, 1.1 eq) was added at 0 °C. The reaction mixture was stirred for 10 min at 0 °C and then allowed to reach RT by removing the ice bath. After 20 h of stirring at RT, the crude product was purified via extensive ultrafiltration (DMF, MWCO < 10 kDa), followed by repeated fractionation. For this purpose, the impure product was dissolved in DCM, which yielded a clear solution. Hexane was added to the clear solution at RT until cloudiness appeared. The cloudy dispersion was heated to obtain a clear solution, followed by the addition of Hexane to obtain a dispersion. The warm solution was allowed to reach RT and centrifuged (1 min, 3900 min−1) to separate into a stable dispersion and sediment. The dispersion was dried and refractionated following the above-described procedure. The progress of purification was monitored using GPC in DMF. After six cycles of refractionation and removal of solvent under reduced pressure, followed by drying at high vacuum, a white solid product was obtained (0.116 g, yield: 12%).
1H NMR (700 MHz, DMSO-d
6): δ = 5.17 (C
HD), 4.24 (C
H2D), 4.12 (C
H2L1,2), 3.98 (C
H2E), 3.59–3.40 (various C
H2A, C
H2A″), 3.24 (PEG–OC
H3), 2.27 (C
H2C, C
H2x), 1.54 (C
H2D1,C
H2D3, C
H2y), 1.29 (C
H2D2) ppm. Abbreviations in accordance with
Figure A5.
13C NMR (500 MHz, DMSO-d
6): δ = 172.7 (–
COOR–), 171.9 (–
COOR–), 78.9, 75.3, 71.3 (
CH
L1,2), 69.8 (PEG backbone), 69.6, 68.22 (various
CH
2,
CH
2), 69.6 (
CH
D), 63.8 (
CH
2L1,2), 63.5 (
CH
2E), 61.8 (
CH
2D), 58.0 (PEG–O
CH
3), 33.3–33.1 (
CH
2C,
CH
2x), 28.5 (
CH
2H1,
CH
2H2), 27.8 (
CH
2D1,
CH
2D3), 24.9 (
CH
2D2), 24.1 (
CH
2y) ppm. Abbreviations in accordance with
Figure A5.
DB: 0.52
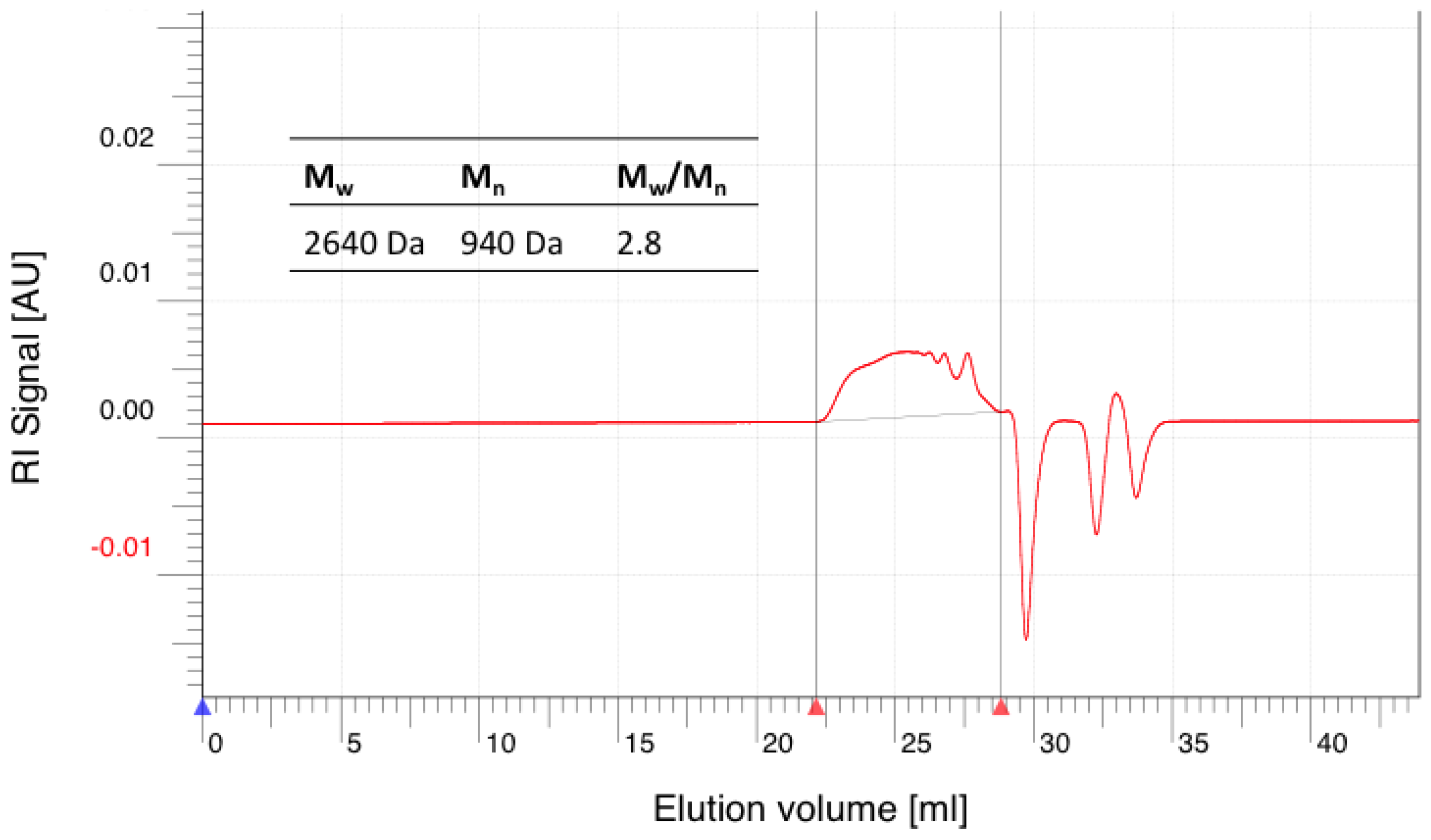
GPC: Mn = 27,900 Da, Mw = 31,100 Da, Mw/Mn = 1.11
Figure A5.
Labelling of atoms and structural units of hPES-OCL9-OH 3 used for NMR description.
Figure A5.
Labelling of atoms and structural units of hPES-OCL9-OH 3 used for NMR description.
A.6. GPC of Hyperbranched Polymers
Figure A6.
GPC elugram of crude hPES 1 in THF. (Top) full range elugram; y-axis: elution volume. (Bottom) detailed elugrams with molecular weights; y-axis: molar mass.
Figure A6.
GPC elugram of crude hPES 1 in THF. (Top) full range elugram; y-axis: elution volume. (Bottom) detailed elugrams with molecular weights; y-axis: molar mass.
Figure A7.
GPC elugram of purified hPES-OH 2 in THF.
Figure A7.
GPC elugram of purified hPES-OH 2 in THF.
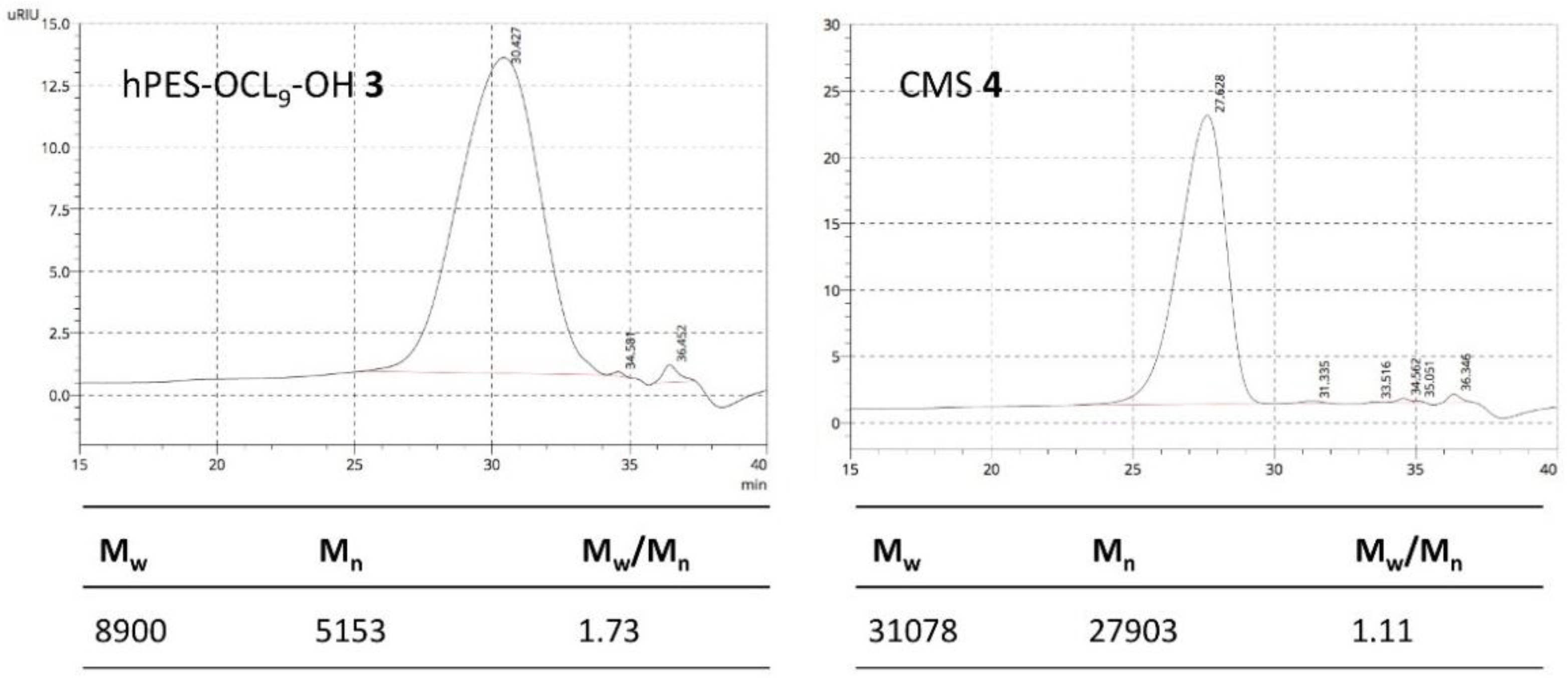
Figure A8.
GPC elugram of purified hPES–OCL9–OH 3 and CMS 4 in DMF.
Figure A8.
GPC elugram of purified hPES–OCL9–OH 3 and CMS 4 in DMF.
A.7. Determination of TAV and THV of hPES 1 and hPES-OH 2
To determine the total acid value (TAV) and total hydroxyl value (THV) it is crucial to know the conversion at the respective point of the reaction. Synthesis of hyperbranched polyester
1 consisting of a diacid (A2) and triol (B3) was stopped before reaching the so-called gel point. According to Flory, the gel point is the point when the conversion reaches its maximum before becoming an infinite network [
14]. The actual gel point can be predicted as a function of the functionality
f and ratio ρ. In our case of adipic acid as A
2 and glycerol as B
3 unit, functionality
f is 3, reflecting three functional groups on the branching unit, while ratio
ρ is the ratio between the number of A functional groups and the number of B functional groups yields:
nCOOH, amount of acid groups in mol; nOH, amount of hydroxyl groups in mol.
This theoretical approach does not take into account the difference in reactivity of primary and secondary functional groups of the B
3 trifunctional branching unit. Functionality
f can be used to determine the critical value α
c of the branching coefficient α for formation of infinite networks:
f, functionality of branching unit; αc, critical value of branching coefficient α
In our case for B
3 monomers,
f is 3 and α
c consequently 0.5. The connection between functionality
f and ratio ρ is as follows:
PA, conversion at gel point; αc, critical value of branching coefficient α; ρ, ratio, see above.
Equation (3) allows for prediction of the theoretical conversion P
A at the gel point. For the synthesis of hPES
1 with a ratio of 1.2:1 (A
2:B
3), the value for P
A is 0.79. The conversion during the reaction was monitored by using
1H NMR spectroscopy and analysis of the ratio of C
H2–COOH (2.3 ppm) versus C
H2–COOR (2.2 ppm). When the reaction approached the theoretical P
A value, the reaction was stopped and the bulk polymer cooled down to stop the polymerization. After evaluation of the real P value, the TAV and THV were calculated for hPES
1 as follows:
Conversion p = (Integral CH2–COOH)/(Integral CH2–COOH + Integral CH2–COOR) = 1/1.21 = 0.83
TAV:
nt(COOH) = (1−P) × n0(COOH) = 94.9 mmol
➔TAV = 94.9 mmol COOH/52.708 g polymer = 1.80 mmol/g polymer
THV:
nt(OH) = (1−P) × n0(OH)= 118.7 mmol
➔THV = 118.7 mmol OH/52.708 g polymer = 2.25 mmol/g polymer
Values of TAV and THV of hPES–OH 2 were determined based on the assumption that COOH reacted with glycidol with a conversion of p = 0.97.so that hPES–OH 2 had 2.9% remaining COOH:
n0(COOH) = 18.7 mmol
n0(OH) = 26.8 mmol
m(polymer) = 11.918 g
TAV:
nt(COOH) = (1−P) × n0(COOH) = 0.5423 mmol
➔TAV = 0.5423 mmol/11.918 g polymer = 0.045 mmol COOH/g polymer
THV:
nt(OH) = n0(OH) + [(n0(COOH) − nt(COOH)) × 2] = 63.1 mmol
➔THV = 63.1 mmol OH/11.918 g polymer = 5.3 mmol OH/g polymer
A.8. Determination of Degree of Branching DB
The degree of branching was calculated based on Equation (4), which is the calculation of the DB as published by Frey
et al. We chose this equation, because Frey states that the original Fréchet Equation (5) overestimates the branching, if small or low branched molecules are considered [
12,
33].
Where: T: relative integral of terminal units of type T1,2, T1,3, and TA.
L: relative integral of linear units of type L1,3 and L1,2
D: relative integral of dendritic unit D.
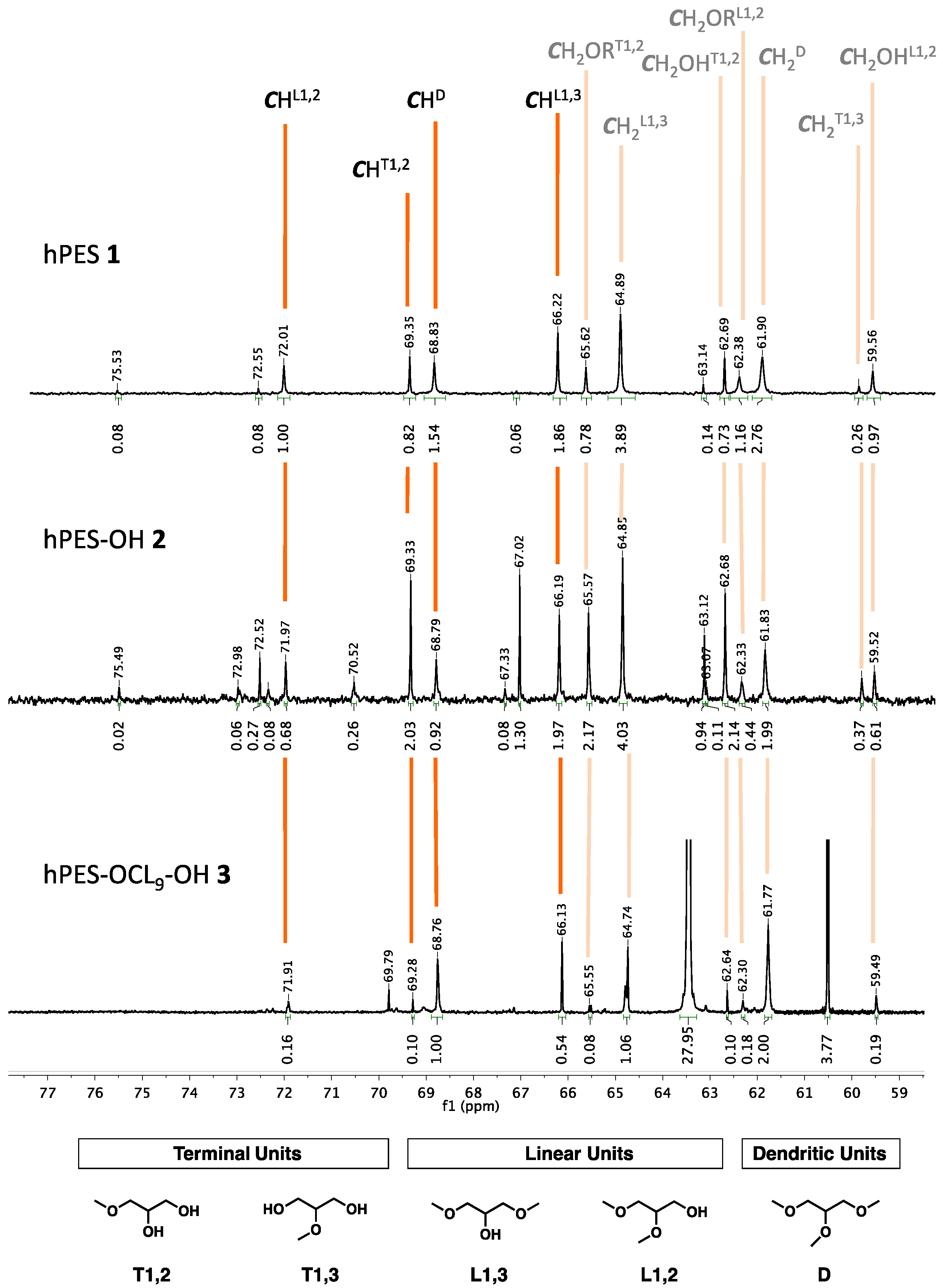
The relative integrals needed for this calculation are the integrals of methine signals of the various glycerol branching units, as depicted in
Figure A9 for the case of hPES 1, hPES-OH 2, and hPES–OCL
9–OH 3. The spectra were measured by inverse-gated
13C spectroscopy, because the obtained carbon peaks were quantifiable.
Table A2 summarizes all the relevant data for the calculation of branched products 1, 2, 3, and 4. In the case of product CMS 4, a quantification of methine and methylene signals was not possible.
Table A2.
Interpretation of inverse-gated (IG) 13C NMR spectra of hyperbranched polyesters in DMSO-d6. Integral values of –CH– signals of relevant glycerol units for evaluating the degree of branching (DB).
Table A2.
Interpretation of inverse-gated (IG) 13C NMR spectra of hyperbranched polyesters in DMSO-d6. Integral values of –CH– signals of relevant glycerol units for evaluating the degree of branching (DB).
| Structure | Integral value | DB |
|---|
| CH L1,2 | CH D | CH L1,3 |
|---|
| hPES 1 | 1.00 | 1.54 | 1.86 | 0.52 |
| hPES-OH 2 | 0.74 | 1.00 | 2.15 | 0.41 |
| hPES–OCL9–OH 3 | 0.16 * | 1.00 * | 0.54 * | 0.74 * |
Figure A9.
Details of 13C NMR spectra of hPES 1, hPES–OH 2, and hPES–OCL9–OH 3 (top to bottom). Depicted are peaks relevant for calculation of Df and DB analysis. Five types of glycerol units can be distinguished in the structure of the hyperbranched polyester. For comparability of signal integrals, the value for diacid peaks was set to be identical in hPES 1 and hPES–OH 2.
Figure A9.
Details of 13C NMR spectra of hPES 1, hPES–OH 2, and hPES–OCL9–OH 3 (top to bottom). Depicted are peaks relevant for calculation of Df and DB analysis. Five types of glycerol units can be distinguished in the structure of the hyperbranched polyester. For comparability of signal integrals, the value for diacid peaks was set to be identical in hPES 1 and hPES–OH 2.
A.9. Determination of Degree of Functionalization Df
A.9.1. hPES-OH 2
Hyperbranched polyester hPES
1 with a total acid value of 1.8 mmol carboxylic acid groups per gram polyester was modified with equimolar amounts of glycidol with respect to the amount of hydroxyl groups of hPES
1, in order to modify one glycidol per carboxylic acid. The reactions were performed at a bath temperature of 110 °C in DMF for two hours; afterwards at room temperature overnight. The reaction progress was monitored by
1H NMR,
i.e., the disappearance of the methylene signal next to the free carboxylic acid as well as the proton signal of terminating carboxylic acid groups. We paid special attention to the expected changes of several structural units as well as the possibility of the side reaction shown in
Scheme A1.
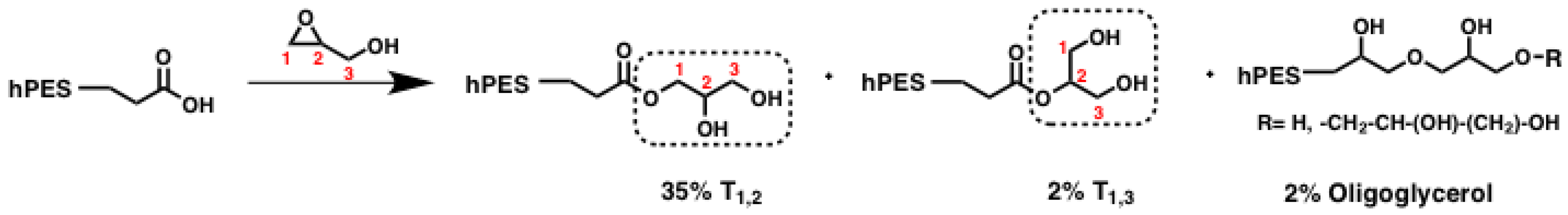
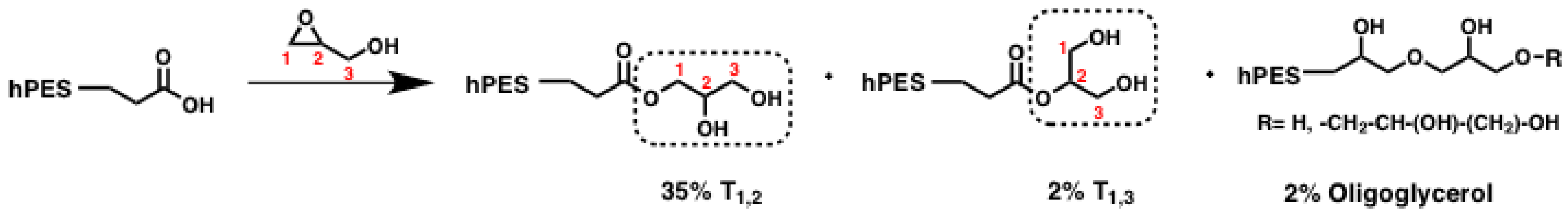
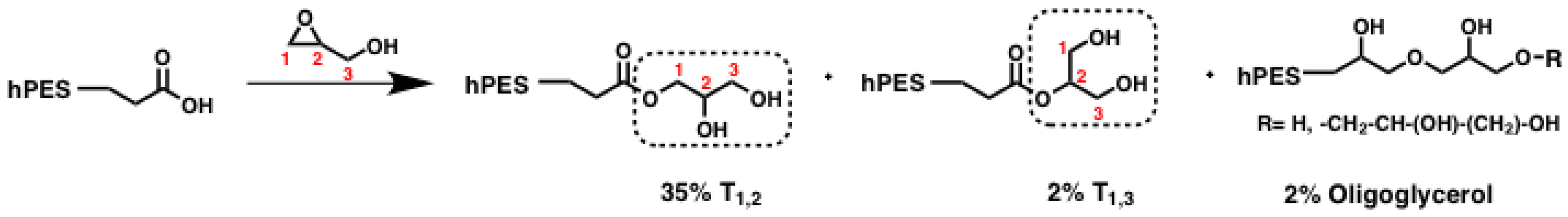
Scheme A1.
Simplified scheme of polyester modification by ring-opening reaction of glycidol; crucial units for NMR evaluation are highlighted in boxes. %: abundance of species relative to abundance of all glycerol units.
Scheme A1.
Simplified scheme of polyester modification by ring-opening reaction of glycidol; crucial units for NMR evaluation are highlighted in boxes. %: abundance of species relative to abundance of all glycerol units.
Even though equimolar amounts of glycidol were used during the reaction, full conversion of all carboxylic acid groups could not be achieved. Evaluation of the methylene signals next to free carboxylic acids versus those next to ester groups after
1H NMR measurements showed that the fraction of carbons from free carboxylic acid decreased from 17.4% before to 2.9% after modification, giving a functionalization degree of D
f (COOH) = 0.83. At the same time, the amount of esterification increased by a factor of 2.5, which was evaluated according to the increased methine peak supplementary inverse-gated
13C NMR spectroscopy. Based on inverse-gated
13C NMR spectroscopy, we can also show that T
1,2 methine and methylene signals increased by a factor of 2.5 as well, indicating that these newly formed T
1,2 units arose from reactions with carboxylic acids at the sterically less-hindered C1 atom of glycidol. We can furthermore observe the increase of T
1,3 methylene signals by a factor of 1.4. Hence, ring-opening of glycidol also occurred on the sterically more-hindered C2 carbon atom, as shown in
Scheme A1. The inverse-gated
13C NMR spectra were also checked for further changes of integrals, as we were also interested in the question whether formation of oligoglycerol formation occurred. If terminal glycerol hydroxyl groups initialized ring-opening of glycidol, new signals should have arisen. In fact, three new signals can be found in the region of methine shifts (70.52–72.98 ppm, see
Figure A1). With regard to the shift of these signals, they could have resulted from the methylene next to the ether bond between two glycerol units. With an average integral of 0.4, the new signals represent only 2% of the amount of overall carbon signals in the region of glycerol’s methine and methylene signals. As this amount is small, the presence of oligoglycerols should not interfere with further reactions and analysis. In summary, terminal carboxylic acid groups of the hyperbranched polyester hPES
1 were modified with glycerol units by in a tin-catalyzed, ring-opening reaction with glycidol. The amount of esterified carboxylic acid units increased by a factor of 2.5 compared to the esterified carboxylic acid units before the modification, while full modification of carboxylic acids could not be achieved. Ring-opening of glycidol did not only take place at C1 carbon atom leading to a new T
1,2 unit but also at the C2 carbon atom, which formed T
1,3 units. Oligoglycerols only formed to a certain extent.
A.9.2. hPES-OCL9-OH 3
Determination of
Df was accomplished based on Equation (6), where the estimated amount of reacted hydroxyl groups of hPES–OCL
9–OH
3 is compared to the estimated amount of theoretically-available hydroxyl groups of hPES–OH
2.
The amounts of hydroxyl groups were estimated based on the integrals in
13C NMR spectra of methine arising from glycerol units, marked with orange lines in
Figure A9. The relative abundance of the methine signals was calculated and normalized according to
Table A2, which gave the amounts of reacted and free OH grous of hPES–OH 2 and hPES–OCL9–-OH 3 that were needed for Equation (A6):
Table A3.
Comparison of
13C NMR spectra of hPES–OH 2 and hPES–OCL
9–OH 3 as shown in
Figure A9 with regard to specific glycerol’s methine signals and calculation of their relative abundance, and absolute and normalized amounts of OH and OR groups for calculation of the degree of functionalization
Df.
Table A3.
Comparison of 13C NMR spectra of hPES–OH 2 and hPES–OCL9–OH 3 as shown in Figure A9 with regard to specific glycerol’s methine signals and calculation of their relative abundance, and absolute and normalized amounts of OH and OR groups for calculation of the degree of functionalization Df.
| | hPES–OH 2 | hPES–OCL9–OH 3 |
|---|
| Signal | CH L1,2 | CH T1,2 | CHD | CHL1,3 | all | CH L1,2 | CH T1,2 | CHD | CHL1,3 | all |
| abs. integral value | 0.74 | 2.21 | 1 | 2.15 | 6.1 | 0.16 | 0.10 | 1 | 0.54 | 1.80 |
| rel. integral value | 12% | 36% | 16% | 35% | 100% | 9% | 6% | 56% | 30% | 100% |
| amount OH a | 12 | 72 | - | 35 | 119 | 6.9 b | 9.2 b | - | 23.1 b | 39.3 b |
| amount OR | - | - | 48 | - | 48 | - | - | 127 b | - | 127 b |
A.9.3. CMS Nanocarrier hPES–OCL9–PEG–OMe 4
Functionalization of terminal caprolactone hydroxyl groups was analyzed via 1H NMR by determining the increase of esterification of caprolactone hydroxyl groups. This was done by comparing the ratio of CH2E and CH2C+x before the reaction to the ratio after the reaction. Since the integral of CH2C+x remained constant upon functionalization with mPEG–COOH, we could fix this integral value and determine the percental increase of CH2E, leading to a value of Df(CL–OH) = 0.7. To cross check this assumption, the integral of methoxy CH3 of mPEG was compared to CH2C+x. This comparison revealed an 80% excess of mPEG chains, which therefore should be attached to internal glycerol hydroxyl groups. The functionalization of internal hydroxyl groups was supported by 13C-NMR of the product, which showed a disappearance of CHL1,2 and CHL1,3 and indicated that the respective hydroxyl groups reacted with mPEG–COOH. As the abundance of internal glycerol units was too small to allow for quantification, we could not estimate the degree of functionalization of internal glycerol hydroxyl groups based on 13C NMR. Nevertheless, 1H NMR analysis led to the assumption, that roughly 55% of present mPEG–COOH chains were attached to 70% of terminal caprolactone units, while 45% of mPEG–COOH reacted with internal glycerol units.
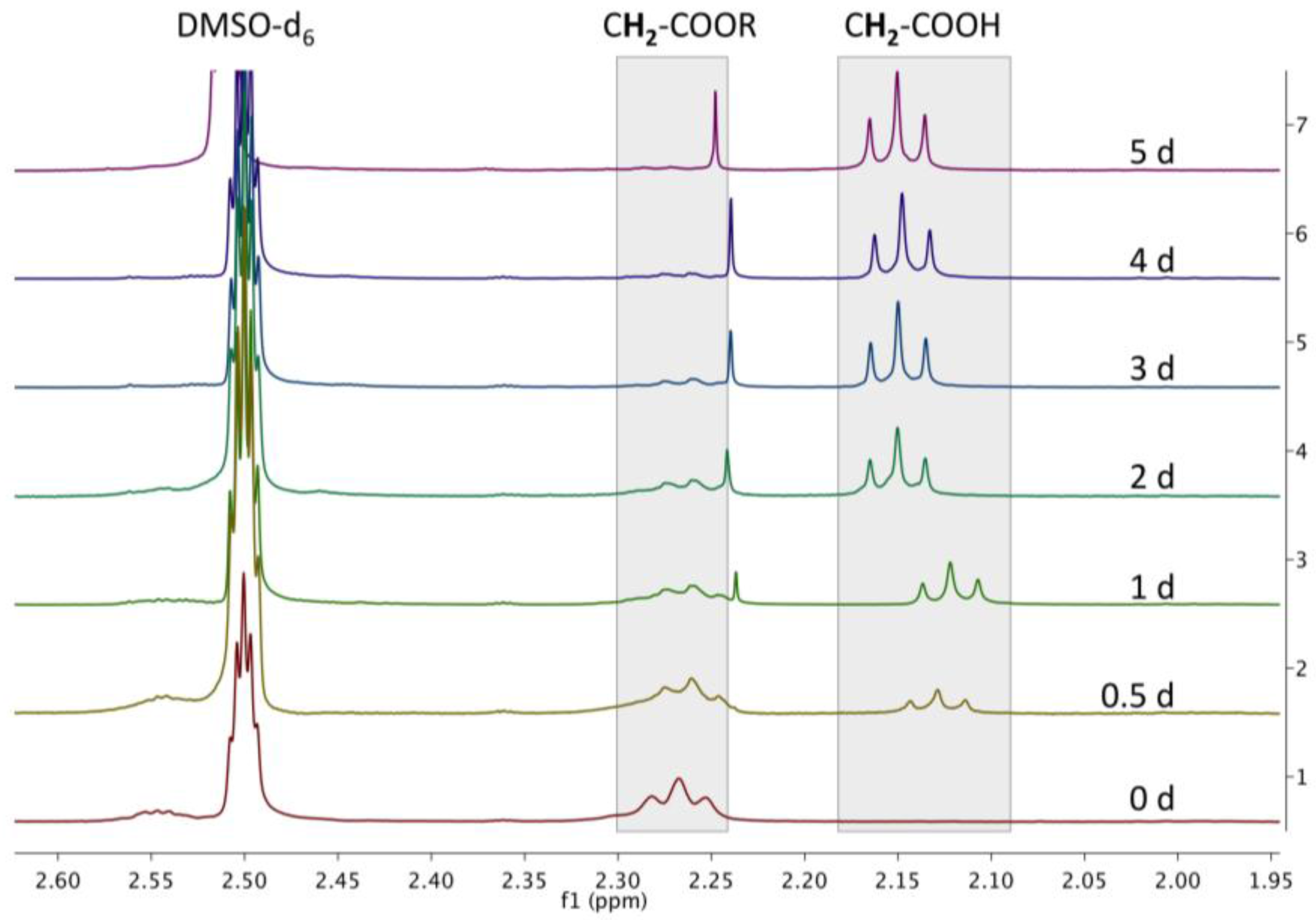
A.10. Enzymatic Degradation—Full Spectrum
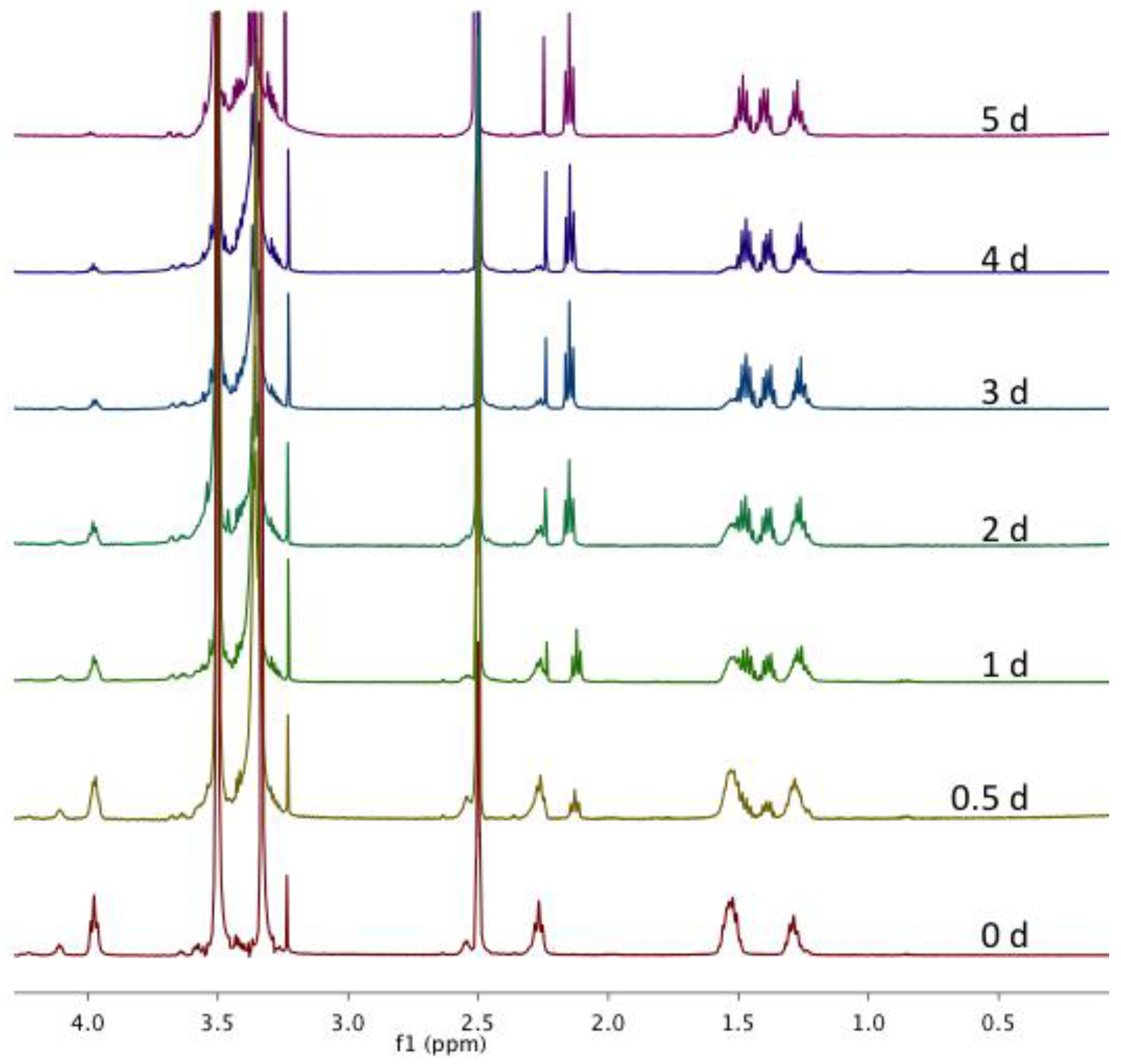
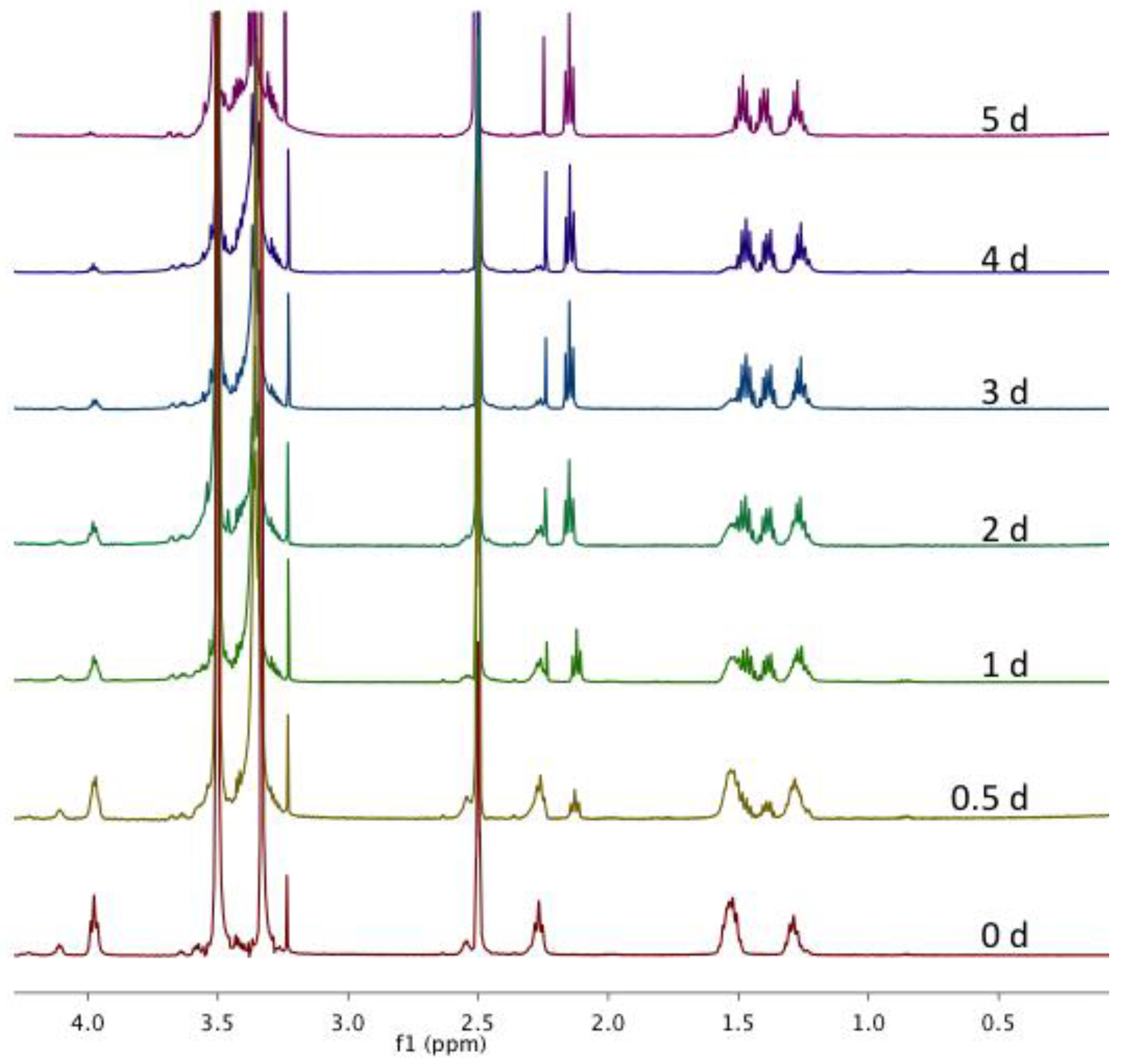
Figure A10.
Overlay of magnified 1H NMR spectra in DMSO-d6 of CMS nanocarriers after incubation with Novozyme® 435 at 32 °C at various time points during a five-day study.
Figure A10.
Overlay of magnified 1H NMR spectra in DMSO-d6 of CMS nanocarriers after incubation with Novozyme® 435 at 32 °C at various time points during a five-day study.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}