1. Introduction
Aescin, a natural mixture of triterpenoid saponins isolated from the horse chestnut tree (
Aesculus hippocastanum (Hippocastanaceae)), possesses diverse biochemical and pharmacological actions. It exists in two forms: α and β, distinguished by differences in melting point, specific rotation, hemolytic index, and solubility in water [
1]. However, the β-aescin form appears as an active component of the mixture and is the molecular form present in the major pharmaceutical products available. In the past few years, the pharmacological profile of β-aescin has received significant attention due to the establishment of its pharmacological basis for the major clinical indication in the treatment of chronic venous insufficiency (CVI) [
1]. There are at least three types of pharmacodynamic actions attributed to β-aescin: anti-edematous and venotonic properties, as well as anti-inflammatory activities [
2]. It is assumed that these actions are based on a molecular mechanism, identified as a selective vascular permeabilization, allowing a higher sensitivity of, e.g., calcium channels, to molecular ions, resulting in increased venous and arterial tone [
3,
4]. These sensitizing effects to ions and other molecules, e.g., 5-HT, result in enhanced venous contractile activity and, consequently, in the anti-edematous property of the molecule [
1]. Therefore, β-aescin is now widely quoted in the literature as a pharmacological tool for assessing the sensitivity of vascular tissues to different agonists, in order to evaluate the mechanism of, for example, hypertension development in animal models [
5]. In addition to pharmacological activity, β-aescin might be used effectively as a macroinitiator of the ring-opening polymerization (ROP) of cyclic esters, due to the presence of hydroxyl groups in the molecule [
6].
β-adrenergic blockers (β-blockers) are an important class of drugs for the treatment of various heart diseases, including high blood pressure, insufficiency of blood flow to the heart muscle (angina pectoris), irregular heartbeat (arrhythmias), thickened heart muscle (hypertrophic cardiomyopathy) and decreased ability of the heart to empty or fill normally (heart failure) [
7]. To the family of β-adrenergic blockers belongs alprenolol (ALP), a non-cardioselective β-blocker, developed in 1966 [
8]. ALP is reported to have intrinsic sympathomimetic activity and some membrane-stabilizing properties. ALP has been given orally, as a benzoate or hydrochloride, in the management of hypertension, angina pectoris, and cardiac arrhythmias. ALP is non-polar and hydrophobic, freely soluble in water and ethanol, with low to moderate lipid solubility, and a half-life of 2–3 h [
9]. When ALP is administered orally, its bioavailability is lowered because it is subjected to the first-pass effect of the liver. For example, it has been reported that the bioavailability of ALP is only 15% in oral administration; thus, multiple dosing is required to maintain therapeutic drug blood levels [
9]. These factors make ALP a suitable candidate for implantable local delivery, which improves the bioavailability of the drug, may maintain a high drug concentration in the blood circulation or prolonging the duration of action, may enhance its physical stability, increase specificity and reduce systemic adverse effects for targeted drug delivery and reduces the frequency of its dosing. In previous years, a series of
o-cyclopropane carboxylic acid ester prodrugs of various β-blocking agents, including ALP, were synthesized. All prodrugs were hydrolyzed to give their parent compounds an aqueous phosphate buffer of pH 7.4 or in 80% human plasma [
10]. It has been found that the formation of the
o-cyclopropane carboxylic acid ester derivatives significantly increased the lipophilicities of the β-blockers, as measured by the distribution coefficient between
n-octanol and an aqueous phosphate buffer of pH 7.4. Prokai
et al. studied ocular delivery of ALP by site-specific bioactivation of its methoxime analogue [
11]. They demonstrated that the benefit of this chemical delivery system approach includes the facile release of a potential anti-glaucoma agent at the site of the action, only.
Over the past few decades, there has been a rapid growth in the medical use of biomaterials in many fields, e.g., tissue engineering, the implantation of medical devices and artificial organs, prostheses, ophthalmology, dentistry and bone repair [
12]. Among all of them, the use of a biodegradable copolymer of poly (lactide-
co-glycolide) (PLGA) has shown immense potential as a drug-delivery carrier and scaffold for tissue engineering [
13]. PLGA belongs to a family of biodegradable polymers approved by the FDA because of its high biocompatibility, favorable degradation characteristics and possibilities for sustained drug delivery. Its use also has the possibility of tuning the overall physical properties of the PLGA-drug matrix by controlling the relevant parameters, such as polymer molecular weight, the ratio of lactide to glycolide, and drug concentration for achieving a desired dosage and release interval depending upon the drug type [
14,
15,
16].
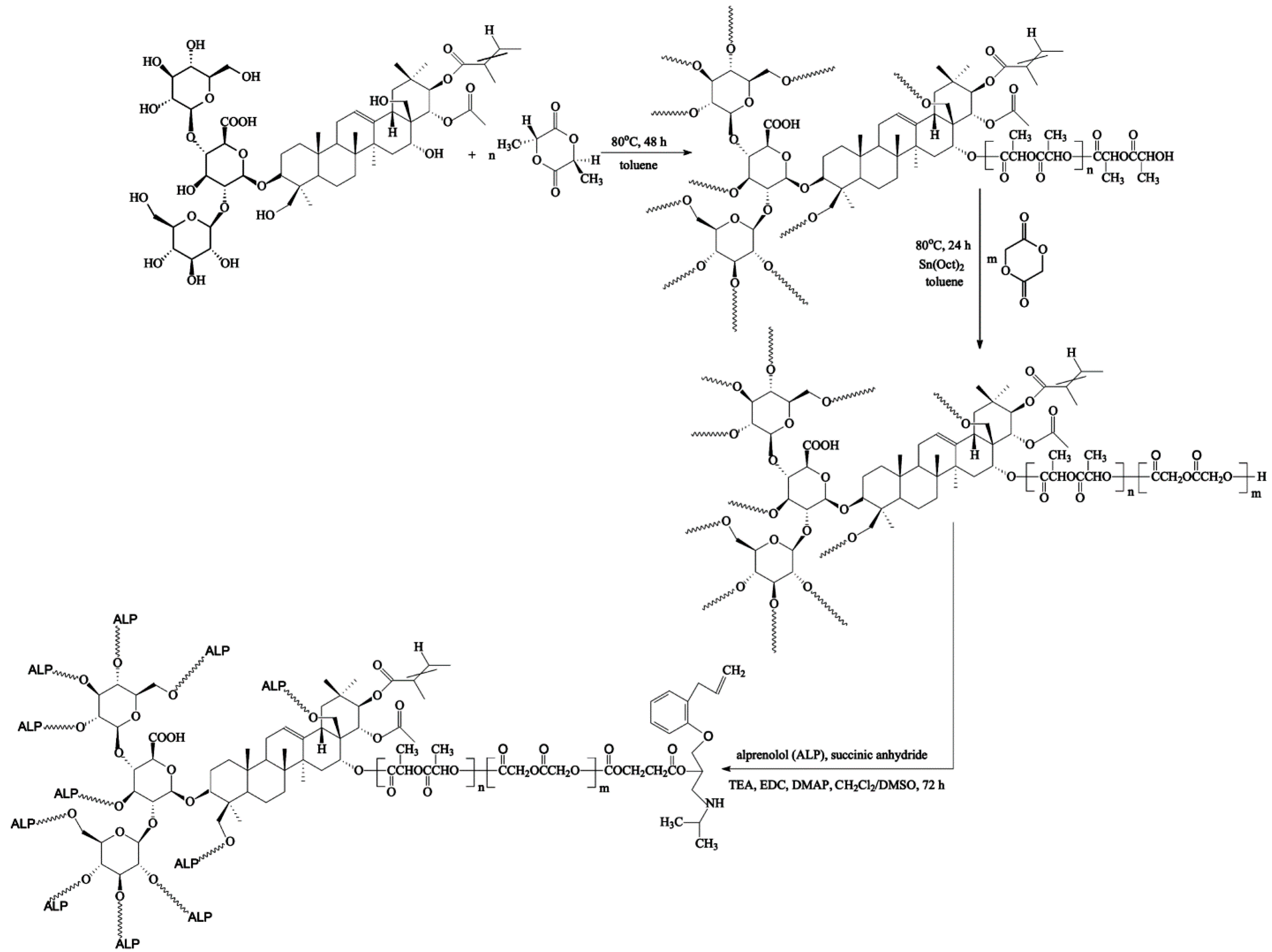
In the present work, we report on the synthesis, characterization and drug-release behavior of a newly formed conjugate of ALP that is bonded to the matrices of the PLGA-aescin for implantable local delivery. To the best of our knowledge, no research group has developed a covalent conjugate of ALP constructed from a biodegradable copolymer with incorporated β-aescin as a central core. To achieve this goal, the copolymers were firstly obtained by the ring-opening polymerization (ROP) of d,l-lactide (LA) and glycolide (GL) using a natural, and therapeutically efficacious, initiator—β-aescin. Different ratios of lactide and glycolide were employed to obtain a suitable copolymer composition for a high drug-release efficiency. The synthesized branched copolymeric matrices have been subjected to cyto- and genotoxicity assays and then bonded to ALP using the EDC (carbodiimide) coupling method. The in vitro release rate of the drug obtained from the synthesized conjugates was analyzed. It was found that the ester-linked ALP conjugate was a promising implantable drug carrier for local delivery, one that might improve the bioavailability and pharmacokinetics of ALP as well as increase its efficiency, resulting from the synergistic action of ALP with β-aescin.
2. Experimental Section
2.1. Materials
Alprenolol hydrochloride (1-(propan-2-ylamino)-3-(2-prop-2-enylphenoxy)propan-2-ol, anhydrous, ≥98%, Sigma Co., Poznan, Poland), succinic anhydride (97%, Aldrich Co. Poznan, Poland), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 98%, Aldrich Co. Poznan, Poland), triethylamine (TEA, ≥99%, Sigma-Aldrich Co., Poznan, Poland) and 4-(dimethylamino)pyridine (DMAP, ≥99%, Aldrich Co., Poznan, Poland) were used without further purification. d,l-lactide (3,6-dimethyl-1,4-dioxane-2,5-dione, 98.0%, Aldrich Co., Poznan, Poland) was recrystallized from dried ethyl acetate in a dry nitrogen atmosphere and then thoroughly dried in a vacuum before use. Glycolide (GL, ≥99%, Sigma Co., Poznan, Poland) and β-aescin (≥95%, MP Biomedicals, Poznan, Poland) were thoroughly dried in a vacuum before use. Stannous octoate (Sn(Oct)2, tin(II) 2-ethylhexanoate, 2-ethylhexanoic acid tin(II) salt, ~95%, Aldrich Co., Poznan, Poland) was used as received. Dichloromethane (DCM, anhydrous, ≥99.8%, POCh, Gliwice, Poland), dimethyl sulfoxide (DMSO, anhydrous, 99%, Aldrich Co., Poznan, Poland), toluene (anhydrous, 99.8%, POCh, Gliwice, Poland) and diethyl ether (anhydrous, 99.8%, POCh, Gliwice, Poland) were used as received. Phosphate buffer solution (pH 7.4 ± 0.05, 0.1 M, PBS, potassium dihydrogen phosphate/di-sodium hydrogen phosphate, 20 °C, Avantor Performance Materials, Gliwice, Poland) was also used as received.
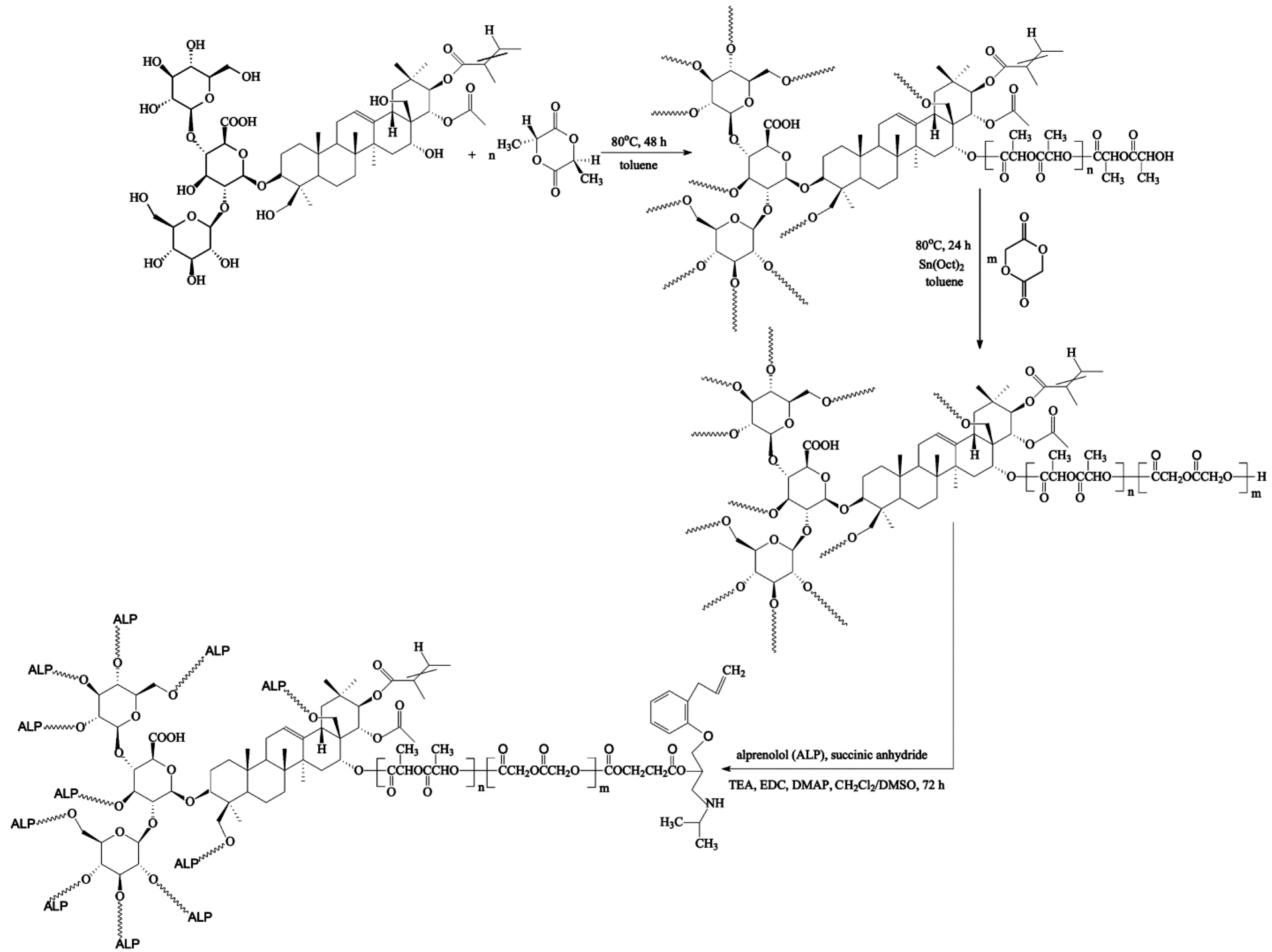
2.2. Copolymerization Procedure
The branched copolymeric materials were prepared using different molar ratios of initiator (aescin) to monomers (
d,
l-lactide (LA) and glycolide (GL)) (
Table 1). The initiator/monomer (LA or GL) feed ratios for the synthesized copolymers were 1/50/50, 1/30/70, and 1/70/30, denoted as aescin/PLA50/PGL50, aescin/PLA30/PGL70 and aescin/PLA70/PGL30, respectively. For the copolymerization, dry aescin and LA were accurately weighed and introduced into a 50 mL polymerization tube. The tube was then connected to a Schlenk line, where exhausting-refilling processes were repeated three times before toluene was added. The tube was immersed in an oil bath at 80 °C under an argon atmosphere for 48 h. After an appropriate time, the precise weight of GL and a catalytic amount of Sn(Oct)
2 (0.02 mol % relative to the total monomer concentration) were added to the mixture and the exhausting-refilling process was carried out again (toluene, 24 h). The resulting products were dissolved in a dry mixture of CH
2Cl
2/CHCl
3, precipitated twice from methanol and dried in a vacuum for 72 h.
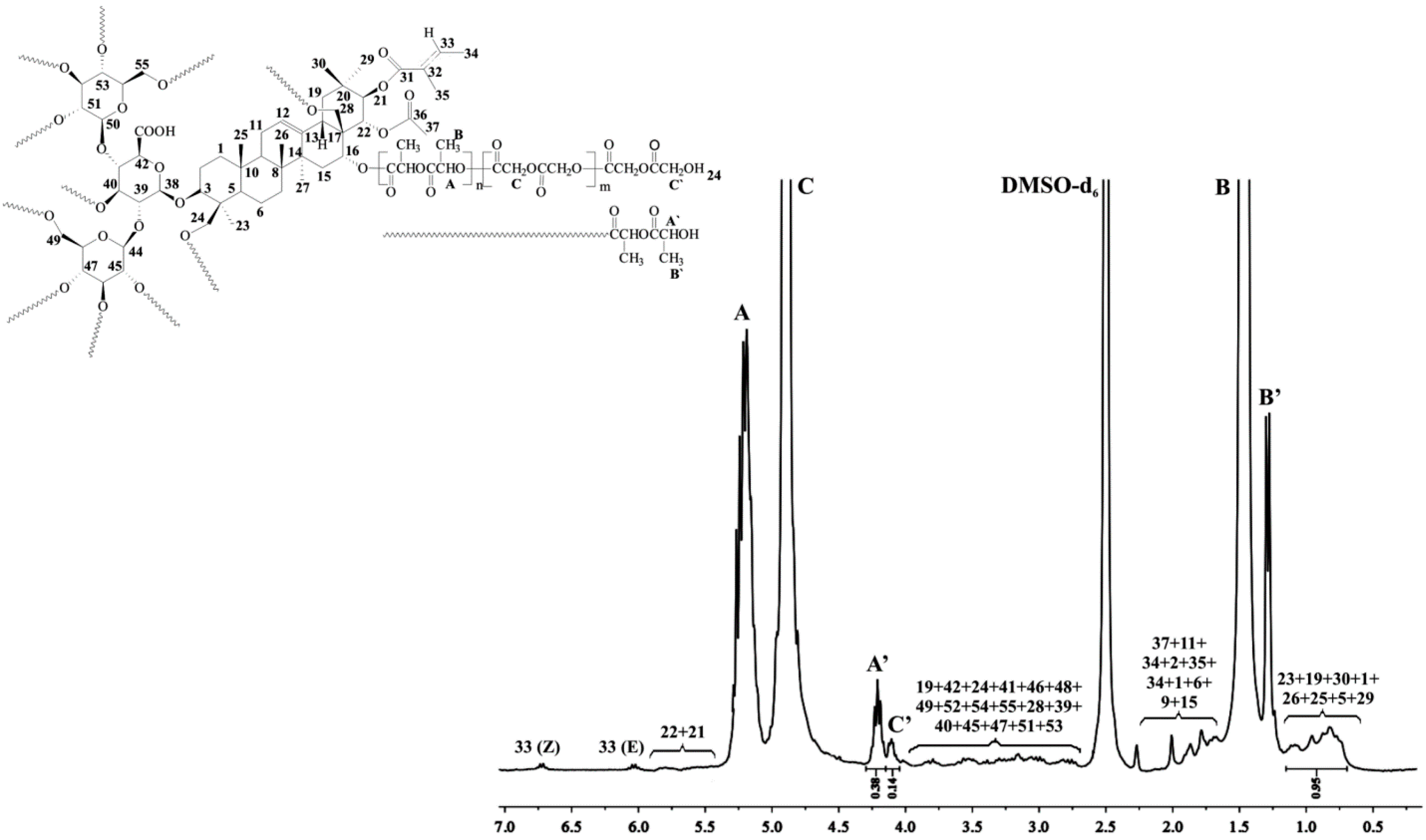
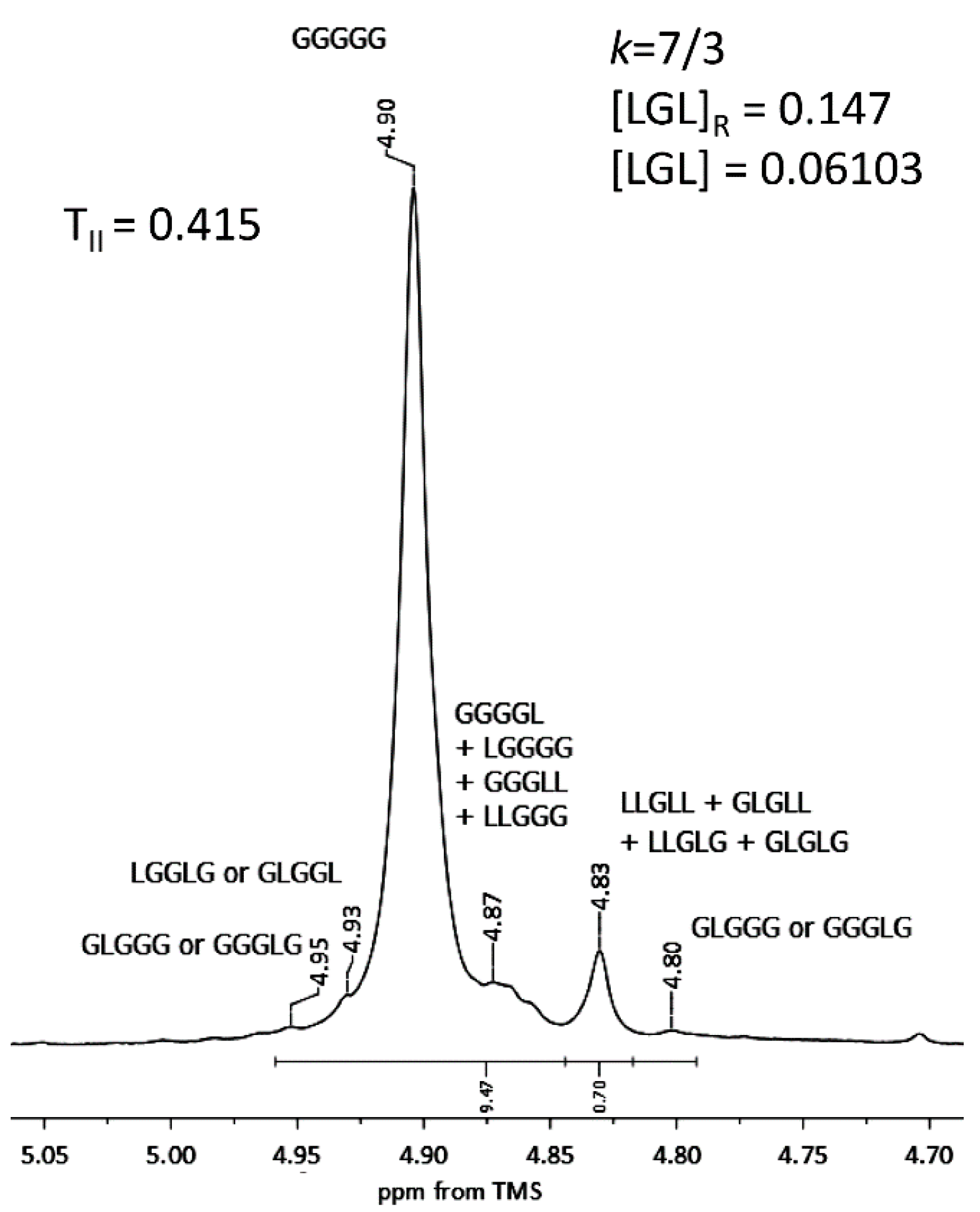
2.2.1. Spectral Characterization of Aescin/PLA50/PGL50
1H NMR (DMSO-d
6, 300 MHz, δ
H, ppm); 0.75–1.09 (18H of aescin,
23,
19,
30,
1,
26,
25,
5,
29), 1.29 (d, –CH
3, end group of PLA (
B’)), 1.47 (d, –CH
3 of PLA (
B)), 1.52–2.01 (17H of aescin,
37,
11,
24,
2,
35,
34,
1,
6,
9,
15), 2.85–3.95 (21H of aescin,
19,
42,
24,
41,
46,
48,
49,
52,
54,
55,
28,
39,
40,
45,
47,
51,
53), 4.10 (d, –C(O)CH
2OH, end group of PGL (
C’)), 4.21 (q, –C(O)CH(CH
3)OH, end group of PLA (
A’)), 4.90 (d, –C(O)CH
2O– of PGL (
C)), 5.20 (q, –C(O)CH(CH
3)O– of PLA (
A)), 5.43–5.74 (2H of aescin,
21,
22), 6.01–6.68 (2H of aescin,
33 (Z, E)) (
Figure 1).
13C NMR (DMSO-d
6, 300 MHz, δ
C, ppm): 15.15 (
C’), 16.49 (
C), 21.9–35.8 (
23,
11,
2,
27,
29,
7,
15,
10,
20), 42.9–46.3 (
4,
17,
19), 59.96 (
E’), 60.76 (
E), 67.93 (
B’), 68.93 (
B), 73.1–76.8 (
21,
51,
42,
45,
52,
48,
46,
53,
47), 102.4–103.0 (
38,
44,
50), 141.9 (
13), 166.76 (
D), 169.58 (
A), 169.77 (
A’) (
Figure S1,
Supplementary Material).
2.2.2. Spectral Characterization of Aescin
1H NMR (DMSO-d
6, 300 MHz, δ
H, ppm); 6.68 (H-33 Z), 6.01 (H-33 E), 5.74 (H-22), 5.43 (H-21), 5.25 (H-12), 4.99 (OH-40, OH-45, OH-51, OH-53), 4.85 (OH-24), 4.78 (OH-16), 4.65 (H-44), 4.56 (OH-47), 4.51 (H-38), 4.48 (OH-46, OH-49, OH-52, OH-55), 4.39 (OH-28), 4.25 (H-50), 3.95 (H-16), 3.91 (H-42), 3.85 (H-24a), 3.69 (H-49a), 3.67 (H-48), 3.62 (H-41), 3.60 (H-46, H-52, H-55a), 3.51 (H-55b), 3.49 (H-54), 3.41 (H-49b), 3.30 (H-3, HDO), 3.24 (H-47), 3.13 (H-24b), 3.03–3.13 (H-39, H-40, H-53) 3.02 (H-28a), 2.99 (H-51), 2.92 (H-45), 2.85 (H-28b), 2.56 (H-18), 2.49 (H-19a), 2.01 (H-37 CH
3), 1.88 (H-34 E CH
3), 1.83 (H-11a), 1.80 (H-35 E CH
3) 1.77 (H-2a, H-11b), 1.74 (H-34 Z CH
3) 1.72 (H-35 Z CH
3), 1.71 (H-2b), 1.59 (H-15a), 1.55 (H-9), 1.52 (H-1a, H-6a, b), 1.46 (H-7a), 1.39 (H-27 CH
3), 1.24 (H-7b), 1.22 (H-15b), 1.09 (H-19b, H-23 CH
3), 0.96 (H-1b, H-30 CH
3), 0.84 (H-5), 0.81 (H-26 CH
3), 0.80 (H-25 CH
3), 0.75 (H-29 CH
3) (
Figure S3a–c,
Supplementary Material).
13C NMR (DMSO-d
6, 300 MHz, δ
C, ppm): 169.8 (C43), 169.5 (C36), 166.7 (C31), 141.9 (C13), 136.3 (C33 E+Z), 128.4 (C32 E+Z), 122.8 (C12), 103.0 (C38), 102.4 (C44, C50), 90.1 (C3), 80.0 (C41), 78.0 (C22), 77.7 (C54), 76.8 (C47), 76.3 (C53), 74.5 (C46, C48, C52), 73.9 (C45), 73.3 (C42, C51), 73.1 (C21), 69.9 (C39), 68.7 (C40), 66.9 (C16), 62.9 (C28), 62.1 (C24), 60.9 (C49), 60.3 (C55), 55.4 (C5), 46.3 (C19), 46.0 (C17), 45.9 (C9), 42.9 (C4), 40.7 (C14), 39.8 (C8), 39.6 (C18), 38.2 (C1), 35.8 (C20), 35.5 (C10), 33.4 (C15), 32.5 (C7), 29.0 (CH
3 29), 26.9 (C27), 25.6 (C2), 23.2 (C11), 21.9 (CH
3 23), 20.8 (CH
3 37), 20.3 (CH
3 35 E), 19.5 (CH
3 30), 18.0 (C6), 16.2 (CH
3 26), 15.2 (CH
3 25, CH
3 34 E), 14.1 (CH
3 34 Z), 12.0 (CH
3 35 Z) (
Figure S4,
Supplementary Material)
2.3. Synthesis of ALP Conjugated the Synthesized Branched Copolymeric Matrices
The synthesized copolymeric matrices—0.5 g of aescin/PLA50/PGL50, aescin/PLA30/PGL70 or aescin/PLA70/PGL30 (the
Mn(GPC) of the copolymers are listed in
Table 1)—were first dissolved in 50 mL anhydrous THF under stirring for 30 min at room temperature. Then, succinic anhydride (the molar ratio of the copolymeric matrices to succinic anhydride was 1:1.5 for each hydroxyl group, see
Table 1) and 0.8 mL of TEA as the catalyst were added to the reaction flask under equal reaction conditions for 24 h. The crude product with carboxyl terminal groups was then precipitated in the cold diethyl ether twice and dried in a vacuum (the yields ranged from 85% to 88%). Subsequently, to a solution of synthesized copolymeric products with carboxyl terminal groups (0.4 g of aescin/PLA50/PGL50, aescin/PLA30/PGL70 or aescin/PLA70/PGL30) in DMSO (20 mL), EDC, and DMAP were added under nitrogen (the molar ratio of copolymeric products with a carboxyl terminal group to EDC was 1:3 for each carboxyl group, and the molar ratio of EDC to DMAP was 1:1.5). The mixture was stirred at room temperature for 2 h and alprenolol hydrochloride (ALP) was added (the molar ratio of copolymeric matrices with a carboxyl terminal group to ALP was 1:1.5 for each carboxyl group). The reaction was stirred for 48 h at room temperature. The crude conjugation products were precipitated in a cold diethyl ether three times and dried in a vacuum. The yields of the conjugation products were 75%, 70% and 78% for aescin/PLA50/PGL50/ALP, aescin/PLA30/PGL70/ALP, and aescin/PLA70/PGL30/ALP, respectively.
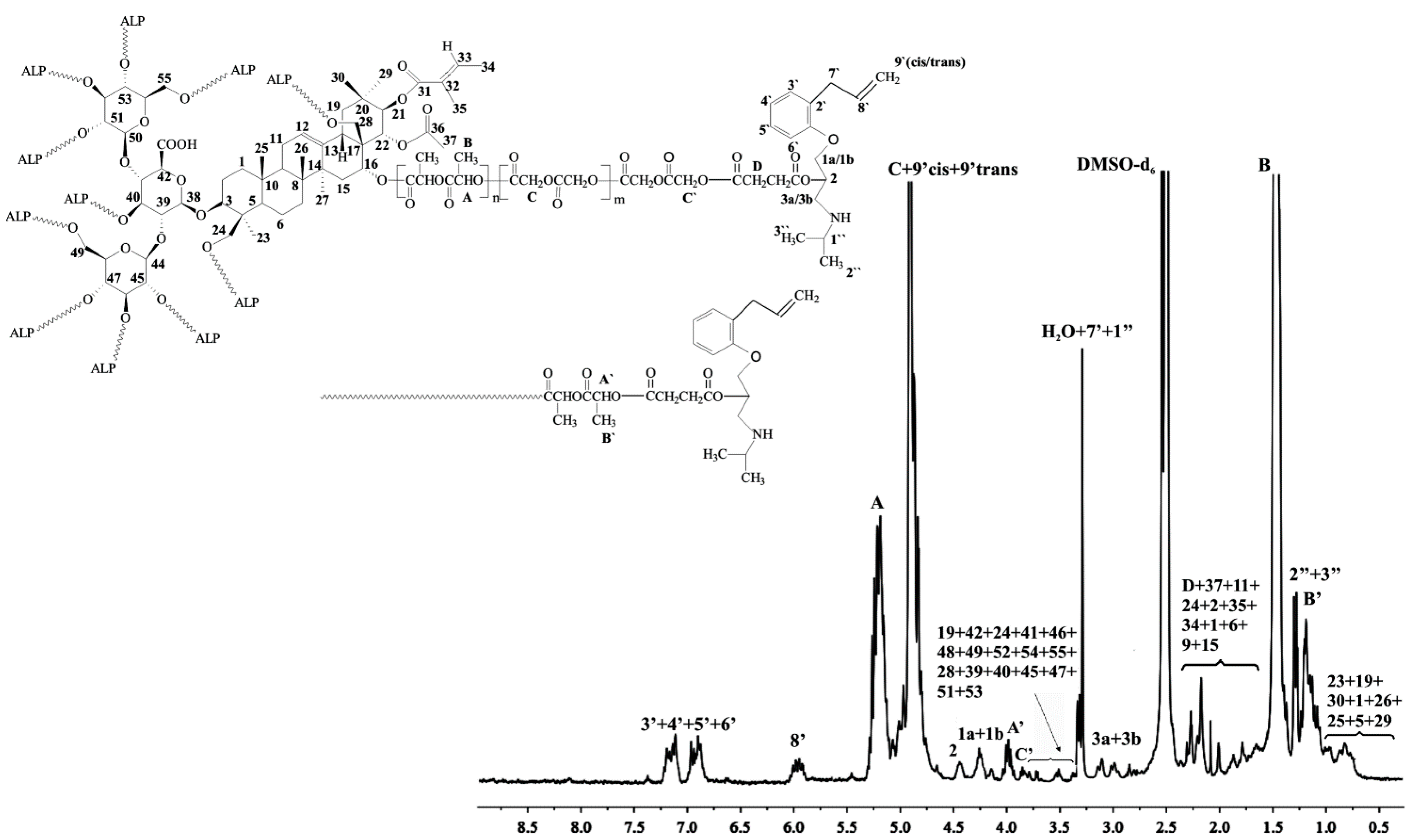
1H NMR of aescin/PLA30/PGL70/ALP (DMSO-d
6, 300 MHz,
δH, ppm): 0.71–1.05 (18H of aescin,
23,
19,
30,
1,
26,
25,
5,
29), 1.17–1.29 (d, –CH
3, end group of PLA (
B’) and (6H, two –CH
3 of ALP (2”, 3”), 1.46 (d, –CH
3 of PLA (
B)), 1.68–2.29 (17H of aescin,
37,
11,
24,
2,
35,
34,
1,
6,
9,
15 and (C(O)OCH
2CH
2– of succinic anhydride (
D)), 2.85–3.93 (21H of aescin,
19,
42,
24,
41,
46,
48,
49,
52,
54,
55,
28,
39,
40,
45,
47,
51,
53) and (2H, –CHCH
2NH– of ALP (3a, 3b)) and (2H, –CH
2=CHCH
2–, –NHCH(CH
3)
2 of ALP (
7’,
1”)); 3.96 (2H, –OCH
2CH– of ALP (1a, 1b)), 4.19 (–C(O)CH
2–, of PGL (
C’)), 4.25 (–C(O)CH(CH
3)– of PLA (
A’)); 4.47 (1H, –CH
2CHCH
2– of ALP (2)), 4.83–4.91 (d, –C(O)CH
2O– of PGL (
C)) and (2H, H
2C=CH– of ALP (
9’cis,
9’trans)), 5.21 (q, –C(O)CH(CH
3)O– of PLA (
A)), 5.47 (1H, H
2C=CH– of ALP (
8’)), 6.92–7.16 (4H of benzyl ring of ALP (
3’,
4’,
5’,
6’)) (
Figure 3).
FTIR (KBr, cm
−1): 3531 (υ
C–Haromatic), 2999–2962 (υ
asCH2), 1759 (υ
C=O), 1453 (υ
C–H), 1396 (δ
asCH3), 1268 (υ
asCOC), 1131 (υ
OC–O), 1091 (υ
C–O–C) (
Figure S7,
Supplementary Material).
2.4. Toxicity Assays
2.4.1. Microtox® Assay
Microtox® assay with the luminescent bacteria Vibrio fischeri was performed with the lyophilized bacteria purchased from Modern Water (New Castle, DE, USA). The metabolism of the bacteria was inhibited by toxic substances, resulted in the luminescence of the samples decreasing. The measurement was made in a luminometer Microtox® M500 after 15 and 30 min incubation of bacteria with the sample. On the basis of obtained results (MicrotoxOmni), the PE as the percent of luminescence changing was calculated and compared to control (non-toxic 2% NaCl solution).
2.4.2. Spirotox Test
Spirotox test with the protozoan
Spirostomum ambiguum was performed according to the standard protocol [
17]. The test was carried out in disposable, polystyrene multiwell plates (24 wells). Ten organisms were added to each well of the multiwell. The samples were incubated in the darkness at 25 °C for 24 h. Afterwards test responses: different deformations such as shortening, bending of the cell,
etc., and lethal response were observed with the use of dissection microscope (magnification of 10). As a diluent and a control, Tyrod solution was used.
Prior to the toxicity test, the materials were pulverized. In the direct contact test, three concentrations of the samples were tested in Spirotox test (1.0, 0.5, and 0.25 mg mL−1) and two in Microtox® assay (0.8 and 0.4 mg mL−1). The tested sample was weighted directly to the test containers and poured with 1 mL of diluent. The test organisms were incubated with the suspension of the tested sample. All the samples were run in triplicate for the toxicity measurements.
2.4.3. The Umu-Test
The
umu-test is a bioassay for evaluating the genotoxic potential of environmental samples and chemical compounds. The test organism is
Salmonella typhimurium TA1535/pSK1002. As a response to different types of DNA damage the
umuC gene in bacterial cells is induced which is a part of the SOS system. The test strain is genetically modified—the
umuC gene activity is linked to the synthesis of β-galactosidase while other DNA regions responsible for this enzyme synthesis were deleted. The transcription of the
umuC gene correlates with the amount of secreted β-galactosidase. The enzyme converts colorless substrate (ortho-nitrophenyl-β-galactoside) into the yellow product which can be quantified colorimetrically at 420 nm. Additionally, the bacteria growth (G) is evaluated by a measurement of an optical density to determine the cytotoxicity of tested samples. The genotoxic potential of the sample is presented as the Induction Ratio (IR)—the β-galactosidase activity ratio of tested sample in comparison to the negative control. Samples with IR ≥ 1.5 are considered as genotoxic [
18]. The
umu-test was performed in 96-well microplates according to the ISO 13829 protocol [
18] with and without metabolic activation (S9 liver fraction from male Sprague–Dawley rats treated five days before the isolation with a single dose of 500 mg kg
−1 body weight of Aroclor 1254 in soya oil). Deionized sterile water was used as a negative control, PBS as solvent control, while 2-aminoanthracene and 4-nitroquinoline
N-oxide were used as positive controls. All tested copolymeric matrices were incubated in phosphate buffered saline (PBS from Gibco, Carlsbad, CA, USA, 1 mg mL
−1) for 24 h, 37 °C with shaking. Before the assay all extracts were sterilized by filtration. All samples were tested in two fold dilution series (six concentrations).
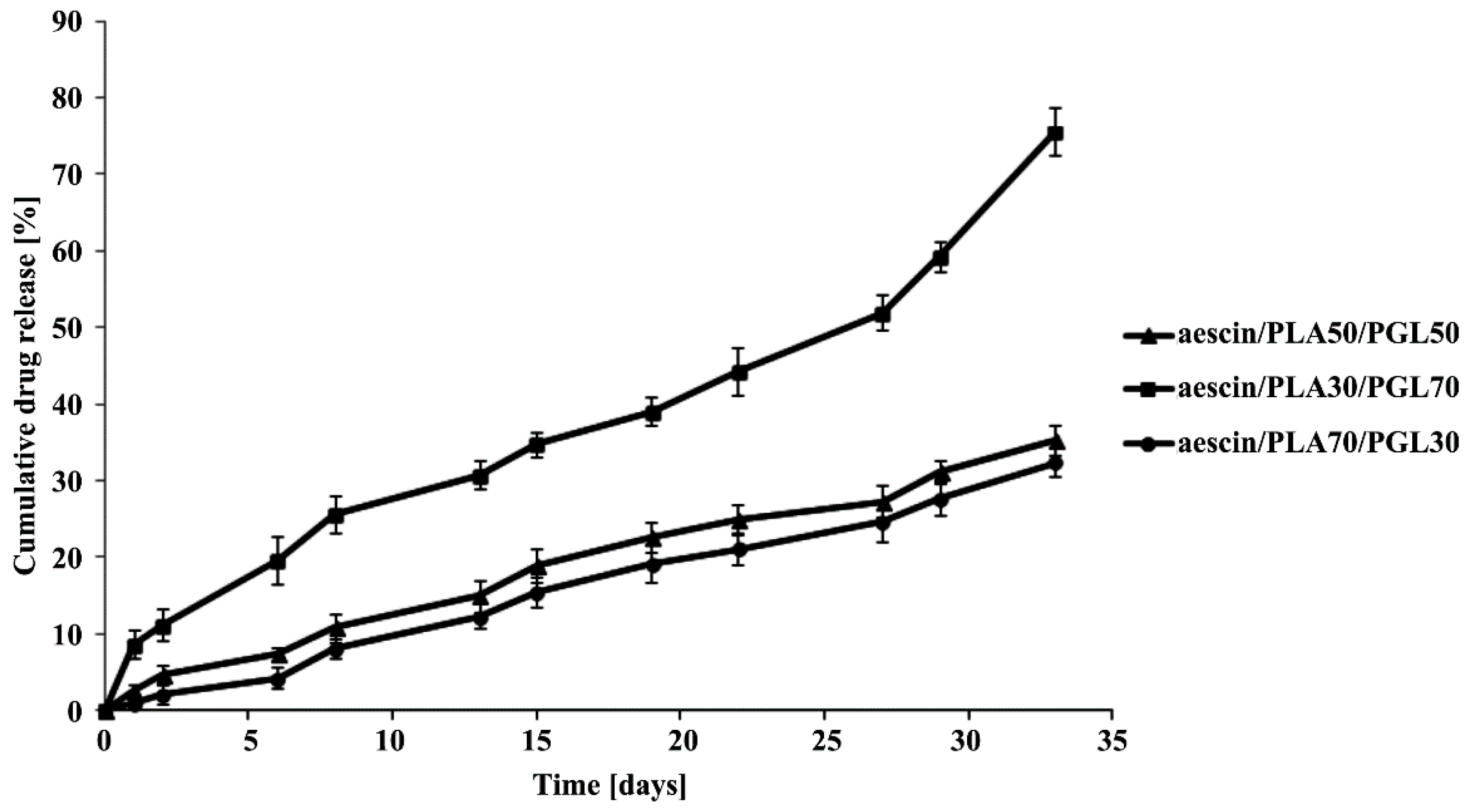
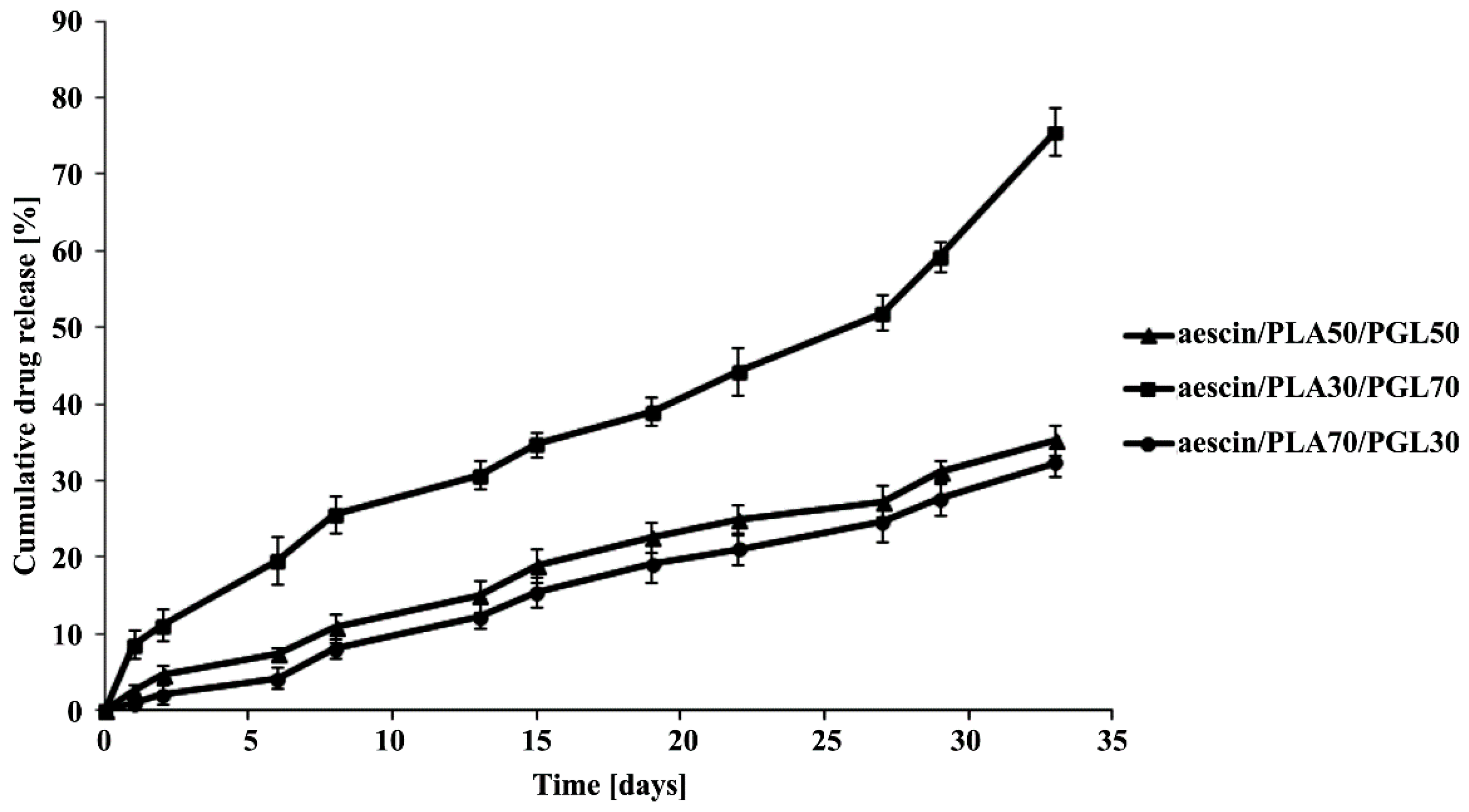
2.5. ALP Release Study from the Copolymeric Conjugates
The
in vitro release study of ALP from the synthesized copolymeric conjugates (aescin/PLA50/PGL50/ALP, aescin/PLA30/PGL70/ALP and aescin/PLA70/PGL30/ALP) were investigated by measuring the concentration of ALP released at pH 7.4 ± 0.05. All experiments were carried out in triplicate. 200 milligram of dried copolymeric conjugates, were immersed into 100 mL of buffer solution (pH 7.4 ± 0.05) and incubated at 37 °C, with continuous orbital rotation at 50 [cycles/min]. At predetermined time intervals, 10 mL samples were withdrawn from the release medium using the filter followed by replacing with 10 mL of a fresh buffer solution. The absorption of buffer solution was determined by a UV-VIS spectrophotometer at the absorbance peak with a wavelength at 274 nm [
19]. The absorbance peak was correlated very well with the concentration of ALP. A linear calibration curve was obtained by measuring the absorption of solutions with predetermined ALP concentrations. For all the measurements in this study, the absorbance readings were within the calibration range.
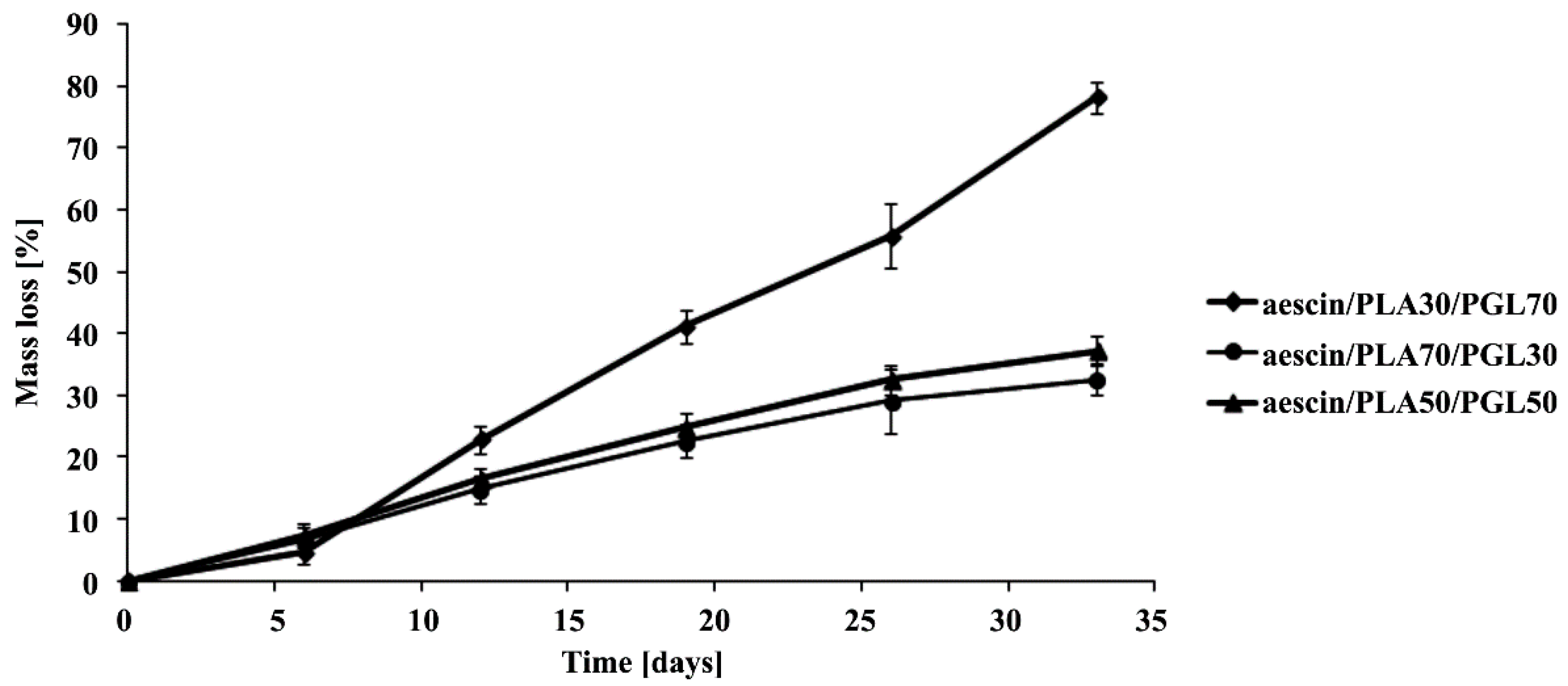
2.6. Hydrolytic Degradation Test
The hydrolytic degradation of the synthesized copolymeric matrices was performed in a 10 mL phosphate buffer solution (pH 7.4 ± 0.05) at 37 °C for 30 days. Following hydrolysis, the polymer samples were washed intensively with distilled water to remove any residual buffer solution, followed by drying under reduced pressure for four days. The degradation rates were estimated by weight loss (
WL, %), calculated with the following equation:
where
W0 is initial weight and
Wt is weight after biodegradation.
Furthermore, there results were evaluated by the ηinh of the copolymeric matrices decreasing. The polymeric matrices viscosity was measured in N,N-dimethylformamide (DMF, at 30 °C) using an Ubbelohde viscometer (on Stabinger Viscometer SVM 3000, Anton Paar’s, Graz, Austria).
2.7. Measurements
The polymerization products were characterized in the DMSO-d6 or CDCl3 solution by means of 1H or 13C NMR (Varian 300 MHz, Palo Alto, CA, USA). The IR spectra were measured from KBr pellets (Perkin–Elmer spectrometer, PerkinElmer, Waltham, MA, USA).
Number-average molecular weights (Mn) and polydispersity indexes (Mw/Mn) were measured using SEC-MALLS instrument (Wyatt Technology Corporation, Santa Barbara, CA, USA) composed of an 1100 Agilent isocratic pump, autosampler, degasser, thermostatic box for columns, a photometer MALLS DAWN EOS (Wyatt Technology Corporation, Santa Barbara, CA, USA) and differential refractometer Optilab Rex (Wyatt Technology Corporation, Santa Barbara, CA, USA). ASTRA 4.90.07 software (Wyatt Technology Corporation, Santa Barbara, CA, USA) was used for data collecting and processing. Two 2× TSKgel MultiporeHXL columns were used for separation. The samples were injected as a solution in methylene chloride. The volume of the injection loop was 100 mL. Methylene chloride was used as a mobile phase at a flow rate of 0.8 mL·min−1.
The amount of the released ALP was quantitatively determined by a UV-VIS spectrophotometry (UV‑1202 Shimadzu, Duisburg, Germany) in aqueous buffered solutions at the adsorption maximum of the free drug (λ = 274 nm) using a 1 cm quartz cell.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}