1. Introduction
Controlled radical polymerization (CRP) (or equivalently, reversible-deactivation radical polymerization, RDRP), such as stable free radical polymerization (SFRP) (or equivalently, aminoxyl-mediated radical polymerization, AMRP) [
1], atom transfer radical polymerization (ATRP) (or equivalently, controlled reversible-deactivation radical polymerization) [
2,
3], and reversible addition fragmentation chain transfer (RAFT) polymerization (or equivalently, degenerate-transfer radical polymerization) [
4], has attracted great attention in synthesis of well-controlled functional polymers. Among different CRP methods, ATRP is of particular interest for its high potential of applications in various areas [
5]. Control over chain microstructure such as molecular weight distribution (MWD) (or equivalently, molar mass distribution) and polymerization system livingness depend on the polymerization recipes and conditions, which determine the rates of radical termination, radical deactivation, dormant chain activation, and monomer propagation. Numerous studies have been carried out to investigate the influences of chemical recipes [
6,
7,
8], catalyst solubility and catalyst supporting [
9,
10,
11,
12], as well as thermal history [
13,
14], on the ATRP kinetics and polymer molecular weight control. There have been also many modeling efforts made to predict the polymerization behaviors [
15,
16,
17,
18,
19,
20].
An important objective of our research program is to tackle the challenges that face commercial exploitations of the CRP processes. Minimizing the catalyst concentration [
21,
22] and maximizing monomer conversion and eliminating organic solvent as much as possible represent some of the major challenges in this area. Both solvent and residual monomer impose high costs and energy consumption in their separation and purification. High monomer conversions and high solid content polymerization systems are thus most desirable. The problem has been attributed to the diffusion-controlled deactivation reaction which occurs at high conversions [
23]. It decreases the radical deactivation rate and causes a “gel effect” in the polymerization rate, leading to the loss of control over polymer molecular weight and resulting in a high polydispersity index (PDI) (or equivalently, dispersity,
Đ) for the synthesized polymers. The most important reactions in an ATRP system are termination, deactivation, activation and propagation as shown in
Scheme 1.
Scheme 1.
Schematic presentation of the most important reactions in atom transfer radical polymerization (ATRP).
Scheme 1.
Schematic presentation of the most important reactions in atom transfer radical polymerization (ATRP).
In
Scheme 1, M, C, L and XC represent the monomer, catalyst, ligand and deactivator; RM
i•, RM
iX, RM
i and RM
i+jR are the living chain, dormant chain, dead chain formed by disproportionation termination and dead chain by combination termination;
i and
j denote the number of monomeric units in the polymer chain;
k is a rate coefficient with subscript ac indicating activation; de, deactivation; p, propagation; t, termination; td, termination by disproportionation and tc, termination by combination, respectively. The reactants are monomer, initiator and catalyst with the initial concentrations of [M]
0, [RX]
0 and [C]
0, respectively.
In our previous study [
13], the bulk ATRP of methyl methacrylate (MMA) was carried out up to very high conversions. The results showed that although the livingness of polymer chains was preserved because of the diffusion-controlled termination, the control over polymer molecular weight was lost because of the diffusion-controlled deactivation. The final products had broad MWDs with high PDIs. In another study [
14], it was found that by elevating polymerization temperature at high conversions, it was possible to postpone the diffusion-controlled deactivation and obtain highly living and well controlled polymer chains at an almost complete conversion.
We focus on developing experimental strategies for high livingness and good control of CRP at high conversions. The molecular processes involved in the high conversion polymerization are indeed very complicated. In general, any reaction could become diffusion controlled. The fast reactions become diffusion controlled earlier. Typically, radical termination is the first reaction that experiences diffusion limitations, because the reaction involves two chain species of low diffusivity. It is also because of the termination rate coefficient which is the highest among all the reactions. Radical deactivation is the second reaction that onsets diffusion control, because it has a lower rate coefficient than termination and also because it involves one chain and the other small catalytic deactivator. In conventional free radical polymerization, diffusion-controlled termination causes an auto-acceleration in the polymerization rate (named Trommsdorff effect or a misnomer “gel effect”). In CRP, it is the diffusion-controlled deactivation that is responsible for the “gel effect” occurring. Diffusion-controlled termination in CRP is considered as an asset that helps the livingness of polymer chains [
13,
16].
By further increasing the conversion, the system becomes more viscous, small reactants such as catalytic activator and deactivator as well as monomer also experience diffusion limitations. This is particularly true when the system approaches its glass state when the glass transition temperature of the synthesized polymer is higher than the temperature of the polymerization. In the conventional free radical polymerization, diffusion-controlled monomer propagation is responsible for the termination of polymerization at an incomplete conversion (termed “glass effect”). However, it is much more complicated in CRP processes. At this stage, all the small molecule reactants are limited by diffusion. Monomer propagation and dormant chain activation reactions also become diffusion controlled. Depending on the relative sizes and diffusivities of monomer and catalytic activator, as well as the rate coefficients of propagation and activation, one can undergo diffusion control earlier than the other. Either diffusion controlled propagation or diffusion controlled activation can stop the polymerization.
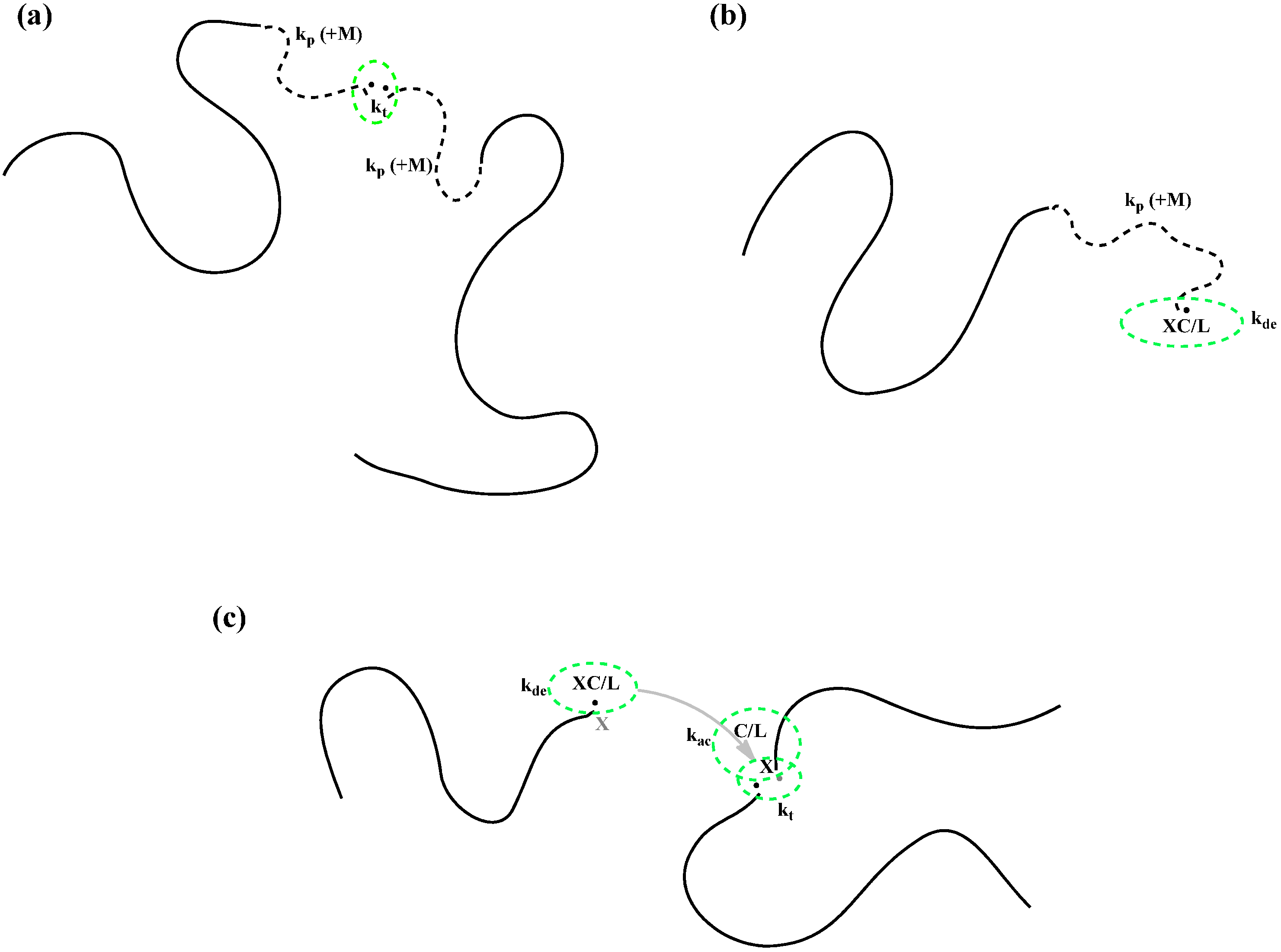
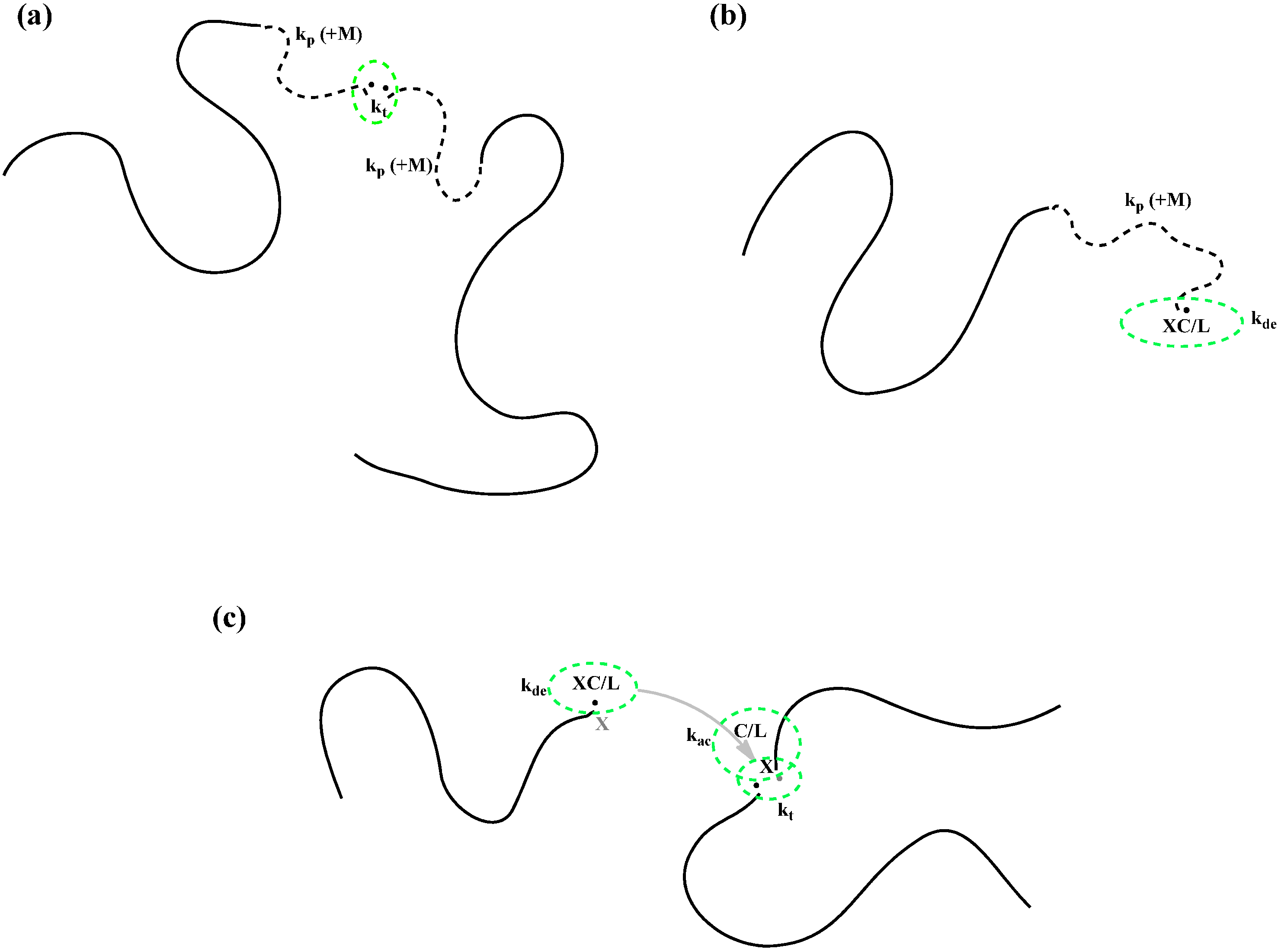
It is also well known that at intermediate and high conversions, while radical chains are limited by diffusion, the radical centers can be still mobile. One mechanism is through monomer propagation. When one monomer is added to a chain end, the radical center moves one step further. This migration of radical center by propagation facilitates residual termination and deactivation. The residual “termination by propagation” has been well studied in the conventional free radical polymerization (
Scheme 2a) [
24]. However, its effect on CRP has not been investigated thoroughly. The residual “deactivation by propagation” is a totally new concept of the present work. Based on intuition, the residual deactivation should help to improve the control at high conversions (
Scheme 2b).
Scheme 2.
Schematic presentation of (a) termination by propagation, (b) deactivation by propagation, and (c) termination by activation/deactivation, which can possibly occur in ATRP at high conversions when small reactants experience diffusion limitations. M, C, L and XC represent the monomer, catalyst, ligand and deactivator; k is a rate coefficient with subscript ac indicating activation; de, deactivation; p, propagation and t, termination, respectively.
Scheme 2.
Schematic presentation of (a) termination by propagation, (b) deactivation by propagation, and (c) termination by activation/deactivation, which can possibly occur in ATRP at high conversions when small reactants experience diffusion limitations. M, C, L and XC represent the monomer, catalyst, ligand and deactivator; k is a rate coefficient with subscript ac indicating activation; de, deactivation; p, propagation and t, termination, respectively.
Furthermore, the recent advent in surface-initiated CRP research reveals additional possible mechanisms of radical center migration. Radicals growing from the surface are constrained by their chains covalently bonded to the surface. The radicals cannot reach each other to be terminated. However, there were some experimental data that supported radical termination. A new “hopping” mechanism was thus proposed by Zhu
et al. [
25,
26]. A radical center can be deactivated in one spot by a catalytic deactivator. The latter quickly moves to another spot to activate a radical center. A net outcome of this deactivation/activation cycle is migration of the radical center from one spot to another, which can facilitate radical termination, if another radical center happens to be at the vicinity. This “hopping” migration can also exist in the bulk or solution CRP at high conversions (
Scheme 2c). The residual termination caused by the radical “hopping” could also affect the control of ATRP, which has not been investigated.
Considering all the complicated mechanisms outlined above, one must resort to the use of modeling approaches to investigate their influences in CRP processes. In this paper, we develop a kinetic model that includes all the possible reactions at high conversions and examine their effects on the polymerization rate and the control of polymer molecular weight in a bulk ATRP using methyl methacrylate (MMA) as a model system.
3. Results and Discussion
The influences of the various diffusion-controlled reactions, residual termination and deactivation on the polymerization rate and polymer molecular weight control are examined using the model simulation, in the order that they could potentially occur during polymerization. The radical termination reaction has the highest rate coefficient and involves two chains, which readily experience diffusion limitations. It thus becomes diffusion controlled at relatively low monomer conversions.
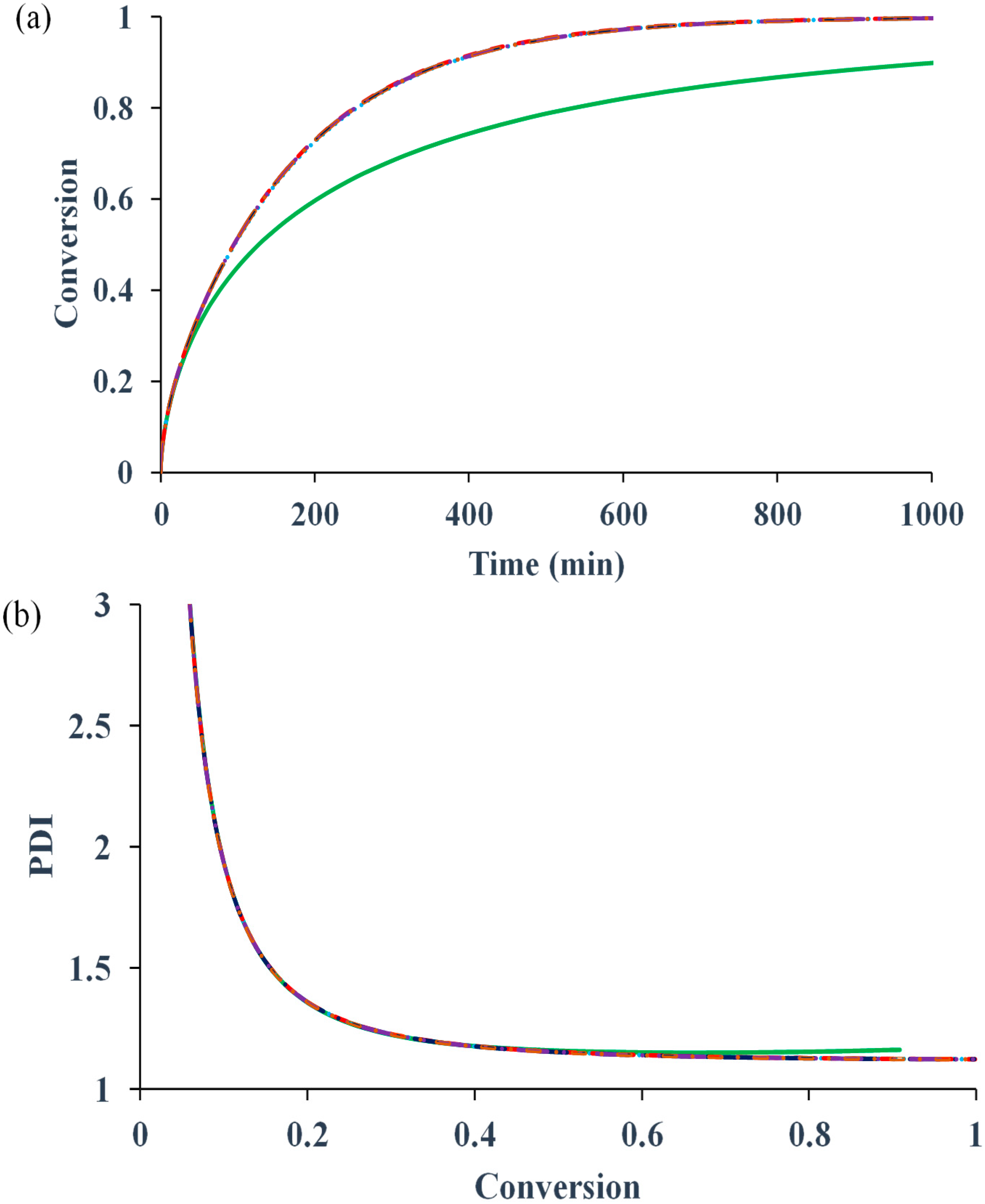
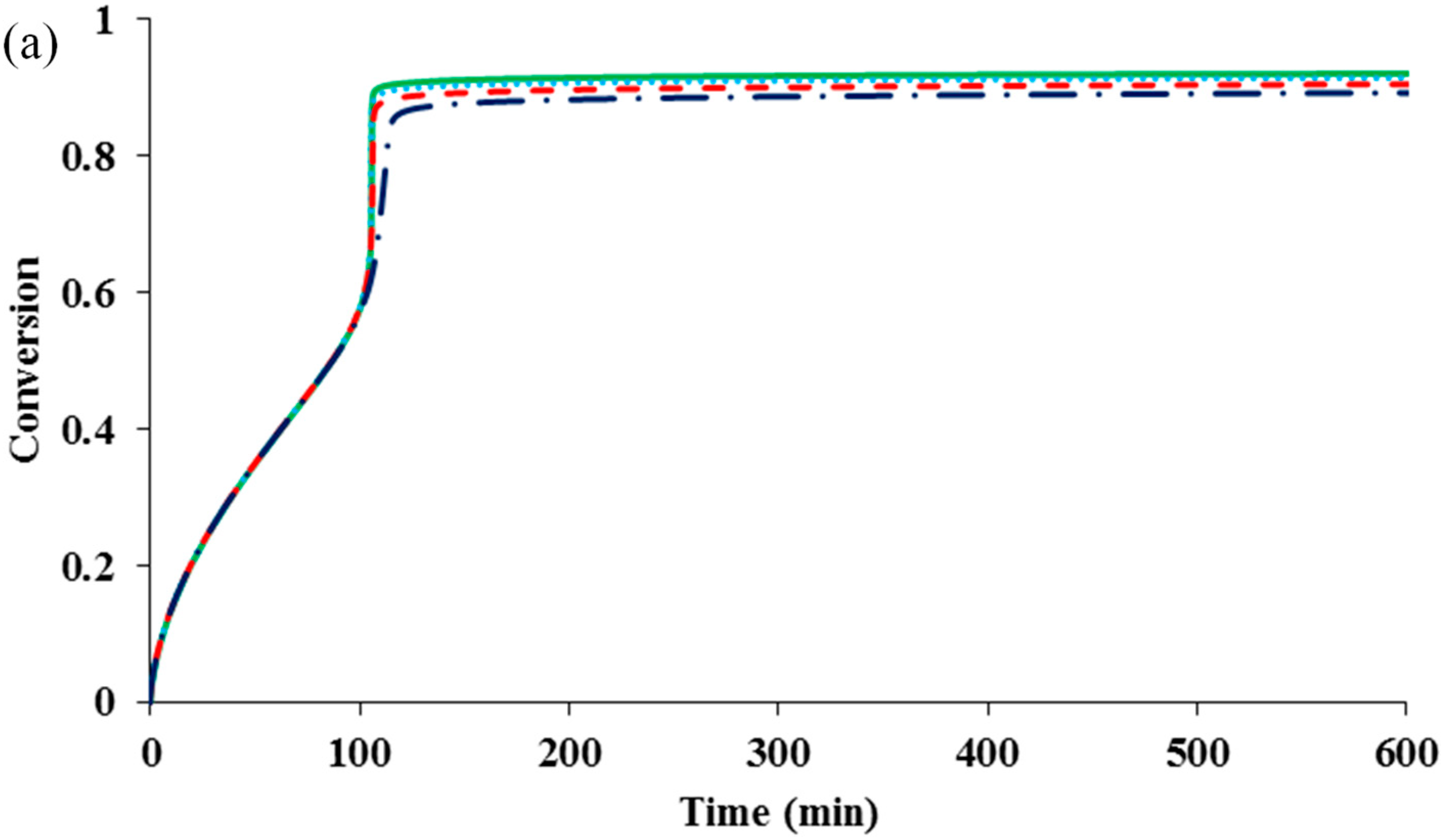
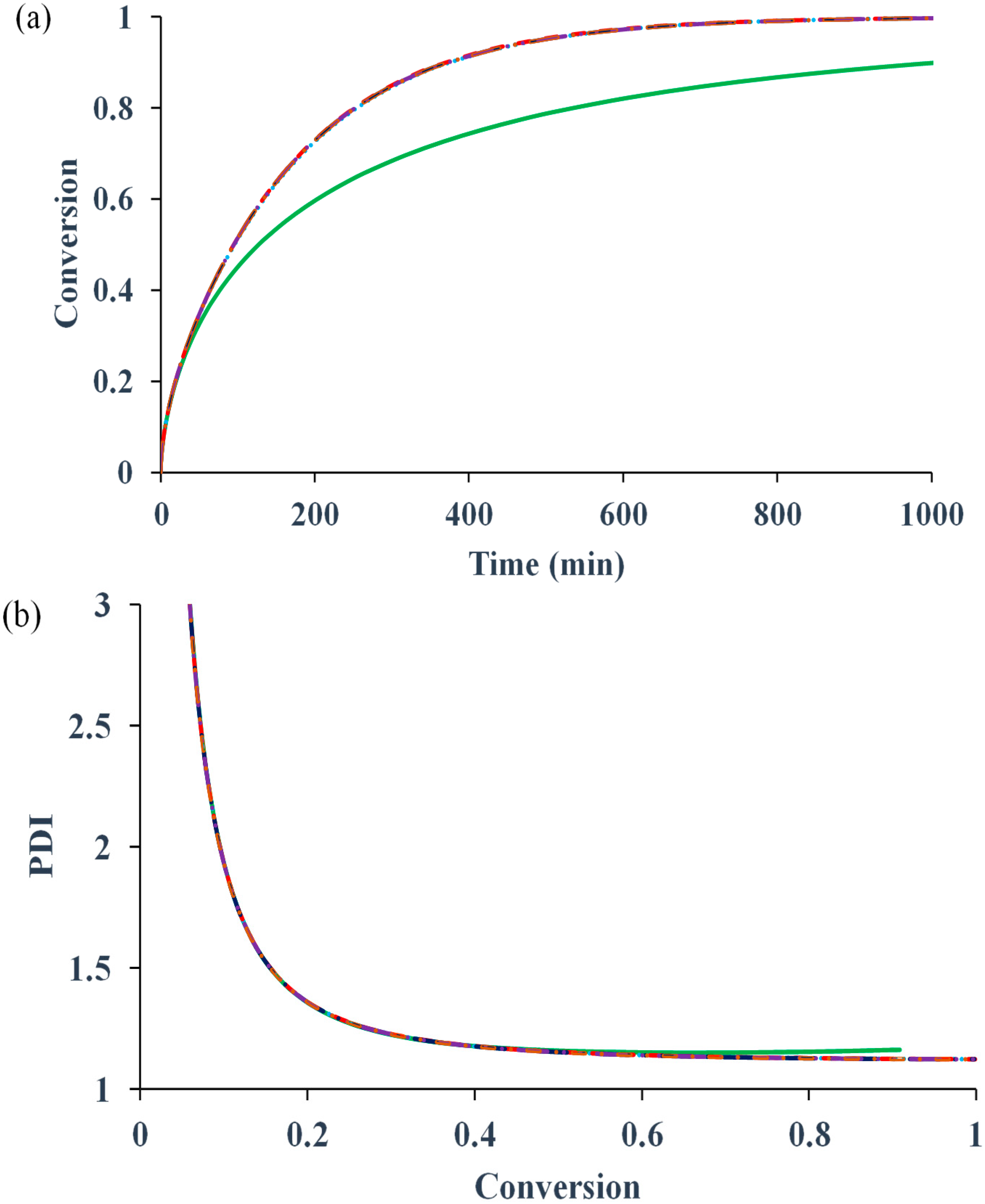
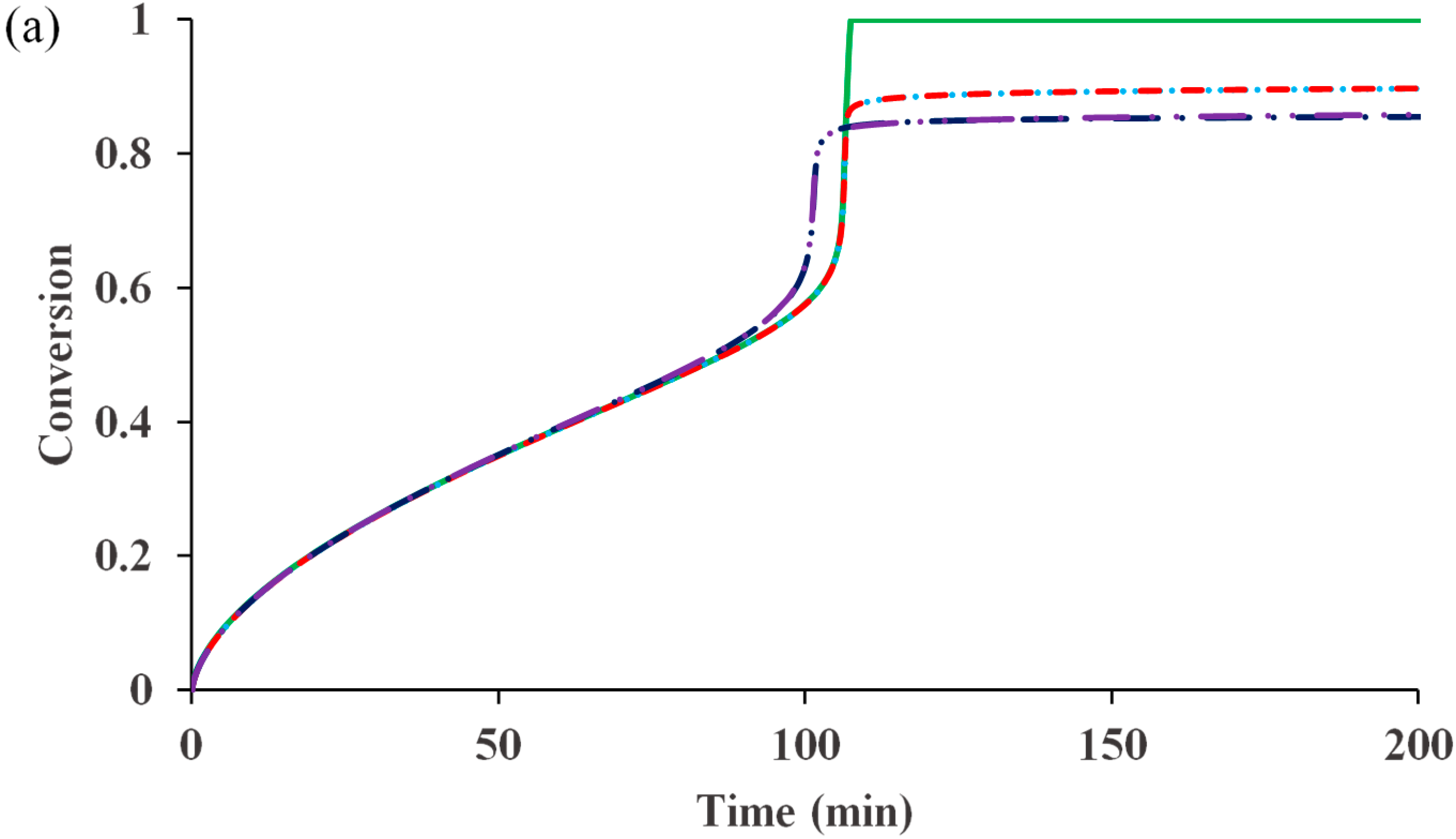
Figure 1 compares the kinetic behaviors of the bulk ATRP of MMA with and without diffusion-controlled termination, with and without residual termination, but excluding the other diffusion-controlled reactions.
Figure 1a shows that for the system without any diffusion-controlled reaction, the polymerization rate is slower. The onset of diffusion-controlled termination slightly increases the radical concentration, resulting in a minor increase in the rate. However, the influences of the residual termination by “hopping” mechanism and that by propagation on the rate are negligible. In contrast to the conventional free radical polymerization, in which diffusion-controlled termination can cause a dramatic auto-acceleration (
i.e., “gel effect”), the diffusion-controlled termination in ATRP does not have as much influence. The reason is that the radical concentration in the former is determined by the initiation and termination reactions, while that in the latter is mainly regulated by the activation and deactivation reactions. The termination does not significantly change the equilibrium of activation and deactivation.
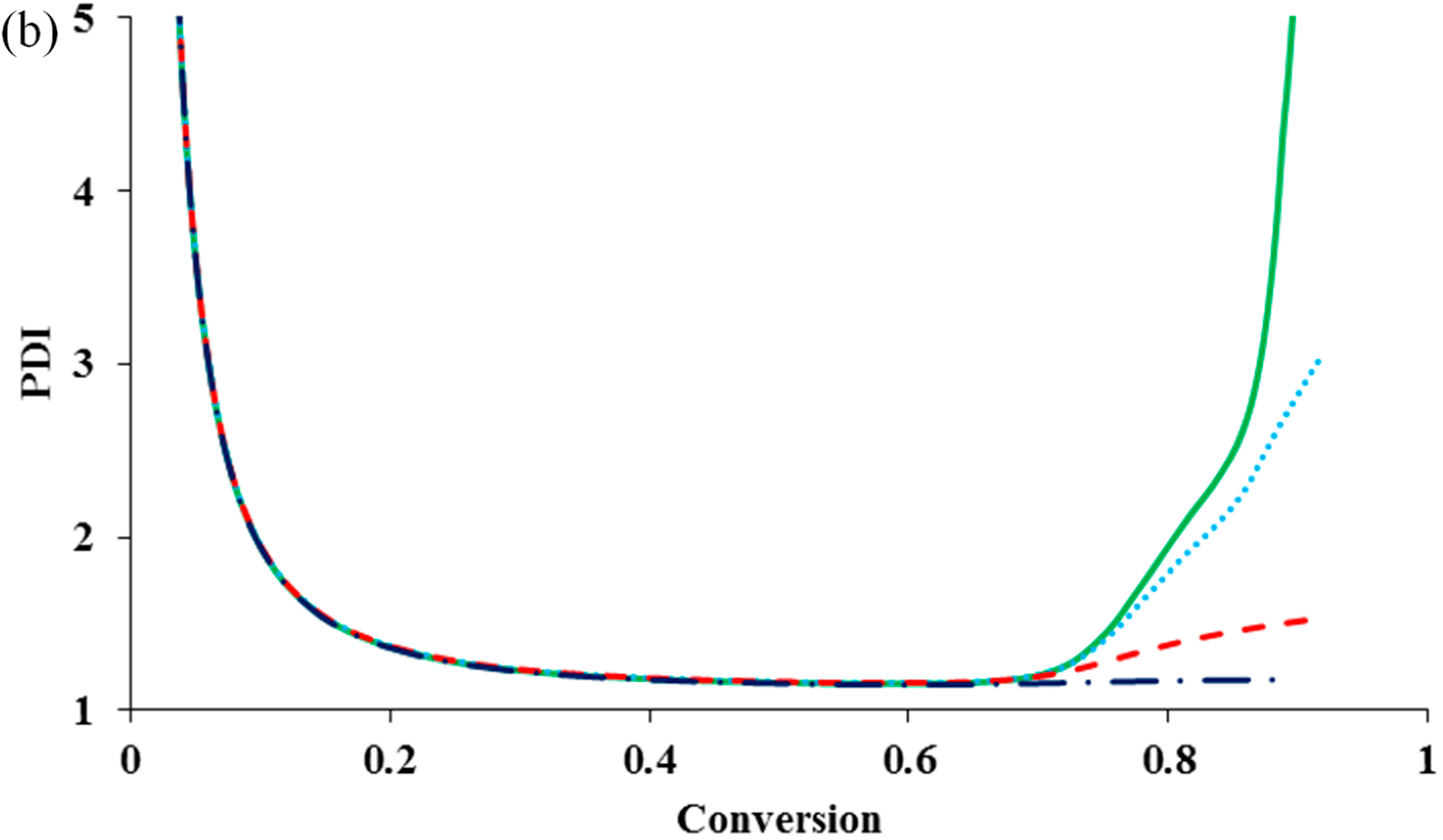
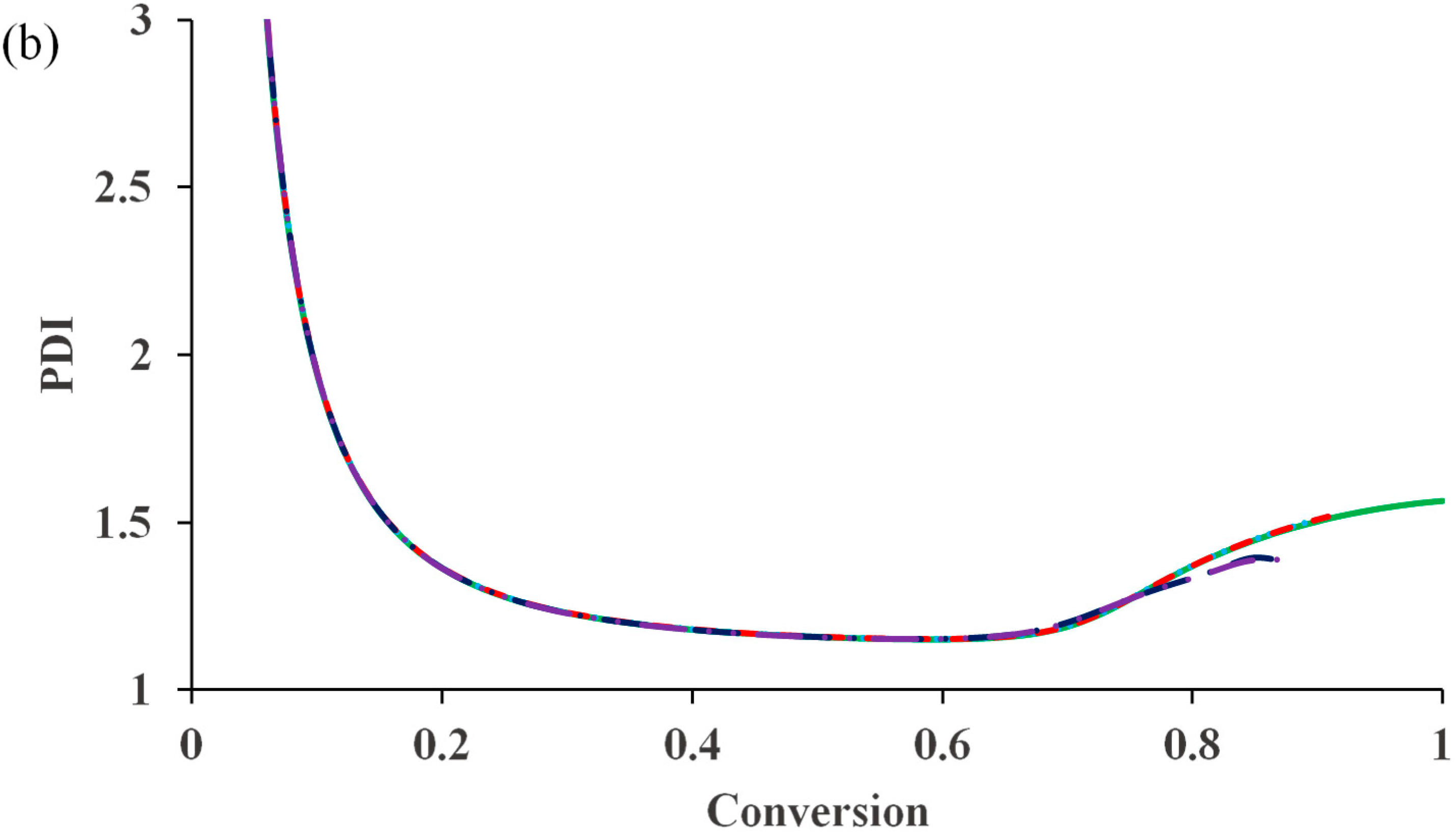
Figure 1b shows that PDI remains lower than 1.2 because of negligible influence of the termination reaction.
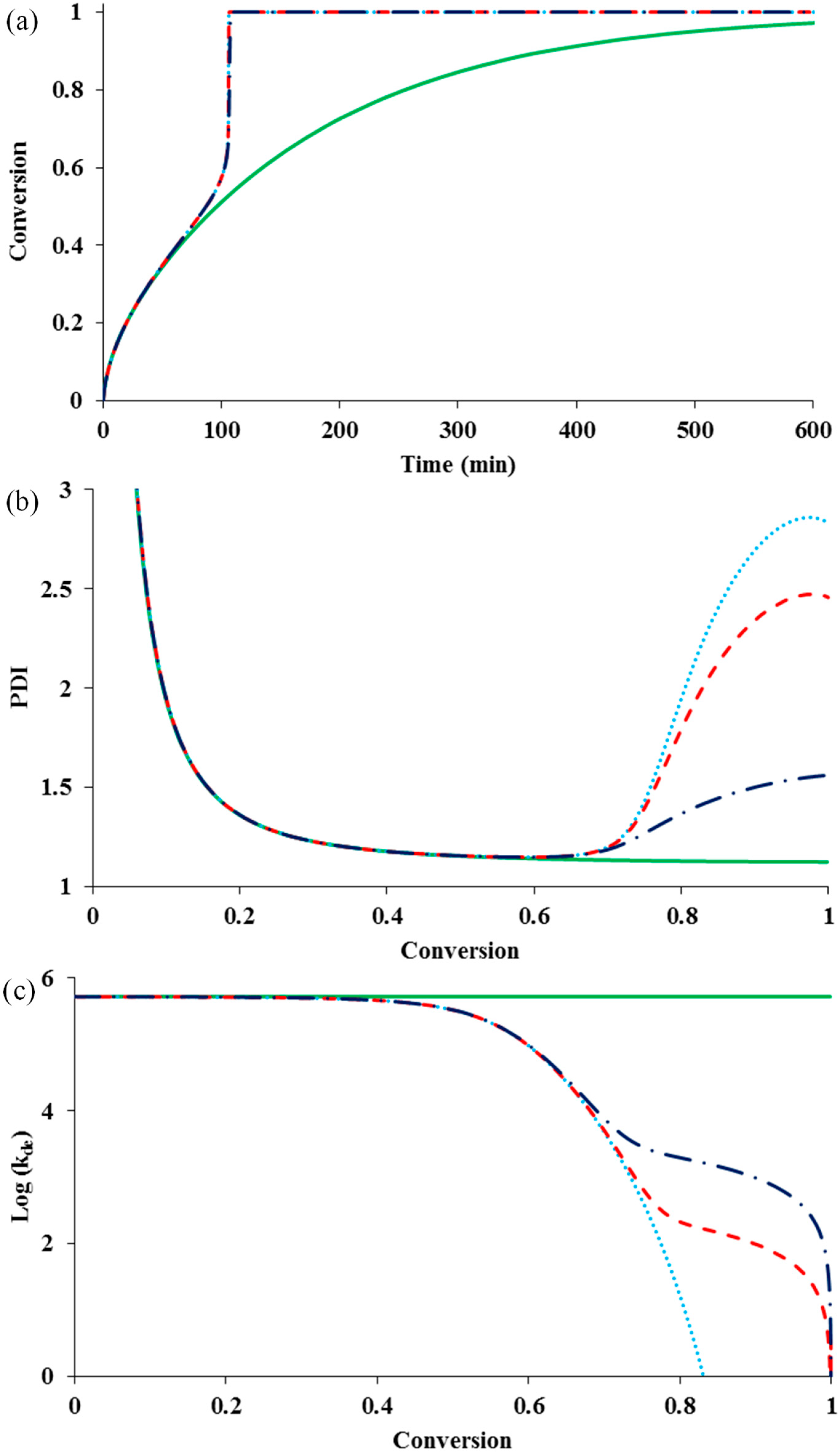
Radical deactivation in ATRP involves a large radical chain and a small catalytic deactivator. Second to termination, it can easily become diffusion controlled at medium conversions (in this case, around 50% conversion). One might argue that activation and deactivation should onset diffusion control at the same time because both reactions are between large chains and small catalytic molecules. However, it is indicated that because of lower diffusivity of larger deactivator, diffusional limitation on deactivation reaction is more severe than activation [
18]. Furthermore, the deactivation rate coefficient is typically much higher than that of activation. A faster reaction becomes diffusion controlled earlier than a slower one, which is evident from Equation 22.
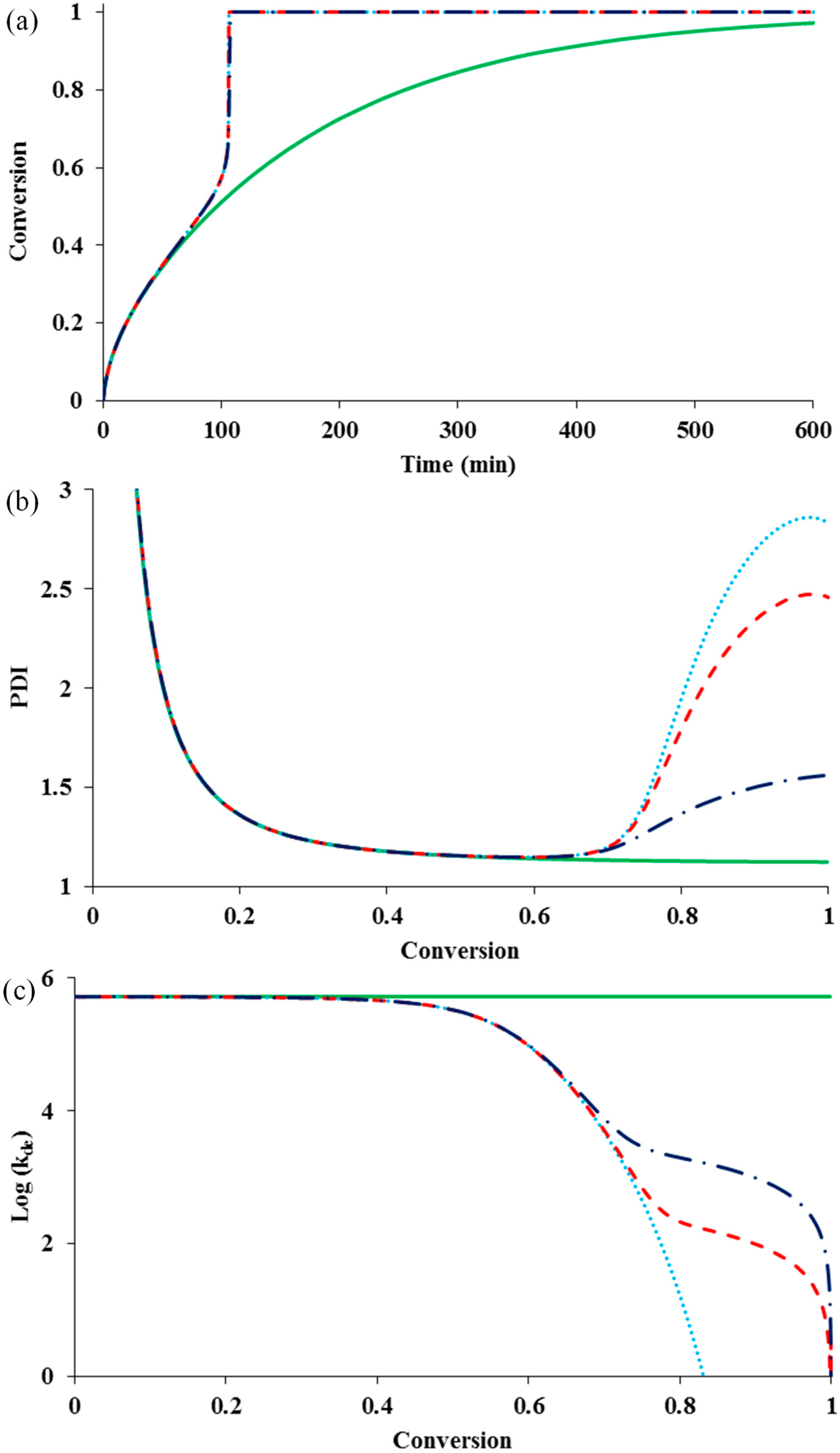
Figure 2 shows the influence of diffusion-controlled deactivation with or without residual deactivation on the rate and control of polymerization, in the presence of diffusion-controlled termination (
= 10
14) and residual termination (z
tde = 100 & z
tp = 10). It also shows the change of
kde in a log scale with conversion.
Figure 1.
Effects of diffusion-controlled termination and residual termination on (
a) conversion
versus time (
b) polydispersity index (PDI)
versus conversion. No diffusion-controlled termination
![Polymers 07 00819 i001]()
, translational diffusion-controlled termination
but no residual termination
, translational diffusion-controlled termination
with residual termination by radical hopping via deactivation/activation reactions
&
, and translational diffusion-controlled termination
with residual termination by radical hopping
and monomer propagation
&
.
Figure 1.
Effects of diffusion-controlled termination and residual termination on (
a) conversion
versus time (
b) polydispersity index (PDI)
versus conversion. No diffusion-controlled termination
![Polymers 07 00819 i001]()
, translational diffusion-controlled termination
but no residual termination
, translational diffusion-controlled termination
with residual termination by radical hopping via deactivation/activation reactions
&
, and translational diffusion-controlled termination
with residual termination by radical hopping
and monomer propagation
&
.
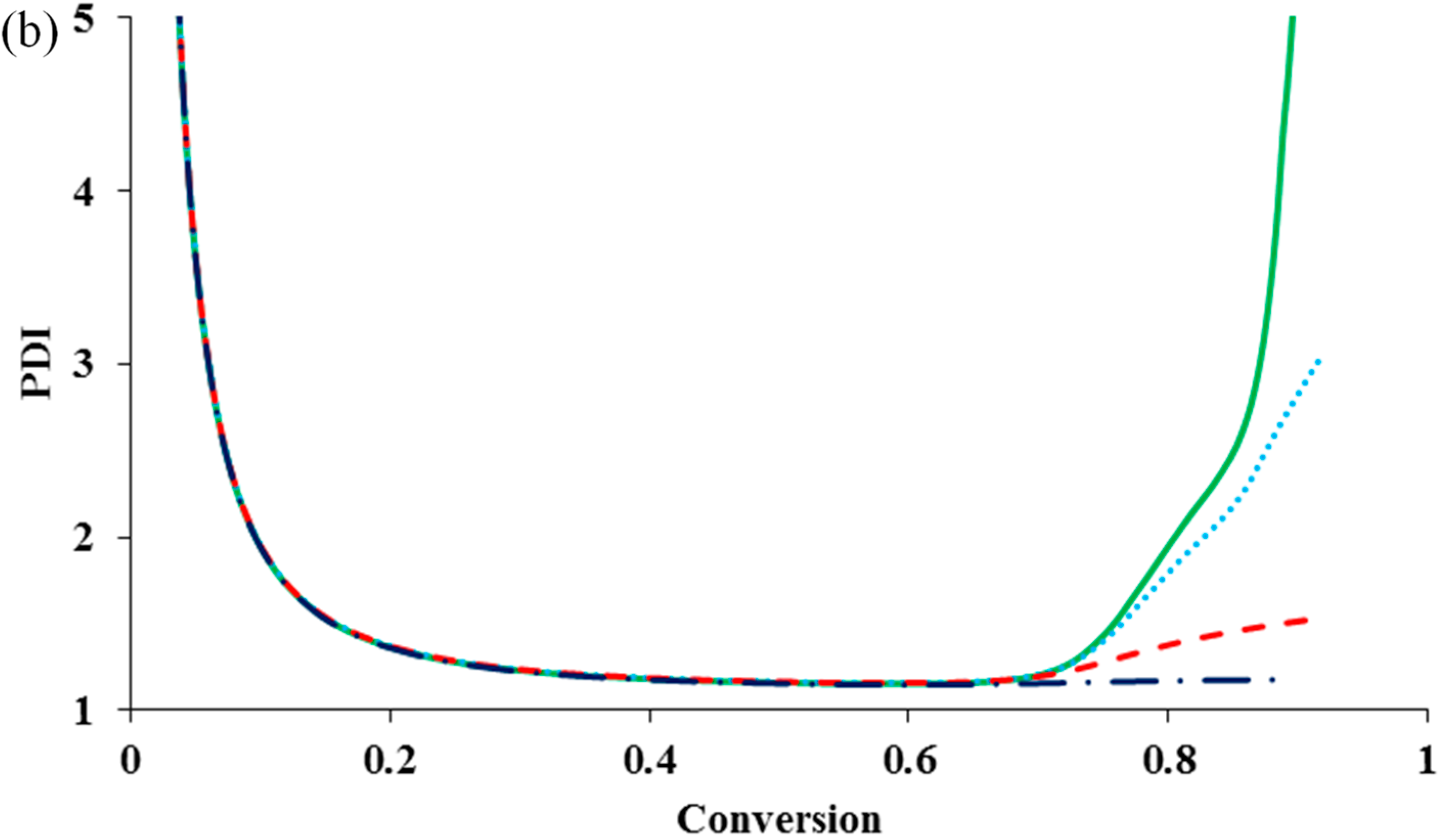
Figure 2.
The influence of diffusion-controlled deactivation and residual deactivation on (
a) conversion
versus time; (
b) PDI
versus conversion; and (
c) log (k
de)
versus conversion for an ATRP system having diffusion-controlled termination
and residual termination
. No diffusion-controlled deactivation
![Polymers 07 00819 i001]()
, diffusion-controlled deactivation
but no residual deactivation
, diffusion-controlled deactivation
with residual deactivation by monomer propagation
.
Figure 2.
The influence of diffusion-controlled deactivation and residual deactivation on (
a) conversion
versus time; (
b) PDI
versus conversion; and (
c) log (k
de)
versus conversion for an ATRP system having diffusion-controlled termination
and residual termination
. No diffusion-controlled deactivation
![Polymers 07 00819 i001]()
, diffusion-controlled deactivation
but no residual deactivation
, diffusion-controlled deactivation
with residual deactivation by monomer propagation
.
It can be seen that the diffusion-controlled deactivation causes a severe auto-acceleration in the polymerization rate (“gel effect”). This is because the radical concentration increases dramatically in the absence of adequate deactivation. The residual deactivation by propagation does not help to compensate the situation. There is no significant difference in the rate profiles with and without the residual deactivation. When the deactivation becomes diffusion controlled, the radical freely propagates with monomer and the chain quickly grows to a high molecular weight, without regulation of the equilibrium of activation and deactivation. This results in the loss of control over polymer molecular weight and leads to the dramatic increase in PDI, as shown in
Figure 2b. It is also clear from the simulation result that the residual deactivation compensates the loss of control to certain extents. Introducing the residual deactivation decreases the PDI at high conversions.
Figure 2c shows the k
de profile during polymerization. The residual deactivation becomes dominant at very high conversions.
After termination and deactivation of radicals, dormant chain activation and monomer propagation can become diffusion controlled as well.
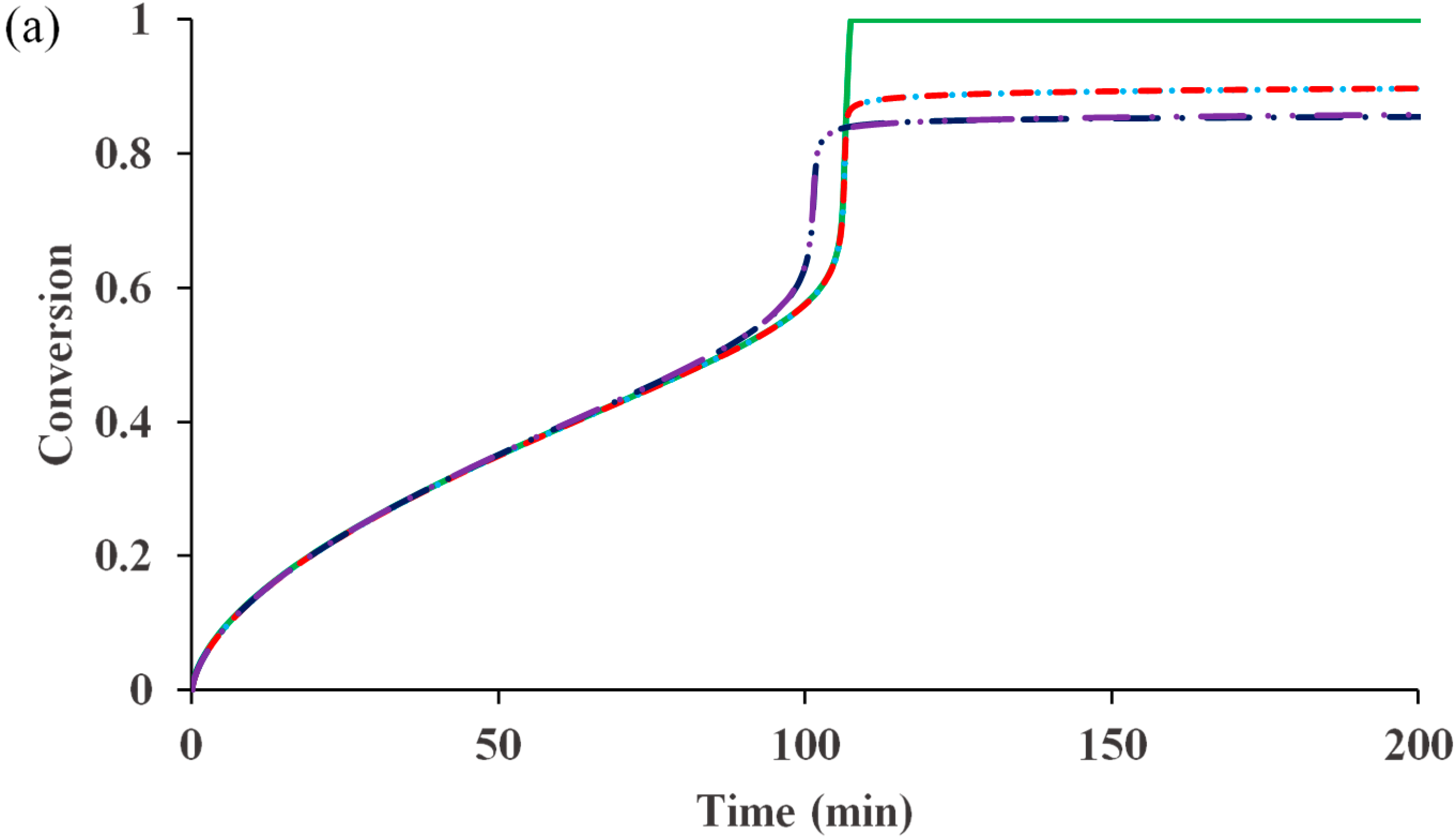
Figure 3 shows the influence of diffusion-controlled activation and propagation on the rate and control of polymerization for the ATRP system having diffusion-controlled termination
= 10
14) and deactivation (
= 10
10), as well as residual termination (z
tde = 100 & z
tp = 10) and deactivation (z
de = 1). It can be clearly seen from
Figure 3a that both diffusion-controlled activation and diffusion-controlled propagation can lead to dead-end polymerization with limited conversion. Depending on their rate coefficients, as well as the relative sizes of monomer and catalyst complex, either activation or propagation can onset diffusion control first and dominate the influence on the high-conversion kinetic behavior. When activation onsets first, it reduces the radical concentration and slows down the polymerization rate. After that, diffusion-controlled propagation cannot make any difference. The opposite is also true, when propagation onsets diffusion control first, the diffusion controlled activation has no further influence. In contrast to the conventional free radical polymerization, in which diffusion-controlled propagation is responsible for the occurrence of “glass effect”, both diffusion-controlled activation and diffusion-controlled propagation can stop the polymerization in ATRP. Furthermore, both diffusion-controlled activation and diffusion-controlled propagation cannot significantly affect the control of molecular weight at high conversions, as evident in
Figure 3b.
Figure 3.
The influence of diffusion-controlled activation and propagation on (
a) conversion
versus time and (
b) PDI
versus conversion for an ATRP system having diffusion-controlled termination
and deactivation
, as well as residual termination (z
tde = 100 & z
tp = 10) and deactivation (z
de = 1). No diffusion-controlled activation and propagation
![Polymers 07 00819 i001]()
, diffusion-controlled activation
, diffusion-controlled activation and propagation
, diffusion-controlled propagation
and diffusion-controlled propagation and activation
Figure 3.
The influence of diffusion-controlled activation and propagation on (
a) conversion
versus time and (
b) PDI
versus conversion for an ATRP system having diffusion-controlled termination
and deactivation
, as well as residual termination (z
tde = 100 & z
tp = 10) and deactivation (z
de = 1). No diffusion-controlled activation and propagation
![Polymers 07 00819 i001]()
, diffusion-controlled activation
, diffusion-controlled activation and propagation
, diffusion-controlled propagation
and diffusion-controlled propagation and activation
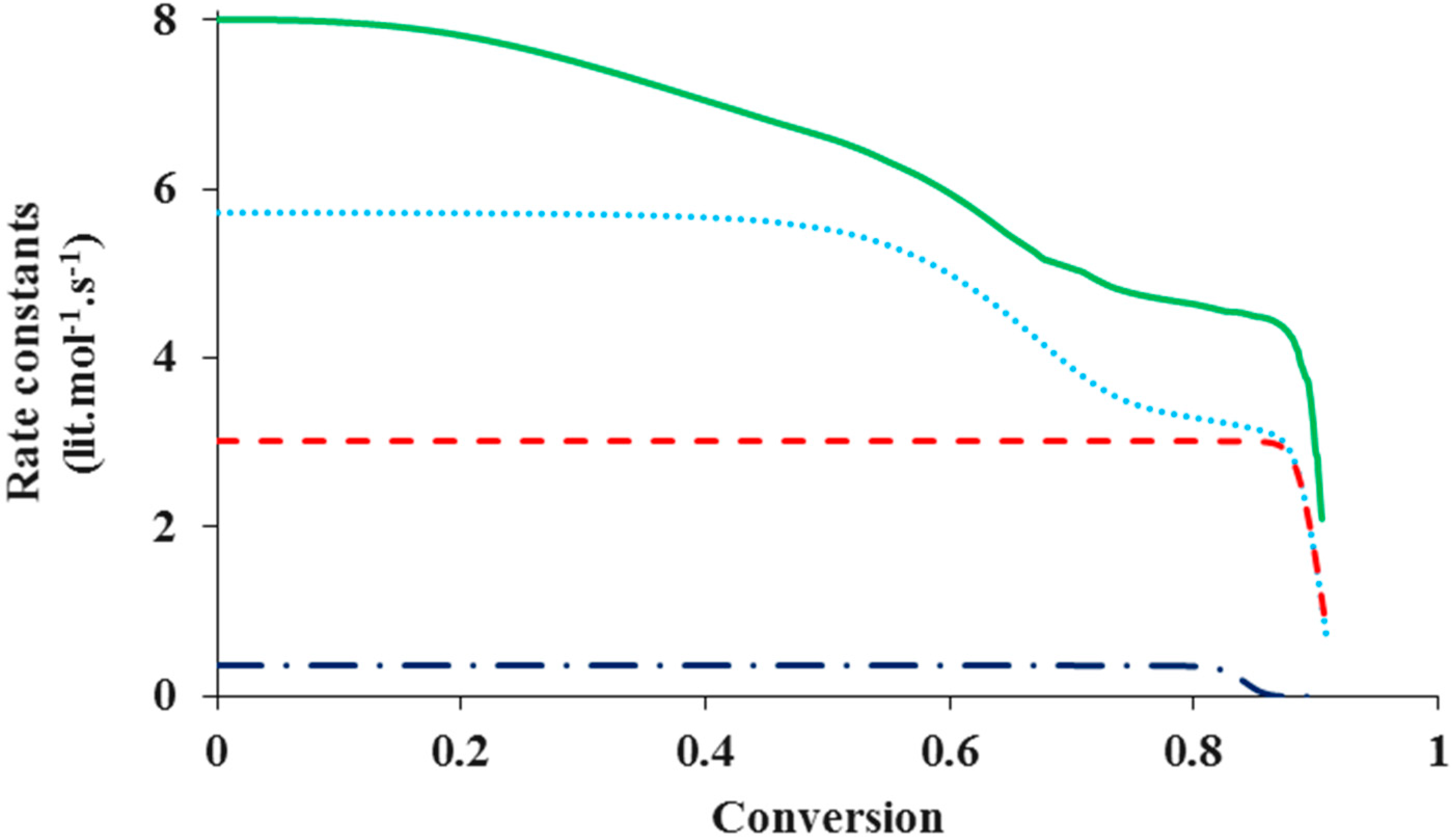
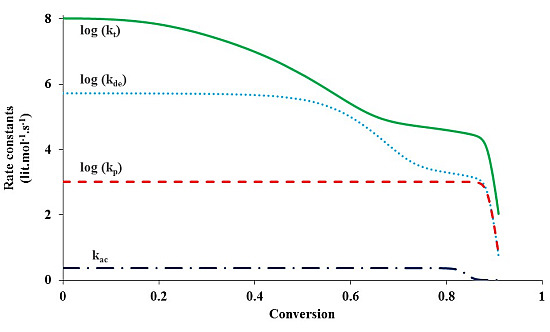
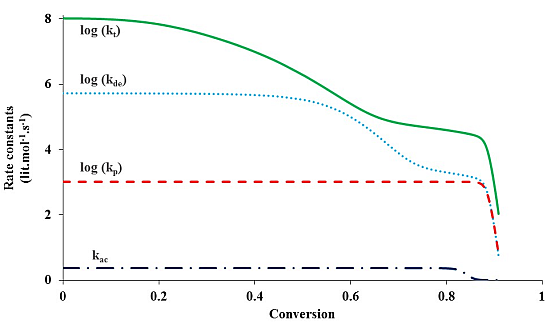
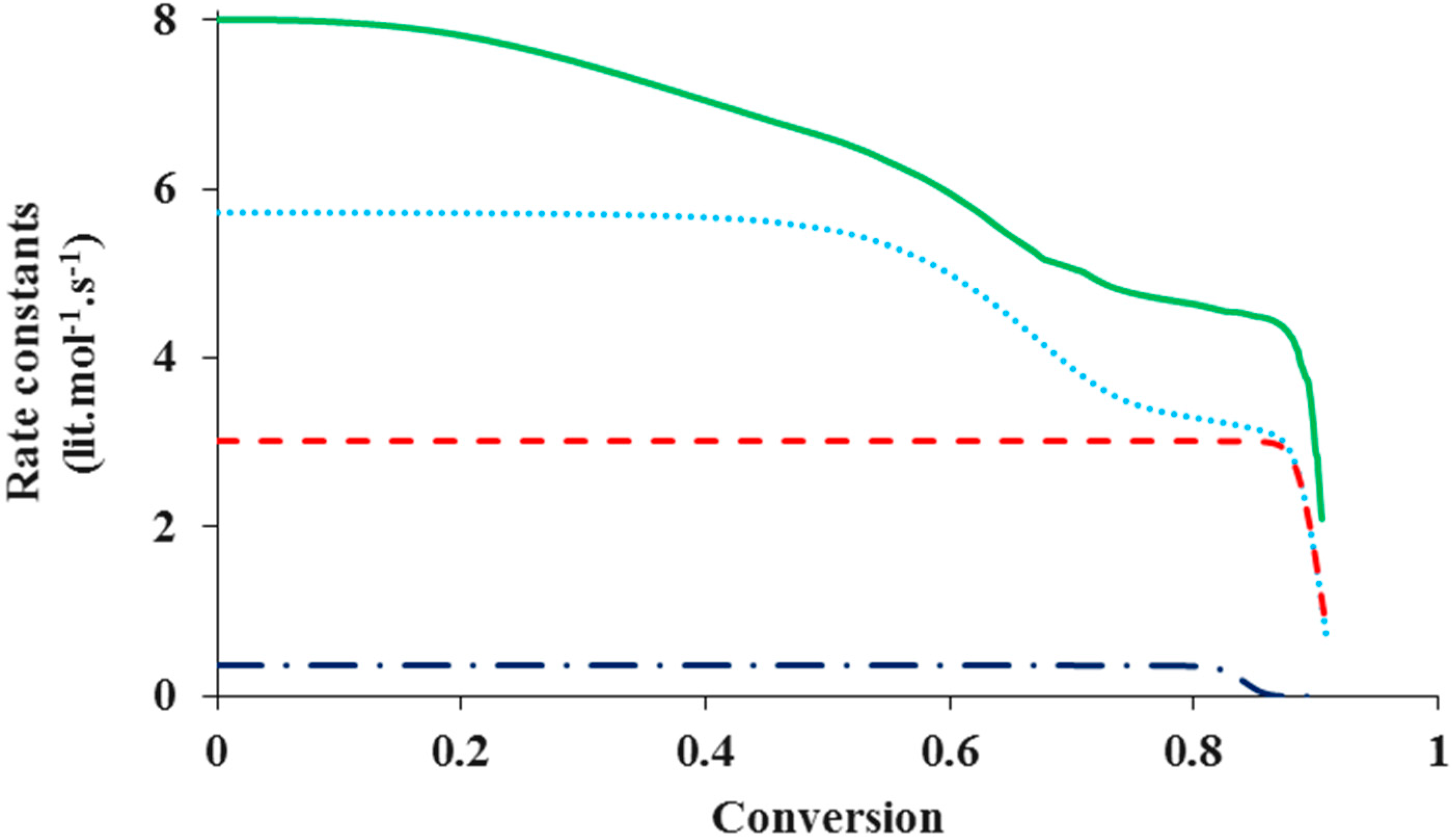
Figure 4 shows the change of various rate coefficients with monomer conversion during the polymerization. The radical termination onsets diffusion control first, at a low monomer conversion. This is because the reaction involves two chains, which easily experience diffusion limitations. It is also because termination has the highest rate coefficient among all the reactions involved in ATRP and the diffusion of reacting species easily becomes a rate-determining step. Diffusion control of radical chains decreases
kt until the “hopping” mechanism through activation/deactivation cycles dominates the radical termination. With further progress of monomer conversion, small catalytic deactivator molecules start to experience diffusion limitations and the deactivation onsets diffusion control. The “hopping” mechanism becomes negligible. At this stage the residual termination is dominated by migration by propagation. This residual termination is at a much lower order of magnitude
and it continues until the propagation onsets diffusion control, which often occurs when the system approaches its glass state. It is evident from
Figure 4 that there exist three steps of reduction in
kt: at a low conversion corresponding to the translational diffusion control of radical chains, at a middle conversion due to the residual termination by “hopping” mechanism, and at a high conversion because of diffusion-controlled propagation.
During the polymerization, the radical termination, radical deactivation and monomer propagation become diffusion-controlled one after another. Similar to termination, diffusion-controlled deactivation decreases at a middle conversion. After that, it remains almost constant up to a high conversion because of residual deactivation by propagation. At the high conversion when the system approaches its glass state and the propagation becomes diffusion-controlled, decreases considerably, in response to kp. The catalytic species for activation and deactivation have similar molecular sizes and thus diffusivities. Because of its much higher rate coefficient, deactivation onsets diffusion control earlier than activation. Activation becomes diffusion-controlled at a very high conversion, similar to propagation.
Figure 4.
The rate coefficients in logarithmic scale of termination (
= 10
14, z
tde = 100 & z
tp = 10)
![Polymers 07 00819 i001]()
, deactivation (
= 10
10 & zde = 1)
and propagation (
=10
16)
, as well as activation (
= 10
10)
, as a function of monomer conversion.
Figure 4.
The rate coefficients in logarithmic scale of termination (
= 10
14, z
tde = 100 & z
tp = 10)
![Polymers 07 00819 i001]()
, deactivation (
= 10
10 & zde = 1)
and propagation (
=10
16)
, as well as activation (
= 10
10)
, as a function of monomer conversion.
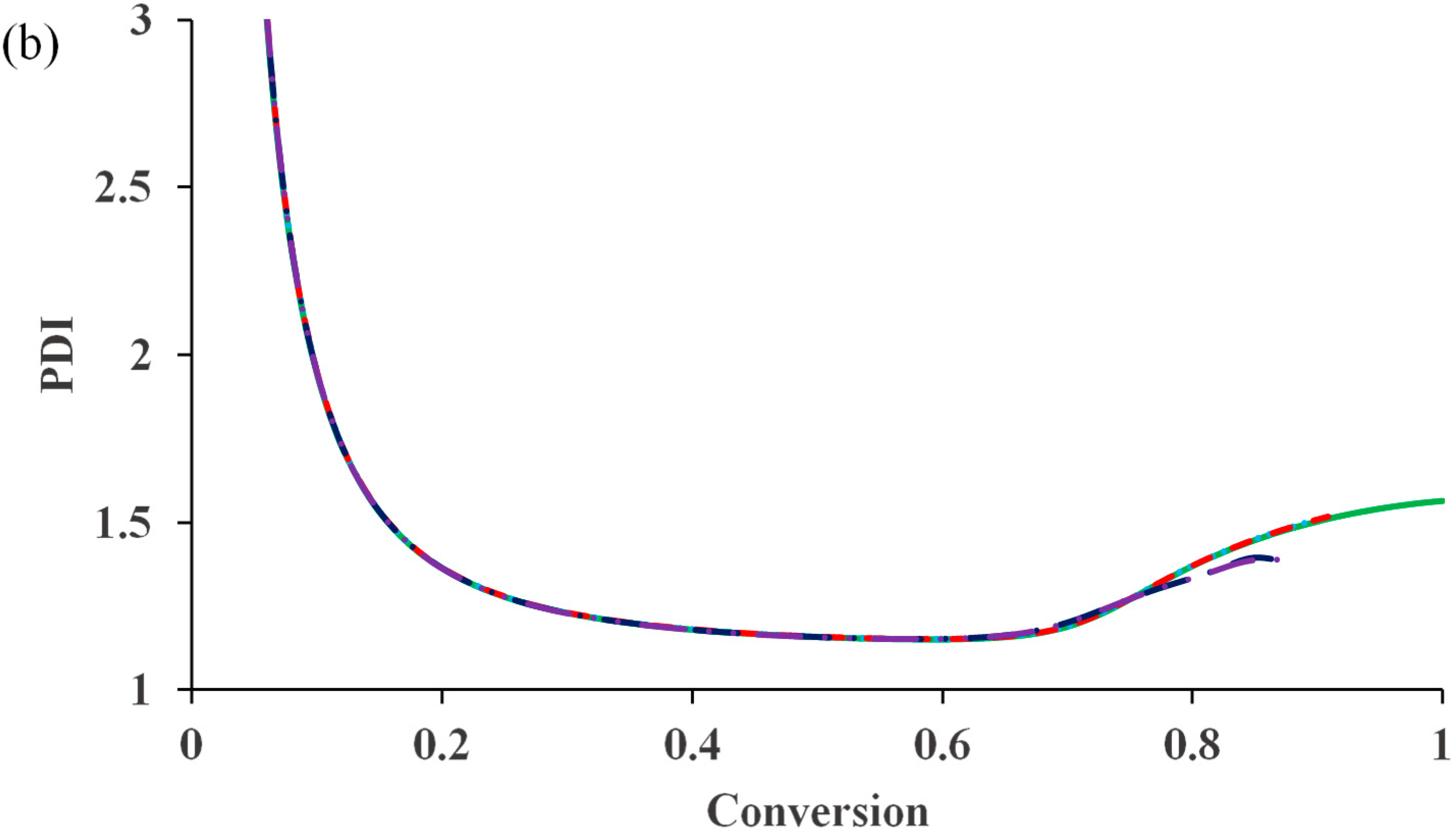
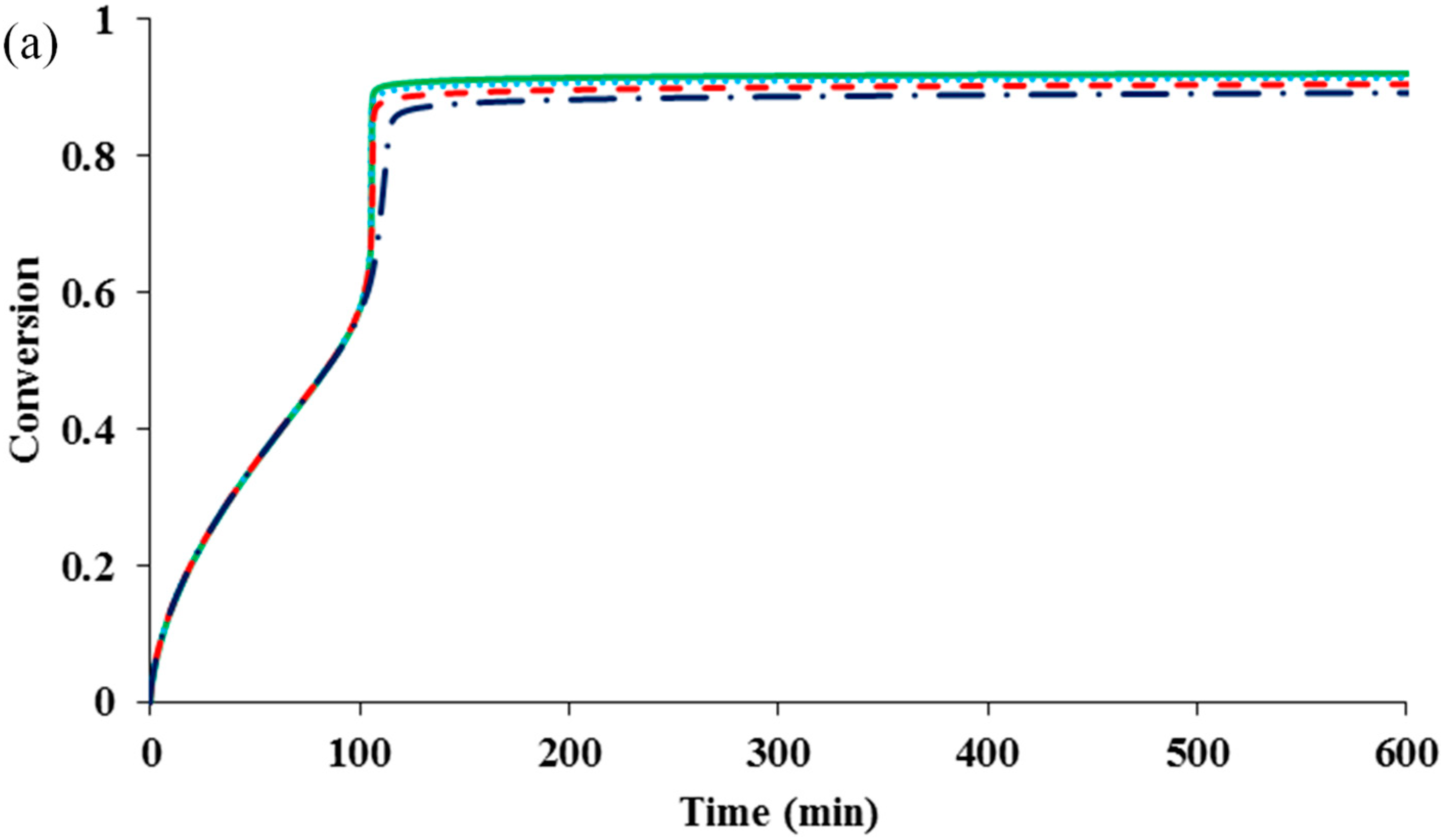
It should be pointed out that the residual deactivation has an important role in keeping the control of polymer molecular weight at high conversions. Without it, the polymerization would be totally out of control.
Figure 5 shows that although its influence on the polymerization rate is negligible, the residual deactivation reduces PDI value at the high conversion considerably. With z
de = 10, the PDI is below 1.2, despite the occurrence of a “gel effect”.
Figure 5.
The influence of residual deactivation (z
de = 0)
![Polymers 07 00819 i001]()
, (z
de = 0.1)
, (z
de = 1)
and (z
de = 10)
on (
a) conversion
versus time and (
b) PDI
versus conversion for a system having diffusion-controlled termination (
= 10
14), deactivation (
= 10
10), activation (
= 10
10), propagation (
= 10
16) and residual termination (z
tde = 100 & z
tp = 10).
Figure 5.
The influence of residual deactivation (z
de = 0)
![Polymers 07 00819 i001]()
, (z
de = 0.1)
, (z
de = 1)
and (z
de = 10)
on (
a) conversion
versus time and (
b) PDI
versus conversion for a system having diffusion-controlled termination (
= 10
14), deactivation (
= 10
10), activation (
= 10
10), propagation (
= 10
16) and residual termination (z
tde = 100 & z
tp = 10).
4. Conclusions
High-conversion bulk controlled radical polymerization such as ATRP represents a great challenge. Various reactions, including radical termination, radical deactivation and activation, monomer propagation, can become diffusion controlled at high conversions. Diffusion-controlled reactions significantly influence the rate of polymerization and the control of polymer molecular weight. Using the modeling and simulation approach, these influences are systematically investigated in this work.
Radical termination onsets diffusion control first, at a relatively low conversion. It is because termination involves two chains, which easily experience diffusion limitations. It is also because termination has the highest rate coefficient among all the reactions, which makes diffusion easily become the rate-determining step. Diffusion-controlled termination increases radical concentration and thus polymerization rate slightly. Unlike conventional radical polymerization where diffusion-controlled termination causes “gel effect”, the radical concentration in ATRP is mainly regulated by the equilibrium of activation and deactivation. At high conversions, translational diffusion of chains may be totally limited but the radical centers can still move around by the “hopping” mechanism via the activation/deactivation cycles, and by propagation. However, influence of the residual termination on polymerization rate and molecular weight control is negligible.
After termination, deactivation becomes diffusion controlled. Although both deactivation and activation reactions involve large chain and small catalyst species, the deactivation rate coefficient is several orders of magnitude higher than that of activation and thus it onsets diffusion control earlier. Diffusion-controlled deactivation dramatically increases the radical concentration, causing a severe auto-acceleration in rate (“gel effect”). It is also responsible for the loss of polymer molecular weight control. However, as along as monomer molecules can still move around, the migration of radical centers through propagation can also facilitate the deactivation to some extent. This residual deactivation helps to lower PDI and improves the control at high conversions. Diffusion-controlled activation and propagation happen at very high conversions when the system approaches to its glassy state. Depending on the relative sizes and diffusivities of monomer and catalytic species, either one can onset diffusion control earlier than the other to stop polymerization, leading to a “dead-end polymerization”.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
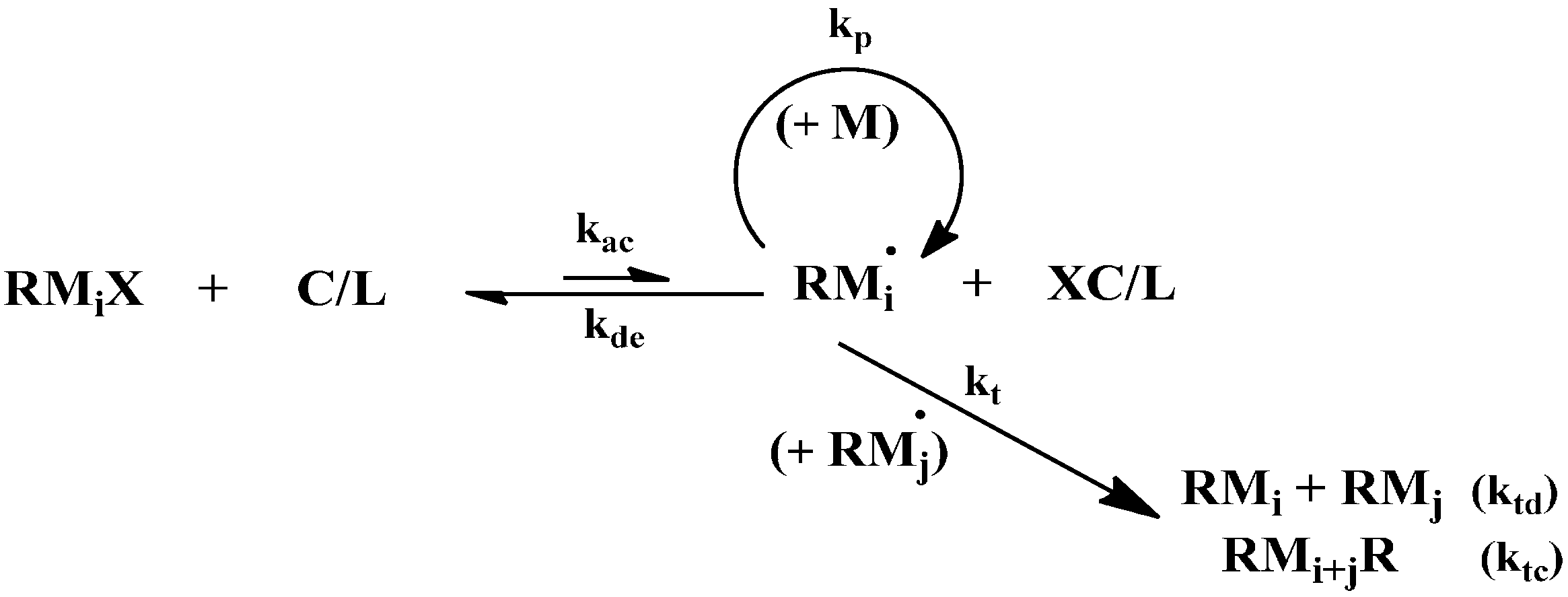{kind=link}
{kind=link}
 , translational diffusion-controlled termination but no residual termination , translational diffusion-controlled termination with residual termination by radical hopping via deactivation/activation reactions & , and translational diffusion-controlled termination with residual termination by radical hopping and monomer propagation & .
, translational diffusion-controlled termination but no residual termination , translational diffusion-controlled termination with residual termination by radical hopping via deactivation/activation reactions & , and translational diffusion-controlled termination with residual termination by radical hopping and monomer propagation & .