
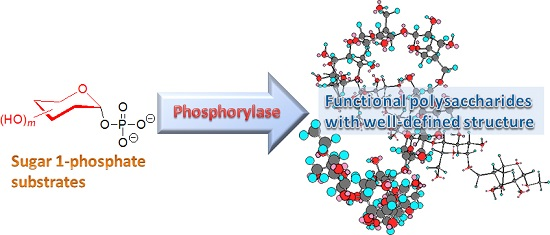
Precision Synthesis of Functional Polysaccharide Materials by Phosphorylase-Catalyzed Enzymatic Reactions


Abstract
:
{kind=link}
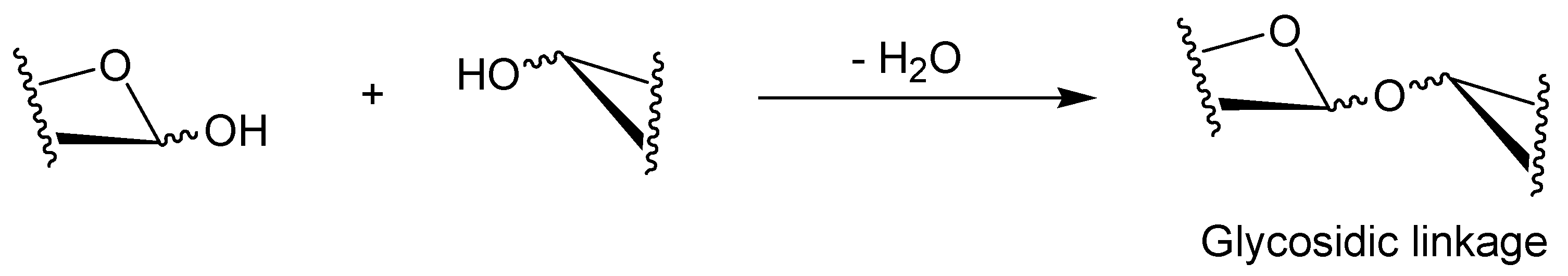{kind=link}
{kind=link}
{kind=link}
{kind=link}
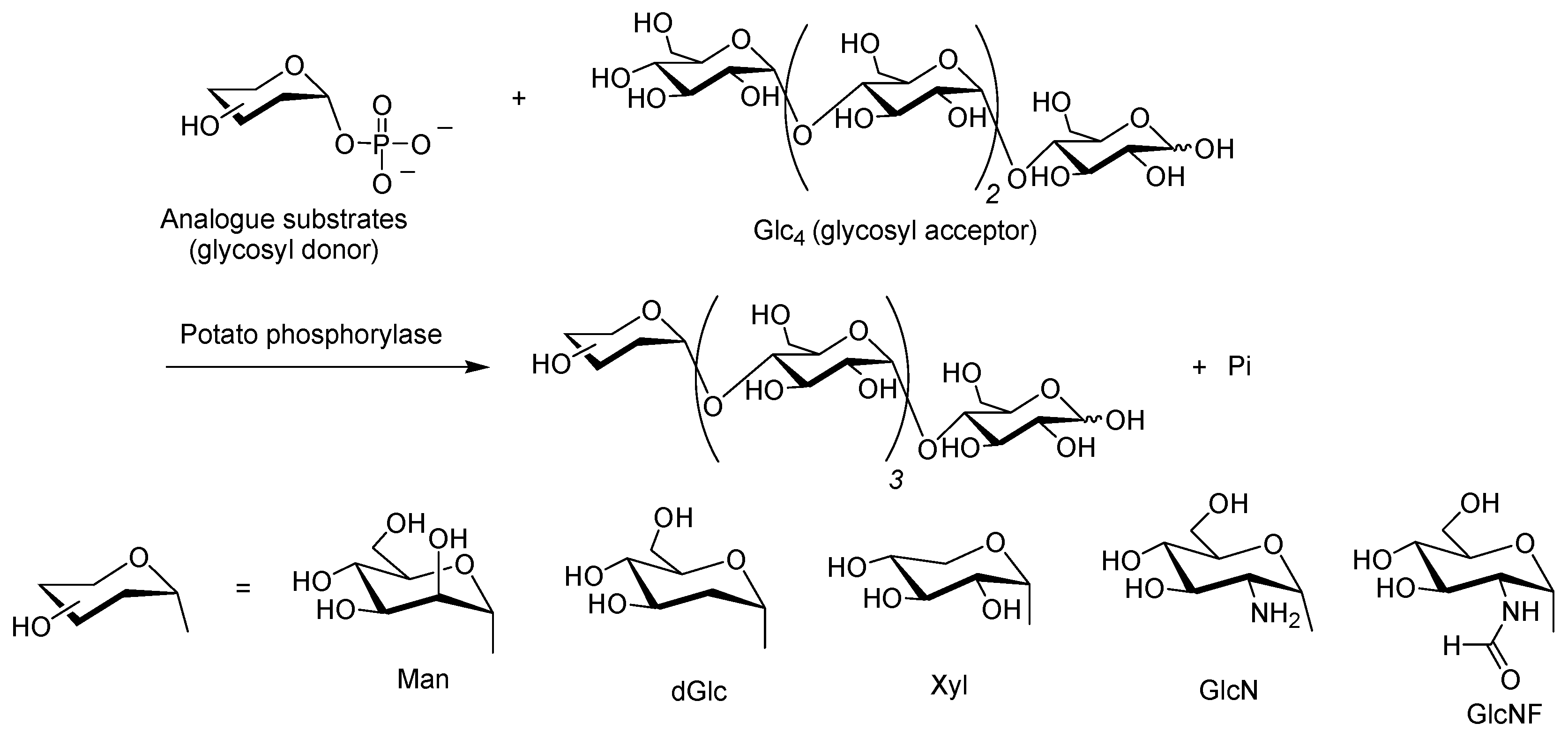{kind=link}
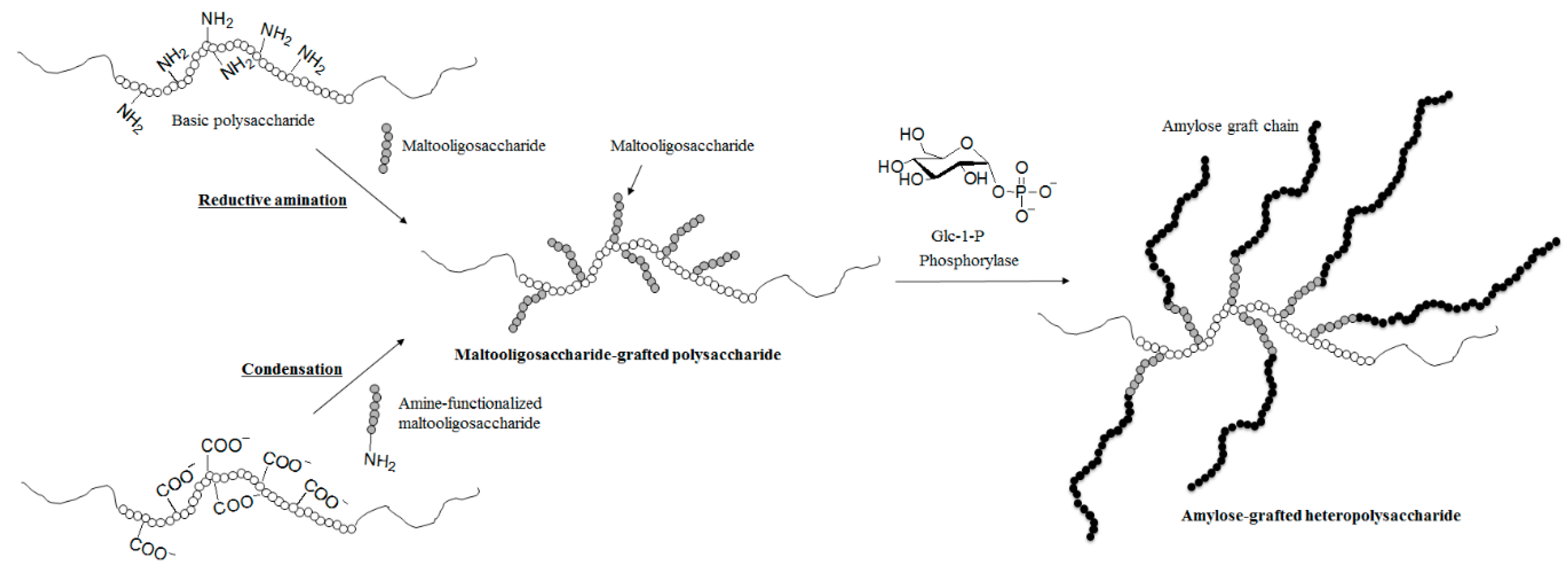{kind=link}
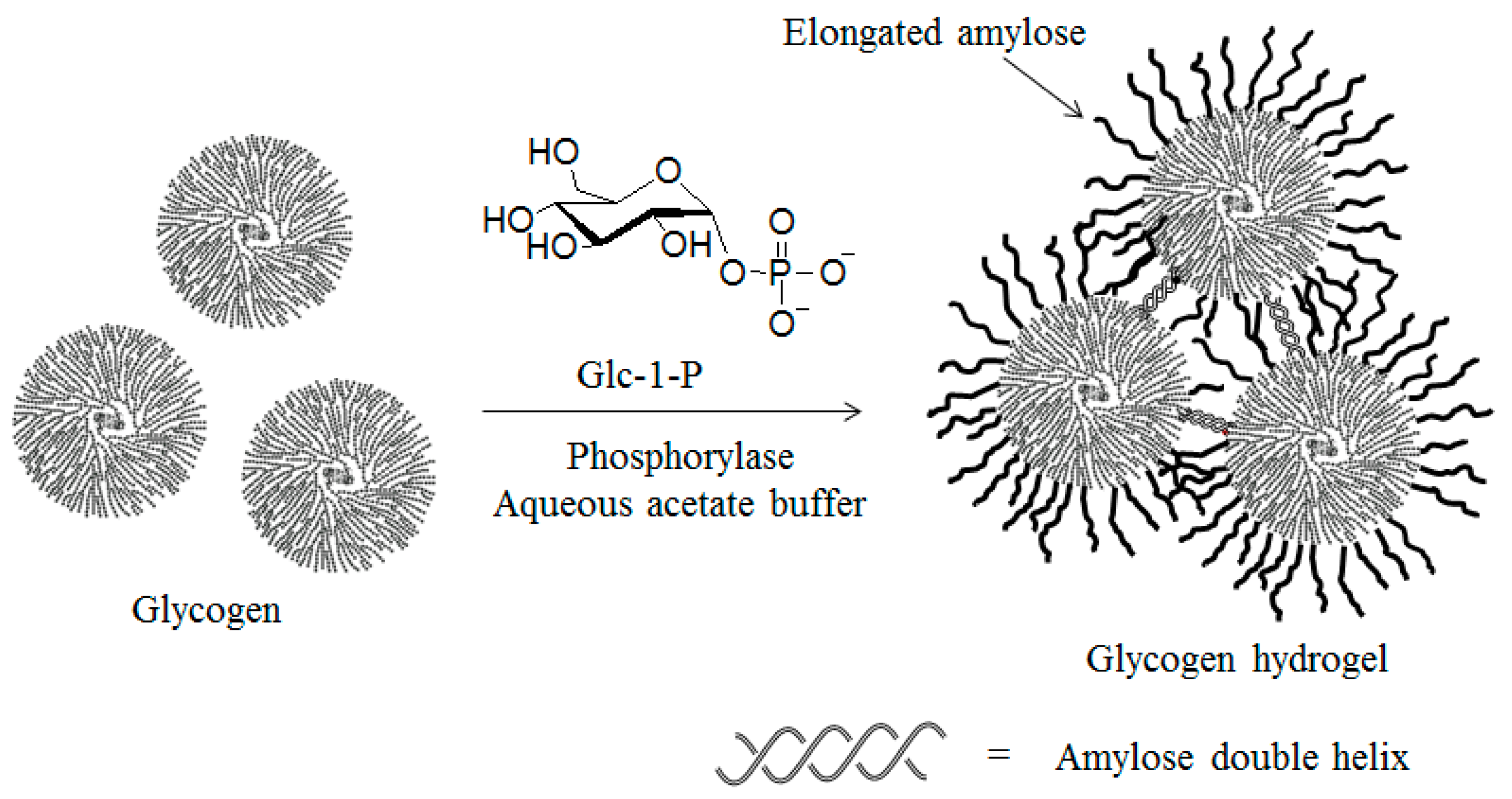{kind=link}
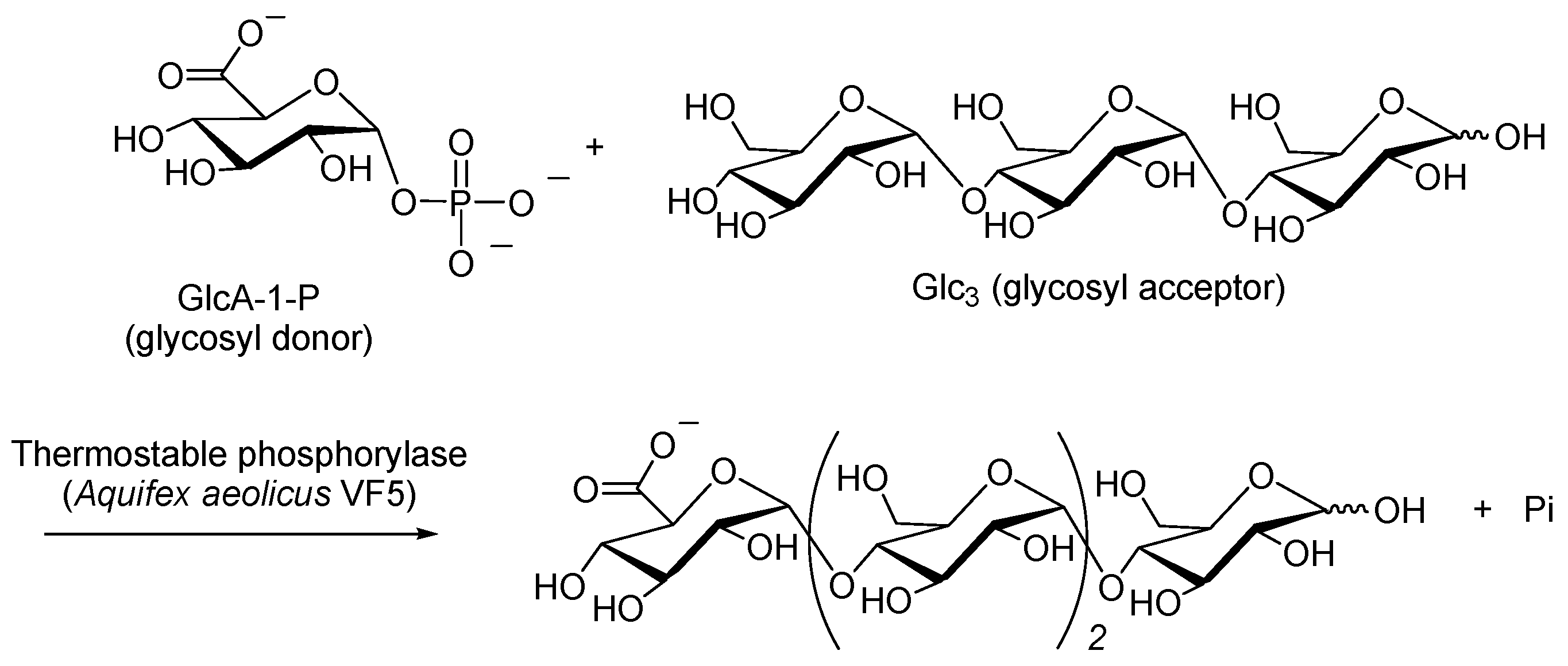{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
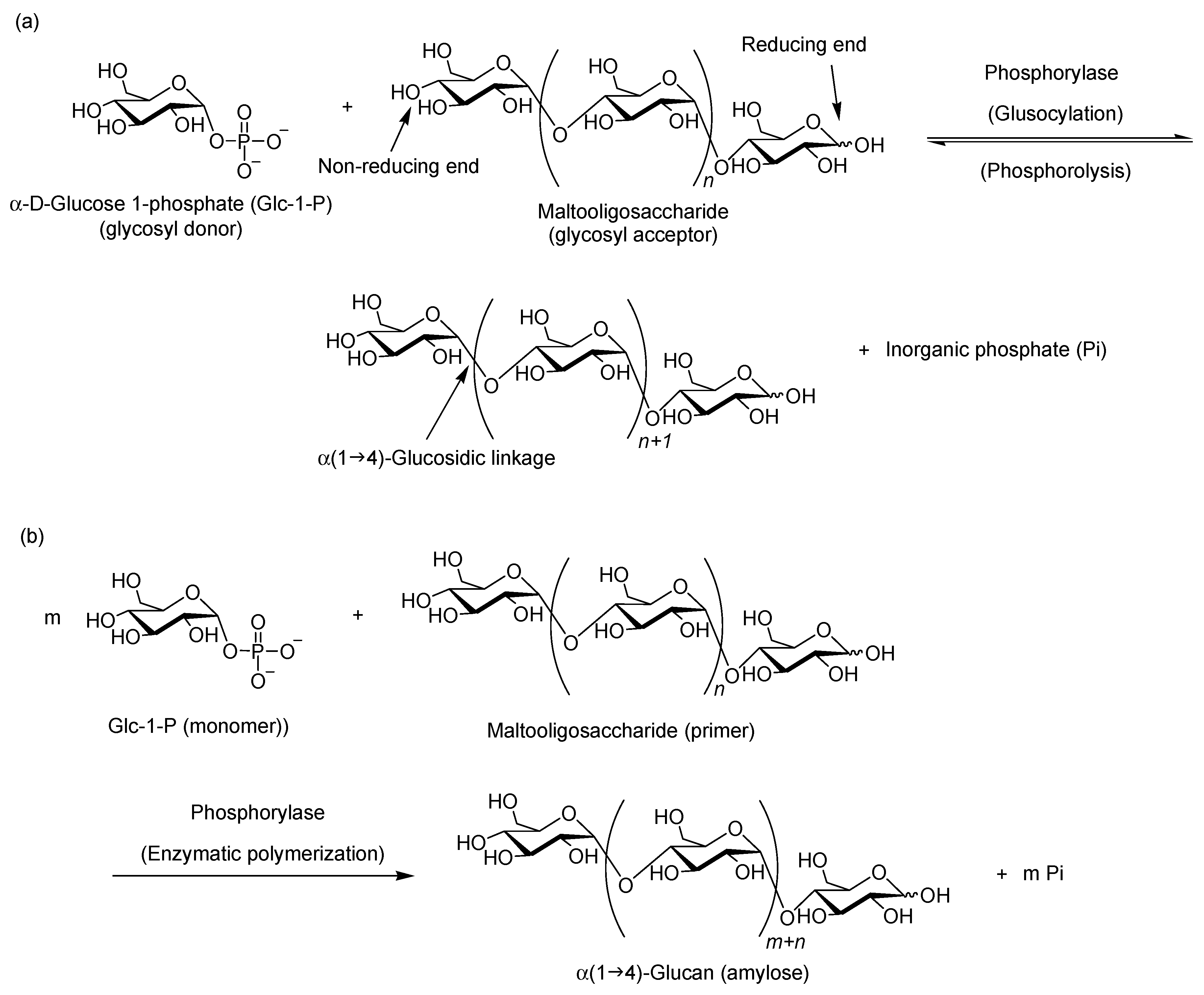
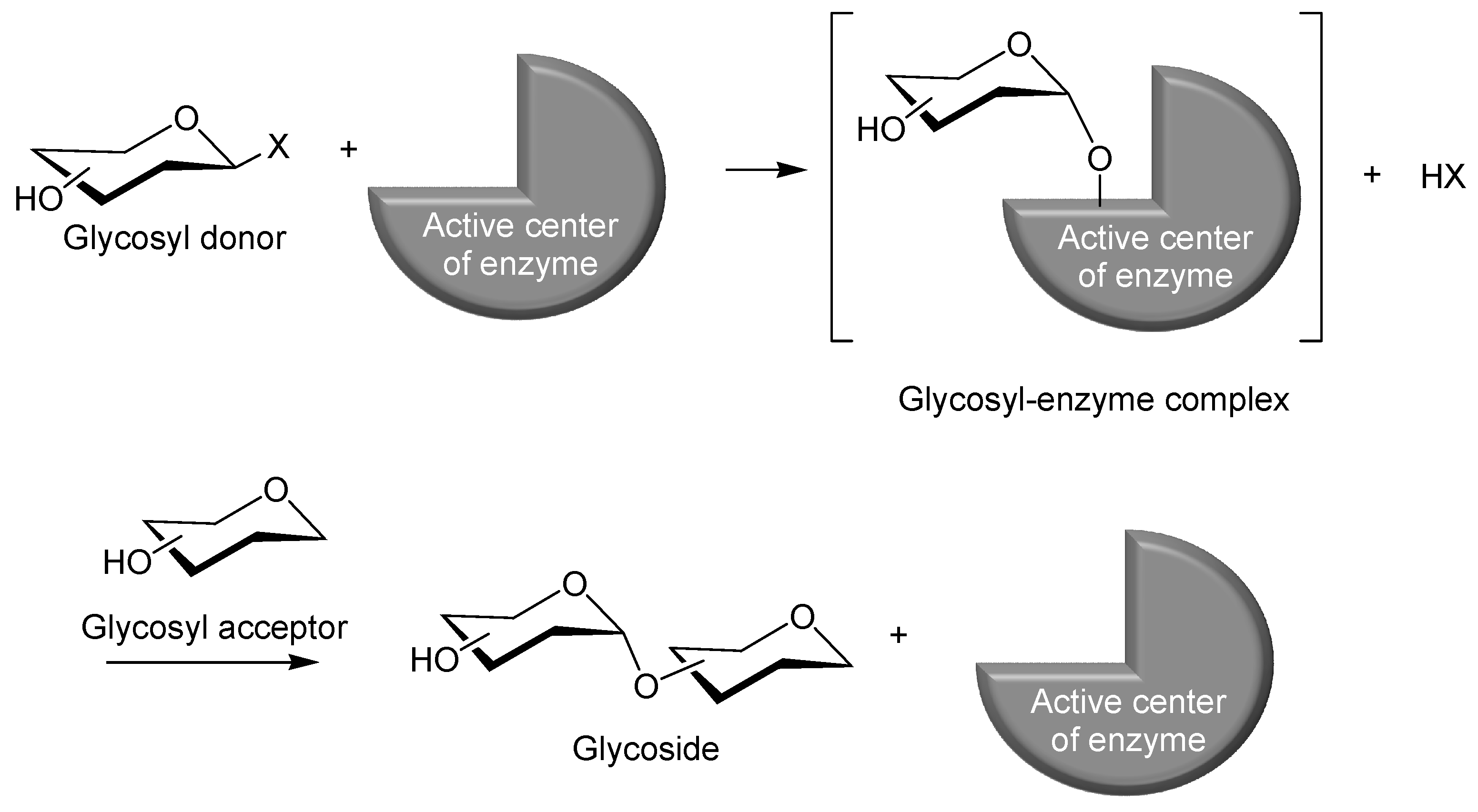
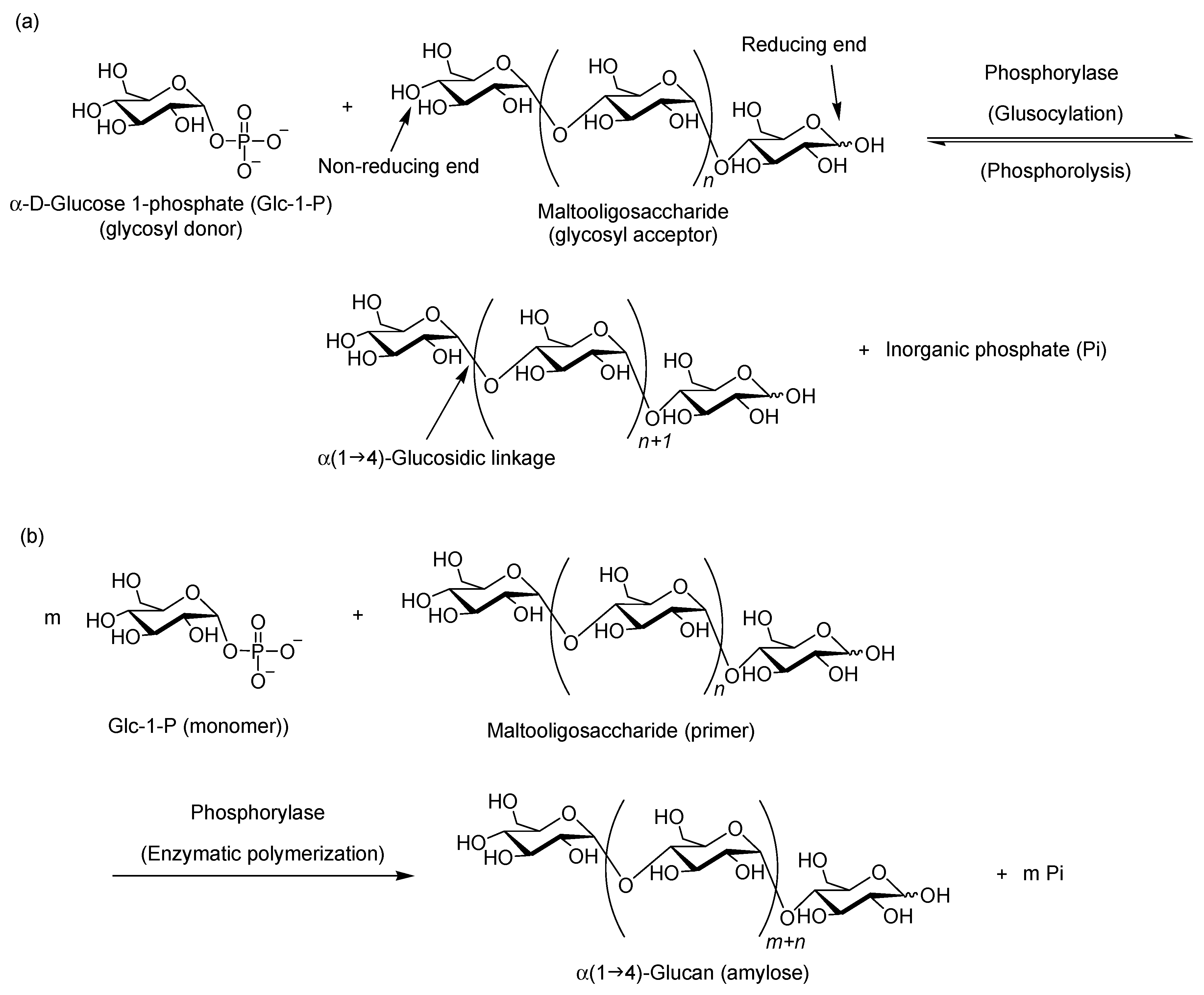
2. Characteristic Features of Phosphorylase Catalysis
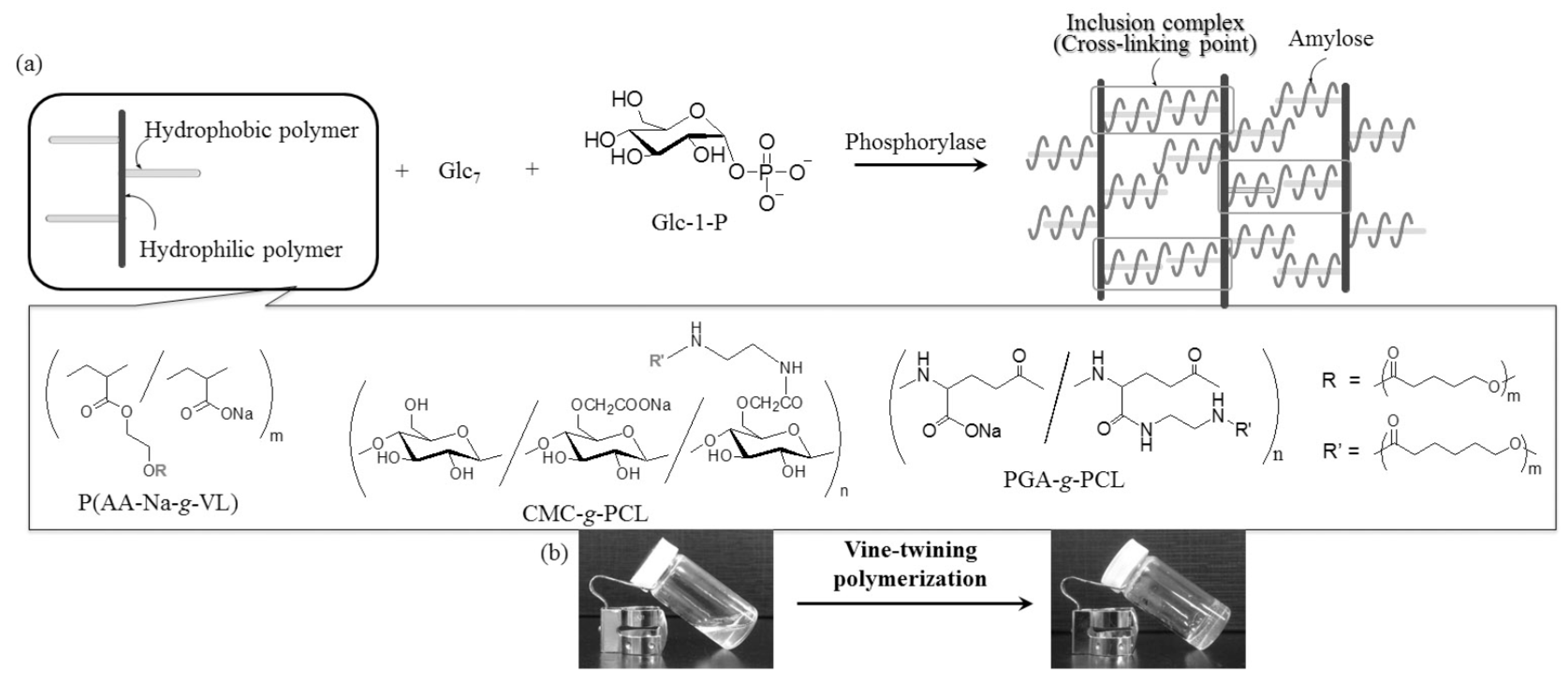
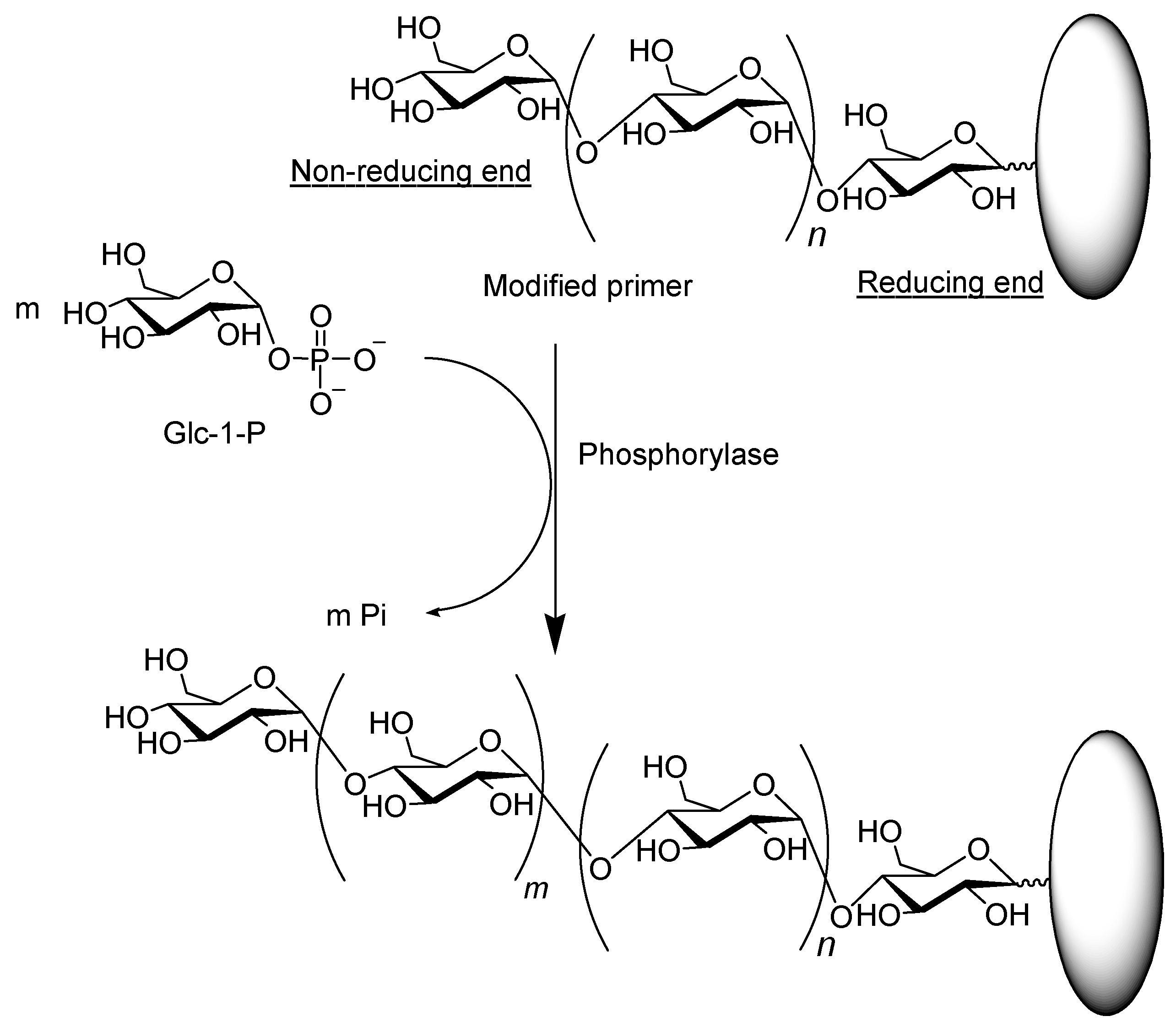
3. Synthesis of Amylose-Containing Functional Polysaccharide Materials by Phosphorylase-Catalyzed Enzymatic Polymerization
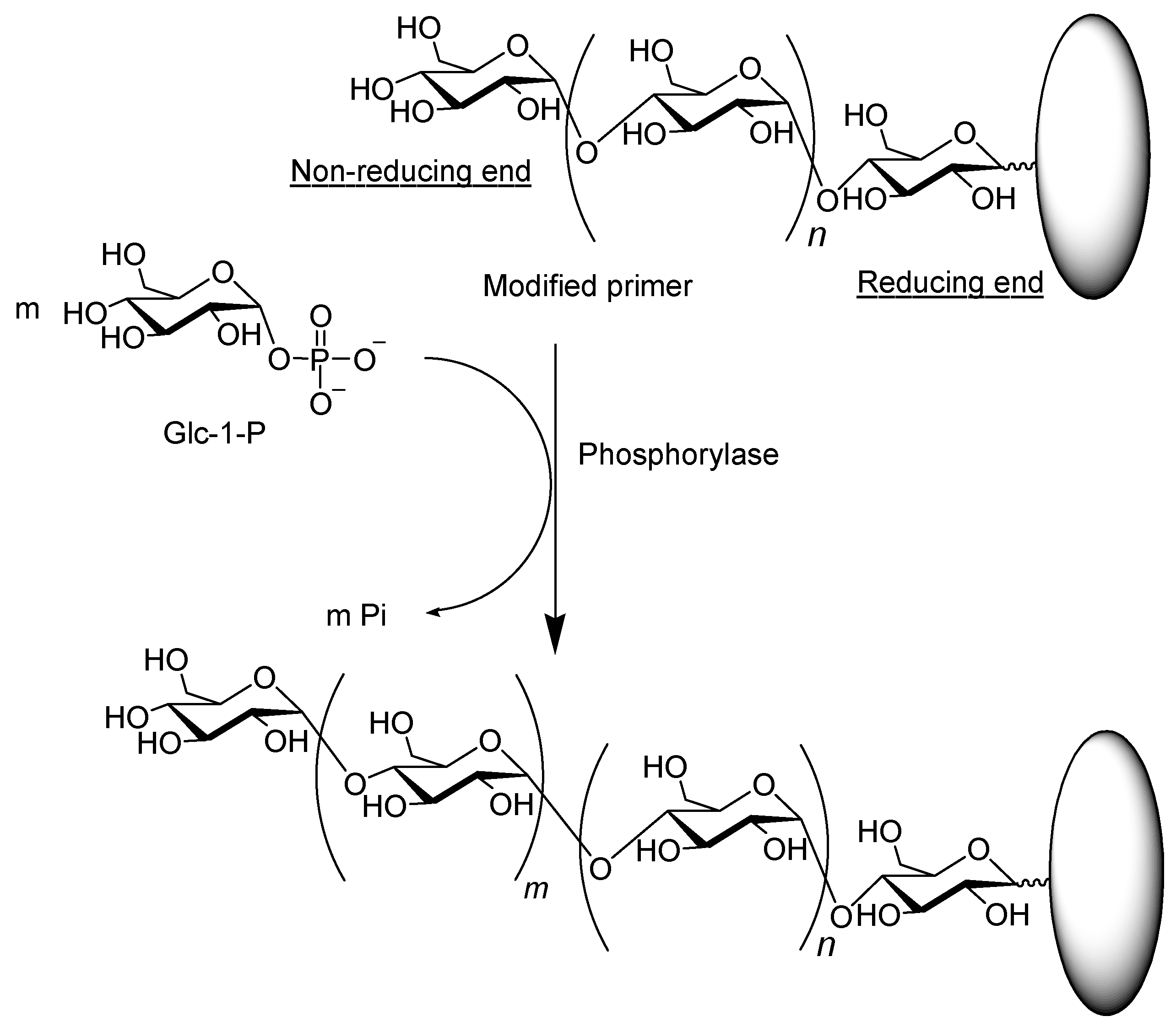
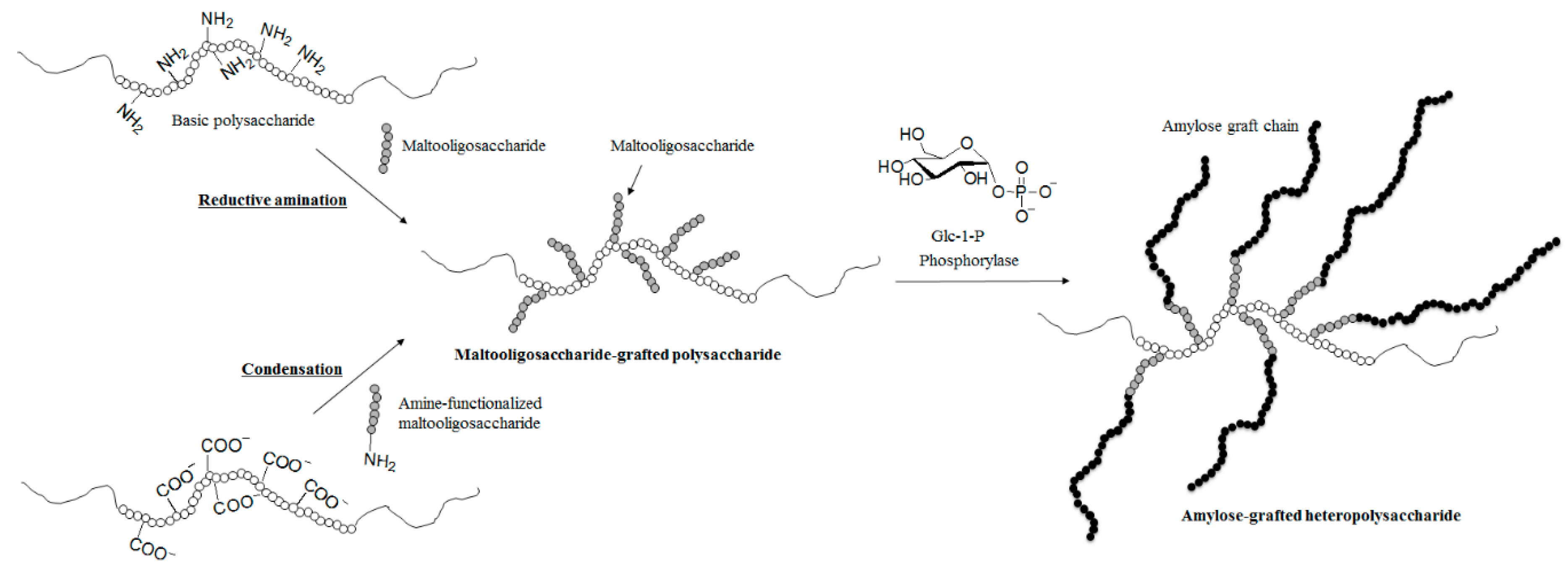
4. Synthesis of Non-Natural Functional Polysaccharide Materials by Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates
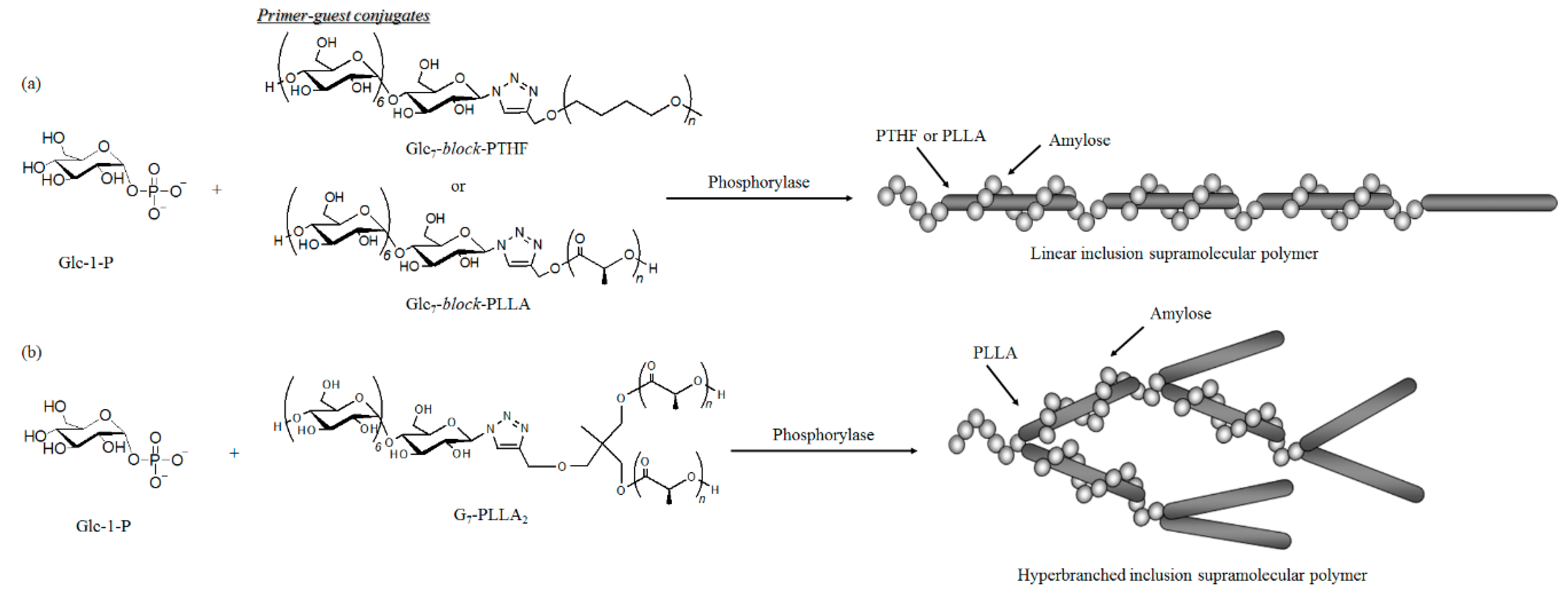
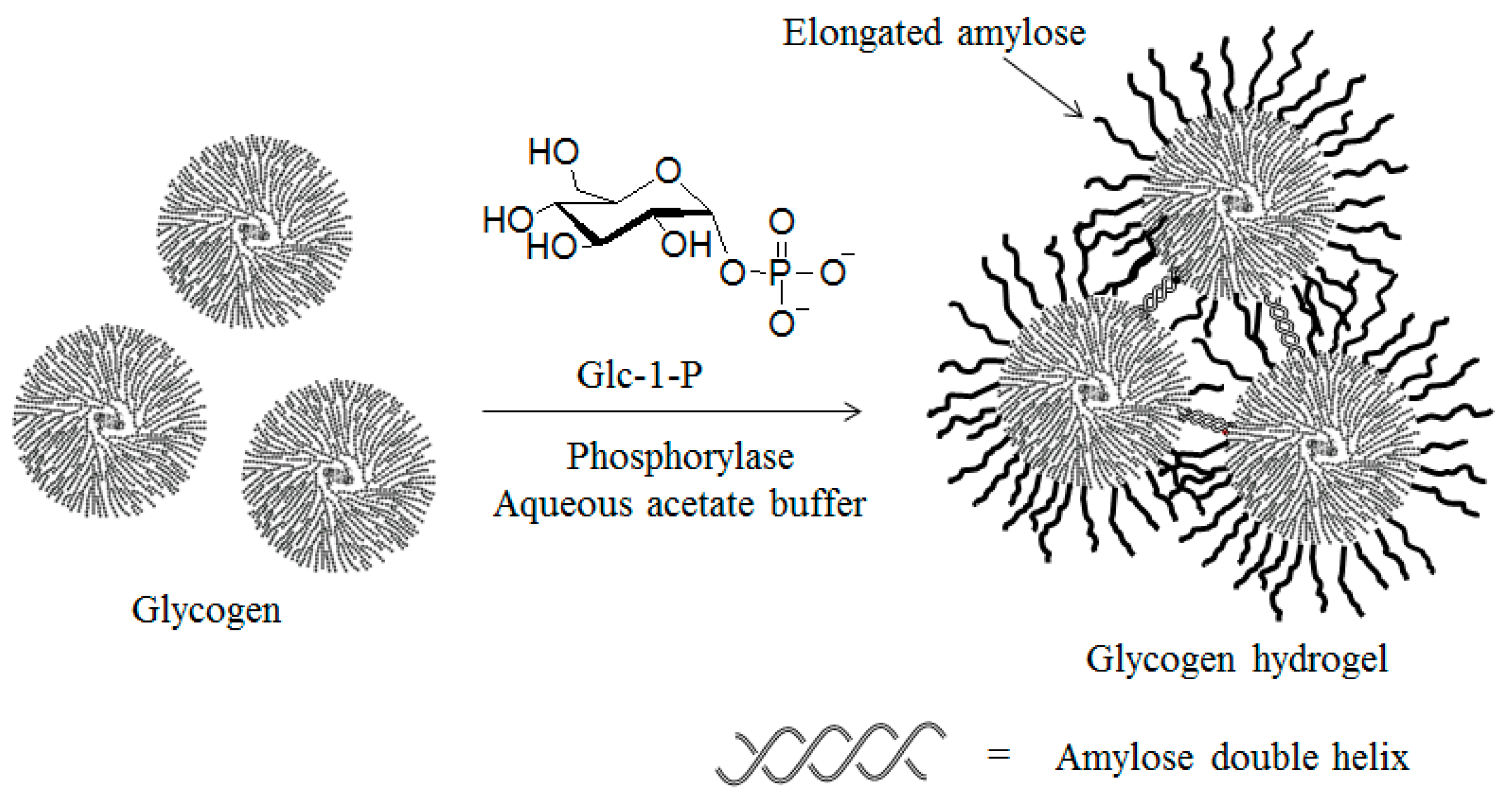
5. Preparation of Amylose Supramolecular Materials by Phosphorylase-Catalyzed Enzymatic Polymerization
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Schuerch, C. Polysaccharides. In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H.F., Bilkales, N., Overberger, C.G., Eds.; John Wiley & Sons: New York, NY, USA, 1986; Volume 13, pp. 87–162. [Google Scholar]
- Paulsen, H. Advances in selective chemical syntheses of complex oligosaccharides. Angew. Chem. Int. Ed. Engl. 1982, 21, 155–173. [Google Scholar] [CrossRef]
- Schmidt, R.R. New methods for the synthesis of glycosides and oligosaccharides—Are there alternatives to the Koenigs-Knorr method? Angew. Chem. Int. Ed. Engl. 1986, 25, 212–235. [Google Scholar] [CrossRef]
- Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem. Rev. 1993, 93, 1503–1531. [Google Scholar] [CrossRef]
- Yalpani, M. Polysaccharides: Syntheses, Modifications, and Structure/Property Relations; Elsevier: Amsterdam, The Neitherland/New York, NY, USA, 1988. [Google Scholar]
- Lenz, R.W. Biodegradable polymers. Adv. Polym. Sci. 1993, 107, 1–40. [Google Scholar]
- Klemm, D.; Heublein, B.; Fink, H.P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Shoda, S. Enzymatic glycosylation. In Glycoscience Chemistry and Chemical Biology; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany/New York, NY, USA, 2001; Volume II, pp. 1465–1496. [Google Scholar]
- Shoda, S.; Izumi, R.; Fujita, M. Green process in glycotechnology. Bull. Chem. Soc. Jpn. 2003, 76, 1–13. [Google Scholar] [CrossRef]
- Blixt, O.; Razi, N. Enzymatic glycosylation by transferases. In Glycoscience Chemistry and Chemical Biology, 2nd ed.; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2008; pp. 1361–1385. [Google Scholar]
- Thiem, J.; Thiem, J. Enzymatic glycosylation by glycohydrolases and glycosynthases. In Glycoscience Chemistry and Chemical Biology, 2nd ed.; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2008; pp. 1387–1409. [Google Scholar]
- Seibel, J.; Beine, R.; Moraru, R.; Behringer, C.; Buchholz, K. A new pathway for the synthesis of oligosaccharides by the use of non-Leloir glycosyltransferases. Biocatal. Biotransform. 2006, 24, 157–165. [Google Scholar] [CrossRef]
- Seibel, J.; Jordening, H.J.; Buchholz, K. Glycosylation with activated sugars using glycosyltransferases and transglycosidases. Biocatal. Biotransform. 2006, 24, 311–342. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J. Precision polysaccharide synthesis catalyzed by enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.; Kaneko, Y. Engineering of Polysaccharide Materials—By Phosphorylase-Catalyzed Enzymatic Chain-Elongation; Pan Stanford Publishing Pte Ltd.: Singapore, 2013. [Google Scholar]
- Kitaoka, M.; Hayashi, K. Carbohydrate-processing phosphorolytic enzymes. Trends Glycosci. Glycotechnol. 2002, 14, 35–50. [Google Scholar] [CrossRef]
- Nakai, H.; Kitaoka, M.; Svensson, B.; Ohtsubo, K. Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr. Opin. Chem. Biol. 2013, 17, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Puchart, V. Glycoside phosphorylases: Structure, catalytic properties and biotechnological potential. Biotechnol. Adv. 2015, 33, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Yanase, M.; Takaha, T.; Kuriki, T. α-Glucan phosphorylase and its use in carbohydrate engineering. J. Sci. Food Agric. 2006, 86, 1631–1635. [Google Scholar] [CrossRef]
- Boeck, B.; Schinzel, R. Purification and characterisation of an α-glucan phosphorylase from the thermophilic bacterium Thermus thermophilus. Eur. J. Biochem. 1996, 239, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Takaha, T.; Yanase, M.; Takata, H.; Okada, S. Structure and properties of Thermus aquaticus α-glucan phosphorylase expressed in Escherichichia coli. J. Appl. Glycosci. 2001, 48, 71–78. [Google Scholar] [CrossRef]
- Yanase, M.; Takata, H.; Fujii, K.; Takaha, T.; Kuriki, T. Cumulative effect of amino acid replacements results in enhanced thermostability of potato type L α-glucan phosphorylase. Appl. Environ. Microbiol. 2005, 71, 5433–5439. [Google Scholar] [CrossRef] [PubMed]
- Ziegast, G.; Pfannemüller, B. Linear and star-shaped hybrid polymers. 4. Phosphorolytic syntheses with di-functional, oligo-functional and multifunctional primers. Carbohydr. Res. 1987, 160, 185–204. [Google Scholar] [CrossRef]
- Fujii, K.; Takata, H.; Yanase, M.; Terada, Y.; Ohdan, K.; Takaha, T.; Okada, S.; Kuriki, T. Bioengineering and application of novel glucose polymers. Biocatal. Biotransform. 2003, 21, 167–172. [Google Scholar] [CrossRef]
- Ohdan, K.; Fujii, K.; Yanase, M.; Takaha, T.; Kuriki, T. Enzymatic synthesis of amylose. Biocatal. Biotransform. 2006, 24, 77–81. [Google Scholar] [CrossRef]
- Kitamura, S. Starch polymers, natural and synthetic. In The Polymeric Materials Encyclopedia, Synthesis, Properties and Applications; Salamone, C., Ed.; CRC Press: New York, NY, USA, 1996; Volume 10, pp. 7915–7922. [Google Scholar]
- Kitamura, S.; Yunokawa, H.; Mitsuie, S.; Kuge, T. Study on polysaccharide by the fluorescence method. 2. Micro-brownian motion and conformational change of amylose in aqueous-solution. Polym. J. 1982, 14, 93–99. [Google Scholar] [CrossRef]
- Kaneko, Y.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted polymers. In Handbook of Carbohydrate Polymers: Development, Properties and Applications; Ito, R., Matsuo, Y., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2009; pp. 671–691. [Google Scholar]
- Izawa, H.; Kadokawa, J. Preparation of functional amylosic materials by phosphorylase-catalyzed polymerization. In Interfacial Researches in Fundamental and Material Sciences of Oligo- and Polysaccharides; Kadoakwa, J., Ed.; Transworld Research Network: Trivandrum, India, 2009; pp. 69–86. [Google Scholar]
- Omagari, Y.; Kadokawa, J. Synthesis of heteropolysaccharides having amylose chains using phosphorylase-catalyzed enzymatic polymerization. Kobunshi Ronbunshu 2011, 68, 242–249. [Google Scholar] [CrossRef]
- Kadoakwa, J. Synthesis of amylose-grafted polysaccharide materials by phosphorylase-catalyzed enzymatic polymerization. In Biobased Monomers, Polymers, and Materials; Smith, P.B., Gross, R.A., Eds.; ACS Symposium Series 1105; American Chemical Society: Washington, DC, USA, 2012; pp. 237–255. [Google Scholar]
- Kadoakwa, J. Synthesis of new polysaccharide materials by phosphorylase-catalyzed enzymatic α-glycosylations using polymeric glycosyl acceptors. In Green Polymer Chemistry: Biocatalysis and Materials II; Cheng, H.N., Gross, R.A., Smith, P.B., Eds.; ACS Symposium Series 1144; American Chemical Society: Washington, DC, USA, 2013; pp. 141–161. [Google Scholar]
- Kadokawa, J. Chemoenzymatic synthesis of functional amylosic materials. Pure Appl. Chem. 2014, 86, 701–709. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kamiya, S.; Enomoto, N. Amylose-carrying styrene macromonomer and its homo- and copolymers: Synthesis via enzyme-catalyzed polymerization and complex formation with iodine. Macromolecules 1996, 29, 8670–8676. [Google Scholar] [CrossRef]
- Narumi, A.; Kawasaki, K.; Kaga, H.; Satoh, T.; Sugimoto, N.; Kakuchi, T. Glycoconjugated polymer 6. Synthesis of poly[styrene-block-(styrene-graft-amylose)] via potato phosphorylase-catalyzed polymerization. Polym. Bull. 2003, 49, 405–410. [Google Scholar] [CrossRef]
- Von Braunmühl, V.; Jonas, G.; Stadler, R. Enzymatic grafting of amylose from poly(dimethylsiloxanes). Macromolecules 1995, 28, 17–24. [Google Scholar] [CrossRef]
- Kadokawa, J.; Nakamura, Y.; Sasaki, Y.; Kaneko, Y.; Nishikawa, T. Chemoenzymatic synthesis of amylose-grafted polyacetylenes. Polym. Bull. 2008, 60, 57–68. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kaneko, Y.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted polyacetylene by polymer reaction manner and its conversion into organogel with DMSO by cross-linking. Polym. Bull. 2009, 62, 291–303. [Google Scholar] [CrossRef]
- Kaneko, Y.; Matsuda, S.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted poly(vinyl alcohol). Polym. Chem. 2010, 1, 193–197. [Google Scholar] [CrossRef]
- Kamiya, S.; Kobayashi, K. Synthesis and helix formation of saccharide–poly(l-glutamic acid) conjugates. Macromol. Chem. Phys. 1998, 199, 1589–1596. [Google Scholar] [CrossRef]
- Mazzocchetti, L.; Tsoufis, T.; Rudolf, P.; Loos, K. Enzymatic synthesis of amylose brushes revisited: Details from X-ray photoelectron spectroscopy and spectroscopic ellipsometry. Macromol. Biosci. 2014, 14, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J. Facile synthesis of unnatural oligosaccharides by phosphorylase-catalyzed enzymatic glycosylations using new glycosyl donors. In Oligosaccharides: Sources, Properties and Applications; Gordon, N.S., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2011; pp. 269–281. [Google Scholar]
- Kadokawa, J. Synthesis of non-natural oligosaccharides by α-glucan phosphorylase-catalyzed enzymatic glycosylations using analogue substrates of α-d-glucose 1-phosphate. Trends Glycosci. Glycotechnol. 2013, 25, 57–69. [Google Scholar] [CrossRef]
- Percival, M.D.; Withers, S.G. Applications of enzymes in the synthesis and hydrolytic study of 2-deoxy-α-d-glucopyranosyl phosphate. Can. J. Chem. 1988, 66, 1970–1972. [Google Scholar] [CrossRef]
- Evers, B.; Mischnick, P.; Thiem, J. Synthesis of 2-deoxy-α-d-arabino-hexopyranosyl phosphate and 2-deoxy-maltooligosaccharides with phosphorylase. Carbohydr. Res. 1994, 262, 335–341. [Google Scholar] [CrossRef]
- Evers, B.; Thiem, J. Further syntheses employing phosphorylase. Bioorg. Med. Chem. 1997, 5, 857–863. [Google Scholar] [CrossRef]
- Nawaji, M.; Izawa, H.; Kaneko, Y.; Kadokawa, J. Enzymatic synthesis of α-d-xylosylated maltooligosaccharides by phosphorylase-catalyzed xylosylation. J. Carbohydr. Chem. 2008, 27, 214–222. [Google Scholar] [CrossRef]
- Nawaji, M.; Izawa, H.; Kaneko, Y.; Kadokawa, J. Enzymatic α-glucosaminylation of maltooligosaccharides catalyzed by phosphorylase. Carbohydr. Res. 2008, 343, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, S.; Izawa, H.; Nawaji, M.; Kaneko, Y.; Kadokawa, J. Phosphorylase-catalyzed N-formyl-α-glucosaminylation of maltooligosaccharides. Carbohydr. Res. 2010, 345, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.M.; Phillips, G.O.; Williams, P.A. Food Polysaccharides and Their Applications, 2nd ed.; CRC/Taylor & Francis: Boca Raton, FL, USA, 2006; p. 733. [Google Scholar]
- Matsuda, S.; Kaneko, Y.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted chitosan. Macromol. Rapid Comm. 2007, 28, 863–867. [Google Scholar] [CrossRef]
- Kaneko, Y.; Matsuda, S.; Kadokawa, J. Chemoenzymatic syntheses of amylose-grafted chitin and chitosan. Biomacromolecules 2007, 8, 3959–3964. [Google Scholar] [CrossRef] [PubMed]
- Omagari, Y.; Matsuda, S.; Kaneko, Y.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted cellulose. Macromol. Biosci. 2009, 9, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Omagari, Y.; Kaneko, Y.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted alginate and its formation of enzymatic disintegratable beads. Carbohydr. Polym. 2010, 82, 394–400. [Google Scholar] [CrossRef]
- Arimura, T.; Omagari, Y.; Yamamoto, K.; Kadokawa, J. Chemoenzymatic synthesis and hydrogelation of amylose-grafted xanthan gums. Int. J. Biol. Macromol. 2011, 49, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.; Arimura, T.; Takemoto, Y.; Yamamoto, K. Self-assembly of amylose-grafted carboxymethyl cellulose. Carbohydr. Polym. 2012, 90, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, D.; Takemoto, Y.; Yamamoto, K.; Kadokawa, J. Hierarchically self-assembled nanofiber films from amylose-grafted carboxymethyl cellulose. Fibers 2013, 2, 34–44. [Google Scholar] [CrossRef]
- Calder, P.C. Glycogen structure and biogenesis. Int. J. Biochem. 1991, 23, 1335–1352. [Google Scholar] [CrossRef]
- Manners, D.J. Recent developments in our understanding of glycogen structure. Carbohydr. Polym. 1991, 16, 37–82. [Google Scholar] [CrossRef]
- Izawa, H.; Nawaji, M.; Kaneko, Y.; Kadokawa, J. Preparation of glycogen-based polysaccharide materials by phosphorylase-catalyzed chain elongation of glycogen. Macromol. Biosci. 2009, 9, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, W.; Buttner, G.; Steifa, M.; Betzel, C.; Zabel, V.; Pfannemuller, B.; Saenger, W. An amylose antiparallel double helix at atomic resolution. Science 1987, 238, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, F.; Schulz, W. Monte-carlo simulation of the hydration shell of double-helical amylose—A left-handed antiparallel double helix fits best into liquid water-structure. Biopolymers 1992, 32, 1643–1664. [Google Scholar] [CrossRef]
- Morimoto, N.; Ogino, N.; Narita, T.; Kitamura, S.; Akiyoshi, K. Enzyme-responsive molecular assembly system with amylose-primer surfactants. J. Am. Chem. Soc. 2007, 129, 458–459. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, N.; Ogino, N.; Narita, T.; Akiyoshi, K. Enzyme-responsive artificial chaperone system with amphiphilic amylose primer. J. Biotechnol. 2009, 140, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, N.; Yamazaki, M.; Tamada, J.; Akiyoshi, K. Polysaccharide-hair cationic polypeptide nanogels: Self-assembly and enzymatic polymerization of amylose primer-modified cholesteryl poly(l-lysine). Langmuir 2013, 29, 7509–7514. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Mukai, S.; Sawada, S.; Akiyoshi, K. Glyco star polymers as helical multivalent host and biofunctional nano-platform. Acs Macro Lett. 2015, 4, 367–371. [Google Scholar] [CrossRef]
- Bhuiyan, S.H.; Rus’d, A.A.; Kitaoka, M.; Hayashi, K. Characterization of a hyperthermostable glycogen phosphorylase from Aquifex aeolicus expressed in Escherichia coli. J. Mol. Catal. B Enzym. 2003, 22, 173–180. [Google Scholar] [CrossRef]
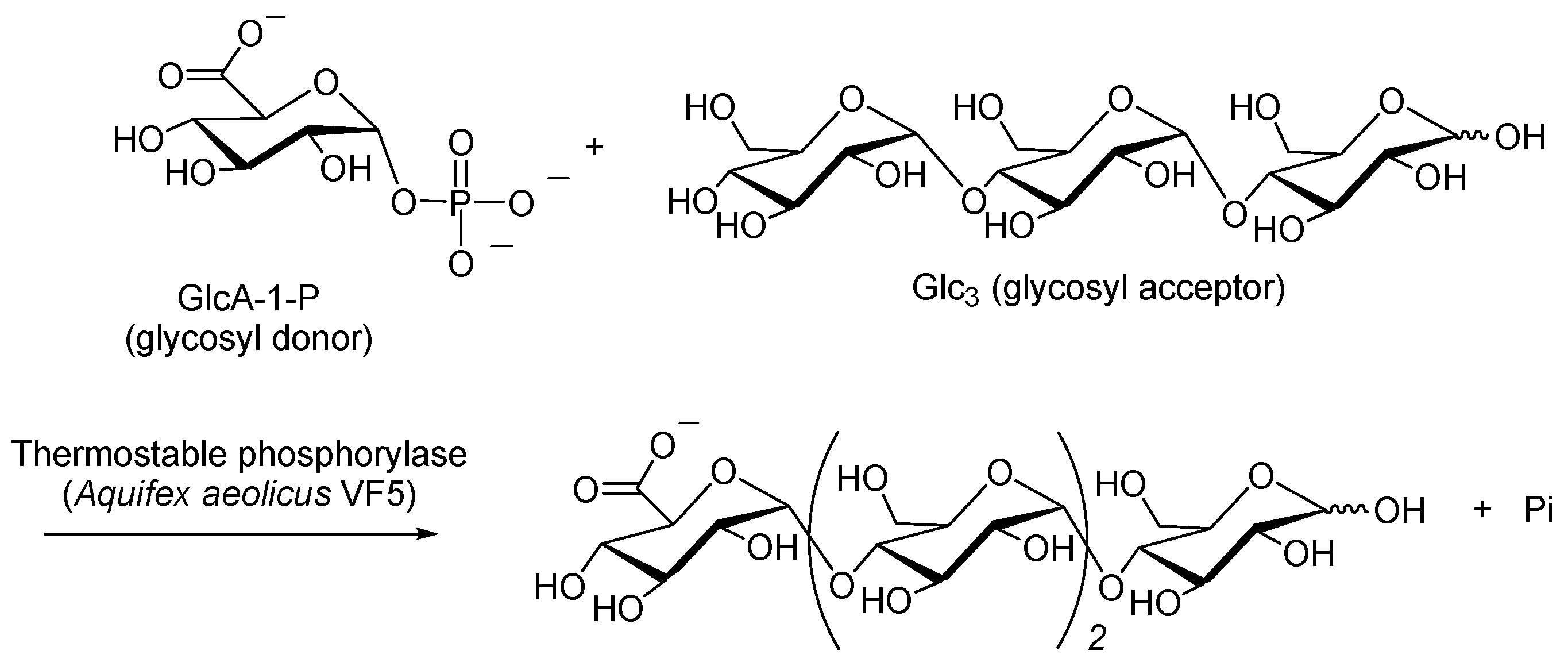
- Umegatani, Y.; Izawa, H.; Nawaji, M.; Yamamoto, K.; Kubo, A.; Yanase, M.; Takaha, T.; Kadokawa, J. Enzymatic α-glucuronylation of maltooligosaccharides using α-glucuronic acid 1-phosphate as glycosyl donor catalyzed by a thermostable phosphorylase from Aquifex aeolicus vf5. Carbohydr. Res. 2012, 350, 81–85. [Google Scholar] [PubMed]
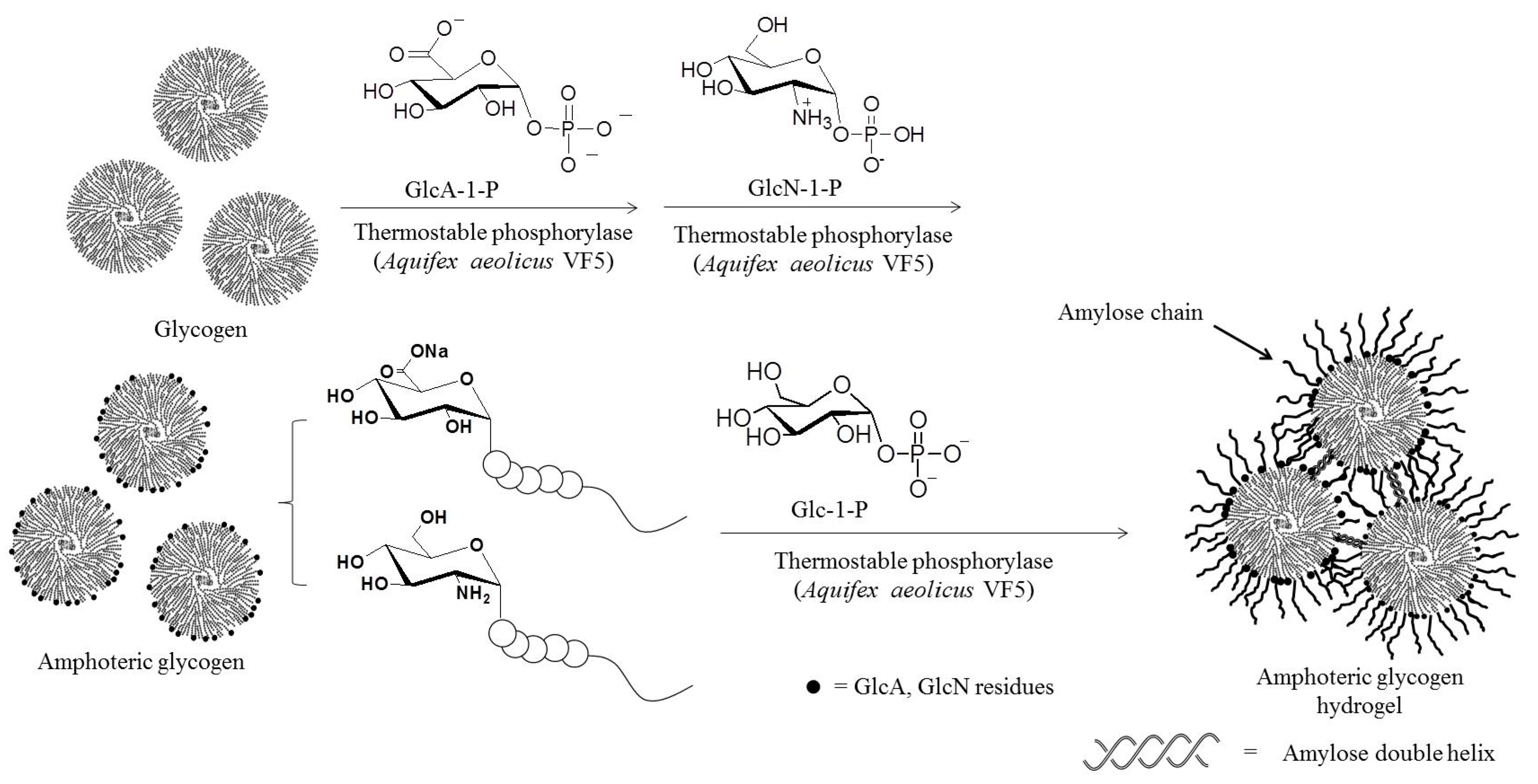
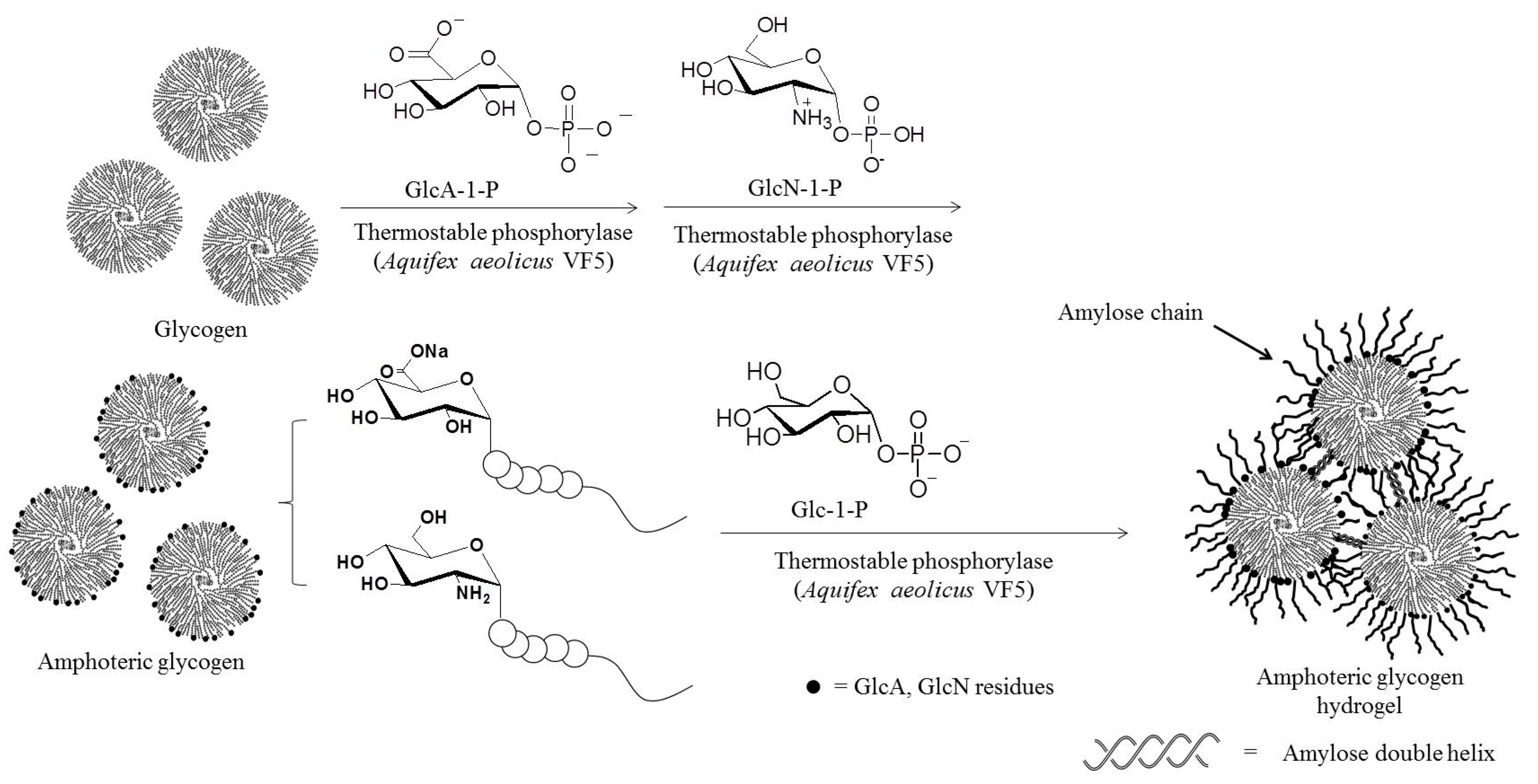
- Takata, Y.; Shimohigoshi, R.; Yamamoto, K.; Kadokawa, J. Enzymatic synthesis of dendritic amphoteric α-glucans by thermostable phosphorylase catalysis. Macromol. Biosci. 2014, 14, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Yamamoto, K.; Kadokawa, J. Preparation of pH-responsive amphoteric glycogen hydrogels by α-glucan phosphorylase-catalyzed successive enzymatic reactions. Macromol. Chem. Phys. 2015, 216, 1415–1420. [Google Scholar] [CrossRef]
- Takemoto, Y.; Izawa, H.; Umegatani, Y.; Yamamoto, K.; Kubo, A.; Yanase, M.; Takaha, T.; Kadokawa, J. Synthesis of highly branched anionic α-glucans by thermostable phosphorylase-catalyzed α-glucuronylation. Carbohydr. Res. 2013, 366, 38–44. [Google Scholar] [CrossRef] [PubMed]
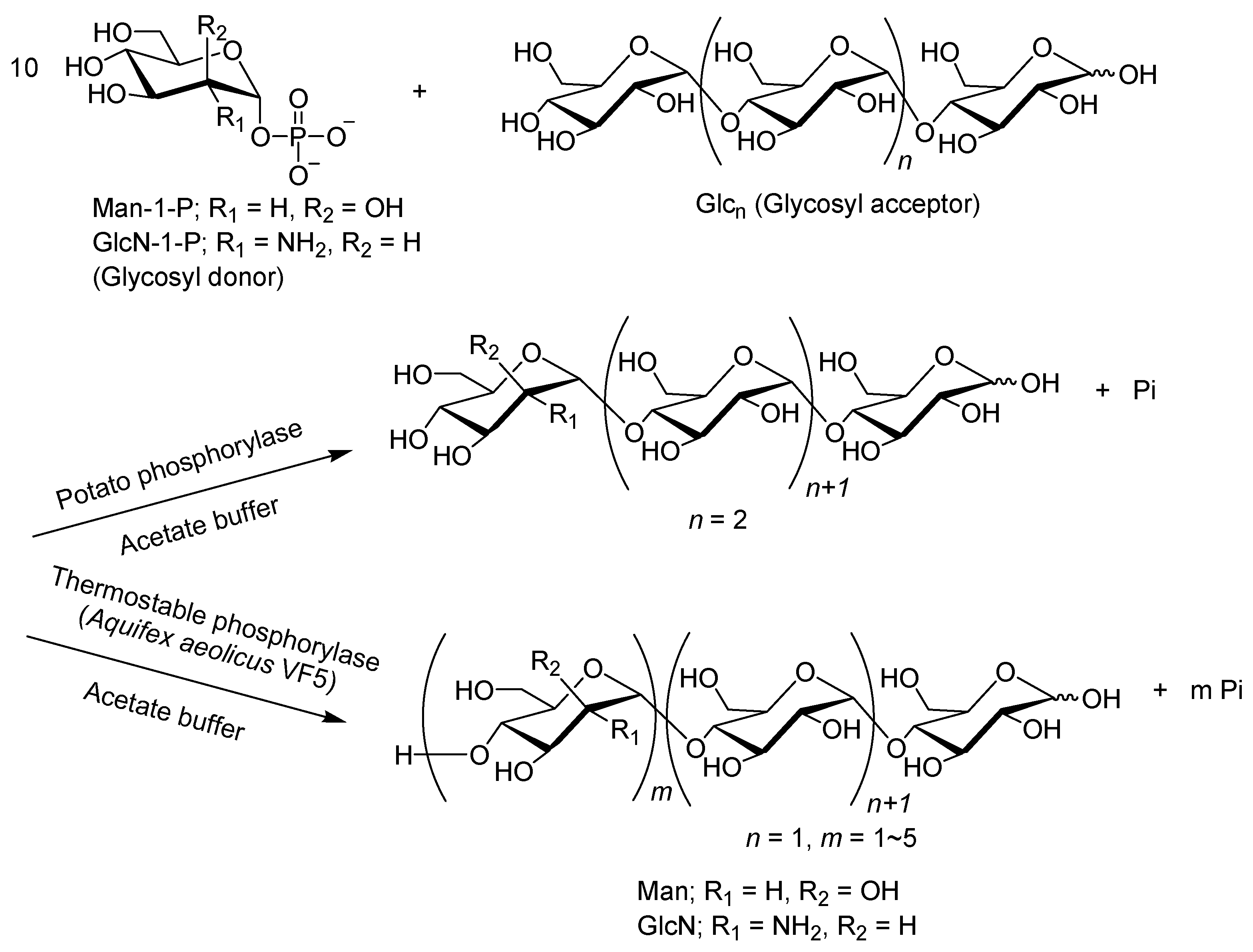
- Shimohigoshi, R.; Takemoto, Y.; Yamamoto, K.; Kadokawa, J. Thermostable α-glucan phosphorylase-catalyzed successive α-mannosylations. Chem. Lett. 2013, 42, 822–824. [Google Scholar] [CrossRef]
- Borgerding, J. Phosphate deposits in digestion systems. J. Water Pollut. Control Fed. 1972, 44, 813–819. [Google Scholar]
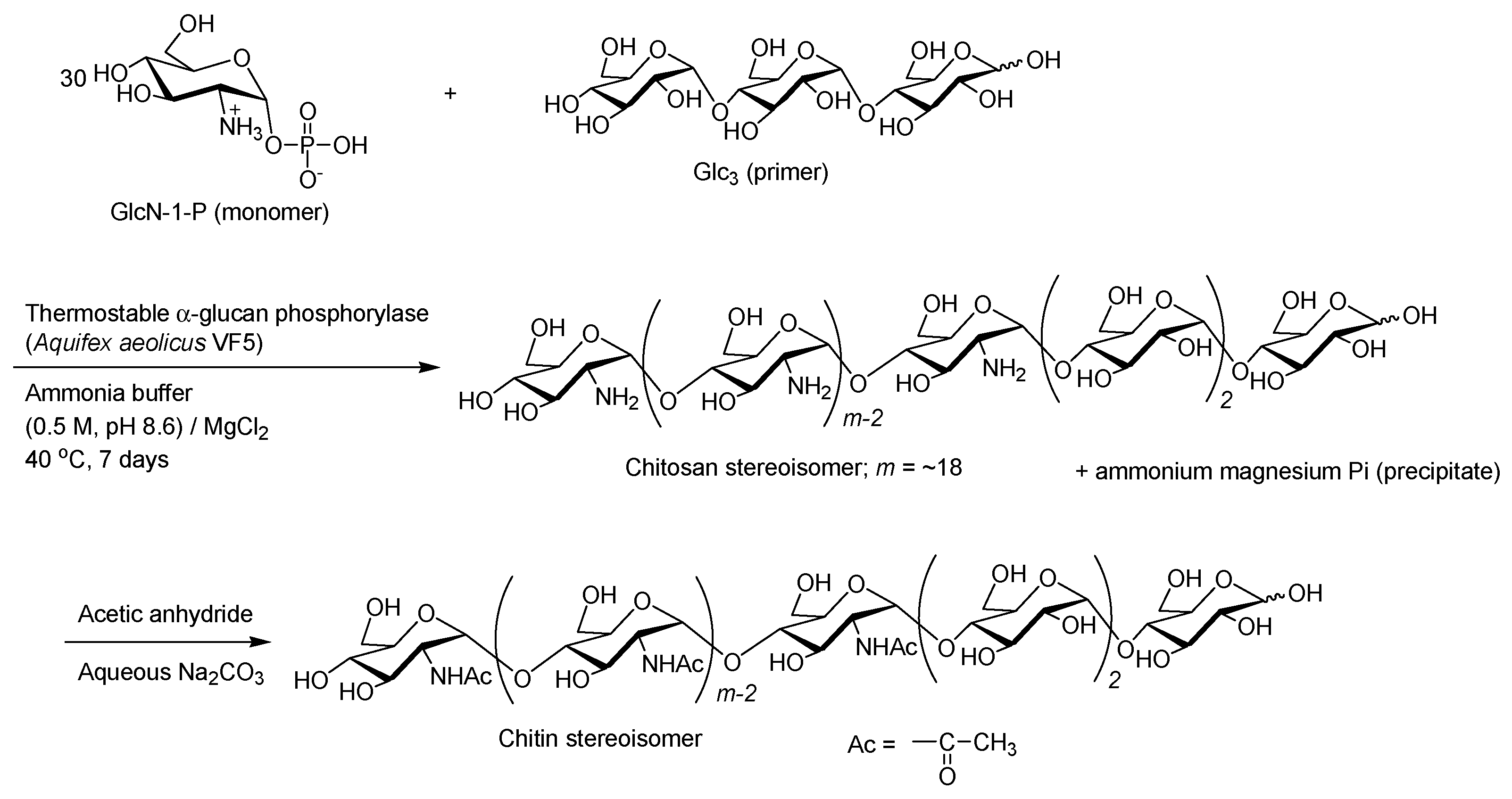
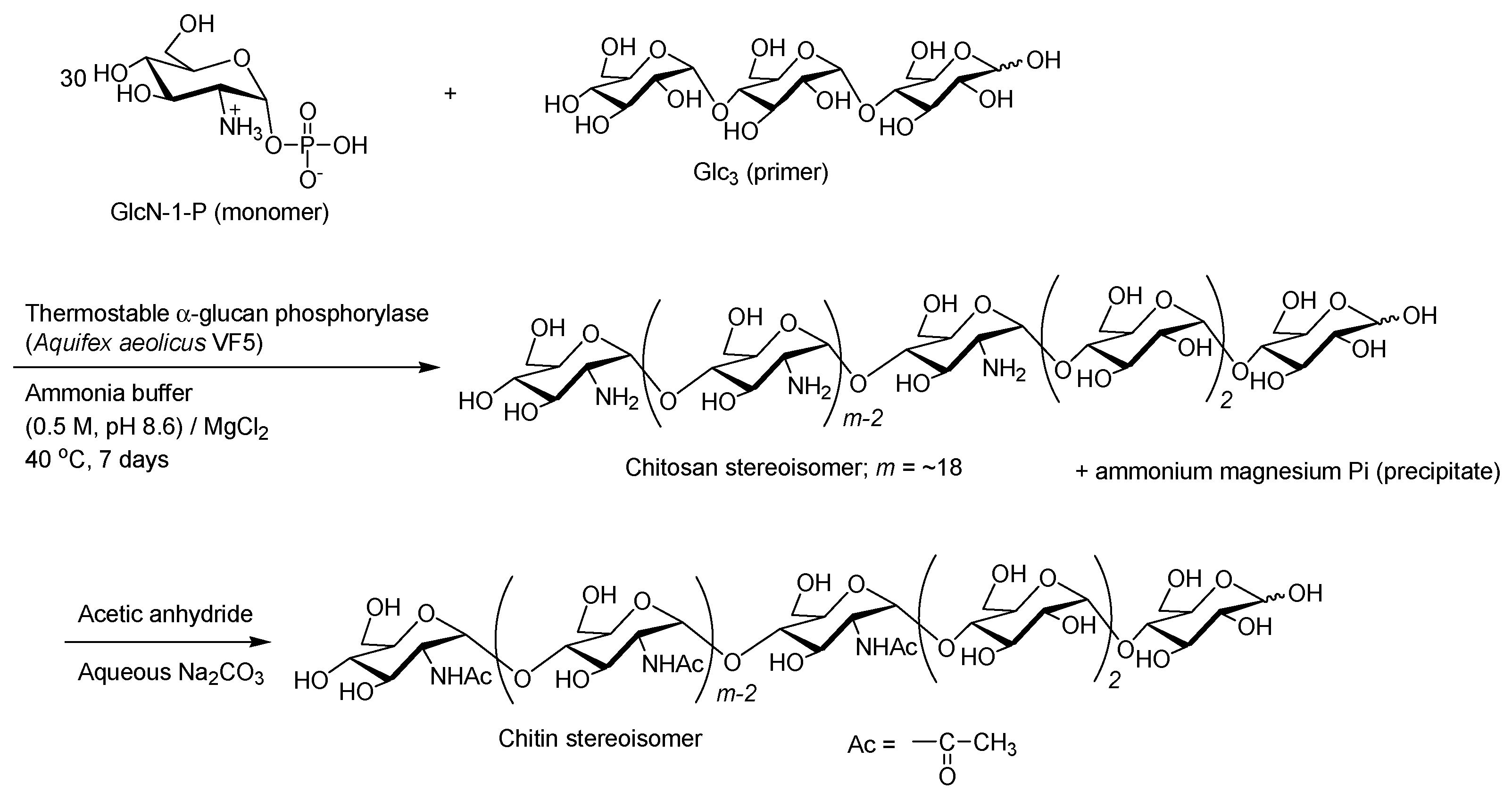
- Kadokawa, J.; Shimohigoshi, R.; Yamashita, K.; Yamamoto, K. Synthesis of chitin and chitosan stereoisomers by thermostable α-glucan phosphorylase-catalyzed enzymatic polymerization of α-d-glucosamine 1-phosphate. Org. Bimol. Chem. 2015, 13, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Yamamoto, K.; Kadoakwa, J. Synthesis of non-natural heteroaminopolysaccharides by α-glucan phosphorylase-catalyzed enzymatic copolymerization: α(1–>4)-linked glucosaminoglucans. Biomacromolecules 2015, 16, 3989–3994. [Google Scholar] [CrossRef] [PubMed]
- Sarko, A.; Zugenmaier, P. Crystal structures of amylose and its derivatives. In Fiber Diffraction Methods; French, A.D., Gardner, K.H., Eds.; ACS Symposium Series 141; American Chemical Society: Washington, DC, USA, 1980; pp. 459–482. [Google Scholar]
- Shogren, R.L.; Greene, R.V.; Wu, Y.V. Complexes of starch polysaccharides and poly(ethylene-co-acrylic acid): Structure and stability in solution. J. Appl. Polym. Sci. 1991, 42, 1701–1709. [Google Scholar] [CrossRef]
- Shogren, R.L. Complexes of starch with telechelic poly(ε-caprolactone) phosphate. Carbohydr. Polym. 1993, 22, 93–98. [Google Scholar] [CrossRef]
- Star, A.; Steuerman, D.W.; Heath, J.R.; Stoddart, J.F. Starched carbon nanotubes. Angew. Chem. Int. Ed. 2002, 41, 2508–2512. [Google Scholar] [CrossRef]
- Ikeda, M.; Furusho, Y.; Okoshi, K.; Tanahara, S.; Maeda, K.; Nishino, S.; Mori, T.; Yashima, E. A luminescent poly(phenylenevinylene)-amylose composite with supramolecular liquid crystallinity. Angew. Chem. Int. Ed. 2006, 45, 6491–6495. [Google Scholar] [CrossRef] [PubMed]
- Kida, T.; Minabe, T.; Okabe, S.; Akashi, M. Partially-methylated amyloses as effective hosts for inclusion complex formation with polymeric guests. Chem. Commun. 2007, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kyutoku, T.; Shimomura, N.; Kadokawa, J. Formation of amylose-poly(tetrahydrofuran) inclusion complexes in ionic liquid media. Chem. Lett. 2011, 40, 31–33. [Google Scholar] [CrossRef]
- Rachmawati, R.; Woortman, A.J.J.; Loos, K. Facile preparation method for inclusion complexes between amylose and polytetrahydrofurans. Biomacromolecules 2013, 14, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Woortman, A.J.J.; Loos, K. Synthesis of amylose-polystyrene inclusion complexes by a facile preparation route. Biomacromolecules 2013, 14, 1955–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmawati, R.; Woortman, A.J.J.; Loos, K. Tunable properties of inclusion complexes between amylose and polytetrahydrofuran. Macromol. Biosci. 2013, 13, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmawati, R.; Woortman, A.J.J.; Loos, K. Solvent-responsive behavior of inclusion complexes between amylose and polytetrahydrofuran. Macromol. Biosci. 2014, 14, 56–68. [Google Scholar] [CrossRef] [PubMed]
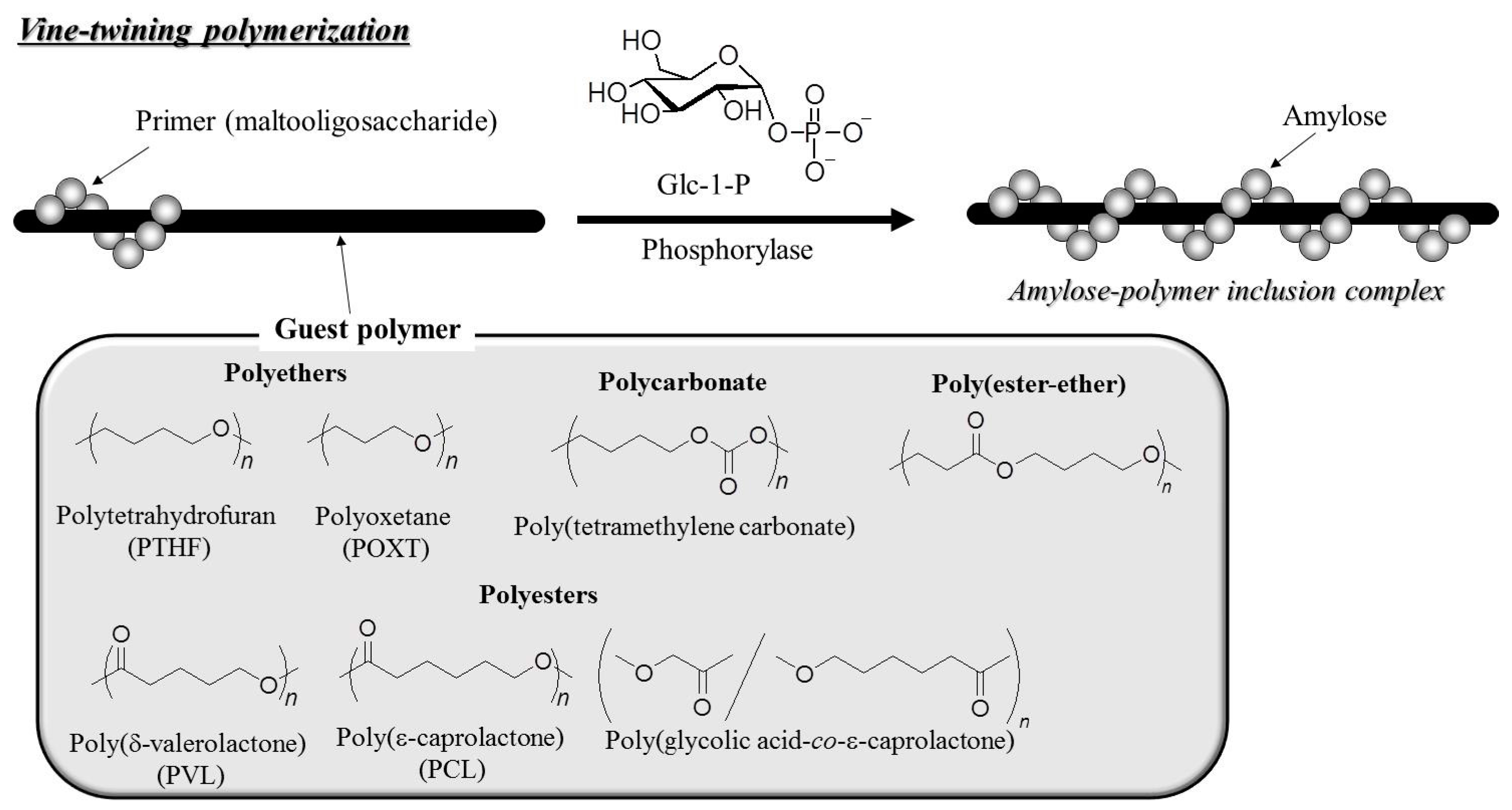
- Kaneko, Y.; Kadokawa, J. Vine-twining polymerization: A new preparation method for well-defined supramolecules composed of amylose and synthetic polymers. Chem. Rec. 2005, 5, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kadokawa, J. Synthesis of nanostructured bio-related materials by hybridization of synthetic polymers with polysaccharides or saccharide residues. J. Biomater. Sci., Polym. Ed. 2006, 17, 1269–1284. [Google Scholar] [CrossRef]
- Kaneko, Y.; Kadokawa, J. Preparation of polymers with well-defined nanostructure in the polymerization field. In Modern Trends in Macromolecular Chemistry; Lee, J.N., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2009; pp. 199–217. [Google Scholar]
- Kaneko, Y.; Kadokawa, J. Preparation method for polysaccharide supramolecules using amylose-forming polymerization field: Vine-twining polymerization. Kobunshi Ronbunshu 2010, 67, 553–559. [Google Scholar] [CrossRef]
- Kadokawa, J. Preparation and applications of amylose supramolecules by means of phosphorylase-catalyzed enzymatic polymerization. Polymers 2012, 4, 116–133. [Google Scholar] [CrossRef]
- Kadokawa, J. Architecture of amylose supramolecules in form of inclusion complexes by phosphorylase-catalyzed enzymatic polymerization. Biomolecules 2013, 3, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.; Kaneko, Y.; Tagaya, H.; Chiba, K. Synthesis of an amylose-polymer inclusion complex by enzymatic polymerization of glucose 1-phosphate catalyzed by phosphorylase enzyme in the presence of polythf: A new method for synthesis of polymer-polymer inclusion complexes. Chem. Commun. 2001, 449–450. [Google Scholar] [CrossRef]
- Kadokawa, J.; Kaneko, Y.; Nagase, S.; Takahashi, T.; Tagaya, H. Vine-twining polymerization: Amylose twines around polyethers to form amylose-polyether inclusion complexes. Chem. Eur. J. 2002, 8, 3321–3326. [Google Scholar] [CrossRef]
- Kadokawa, J.; Kaneko, Y.; Nakaya, A.; Tagaya, H. Formation of an amylose-polyester inclusion complex by means of phosphorylase-catalyzed enzymatic polymerization of α-d-glucose 1-phosphate monomer in the presence of poly(ε-caprolactone). Macromolecules 2001, 34, 6536–6538. [Google Scholar] [CrossRef]
- Kadokawa, J.; Nakaya, A.; Kaneko, Y.; Tagaya, H. Preparation of inclusion complexes between amylose and ester-containing polymers by means of vine-twining polymerization. Macromol. Chem. Phys. 2003, 204, 1451–1457. [Google Scholar] [CrossRef]
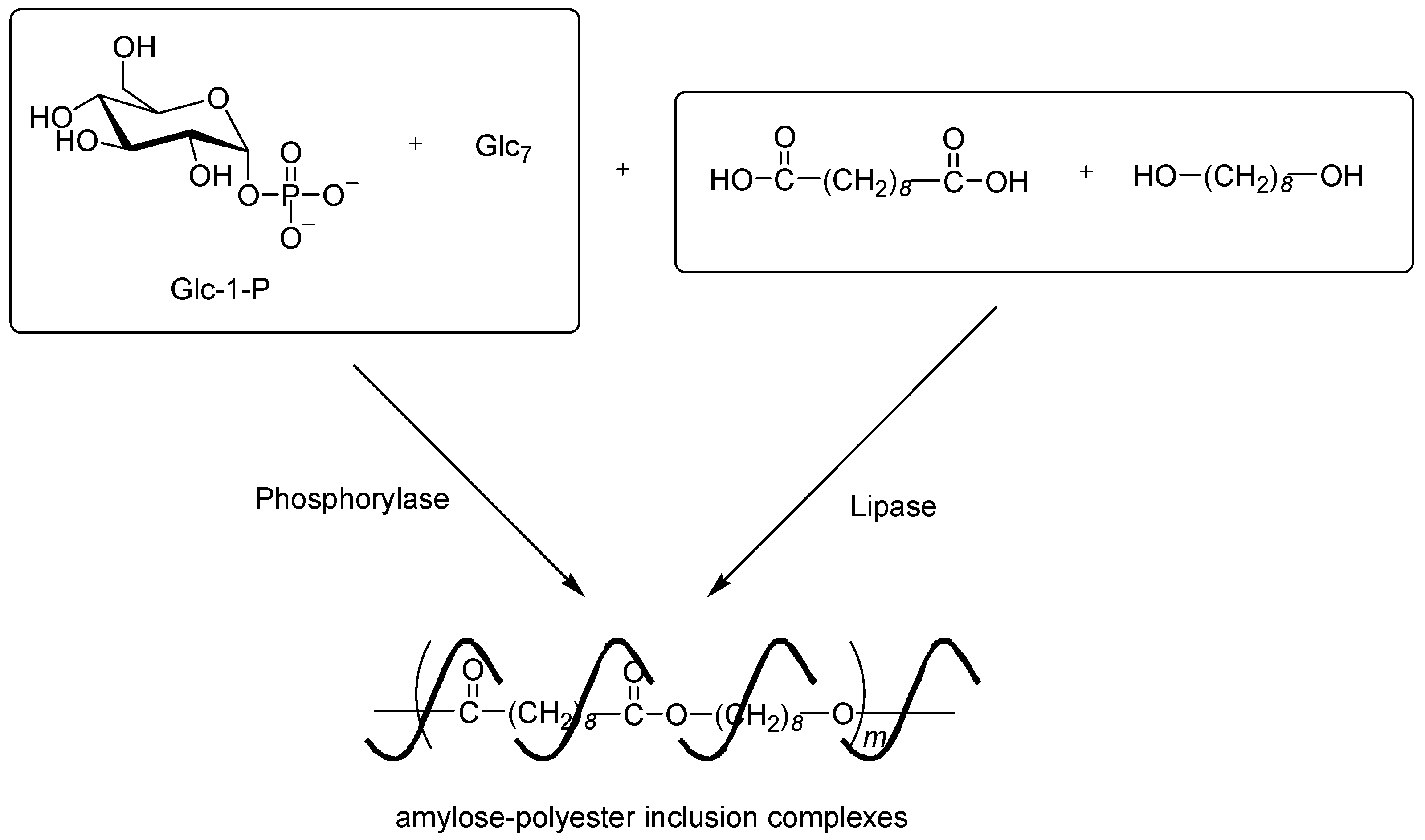
- Nomura, S.; Kyutoku, T.; Shimomura, N.; Kaneko, Y.; Kadokawa, J. Preparation of inclusion complexes composed of amylose and biodegradable poly(glycolic acid-co-ε-caprolactone) by vine-twining polymerization and their lipase-catalyzed hydrolysis behavior. Polym. J. 2011, 43, 971–977. [Google Scholar] [CrossRef]
- Kaneko, Y.; Beppu, K.; Kadokawa, J. Preparation of amylose/polycarbonate inclusion complexes by means of vine-twining polymerization. Macromol. Chem. Phys. 2008, 209, 1037–1042. [Google Scholar] [CrossRef]
- Kaneko, Y.; Saito, Y.; Nakaya, A.; Kadokawa, J.; Tagaya, H. Preparation of inclusion complexes composed of amylose and strongly hydrophobic polyesters in parallel enzymatic polymerization system. Macromolecules 2008, 41, 5665–5670. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Suda, S.; Namekawa, S. Dehydration polymerization in aqueous medium catalyzed by lipase. Chem. Lett. 1997, 105. [Google Scholar] [CrossRef]
- Suda, S.; Uyama, H.; Kobayashi, S. Dehydration polycondensation in water for synthesis of polyesters by lipase catalyst. Proc. Jpn. Acad. B, Phys. Biol. Sci. 1999, 75, 201–206. [Google Scholar] [CrossRef]
- Kaneko, Y.; Beppu, K.; Kadokawa, J. Amylose selectively includes one from a mixture of two resemblant polyethers in vine-twining polymerization. Biomacromolecules 2007, 8, 2983–2985. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Beppu, K.; Kyutoku, T.; Kadokawa, J. Selectivity and priority on inclusion of amylose toward guest polyethers and polyesters in vine-twining polymerization. Polym. J. 2009, 41, 279–286. [Google Scholar] [CrossRef]
- Kaneko, Y.; Beppu, K.; Kadokawa, J. Amylose selectively includes a specific range of molecular weights in poly(tetrahydrofuran)s in vine-twining polymerization. Polym. J. 2009, 41, 792–796. [Google Scholar] [CrossRef]
- Kaneko, Y.; Ueno, K.; Yui, T.; Nakahara, K.; Kadokawa, J. Amylose’s recognition of chirality in polylactides on formation of inclusion complexes in vine-twining polymerization. Macromol. Biosci. 2011, 11, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Fujisaki, K.; Kyutoku, T.; Furukawa, H.; Kadokawa, J. Preparation of enzymatically recyclable hydrogels through the formation of inclusion complexes of amylose in a vine-twining polymerization. Chem. Asian J. 2010, 5, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.; Nomura, S.; Hatanaka, D.; Yamamoto, K. Preparation of polysaccharide supramolecular films by vine-twining polymerization approach. Carbohydr. Polym. 2013, 98, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.; Tanaka, K.; Hatanaka, D.; Yamamoto, K. Preparation of multiformable supramolecular gels through helical complexation by amylose in vine-twining polymerization. Polym. Chem. 2015, 6, 6402–6408. [Google Scholar] [CrossRef]
- Tanaka, T.; Sasayama, S.; Nomura, S.; Yamamoto, K.; Kimura, Y.; Kadokawa, J. An amylose-poly(l-lactide) inclusion supramolecular polymer: Enzymatic synthesis by means of vine-twining polymerization using a primer–guest conjugate. Macromol. Chem. Phys. 2013, 214, 2829–2834. [Google Scholar] [CrossRef]
- Tanaka, T.; Tsutsui, A.; Gotanda, R.; Sasayama, S.; Yamamoto, K.; Kadokawa, J. Synthesis of amylose-polyether inclusion supramolecular polymers by vine-twining polymerization using maltoheptaose-functionalized poly(tetrahydrofuran) as a primer–guest conjugate. J. Appl. Glycosci. 2015, 62, 135–141. [Google Scholar] [CrossRef]
- Tanaka, T.; Gotanda, R.; Tsutsui, A.; Sasayama, S.; Yamamoto, K.; Kimura, Y.; Kadokawa, J. Synthesis and gel formation of hyperbranched supramolecular polymer by vine-twining polymerization using branched primer-guest conjugate. Polymer 2015, 73, 9–16. [Google Scholar] [CrossRef]
- Tanaka, T.; Sasayama, S.; Yamamoto, K.; Kimura, Y.; Kadokawa, J. Evaluating relative chain orientation of amylose and poly(l-lactide) in inclusion complexes formed by vine-twining polymerization using primer–guest conjugates. Macromol. Chem. Phys. 2015, 216, 794–800. [Google Scholar] [CrossRef]
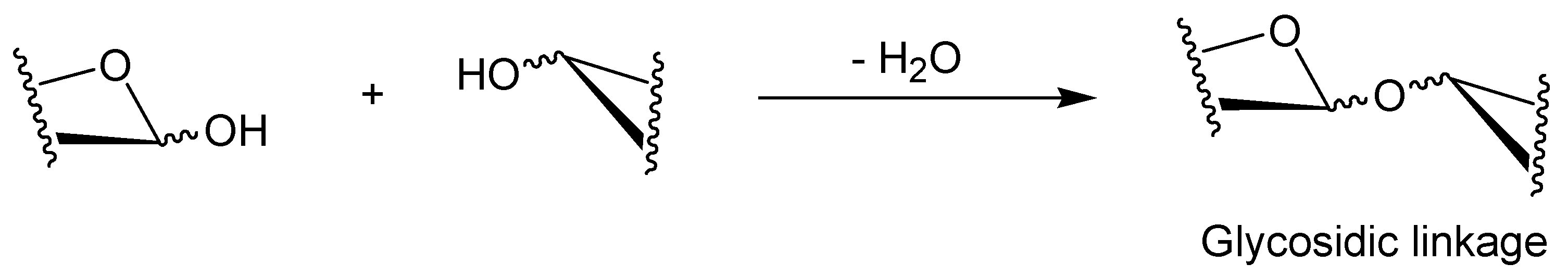


© 2016 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadokawa, J.-i. Precision Synthesis of Functional Polysaccharide Materials by Phosphorylase-Catalyzed Enzymatic Reactions. Polymers 2016, 8, 138. https://doi.org/10.3390/polym8040138
Kadokawa J-i. Precision Synthesis of Functional Polysaccharide Materials by Phosphorylase-Catalyzed Enzymatic Reactions. Polymers. 2016; 8(4):138. https://doi.org/10.3390/polym8040138
Chicago/Turabian StyleKadokawa, Jun-ichi. 2016. "Precision Synthesis of Functional Polysaccharide Materials by Phosphorylase-Catalyzed Enzymatic Reactions" Polymers 8, no. 4: 138. https://doi.org/10.3390/polym8040138