
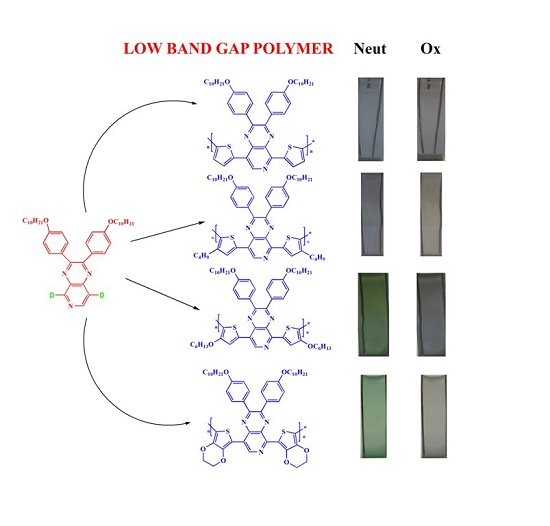
Low Band Gap Donor–Acceptor Type Polymers Containing 2,3-Bis(4-(decyloxy)phenyl)pyrido[4,3-b]pyrazine as Acceptor and Different Thiophene Derivatives as Donors


Abstract
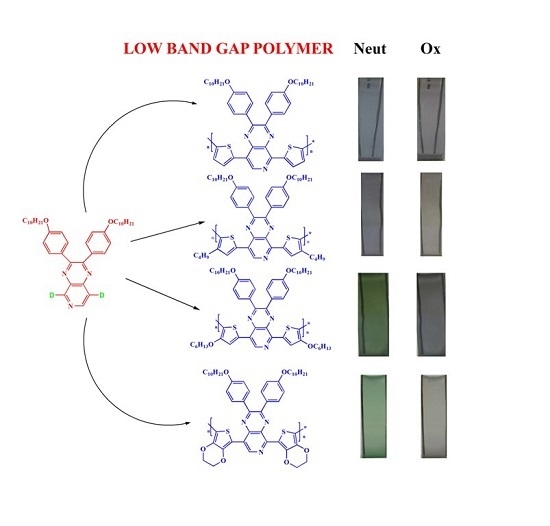
:
1. Introduction
2. Materials and Methods
2.1. Materials
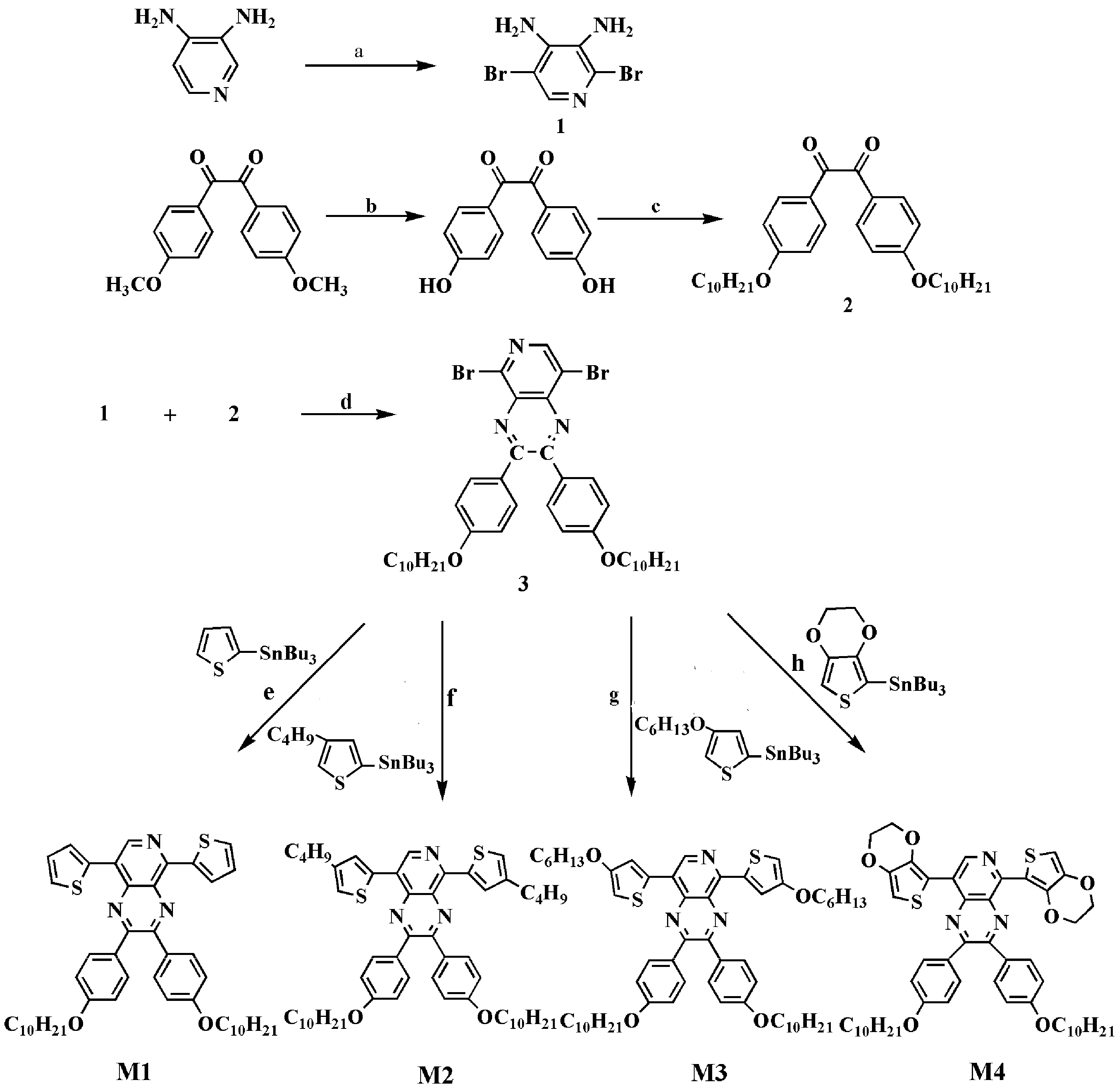
2.2. Monomer Syntheses
2.2.1. Process of Synthesis
2.2.2. Syntheses of M1, M2, M3 and M4
2.3. Characterization of the Monomers and Polymers
3. Results and Discussion
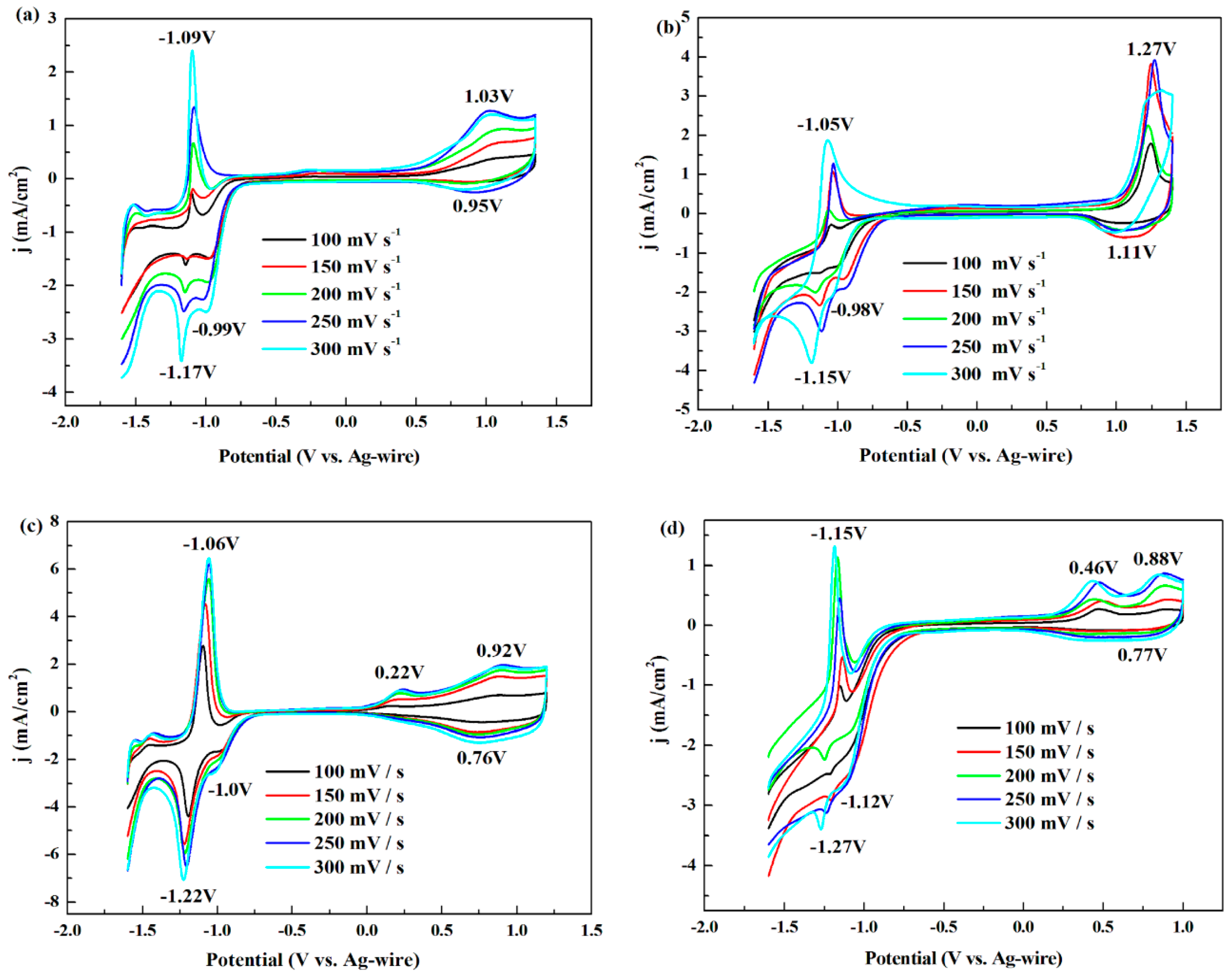
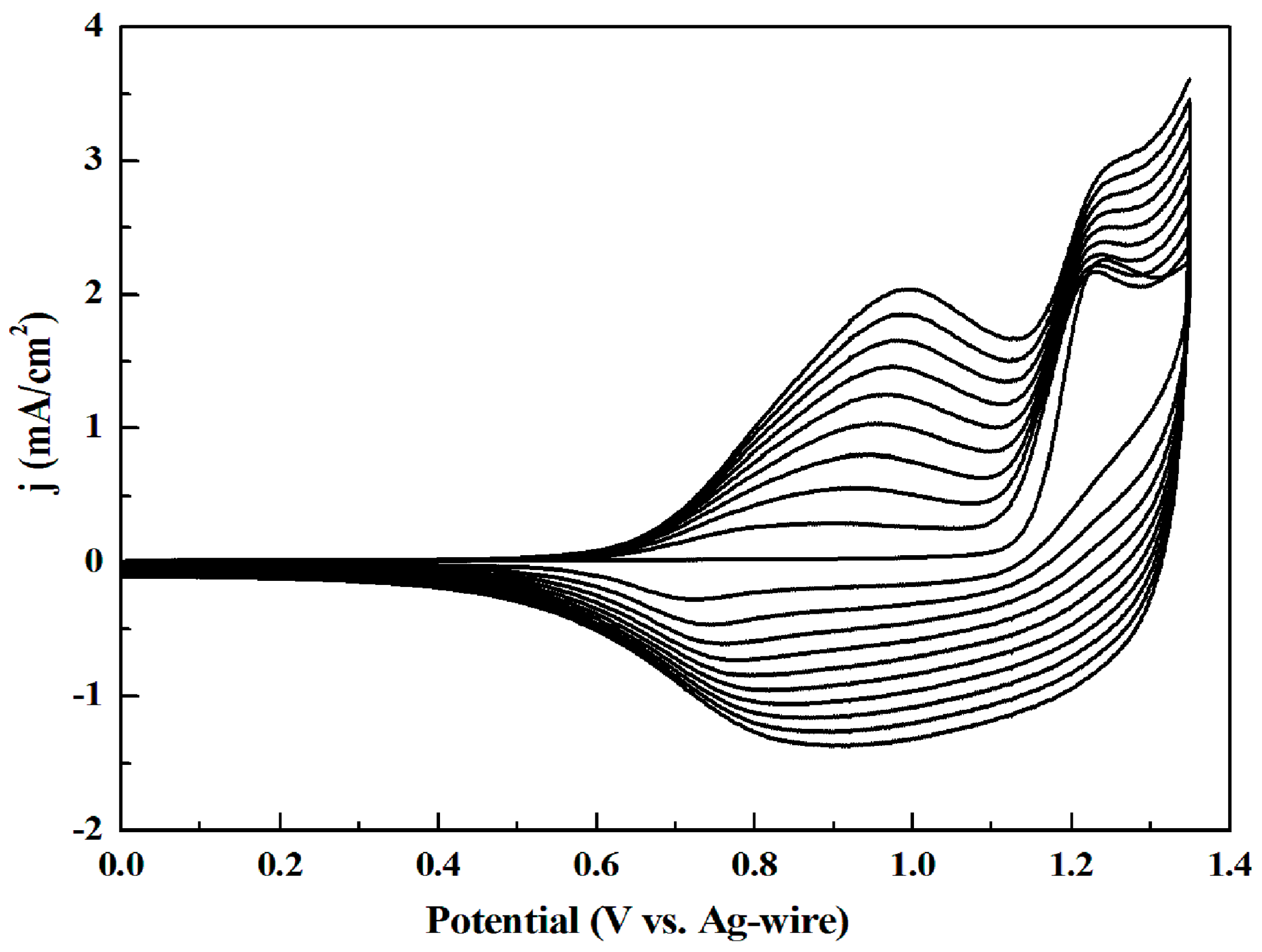
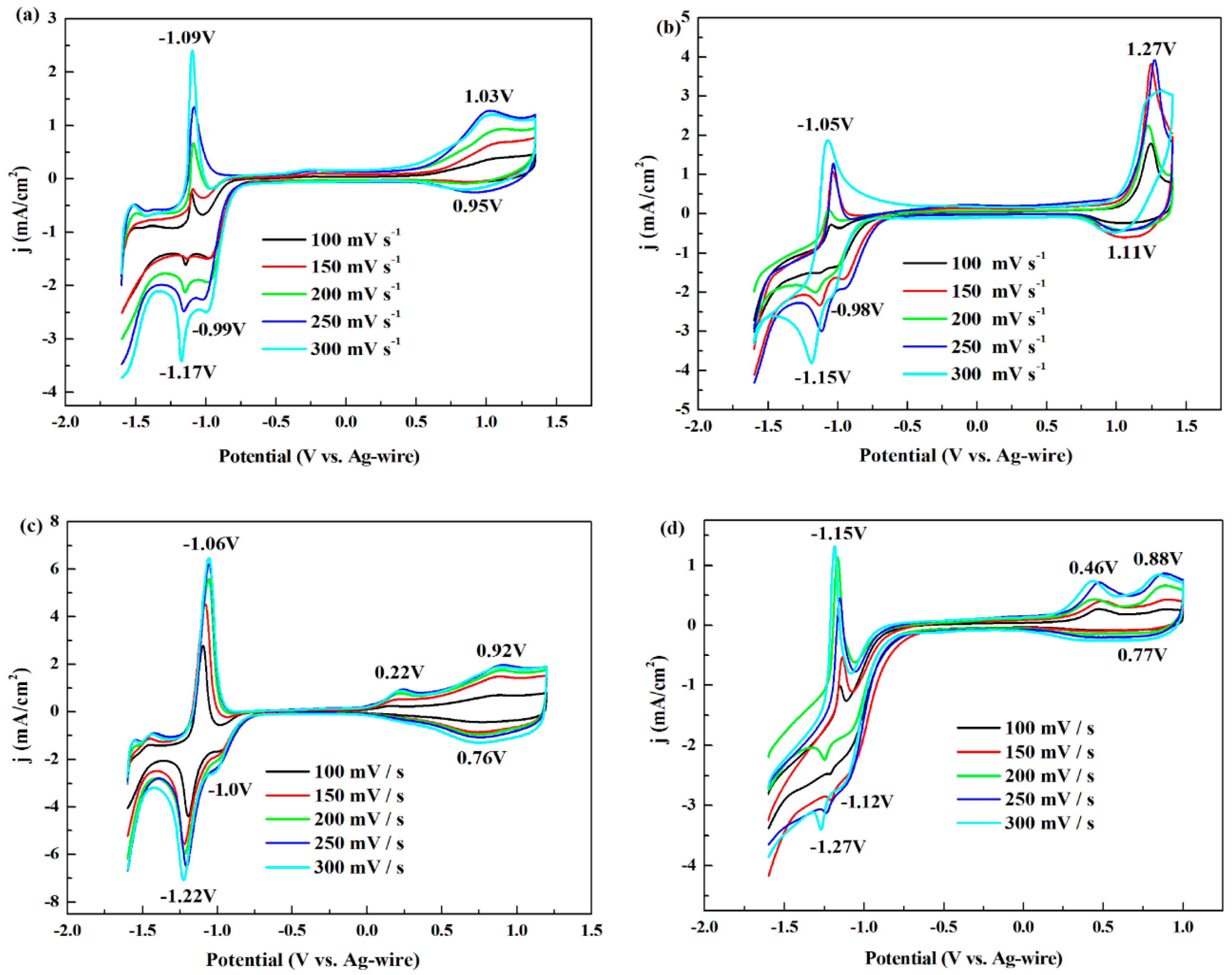
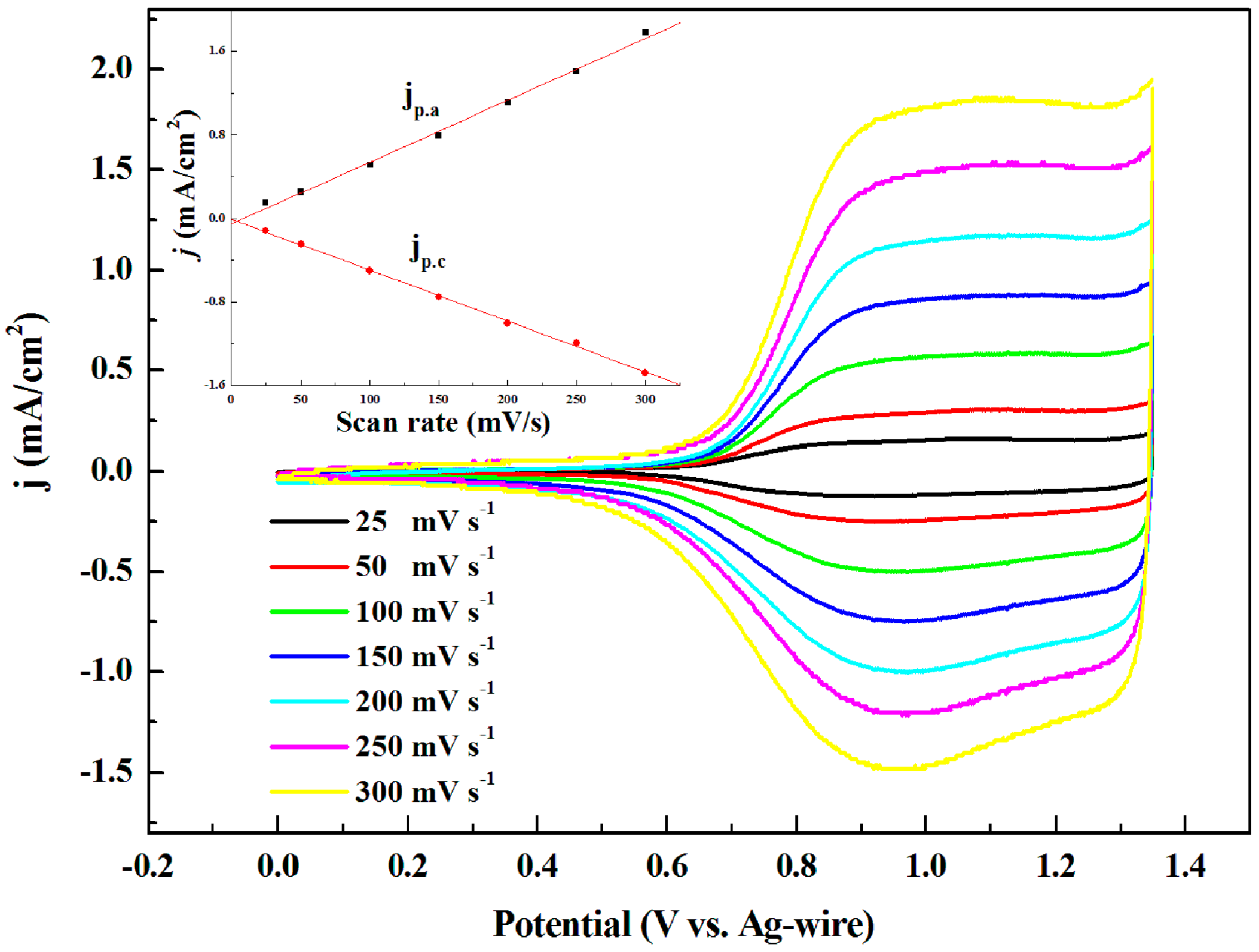
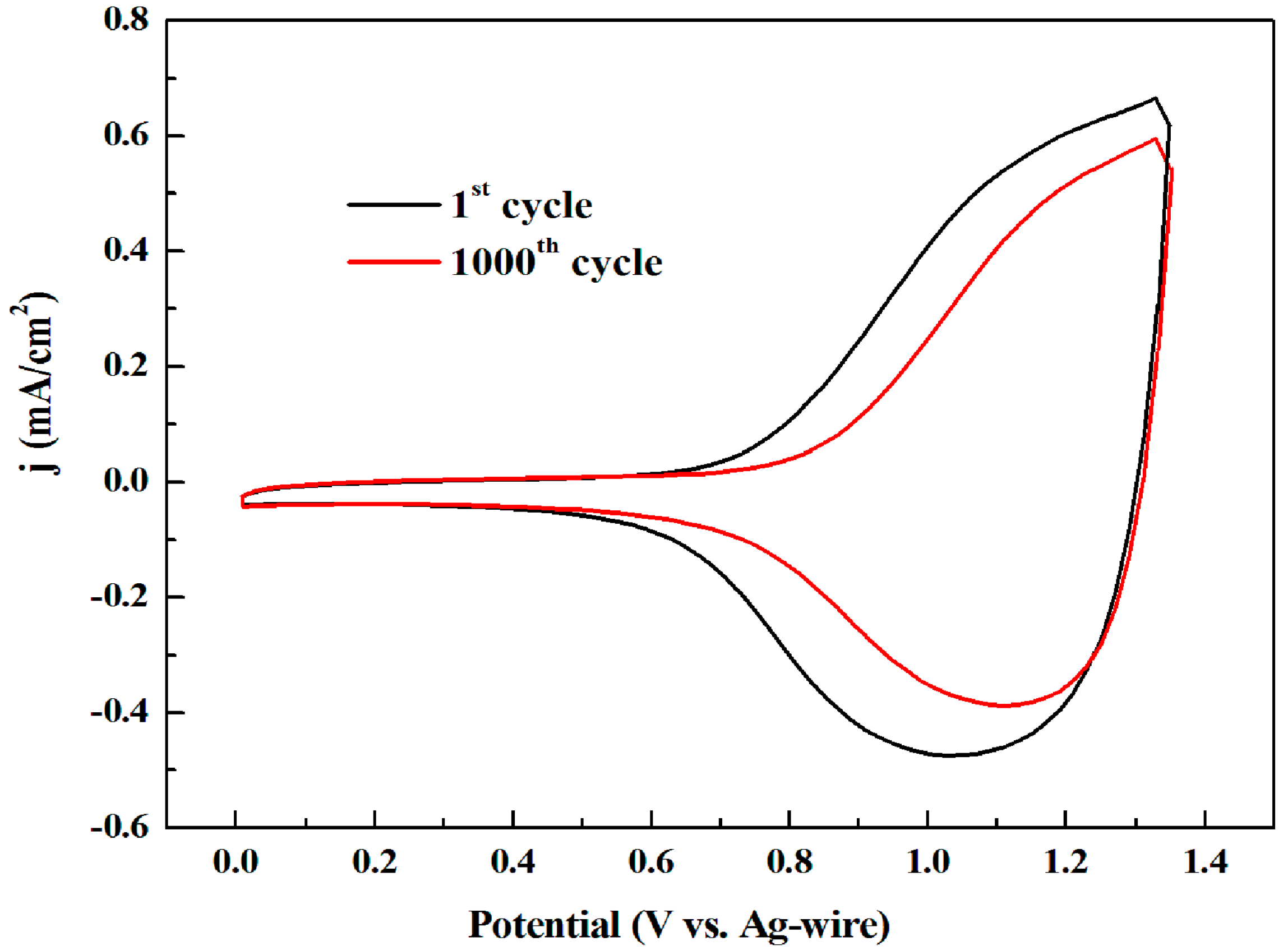
3.1. Electrochemistry
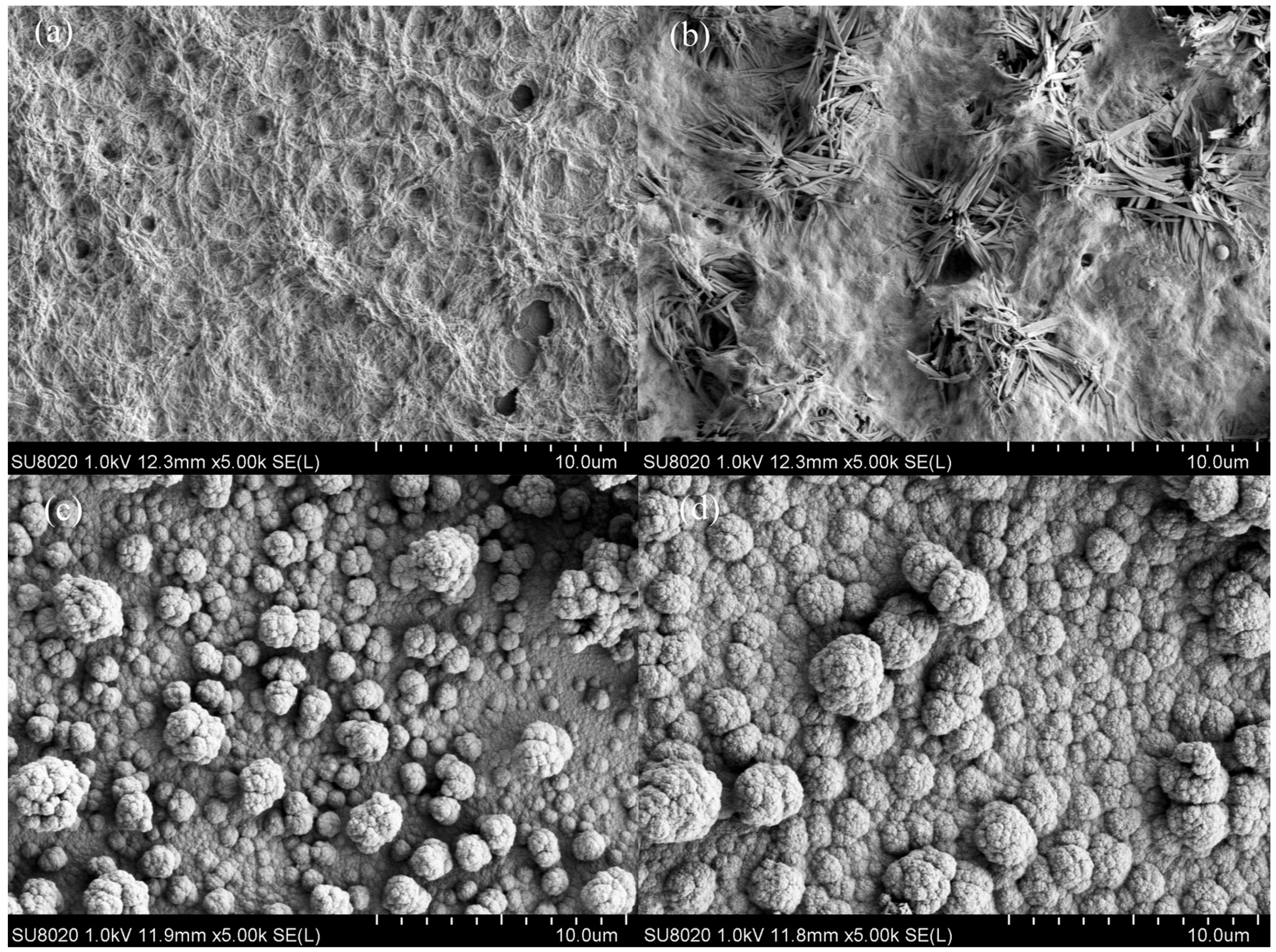
3.2. Morphology
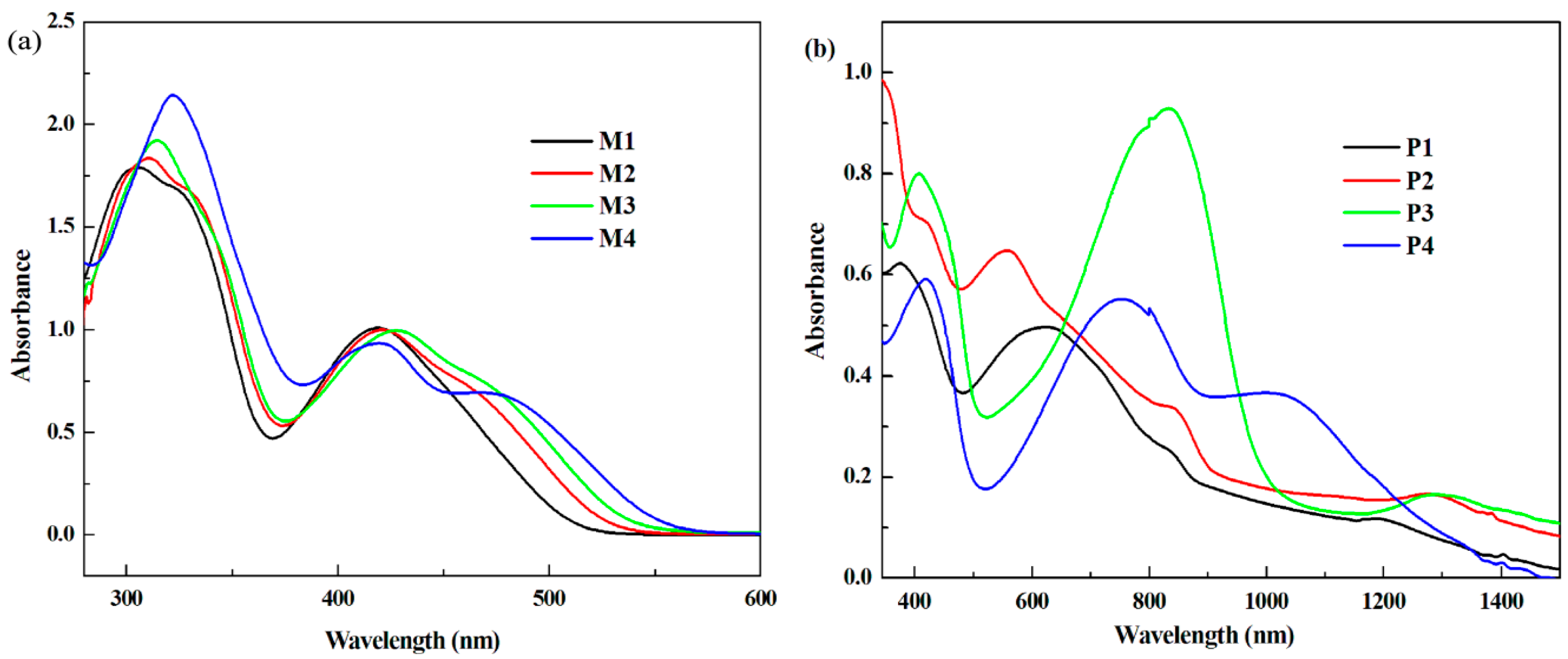
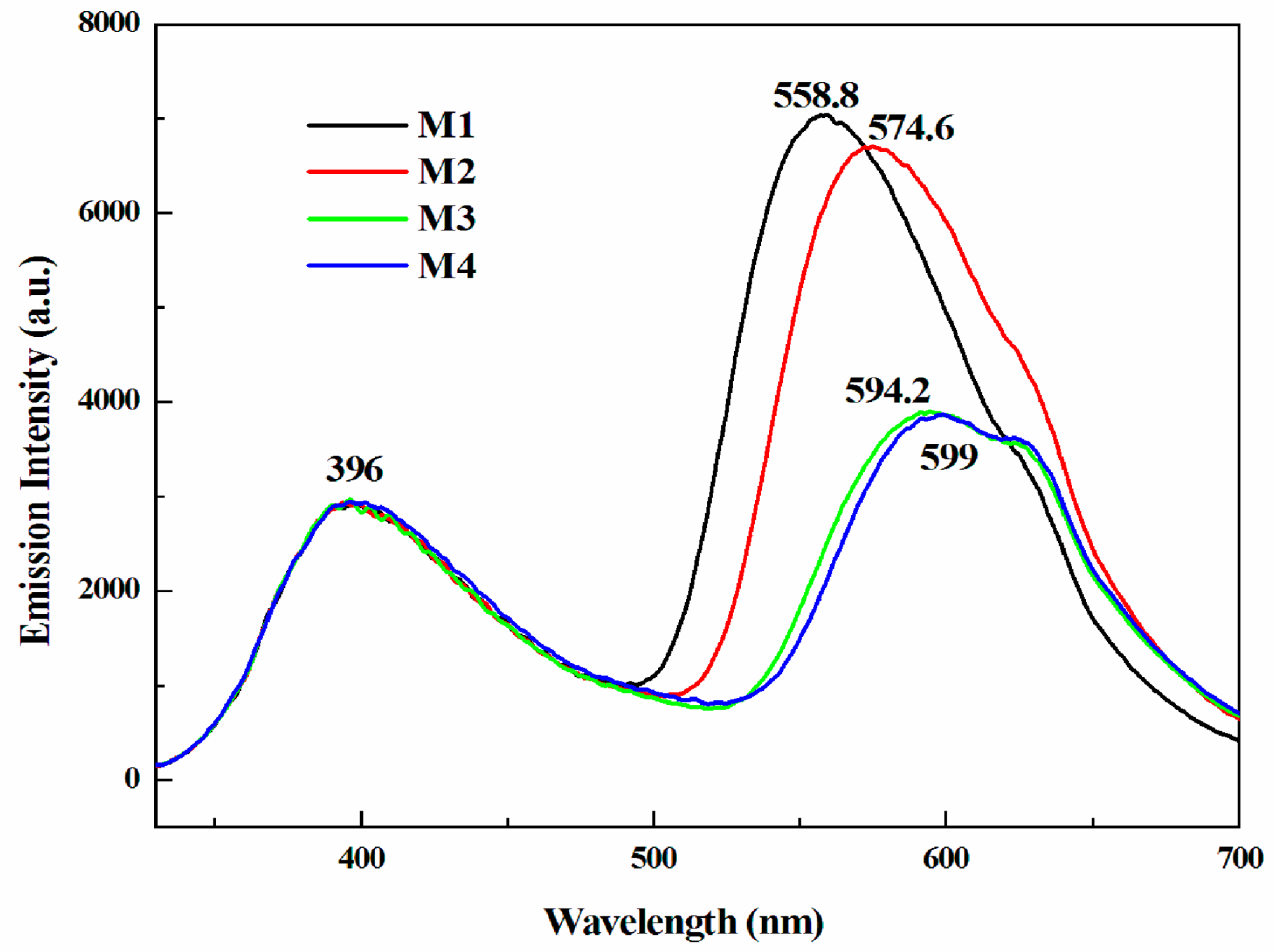
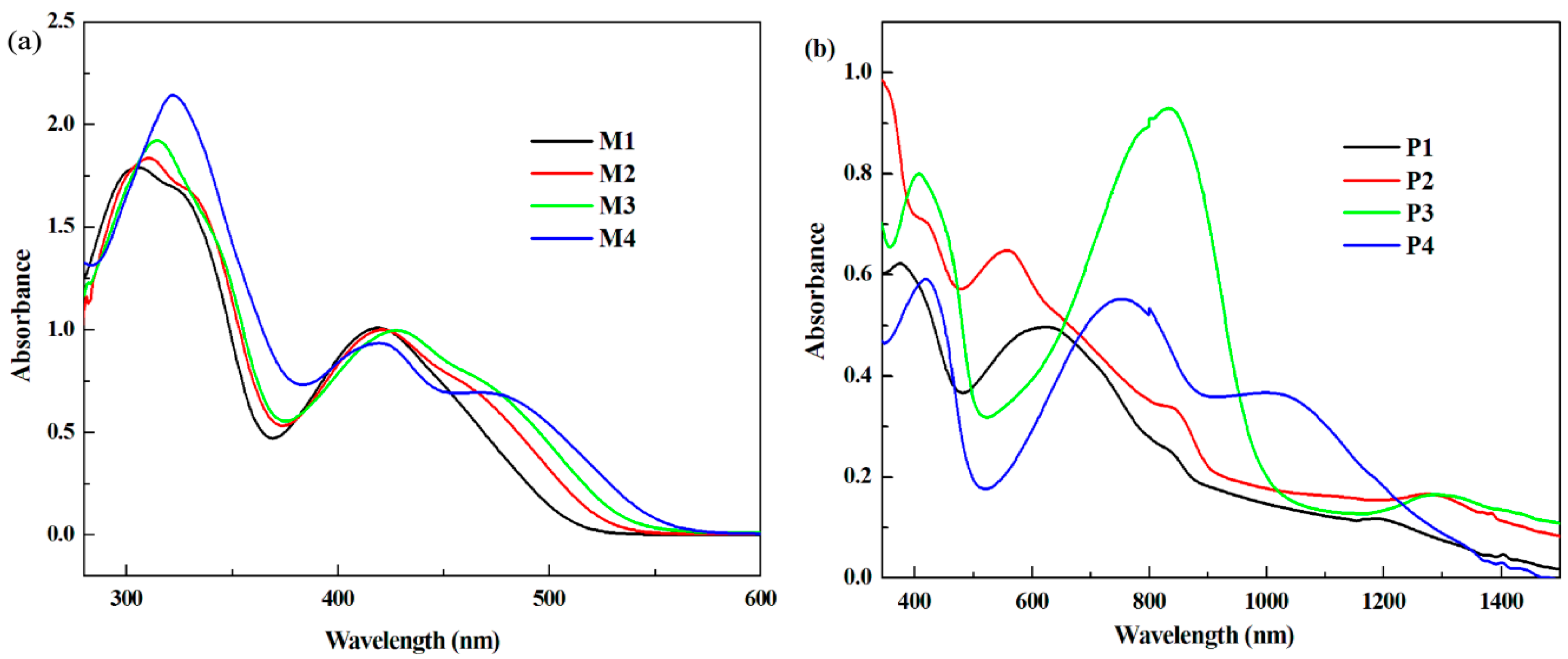
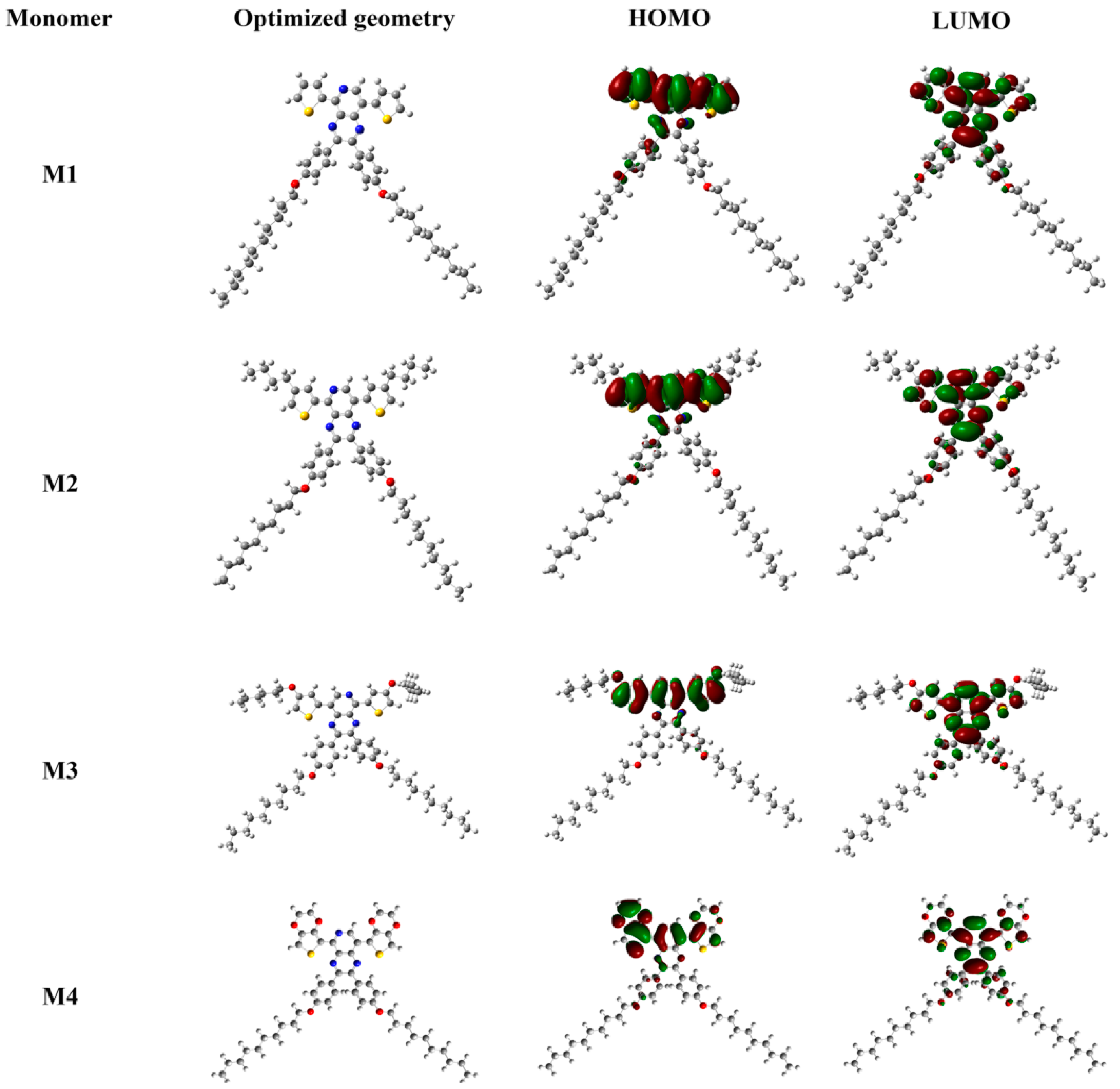
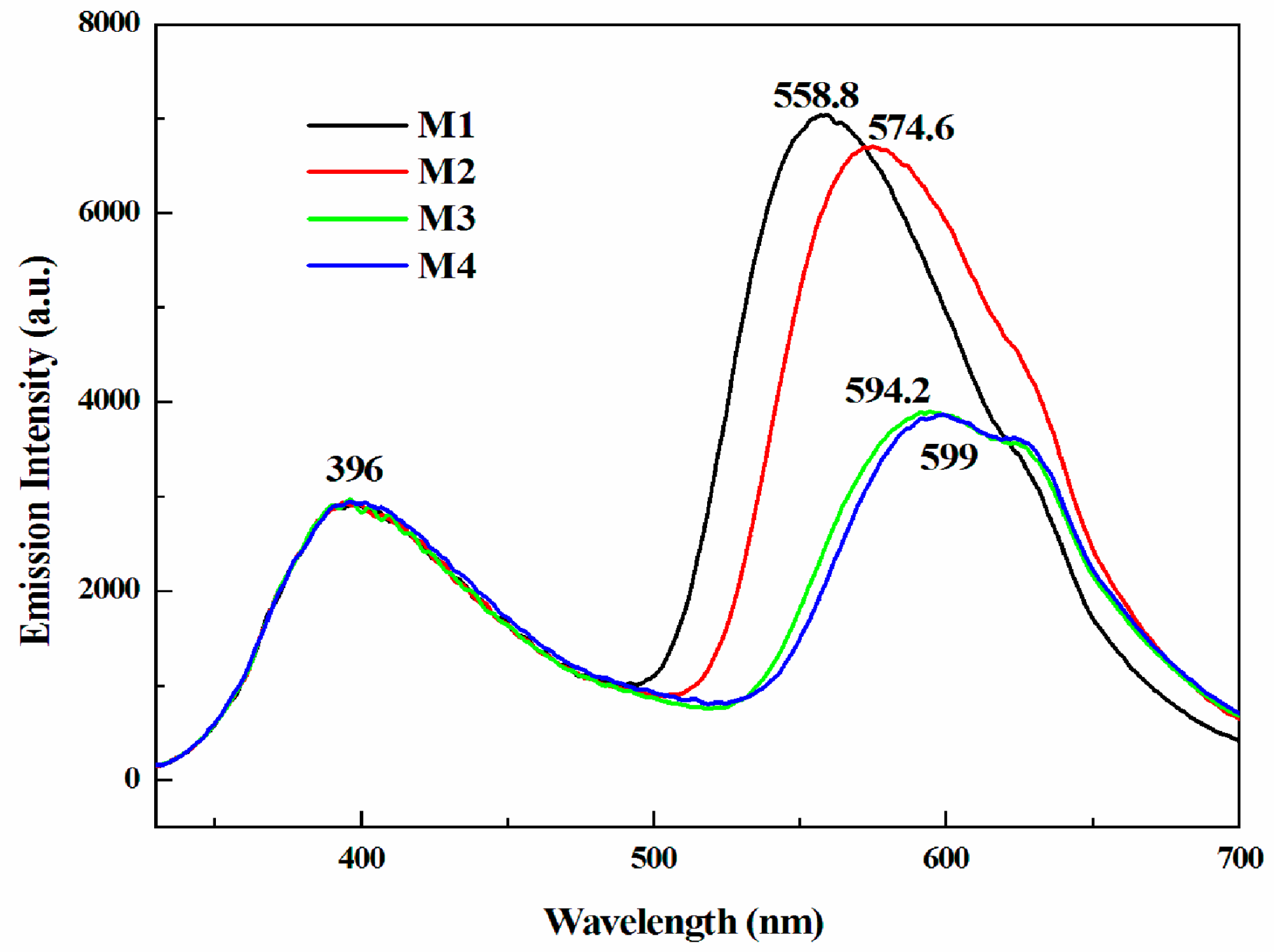
3.3. Optical Properties
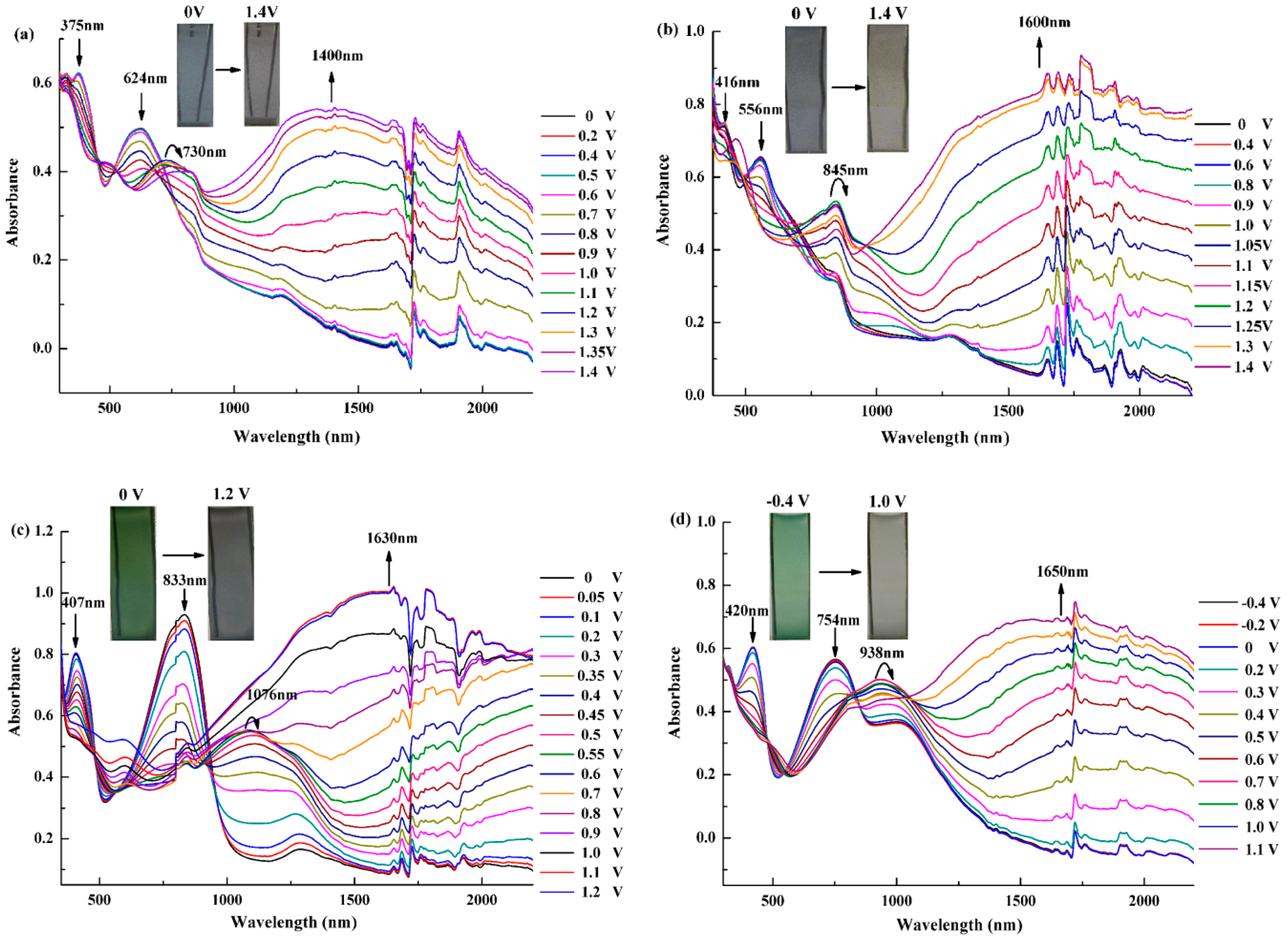
3.4. Electrochromic Properties of the Polymers
3.5. Electrochromic Switching
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jensen, J.; Hösel, M.; Dyer, A.L.; Krebs, F.C. Development and manufacture of polymer-based electrochromic devices. Adv. Funct. Mater. 2015, 25, 2073–2090. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, C.; Niu, H.; Zhao, X.; Wang, C.; Qin, C.; Wang, W.; Bai, X. Preparation and electrochromic properties of two series of polyurethanes containing separated triphenylamine moiety with different blocks. Dyes Pigment. 2016, 125, 106–115. [Google Scholar] [CrossRef]
- Qin, C.; Fu, Y.; Chui, C.; Kan, C.W.; Xie, Y.; Wang, L.; Wong, W.Y. Tuning the donor-acceptor strength of low-band gap platinum-acetylide polymers for near-infrared photovoltaic applications. Macromol. Rapid Commun. 2011, 32, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Zhong, Y.; Yao, J. Charge delocalization in a cyclometalated bisruthenium complex bridged by a noninnocent 1,2,4,5-Tetra(2-pyridyl)benzene Ligand. J. Am. Chem. Soc. 2011, 133, 15697–15706. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, J.; Zeng, X.; Cheng, D.; Huang, H.; Cao, D. Enhanced near-infrared shielding ability of (Li,K)-codoped WO3 for smart windows: DFT prediction validated by experiment. Nanotechnology 2016, 27, 075203. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.H.; Wang, H.M.; Liao, S.H. Redox-stable and visible/near-infrared electrochromic aramids with main-chain triphenylamine and pendent 3,6-di-tert-butylcarbazole units. Polym. Chem. 2014, 5, 2473–2483. [Google Scholar] [CrossRef]
- Chuang, Y.W.; Yen, H.J.; Wu, J.H.; Liou, G.S. Colorless triphenylamine-based aliphatic thermoset epoxy for multicolored and near-infrared electrochromic applications. ACS Appl. Mater. Interfaces 2014, 6, 3594–3599. [Google Scholar] [CrossRef] [PubMed]
- Qiao, W.; Zheng, J.; Wang, Y.; Zheng, Y.; Song, N.; Wan, X.; Wang, Z. Efficient synthesis and properties of novel near-infrared electrochromic anthraquinone imides. Org. Lett. 2008, 10, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Kavak, E.; Us, C.N.; Yavuz, E.; Kivrak, A.; İçli Özkut, M. A camouflage material: p- and n-type dopable furan based low band gap electrochromic polymer and its EDOT based copolymer. Electrochim. Acta 2015, 182, 537–543. [Google Scholar] [CrossRef]
- Yen, H.J.; Lin, H.Y.; Liou, G.S. Novel starburst triarylamine-containing electroactive aramids with highly stable electrochromism in near-infrared and visible light regions. Chem. Mater. 2011, 23, 1874–1882. [Google Scholar] [CrossRef]
- Qian, G.; Abua, H.; Wang, Z.Y. A precursor strategy for the synthesis of low band-gap polymers: An efficientroute to a series of near-infrared electrochromic polymers. J. Mater. Chem. 2011, 21, 7678–7685. [Google Scholar]
- Atwani, O.; Baristiran, C.; Erden, A.; Sonmez, G. A stable, low band gap electroactive polymer: Poly(4,7-dithien-2-yl-2,1,3-benzothiadiazole). Synth. Met. 2008, 158, 83–89. [Google Scholar] [CrossRef]
- Li, H.; Tam, T.L.; Lam, Y.M.; Mhaisalkar, S.G.; Grimsdale, A.C. Synthesis of low band gap [1,2,5]-thiadiazolo[3,4-g] quinoxaline and pyrazino [2,3-g] quinoxaline derivatives by selective reduction of benzo [1,2-c;4,5-c′] bis[1,2,5] thiadiazole. Org. Lett. 2011, 13, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Akpınar, H.; Balan, A.; Baran, D.; Ünver, E.K.; Toppare, L. Donor-acceptor-donor type conjugated polymers for electrochromic applications: Benzimidazole as the acceptor unit. Polymer 2010, 51, 6123–6131. [Google Scholar] [CrossRef]
- Balan, A.; Gunbas, G.; Durmus, A.; Toppare, L. Donor-acceptor polymer with benzotriazole moiety: Enhancing the electrochromic properties of the “donor unit”. Chem. Mater. 2008, 20, 7510–7513. [Google Scholar] [CrossRef]
- Durmus, A.; Gunbas, G.E.; Toppare, L. New, highly stable electrochromic polymers from 3,4-ethylenedioxythiophene-bis-substituted quinoxalines toward green polymeric materials. Chem. Mater. 2007, 19, 6247–6251. [Google Scholar] [CrossRef]
- Zhao, H.; Wei, Y.; Zhao, J.; Wang, M. Three donor-acceptor polymeric electrochromic materials employing 2,3-bis(4-(decyloxy)phenyl)pyrido[4,3-b]pyrazine as acceptor unit and thiophene derivatives as donor units. Electrochim. Acta 2014, 146, 231–241. [Google Scholar] [CrossRef]
- Nikolou, M.; Dyer, A.L.; Steckler, T.T.; Donoghue, E.P.; Wu, Z.; Heston, N.C.; Rinzler, A.G.; Tanner, D.B.; Reynolds, J.R. Dual n-and p-type dopable electrochromic devices employing transparent carbon nanotube electrodes. Chem. Mater. 2009, 21, 5539–5547. [Google Scholar] [CrossRef]
- Çetin, G.A.; Balan, A.; Durmuş, A.; Günbaş, G.; Toppare, L. A new p-and n-dopable selenophene derivative and its electrochromic properties. Org. Electron. 2009, 10, 34–41. [Google Scholar] [CrossRef]
- Richard, C.A.; Pan, Z.; Hsu, H.Y.; Cekli, S.; Schanze, K.S.; Reynolds, J.R. Effect of isomerism and chain length on electronic structure, photophysics, and sensitizer efficiency in quadrupolar (donor)2-acceptor systems for application in dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2014, 6, 5221–5227. [Google Scholar] [CrossRef] [PubMed]
- Stalder, R.; Puniredd, S.R.; Hansen, M.R.; Koldemir, U.; Grand, C.; Zajaczkowski, W.; Müllen, K.; Pisula, W.; Reynolds, J.R. Ambipolar charge transport in isoindigo-based donor-acceptor polymers. Chem. Mater. 2016, 28, 1286–1297. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Vasilyeva, S.V.; Liu, D.Y.; Ellinger, S.; McCarley, T.D.; Reynolds, J.R. Structure-performance correlations in spray-processable green dioxythiophene-benzothiadiazole donor-acceptor polymer electrochromes. Chem. Mater. 2012, 24, 255–268. [Google Scholar] [CrossRef]
- Lee, B.L.; Yamamoto, T. Syntheses of new alternating CT-type copolymers of thiophene and pyrido[3,4-b]pyrazine units: Their optical and electrochemical properties in comparison with similar CT copolymers of thiophene with pyridine and quinoxaline. Macromolecules 1999, 32, 1375–1382. [Google Scholar] [CrossRef]
- Yuan, M.-C.; Chiu, M.-Y.; Chiang, C.-M.; Wei, K.-H. Synthesis and characterization of pyrido[3,4-b]pyrazine-based low-bandgap copolymers for bulk heterojunction solar cells. Macromolecules 2010, 43, 6270–6277. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, M.; Fan, W.; Zhao, J.; Wang, H. The synthesis of new donor–acceptor polymers containing the 2,3-di(2-furyl) quinoxaline moiety: Fast-switching, low-band-gap, p-and n-dopable, neutral green-colored materials. Electrochim. Acta 2015, 160, 271–280. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Wang, M.; Zhang, Y.; Zhao, J.; Fu, C. Two novel D-A type polymers employing 2,3-(4-(decyloxy)phenyl) quinoxaline as the acceptor unit, thiophene and selenophene as the donor units. Int. J. Electrochem. Sci. 2016, 11, 1581–1600. [Google Scholar]
- Cihaner, A.; Algı, F. A novel neutral state green polymeric electrochromic with superior n- and p- doping qrocesses: Closer to red-blue-green (RGB) display realization. Adv. Funct. Mater. 2008, 18, 3583–3589. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, J.; Dou, L.; Qiao, W.; Wan, X. Synthesis and characterization of a novel kind of near-infrared electrochromic polymers containing an anthraquinone imide group and ionic moieties. J. Mater. Chem. 2009, 19, 8470–8477. [Google Scholar] [CrossRef]
- Xu, L.; Su, C.; Zhang, C.; Ma, C. Electrosynthesis and characterization of a novel electrochromic copolymer based on 1,4-bis(3-hexylthiophen-2-yl)benzene and perylene dye. Synth. Met. 2011, 161, 1856–1860. [Google Scholar] [CrossRef]
- Lei, T.; Dou, J.H.; Cao, X.Y.; Wang, J.Y.; Pei, J. A BDOPV-based donor-acceptor polymer for high-performance n-type and oxygen-doped ambipolar field-effect transistors. Adv. Mater. 2013, 25, 6589–6593. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Chen, Z.; Zheng, Y.; Newman, C.; Quinn, J.R.; Dötz, F.; Kastler, M.; Facchetti, A. A high-mobility electron-transporting polymer for printed transistors. Nature 2009, 457, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chen, Z.; Jing, Y.; Rong, Y.; Facchetti, A.; Yao, Y. Heavily n-dopable π-donjugated redox polymers with ultrafast energy storage capability. J. Am. Chem. Soc. 2015, 137, 4956–4959. [Google Scholar] [CrossRef] [PubMed]
- DuBois, C.J.; Abboud, K.A.; Reynolds, J.R. Electrolyte-controlled redox conductivity and n-type doping in poly (bis-EDOT-pyridine)s. J. Phys. Chem. B 2004, 108, 8550–8557. [Google Scholar] [CrossRef]
- Tarkuc, S.; Udum, Y.A.; Toppare, L. Tuning of the neutral state color of the π–conjugated donor-acceptor-donor type polymer from blue to green via changing the donor strength on the polymer. Polymer 2009, 50, 3458–3464. [Google Scholar] [CrossRef]
- Borjas, R.; Buttry, D.A. EQCM studies of film growth, redox cycling, and charge trapping of n-doped and p-doped poly(thiophene). Chem. Mater. 1991, 3, 872–878. [Google Scholar] [CrossRef]
- Zotti, G.; Schiavon, G.; Zecchin, S. Irreversible processes in the electrochemical reduction of polythiophenes. Chemical modifications of the polymer and charge-trapping phenomena. Synth. Met. 1995, 72, 275–281. [Google Scholar] [CrossRef]
- Hillman, A.R.; Daisley, S.J.; Bruckenstein, S. Ion and solvent transfers and trapping phenomena during n-doping of PEDOT films. Electrochim. Acta 2008, 53, 3763–3771. [Google Scholar] [CrossRef]
- Havinga, E.E.; Ten Hoeve, W.; Wynberg, H. Alternate donor-acceptor small-band-gap semiconducting polymers; Polysquaraines and polycroconaines. Synth. Met. 1993, 55, 299–306. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, J.; Cui, C.; Wang, M.; Kong, Y.; Zhang, X. Triphenylamine-based multielectrochromic material and its neutral green electrochromic devices. J. Electroanal. Chem. 2012, 682, 29–36. [Google Scholar] [CrossRef]
- Ma, C.Q.; Fonrodona, M.; Schikora, M.C.; Wienk, M.M.; Janssen, R.A.; Bäuerle, P. Solution-processed bulk-heterojunction solar cells based on monodisperse dendritic oligothiophenes. Adv. Funct. Mater. 2008, 18, 3323–3331. [Google Scholar] [CrossRef]
- Gunbas, G.E.; Camurlu, P.; Akhmedov, İ.M.; Tanyeli, C.; Önal, A.M.; Toppare, L. A fast switching, low band gap, p-and n-dopable, donor-acceptor type polymer. J. Electroanal. Chem. 2008, 615, 75–83. [Google Scholar] [CrossRef]
- Neo, W.T.; Cho, C.M.; Shi, Z.; Chua, S.J.; Xu, J. Modulating high-energy visible light absorption to attain neutral-state black electrochromic polymers. J. Mater. Chem. C 2016, 4, 28–32. [Google Scholar] [CrossRef]
- Baycan Koyuncu, F.; Koyuncu, S.; Ozdemir, E. A new multi-electrochromic 2,7 linked polycarbazole derivative: Effect of the nitro subunit. Org. Electron. 2011, 12, 1701–1710. [Google Scholar] [CrossRef]



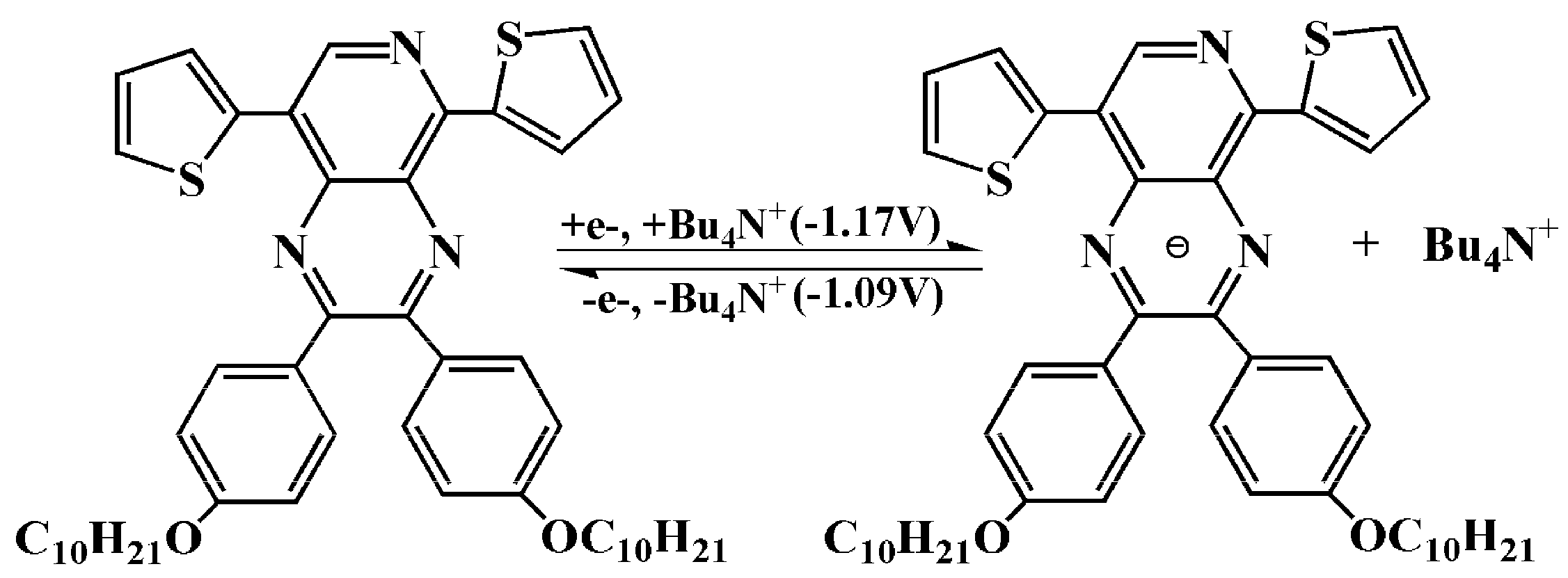






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
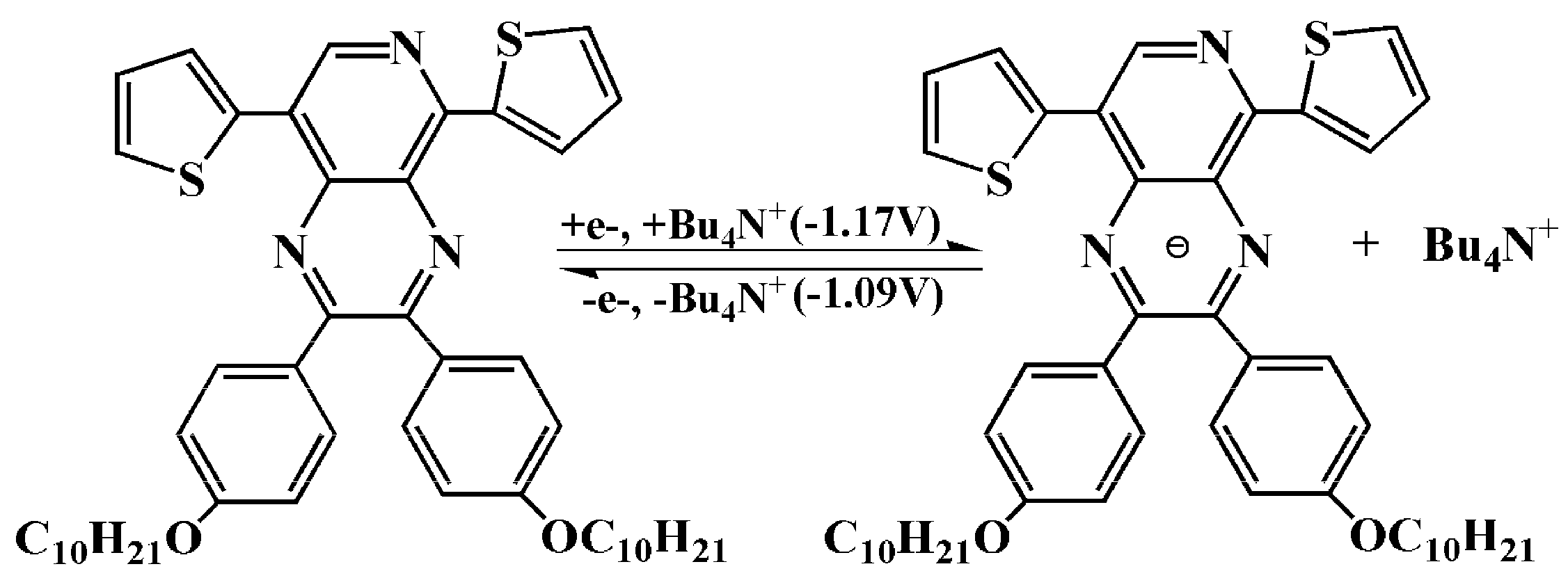{kind=link}
| Polymers | Redox Potential vs. Ag (V) | Redox Potential vs. Ag (V) | ||||
|---|---|---|---|---|---|---|
| p-Doping | p-De-doping | n-Doping | n-De-doping | |||
| P1 | 1.03 | 0.95 | 0.99 | −1.17 | −1.09 | −1.13 |
| - | - | −0.99 | - | |||
| P2 | 1.27 | 1.11 | 1.19 | −1.15 | −1.05 | −1.10 |
| - | - | −0.98 | - | |||
| P3 | 0.92 | 0.76 | 0.84 | −1.22 | −1.06 | −1.14 |
| 0.22 | - | −1.00 | - | |||
| P4 | 0.88 | 0.77 | 0.825 | −1.27 | −1.15 | −1.21 |
| 0.46 | - | −1.12 | - | |||
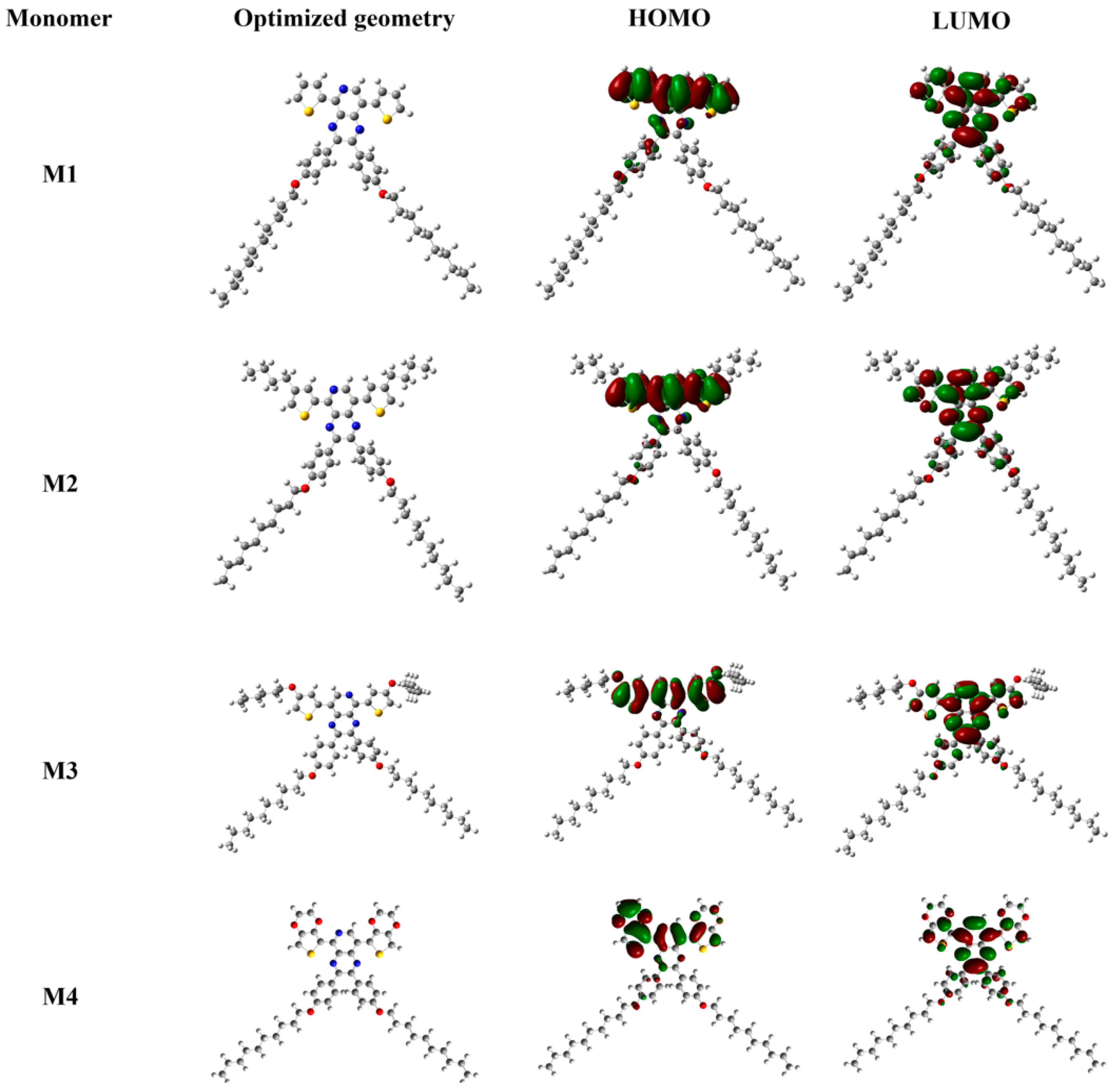
| Compound | Eonset vs. Ag (V) | λonset (nm) | Eg,op (a) (eV) | HOMO (b) (eV) | LUMO (c) (eV) | Eg,cal (d) (eV) | Eg,ec (e) (eV) |
|---|---|---|---|---|---|---|---|
| M1 | 1.15 | 523 | 2.37 | −5.57 | −3.20 | 2.84 | - |
| M2 | 1.15 | 543 | 2.28 | −5.57 | −3.29 | 2.79 | - |
| M3 | 0.95 | 550 | 2.25 | −5.37 | −3.12 | 2.73 | - |
| M4 | 0.77 | 576 | 2.15 | −5.19 | −3.04 | 2.70 | - |
| P1 | 0.69 | 844 | 1.47 | −5.11 | −3.64 | - | 1.48 |
| P2 | 0.93 | 870 | 1.42 | −5.35 | −3.93 | - | 1.87 |
| P3 | 0.02 | 1002 | 1.24 | −4.44 | −3.20 | - | 1.27 |
| P4 | 0.15 | 1342 | 0.92 | −4.57 | −3.65 | - | 1.45 |
| P(A) (f) | - | - | 1.14 | - | - | - | - |
| P(B) (g) | - | - | 1.4 | - | - | - | - |
| Polymers | E, vs. Ag (V) | L* | a* | b* | Color |
|---|---|---|---|---|---|
| P1 | 0 | 46 | −2 | −7 |  |
| 1.4 | 49 | −1 | 2 |  | |
| P2 | 0 | 37 | −8 | −40 |  |
| 1.4 | 60 | −12 | 8 |  | |
| P3 | 0 | 44 | −62 | −20 |  |
| 1.2 | 56 | −2 | 3 |  | |
| P4 | −0.4 | 60 | −40 | 16 |  |
| 1.1 | 62 | −12 | 8 |  |
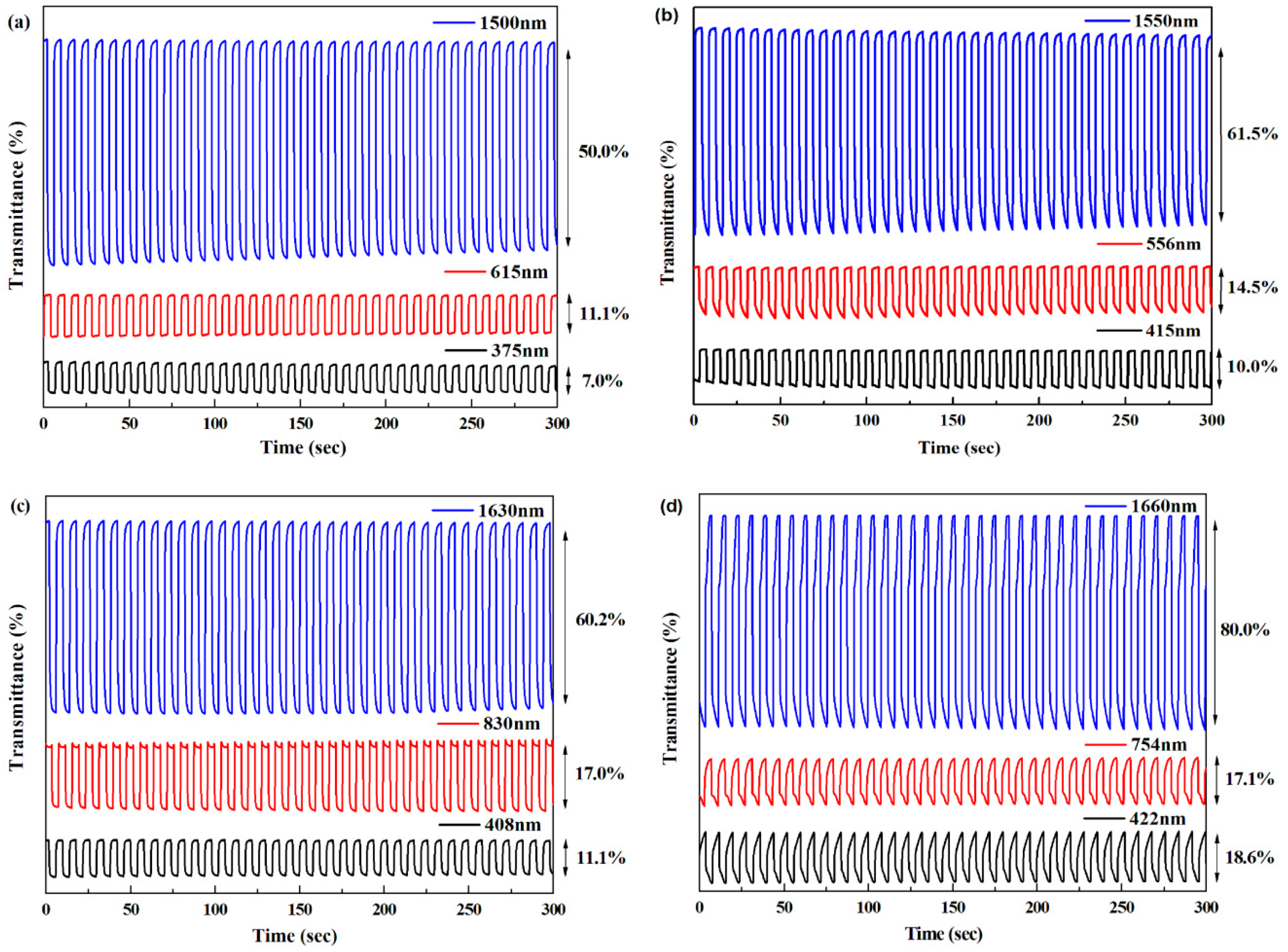
| Polymer | λ (nm) | Optical contrast (%) | Response time (s) | Coloration efficiency (cm2·C−1) |
|---|---|---|---|---|
| P1 | 375 | 7.0 | 0.92 | 77.00 |
| 615 | 11.1 | 0.70 | 72.97 | |
| 1,500 | 50.0 | 1.35 | 297.61 | |
| P2 | 415 | 10.0 | 0.62 | 78.45 |
| 556 | 14.5 | 0.70 | 82.67 | |
| 1,550 | 61.5 | 1.80 | 323.25 | |
| P3 | 408 | 11.1 | 1.30 | 80.65 |
| 830 | 17.0 | 0.81 | 98.88 | |
| 1,630 | 60.2 | 1.40 | 279.50 | |
| P4 | 422 | 18.6 | 3.33 | 106.76 |
| 754 | 17.1 | 2.42 | 106.72 | |
| 1,660 | 80.0 | 2.20 | 340.15 | |
| P(A) (a) | 408 | 16.7 | 0.61 | 87 |
| 820 | 25.9 | 0.55 | 161.8 | |
| 2,000 | 61.3 | 0.35 | 309.6 | |
| P(B) (b) | 1,490 | 50.0 | - | - |
| 2,000 | 18.0 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Liu, X.; Wang, M.; Liu, X.; Zhao, J. Low Band Gap Donor–Acceptor Type Polymers Containing 2,3-Bis(4-(decyloxy)phenyl)pyrido[4,3-b]pyrazine as Acceptor and Different Thiophene Derivatives as Donors. Polymers 2016, 8, 377. https://doi.org/10.3390/polym8100377
Zhang Y, Liu X, Wang M, Liu X, Zhao J. Low Band Gap Donor–Acceptor Type Polymers Containing 2,3-Bis(4-(decyloxy)phenyl)pyrido[4,3-b]pyrazine as Acceptor and Different Thiophene Derivatives as Donors. Polymers. 2016; 8(10):377. https://doi.org/10.3390/polym8100377
Chicago/Turabian StyleZhang, Yan, Xuezhong Liu, Min Wang, Xiaoli Liu, and Jinsheng Zhao. 2016. "Low Band Gap Donor–Acceptor Type Polymers Containing 2,3-Bis(4-(decyloxy)phenyl)pyrido[4,3-b]pyrazine as Acceptor and Different Thiophene Derivatives as Donors" Polymers 8, no. 10: 377. https://doi.org/10.3390/polym8100377