1. Introduction
Poly(lactic acid) (PLA) is one of the few bio-based polymers currently being produced on a commercial scale with an attractive cost structure [
1,
2]. This linear, aliphatic thermoplastic polyester possesses many attractive characteristics, including biocompatibility, high modulus and the ability to be melt processed using conventional processing techniques such as extrusion and injection molding [
3,
4]. Despite all of its advantages, there are limitations such as its inherent brittleness, poor melt strength, narrow processing window and low thermal stability [
4]. It is also well known that thermal processing of PLA is a challenge because of hydrolytic degradation during processing at elevated temperatures. The drop in the molecular weight adversely affects the rheological properties, mechanical properties and durability of the PLA based materials [
5].
In order to overcome these limitations, the use of plasticizers, blends, copolymerization, micro and nanocomposites have been investigated [
6,
7,
8,
9,
10,
11]. It has been shown that these modifications result in PLA based materials with improved elongation at break, impact strength, toughness and barrier properties. However, similar to unmodified PLA, hydrolytic degradation remains a concern [
12,
13,
14]. In fact depending on the hydrophilicity of the modifier used, the degradation rate of PLA has been found to increase in modified materials significantly affecting their processability [
11,
14].
By introducing a polymer consisting of all D isomer (PDLA) based triblock copolymer it is possible to convert PLLA from a stiff, brittle material to a soft, flexible rubbery material [
15,
16,
17]. In these blends the slow quiescent crystallization of poly(
l-lactic acid) (PLLA) and the preferential crystallization of the PLA stereocomplex resulted in the formation of a morphology that can be described as stereocomplex crystals dispersed in a soft continuous amorphous phase when the softblock used in the triblock copolymer was miscible with PLLA [
15]. In addition it was found that on addition of a molecular plasticizer to these blends, further improvement in the rubbery property of the blend was obtained [
17]. Despite the attractive mechanical properties achieved, it was found that because of the hydrophilic nature of the soft mid-block used in the triblock copolymer, the degradation of PLLA was accelerated in these blends during melt processing leading to a significant drop in the molecular weight of PLLA. This negatively affected the rheological properties and durability of these blends.
“Chain extenders” are typically introduced to minimize the drop in molecular weight during processing of condensation polymers such as polyesters and polyamides [
18]. In addition, chain extenders historically are used to recover the molecular weight associated with polyester recycling efforts. The chain extenders have two or more functional groups that react with the chemical groups formed during the degradation reaction. It is possible to re-link the degraded chains, thus restoring the polymer molecular weight and countering the effects of molecular weight degradation. In theory, di-functional molecules should be sufficient to function as chain extenders. However, in practice, multifunctional chain extenders are often used as they provide a higher efficiency compared to di-functional chain extenders [
18]. In fact, multifunctional epoxy based reactive oligomers have been commercialized as efficient chain extenders that can maximize chain extension while delaying the incidence of gelation [
19].
Commercially available epoxy based chain extenders, especially Joncryl 4368, have proven to be extremely effective for maintaining molecular weight during polyester processing [
14,
20,
21,
22]. Joncryl 4368 is an oligomeric copolymer based on glycidyl methacrylate, styrene and other acrylates with a functionality described only as “greater than four” [
19]. To our knowledge, an apparent gap in the open literature is that no experimental characterization of the functionality has been reported for any of these additives. Furthermore, other reactive oligomers, with lower or higher average functionalities than Joncryl 4368 have not been explored. Hence, an understanding of the effect of functionality of the reactive oligomers on the chain extension process in PLA is unavailable. Thus a systematic study on the effect of the functionality of the reactive oligomers on the chain extension in PLA has been performed by studying varying functionality reactive oligomers as chain extenders for PLA.
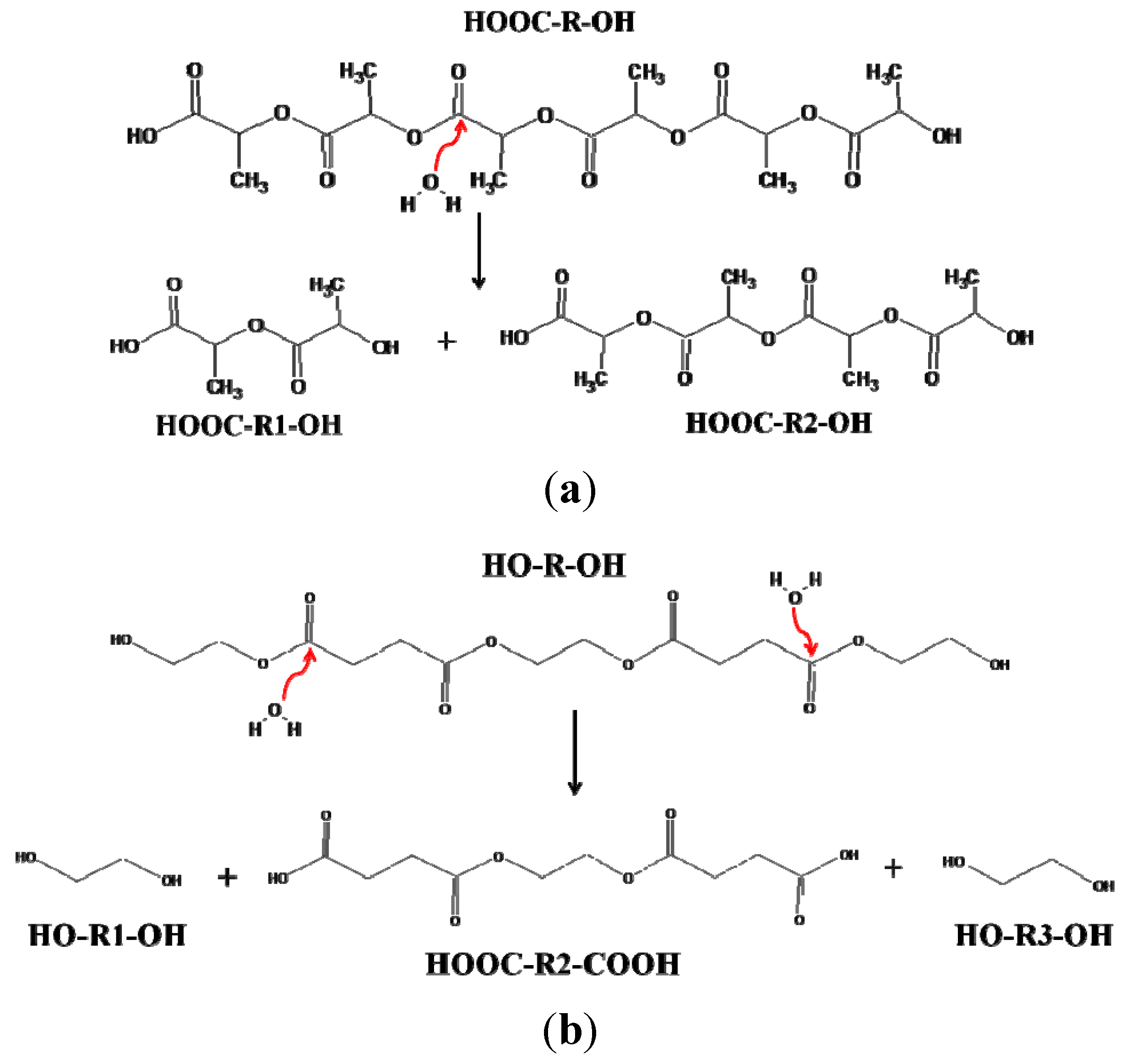
PLA degrades during melt processing (180 °C and above) because of the hydrolysis of the ester linkages or because of main chain scission by a β C–H transfer reaction [
20]. As PLA is a hydroxy-acid based polyester, upon hydrolysis of a poly(lactic acid) chain, the two newly created chains each have a carboxylic acid group at one end and a hydroxyl group at the other end (
Figure 1a). This is in contrast to other common polyesters such as poly(ethylene terephthalate) (PET), where, because of their diol-diacid nature, the hydrolytic degradation pathways can lead to chains with that have both ends with acid groups, or both ends with hydroxyl groups, or one end with acid and the other end with hydroxyl group depending on the site of attack of the water molecules [
20] (
Figure 1b).
Figure 1.
(a) Hydrolytic degradation pathway for poly(l-lactic acid) (PLA) (hydroxyl acid based polyesters); (b) Hydrolytic degradation pathway for diol-diacid based polyesters.
Figure 1.
(a) Hydrolytic degradation pathway for poly(l-lactic acid) (PLA) (hydroxyl acid based polyesters); (b) Hydrolytic degradation pathway for diol-diacid based polyesters.
Even in other modes of PLA degradation, such as random main chain scission due to a β C–H transfer reaction, the degraded chains can never possess carboxylic acid groups on both chain ends [
20]. It is well known that the electrophilic epoxy group reacts preferentially with the carboxylic acid group as compared to the hydroxyl group [
23]. In an epoxy-polyester blend, it has been shown that the glycidyl to carboxyl end group esterification reaction precedes the slower glycidyl to hydroxyl group etherification reaction. The reactions of glycidyl to secondary hydroxyl etherification and the transesterification of primary and secondary hydroxyl chain ends onto polyester chains are slower reactions than the corresponding primary reactions [
21,
24]. In fact, model compounds studies have shown that no reaction occurs between the hydroxyl group and the epoxy group up to 220 °C. In contrast, rapid reaction always occurs between the carboxylic acid group and the epoxy group at temperatures lower than 200 °C [
24]. The epoxy group clearly exhibits different reactivity with the functional groups present at the two ends of a degraded PLA chain.
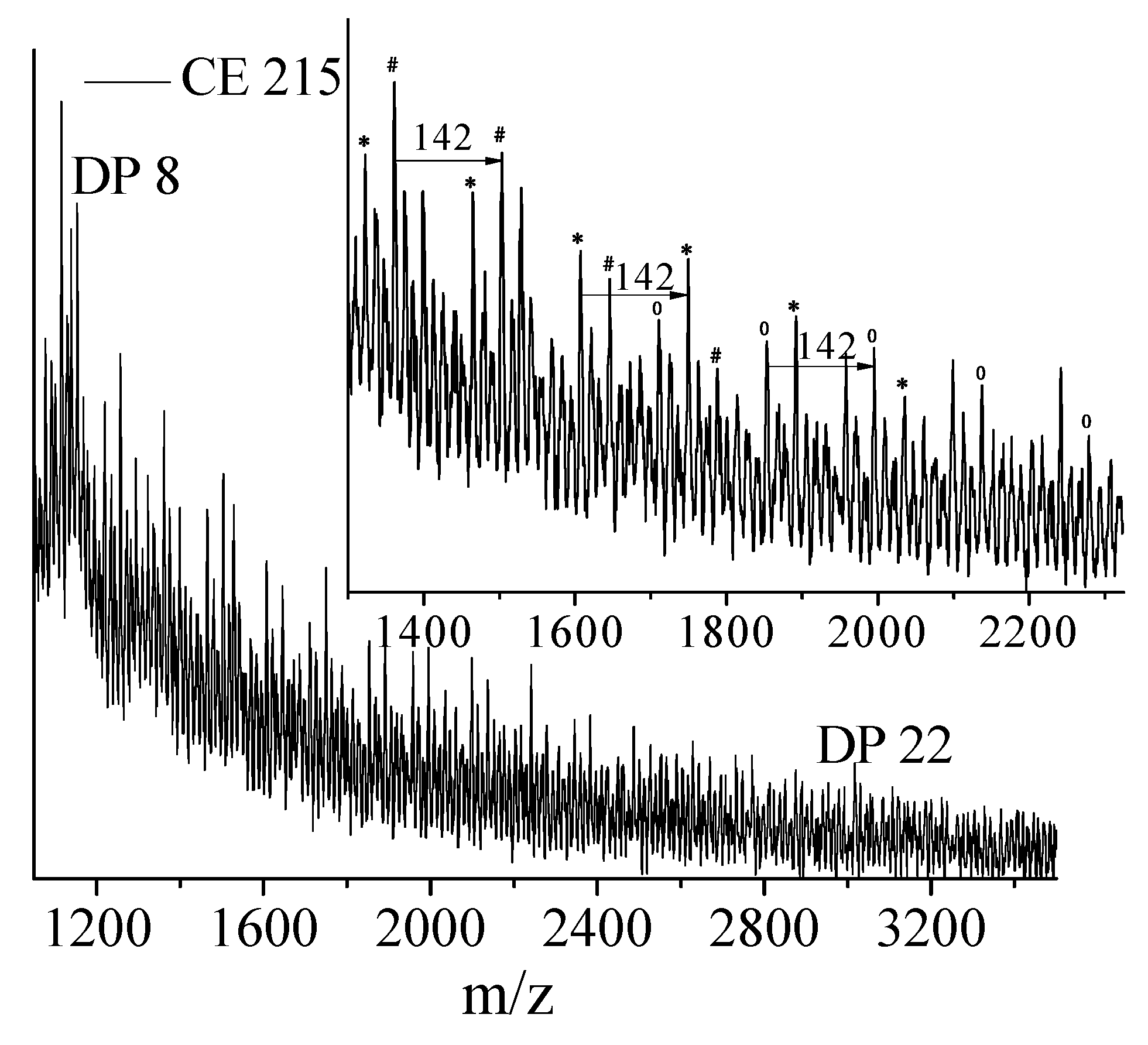
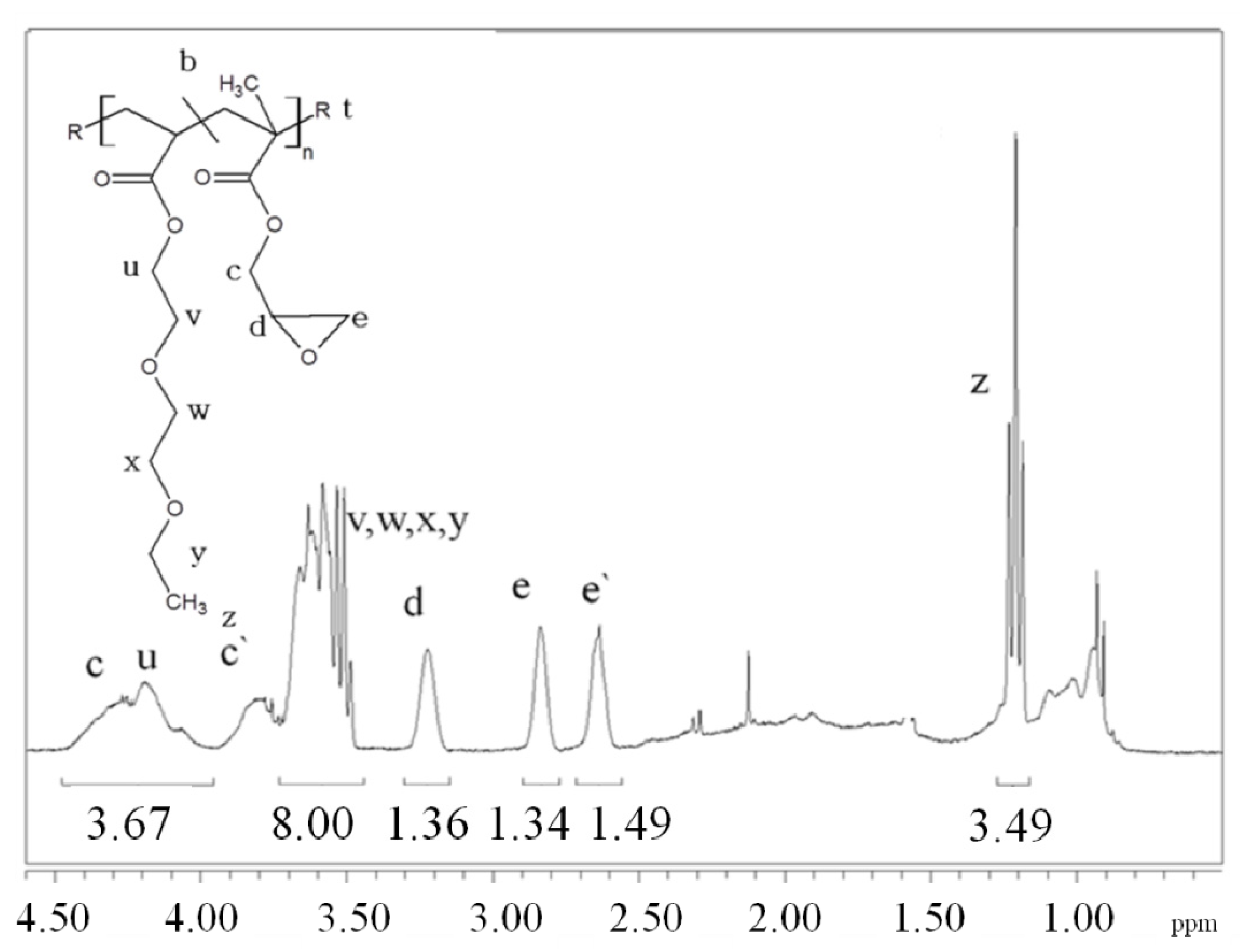
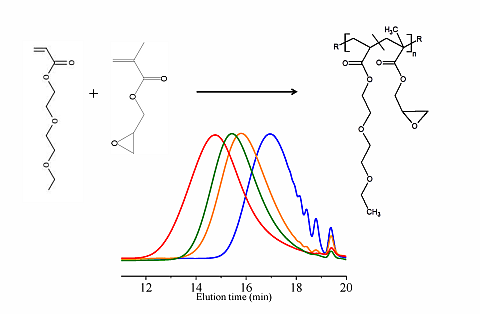
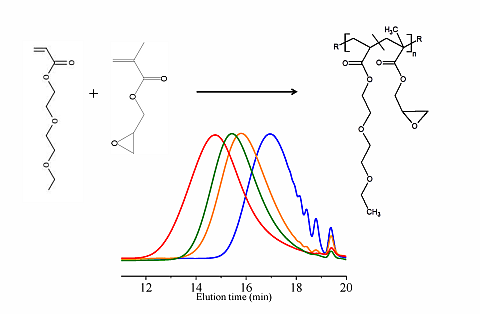
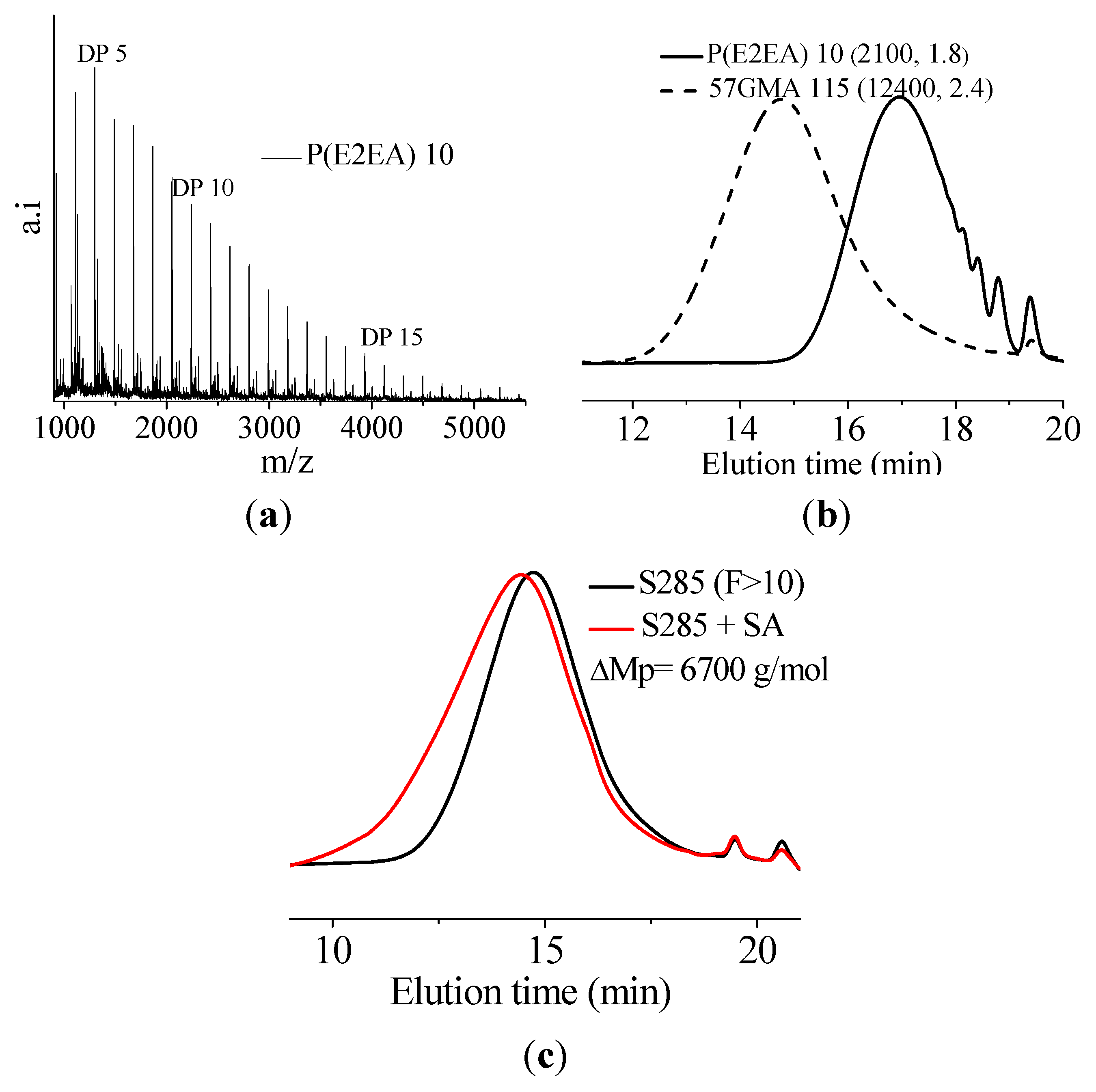
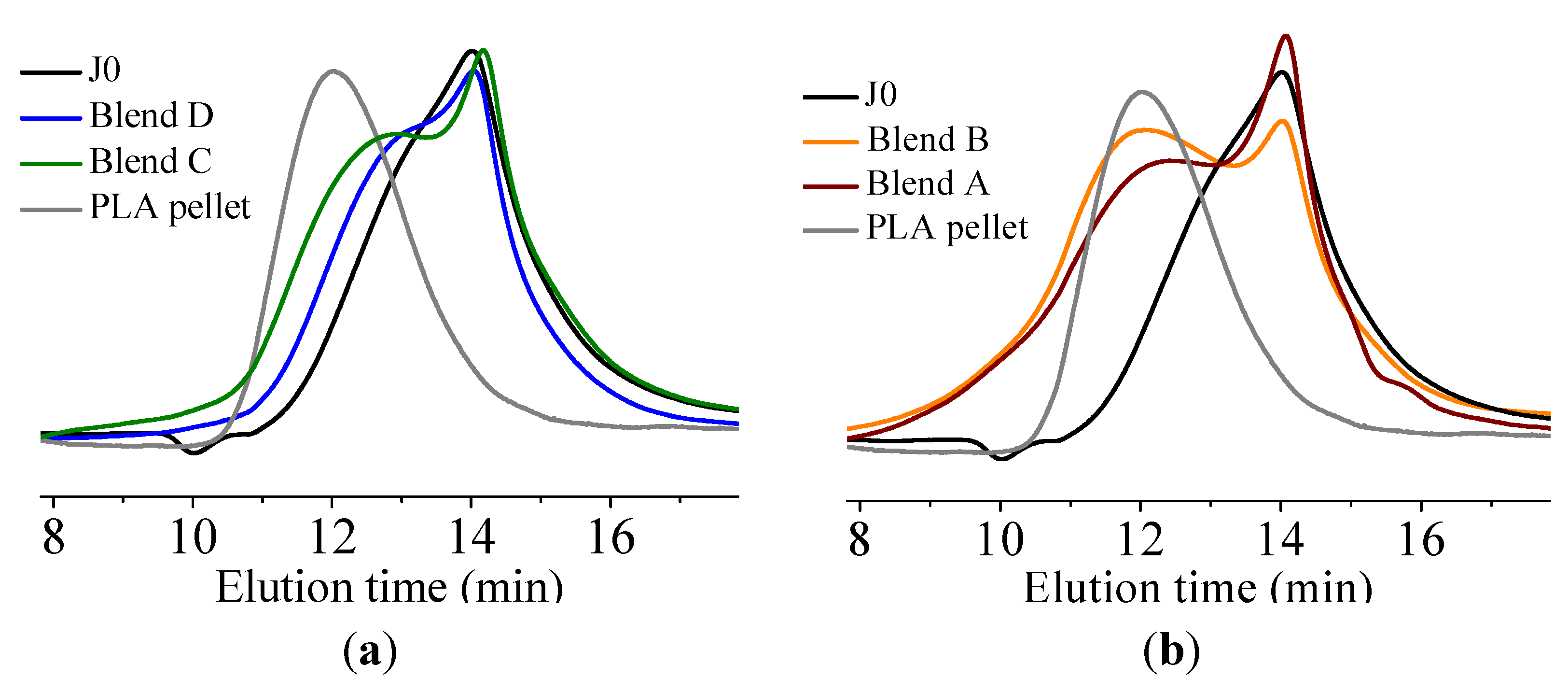
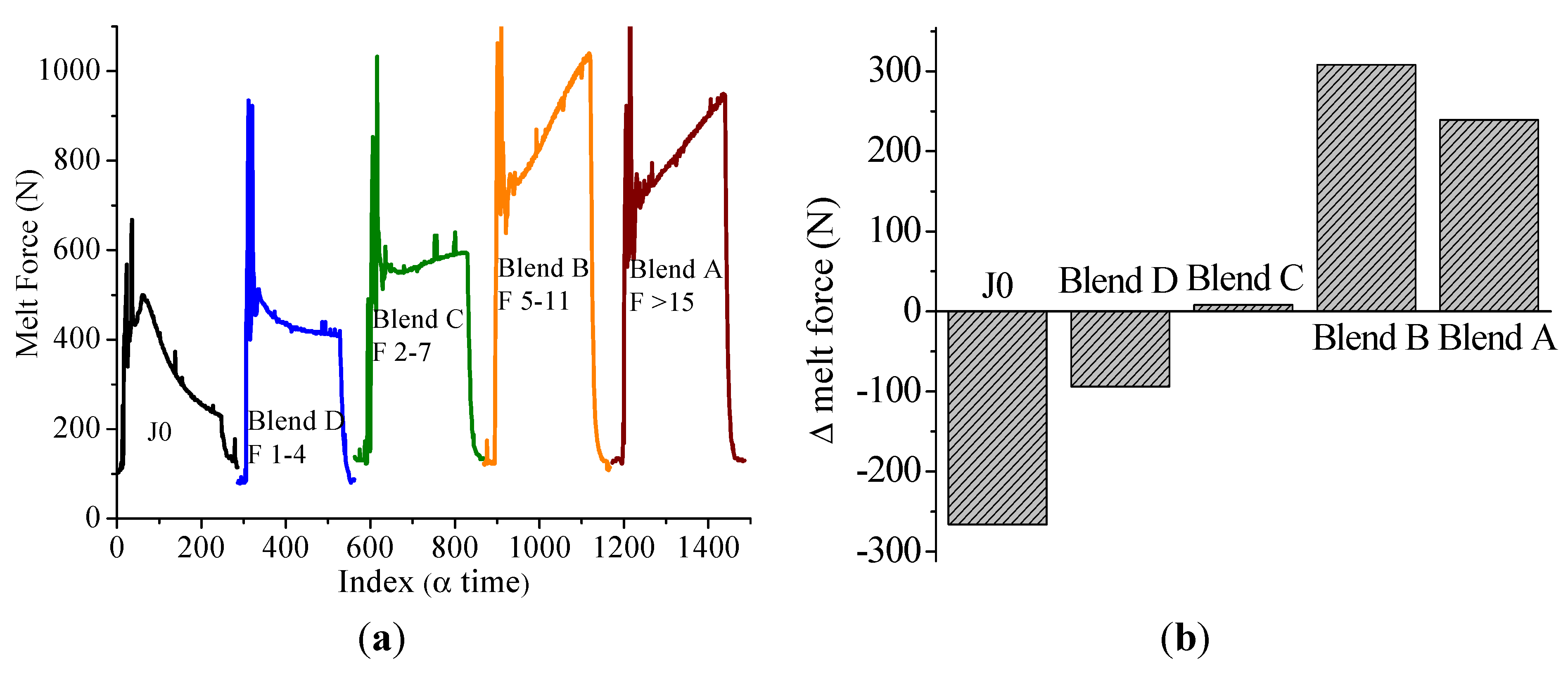
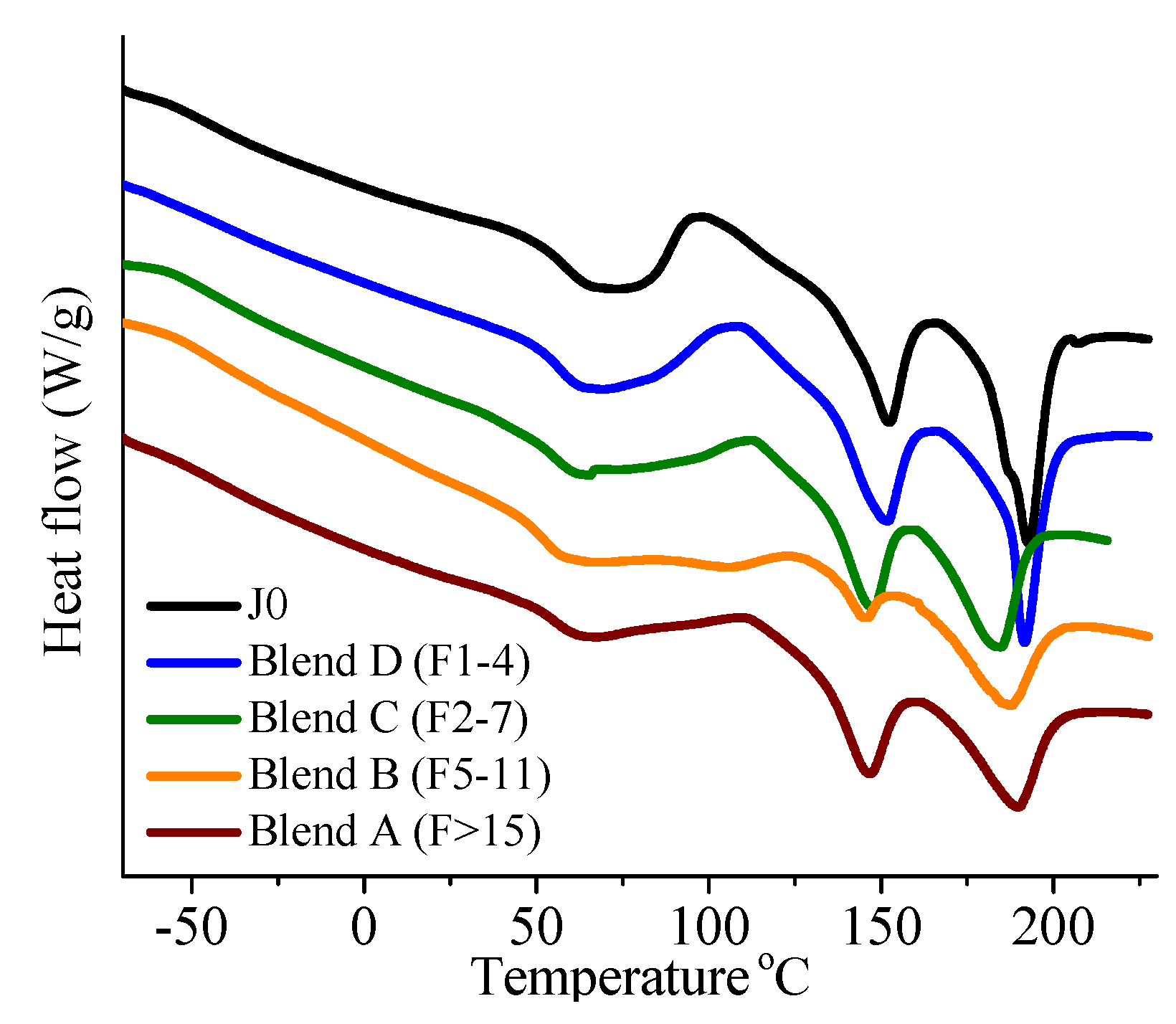
In this paper, apart from the commercially available Joncryl 4368, three other reactive random copolymers based on glycidyl methacrylate, styrene and other acrylates were studied. They possess, on average, functionality that is either lower or higher than that of Joncryl 4368. The increase in functionality can have two effects. First, because of the preferential reaction of the epoxy group with the carboxylic acid group, more PLA chains will be grafted onto the reactive oligomer chains leading to a larger increase in molecular weight, or second, cross linking may take place with increasing functionality of the chain extender if sufficient hydroxyl groups react with the epoxy groups. Therefore, in this study, we have attempted to quantify the functionality of various reactive oligomers and characterize their effects on the molecular weight increase of PLLA, and the stereocomplex crystallization, mechanical properties and hydrolytic stability of triblock copolymer/PLLA/Plasticizer blends. For the first time, this study demonstrates that the functionality of the reactive oligomers is an important parameter that affects the efficiency of the chain extension reaction of PLLA based materials. The resultant effects on hydrolytic stability, crystallinity and mechanical properties are also reported.
4. Conclusions
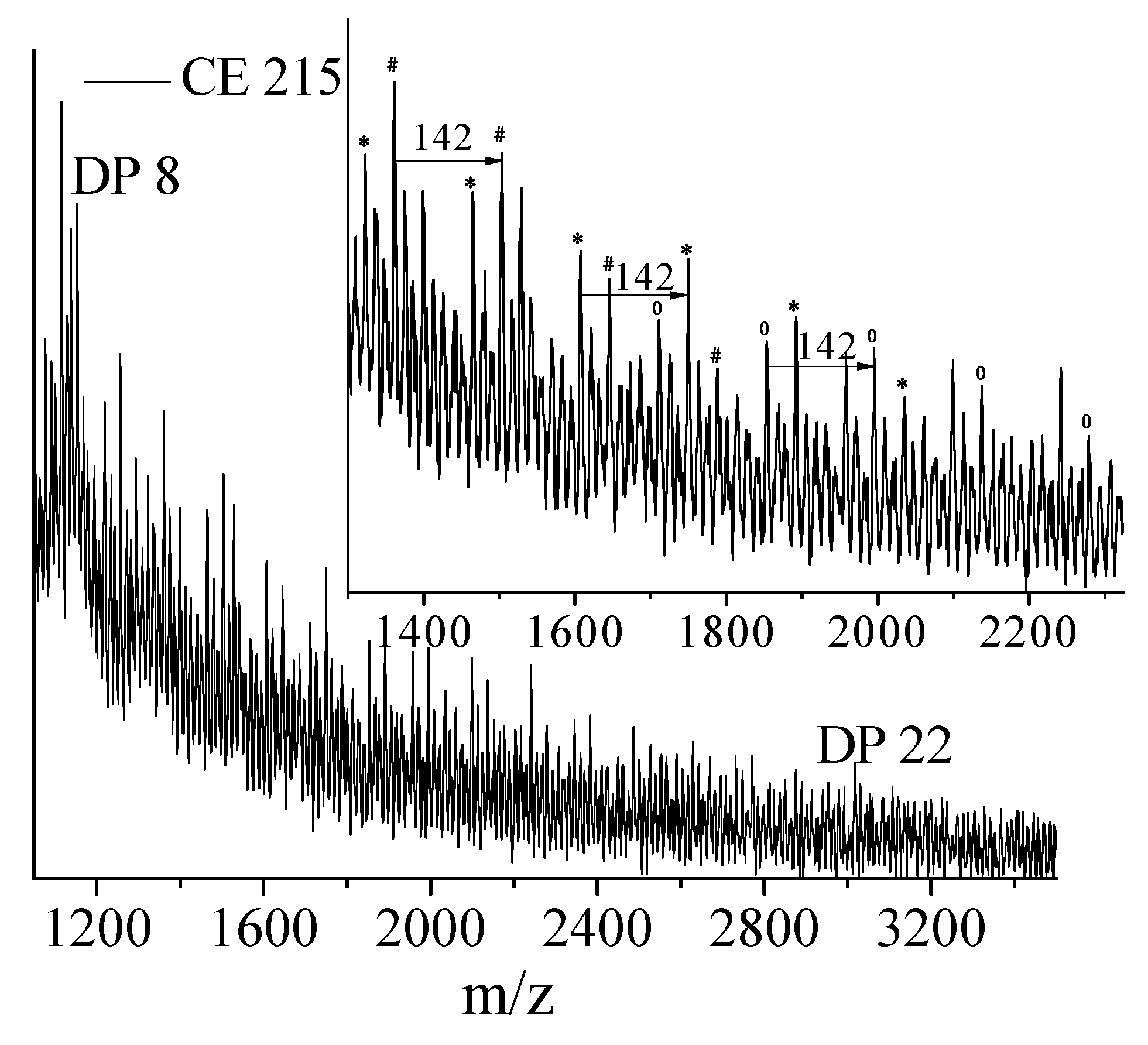
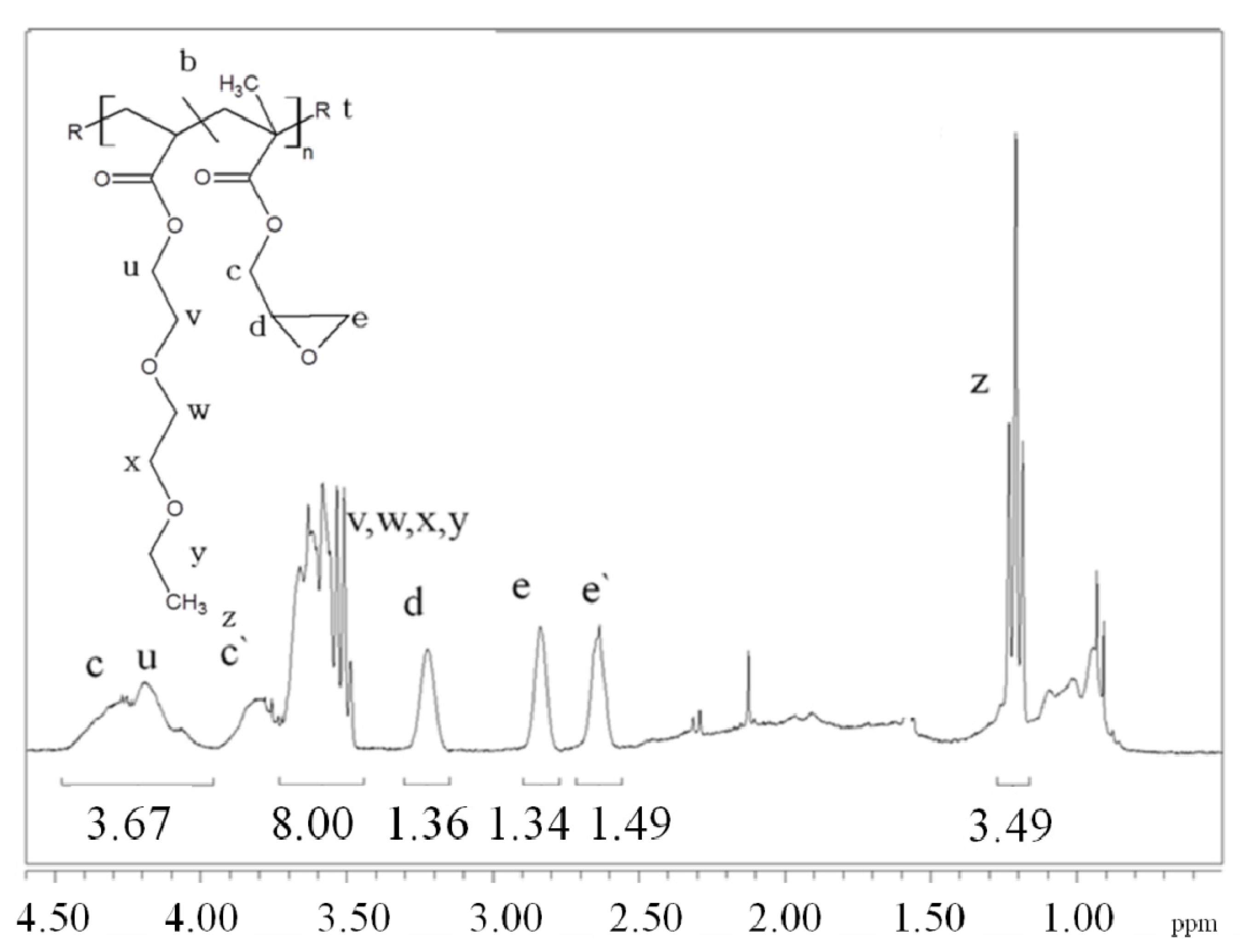
For the first time, the effect of functionality of the epoxy functional reactive oligomers on the molecular weight increase of PLLA in PLLA/PDLA-softblock-PDLA/Plasticizer based blends has been investigated. Three reactive oligomers with low, medium and high epoxy functionality were studied. In addition, a reactive oligomer with very high functionality was synthesized and studied. The functionality of these reactive oligomers was estimated using MALDI, GPC and epoxy equivalent weight data, and GPC studies on reactive oligomers grafted with a model monofunctional acid.
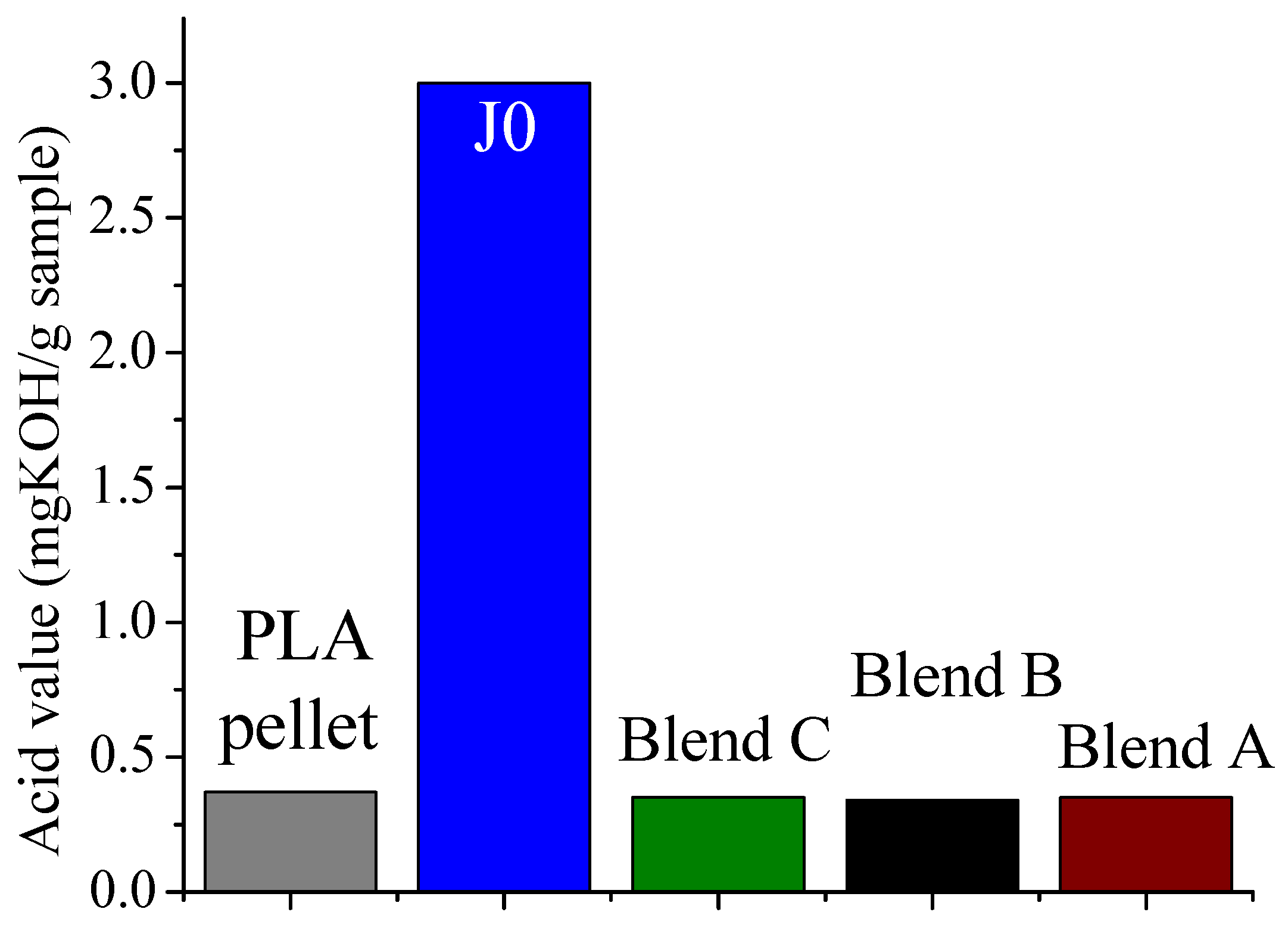
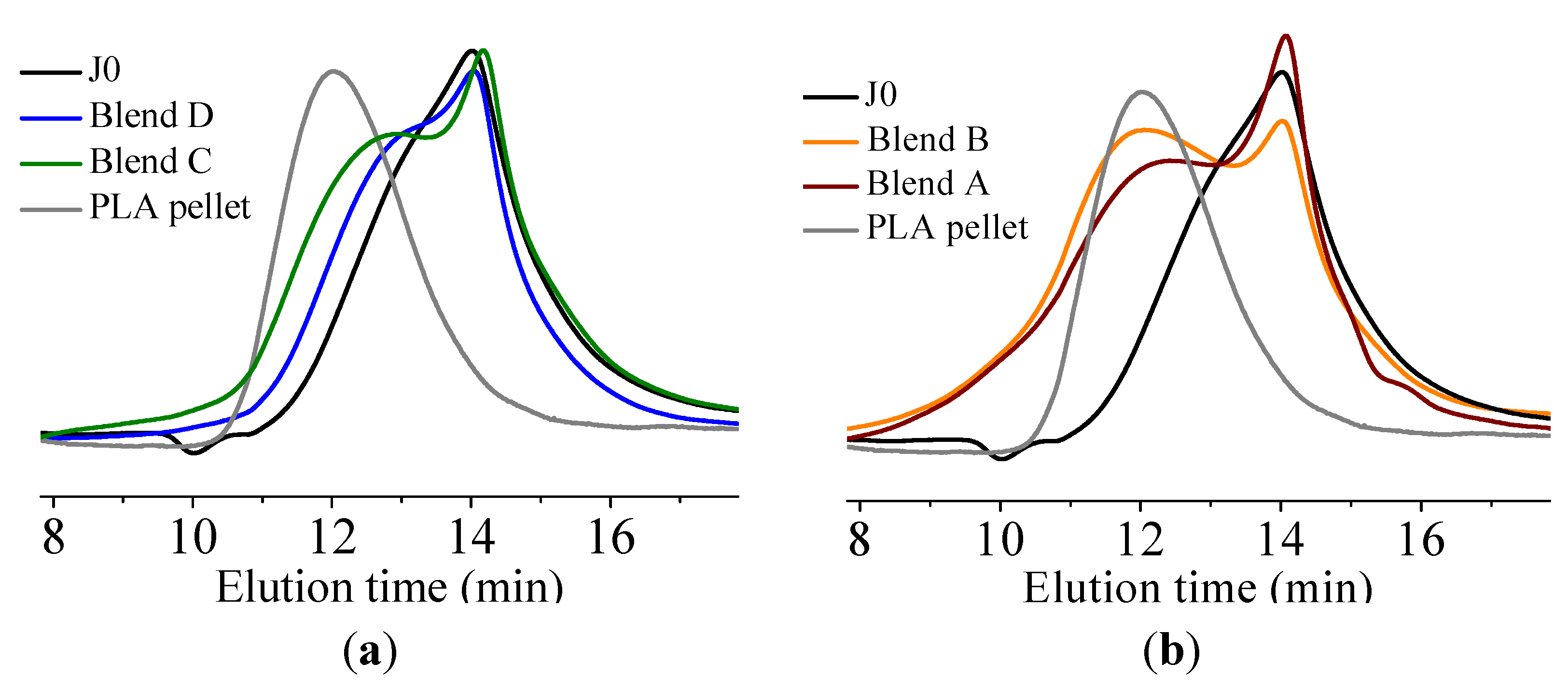
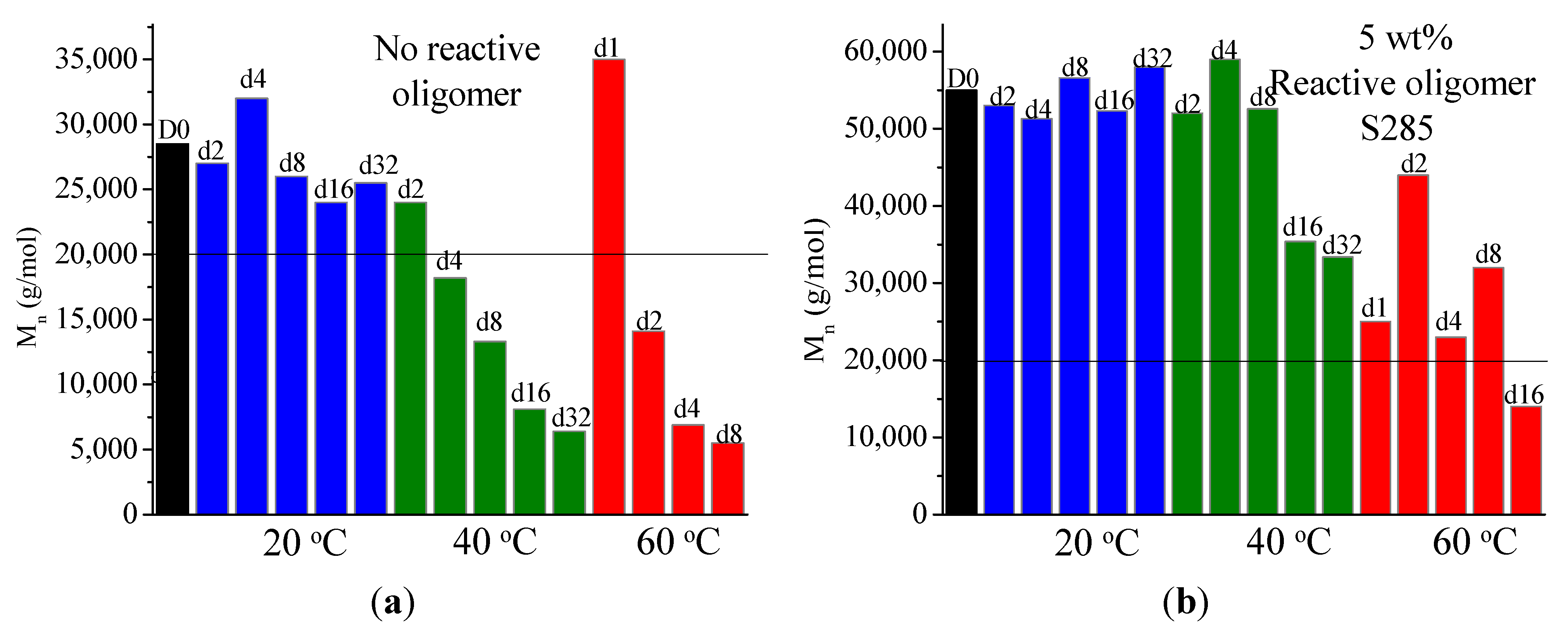
It was found that acid groups were consumed in all the blends but the molecular weight increase depended on the functionality of the reactive oligomer. The molecular weight of the degraded PLLA chains increased with an increase in the functionality of the reactive oligomer up-to a functionality of 5–11. Further increase in functionality leads to a plateau in the molecular weight increase possibly because of steric effects. No gel formation occurred in these blends despite the high functionality of the reactive oligomers. These results indicate that because of the negligible reactivity of the hydroxyl groups with the epoxy groups under PLA processing conditions, it is possible to kinetically preventing gelation from occurring. Because of the hydroxy-acid nature of PLA, the degraded chains necessarily have only one chain end with the carboxylic acid group. This makes the “reactive functionality” of the PLA chains with epoxy groups to be 1 under normal PLA processing conditions.
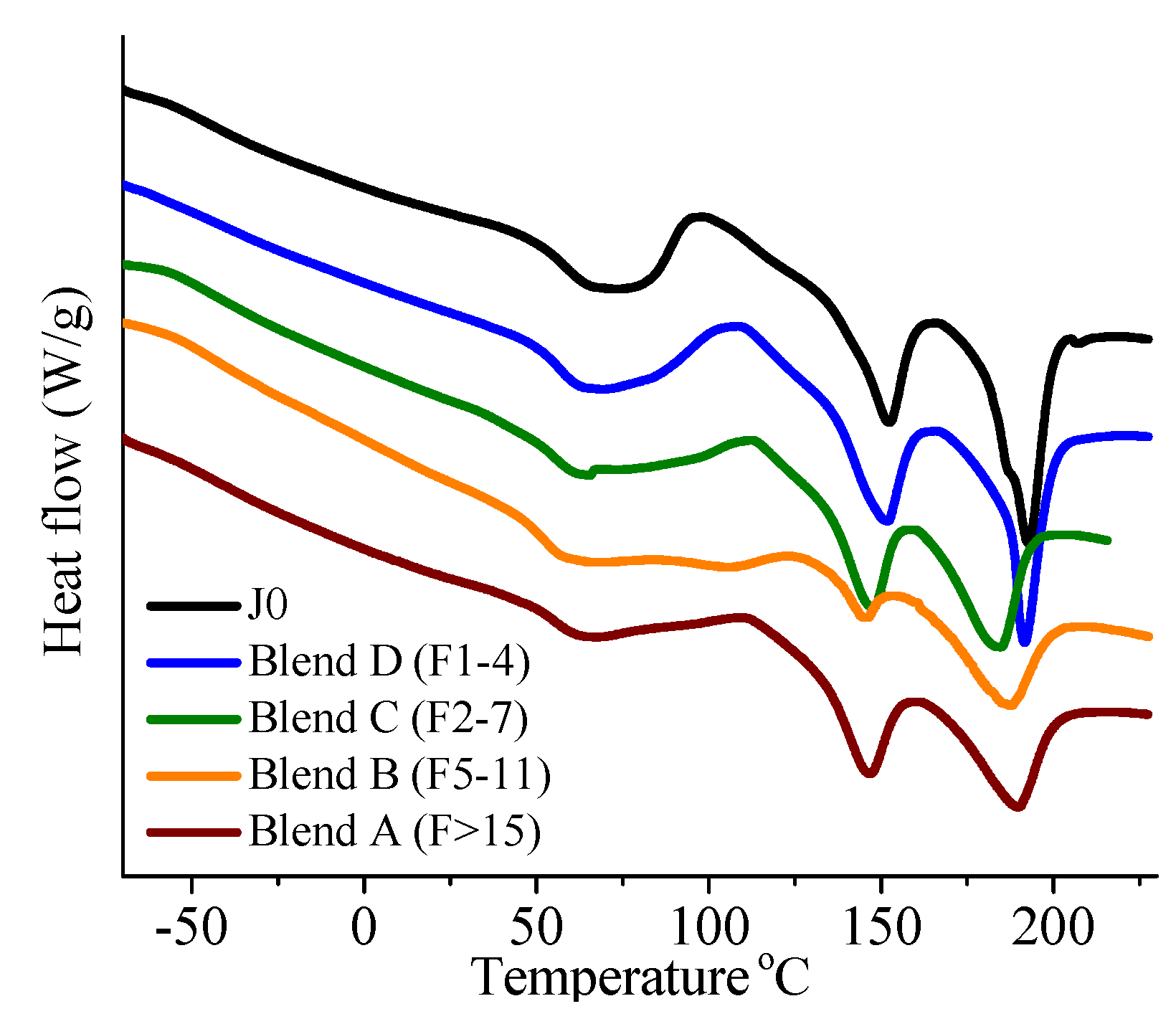
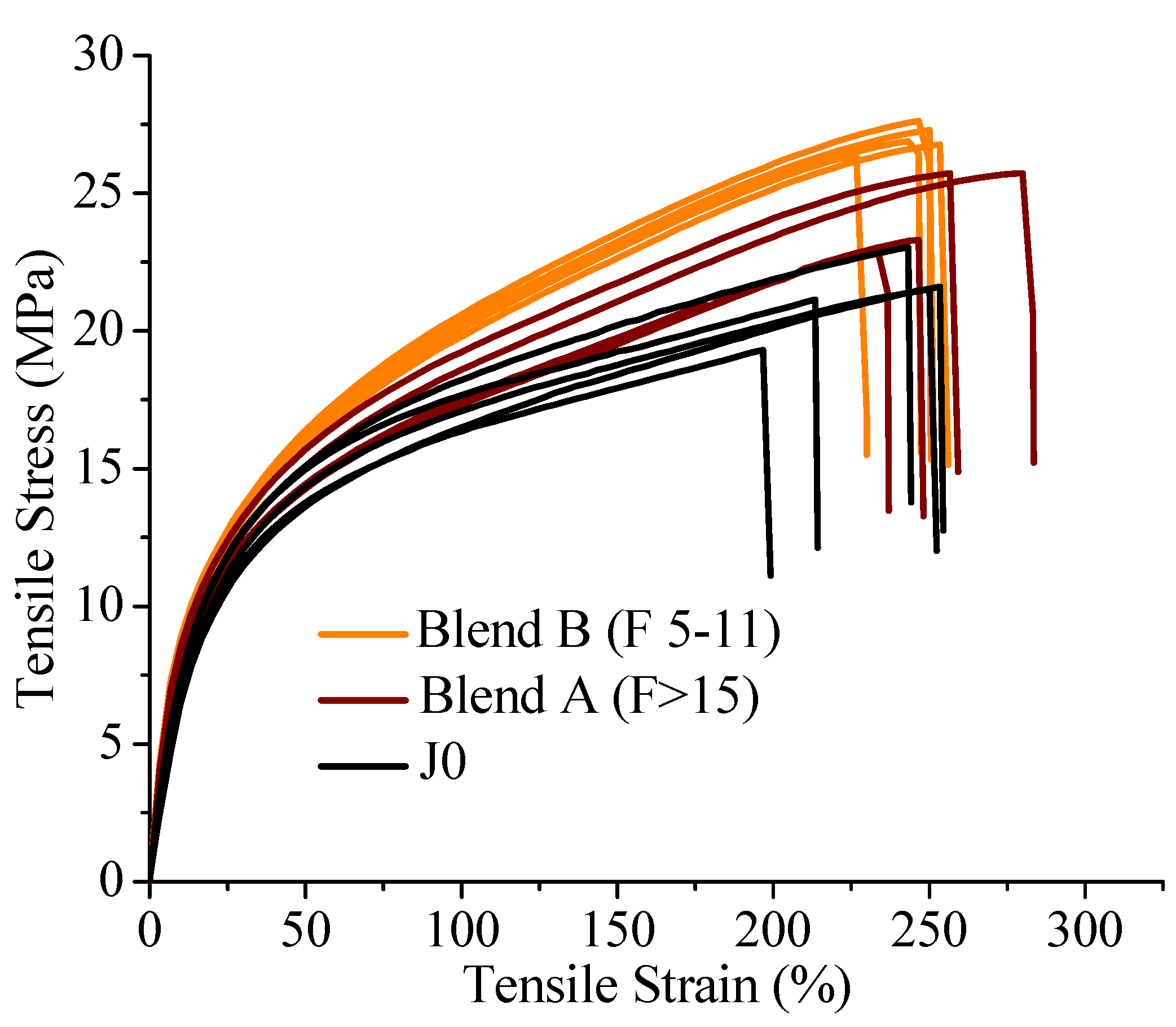
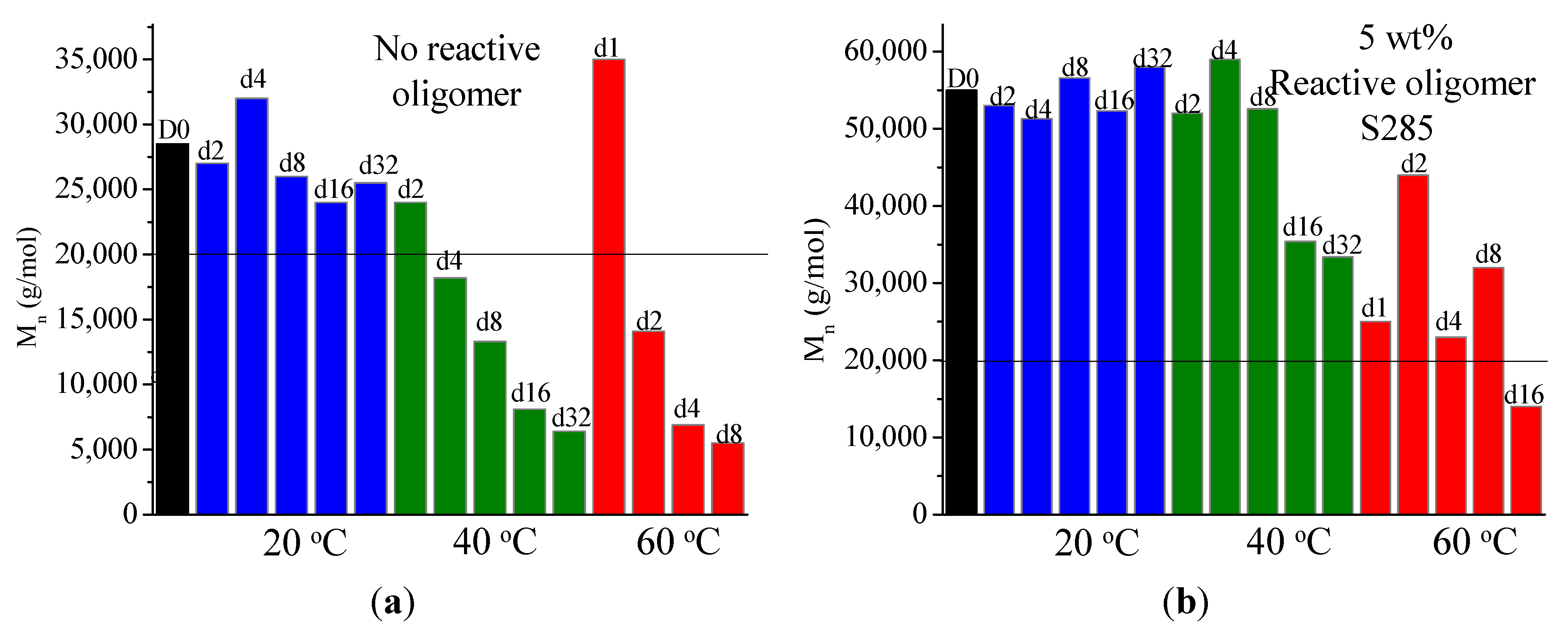
On investigation of the hydrolytic stability, it was found that the high functionality reactive oligomer S285 caused a significant improvement in the hydrolytic stability of the blend, as the molecular weight did not fall below the critical molecular weight (20,000 g/mol) even after 32 days at 40 °C and 90% relative humidity. The molecular weight increase caused a slight reduction in the co-crystallization between the PLLA and the triblock copolymer. The increased molecular weight caused an increase in the ultimate tensile strength of the blends. The use of low Tg reactive oligomer did not have any significant effect on the modulus of the blend because of its use in small amounts.
This study provides a better understanding of the degradation and chain extension reaction of PLA with epoxy functional reactive oligomers and shows how the functionality of the reactive oligomers controls the molecular weight increase of the degraded PLA chains. We showed that in modified PLA based systems where the rate of degradation is increased, higher functionality epoxy oligomers, at higher concentrations, than those used for unmodified PLA need to be used to bring about a sufficient increase in the molecular weight. Use of the high functionality reactive oligomers causes a significant increase in the blend molecular weight and hence the hydrolytic stability and processability. These findings will be useful for increasing the hydrolytic stability and durability of PLA based blends and composites.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}