Design and Synthesis of Cross-Linked Copolymer Membranes Based on Poly(benzoxazine) and Polybenzimidazole and Their Application to an Electrolyte Membrane for a High-Temperature PEM Fuel Cell

Abstract
:1. Introduction
2. Core Technologies and Issues
2.1. Catalyst
2.2. Membrane
2.3. MEA
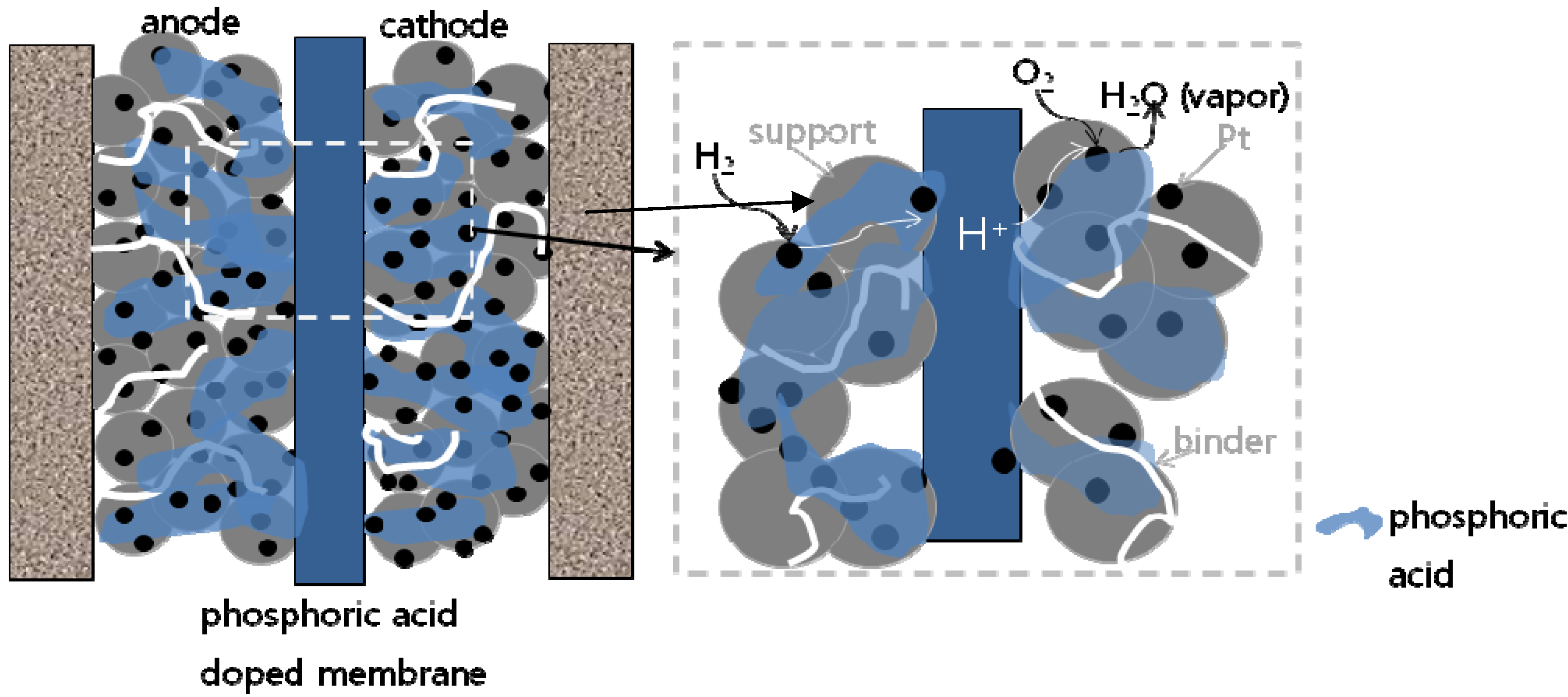
2.3.1. MEA Background
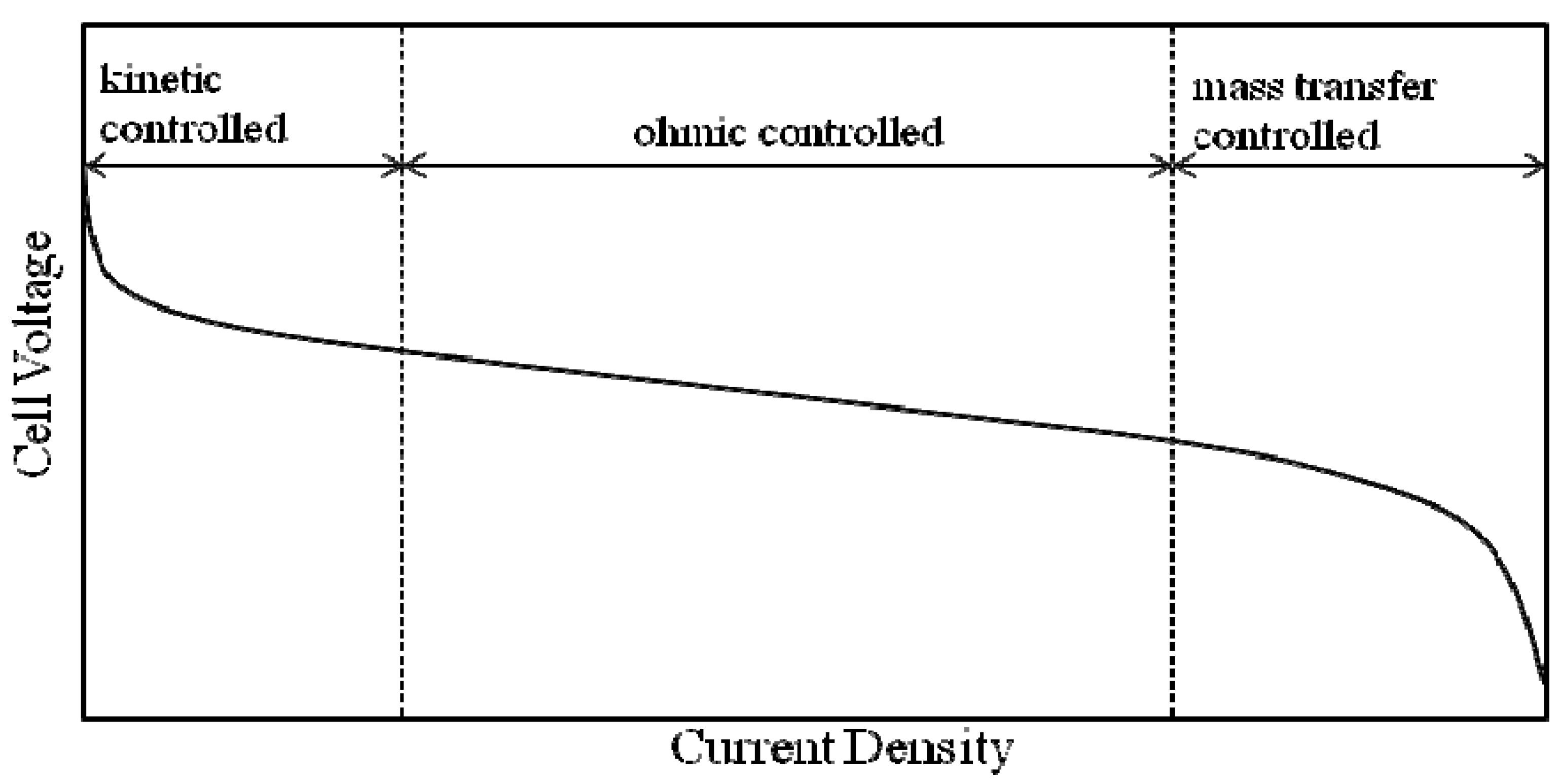
2.3.2. MEA Performance
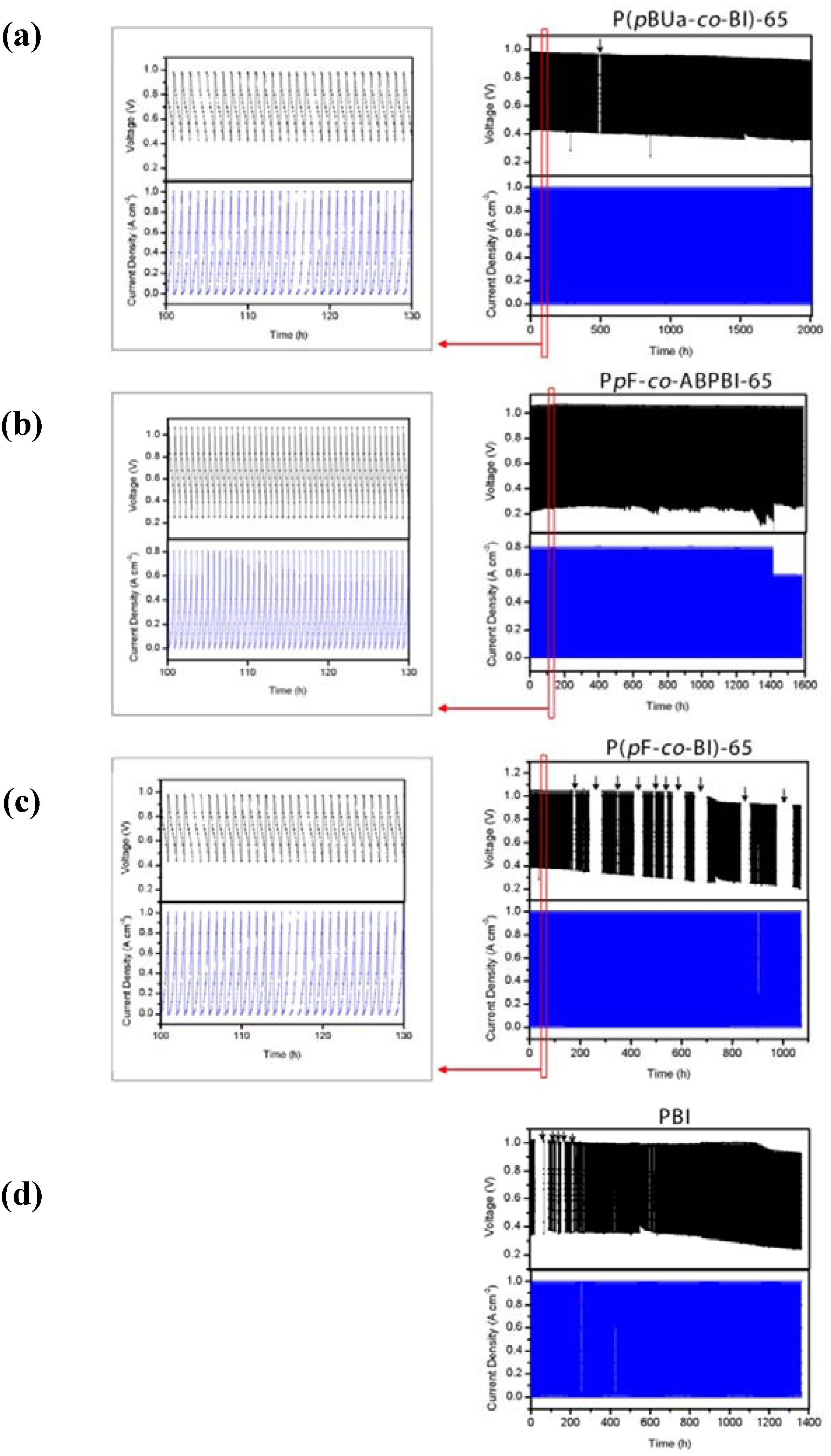
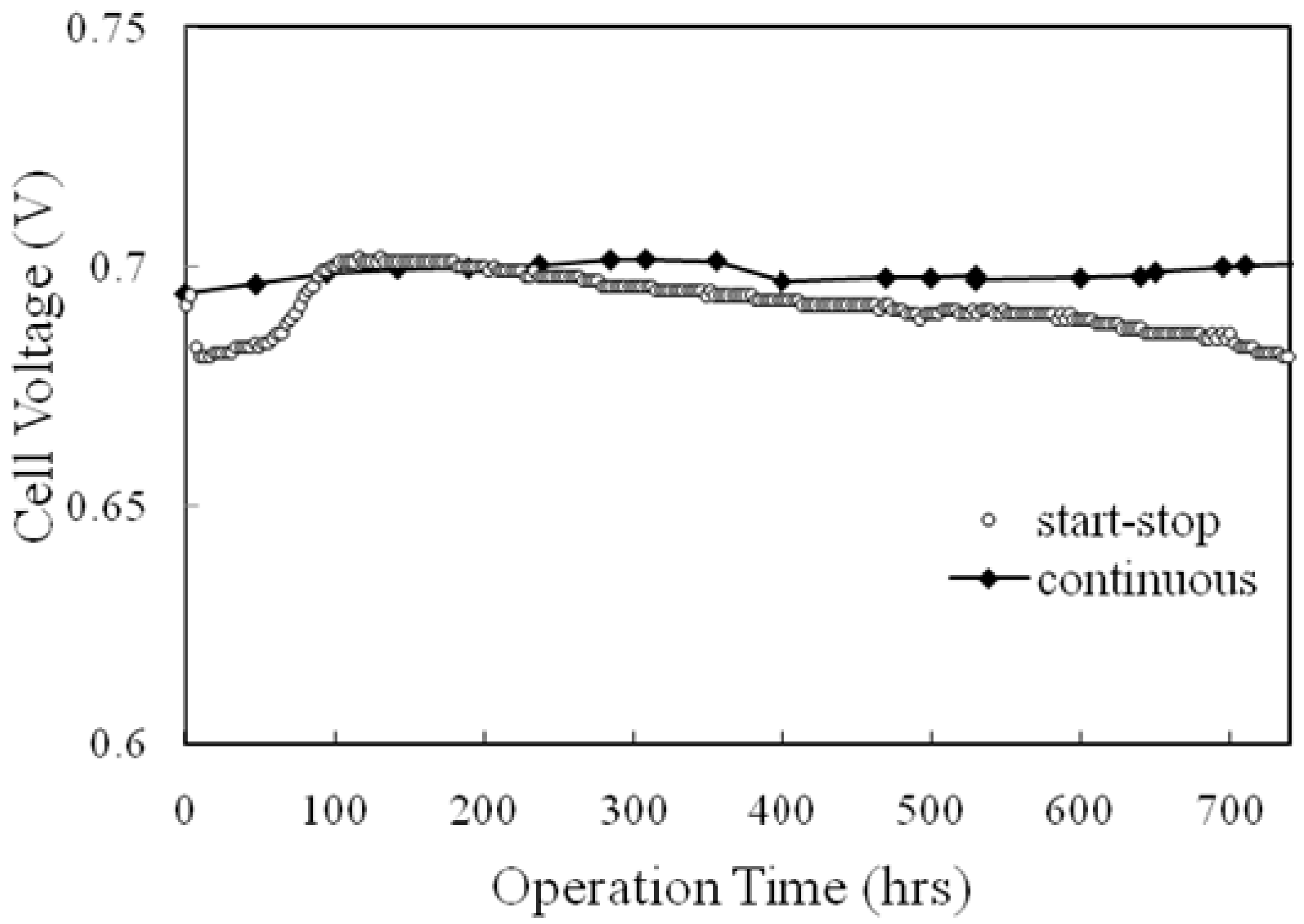
2.3.3. Durability of the MEA
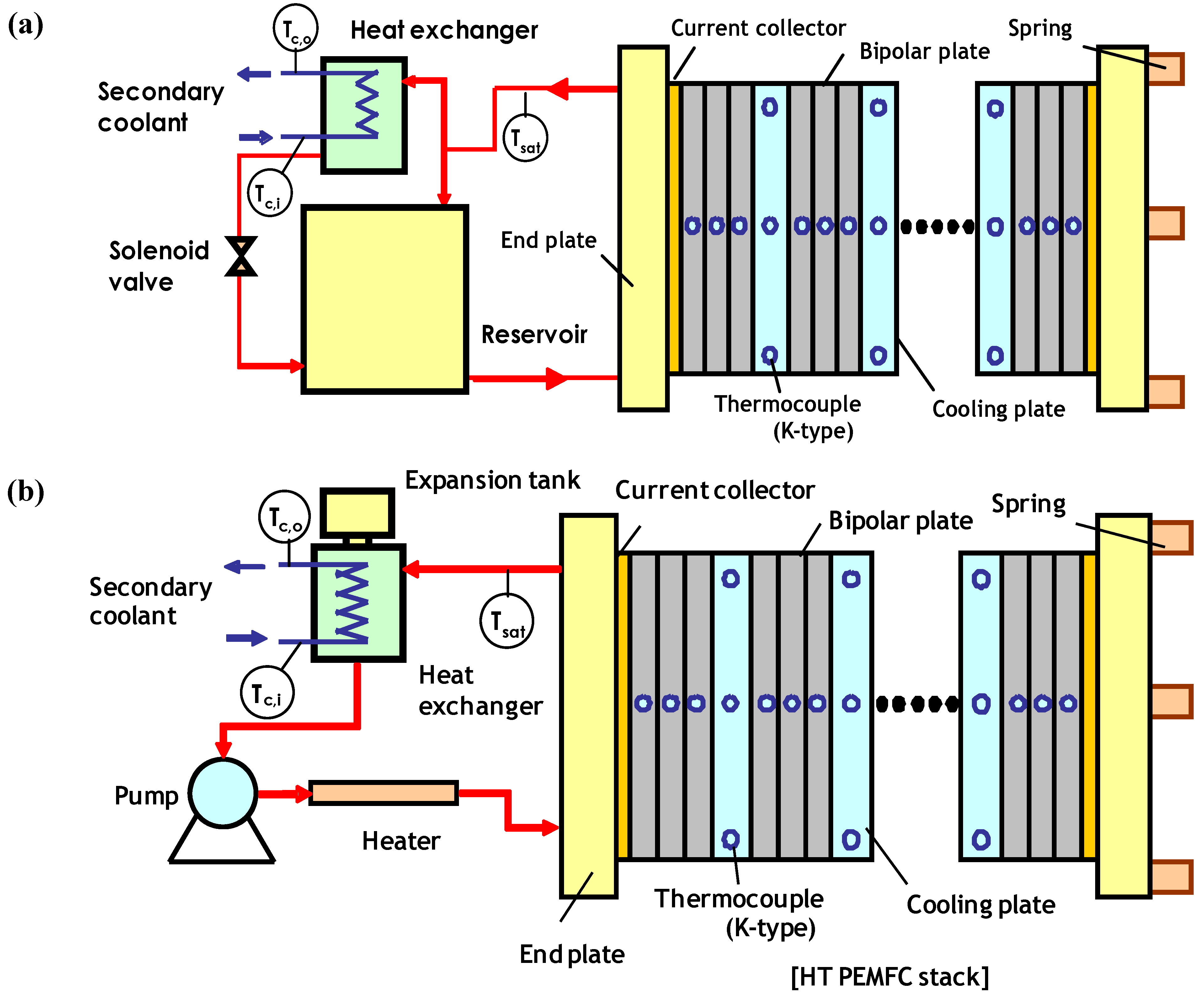
2.4. Stack

3. High-Temperature PEM
3.1. Design Rules of the Membrane
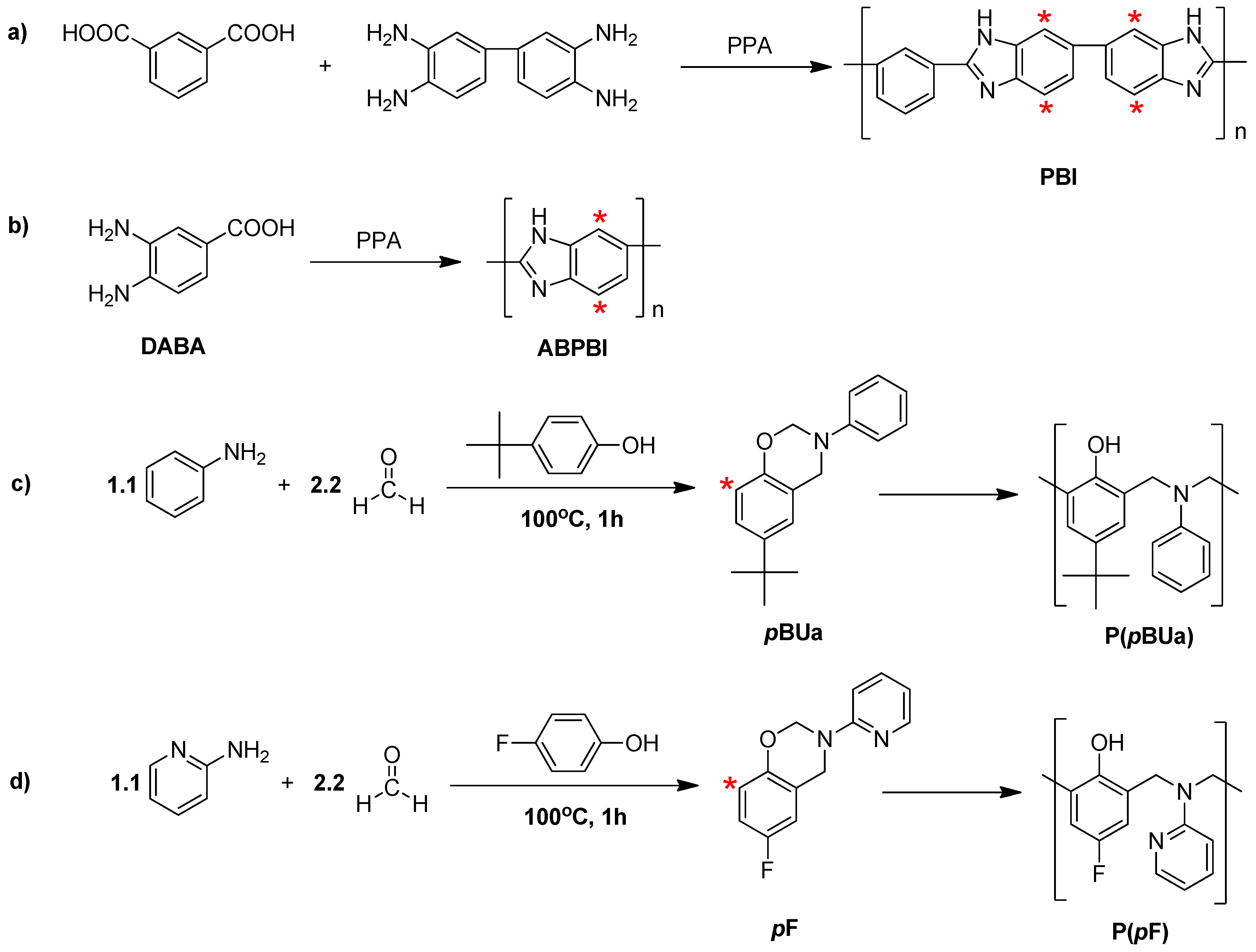
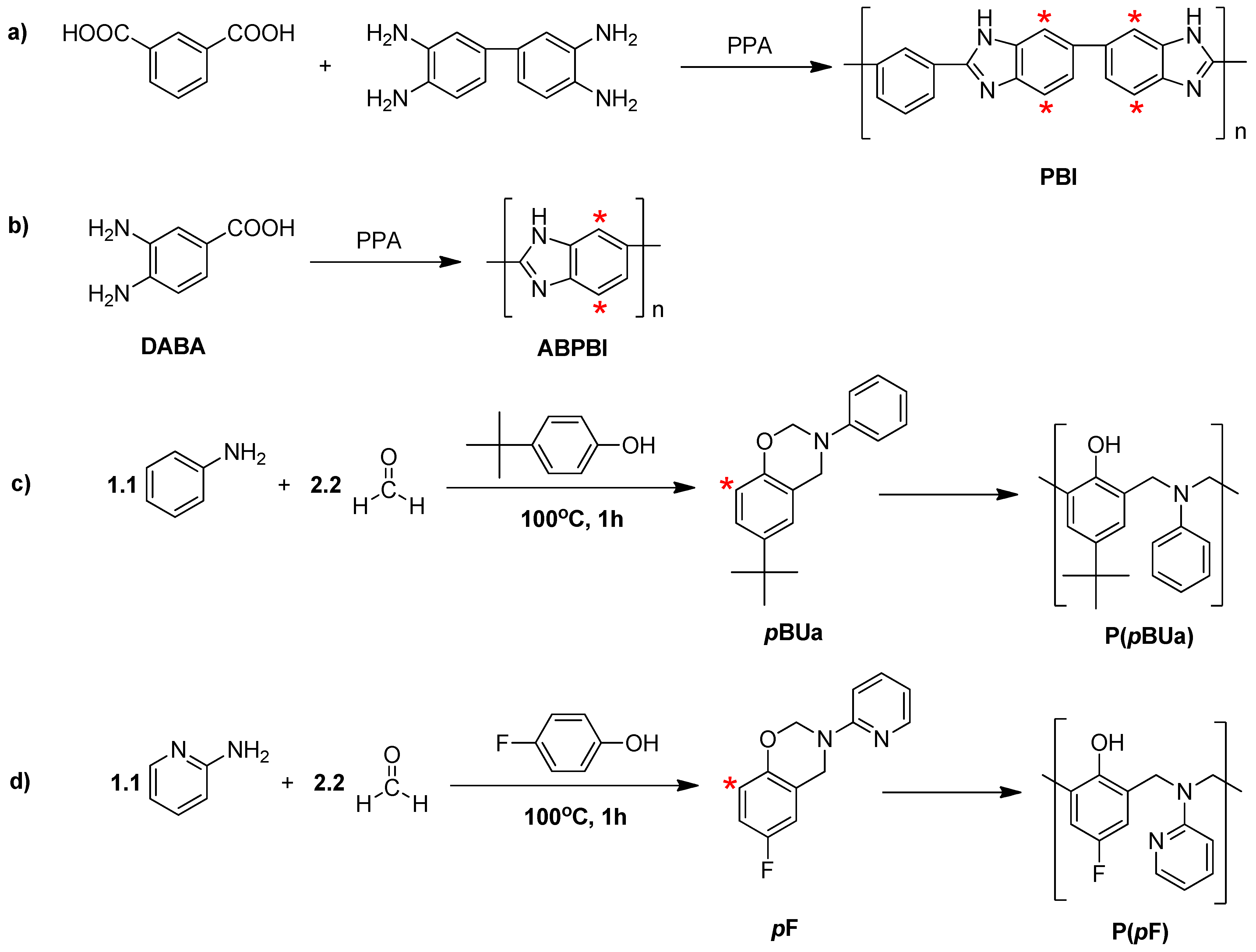
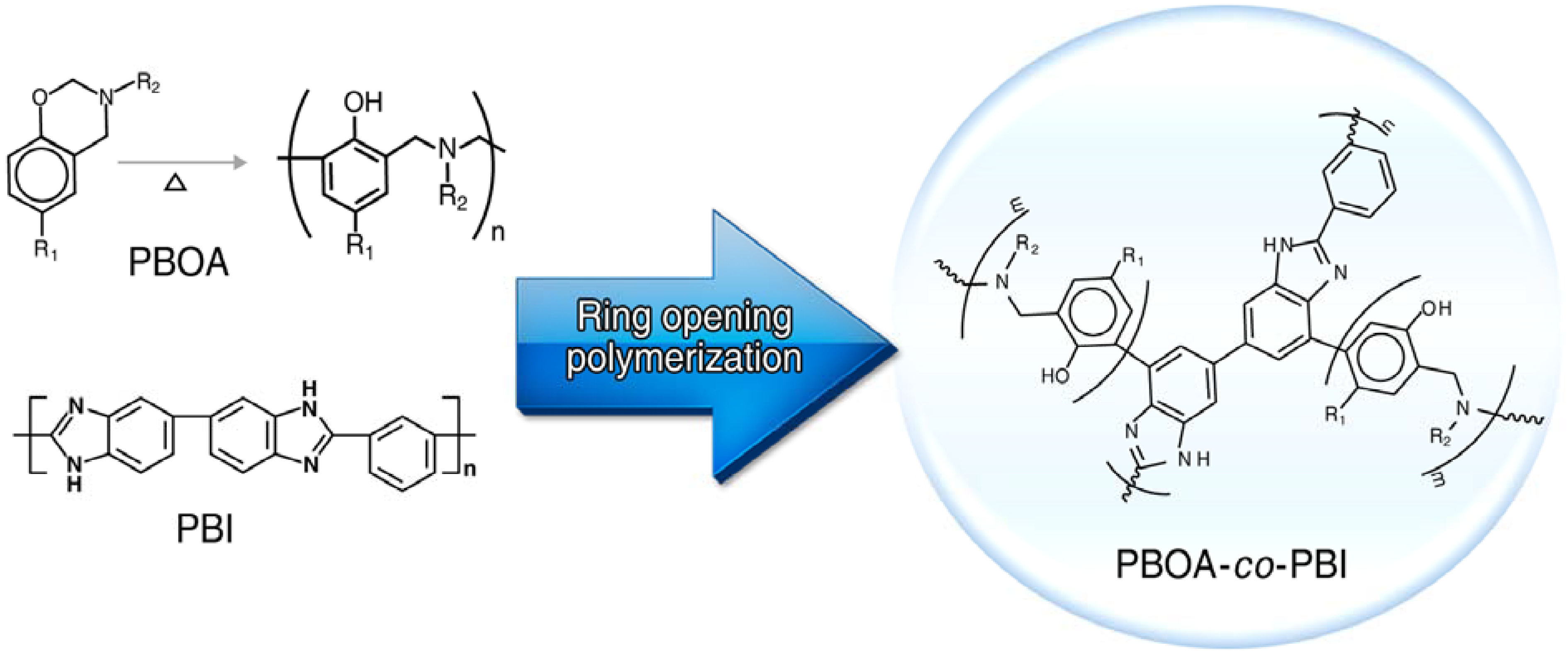
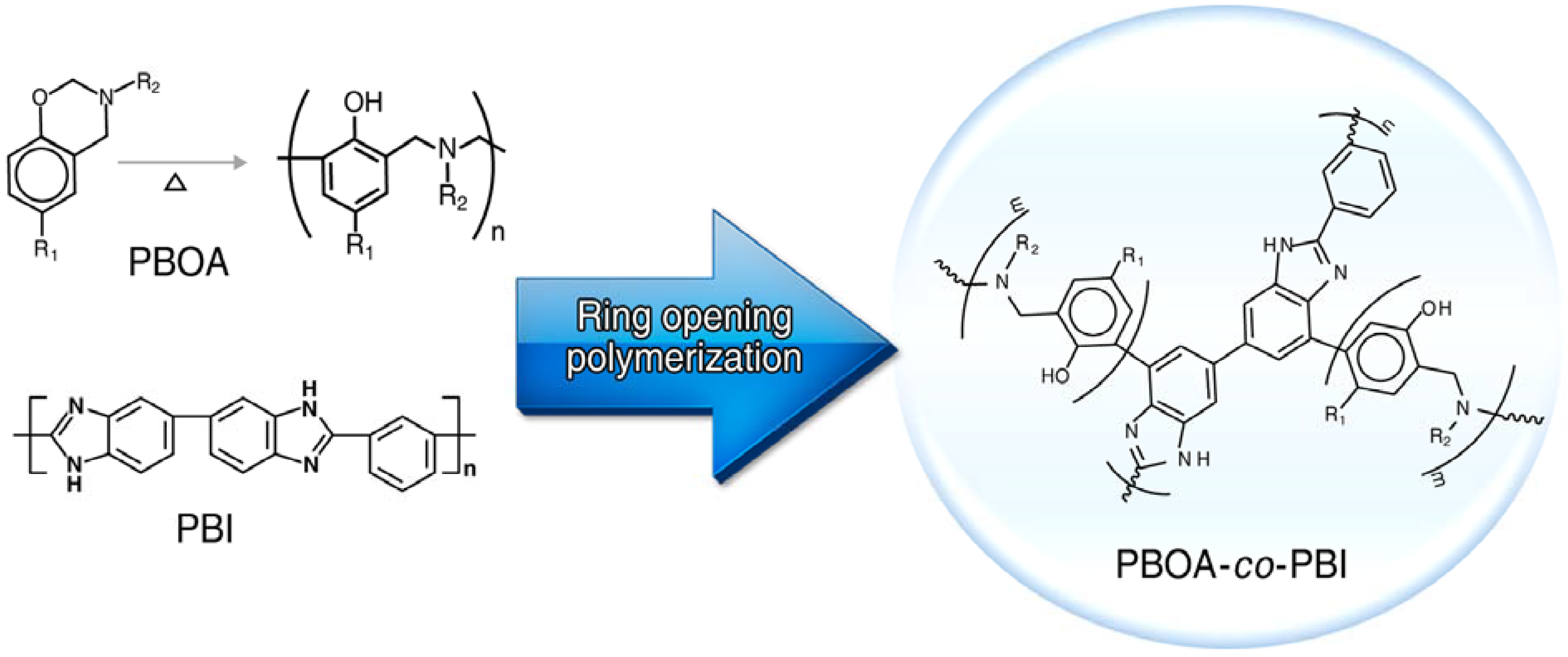
3.2. Synthesis of Poly(benzoxazine), Polybenzimidazole and Its Membrane
3.2.1. Synthesis of Benzoxazine Monomer, 6-tert-butyl-3-phenyl-3,4-dihydro-2H-benzo[e][1,3]oxazine (pBUa) and 6-Fluoro-3-(pyridine-2-yl)-3,4-dihydro-2H-benzo[e][1,3]oxazine (pF)
3.2.2. Synthesis of Poly[2,2'-(m-phenylene)-5,5'-bibenzimidazole] (PBI)
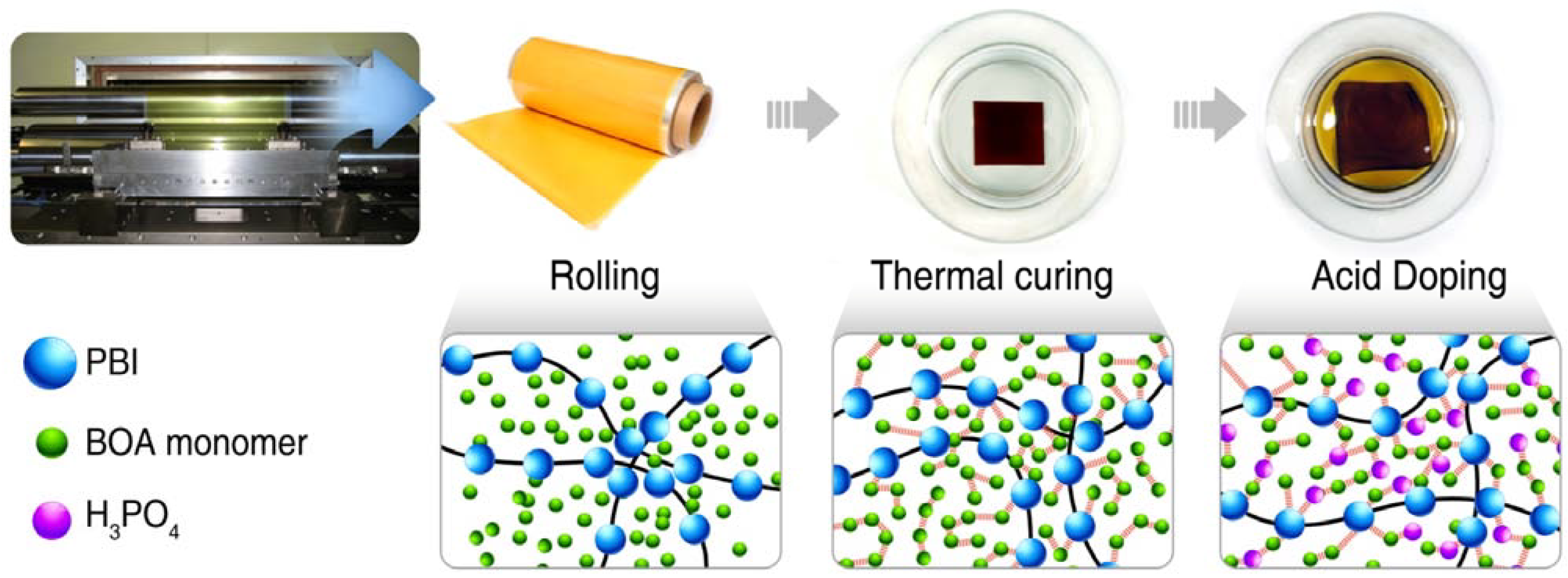
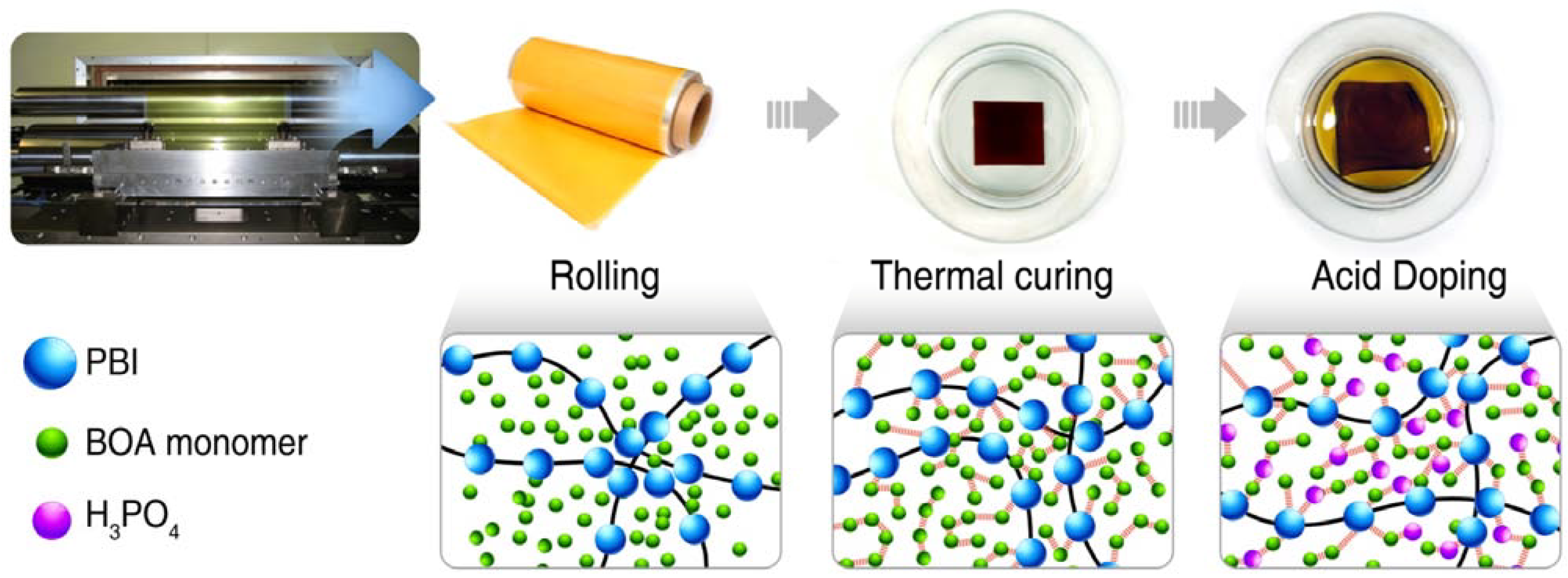
3.2.3. Preparation of the PA-Doped P(pBUa-co-BI) Membranes
3.2.4. Preparation of PA-Doped PpF-co-ABPBI
3.2.5. Preparation of Membrane-Electrode Assembly (MEA)
3.2.6. Cell Performance Measurement and Acceleration Lifetime Test (ALT)
3.3. Characterization
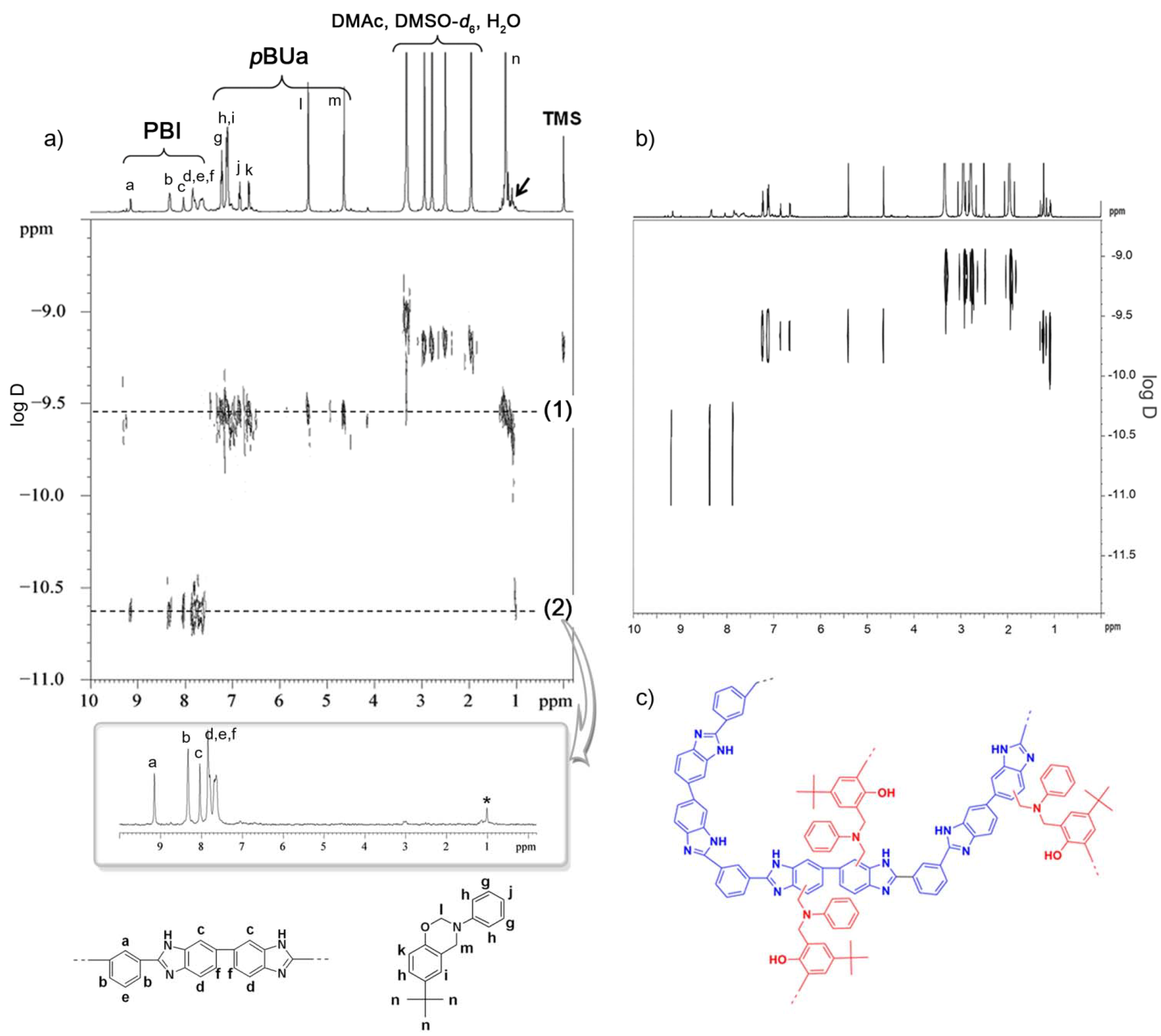
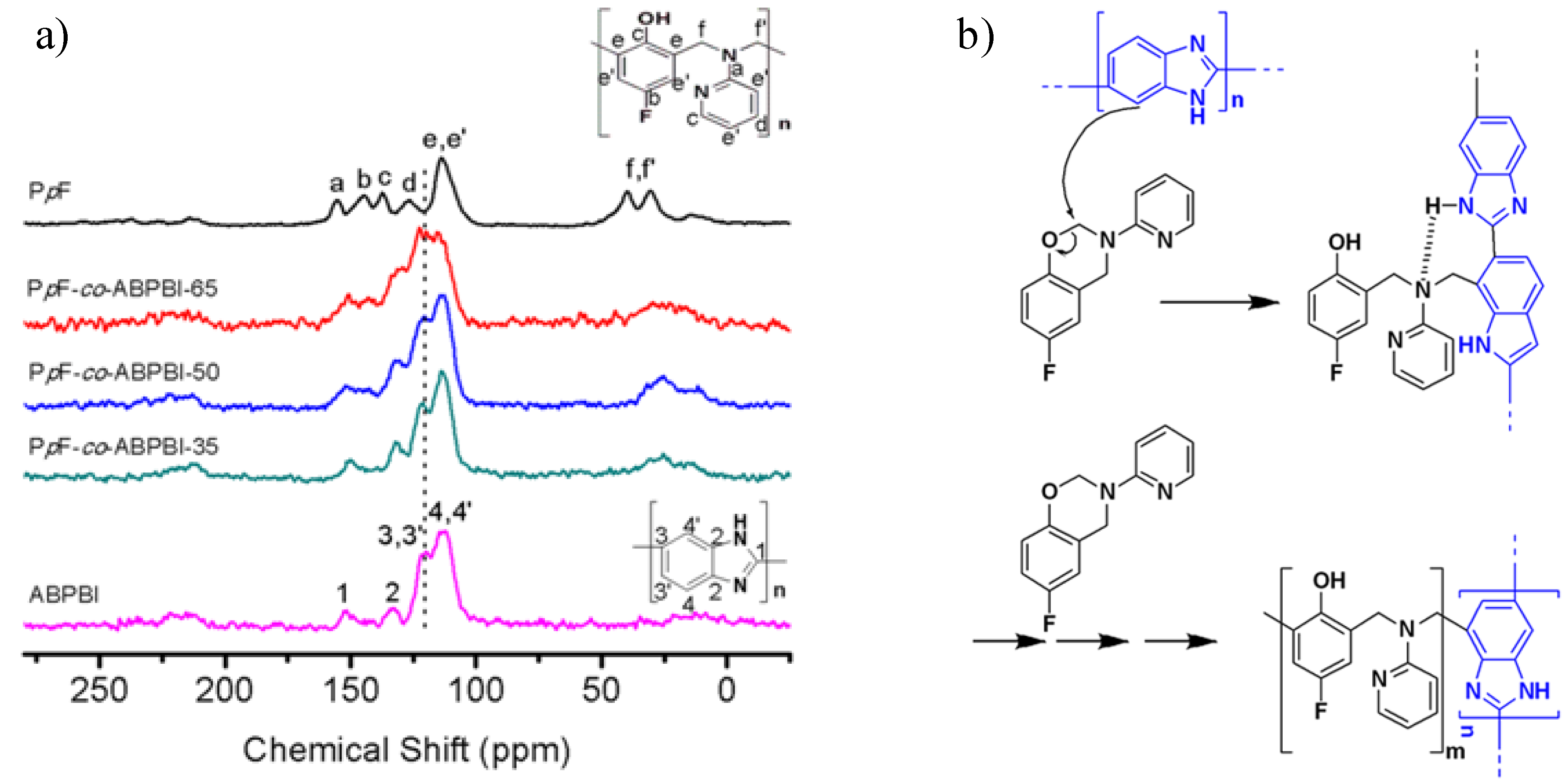
3.3.1. Copolymer Formation
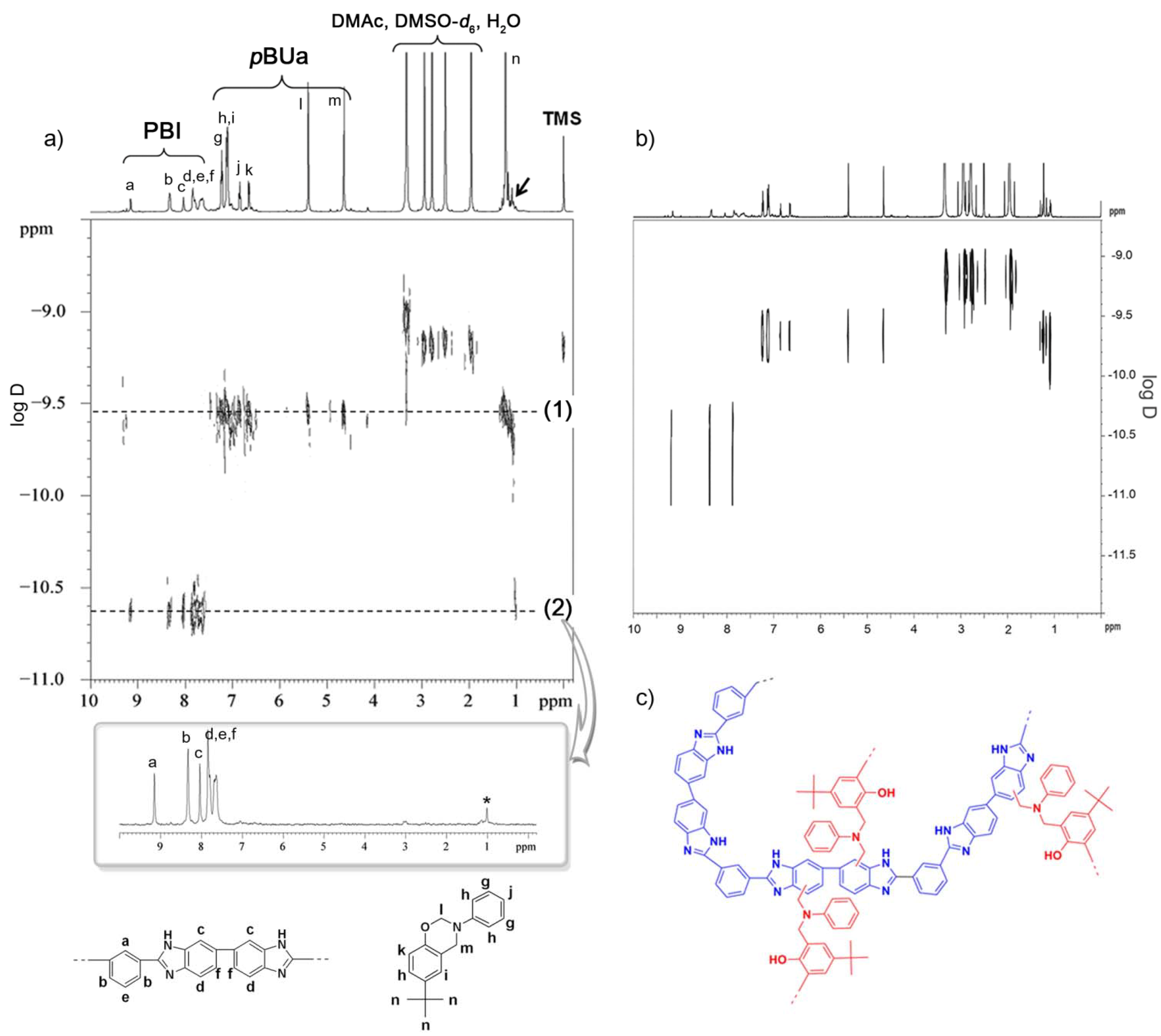
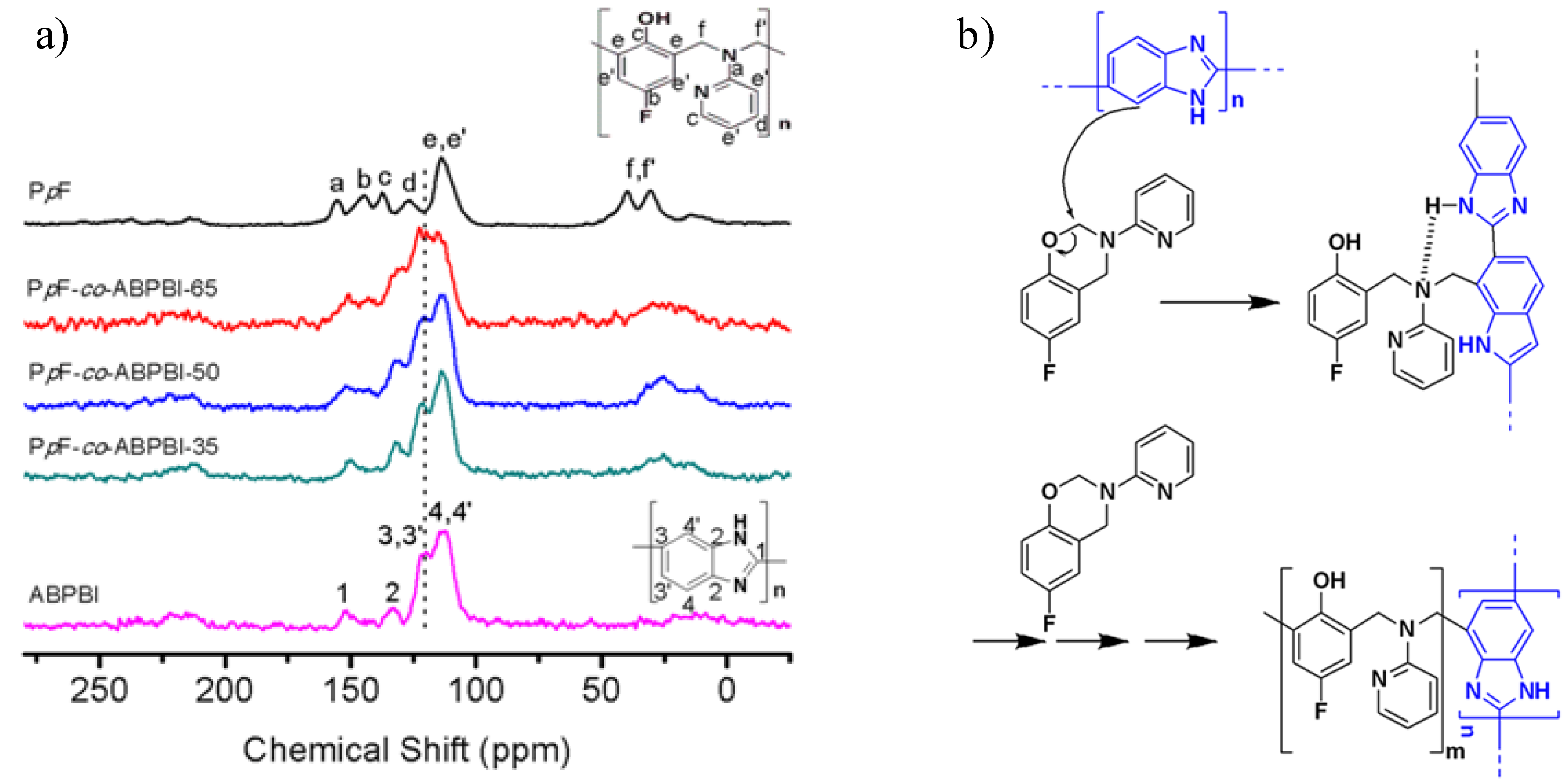
3.3.2. Chemical Stability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membranes | PA content (wt %) | Proton Conductivity (S cm−1) |
|---|---|---|
| P(pBUa-co-BI)-65 | 88.5 | 0.1206 |
| P(pBUa-co-BI)-50 | 83.9 | 0.0922 |
| P(pBUa-co-BI)-35 | 83.0 | 0.0792 |
| P(pF-co-BI)-65 | 86.6 | 0.1267 |
| P(pF-co-BI)-50 | 84.9 | 0.1327 |
| P(pF-co-BI)-35 | 83.0 | 0.1091 |
| PpF-co-ABPBI-65 | 73.0 | 0.1015 |
| 77.1 | 0.1472 | |
| PpF-co-ABPBI-50 | 74.4 | 0.0983 |
| 78.3 | 0.1098 | |
| 81.0 | 0.1315 | |
| 84.4 | 0.1434 | |
| PpF-co-ABPBI-35 | 71.9 | 0.0656 |
| 75.6 | 0.0889 | |
| 84.1 | 0.1449 | |
| ABPBI a | 75.3 b | 0.0832 |
| PBI a | 81.2 b | 0.0951 |
| Membranes | PA content (wt %) | Tensile strength (MPa) | Elongation at break (%) | Modulus (MPa) |
|---|---|---|---|---|
| P(pBUa-co-BI)-65 | 88.5 | 2.19 | 72.3 | 5.70 |
| P(pBUa-co-BI)-50 | 83.9 | 2.69 | 54.8 | 14.5 |
| P(pBUa-co-BI)-35 | 83.0 | 4.25 | 38.1 | 41.4 |
| PpF-co-ABPBI-65 | 62.8 | 24.5 | 48.9 | 34.8 |
| 72.9 | 11.5 | 73.9 | 17.1 | |
| PpF-co-ABPBI-50 | 76.6 | 14.9 | 70.6 | 21.4 |
| 79.3 | 13.4 | 113 | 11.2 | |
| PpF-co-ABPBI-35 | 67.1 | 25.4 | 48.5 | 47.2 |
| 77.4 | 18.1 | 78.9 | 25.3 | |
| 82.5 | 11.1 | 61.5 | 23.3 | |
| ABPBI a | 67.1 | 39.5 | 247 | 260 |
| PBI a | 76.8 | 19.5 | 143 | 25.1 |
| 81.2 | 5.34 | 47.6 | 43.2 |
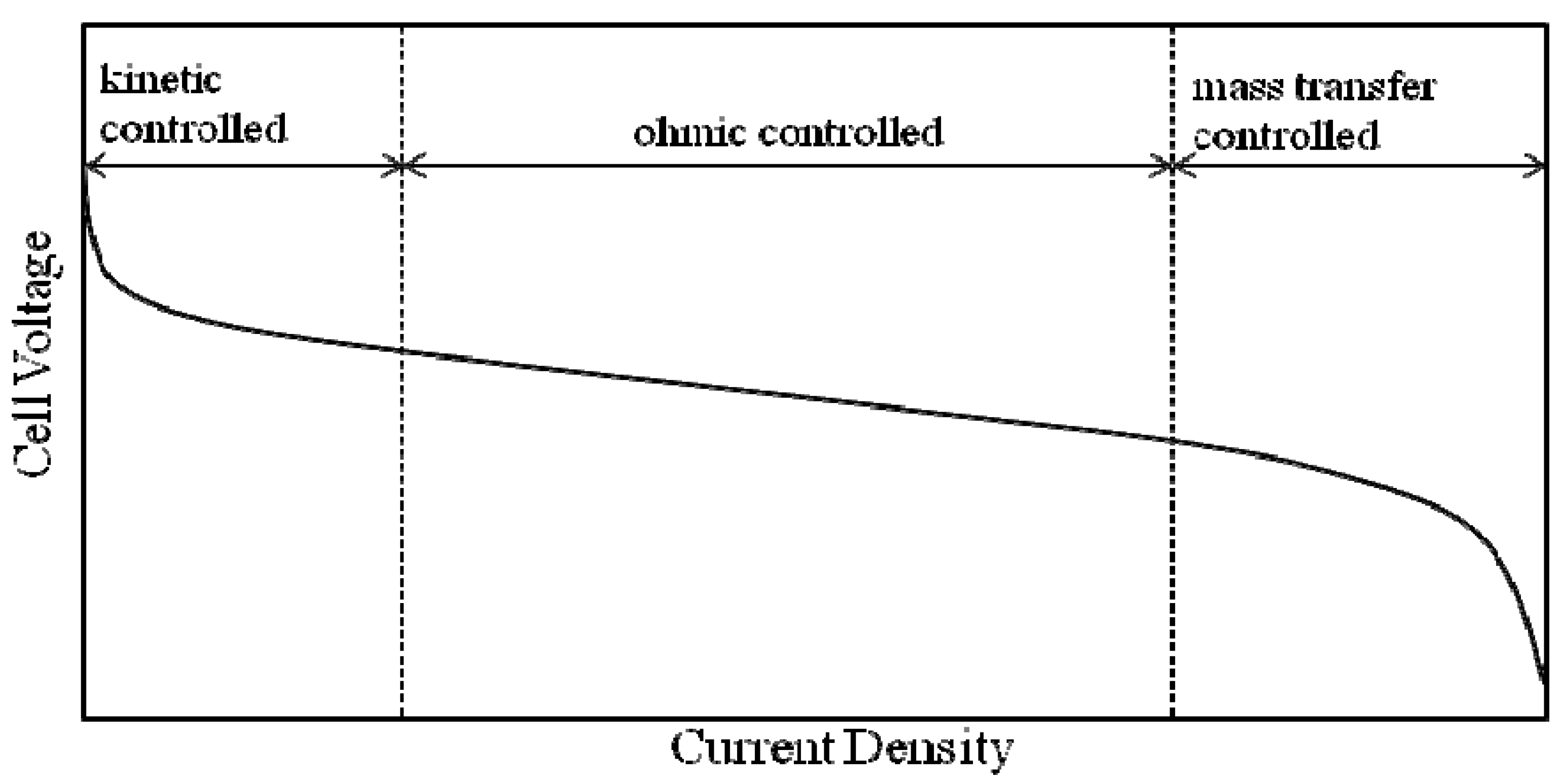
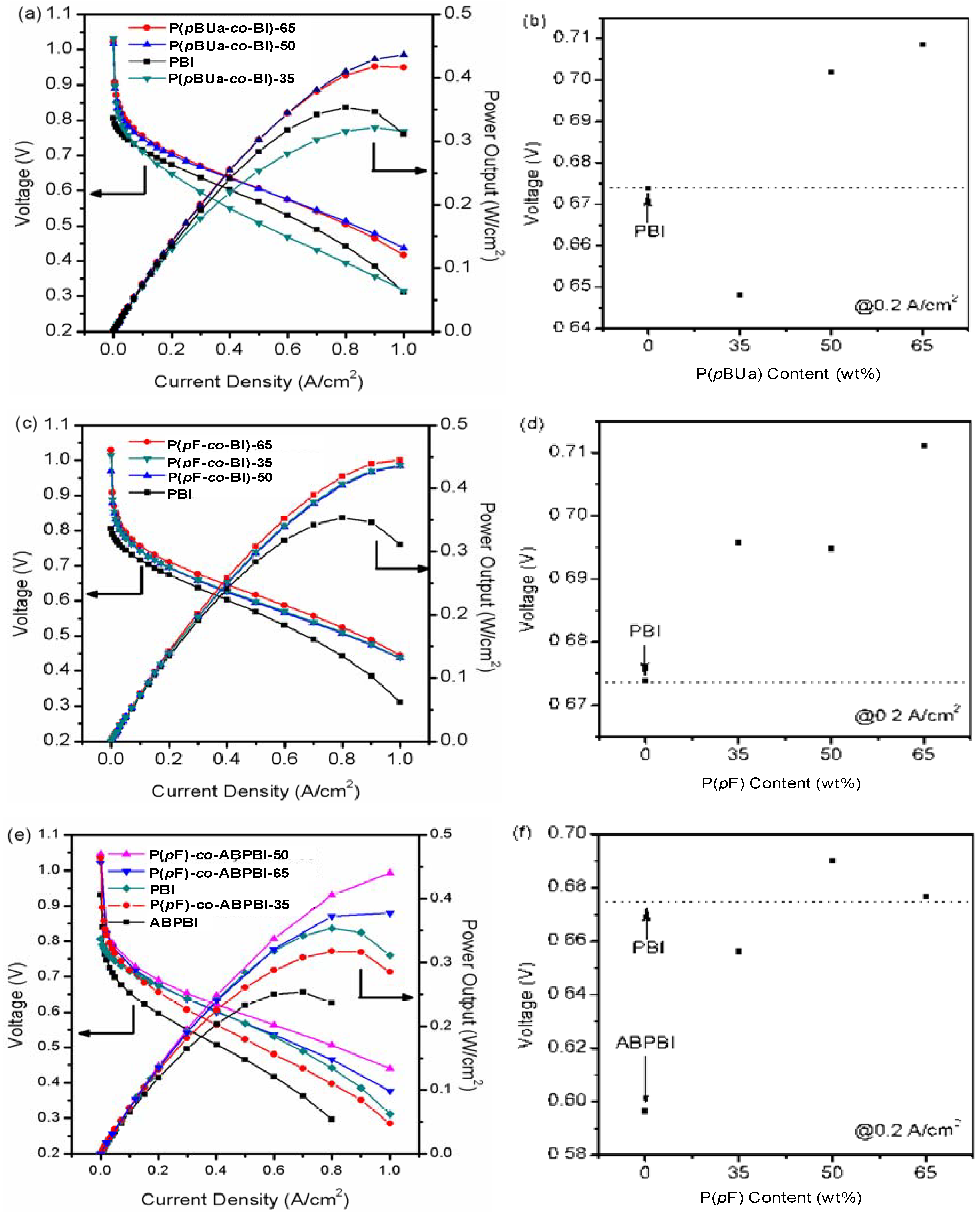
3.3.3. The Proton Conductivity
3.3.4. Fuel Cell Evaluation
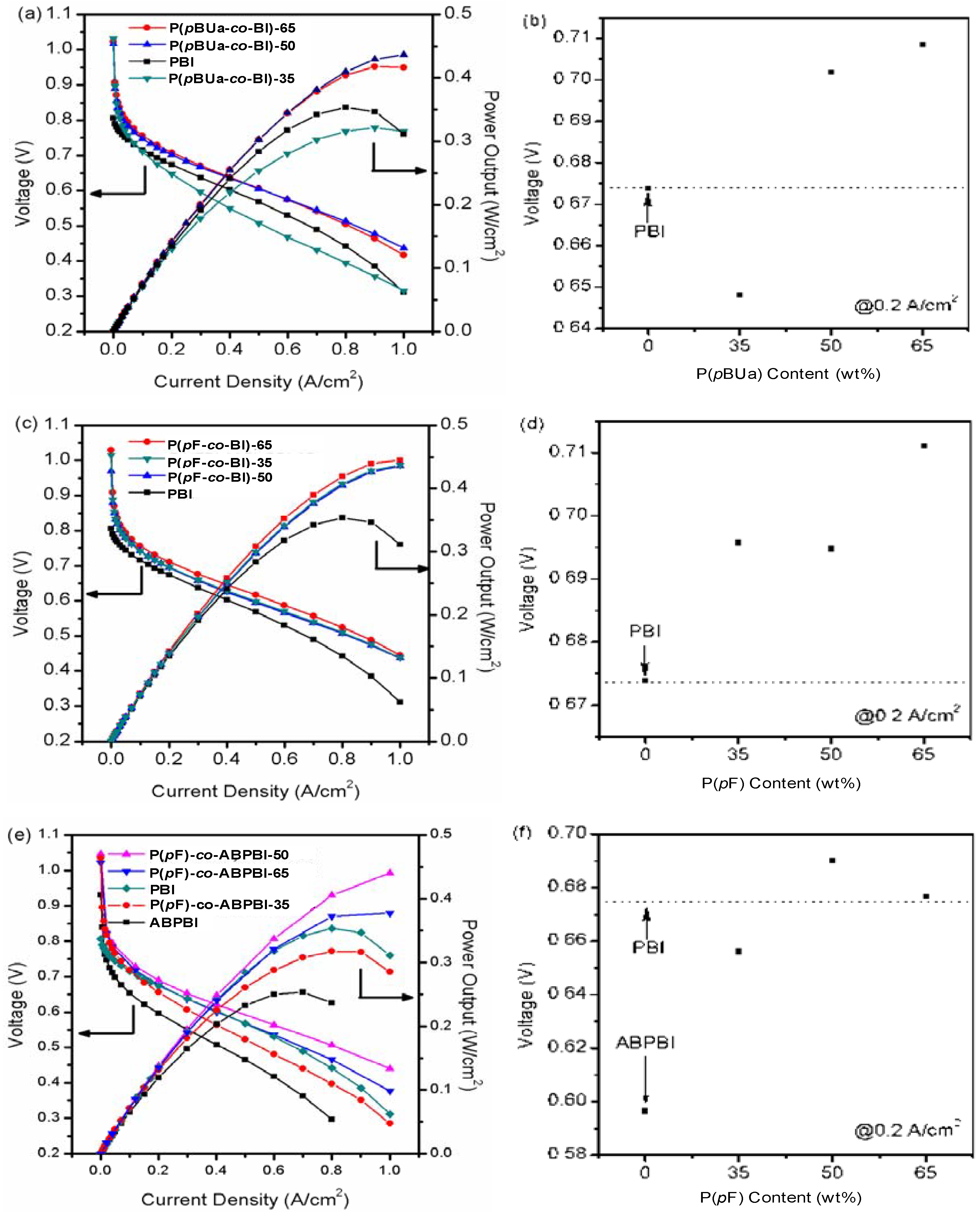
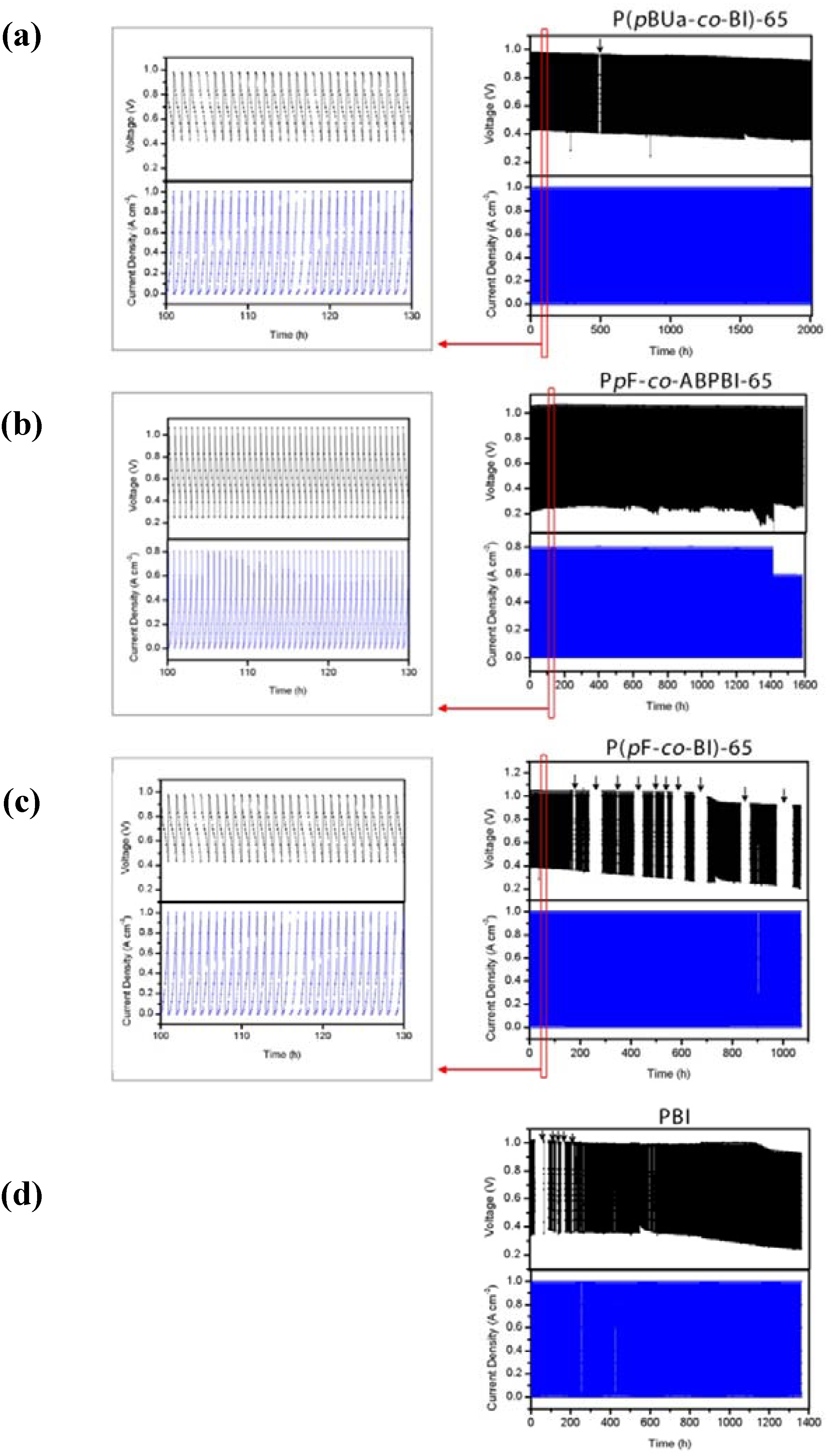
4. High Temperature MEA
4.1. MEA Design and Characteristics

4.2. MEA Performance on System Level Requirements
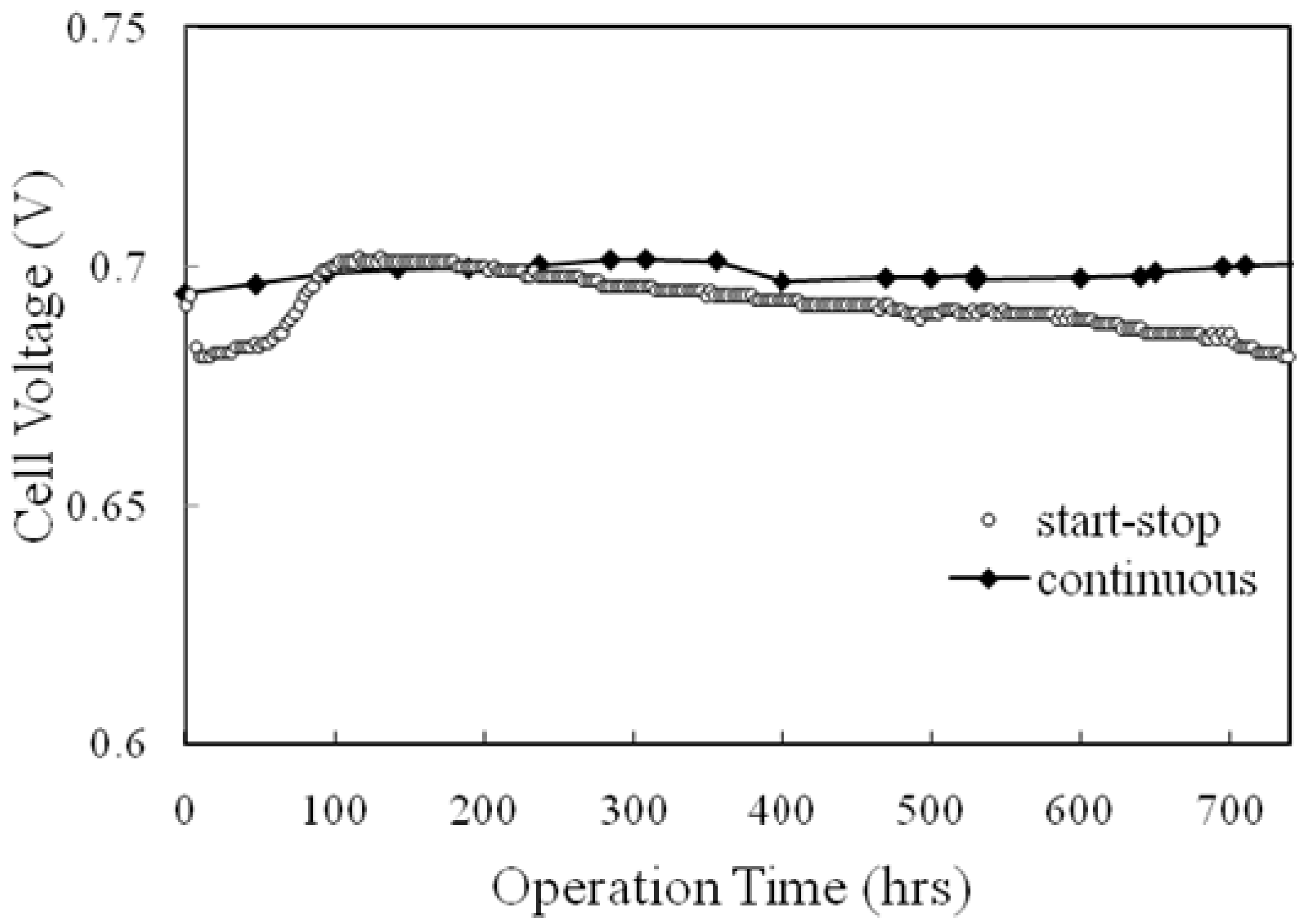
5. Summary and Further Studies
References
- Mehta, V.; Cooper, J.S. Review and analysis of PEM fuel cell design and manufacturing. J. Power Sources 2003, 114, 32–53. [Google Scholar] [CrossRef]
- Ahn, S.-Y.; Shin, S.-J.; Ha, H.Y.; Hong, S.-A.; Lee, Y.-C.; Lim, T.W.; Oh, I.-H. Performance and lifetime analysis of the kW-class PEMFC stack. J. Power Sources 2002, 106, 295–303. [Google Scholar] [CrossRef]
- Mallant, R.K.A.M. PEMFC systems: The need for high temperature polymers as a consequence of PEMFC water and heat management. J. Power Sources 2003, 118, 424–429. [Google Scholar] [CrossRef]
- Li, Q.; Jensen, J.O.; Savinell, R.F.; Bjerrum, N.J. High temperature proton exchange membranes based on polybenzimidazoles for fuel cells. Prog. Polym. Sci. 2009, 34, 449–477. [Google Scholar] [CrossRef]
- Kwon, K.; Kim, T.Y.; Yoo, D.Y.; Hong, S.-G.; Park, J.O. Maximization of high-temperature proton exchange membrane fuel cell performance with the optimum distribution of phosphoric acid. J. Power Sources 2009, 188, 463–467. [Google Scholar] [CrossRef]
- Wannek, C.; Konradi, I.; Mergel, J.; Lehnert, W. Redistribution of phosphoric acid in membrane electrode assemblies for high-temperature polymer electrolyte fuel cells. Int. J. Hydrog. Energy 2009, 34, 9479–9485. [Google Scholar] [CrossRef]
- He, R.; Li, Q.; Bach, A.; Jensen, J.O.; Bjerrum, N.J. Physicochemical properties of phosphoric acid doped polybenzimidazole membranes for fuel cells. J. Membr. Sci. 2006, 277, 38–45. [Google Scholar] [CrossRef]
- Park, J.O.; Kwon, K.; Cho, M.D.; Hong, S.-G.; Kim, T.Y.; Yoo, D.Y. Role of binders in high-temperature PEMFC electrode. J. Electrochem. Soc. 2011, 158, B675–B681. [Google Scholar] [CrossRef]
- Mamlouk, M.; Scott, K. An investigation of Pt alloy oxygen reduction catalysts in phosphoric acid doped PBI fuel cells. J. Power Sources 2011, 196, 1084–1089. [Google Scholar] [CrossRef]
- Andujar, J.M.; Segura, F. Fuel cells: History and updating. A walk along two centuries. Renew. Sustain. Energy Rev. 2009, 13, 2309–2322. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Steele, B.C.H. Material science and engineering: The enabling technology for the commercialization of fuel cell systems. J. Mater. Sci. 2001, 36, 1053–1068. [Google Scholar]
- You, D.J.; Kwon, K.; Joo, S.H.; Kim, J.H.; Kim, J.M.; Pak, C.; Chang, H. Carbon-supported ultra-high loading Pt nanoparticle catalyst by controlled overgrowth of Pt: Improvement of Pt utilization leads to enhanced direct methanol fuel cell performance. Int. J. Hydrog. Energy 2012, 37, 6880–6885. [Google Scholar] [CrossRef]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef]
- Petrii, O.A. Pt-Ru electrocatalysts for fuel cells: A representative review. J. Solid State Electrochem. 2008, 12, 609–642. [Google Scholar] [CrossRef]
- Yang, H. Platinum-based electrocatalysts with core-shell nanostructures. Angew. Chem. Int. Ed. 2011, 50, 2674–2676. [Google Scholar] [CrossRef]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; Toney, M.F.; Nilsson, A. Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Markovic, N. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef]
- Wang, J.X.; Inada, H.; Wu, L.; Zhu, Y.; Choi, Y.; Liu, P.; Zhou, W.-P.; Adzic, R.R. Oxygen reduction on well-defined core-shell nanocatalysts: particle size, facet, and Pt shell thickness effects. J. Am. Chem. Soc. 2009, 131, 17298–17302. [Google Scholar]
- Sasaki, K.; Naohara, H.; Cai, Y.; Choi, Y.M.; Liu, P.; Vukmirovic, M.B.; Wang, J.X.; Adzic, R.R. Core-protected platinum monolayer shell high-stability electrocatalysts for fuel-cell cathodes. Angew. Chem. Int. Ed. 2010, 49, 8602–8607. [Google Scholar] [CrossRef]
- Sasaki, K.; Naohara, H.; Choi, Y.M.; Cai, Y.; Chen, W.-F.; Liu, P.; Adzic, R.R. Highly stable Pt monolayer on PdAu nanoparticle electrocatalysts for the oxygen reduction reaction. Nat. Commun. 2012, 3, 1115–1119. [Google Scholar] [CrossRef]
- Wang, J.X.; Ma, C.; Choi, Y.M.; Su, D.; Zhu, Y.; Liu, P.; Si, R.; Vukmirovic, M.B.; Zhang, Y.; Adzic, R.R. Kirkendall effect and lattice contraction in nanocatalysts: A new strategy to enhance sustainable activity. J. Am. Chem. Soc. 2011, 133, 13551–13557. [Google Scholar]
- Watanabe, M.; Uchida, M.; Motoo, S. Preparation of Highly Dispersed Pt + Ru Clusters and the Activity for the Electrooxidation of Methanol. J. Electroanal. Chem. 1987, 229, 395–406. [Google Scholar] [CrossRef]
- Wakisaka, M.; Mitsui, S.; Hirose, Y.; Kawashima, K.; Uchida, H.; Watanabe, M. Electronic Structures of Pt-Co and Pt-Ru Alloys for CO-Tolerant Anode Catalysts in Polymer Electrolyte Fuel Cells Studied by EC-XPS. J. Phys. Chem. B 2006, 110, 23489–23496. [Google Scholar]
- Li, Q.; He, R.; Gao, J.-A.; Jensen, J.O.; Bjerrum, N.J. The CO Poisoning Effect in PEMFCs Operational at Temperatures up to 200°C. J. Electrochem. Soc. 2003, 150, A1599–A1605. [Google Scholar] [CrossRef]
- Skulason, E.; Tripkovic, V.; Bjorketun, M.E.; Gudmundsdottir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jonsson, H.; Norskov, J.K. Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar]
- Markovic, N.M.; Grgur, B.N.; Ross, P.N. Temperature-dependent hydrogen electrochemistry on platinum low-index single-crystal surfaces in acid solutions. J. Phys. Chem. B 1997, 101, 5405–5413. [Google Scholar]
- Murthi, V.S.; Urian, R.C.; Mukerjee, S. Oxygen reduction kinetics in low and medium temperature acid environment: Correlation of water activation and surface properties in supported Pt and Pt alloy electrocatalysts. J. Phys. Chem. B 2004, 108, 11011–11023. [Google Scholar] [CrossRef]
- Suzuki, A.; Oono, Y.; Williams, M.C.; Miura, R.; Inaba, K.; Hatakeyama, N.; Takaba, H.; Hori, M.; Miyamoto, A. Evaluation for sintering of electrocatalysts and its effect on voltage drops in high-temperature proton exchange membrane fuel cells (HT-PEMFC). Int. J. Hydrog. Energy 2012, 37, 18272–18289. [Google Scholar]
- Shao, M. Palladium-based electrocatalysts for hydrogen oxidation and oxygen reduction reactions. J. Power Sources 2011, 196, 2433–2444. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Serov, A.; Kwak, C. Review of non-platinum anode catalysts for DMFC and PEMFC application. Appl. Catal. B Environ. 2009, 90, 313–320. [Google Scholar] [CrossRef]
- Kwon, K.; Lee, K.H.; Jin, S.-A.; You, D.J.; Pak, C. Ceria-promoted oxygen reduction reaction in Pd-based electrocatalysts. Electrochem. Commun. 2011, 13, 1067–1069. [Google Scholar] [CrossRef]
- Jin, S.-A.; Kwon, K.; Pak, C.; Chang, H. The oxygen reduction electrocatalytic activity of intermetallic compound of palladium-tin supported on tin oxide-carbon composite. Catal. Today 2011, 164, 176–180. [Google Scholar]
- You, D.J.; Jin, S.-A.; Lee, K.H.; Pak, C.; Choi, K.H.; Chang, H. Improvement of activity for oxygen reduction by decoration of Ir on PdCu/C catalyst. Catal. Today 2012, 185, 138–142. [Google Scholar]
- Antolini, E. Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B Environ. 2009, 88, 1–24. [Google Scholar] [CrossRef]
- Yang, Y.; Chiang, K.; Burke, N. Porous carbon-supported catalysts for energy and environmental application: A short review. Catal. Today 2011, 178, 197–205. [Google Scholar]
- Dicks, A.L. The role of carbon in fuel cells. J. Power Sources 2006, 156, 128–141. [Google Scholar] [CrossRef]
- Lee, H.I.; Stucky, G.D.; Kim, J.H.; Pak, C.; Chang, H.; Kim, J.M. Spontaneous phase separation mediated synthesis of 3D mesoporous carbon with controllable cage and window size. Adv. Mater. 2011, 23, 2357–2361. [Google Scholar] [CrossRef]
- Pak, C.; Kim, J.M.; Chang, H. Mesoporous carbon-supported catalysts for direct methanol fuel cells. In Electrocatalysis of Direct Methanol Fuel Cells, 1st; Liu, H., Zhang, J., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 355–378. [Google Scholar]
- Chang, H.; Joo, S.H.; Pak, C. Synthesis and characterization of mesoporous carbon for fuel cell applications. J. Mater. Chem. 2007, 17, 3078–3088. [Google Scholar]
- Kwon, K.; Jin, S.-A.; Pak, C.; Chang, H.; Joo, S.H.; Lee, H.I.; Kim, J.H.; Kim, J.M. Enhancement of electrochemical stability and catalytic activity of Pt nanoparticles via strong metal-support interaction with sulfur-containing ordered mesoporous carbon. Catal. Today 2011, 164, 186–189. [Google Scholar]
- Lee, H.I.; Joo, S.H.; Kim, J.H.; You, D.J.; Kim, J.M.; Park, J.-N.; Chang, H.; Pak, C. Ultrastable Pt nanoparticles supported on sulfur-containing ordered mesoporous carbon via strong metal-support interaction. J. Mater. Chem. 2009, 19, 5934–5939. [Google Scholar] [CrossRef]
- Joo, S.H.; Pak, C.; You, D.J.; Lee, S.-A.; Lee, H.I.; Kim, J.M.; Chang, H.; Seung, D. Ordered mesoporous carbons (OMC) as supports of electrocatalysts for direct methanol fuel cells (DMFC): Effect of carbon precursors of OMC on DMFC performances. Electrochim. Acta 2006, 52, 1618–1626. [Google Scholar] [CrossRef]
- Joo, S.H.; Kwon, K.; You, D.J.; Pak, C.; Chang, H.; Kim, J.M. Preparation of high loading Pt nanoparticles on ordered mesoporous carbon with a controlled Pt size and its effect on oxygen reduction and methanol oxidation reactions. Electrochim. Acta 2009, 54, 5746–5753. [Google Scholar] [CrossRef]
- Joo, S.H.; Lee, H.I.; You, D.J.; Kwon, K.; Kim, J.H.; Choi, Y.S.; Kang, M.; Kim, J.M.; Pak, C.; Chang, H.; Seung, D. Ordered mesoporous carbons with controlled particle sizes as catalyst supports for direct methanol fuel cell cathodes. Carbon 2008, 46, 2034–2045. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Wang, Y.J.; Wilkinson, D.; Zhang, J. Noncarbon support materials for polymer electrolyte membrane fuel cell electrocatalysts. Chem. Rev. 2011, 111, 7625–7651. [Google Scholar] [CrossRef]
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef]
- Kreuer, K.D. On the development of proton conducting polymer membranes for hydrogen andmethanol fuel cells. J. Membr. Sci. 2001, 185, 29–39. [Google Scholar] [CrossRef]
- Miyatake, K.; Chikashige, Y.; Higuchi, E.; Watanabe, M. Tuned polymer electrolyte membranes based on aromatic polyethers for fuel cell applications. J. Am. Chem. Soc. 2007, 129, 3879–3887. [Google Scholar]
- Savadogo, O. Emerging membranes for electrochemical systems: Part II. High temperature composite membranes for polymer electrolyte fuel cell (PEFC) applications. J. Power Sources 2004, 127, 135–161. [Google Scholar] [CrossRef]
- Mustarelli, P.; Quartarone, E.; Grandi, S.; Carollo, A.; Magistris, A. Polybenzimidazole-based membranes as a real alternative to Nafion for fuel cells operating at low temperature. Adv. Mater. 2008, 20, 1339–1343. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Ogawa, A.; Kanno, M.; Nakamoto, H.; Yasuda, T.; Watanabe, M. Nonhumidified intermediate temperature fuel cells using protic ionic liquids. J. Am. Chem. Soc. 2010, 132, 9764–9773. [Google Scholar]
- Wainright, J.S.; Wang, J.-T.; Weng, D.; Savinell, R.F.; Litt, M. Acid-doped polybenzimidazoles: A new polymer electrolyte. J. Electrochem. Soc. 1995, 142, L121–L123. [Google Scholar]
- Li, Q.; He, R.; Jensen, J.O.; Bjerrum, N.J. Approaches and recent development of polymer electrolyte membranes for fuel cells operating above 100 °C. Chem. Mater. 2003, 15, 4896–4915. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, Z.; Zhang, J.; Tang, Y.; Song, C.; Navessin, T.; Shi, Z.; Song, D.; Wang, H.; Wilkinson, D.P.; Liu, Z.-S.; Holdcroft, S. High temperature PEM fuel cells. J. Power Sources 2006, 160, 872–891. [Google Scholar] [CrossRef]
- Hogarth, W.H.J.; Diniz da Costa, J.C.; Lu, G.Q. Solid acid membranes for high temperature (at 140°C) proton exchange membrane fuel cells. J. Power Sources 2005, 142, 223–237. [Google Scholar] [CrossRef]
- Shao, Y.; Yin, G.; Wang, Z.; Gao, Y. Proton exchange membrane fuel cell from lowtemperature to high temperature: Material challenges. J. Power Sources 2007, 167, 235–242. [Google Scholar] [CrossRef]
- Li, Q.; He, R.; Jenson, J.O.; Bjerrum, N.J. PBI-based polymer membranes for high temperature fuel cells—Preparation, Characterization and Fuel Cell Demonstration. Fuel Cells 2004, 4, 147–159. [Google Scholar] [CrossRef]
- Lobato, J.; Cañizares, P.; Rodrigo, M.A.; Linares, J.J.; Manjavacas, G. Synthesis and characterization ofpoly[2,2-(m-phenylene)-5,5-bibenzimidazole] as polymer electrolyte membrane for high-temperature PEMFCs. J. Membr. Sci. 2006, 280, 351–362. [Google Scholar] [CrossRef]
- He, R.; Li, Q.; Bach, A.; Jensen, J.O.; Bjerrum, N.J. Doping phosphoric acid in polybenzimidazole membranes for high temperature proton exchange membrane fuel cells. J. Polym. Sci. A Polym. Chem. 2007, 45, 2989–2997. [Google Scholar] [CrossRef]
- He, R.; Li, Q.; Xiao, G.; Bjerrum, N.J. Proton conductivity of phosphoric acid doped polybenzimidazole and its composites with inorganic proton conductors. J. Membr. Sci. 2003, 226, 169–184. [Google Scholar] [CrossRef]
- Pu, H.; Meyer, W.H.; Wegner, G. Proton transport in polybenzimidazole blended with H3PO4 or H2SO4. J. Polym. Sci. B Polym. Phys. 2002, 40, 663–669. [Google Scholar] [CrossRef]
- Li, Q.; Hjuler, H.A.; Bjerrum, N.J. Phosphoric acid doped polybenzimidazole membranes: Physiochemical characterization and fuel cell applications. J. Appl. Electrochem. 2001, 31, 773–779. [Google Scholar] [CrossRef]
- Weng, D.; Wainright, J.S.; Landau, U.; Savinell, R.F. Electro-osmotic drag coefficient of water and methanol in polymer electrolytes at elevated temperatures. J. Electrochem. Soc. 1996, 143, 1260–1263. [Google Scholar] [CrossRef]
- Xiao, L.; Zhang, H.; Scanlon, E.; Ramanathan, L.S.; Choe, E.W.; Rogers, D.; Apple, T.; Benicewicz, B.C. High-temperature polybenzimidazole fuel cell membranes via a sol-gel process. Chem. Mater. 2005, 17, 5328–5333. [Google Scholar]
- Kim, T.-H.; Lim, T.-W.; Lee, J.-C. High temperature fuel cell membranes based on mechanically stable para-ordered polybenzimidazole prepared by direct casting. J. Power Sources 2007, 172, 172–179. [Google Scholar] [CrossRef]
- Wang, J.-T.; Savinell, R.F.; Wainright, J.; Litt, M.; Yu, H. A H2O2 fuel cell using acid doped polybenzimidazole as polymer electrolyte. Electrochim. Acta 1996, 41, 193–197. [Google Scholar] [CrossRef]
- Samms, S.R.; Wasmus, S.; Savinell, R.F. Thermal stability of proton conducting acid doped polybenzimidazole in simulated fuel cell environments. J. Electrochem. Soc. 1996, 143, 1225–1232. [Google Scholar] [CrossRef]
- Asensio, J.A.; Gómez-Romero, P. Recent developments on proton conducting poly(2,5-benzimidazole) (ABPBI) membranes for high temperature polymer electrolyte membrane fuel cells. Fuel Cells 2005, 5, 336–343. [Google Scholar] [CrossRef]
- Kim, H.J.; Lim, T.H. PBI derivatives: Polymer electrolyte fuel cell membrane for high temperature operation. J. Ind. Eng. Chem. 2004, 10, 1081–1085. [Google Scholar]
- Kim, S.-K.; Kim, T.-H.; Ko, T.; Lee, J.-C. Cross-linked poly(2,5-benzimidazole) consisting of wholly aromatic groups for high-temperature PEM fuel cell applications. J. Membr. Sci. 2011, 373, 80–88. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cho, S.Y.; An, S.J.; Eun, Y.C.; Kim, J.-Y.; Yoon, H.-K.; Kweon, H.-J.; Yew, K.H. Synthesis of poly(2,5-benzimidazole) for use as a fuel cell membrane. Macromol. Rapid Commun. 2004, 25, 894–897. [Google Scholar] [CrossRef]
- Asensio, J.A.; Borrós, S.; Gómez-Romero, P. Polymer electrolyte fuel cells based on phosphoric acid impregnated poly(2,5-benzimidazole) membranes. J. Electrochem. Soc. 2004, 151, A304–A310. [Google Scholar] [CrossRef]
- Kim, T.-H.; Lim, T.-W.; Park, Y.-S.; Shin, K.; Lee, J.-C. Proton-conducting zirconium pyrophosphate/poly(2,5-benzimidazole) composite membranes prepared by a PPA direct casting method. Macromol. Chem. Phys. 2007, 208, 2293–2302. [Google Scholar] [CrossRef]
- Wannek, C.; Lehnert, W.; Mergel, J. Membrane electrode assemblies for high-temperature polymer electrolyte fuel cells based on poly(2,5-benzimidazole) membranes with phosphoric acid impregnation via the catalyst layers. J. Power Sources 2009, 192, 258–266. [Google Scholar] [CrossRef]
- Burke, W.J. 3,4-Dihydro-1,3,2H-Benzoxazines. Reaction of p-substituted phenols with N,N-dimethylolamines. J. Am. Chem. Soc. 1949, 71, 609–612. [Google Scholar] [CrossRef]
- Burke, W.J.; Kolbezen, M.J.; Stephens, C.W. Condensation of naphthols with formaldehyde and primary amines. J. Am. Chem. Soc. 1952, 74, 3601–3605. [Google Scholar] [CrossRef]
- Burke, W.J.; Hammer, C.R.; Weatherbee, C.J. Bis-m-oxazines from Hydroquinone. J. Org. Chem. 1961, 26, 4403–4407. [Google Scholar] [CrossRef]
- Burke, W.J.; Glennie, E.L.M.; Weatherbee, C. Condensationof halophenols with formaldehyde and primary amines. J. Org. Chem. 1964, 29, 909–912. [Google Scholar] [CrossRef]
- Burke, W.J.; Bishop, J.L.; Glennie, E.L.M.; Bauer, W.N., Jr. A new aminoalkylation reaction. Condensation of phenols with dihydro-1,3-aroxazines. J. Org. Chem. 1965, 30, 3423–3427. [Google Scholar]
- Gardziella, A.; Pilato, L.; Knop, A. Pilato, Phenolic Resins, Chemistry, Applications, Standardization, Safety, and Ecology and Performance, Future Directions, 2nd ed; Springer-Verlag: Heidelberg, Germany, 2000. [Google Scholar]
- Ning, X.; Ishida, H. Phenolic materials via ring-opening polymerization of benzoxazines: Effect of molecular structure on mechanical and dynamic mechanical properties. J. Polym. Sci. B Polym. Phys. 1994, 32, 921–927. [Google Scholar] [CrossRef]
- Ning, X.; Ishida, H. Phenolic materials via ring-opening polymerization: Synthesis and characterization of bisphenol—A based benzoxazines and their polymers. J. Polym. Sci. A Polym. Chem. 1994, 32, 1121–1129. [Google Scholar] [CrossRef]
- Ishida, H.; Rodriguez, Y. Curing kinetics of a new benzoxazine-based phenolic resin by differential scanning calorimetry. Polymer 1995, 36, 3151–3158. [Google Scholar] [CrossRef]
- Ishida, H.; Allen, D.J. Physical and mechanical characterization of near-zero shrinkage polybenzoxazines. J. Polym. Sci. B Polym. Phys. 1996, 34, 1019–1030. [Google Scholar] [CrossRef]
- Ishida, H. Process for Preparation of Benzoxazine Compounds in Solventless Systems. U.S. Patent 5,543,516, 6 August 1996. [Google Scholar]
- Ghosh, N.N.; Kiskan, B.; Yagci, Y. Polybenzoxazines—New high performance thermosetting resins: synthesis and properties. Prog. Polym. Sci. 2007, 32, 1344–1391. [Google Scholar] [CrossRef]
- Lee, M.-J.; Choi, S.-W.; Sun, H.-Y.; Jeon, W.-S. Electrolyte Membrane using Polybenzoxazine Based Compound and Method of Manufacturing the Same. U.S. Patent 7,858,668, 5 September 2006. [Google Scholar]
- Choi, S.-W.; Sun, H.-Y.; Lee, M.-J.; Jeon, W.-S. Polybenzoxazine-Based Compound, Electrolyte Membrane Including the Same, and Fuel Cell Employing the Electrolyte Membrane. U.S. Patent 8,034,508, 1 September 2006. [Google Scholar]
- Choi, S.-W.; Sun, H.-Y.; Jeon, W.-S. Polybenzoxazine-Based Compound, Electrolyte Membrane Including the Same, and Fuel Cell Employing the Electrolyte Membrane. U.S. Patent 8,148,028, 3 May 2007. [Google Scholar]
- Choi, S.-W.; Lee, J.-C.; Park, J.O. Cross-Linked Polyazole, Method of Preparing the Polyazole, Electrode for Fuel Cell including the Cross-Linked Polyazole, Electrolyte Membrane for Fuel Cell. U.S. Patent 20100273087A1, 23 April 2010. [Google Scholar]
- Kim, S.-K.; Choi, S.-W.; Jeon, W.S.; Park, J.O.; Ko, T.; Pak, C.; Chang, H.; Lee, J.-C. Cross-linked benzoxazine-benzimidazole copolymer electrolyte membranes for fuel cells at elevated temperature. Macromolecules 2012, 45, 1438–1446. [Google Scholar] [CrossRef]
- Seel, D.C.; Benicewicz, B.C.; Xiao, L.; Schmidt, T.J. High-temperature polybenzimidazole-based membranes. In Handbook of Fuel Cells—Advances in Electorocatalysis, Materials, Diagnostics and Durability; Vielstich, W., Yokokawa, H., Gasteiger, H.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 5, pp. 1–13. [Google Scholar]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Janssen, G.J.M.; Sittersa, E.F.; Pfrang, A. Proton-exchange-membrane fuel cells durability evaluated by load-on/off cycling. J. Power Sources 2009, 191, 501–509. [Google Scholar] [CrossRef]
- Asghari, S.; Akhgar, H.; Imani, B. Design of thermal management subsystem for a 5kW polymer electrolyte membrane fuel cell system. J. Power Sources 2011, 196, 3141–3148. [Google Scholar] [CrossRef]
- Andreasen, S.; Kær, S. Modelling and evaluation of heating strategies for high temperature polymer electrolyte membrane fuel cell stacks. Int. J. Hydrog. Energy 2008, 33, 4655–4664. [Google Scholar] [CrossRef]
- Pfeifer, P.; Wall, C.; Jensen, O.; Hahn, H.; Fichtner, M. Thermal coupling of a high temperature PEM fuel cell with a complex hydride tank. Int. J. Hydrog. Energy 2009, 34, 3457–3466. [Google Scholar] [CrossRef]
- Song, T.W.; Choi, K.H.; Kim, J.R.; Yi, J.S. Pumpless thermal management of water-cooled high-temperature proton exchange membrane fuel cell. J. Power Sources 2011, 196, 4671–4679. [Google Scholar] [CrossRef]
- Asensio, J.A.; Sánchez, E.M.; Gómez-Romero, P. Proton-conducting membranes based on benzimidazole polymers for high-temperature PEM fuel cells. A chemical quest. Chem. Soc. Rev. 2010, 39, 3210–3239. [Google Scholar] [CrossRef]
- Kim, S.-K.; Ko, T.; Choi, S.-W.; Park, J.O.; Kim, K.-H.; Pak, C.; Chang, H.; Lee, J.-C. Durable cross-linked copolymer membranes based on poly(benzoxazine) and poly(2,5-benzimidazole) for use in fuel cells at elevated temperatures. J. Mater. Chem. 2012, 22, 7194–7205. [Google Scholar]
- Kim, S.-K.; Kim, T.-H.; Jung, J.-W.; Lee, J.-C. Copolymers of poly(2,5-benzimidazole) and poly[2,2-bep-phenylene)-5,5'-bibenzimidazole] for high-temperature fuel cell applications. Macromol. Mater. Eng. 2008, 293, 914–921. [Google Scholar] [CrossRef]
- Antalek, B. Using pulsed gradient spin echo NMR for chemical mixture analysis: How to obtain optimum results. Concepts Magn. Reson. 2002, 14, 225–258. [Google Scholar] [CrossRef]
- Chen, A.; Wu, D.; Johnson, C.S., Jr. Determination of molecular weight distributions for polymers by diffusion-ordered NMR. J. Am. Chem. Soc. 1995, 117, 7965–7970. [Google Scholar]
- Viel, S.; Capitani, D.; Mannina, L.; Segre, A. Diffusion-ordered NMR spectroscopy: Aversatile tool for the molecular weight determination of uncharged polysaccharides. Biomacromolecules 2003, 4, 1843–1847. [Google Scholar] [CrossRef]
- Jayawickrama, D.A.; Larive, C.K.; Macord, E.F.; Roe, D.C. Polymer additives mixture analysis using pulsed-field gradient NMR spectroscopy. Magn. Reson. Chem. 1998, 36, 755–760. [Google Scholar] [CrossRef]
- Nishinari, K.; Kohyama, K.; Williams, P.A.; Phillips, G.O.; Burchard, W.; Ogino, K. Solution properties of pullulan. Macromolecules 1991, 24, 5590–5593. [Google Scholar] [CrossRef]
- Gorkom, V.; Leon, C.M.; Hancewicz, T.M. Analysis of DOSY and GPC-NMR experiments on polymers by multivariate curve resolution. J. Magn. Reson. 1998, 130, 125–130. [Google Scholar]
- Bovey, F.A.; Mirau, P.A. The solution characterization of polymers. In NMR of Polymers, 1st ed; Academic Press, Inc.: San Diego, CA, USA, 1996; Volume 3, pp. 155–242. [Google Scholar]
- Wang, Y.-X.; Ishida, H. Synthesis and properties of new thermoplastic polymers from substituted 3,4-dihydro-2H-1,3-benzoxazines. Macromolecules 2000, 33, 2839–2847. [Google Scholar] [CrossRef]
- Leykin, A.Y.; Fomenkov, A.I.; Galpern, E.G.; Stankevich, I.V.; Rusanov, A.L. Some aspects of polybenzimidazoles’ synthesis in P2O5 containing condensation media. Polymer 2010, 51, 4053–4057. [Google Scholar] [CrossRef]
- Oh, S.-Y.; Yoshida, T.; Kawamura, G.; Muto, H.; Sakai, M.; Matsuda, A. Inorganic-organic composite electrolytes consisting of polybenzimidazole and Cs-substituted heteropoly acids and their application for medium temperature fuel cells. J. Mater. Chem. 2010, 20, 6359–6366. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, Y.; Song, C.; Zhang, J. Polybenzimidazole-membrane-based PEM fuel cell in the temperature range of 120-200 °C. J. Power Sources 2007, 172, 163–171. [Google Scholar] [CrossRef]
- Li, Q.; Pan, C.; Jensen, J.O.; Noyé, P.; Bjerrum, N.J. Cross-linked polybenzimidazole membranes for fuel cells. Chem. Mater. 2007, 19, 350–352. [Google Scholar] [CrossRef]
- Marrony, M.; Barrer, R.; Quenet, S.; Ginocchio, S.; Montelatici, L.; Aslanides, A. Durability study and lifetime prediction of baseline proton exchange membrane fuel cell under severe operating conditions. J. Power Sources 2008, 182, 469–475. [Google Scholar] [CrossRef]
- Pei, P.; Chang, Q.; Tang, T. A quick evaluating method for automotive fuel cell lifetime. Int. J. Hydrog. Energy 2008, 33, 3829–3836. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, X.; Wang, H.; Mérida, W.; Zhu, H.; Shen, J.; Wu, S.; Zhang, J. A review of accelerated stress tests of MEA durability in PEM fuel cells. Int. J. Hydrog. Energy 2009, 34, 388–404. [Google Scholar] [CrossRef]
- Zelenay, P.; Scharifker, B.R.; O’M Bockris, J.; Gervasio, D. A comparison of the properties of CF3SO3H and H3PO4 in relation to fuel cells. J. Electrochem. Soc. 1986, 133, 2262–2267. [Google Scholar] [CrossRef]
- Razaq, M.; Razaq, A.; Yeager, E.; DesMarteau, D.D.; Singh, S. Perfluorosulfonimide as an additive in phosphoric acid fuel cell. J. Electrochem. Soc. 1989, 136, 385–390. [Google Scholar] [CrossRef]
- Hsueh, K.-L.; Gonzolez, E.R.; Srinivasan, S. Effects of phosphoric acid concentration on oxygen reduction kinetics at platinum. J. Electrochem. Soc. 1984, 131, 823–828. [Google Scholar] [CrossRef]
- Qingfeng, L.; Hjuler, H.A.; Bjerrum, N.J. Oxygen reduction on carbon supported platinum catalysts in high temperature polymer electrolytes. Electrochim. Acta 2000, 45, 4219–4226. [Google Scholar] [CrossRef]
- Kanamura, K.; Tanaka, A.; Gervasio, D.; Kennedy, V.; Adzic, R.; Yeager, E.B. Perfluoro-ethylene-1,2-bis-phosphonic acid fuel cell electrolyte. J. Electrochem. Soc. 1996, 143, 2765–2770. [Google Scholar] [CrossRef]
- Seland, F.; Berning, T.; Borresen, B.; Tunold, R. Improving the performance of high-temperature PEM fuel cells based on PBI electrolyte. J. Power Sources 2006, 160, 27–36. [Google Scholar] [CrossRef]
- Zecevic, S.K.; Wainright, J.S.; Litt, M.H.; Gojkovic, S.L.; Savinell, R.F. Kinetics of O2 reduction on a Pt electrode covered with a thin film of solid polymer electrolyte. J. Electrochem. Soc. 1997, 144, 2973–2982. [Google Scholar] [CrossRef]
- Liu, Z.; Wainright, J.S.; Litt, M.H.; Savinell, R.F. Study of the oxygen reduction reaction (ORR) at Pt interfaced with phosphoric acid doped polybenzimidazole at elevated temperature and low relative humidity. Electrochim. Acta 2006, 51, 3914–3923. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Baurmeister, J. Properties of high-temperature PEFC Celtec®-P 1000 MEAs in start/stop operation mode. J. Power Sources 2008, 176, 428–434. [Google Scholar] [CrossRef]
- Park, J.O.; Choi, S.-W.; Jeon, W.-S.; Yi, J.-S.; Oemer, U. A Composition Containing a Uniformly Dispersed Polyoxazine-Based Compound, a Method of Preparing the Composition, an Electrode Including the Composition, and a Fuel Cell Including the Electrode. U.S. Patent 0217627A1, 3 March 2011. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choi, S.-W.; Park, J.O.; Pak, C.; Choi, K.H.; Lee, J.-C.; Chang, H. Design and Synthesis of Cross-Linked Copolymer Membranes Based on Poly(benzoxazine) and Polybenzimidazole and Their Application to an Electrolyte Membrane for a High-Temperature PEM Fuel Cell. Polymers 2013, 5, 77-111. https://doi.org/10.3390/polym5010077
Choi S-W, Park JO, Pak C, Choi KH, Lee J-C, Chang H. Design and Synthesis of Cross-Linked Copolymer Membranes Based on Poly(benzoxazine) and Polybenzimidazole and Their Application to an Electrolyte Membrane for a High-Temperature PEM Fuel Cell. Polymers. 2013; 5(1):77-111. https://doi.org/10.3390/polym5010077
Chicago/Turabian StyleChoi, Seong-Woo, Jung Ock Park, Chanho Pak, Kyoung Hwan Choi, Jong-Chan Lee, and Hyuk Chang. 2013. "Design and Synthesis of Cross-Linked Copolymer Membranes Based on Poly(benzoxazine) and Polybenzimidazole and Their Application to an Electrolyte Membrane for a High-Temperature PEM Fuel Cell" Polymers 5, no. 1: 77-111. https://doi.org/10.3390/polym5010077