
Synthesis of β-cyclodextrin-Based Star Block Copolymers with Thermo-Responsive Behavior


Abstract
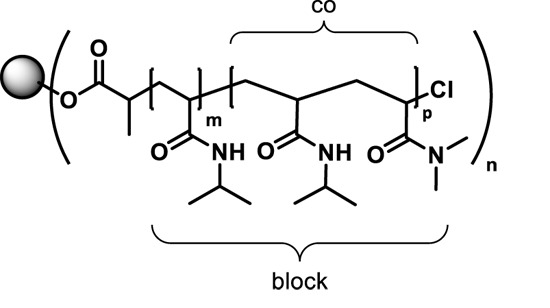
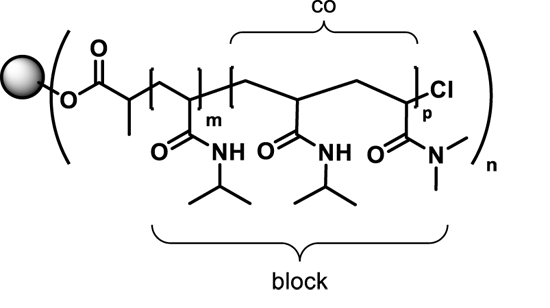
:
1. Introduction
2. Experimental Section
2.1. Materials
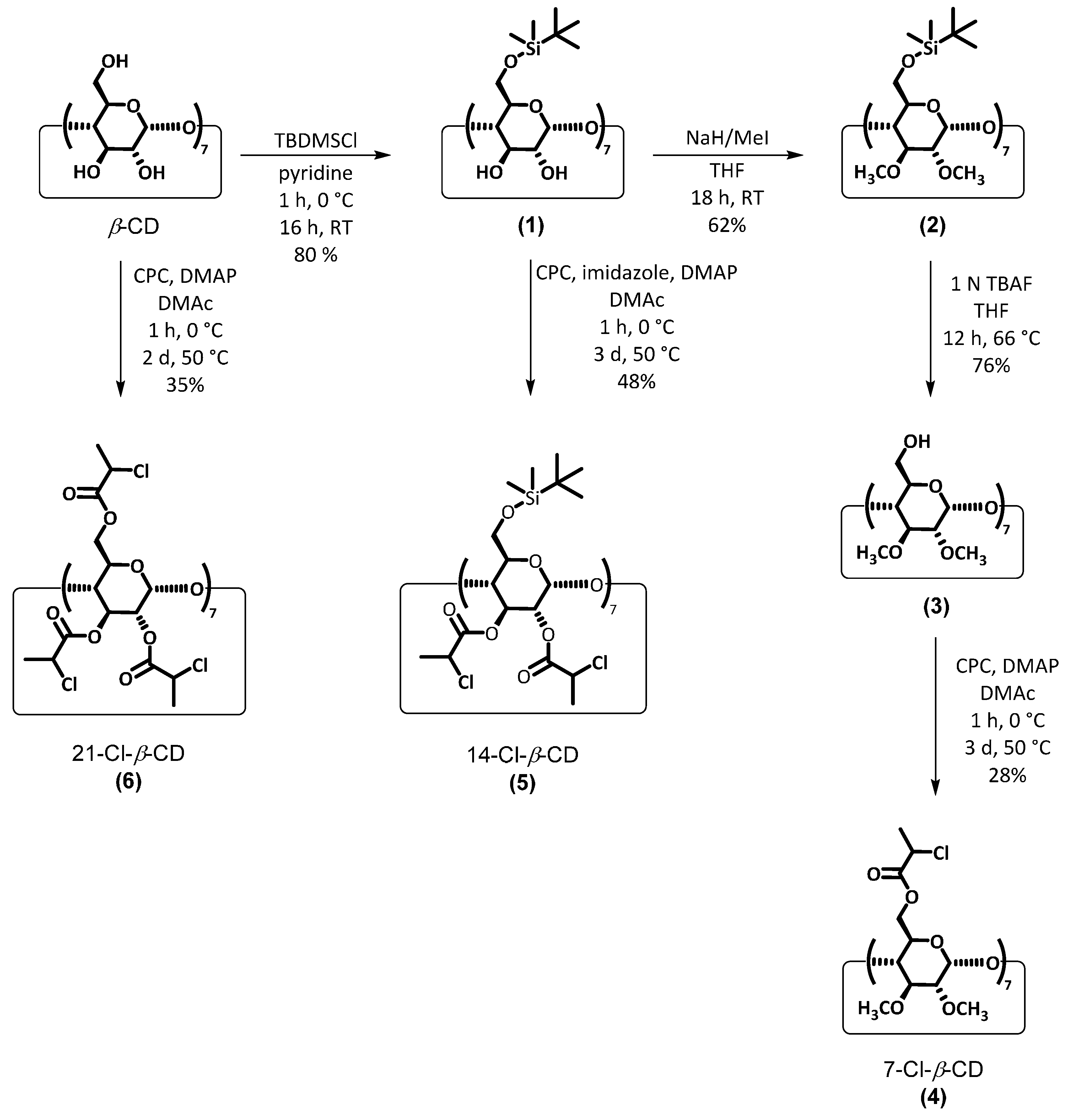
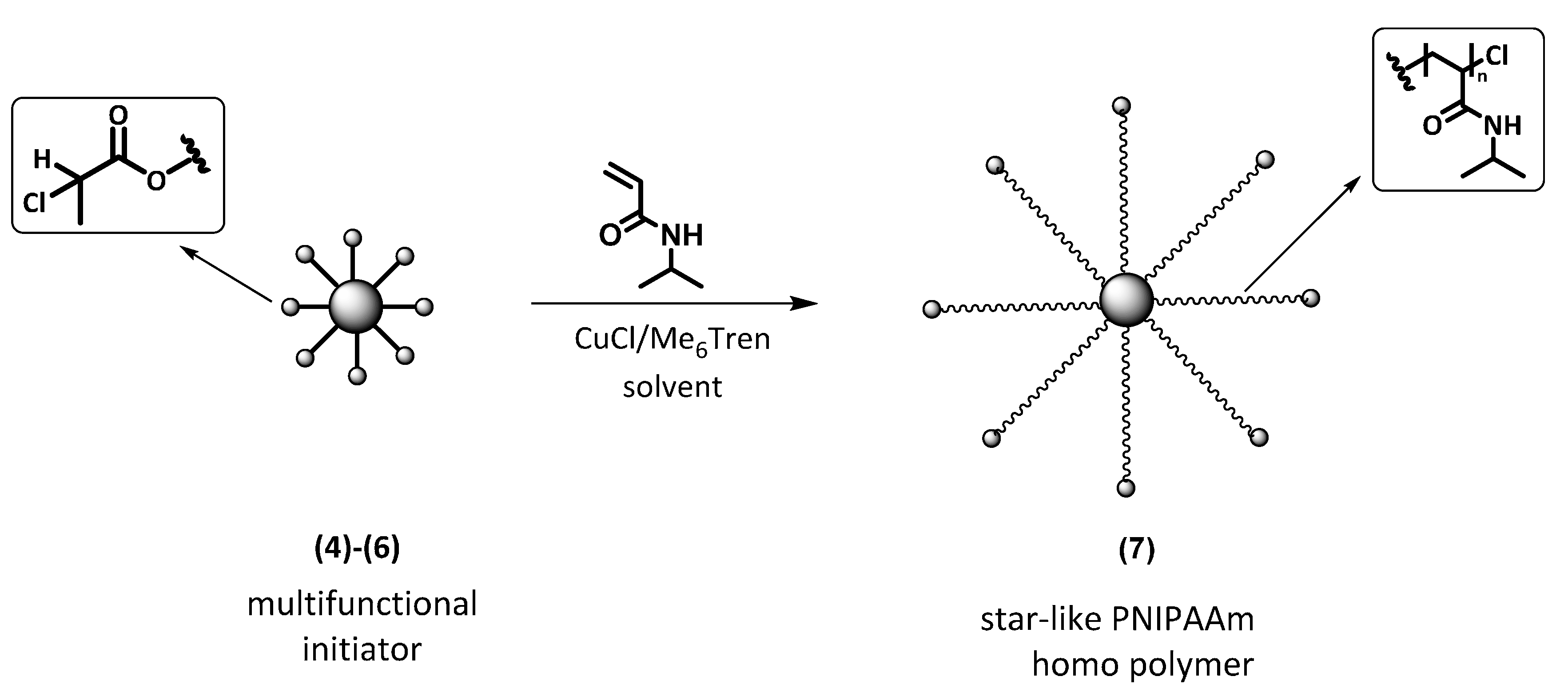
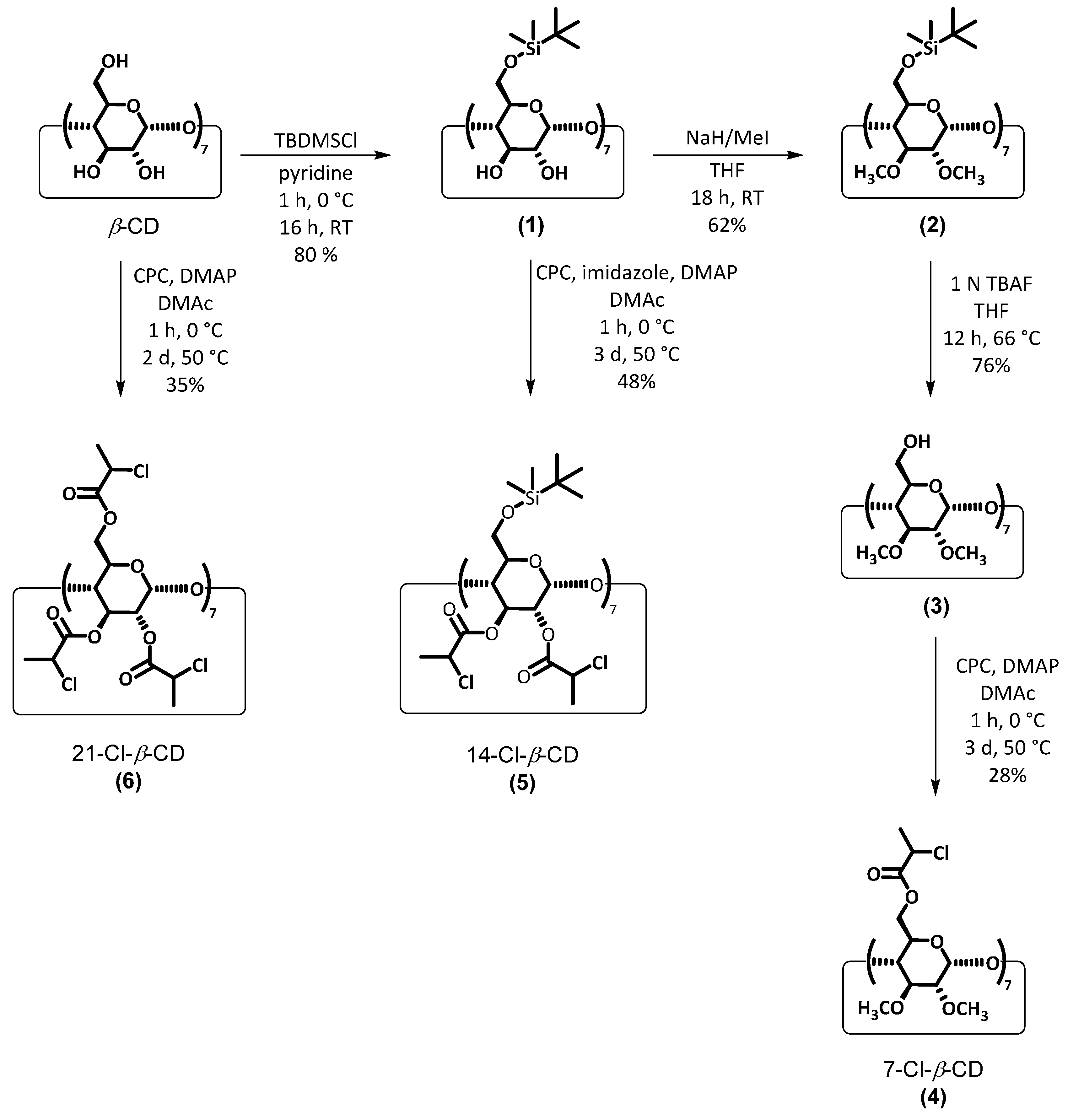
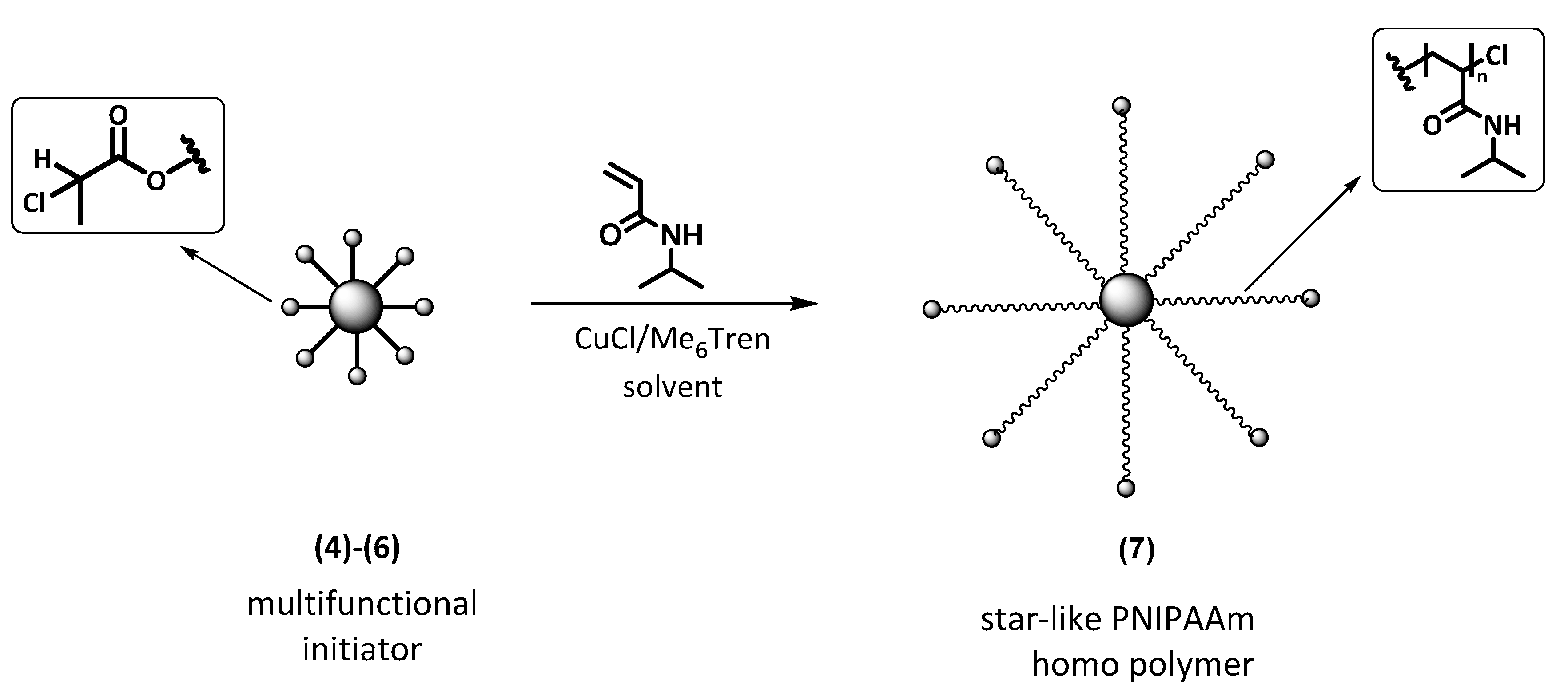
2.2. Synthesis of Multifunctional ATRP Initiators
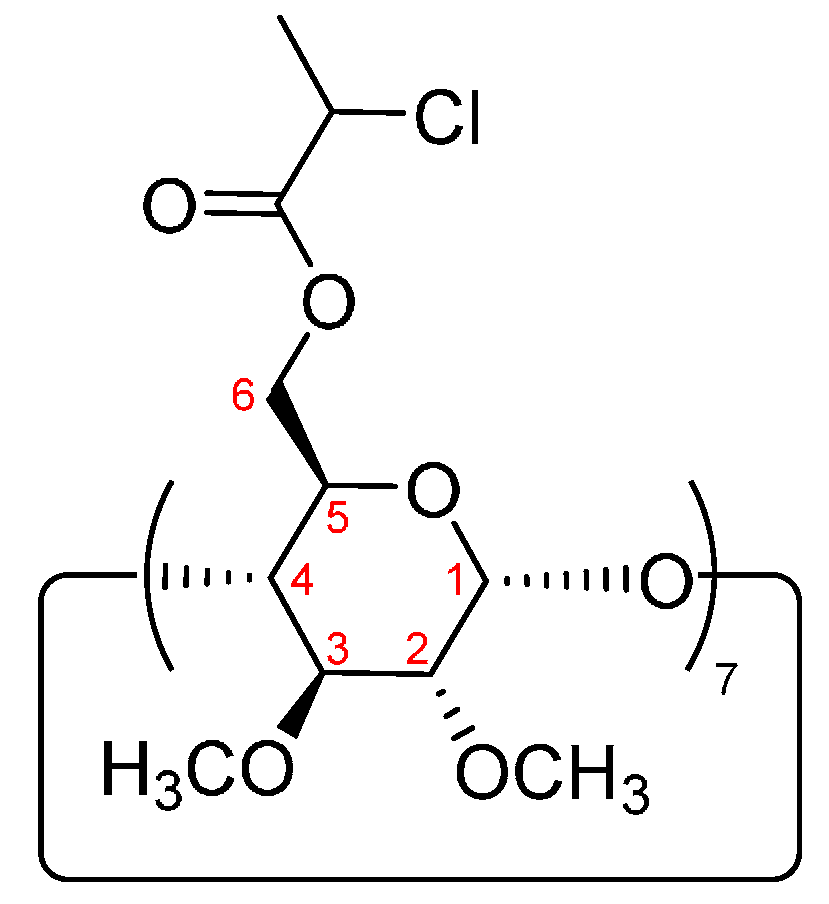
2.3. Synthesis of Heptakis-(6-O-chloropropionyl-2,3-di-O-methyl)-β-cyclodextrin (4) (7-Cl-β-CD)
2.4. Synthesis of Heptakis-(6-(tert-butyldimethysilyl)oxy-2,3-di-O-chloropropionyl)-β-cyclodextrin (5) (14-Cl-β-CD)
2.5. Synthesis of Heptakis-(2,3,6-tri-O-chloropropionyl)-β-cyclodextrin (6) (21-Cl-β-CD)
2.6. Synthesis of Star-like PNIPAAm Homopolymers by ATRP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initiator | M/I/Cu(I)/Cu(II)/L a | Solvent | T (°C) | Time (h) | convNMR (%) | Mn,SEC (g mol−1) | PDSEC |
|---|---|---|---|---|---|---|---|---|
| 7a | 4 | 200/1/1/0/1 | DMSO | 20 | 1.5 | 68 | 152,000 | 1.24 |
| 7b | 4 | 100/1/1.6/0.4/2 | DMSO | 20 | 18 | 68 | 94,000 | 1.20 |
| 8a | 5 | 100/1/1/0/1 | AN | 30 | 18 | 82 | 146,000 | 1.23 |
| 8b | 5 | 100/1/1/0/1 | DMSO | 20 | 18 | 58 | 96,000 | 1.22 |
| 9a | 6 | 50/1/1/0/1 | AN | 30 | 19 | 95 | 61,400 | 1.10 |
| 9b | 6 | 100/1/1.6/0.4/2 | DMSO | 20 | 6.5 | 67 | 120,000 | 1.30 |
| 9c | 6 | 150/1/1/0/1 | AN | 30 | 20 | 99 | 231,000 | 1.20 |
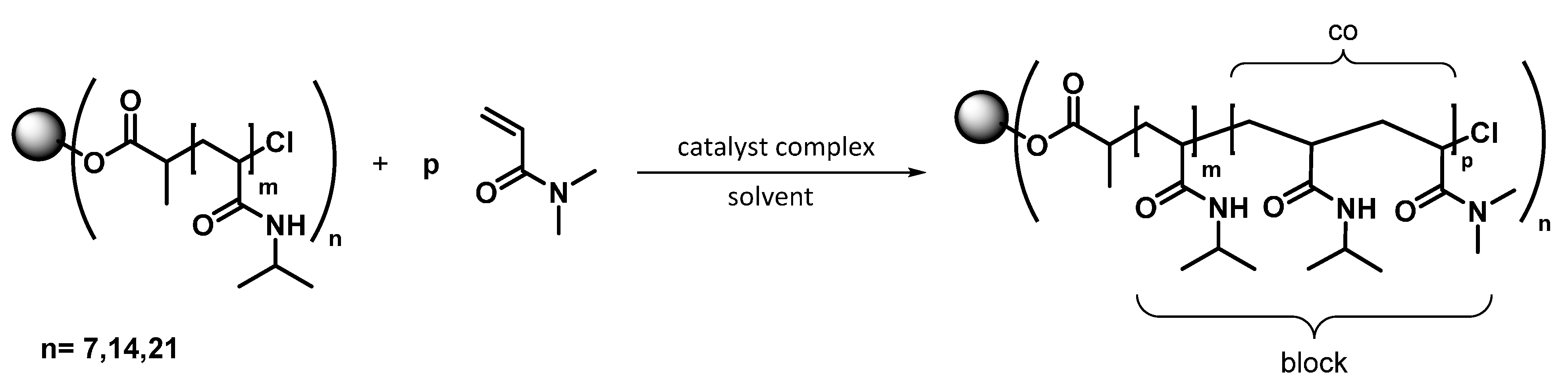
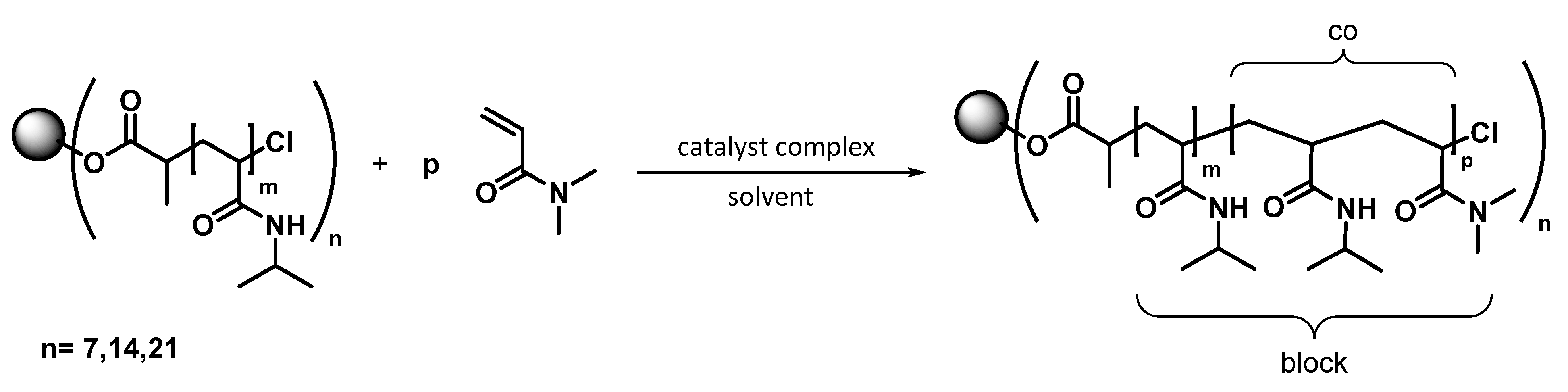
2.7. Synthesis of the Star Block Copolymers
2.8. Characterization
| Sample | Initiator | A/B/I/Cu(I)/Cu(II)/L a | Solvent | Time (h) | convNMR (A %) | Time (h) | convNMR (A %/B %) |
|---|---|---|---|---|---|---|---|
| 10a | 4 | 150/450/1/1/0/1 | DMSO | 2 | 62 | 20 | 70/24 |
| 10b | 4 | 150/900/1/1/0/1 | DMSO | 2 | 66 | 15.5 | 61/23 |
| 11a | 5 | 100/450/1/1/0/1 | DMSO | 2 | 53 | 19 | 57/24 |
| 11b | 5 | 100/500/1/1/0/1 | DMSO | 2 | 60 | 17 | 65/21 |
| 11c | 5 | 100/1000/1/1.6/0.4/1 | DMSO | 2.5 | 47 | 16 | 47/19 |
| 12a | 6 | 150/440/1/2/0/2 | AN | 7 | 64 | 17 | 64/12 |
| 12b | 6 | 100/500/1/1.6/0.4/2 | DMSO | 3 | 59 | 19 | 50/19 |
| 12c | 6 | 100/1000/1.6/0.4/2 | DMSO | 3.5 | 49 | 13 | 48/19 |
3. Results and Discussion
3.1. Synthesis of Multifunctional ATRP Initiators

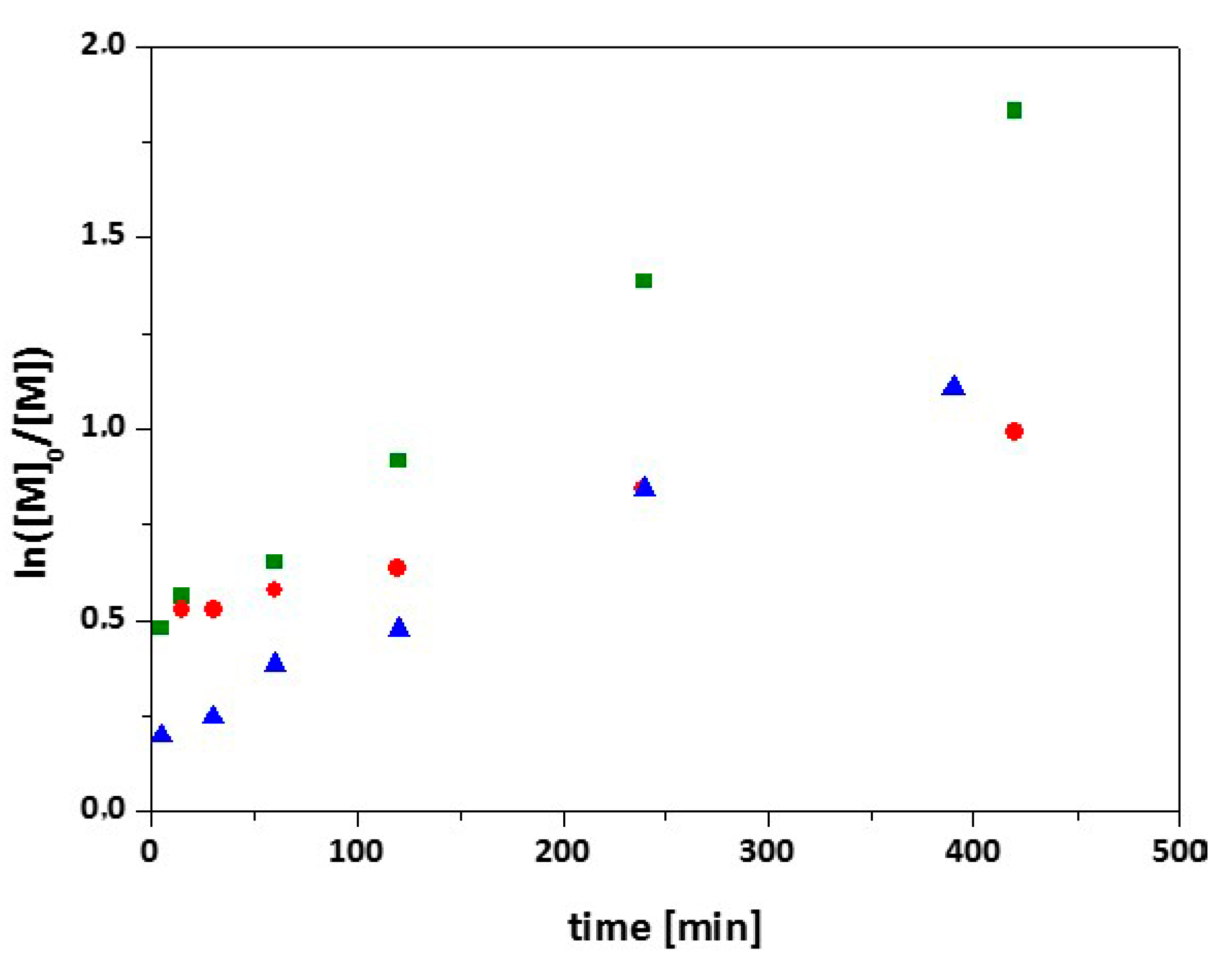
3.2. Synthesis of 7-Arm, 14-Arm and 21-Arm Star-like PNIPAAm Homo Polymers
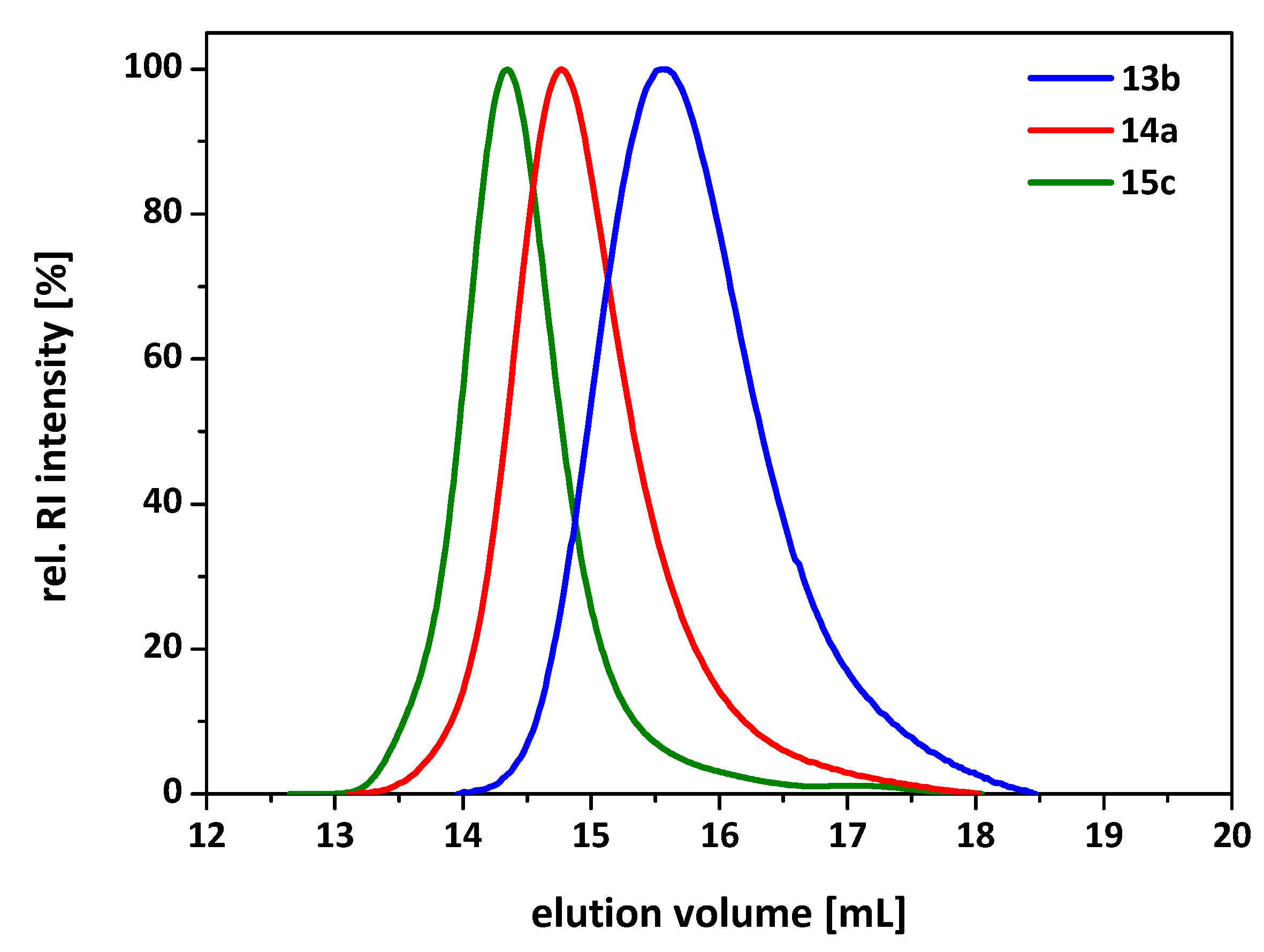
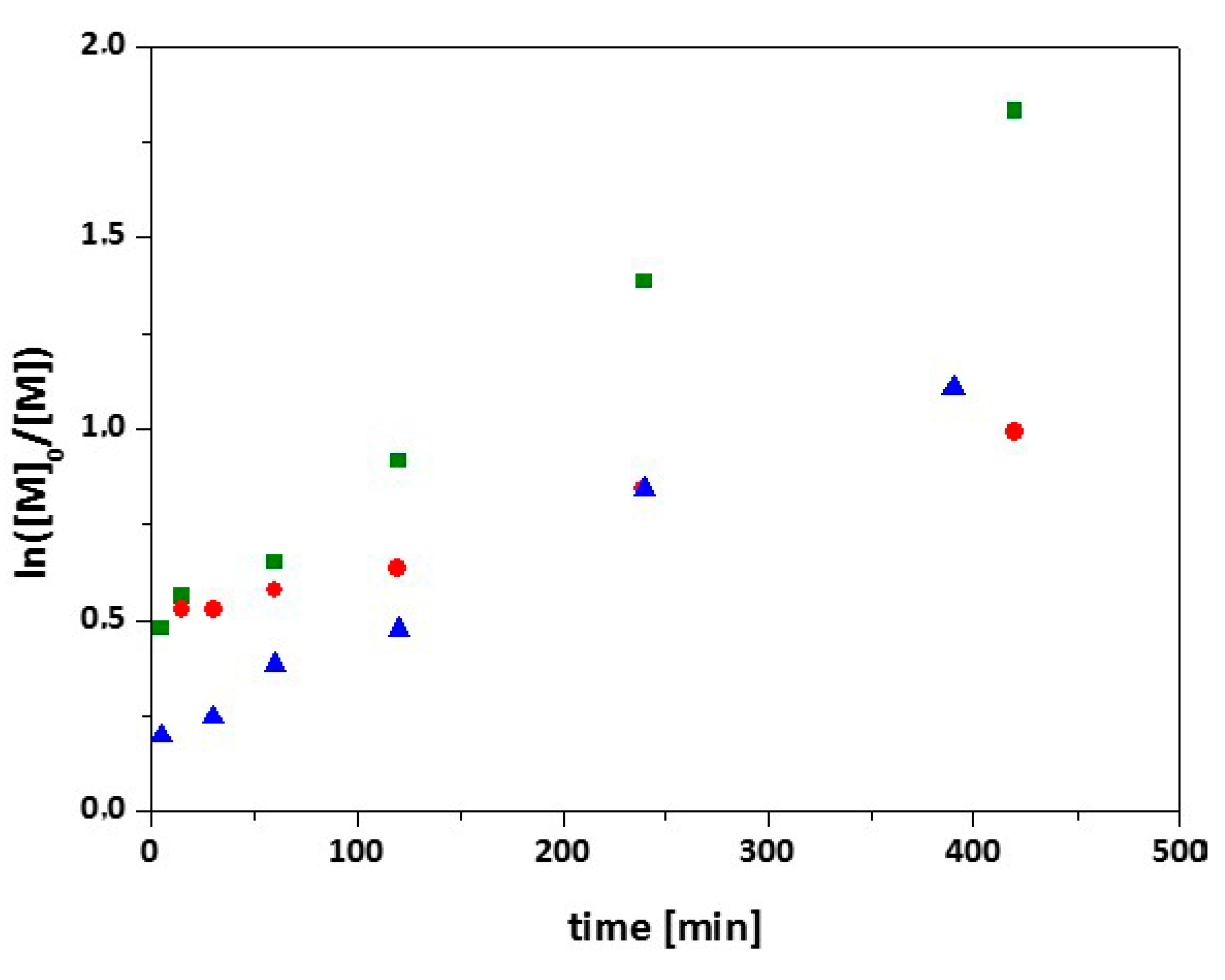
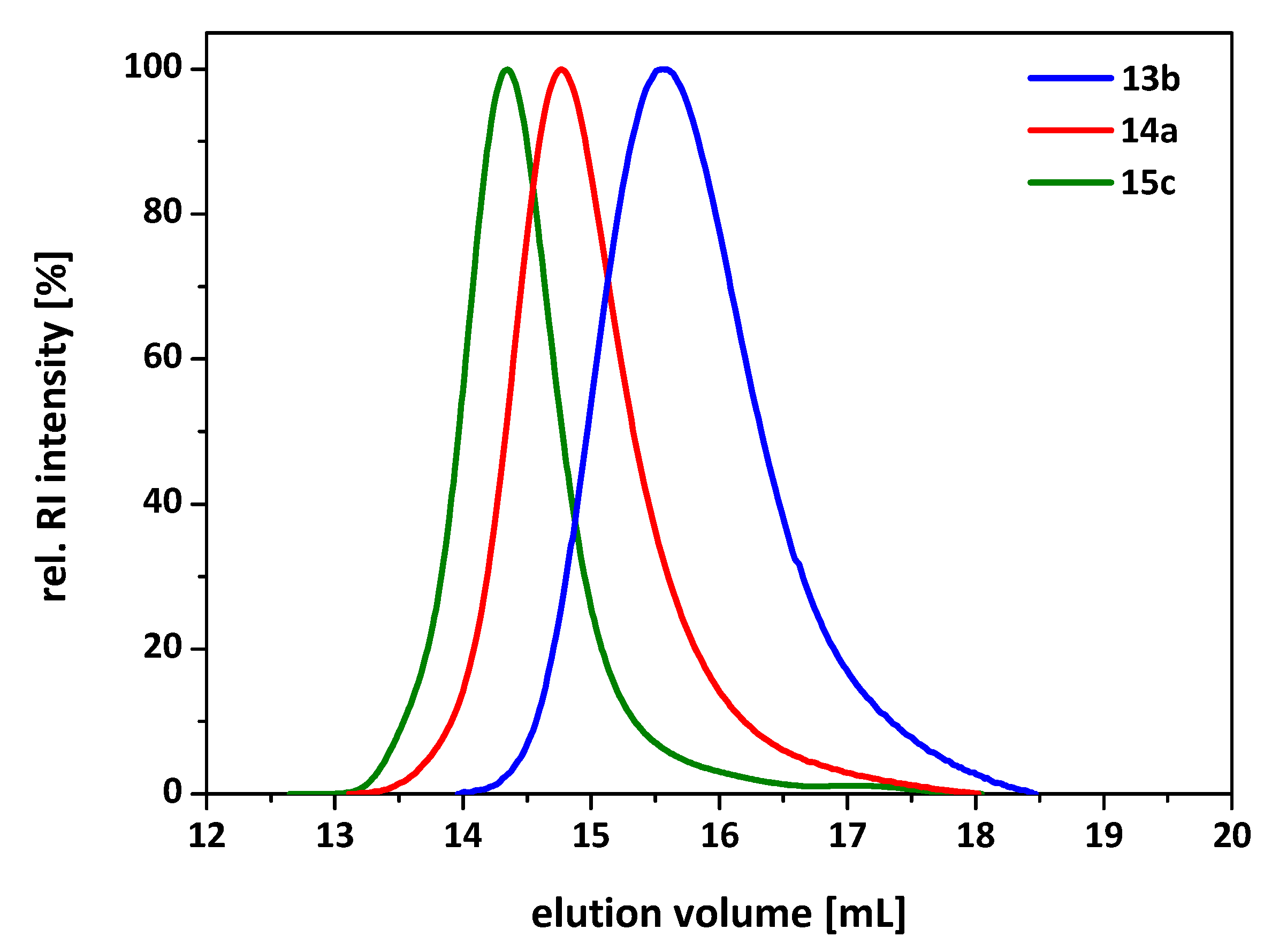
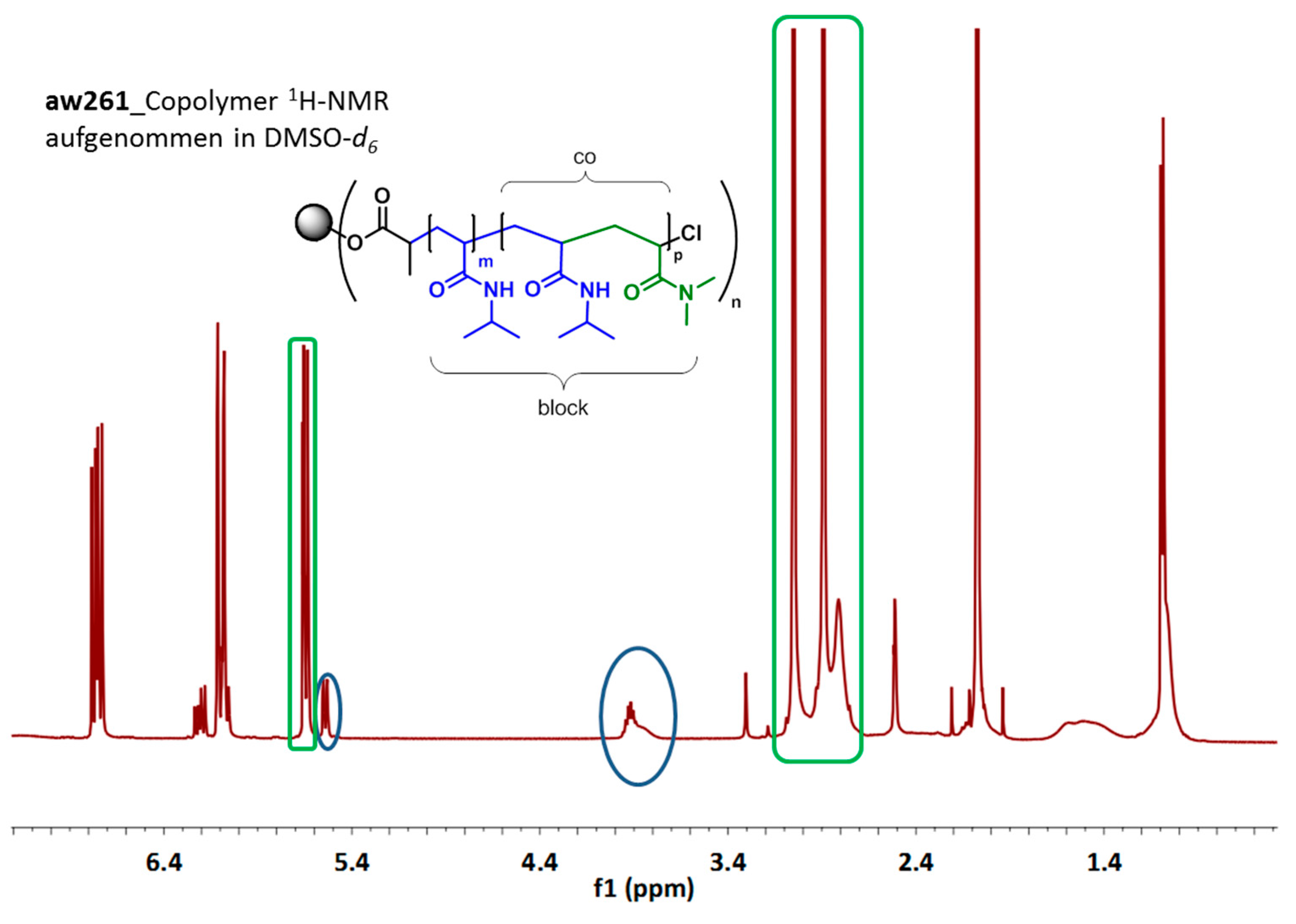
3.3. Synthesis of 7-Arm, 14-Arm and 21-Arm Star-like PNIPAAM-b-PDMAAm Diblock Copolymers
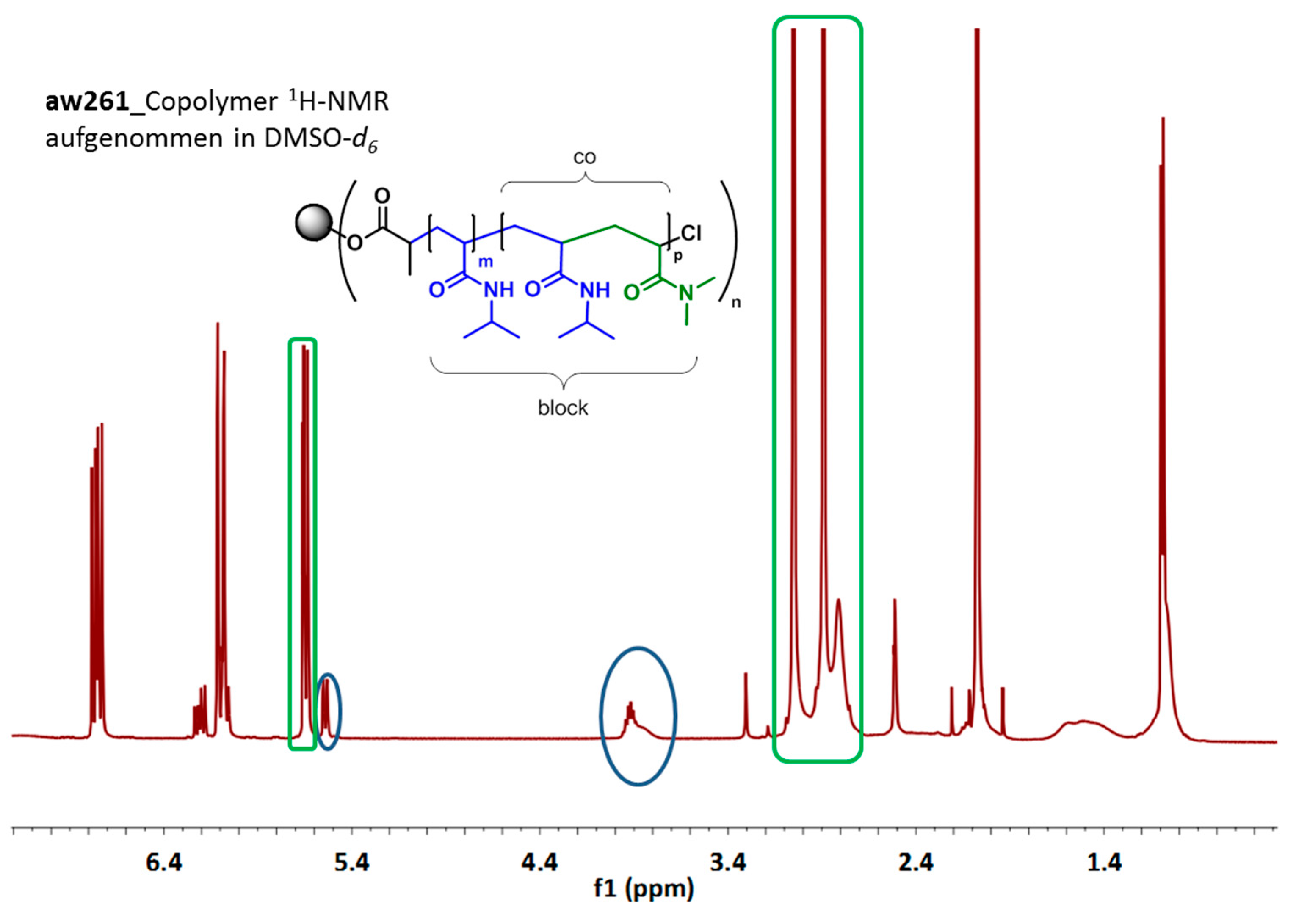

| Sample | Initiator | Mn,SEC (g/mol) | PDSEC | Mn,SEC (g/mol) | PDSEC | Polymer Composition a,b |
|---|---|---|---|---|---|---|
| 10a | 4 | 143,000 | 1.33 | 189,000 | 1.77 | A93-b-(A12-co-B108) |
| 10b | 4 | 138,000 | 1.21 | 161,000 | 1.85 | A99-co-B207 |
| 11a | 5 | 81,000 | 1.19 | 153,000 | 1.62 | A74-b-B99 |
| 11b | 5 | 120,000 | 1.14 | 161,000 | 1.56 | A60-b-(A5-co-B189) |
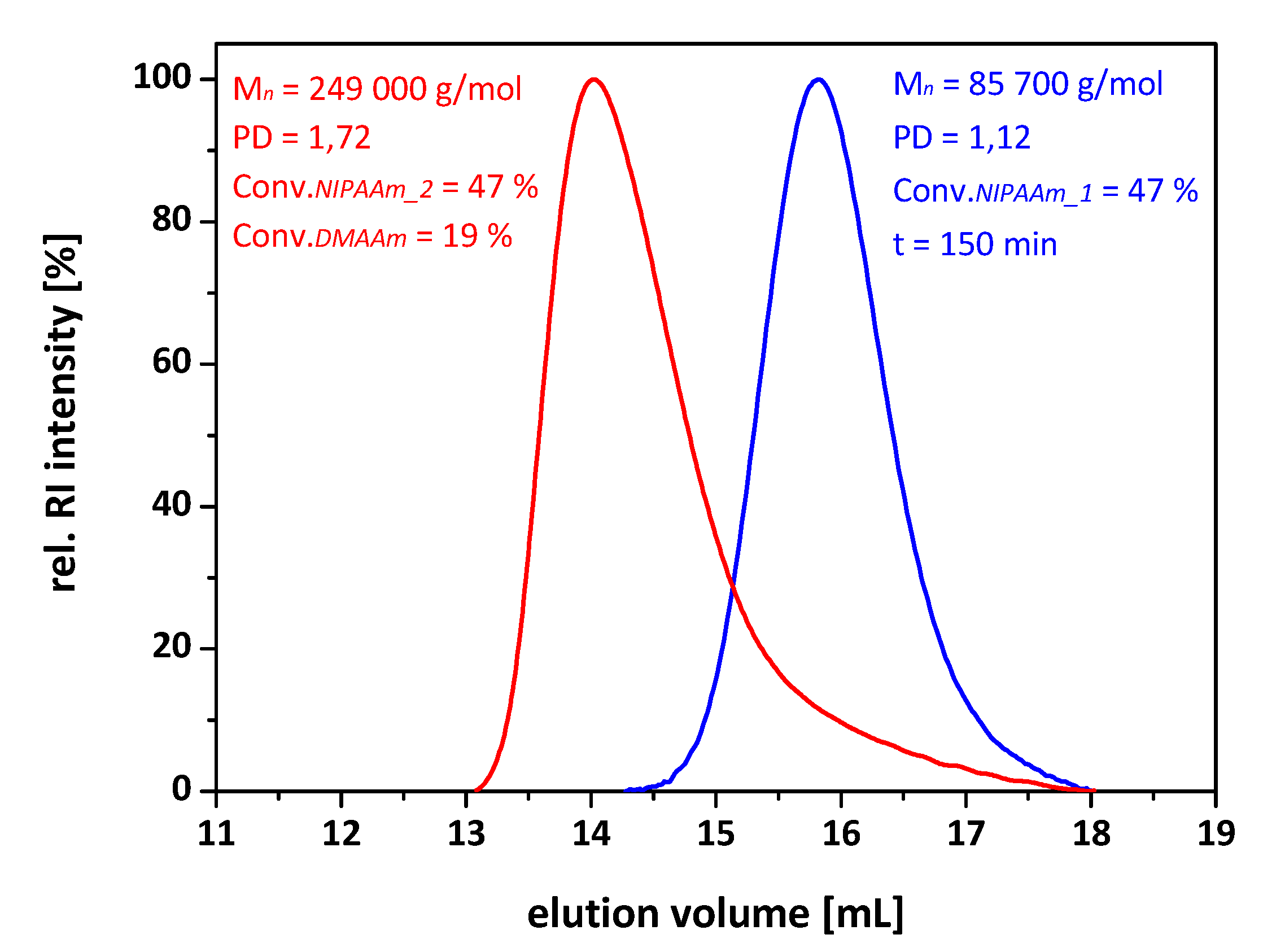
| 11c | 5 | 85,700 | 1.12 | 249,000 | 1.72 | A47-b-B190 |
| 12a | 6 | 192,000 | 1.31 | 254,000 | 1.35 | A96-b-B50 |
| 12b | 6 | 124,000 | 1.15 | 209,000 | 1.70 | A60-b-B95 |
| 12c | 6 | 121,000 | 1.16 | 272,000 | 1.99 | A49-b-B190 |
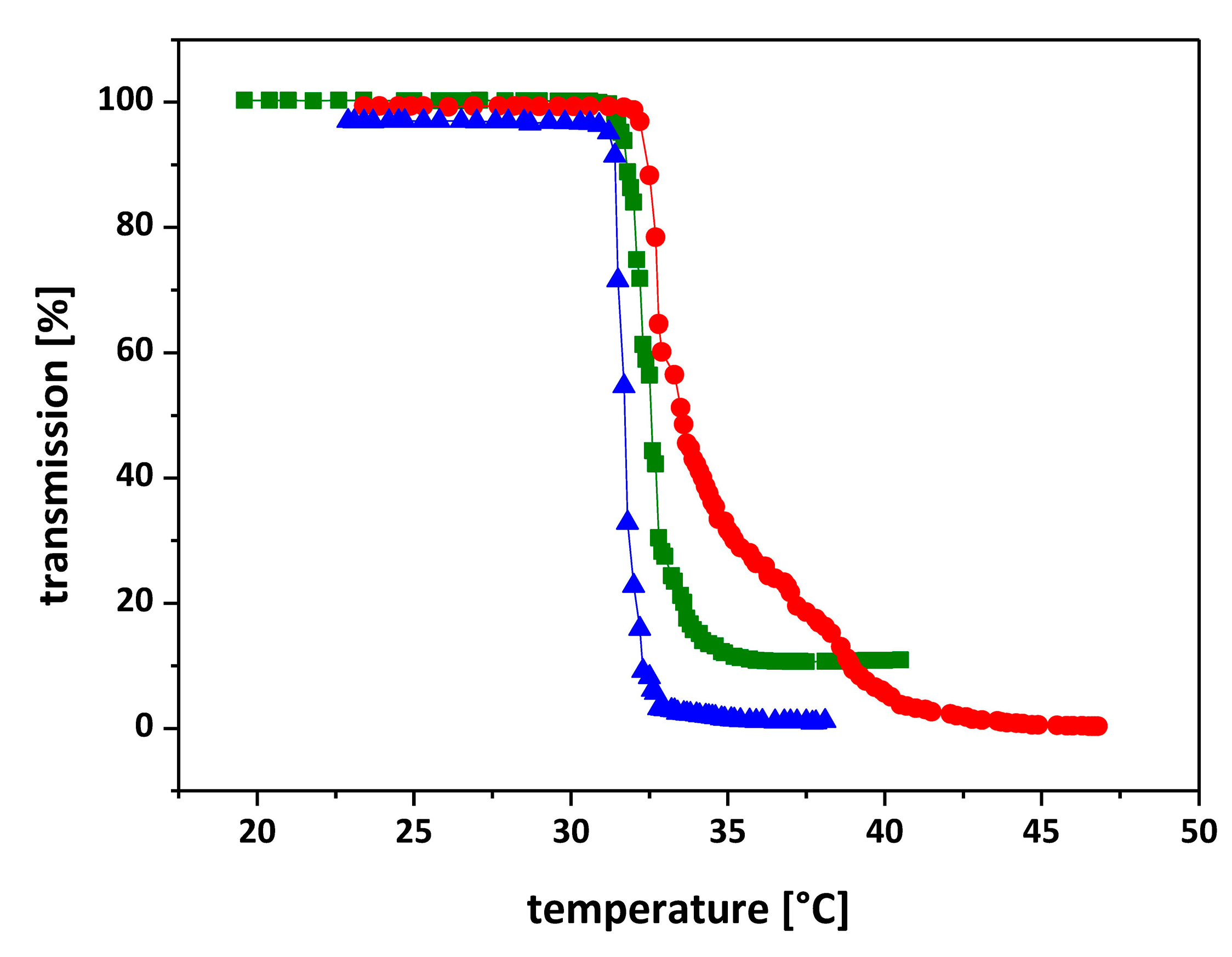
3.4. Thermal Phase Transition Behavior of Star-like PNIPAAm Homo Polymers
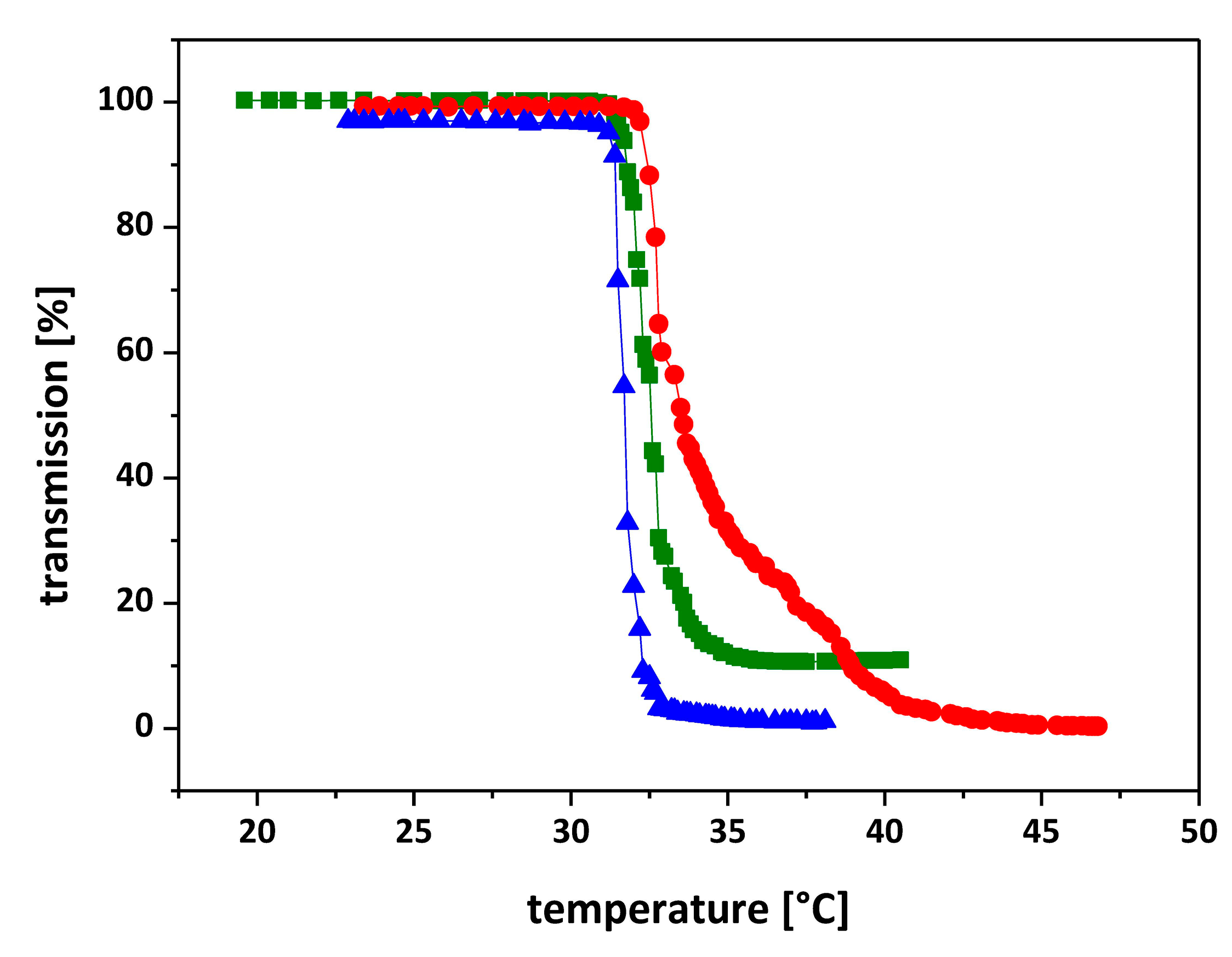
| Sample | Composition of Arms a | Tc (°C) UV-Vis b | Tc (°C) DSC c |
|---|---|---|---|
| 7b | A68 | 32.0 | – |
| 8a | A88 | 33.5 | – |
| 9a | A48 | 32.5 | – |
| 9b | A67 | 31.2 | – |
| 9c | n.d. | 31.8 | – |
| 10a | A93-b-(A12-co-B108) | – | 21.9 (31.3) |
| 10b | A99-b-B207 | – | 23.0 (31.8) |
| 11b | A60-b-(A5-co-B189) | – | 19.6 (30.7) |
| 11c | A47-b-B190 | – | 24.6 (32.6) |
| 12a | A96-b-B50 | – | 31.0 (33.2) |
| 12b | A60-b-B95 | – | 21.9 (32.8) |
| 12c | A49-b-B190 | – | 22.8 (31.8) |
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Kuckling, D.; Wycisk, A. Stimuli-responsive star polymers. J. Polym. Sci. A Polym. Chem. 2013, 51, 2980–2994. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Polymers with complex architecture by living anionic polymerization. Chem. Rev. 2001, 101, 3747–3792. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Dairoku, M. Synthesis and characterization of 6- and 12-arm star polymers by nitroxide-mediated radical polymerization of St and MA from dendritic TIPNO-based hexafunctional and dodecafunctional macroinitiators. J. Polym. Sci. A Polym. Chem. 2007, 45, 4364–4376. [Google Scholar] [CrossRef]
- Hawker, C.J. Architectural control in “living” free radical polymerizations: Preparation of star and graft polymers. Angew. Chem. Int. Ed. Engl. 1995, 34, 1456–1459. [Google Scholar] [CrossRef]
- Chong, B.Y.K.; Le, T.P.T.; Moad, G.; Rizzardo, E.; Thang, S.H. A more versatile route to block copolymers and other polymers of complex architecture by living radical polymerization: The RAFT process. Macromolecules 1999, 32, 2071–2074. [Google Scholar] [CrossRef]
- Liu, J.; Tao, L.; Xu, J.; Jia, Z.; Boyer, C.; Davis, T.P. RAFT controlled synthesis of six-armed biodegradable star polymeric architectures via a “core-first” methodology. Polymer 2009, 50, 4455–4463. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef] [PubMed]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” atom transfer radical polymerization: From process design to preparation of well-defined environmentally friendly polymeric materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xia, J.; Matyjaszewski, K. End-functional poly(tert-butyl acrylate) star polymers by controlled radical polymerization. Macromolecules 2000, 33, 2340–2345. [Google Scholar] [CrossRef]
- Ohno, S.; Gao, H.; Cusick, B.; Kowalewski, T.; Matyjaszewski, K. Methacryloyl and/or hydroxyl end-functional star polymers synthesized by ATRP using the arm-first method. Macromol. Chem. Phys. 2009, 210, 421–430. [Google Scholar] [CrossRef]
- Gao, H.; Matyjaszewski, K. Synthesis of star polymers by a combination of ATRP and the “click” coupling method. Macromolecules 2006, 39, 4960–4965. [Google Scholar] [CrossRef]
- Altintas, O.; Yankul, B.; Hizal, G.; Tunca, U. A3-type star polymers via click chemistry. J. Polym. Sci. A Polym. Chem. 2006, 44, 6458–6465. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Miller, P.J.; Pyun, J.; Kickelbick, G.; Diamanti, S. Synthesis and characterization of star polymers with varying arm number, length, and composition from organic and hybrid inorganic/organic multifunctional initiators. Macromolecules 1999, 32, 6526–6535. [Google Scholar] [CrossRef]
- Angot, S.; Murthy, K.S.; Taton, D.; Gnanou, Y. Atom transfer radical polymerization of styrene using a novel octafunctional initiator: Synthesis of well-defined polystyrene stars. Macromolecules 1998, 31, 7218–7225. [Google Scholar] [CrossRef]
- Lecolley, F.; Waterson, C.; Carmichael, A.J.; Mantovani, G.; Harrisson, S.; Chappell, H.; Limer, A.; Williams, P.; Ohno, K.; Haddleton, D.M. Synthesis of functional polymers by living radical polymerization. J. Mater. Chem. 2003, 13, 2689–2695. [Google Scholar] [CrossRef]
- Strandman, S.; Tenhu, H. Star polymers synthesised with flexible resorcinarene-derived ATRP initiators. Polymer 2007, 48, 3938–3951. [Google Scholar] [CrossRef]
- Haddleton, D.M.; Waterson, C. Phenolic ester-based initiators for transition metal mediated living polymerization. Macromolecules 1999, 32, 8732–8739. [Google Scholar] [CrossRef]
- Ohno, K.; Wong, B.; Haddleton, D.M. Synthesis of well-defined cyclodextrin-core star polymers. J. Polym. Sci. A Polym. Chem. 2001, 39, 2206–2214. [Google Scholar] [CrossRef]
- Karaky, K.; Reynaud, S.; Billon, L.; François, J.; Chreim, Y. Organosoluble star polymers from a cyclodextrin core. J. Polym. Sci. A Polym. Chem. 2005, 43, 5186–5194. [Google Scholar] [CrossRef]
- Adeli, M.; Zarnegar, Z.; Kabiri, R. Amphiphilic star copolymers containing cyclodextrin core and their application as nanocarrier. Eur. Polym. J. 2008, 44, 1921–1930. [Google Scholar] [CrossRef]
- Pang, X.; Zhao, L.; Akinc, M.; Kim, J.K.; Lin, Z. Novel amphiphilic multi-arm, star-like block copolymers as unimolecular micelles. Macromolecules 2011, 44, 3746–3752. [Google Scholar] [CrossRef]
- Gou, P.-F.; Zhu, W.-P.; Xu, N.; Shen, Z.-Q. Synthesis and self-assembly of well-defined cyclodextrin-centered amphiphilic A14B7 multimiktoarm star copolymers based on poly(ε-caprolactone) and poly(acrylic acid). J. Polym. Sci. A Polym. Chem. 2010, 48, 2961–2974. [Google Scholar] [CrossRef]
- Döring, A.; Birnbaum, W.; Kuckling, D. Responsive hydrogels—Structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science. Chem. Soc. Rev. 2013, 42, 7391–7420. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, S. Synthesis of well-defined 7-arm and 21-arm poly(N-isopropylacrylamide) star polymers with β-cyclodextrin cores via click chemistry and their thermal phase transition behavior in aqueous solution. J. Polym. Sci. A Polym. Chem. 2009, 47, 404–419. [Google Scholar] [CrossRef]
- Mauricio, M.R.; Otsuka, I.; Borsali, R.; Petzhold, C.L.; Cellet, T.S.P.; de Carvalho, G.M.; Rubira, A.F. Synthesis of star poly(N-isopropylacrylamide) by β-cyclodextrin core initiator via ATRP approach in water. React.Func. Polym. 2011, 71, 1160–1165. [Google Scholar] [CrossRef]
- Ciampolini, M.; Nardi, N. Five-coordinated high-spin complexes of bivalent cobalt, nickel, and copper with tris(2-dimethylaminoethyl)amine. Inorg. Chem. 1966, 5, 41–45. [Google Scholar] [CrossRef]
- Zhang, P.; Ling, C.C.; Coleman, A.W.; Parrot-Lopez, H.; Galons, H. Formation of amphiphilic cyclodextrins via hydrophobic esterification at the secondary hydroxyl face. Tetrahedron Lett. 1991, 32, 2769–2770. [Google Scholar] [CrossRef]
- Ashton, P.R.; Königer, R.; Stoddart, J.F. Amino acid derivatives of β-cyclodextrin. J. Org. Chem. 1996, 61, 903–908. [Google Scholar] [CrossRef]
- Takeo, K.; Mitoh, H.; Uemura, K. Selective chemical modification of cyclomalto-oligosaccharides via tert-butyldimethylsilylation. Carbohydr. Res. 1988, 187, 203–221. [Google Scholar] [CrossRef]
- Bontempo, D.; Li, R.C.; Ly, T.; Brubaker, C.E.; Maynard, H.D. One-step synthesis of low polydispersity, biotinylated poly(N-isopropylacrylamide) by ATRP. Chem. Commun. 2005, 4702–4704. [Google Scholar] [CrossRef]
- Li, J.; Xiao, H. An efficient synthetic-route to prepare [2,3,6-tri-O-(2-bromo-2-methylpropionyl]-β-cyclodextrin). Tetrahedron Lett. 2005, 46, 2227–2229. [Google Scholar] [CrossRef]
- Carpov, A.; Mocanu, G.; Vizitiu, D. Functional cyclodextrins. 1. Chloroacetylated cyclodextrins. Angew. Makromol. Chem. 1998, 256, 75–79. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, X.; Zhang, X.; Xin, J.; Li, J.; Xiao, H. Effective syntheses of per-2,3-di- and per-3-O-chloroacetyl-β-cyclodextrins: A new kind of ATRP initiators for star polymers. Tetrahedron Lett. 2010, 51, 2351–2353. [Google Scholar] [CrossRef]
- Birnbaum, W.; Kuckling, D. Synthesis of α-biotinyl poly(ethylene glycol-b-N-isopropylacrylamide) block copolymers with different fluorescent dyes at the ω-side. Polym. Chem. 2012, 3, 2039–2049. [Google Scholar] [CrossRef]
- Pintauer, T.; Matyjaszewski, K. Structural aspects of copper catalyzed atom transfer radical polymerization. Coord. Chem. Rev. 2005, 249, 1155–1184. [Google Scholar] [CrossRef]
- Teodorescu, M.; Matyjaszewski, K. Atom transfer radical polymerization of (meth)acrylamides. Macromolecules 1999, 32, 4826–4831. [Google Scholar] [CrossRef]
- Rademacher, J.T.; Baum, M.; Pallack, M.E.; Brittain, W.J. Atom transfer radical polymerization of N,N-dimethylacrylamide. Macromolecules 2000, 33, 284–288. [Google Scholar] [CrossRef]
- Teodorescu, M.; Matyjaszewski, K. Controlled polymerization of (meth)acrylamides by atom transfer radical polymerization. Macromolecules 2000, 21, 190–194. [Google Scholar]
- Neugebauer, D.; Matyjaszewski, K. Copolymerization of N,N-dimethylacrylamide with N-butyl acrylate via atom transfer radical polymerization. Macromolecules 2003, 36, 2598–2603. [Google Scholar] [CrossRef]
- Xia, A.; Burke, N.A.D.; Stöver, H.D.H. End group effect on the thermal response of narrow-disperse poly(N-isopropylacrylamide) prepared by atom transfer radical polymerization. Macromolecules 2006, 39, 2275–2283. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. Chem. A 1968, 8, 1441–1455. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Plummer, R.; Hill, D.T.J.; Whittaker, A.K. Solution properties of star and linear poly(N-isopropylacrylamide). Macromolecules 2006, 39, 8379–8388. [Google Scholar] [CrossRef]
- Pamies, R.; Zhu, K.; Kjoniksen, A.-L.; Nyström, B. Thermal response of low molecular weight poly-(N-isopropylacrylamide) polymers in aqueous solution. Polym. Bull. 2009, 62, 487–502. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wycisk, A.; Döring, A.; Schneider, M.; Schönhoff, M.; Kuckling, D. Synthesis of β-cyclodextrin-Based Star Block Copolymers with Thermo-Responsive Behavior. Polymers 2015, 7, 921-938. https://doi.org/10.3390/polym7050921
Wycisk A, Döring A, Schneider M, Schönhoff M, Kuckling D. Synthesis of β-cyclodextrin-Based Star Block Copolymers with Thermo-Responsive Behavior. Polymers. 2015; 7(5):921-938. https://doi.org/10.3390/polym7050921
Chicago/Turabian StyleWycisk, Agnes, Artjom Döring, Martin Schneider, Monika Schönhoff, and Dirk Kuckling. 2015. "Synthesis of β-cyclodextrin-Based Star Block Copolymers with Thermo-Responsive Behavior" Polymers 7, no. 5: 921-938. https://doi.org/10.3390/polym7050921