
Preparation and Characterization of High Surface Area Activated Carbon Fibers from Lignin

Abstract
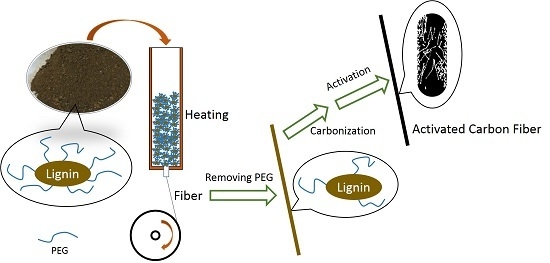
:
1. Introduction
2. Materials and Methods
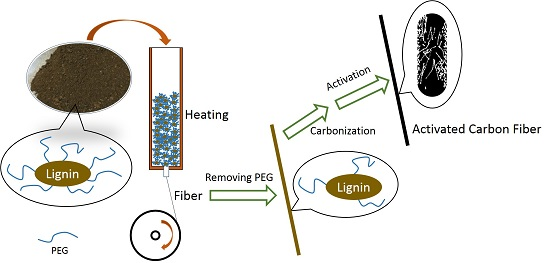
2.1. ACFs Preparation
2.2. Characterization
2.2.1. Scanning Electronic Microscopy (SEM)
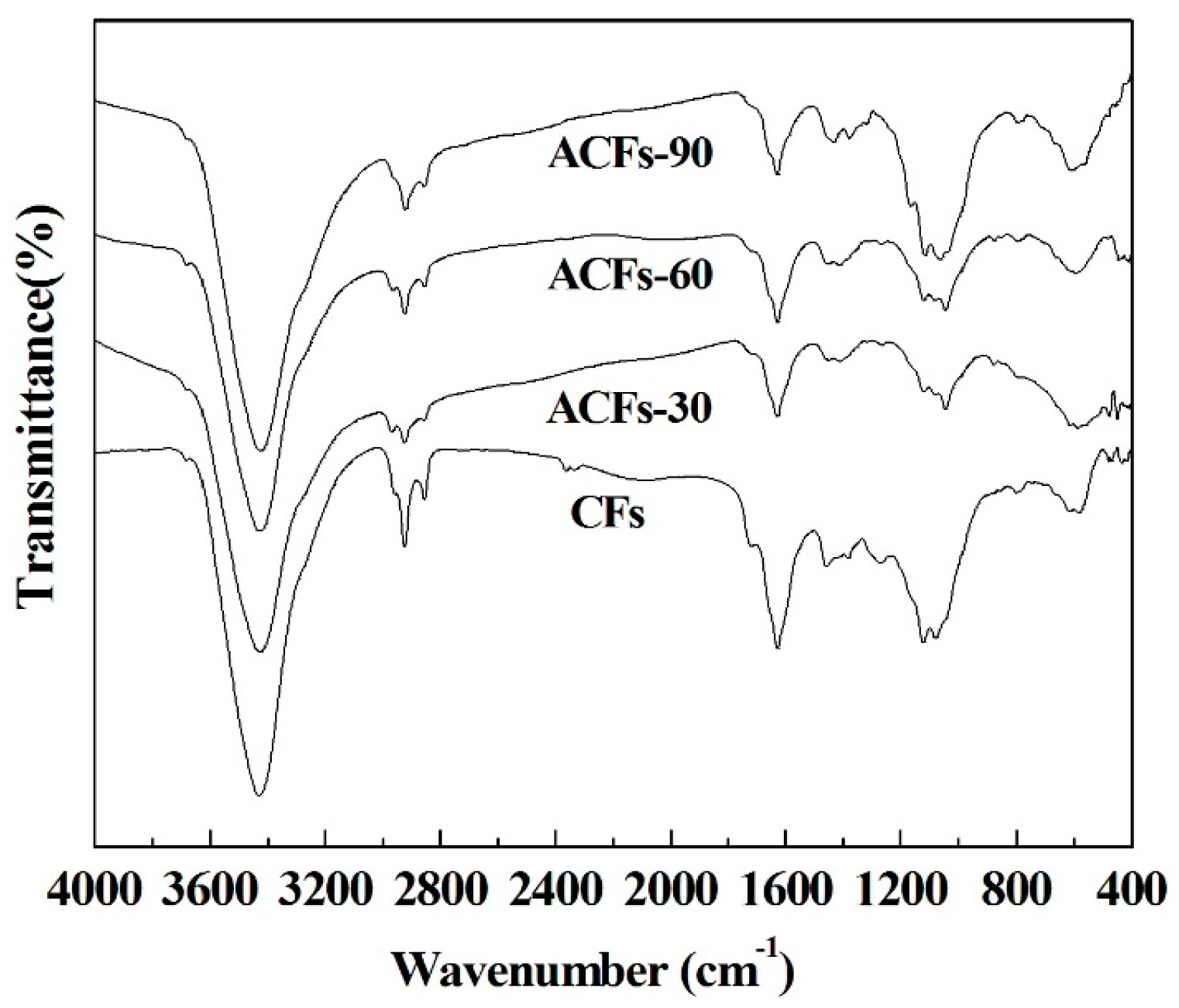
2.2.2. Fourier Transform Infrared Spectroscopy (FTIR)
2.2.3. Porous Measurement
2.2.4. X-ray Photoelectron Spectroscopy (XPS)
2.2.5. Methylene Blue Adsorption Capacity
3. Results
3.1. Yield and Morphologies of ACFs
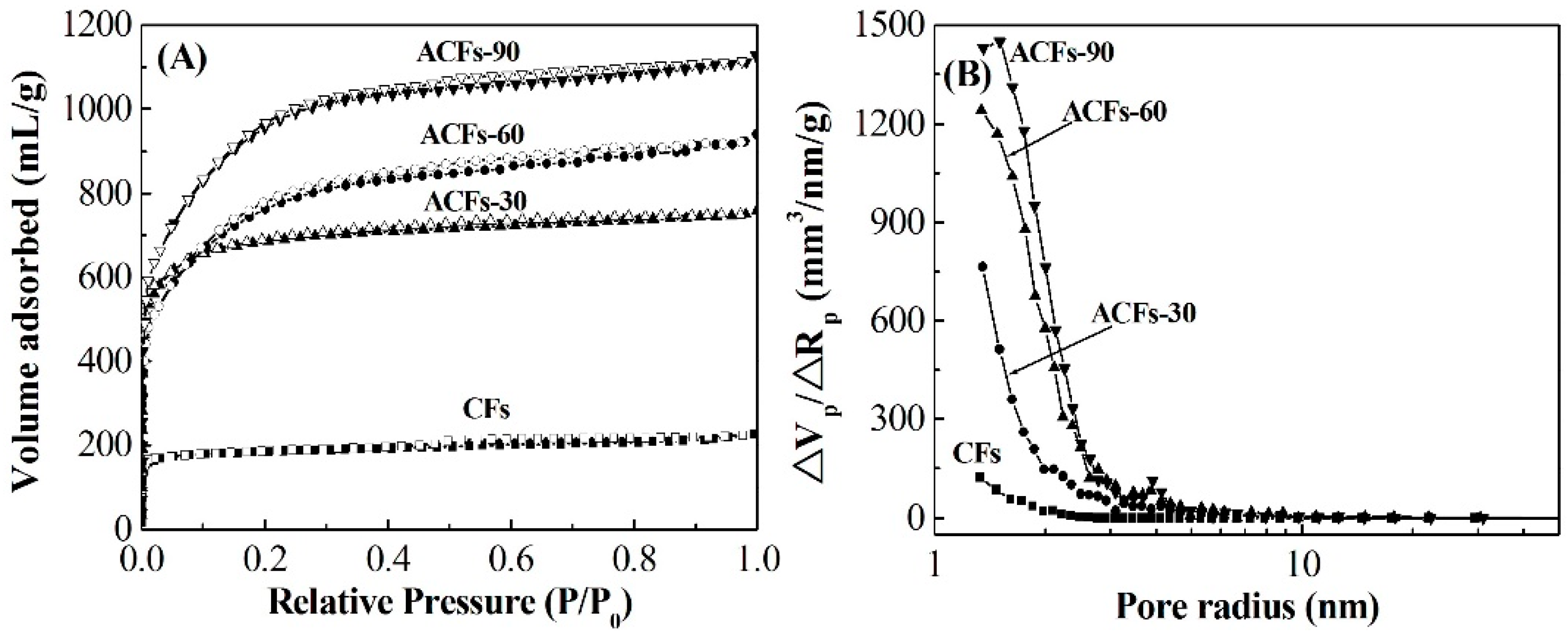
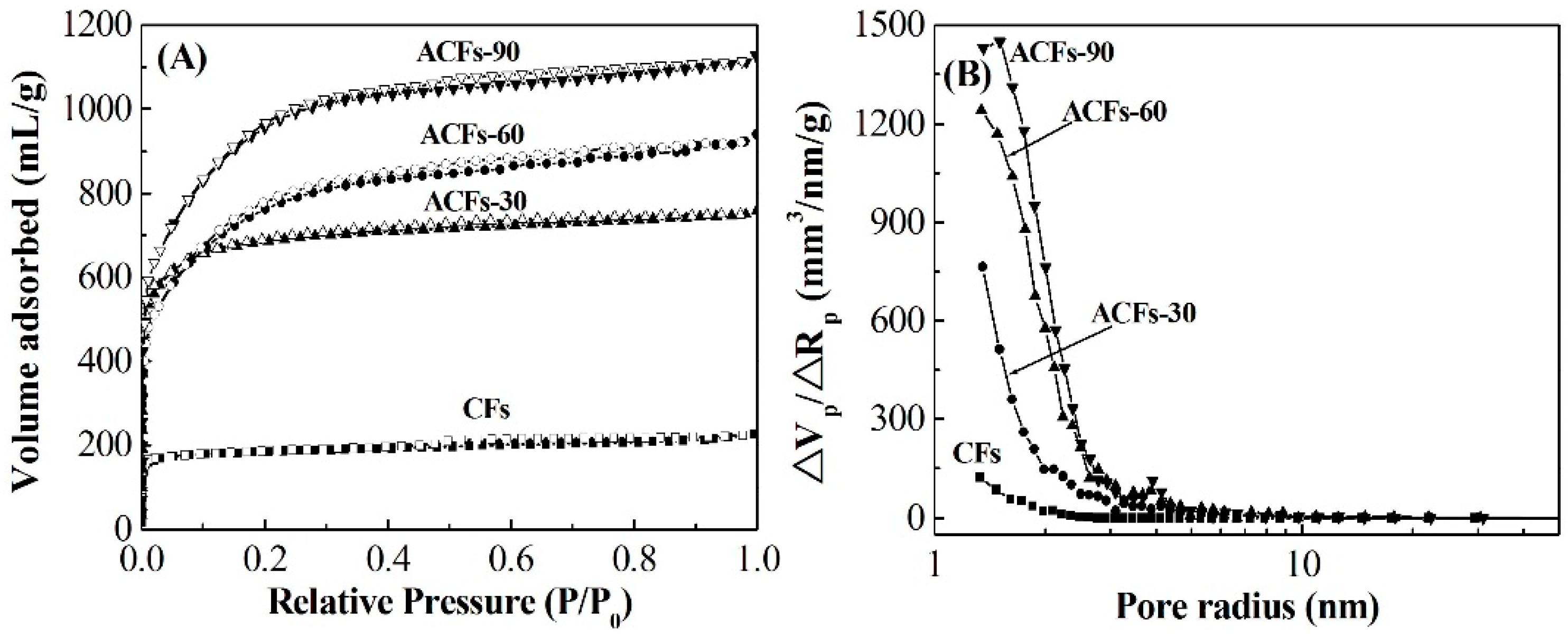
3.2. Pore Structure of ACFs
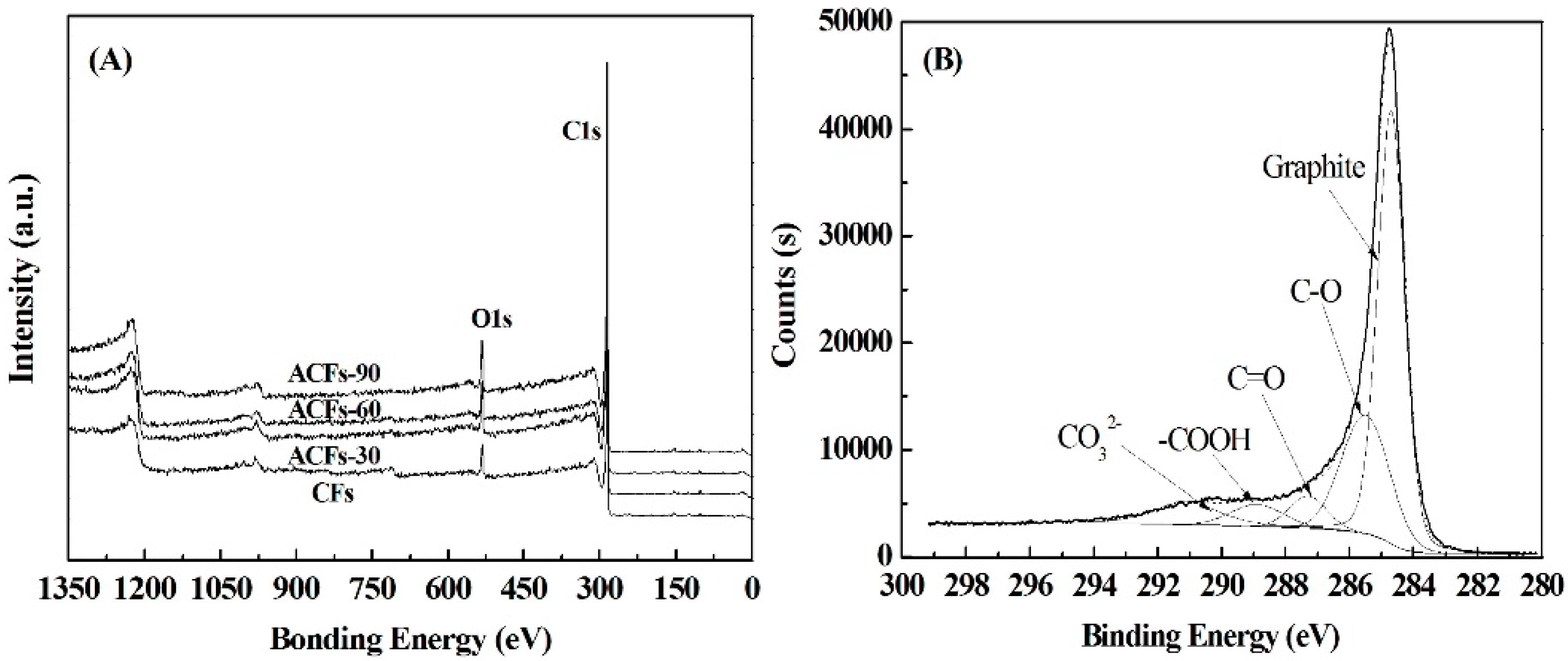
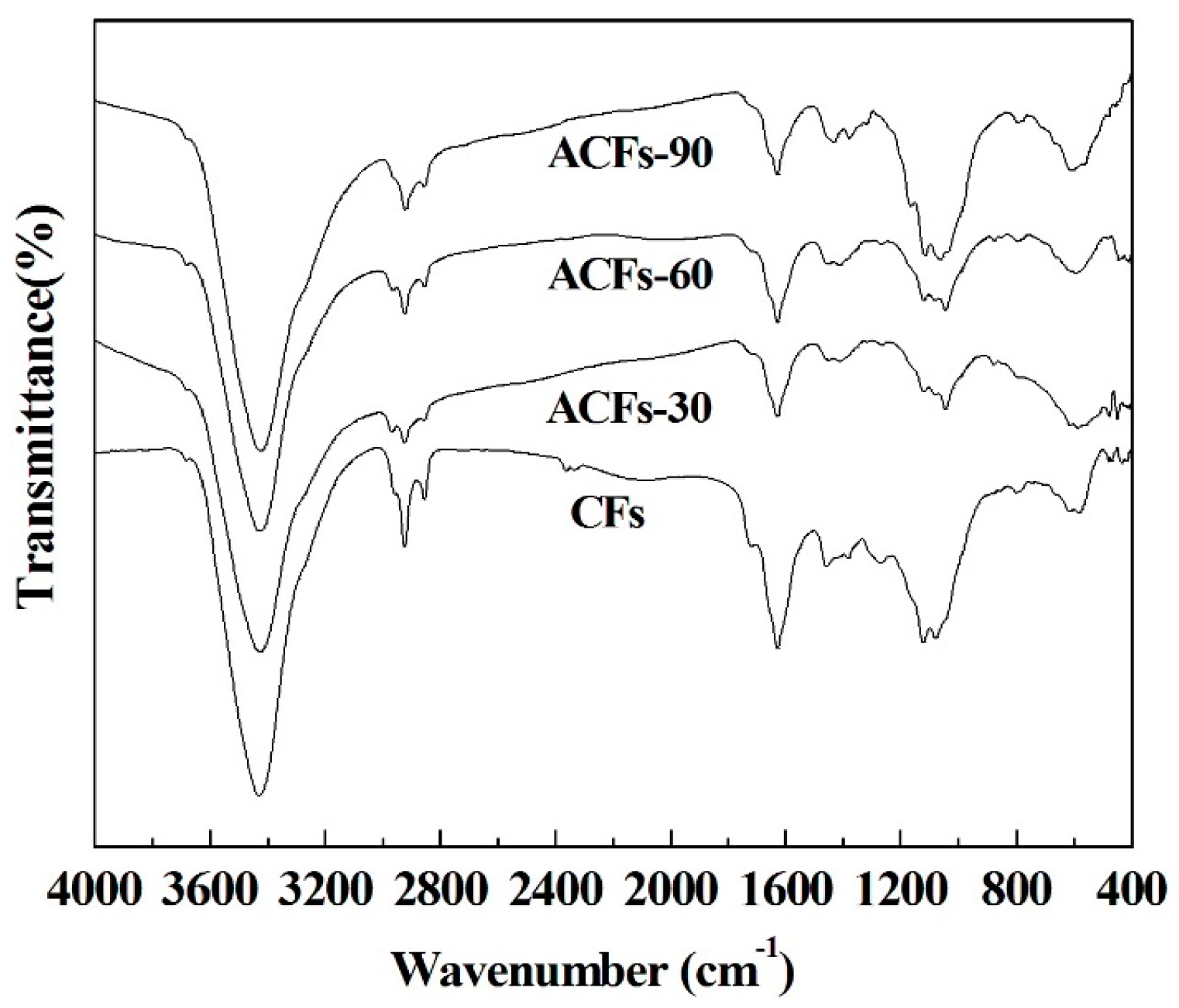
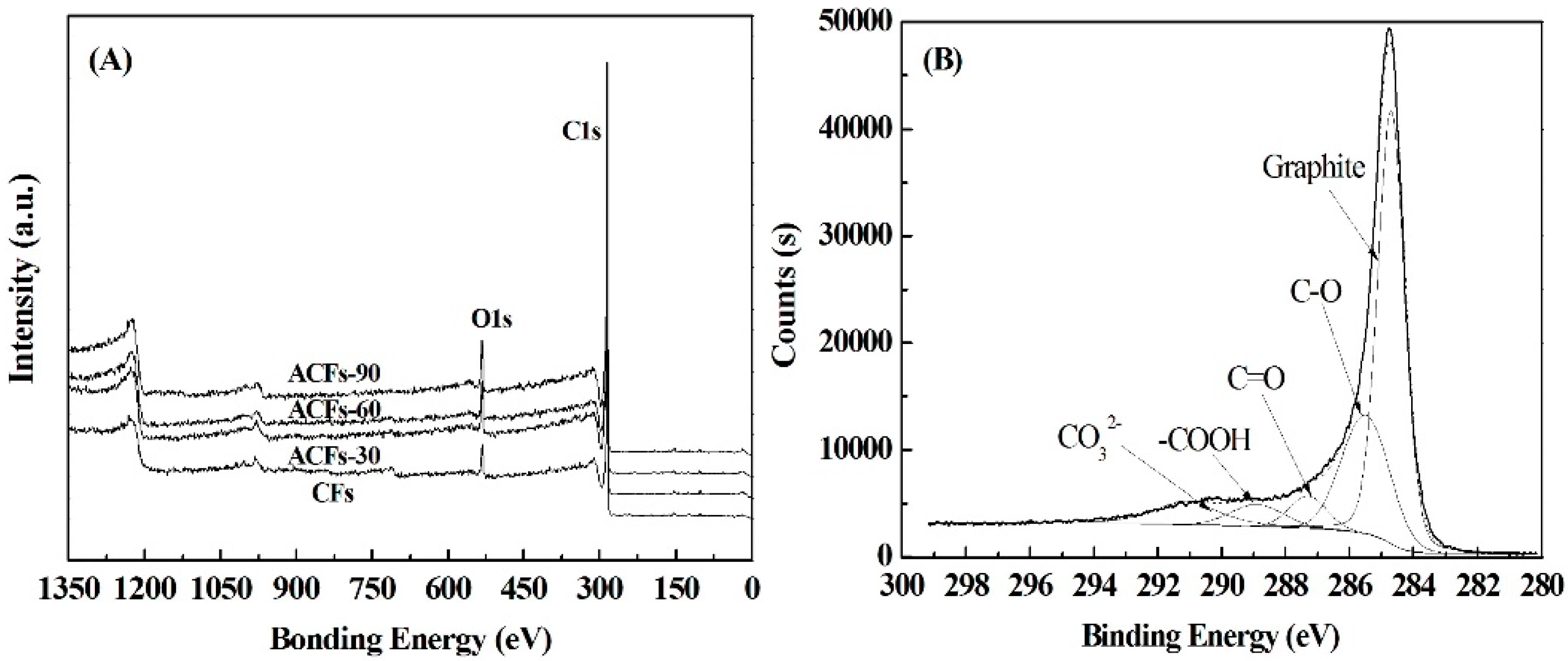
3.3. Chemical Compositions of ACFs
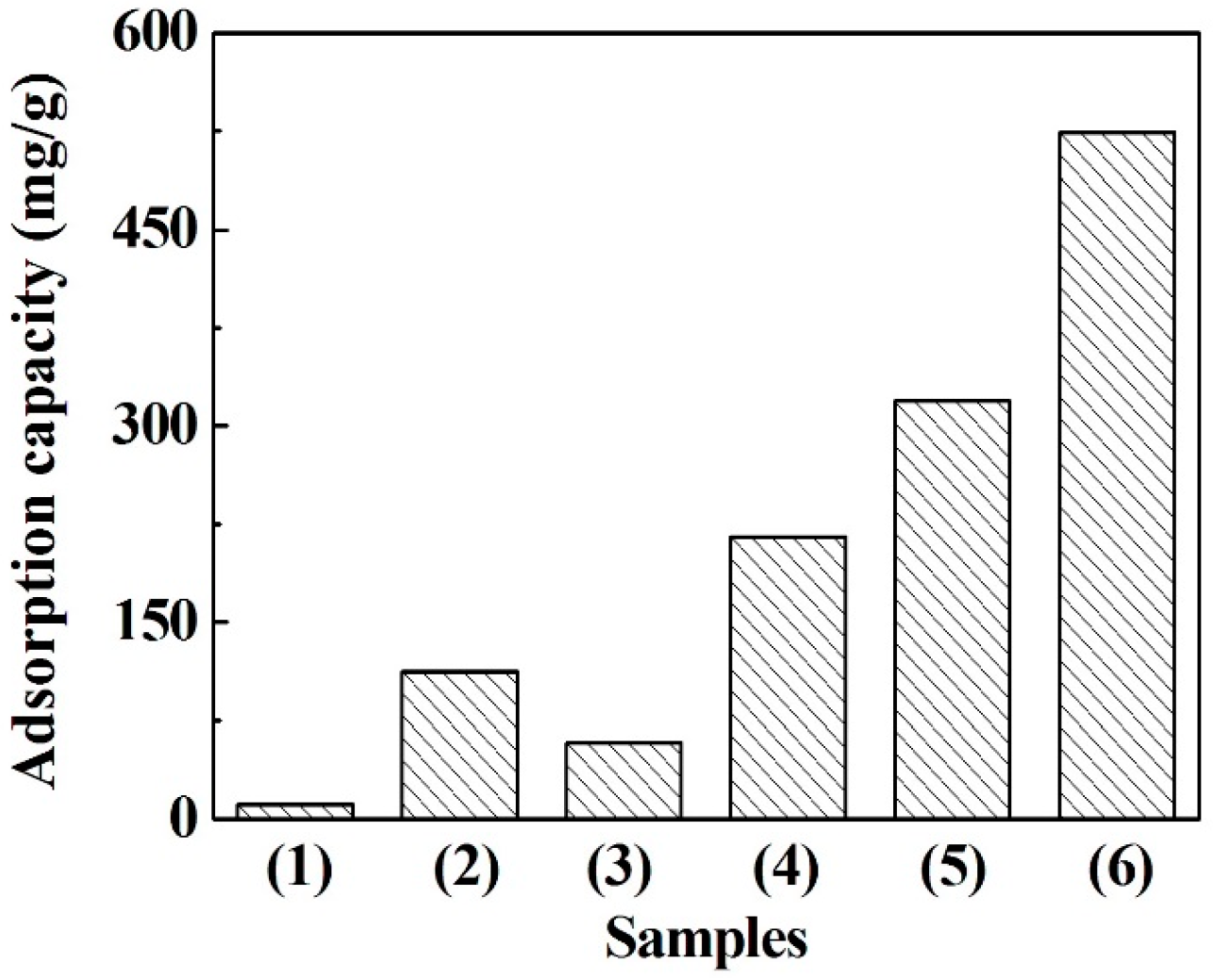
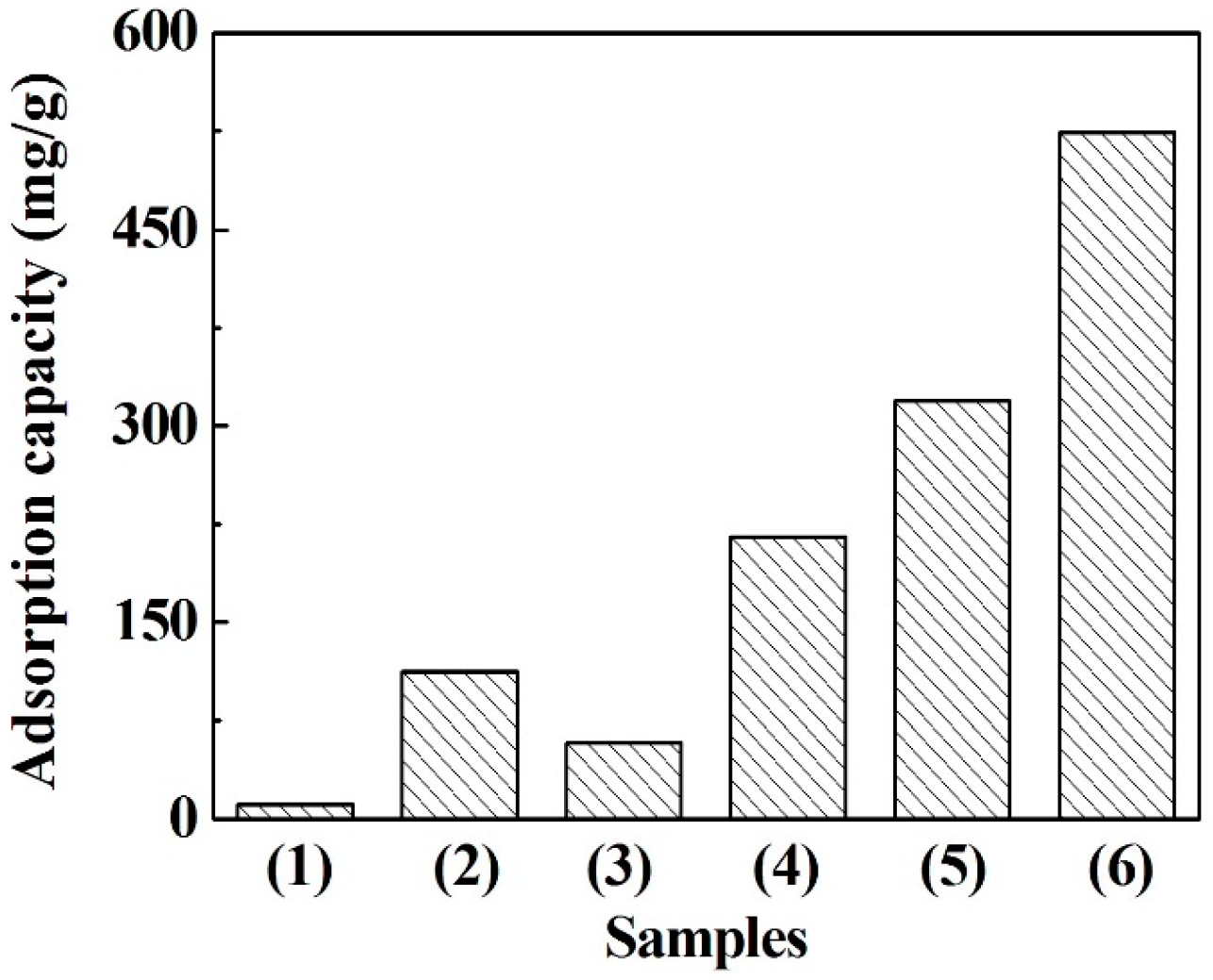
3.4. Adsorption of Methylene Blue
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Suzuki, M. Activated carbon fiber: Fundamentals and applications. Carbon 1994, 32, 577–586. [Google Scholar] [CrossRef]
- Jain, A.; Balasubramanian, R.; Srinivasan, M.P. Hydrothermal conversion of biomass waste to activated carbon with high porosity: A review. Chem. Eng. J. 2016, 283, 789–805. [Google Scholar] [CrossRef]
- Ryu, S.K.; Kim, S.Y.; Gallego, N.; Edie, D.D. Physical properties of silver-containing pitch-based activated carbon fibers. Carbon 1999, 37, 1619–1625. [Google Scholar] [CrossRef]
- Sun, J.F.; He, C.J.; Zhu, S.J.; Wang, Q.R. Effects of oxidation time on the structure and properties of polyacrylonitrile-based activated carbon hollow fiber. J. Appl. Polym. Sci. 2007, 106, 470–474. [Google Scholar] [CrossRef]
- Zeng, F.L.; Pan, D. The structural transitions of rayon under the promotion of a phosphate in the preparation of ACF. Cellulose 2008, 15, 91–99. [Google Scholar] [CrossRef]
- Mangun, C.L.; Benak, K.R.; Economy, J.; Foster, K.L. Surface chemistry, pore sizes and adsorption properties of activated carbon fibers and precursors treated with ammonia. Carbon 2001, 39, 1809–1820. [Google Scholar] [CrossRef]
- Kubo, S.; Kadla, J.F. Lignin-based carbon fibers: Effect of synthetic polymer blending on fiber properties. J. Polym. Environ. 2005, 13, 97–104. [Google Scholar] [CrossRef]
- Kadla, J.F.; Kubo, S.; Venditti, R.A.; Gilbert, R.D. Novel hollow core fibers prepared from lignin polypropylene blends. J. Appl. Polym. Sci. 2002, 85, 1353–1355. [Google Scholar] [CrossRef]
- Kadla, J.F.; Kubo, S.; Venditti, R.A.; Gilbert, R.D.; Compere, A.L.; Griffith, W. Lignin-based carbon fiber for composite fiber applications. Carbon 2002, 40, 2913–2920. [Google Scholar] [CrossRef]
- Baker, D.A.; Gallego, N.C.; Baker, F.S. On the characterization and spinning of an organic-purified lignin toward the manufacture of low-cost carbon fiber. J. Appl. Polym. Sci. 2012, 124, 227–234. [Google Scholar] [CrossRef]
- Norberg, I.; Nordstrom, Y.; Drougge, R.; Gellerstedt, G.; Sjoholm, E. A new method for stabilizing softwood kraft lignin fibers for carbon fiber production. J. Appl. Polym. Sci. 2013, 128, 3824–3830. [Google Scholar] [CrossRef]
- Sudo, K.; Shimizu, K. A new carbon fiber from lignin. J. Appl. Polym. Sci. 1992, 44, 127–134. [Google Scholar] [CrossRef]
- Sudo, K.; Shimizu, K.; Nakashima, N.; Yokoyama, A. A new modification method of exploded lignin for the preparation of a carbon fiber precursor. J. Appl. Polym. Sci. 1993, 48, 1485–1491. [Google Scholar] [CrossRef]
- Uraki, Y.; Kubo, S.; Nigo, N.; Sano, Y.; Sasaya, T. Preparation of carbon fibers from organosolv lignin obtained by aqueous acetic acid pulping. Holzforschung 1995, 49, 343–350. [Google Scholar] [CrossRef]
- Kubo, S.; Uraki, Y.; Sano, Y. Preparation of carbon fibers from softwood lignin by atmospheric acetic acid pulping. Carbon 1998, 36, 1119–1124. [Google Scholar] [CrossRef]
- Lin, J.; Kubo, S.; Yamada, T.; Koda, K.; Uraki, Y. Chemical thermostabilization for the preparation of carbon fibers from softwood lignin. BioResources 2012, 7, 5634–5646. [Google Scholar] [CrossRef]
- Uraki, Y.; Kubo, S.; Kurakami, H.; Sano, Y. Activated carbon fibers from acetic acid lignin. Holzforschung 1997, 51, 188–192. [Google Scholar] [CrossRef]
- Uraki, Y.; Nakatani, A.; Kubo, S.; Sano, Y. Preparation of activated carbon fibers with large specific surface area from softwood acetic acid lignin. J. Wood Sci. 2001, 47, 465–469. [Google Scholar] [CrossRef]
- Yamada, T.; Aratani, M.; Kubo, S. Chemical analysis of the product in acid-catalyzed solvolysis of cellulose using polyethylene glycol and ethylene carbonate. J. Wood Sci. 2007, 53, 487–493. [Google Scholar] [CrossRef]
- Okada, K.; Yamamoto, N.; Kameshima, Y.; Yasumori, A. Porous properties of activated carbons from waste newspaper prepared by chemical and physical activation. J. Colloid Interface Sci. 2003, 262, 179–193. [Google Scholar] [CrossRef]
- Azargohar, R.; Dalai, A.K. Steam and KOH activation of biochar: Experimental and modeling studies. Microporous Mesoporous Mater. 2008, 110, 413–421. [Google Scholar] [CrossRef]
- Shimada, M.; Iida, T.; Kawarada, K.; Chiba, Y.; Mamoto, T.; Okayama, T. Porous structure of activated carbon prepared from waste newspaper. J. Mater. Cycles Waste Manag. 2000, 2, 100–108. [Google Scholar]
- Rodriguez-Reinoso, F.; Lopez-Gonzalez, J.D.; Berenguer, C. Activated carbon from almost shells. Carbon 1982, 20, 513–518. [Google Scholar] [CrossRef]
- Lastoskie, C.; Gubbins, K.E.; Quirke, N. Pore size distribution analysis of microporous carbon: A density functional theory approach. J. Phys. Chem. 1993, 97, 4786–4796. [Google Scholar] [CrossRef]
- Christina, C.; Small, Z.H.; Ulrich, A.C. Preparation and characterization of activated carbon from oil sands coke. Fuel 2012, 92, 69–76. [Google Scholar]
- Shimada, M.; Hamabe, H.; Iida, T.; Kawarada, K.; Okayama, T. The properties of activated carbon made from waste newsprint paper. J. Porous Mater. 1999, 6, 191–196. [Google Scholar] [CrossRef]
- Qin, W.; Kadla, J.F. Carbon fibers based on pyrolytic lignin. J. Appl. Polym. Sci. 2011, 126, E203–E212. [Google Scholar] [CrossRef]
- Ishii, C.; Suzuki, T.; Shindo, N.; Kaneko, K. Structural characterization of heat-treated activated carbon fibers. J. Porous Mater. 1997, 4, 181–186. [Google Scholar] [CrossRef]
- Sánchez-Montero, M.J.; Salvador, F.; Izquierdo, C. Reactivity and porosity of a carbon fiber activated with supercritical CO2. J. Phys. Chem. C 2008, 112, 4991–4999. [Google Scholar] [CrossRef]
- Rong, H.Q.; Ryu, Z.Y.; Zheng, J.T.; Zhang, Y.L. Effect of air oxidation of rayon-based activated carbon fibers on the adsorption behavior for formaldehyde. Carbon 2002, 40, 2291–2300. [Google Scholar] [CrossRef]
- Tavanai, H.; Jalili, R.; Morshed, M. Effects of fiber diameter and CO2 activation temperature on the pore characteristics of polyacrylonitrile based activated carbon nanofibers. Surface Interface Anal. 2009, 41, 814–819. [Google Scholar] [CrossRef]
- Alcaniz-monge, J.; De La Casa-Lillo, M.A.; Cazorla-amoros, D.; Linares-solano, A. Methane storage in activated carbon fibers. Carbon 1997, 35, 291–297. [Google Scholar] [CrossRef]
- Shimada, M.; Takigami, S.; Nakamura, Y.; Abe, Y.; Iizuka, T.; Makiyama, N. Efficiently activated carbon fiber derived from grafted novoloid fiber. J. Appl. Polym. Sci. 1993, 48, 1121–1126. [Google Scholar] [CrossRef]
- Chiang, Y.; Lee, C.; Lee, H. Characterization of microstructure and surface properties of heat-treated PAN-and rayon-based activated carbon fibers. J. Porous Mater. 2007, 14, 227–237. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Infrared Spectrometry. In Spectrometric Identification of Organic Compounds; Robert, M.S., Ed.; Wiley: Hoboken, NJ, USA, 2005; pp. 72–126. [Google Scholar]
- Myglovets, M.; Poddubnaya, O.I.; Sevastyanova, O.; Lindstrom, M.E.; Gawdzik, B.; Sobiesiak, M.; Tsyba, M.M.; Sapsay, V.I.; Klymchuk, D.O.; Puziy, A.M. Preparation of carbon adsorbents from lignosulfonate by phosphoric acid activation for the adsorption of metal ions. Carbon 2014, 80, 771–783. [Google Scholar] [CrossRef]
- Yacob, A.R.; Majid, Z.A.; Dasril, R.S.D.; Inderan, V. Comparison of various sources of high surface area carbon prepared by different types of activation. Malays. J. Anal. Sci. 2008, 12, 264–271. [Google Scholar]
- Ma, X.J.; Yang, H.M.; Yu, L.L.; Chen, Y.; Li, Y. Preparation, surface and pore structure of high surface area activated carbon fibers from bamboo by steam activation. Materials 2014, 7, 4431–4441. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Yield 1 (%) | SBET (m2/g) | Smicro (m2/g) | Smeso (m2/g) | Vtot (mL/g) | D 2 (nm) |
|---|---|---|---|---|---|---|
| CFs | 100 | 775 | 741 | 34 | 0.410 | 1.9 |
| ACFs-30 | 54.4 | 2064 | 1910 | 127 | 1.029 | 2.3 |
| ACFs-60 | 19.5 | 2490 | 2200 | 246 | 1.153 | 2.4 |
| ACFs-90 | 12.9 | 3110 | 2830 | 231 | 1.472 | 2.3 |
| Samples | C (%) | O (%) | N (%) | S (%) | O/C (%) |
|---|---|---|---|---|---|
| CFs | 93.5 | 5.7 | 0.7 | 0.1 | 6.1 |
| ACFs-30 | 93.9 | 5.6 | 0.5 | 0.1 | 6.0 |
| ACFs-60 | 94.3 | 5.1 | 0.6 | 0.1 | 5.4 |
| ACFs-90 | 93.5 | 6.0 | 0.5 | 0.1 | 6.4 |
| Samples | Graphite C–C (CP1) | C–OH (CP2) | C=O (CP3) | C–OOH (CP4) | CO32− (CP5) |
|---|---|---|---|---|---|
| CFs | 68.5 | 20.2 | 8.0 | 1.9 | 1.4 |
| ACFs-30 | 58.7 | 21.2 | 8.1 | 3.6 | 8.3 |
| ACFs-60 | 57.2 | 21.7 | 7.7 | 4.0 | 9.4 |
| ACFs-90 | 55.7 | 25.2 | 5.4 | 5.2 | 8.4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Zhao, G. Preparation and Characterization of High Surface Area Activated Carbon Fibers from Lignin. Polymers 2016, 8, 369. https://doi.org/10.3390/polym8100369
Lin J, Zhao G. Preparation and Characterization of High Surface Area Activated Carbon Fibers from Lignin. Polymers. 2016; 8(10):369. https://doi.org/10.3390/polym8100369
Chicago/Turabian StyleLin, Jian, and Guangjie Zhao. 2016. "Preparation and Characterization of High Surface Area Activated Carbon Fibers from Lignin" Polymers 8, no. 10: 369. https://doi.org/10.3390/polym8100369