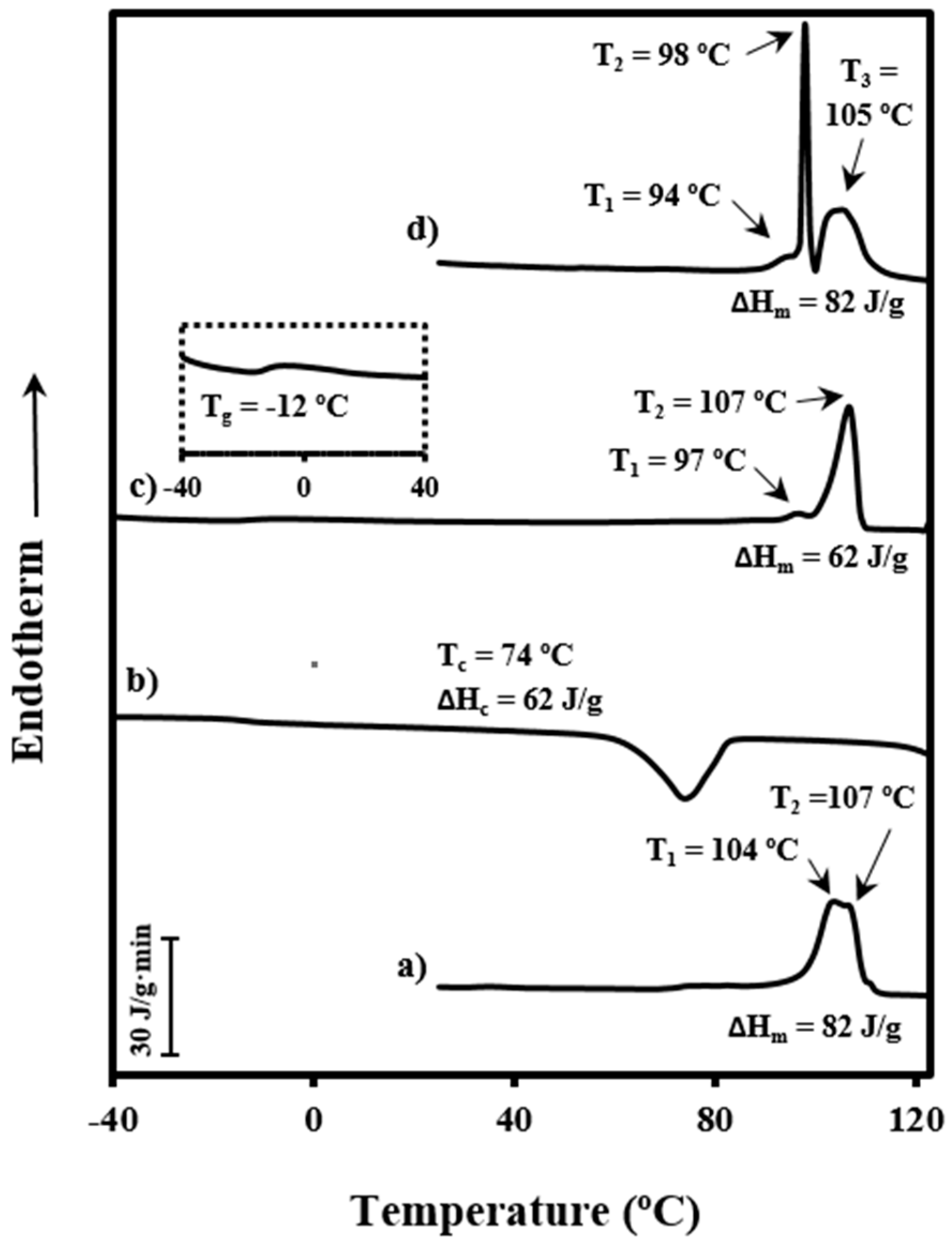
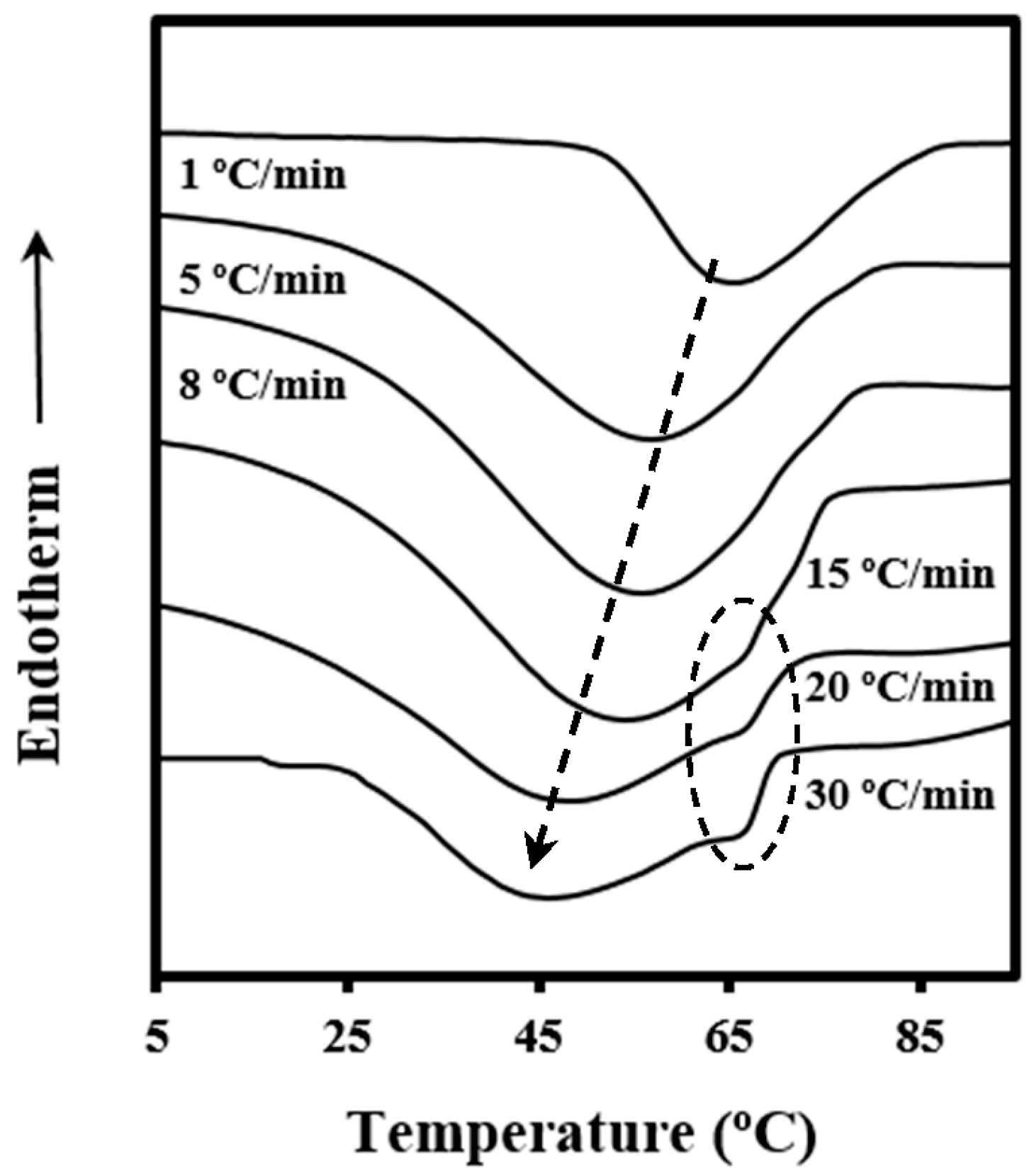
3.2. Non-Isothermal Kinetic Analysis of Poly(p-dioxanone) Melt Crystallization from DSC Data
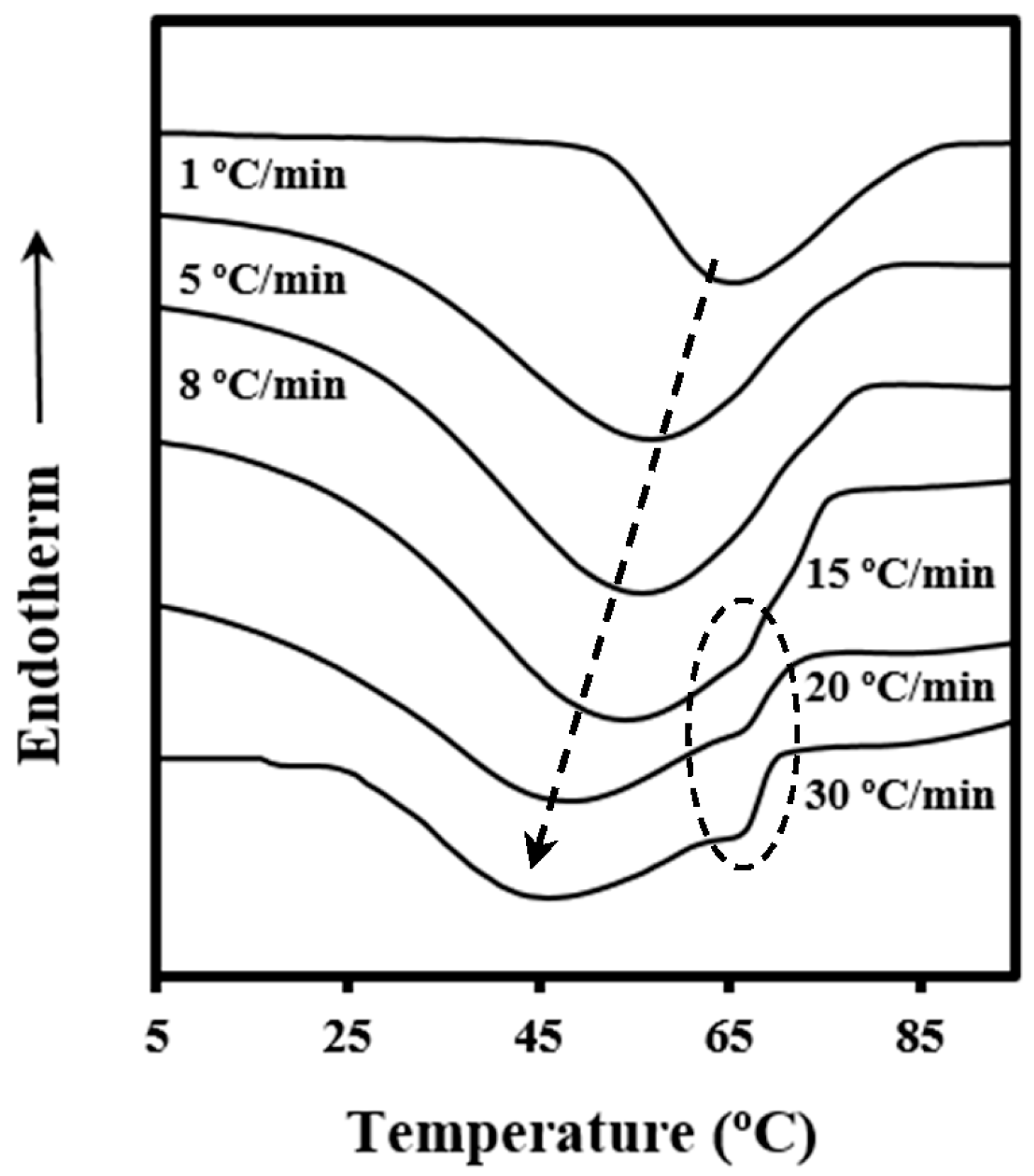
The process of crystallization under non-isothermal conditions is rather complicated to be analyzed since crystallization from the melt takes place under different degrees of supercooling, and therefore caution should be taken when interpreting experimental results.
Calorimetric data were used to determine the relative degree of crystallinity at any temperature,
χ(
T), for all cooling rates by the expression:
where d
Hc is the enthalpy of crystallization released within an infinitesimal temperature range d
T,
T0 denotes the initial crystallization temperature and
Tc and
T∞ are the crystallization temperatures at time
t and after completion of the crystallization process, respectively. Thus, the denominator corresponds to the overall enthalpy of crystallization for specific heating/cooling conditions.
The relative degree of crystallinity was calculated as a function of time by the relationship
where
T0 is the temperature at which crystallization begins (
t =
t0) and
φ is the value of the cooling rate.
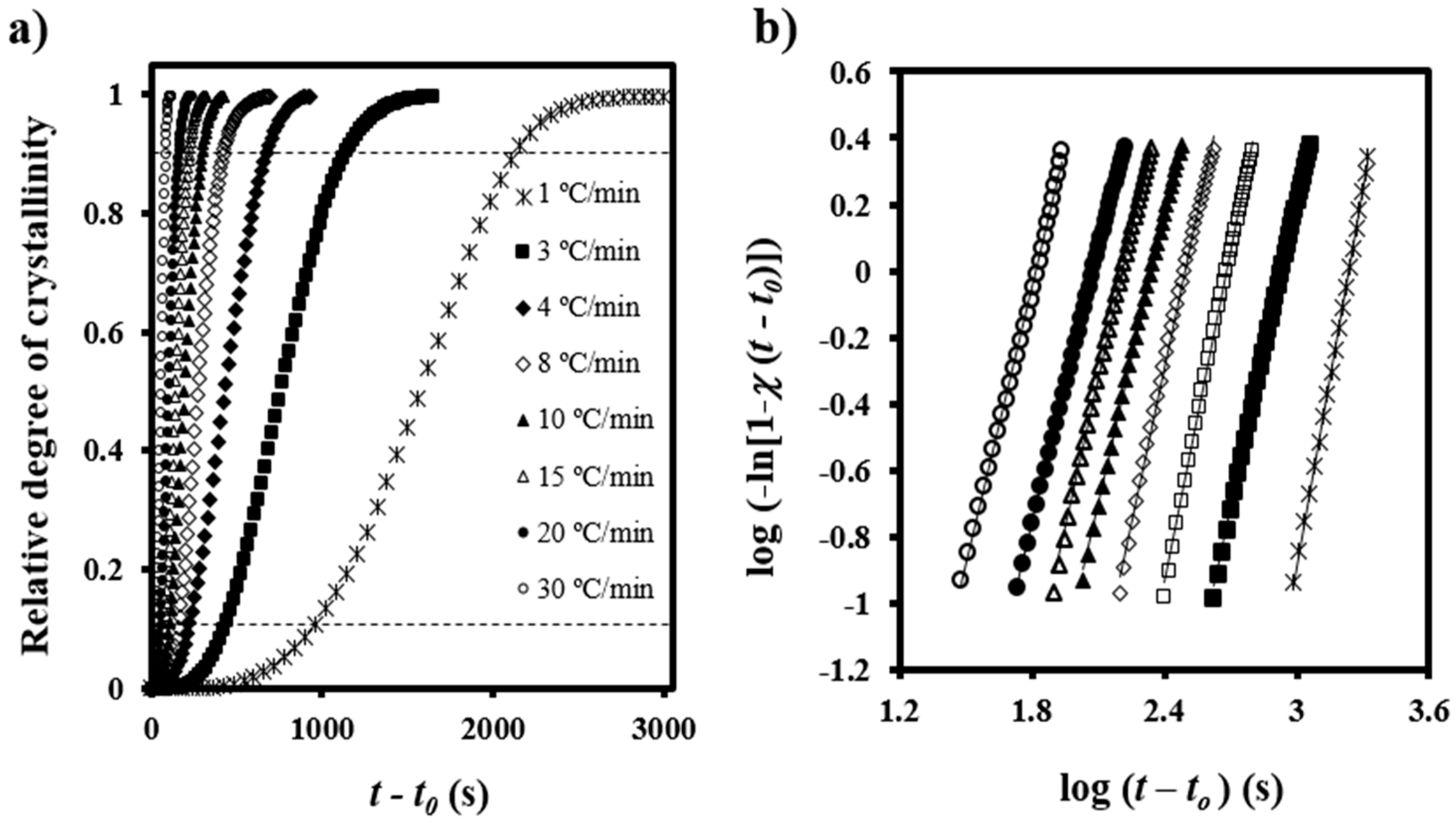
Figure 3a illustrates the variation of the time-dependent degree of crystallinity,
χ(
t −
t0), at different cooling rates, which allows a typical Avrami analysis to be performed [
20] according to the equation
where
Z is the rate constant and
n is the Avrami exponent. A normalized rate constant,
k =
Z1/n, is however more useful for comparison because its dimension (time
−1) becomes independent of the Avrami exponent.
Figure 3b shows the plots of log{−ln[1 −
χ(
t −
t0)]} versus log(
t − t0) at different cooling rates. A good linearity was observed between the relative degree of crystallinities of 0.10 and 0.90, that is, after formation of well-defined spherulitic morphologies and before occurrence of a secondary crystallization caused by the impingement of spherulites (see dashed lines in
Figure 3a).
Table 1 summarizes the main kinetic parameters calculated by the Avrami analysis. As known from isothermal studies, the normalized rate constant was low (i.e., between 0.58 × 10
−3 s
−1 and 15.26 × 10
−3 s
−1) and increased with the cooling rate. Avrami exponents showed a moderate variation (i.e., between 3.76 and 2.77), with the lowest values being determined for high cooling rates and the average value being close to 3.0. These exponents are lower than those previously reported by Zhang et al. [
24] (i.e., 4.26–3.40) and in good agreement with those given by Andjelic et al. [
4] for low crystallization rates (i.e., 3.0), although in this case a value of 1.1 was found for high crystallization rates. Isothermal crystallization studies also indicate slightly contradictory values. Thus, minimum changes with crystallization temperature were determined by Andjelic et al. (i.e., exponents varied between 2.22 and 2.62, with 2.5 being the average value) [
4], but a systematic increase (i.e., from approximately 2 to 3.8) was also reported [
6] for higher isothermal crystallization temperatures (i.e., from 30 to 80 °C). The last behavior was interpreted as a consequence of a change from instantaneous to sporadic nucleation as
Tc was increased [
6].
It can be well stated that application of the Avrami equation under non-isothermal conditions merely corresponds to a mathematical fitting that allows appropriate values of the rate constant to be derived [
26,
27,
28]. In this case, it should be pointed out that the determined exponents may even have a physical meaning since they suggest three-dimensional spherulitic growth and instantaneous nucleation, as postulated from isothermal studies [
4,
6]. Furthermore, the sporadic nucleation detected at high isothermal crystallization temperatures [
6] is in agreement with the increase of the exponent observed at low crystallization rates (i.e., 3.76 at 1 °C/min) and supports DSC evidence of the occurrence of two crystallization processes.
The values of the corresponding reciprocal crystallization half times (1/
τ1/2), calculated as the inverse of the difference between crystallization starting time and crystallization half time, are also given in
Table 1. This parameter is a direct measure of the crystallization process, and could therefore be used to check the accuracy of Avrami analyses, as demonstrated by the excellent agreement with the theoretical kinetic value (i.e., 1/
τ1/2 = (
Z/ln2)
1/n). In conclusion, the deduced Avrami parameters are completely appropriate to simulate the non-isothermal crystallization process.
A kinetic equation that combines the Avrami [
20] and Ozawa [
17] expressions has been derived and applied in different non-isothermal studies [
29]:
where
F(
T) is a new kinetic parameter referring to the cooling rate which must be chosen at a unit crystallization time when the system reaches a certain crystallinity, and
a is the ratio of apparent Avrami and Ozawa exponents.
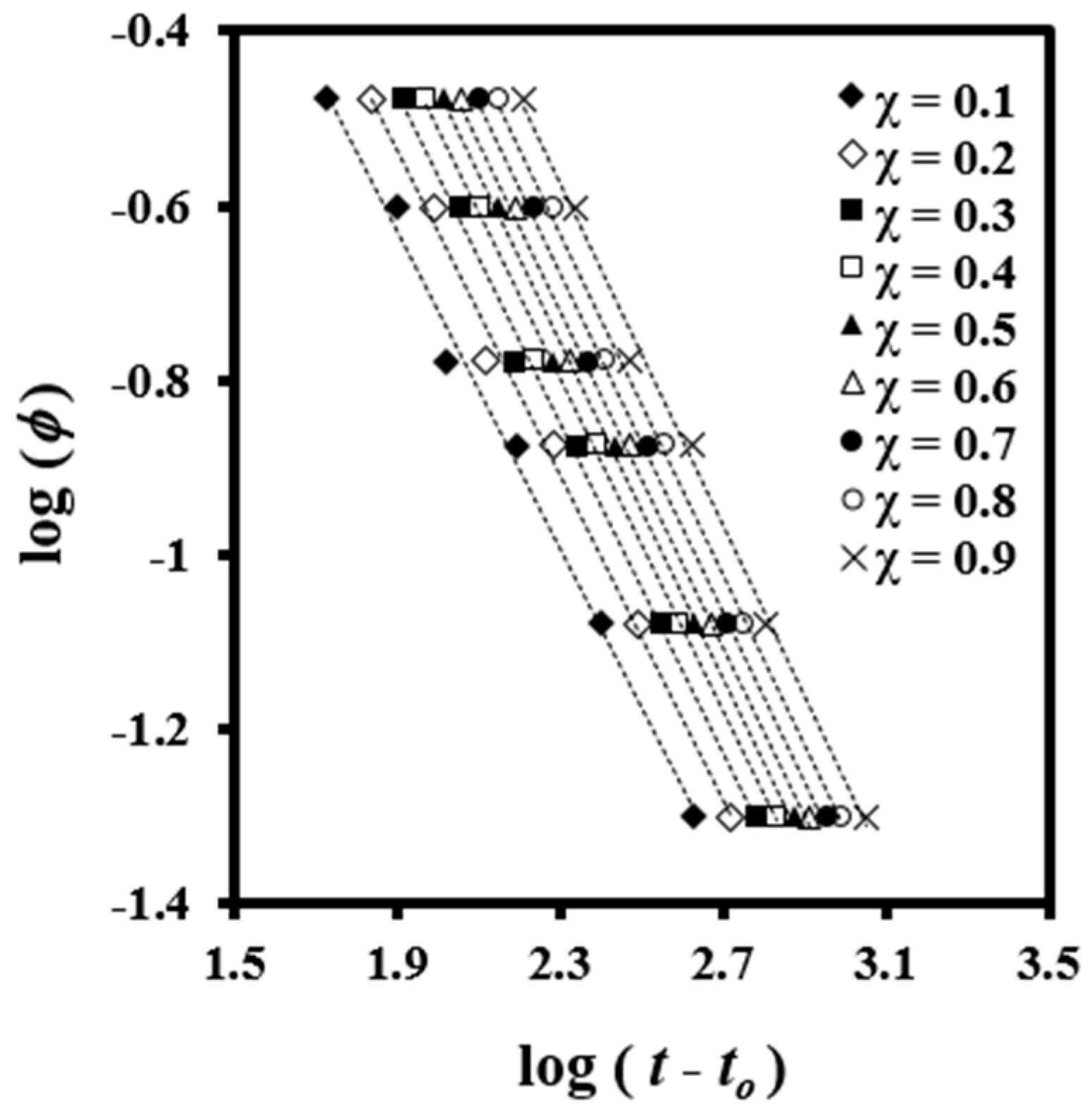
A plot of log
φ versus log (
t −
t0) yields a series of straight lines at a given value of
χ(
T) (
Figure 4), which suggest the validity of the combined equation for this system. Kinetic parameters can be estimated by the intercept and slope of these lines.
Results showed that
F(
T) values increased with crystallinity (
Table 2), which has a physical sense because the motion of the molecular chains became slower as the material crystallized and formation of new crystals was hindered. The values of
a were almost constant between 0.91 and 0.98 although slightly increased with the relative degree of crystallinity.
The crystallization process has non-Arrhenius behavior, and therefore a temperature-dependent
effective activation energy needs to be defined. The value corresponding to a given degree of crystallinity,
Eχ, can be determined by the Friedman isoconversional method [
30]:
where
A is a pre-exponential factor and
f[
χ] is the crystallization model. Values of ln[d
χ/d
t]
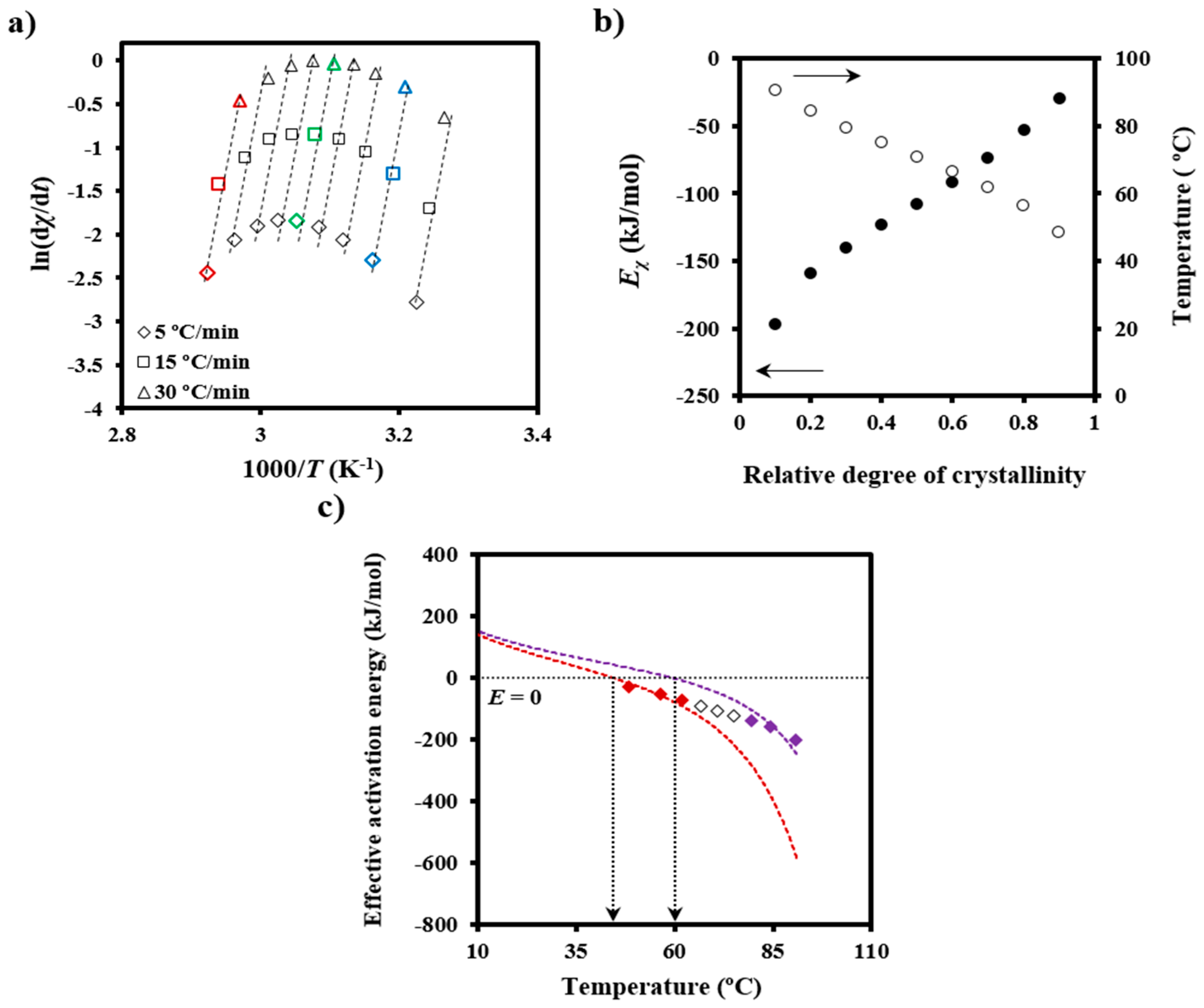
χ at different temperatures and degrees of crystallization can be obtained from crystallization experiments performed at different cooling rates. In this way, the slopes of the linear plots of ln[d
χ/d
t]
χ versus 1/
T (
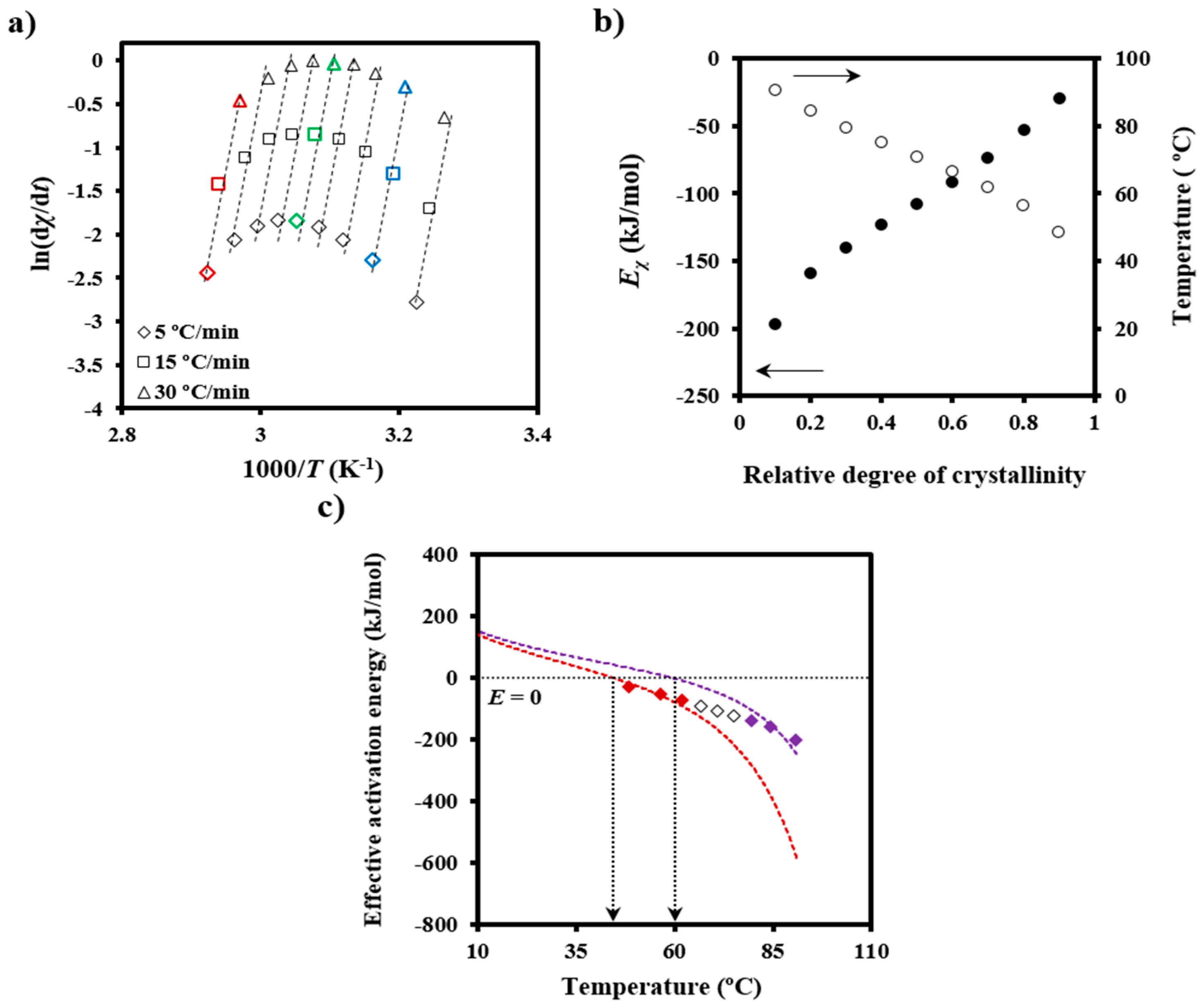
Figure 5a) allow
Eχ to be determined (
Figure 5b). The temperature dependence of the effective activation energy (
Figure 5c) could finally be derived by considering also the average temperature associated with a given conversion (
Figure 5b).
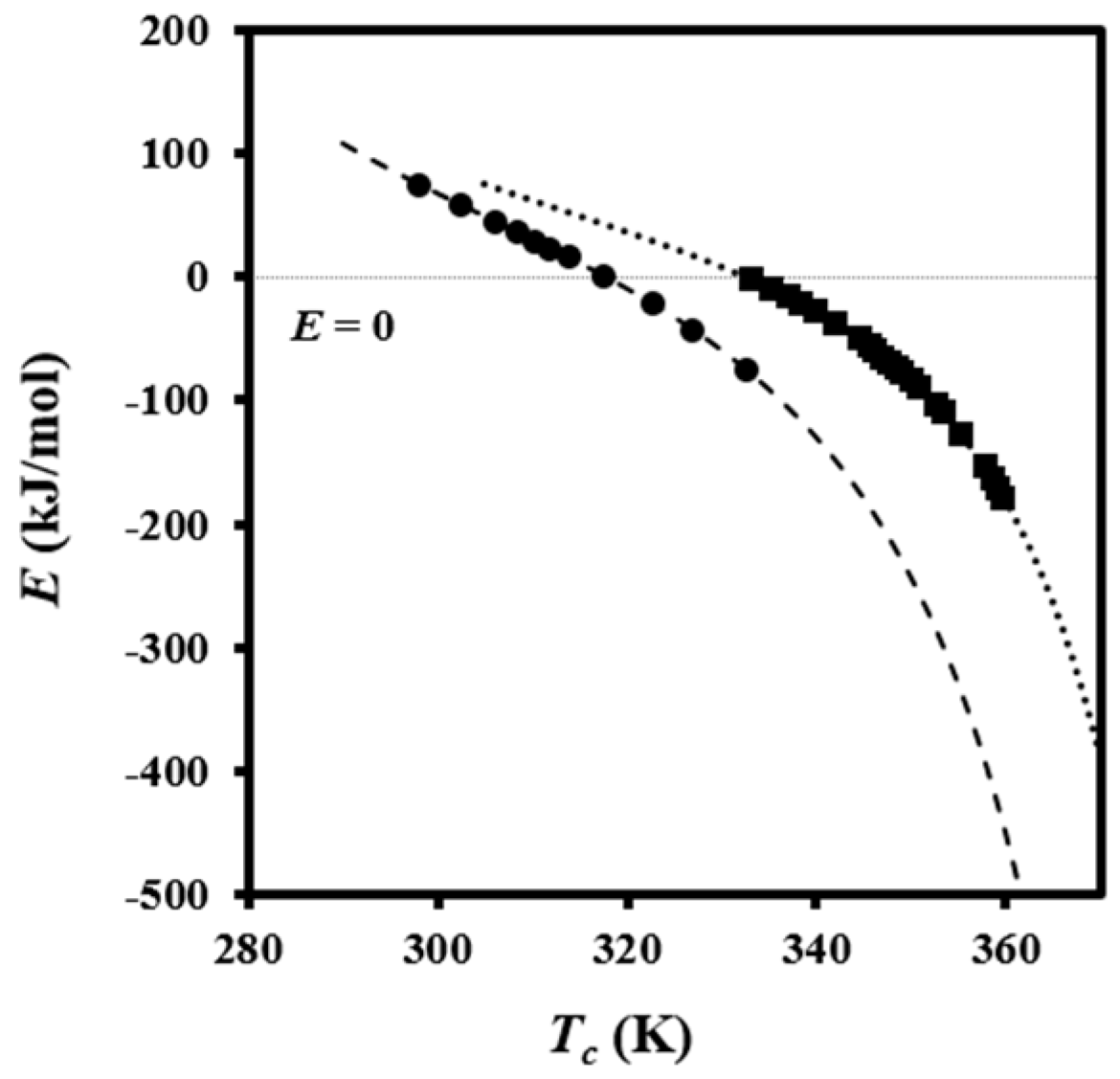
The effective activation energy was negative for crystallization experiments performed from the melt state and at low supercooling degrees (i.e., the temperature range where secondary nucleation plays a fundamental role), as shown in
Figure 5c. This energy increased progressively (i.e., the crystallization rate increased) as the temperature decreased, as discussed at length by Vyazovkin and Dranca [
31], reflecting the expected behavior for crystallizations performed at temperatures higher than those associated with the maximum crystallization rate.
Vyazovkin and Sbirrazzuoli proposed that crystallization parameters like the secondary nucleation constant should be derived through the effective activation energies [
31,
32,
33]:
where
G is the crystal growth rate,
U* represents the activation energy characteristic of the transport of crystallizing segments across the liquid-crystal interface,
T∞ is the temperature below which such motion ceases,
R is the gas constant,
Kg is the secondary nucleation constant, Δ
T is the degree of supercooling measured as
Tm −
Tc (where
Tm is the equilibrium melting temperature and
Tc is the crystallization temperature), and
f is a correction factor accounting for the variation in the bulk melting enthalpy per unit volume with temperature (
f = 2
Tc/(
Tm +
Tc)).
Figure 5c also compares the experimental
Eχ −
T plot with simulated ones using Equation (6),
U* and
T∞ values of 1600 cal/mol and
Tg-35 K, respectively (i.e., close to the universal values reported by Suzuki and Kovacs [
34]), the equilibrium melting temperature of 127 °C, as previously determined for PDO [
3], and representative
Kg values. In fact,
U* and
T∞ have little influence on a temperature range that is far from the glass transition temperature. The best fit between experimental and theoretical data was obtained considering two
Kg parameters (i.e., 3.07 × 10
5 K
2 and 1.42 × 10
5 K
2), which are in agreement with the two crystallization regimes reported for PDO from isothermal crystallization experiments [
3]. Note that the isoconversional analysis was able to detect the existence of several crystallization regimes and also to predict two maximum crystallization rates at temperatures of 45 and 60 °C, as deduced from the temperatures for each simulated curve where the effective activation energy was zero.
3.3. Non-Isothermal Kinetic Analysis of Poly(p-dioxanone) Melt Crystallization from Optical Microscopy Data
Non-isothermal crystallization of PDO rendered double banded spherulites with progressively decreasing periodicity (
Figure 6), which is in accordance with the continuous temperature decrease of a non-isothermal crystallization. In fact, morphologies obtained under isothermal conditions have been extensively studied [
3,
5,
6], and it was assumed that interband spacing decreased when crystallization temperature was lowered. Actually, two different banding periodicities could be detected where the broader bands had a negative birefringence. These kinds of double bands with uneven spacings are characteristic of spherulites having a biaxial indicatrix twisted about the optic normal [
6,
32,
35]. PDO spherulites were also characterized by their big size, which led to poor nucleation and slow growth rate.
Figure 6 also shows that the number of active nuclei increased during cooling, and consequently the size and morphology of the spherulites were not identical. Logically smaller spherulites with a practically indistinguishable double band texture were formed at lower temperatures (see yellow arrows).
Spherulitic growth rates (
G) were also determined for non-isothermal crystallization by measuring the change of the spherulite radius (
R) with temperature (
T) at a constant cooling/heating rate (d
T/d
t) [
36,
37]:
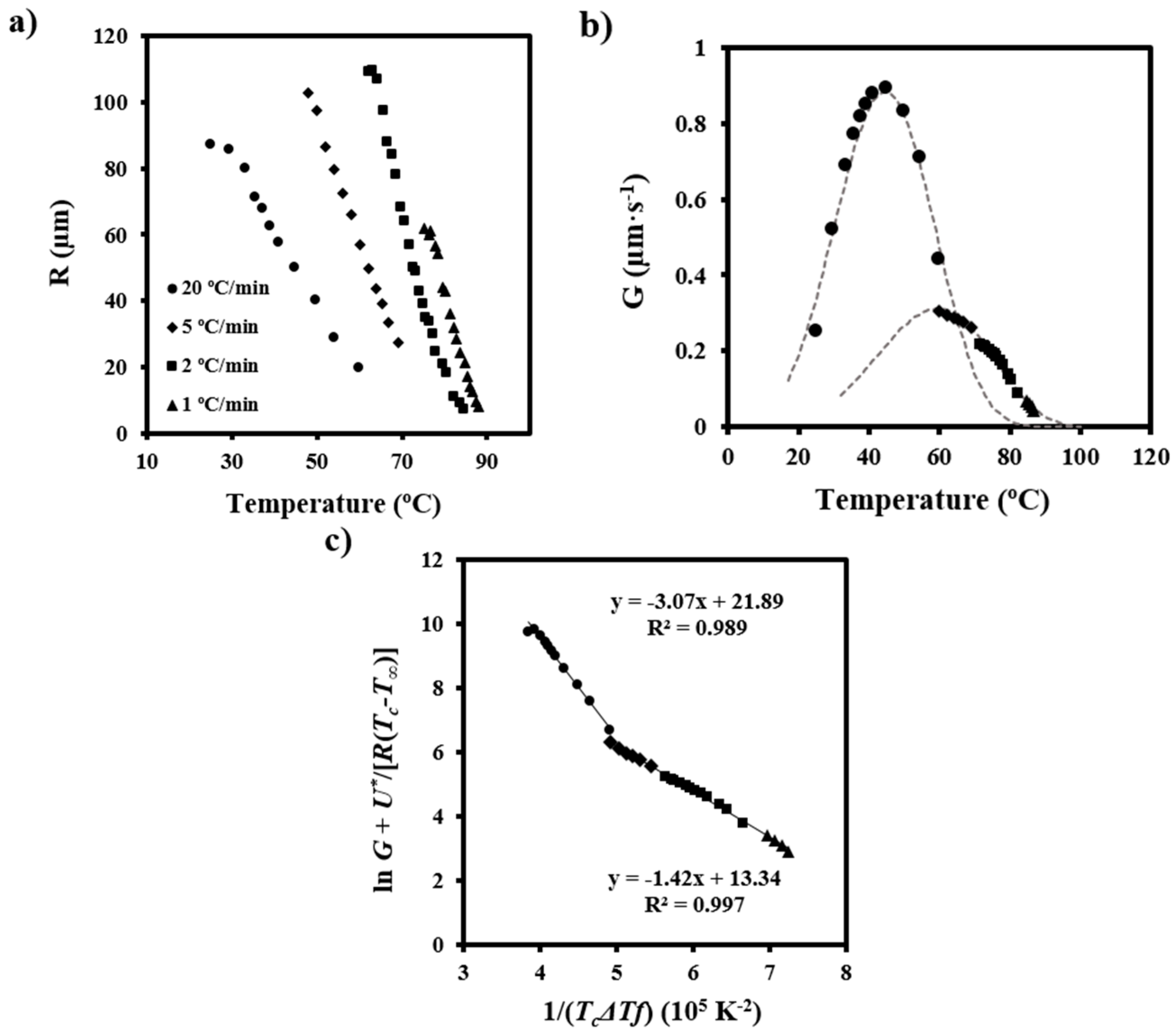
Plots showing the variation of the spherulitic radius with crystallization temperature could be adjusted to third order equations with good regression coefficients (i.e., higher than 0.990) (
Figure 7a). These coefficients were significantly better than those calculated for second order equations and remained constant for higher orders. Therefore, third order equations were employed to determine d
R/d
T as a function of the crystallization temperature. The corresponding crystal growth rate versus crystallization temperature curves are displayed in
Figure 7b. Note that data were obtained at different cooling rates in order to maximize the crystallization temperature range where radii could be well measured.
Two bell-shaped curves with maximums of 45 and 60 °C reflected the temperature dependence of
G, and therefore the existence of two crystallization regimes with different secondary nucleation constants. These were determined by the Lauritzen-Hoffman equation [
38]:
where
G0 is the constant pre-exponential factor and the other parameters as previously defined.
Figure 7c shows the linear plots obtained using
U* and
T∞ parameters of 1600 cal/mol and
Tg-35 K, respectively. It is clear that two crystallization regimes defined by secondary nucleation constants of 3.07 × 10
5 K
2 and 1.42 × 10
5 K
2 fit the experimental data. Furthermore, regimes III and II could be assumed since the experimental ratio between slopes (2.16) was close to the theoretical
KgIII/
KgII value of 2.
Figure 7b also shows that the two bell-shaped curves calculated by Equation (8), the estimated
U* and
T∞ parameters, and the deduced values of ln
G0 and
Kg for each regime fit well with the experimental spherulitic growth data. The maximum growth rate was found in regime III and corresponded to a temperature of 45 °C. Our observations are in agreement with the same crystallization regimes determined from isothermal crystallization although the nucleation constant becomes slightly higher than those previously reported (i.e., 2.49 × 10
5 K
2 and 1.19 × 10
5 K
2) [
3]. Note also that the deduced data support the results of the calorimetric study.
The fact that the crystallization rate is governed by two different processes makes it unfeasible to determine a single activation energy for the entire
Tc range. Instead, an effective activation energy (
E) dependent on
Tc was evaluated by Equation (9) [
32]:
The calculated effective activation energies are plotted in
Figure 8 and show non-Arrhenius behavior as expected. Different
Kg values were used according to the crystallization regime. The effective activation energy is zero at the maximum crystallization rate for regimes III and II, which corresponds to temperatures of 45 and 60 °C and agree again with those deduced by isoconversional analysis. In each case, positive values are found for temperatures lower than the corresponding maxima because the crystallization rate increases with increasing temperature, whereas negative values are determined for higher temperatures characterized by a decrease of the crystallization rate with increasing temperature.
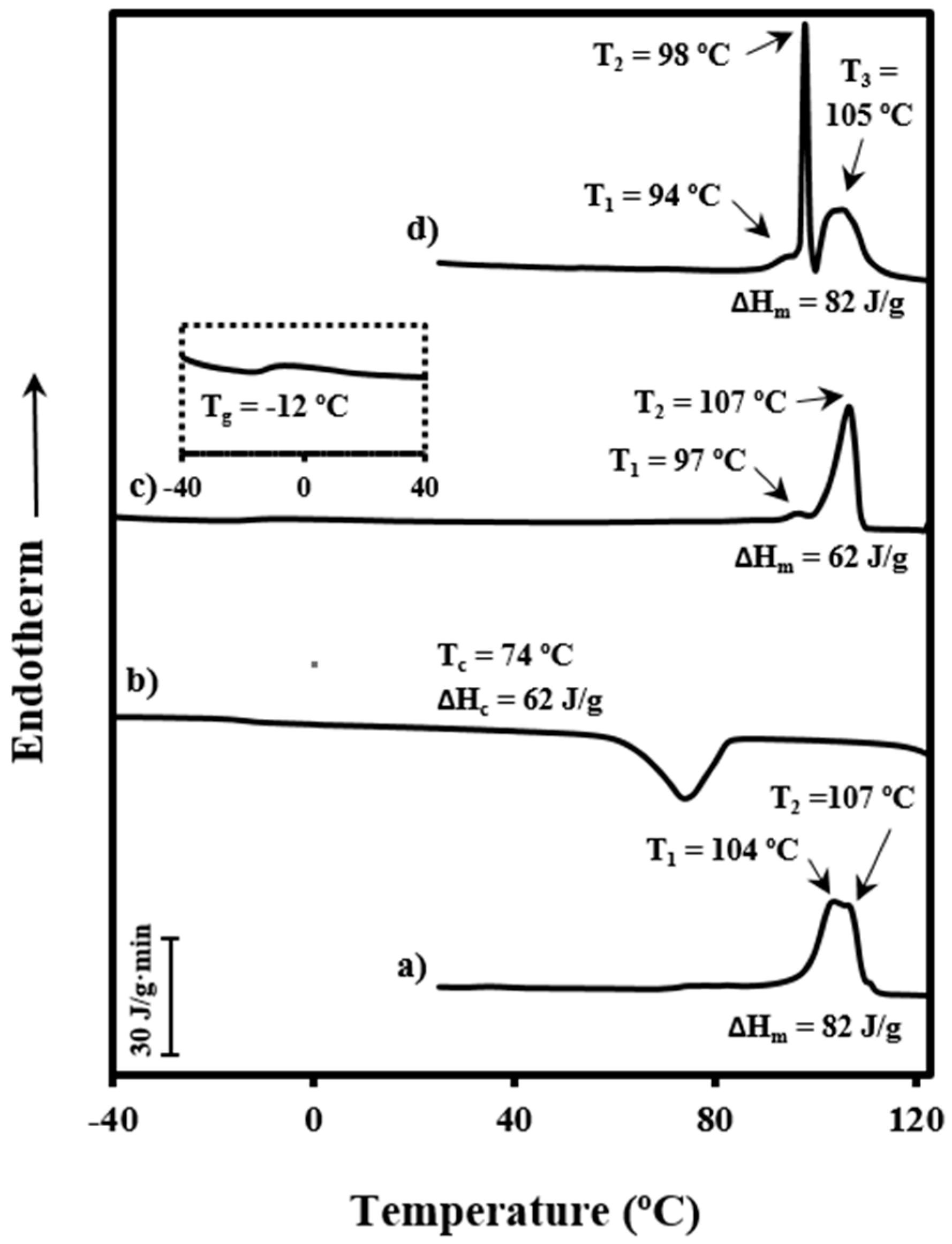
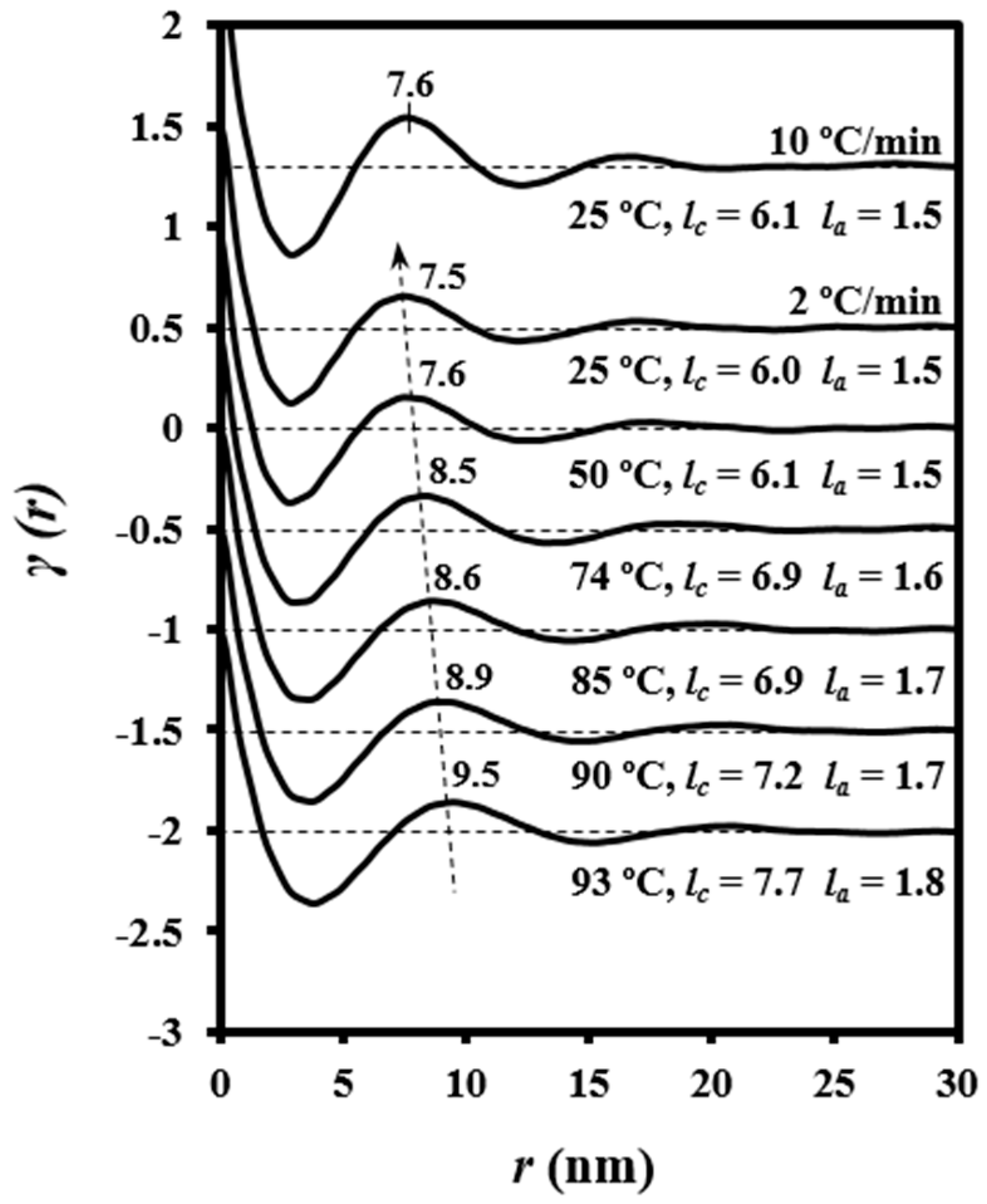
3.4. Evolution of Morphologic Parameters during Heating of Poly(p-dioxanone) Samples
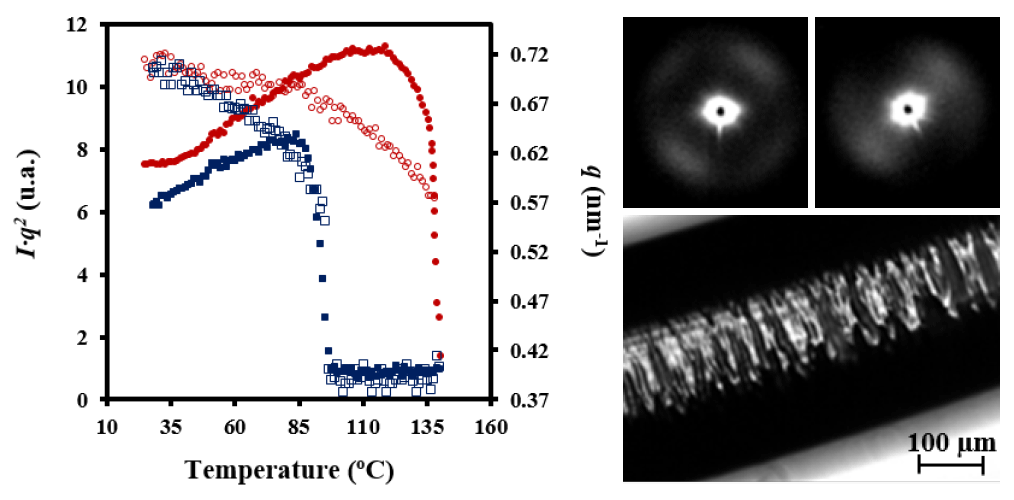
Figure 9 shows the evolution of the intensity of the peak detected in SAXS patterns during heating and cooling processes of the granulated PDO sample. In the first case, a recrystallization process that led to thicker lamellae can be deduced from the increase in SAXS peak intensity and its shift towards lower values of the scattering vector (
q = [4π/
λ] senθ). In the second case, a shift of the peak once samples were crystallized towards higher
q values and a slight decrease on its intensity was observed. These features can be explained considering that a lamellar insertion mechanism took place at low temperatures together with a densification of the amorphous phase (i.e., a smaller difference between the density of amorphous and crystalline phases).
Characteristic lamellar parameters (i.e., long period,
Lγ, amorphous layer thickness,
la, and crystalline lamellar thickness,
lc) and crystallinity (i.e., crystallinity within the lamellar stacks,
XcSAXS =
lc/
Lγ) were determined by the normalized one-dimensional correlation function [
35],
γ(
r):
where
I(
q) is the intensity at each value of the scattering vector.
SAXS data were collected within a limited angular range, with application of the Vonk’s model [
39] and Porod’s law to perform extrapolations to low and high
q values.
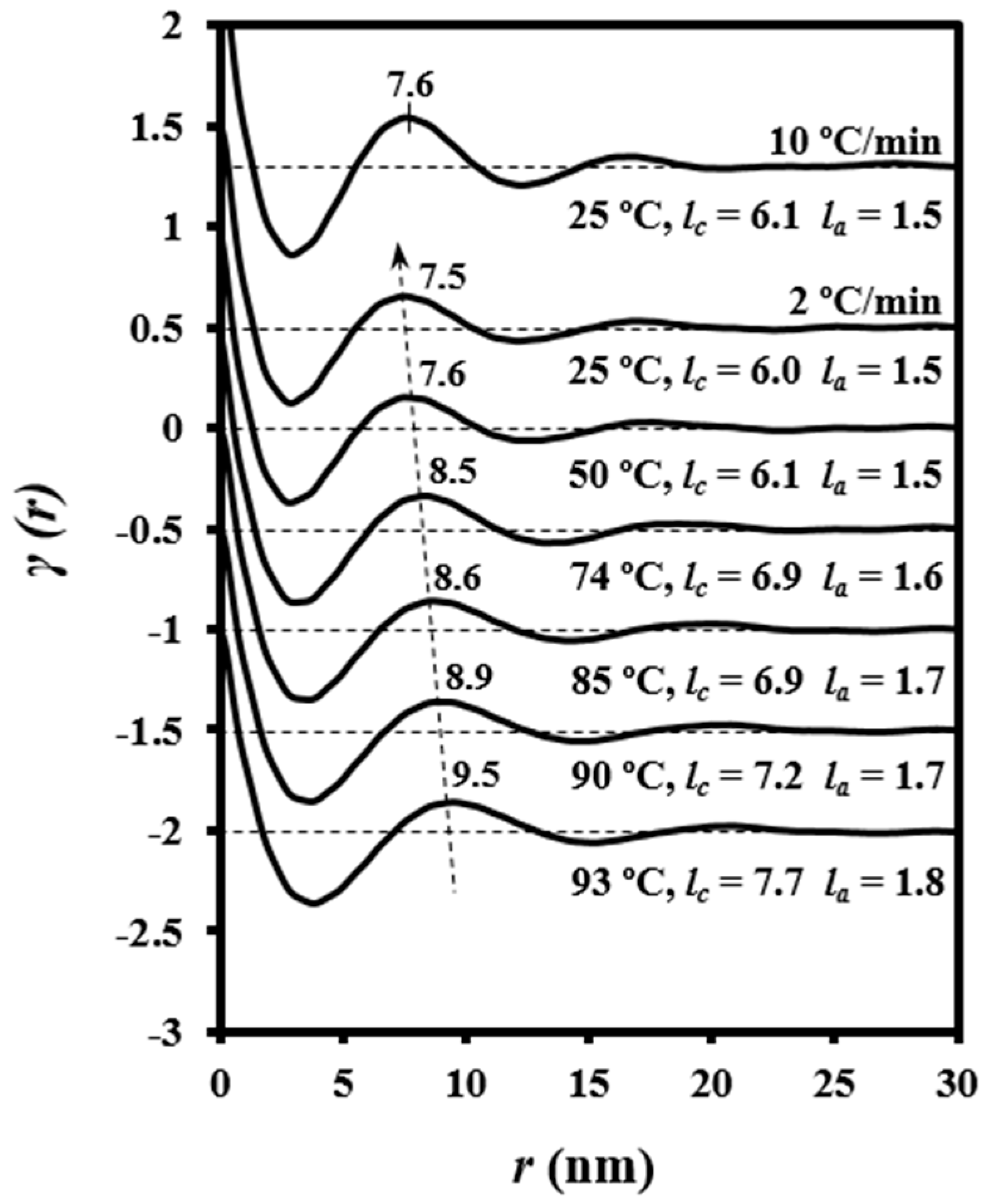
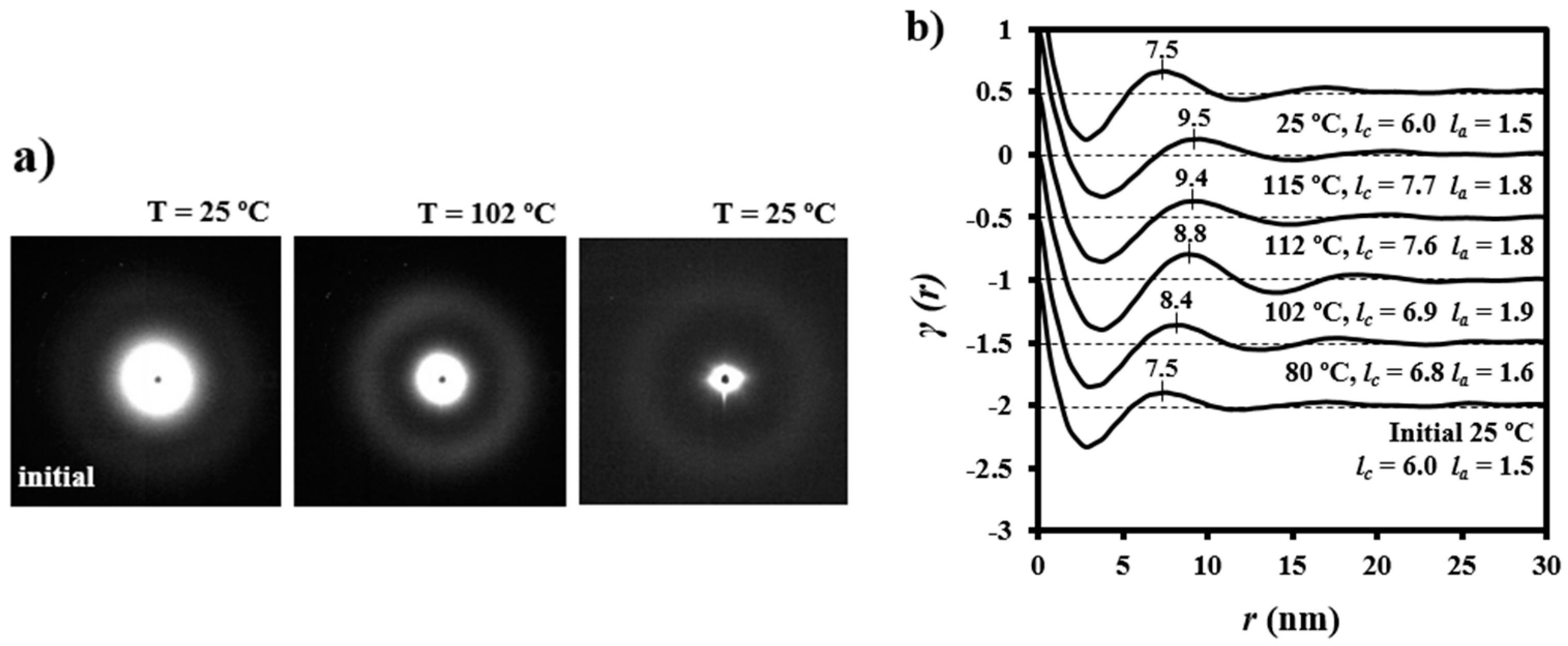
Figure 10 illustrates representative correlation functions obtained from patterns acquired during the heating of granulated PDO. Lamellar thickening was due to the increase in crystalline lamellar thickness (i.e., from 6.0 to 7.7 nm) and amorphous layer thickness (i.e., from 1.5 to 1.8 nm). In this way, the reordering process led to a minimum increase of the crystallinity within lamellar stacks (i.e., from 0.80 to 0.81). However, it should be pointed out that the correlation function of the sample heated up to 102 °C (i.e., just before the first melting peak observed in the DSC trace) exhibited more defined peaks (see also the diffraction patterns in
Figure 10) that were indicative of a high contrast between electronic densities of amorphous and crystalline phases. Basically, the amorphous phase became less dense in agreement with the maximum value detected for
la (i.e., 1.9 nm) and the minimum value of crystallinity (i.e., 0.78).
Figure 10 also shows that morphological features were completely recovered after cooling the sample and specifically the correlation functions and the X-ray diffraction pattern were identical to those obtained from the initial sample.
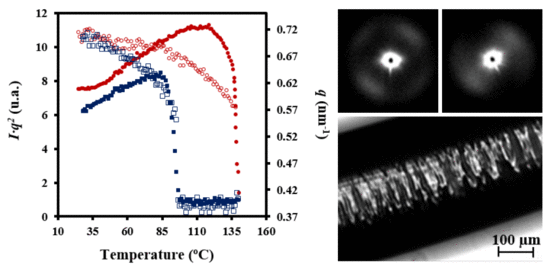
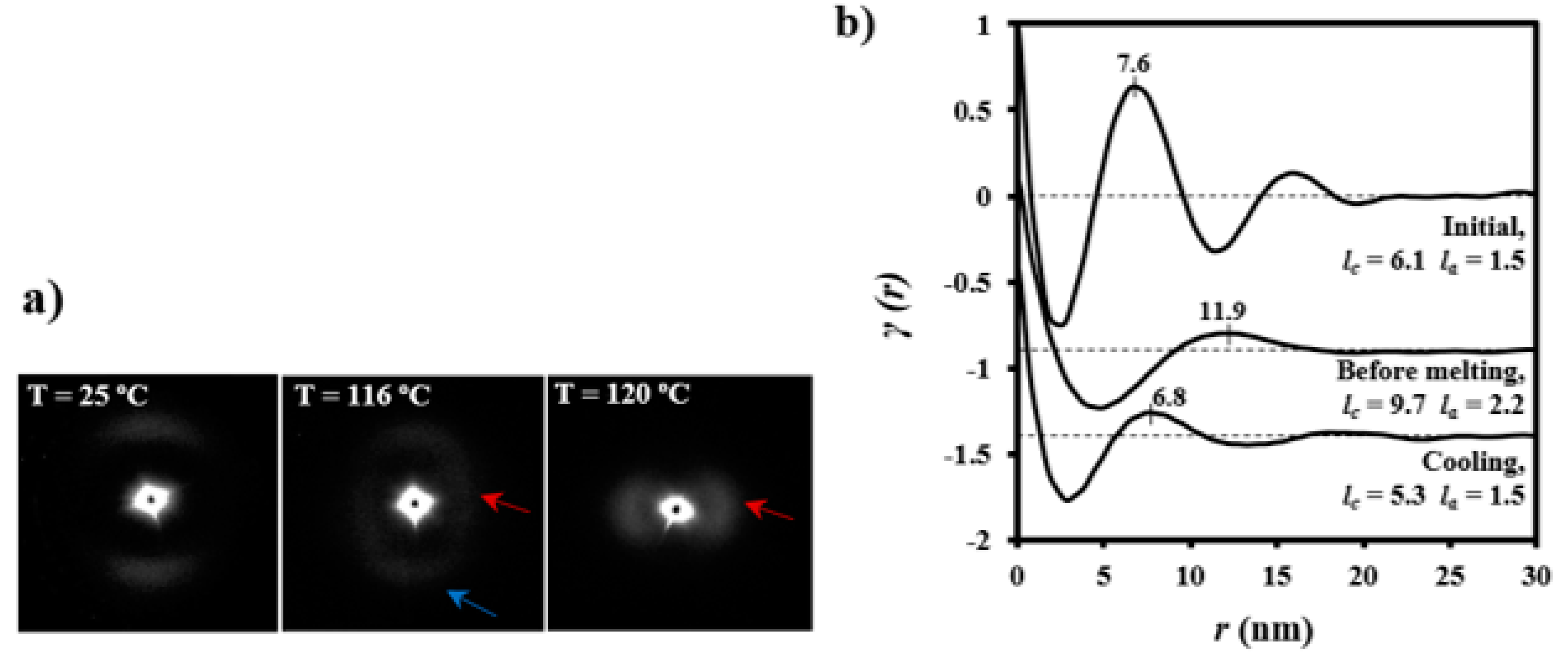
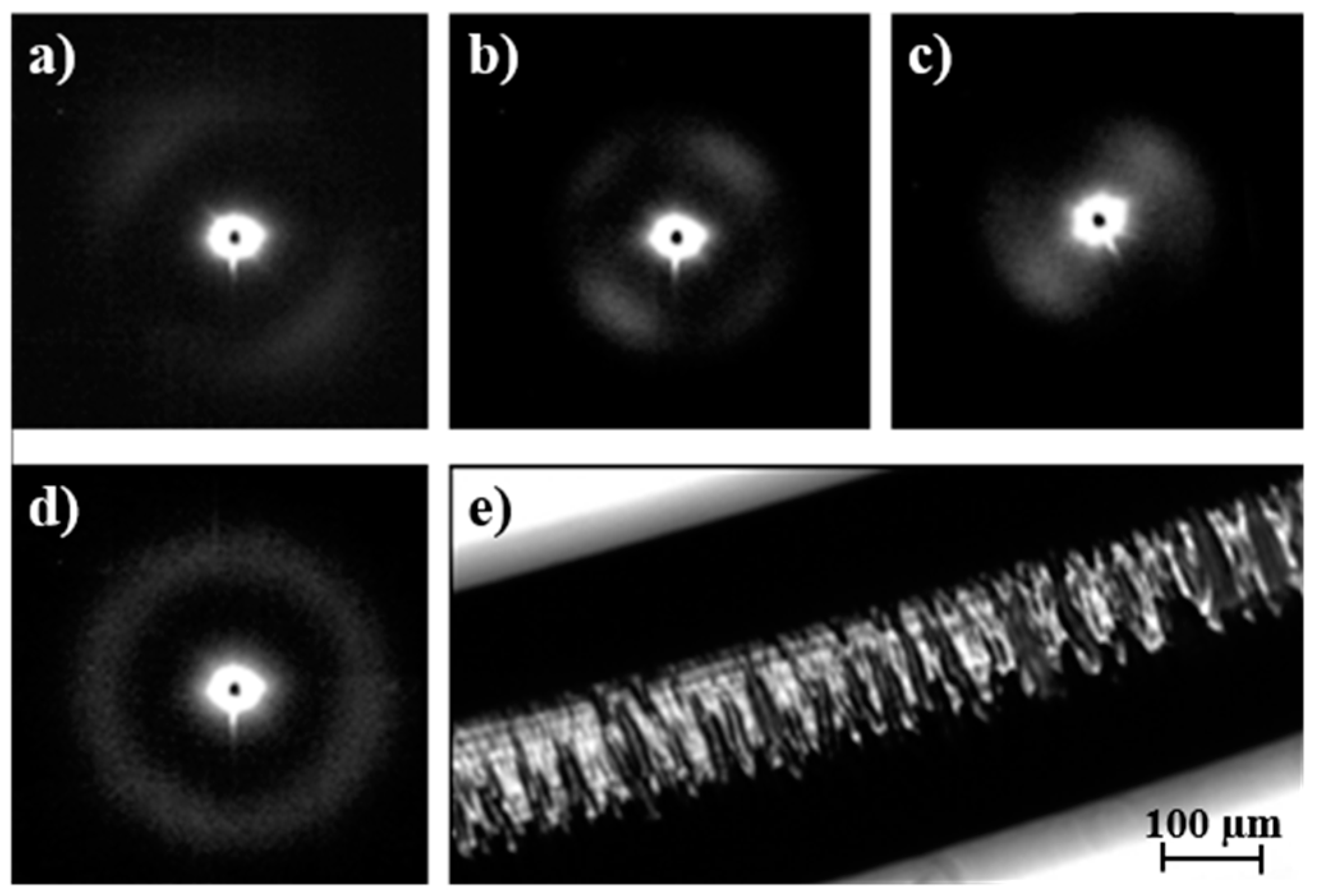
The evolution of SAXS patterns of a PDO thread on heating (
Figure 11) was very different because they reflect the microstructure of the processed sample.
Some observations can be made: (a) The initial pattern was characterized by a meridional reflection that indicates a stacking of lamellae perpendicularly oriented to the fiber axis. Peaks observed in the corresponding correlation function were highly prominent suggesting a tie molecular arrangement in the dense crystalline phase. However, lc and la values (6.1 nm and 1.5 nm) were close to those determined for the granulated sample; (b) As the temperature was increased the interlamellar reflection decreased in intensity while a new perpendicular reflection appeared and progressively increased in intensity. This new equatorial reflection could be associated with the existence of interfibrillar amorphous domains that correspond to the regions placed on the lateral sides of lamellae. Note that molecular chains in these domains may have had a partial orientation at the beginning of the heating (i.e., the as processed thread) but became more randomly distributed as the temperature was increased. Therefore, a decrease in electronic density of the amorphous phase and an enhancement of the intensity of the SAXS reflection were derived. The correlation function of the interfibrillar reflection observed in the pattern taken just before fusion gave lc and la values (i.e., 9.7 nm and 2.2 nm, respectively), which were clearly different from those determined for the interlamellar reflection. In any case, XcSAXS was again close to 0.81; (c) The pattern and correlation function of the sample after being cooled to room temperature from the melt state were similar to those determined from the initial sample and indicated a similar lamellar organization. Nevertheless, a slight decrease of lc was detected (i.e., 5.3 nm as opposed to 6.1 nm) as well as a decrease of crystallinity within lamellar stacks (i.e., 0.78 as opposed to 0.80), in agreement with the lack of an annealing process for the melt crystallized sample.
3.6. Changes in Microstructure of Poly(p-dioxanone) Degraded Samples during Heating
Insights on the crystalline microstructure of sutures can be obtained following the evolution of SAXS patterns of degraded samples during a subsequent heating process [
25]. To this end, PDO sutures were exposed to hydrolytic degradation media at pHs 7 and 11 and at 37 °C for 36 days to analyze samples with clear differences in their degree of hydrolysis. Specifically,
Mw values of 117,700 and 83,800 g/mol were determined after exposure to pHs 7 and 11, respectively. Weight losses were close to 3% (pH 7) and 11% (pH 11). Micrographs in
Figure 13 and
Figure 14 reveal the greater morphological changes occurred under basic conditions and specifically the appearance of deep transversal cracks that led to narrow disks and tortuous suture surfaces (
Figure 13). This degradation can be interpreted as a consequence of greater hydrolysis of interlamellar amorphous regions, which are depleted and dissolved in the medium [
11]. The morphology of sutures exposed to neutral pH for a relatively short time is quite different because in this case smoother surfaces and numerous longitudinal cracks formed (
Figure 14). In fact, it has been reported that hydrated, interfibrillar amorphous regions swell more easily than interlamellar amorphous regions [
12]. Note that the latter are constituted by tie chains, which connect the lamellae in each fibril whereas fewer tie chains are expected to connect adjacent fibrils. Thus, water diffusion and formation of longitudinal cracks seem to be favored.
SAXS patterns of degraded samples showed a slight decrease of meridional interlamellar spacing, LB, because of the more compact structure achieved after scission of chains belonging to the amorphous regions. Therefore, the initial spacing of 9.6 nm decreased to 9.2 and 9.3 nm after 36 days of degradation in basic and neutral pH, respectively. Analysis of correlation functions (not shown) indicated that lc and la values decreased from 6.1 to 1.5 nm, respectively, to 5.3 and 1.3 nm for the basic pH. Logically, SAXS crystallinity increased during degradation since the main change occurred in the amorphous layer.
Lamellar crystals in degraded samples were able to recrystallize and even reorient during subsequent heating runs in an easier way than that observed for the initial suture. The increased freedom resulting from scission of tie chains belonging to interfibrillar and interlamellar amorphous regions should play a fundamental role. Diffraction patterns during subsequent heating showed clear differences between highly and scarcely degraded samples. Thus, in the first case, an increase in lamellar thickness, together with the appearance of an intense equatorial reflection associated with the interfibrillar spacing previous to the disappearance of the meridional reflection, was observed. Specifically, LB values of 13.2 and 11.7 nm were determined at temperatures close to fusion (i.e., 102 °C). This behavior was similar to that observed for the initial suture taking into account the differences in spacings and intensities of reflections. Logically, greater spacings and intensities were detected for degraded samples as a consequence, in the first case, of an enhanced reorganization when tie interconnecting chains were cleaved and, in the second case, of a higher contrast between amorphous/crystalline regions.
Figure 14 reveals a different evolution when samples were degraded in the pH 7 medium because the great thickening of lamellae was hindered due to a still compact chain packing (note that weight loss was minimal). Therefore, the thickened lamellae (
LB = 11.4 nm) tilted with respect to the fiber axis gave rise to a second reflection. The breadth of reorganized crystals was also clearly increased, as could be deduced from the observed spot like reflections. Finally,
Figure 13 and
Figure 14 show the reversibility of the thermal process for degraded samples since the diffraction patterns obtained after cooling to room temperature are identical to those obtained from the initial sample before any thermal treatment.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}