Controlled Release of Damascone from Poly(styrene-co-maleic anhydride)-based Bioconjugates in Functional Perfumery

Abstract
:1. Introduction
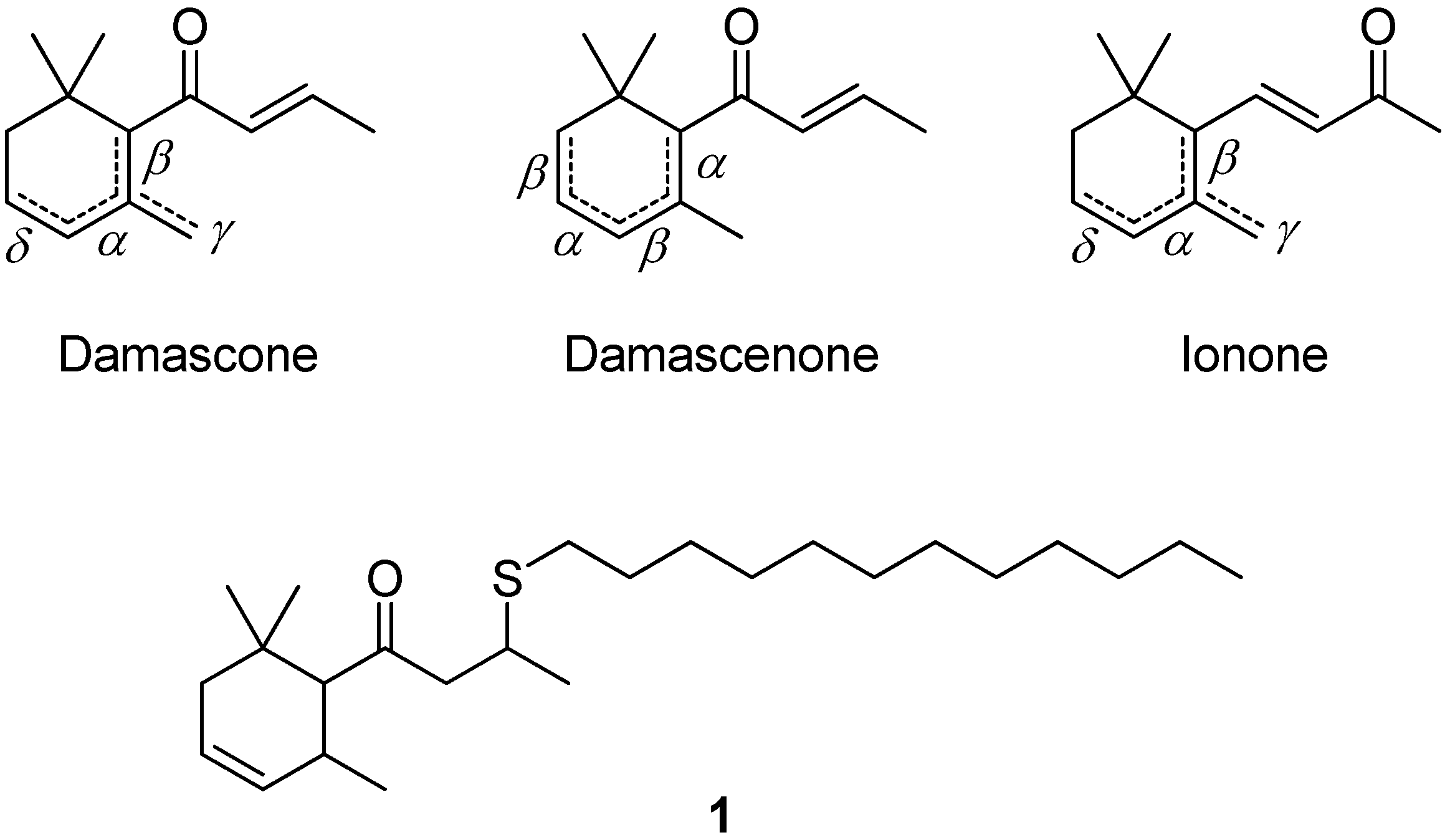
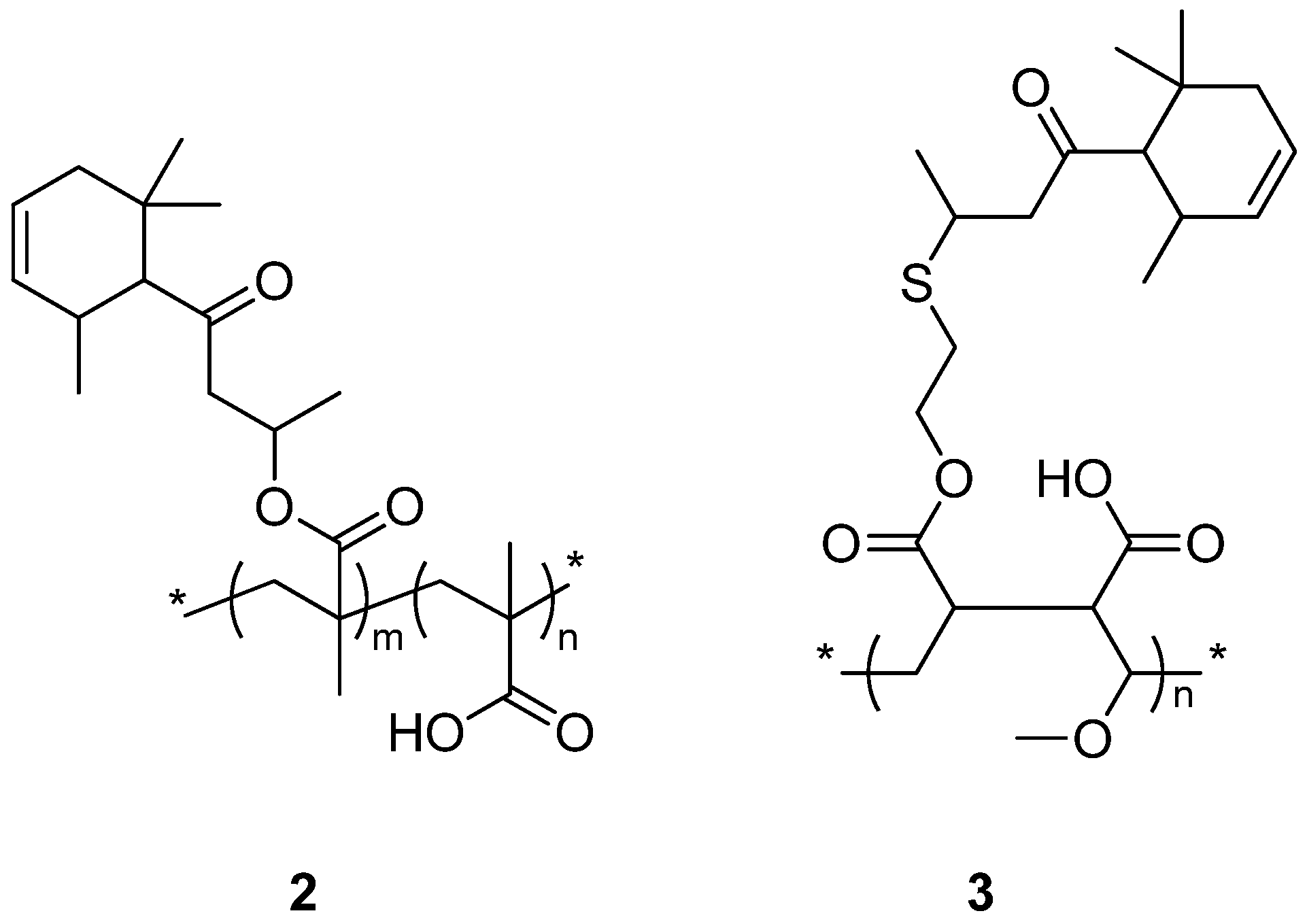
2. Experimental Section
2.1. General
 of weak (w), medium (m) or strong (s) bands in cm−1. 1H- and 13C-nuclear magnetic resonance (NMR) spectra: Bruker 400 MHz Avance III spectrometer; δ in ppm downfield from Me4Si as internal standard. Standard pulse sequences and parameters were used for one-dimensional 1H- and 13C-NMR experiments and for two-dimensional, gradient selected correlation spectroscopy (COSY), 1H,13C-HSQC and 1H,13C-heteronuclear multiple bond correlation (HMBC) experiments, respectively.
of weak (w), medium (m) or strong (s) bands in cm−1. 1H- and 13C-nuclear magnetic resonance (NMR) spectra: Bruker 400 MHz Avance III spectrometer; δ in ppm downfield from Me4Si as internal standard. Standard pulse sequences and parameters were used for one-dimensional 1H- and 13C-NMR experiments and for two-dimensional, gradient selected correlation spectroscopy (COSY), 1H,13C-HSQC and 1H,13C-heteronuclear multiple bond correlation (HMBC) experiments, respectively. 2.2. Analytical Size-Exclusion Chromatography (SEC)
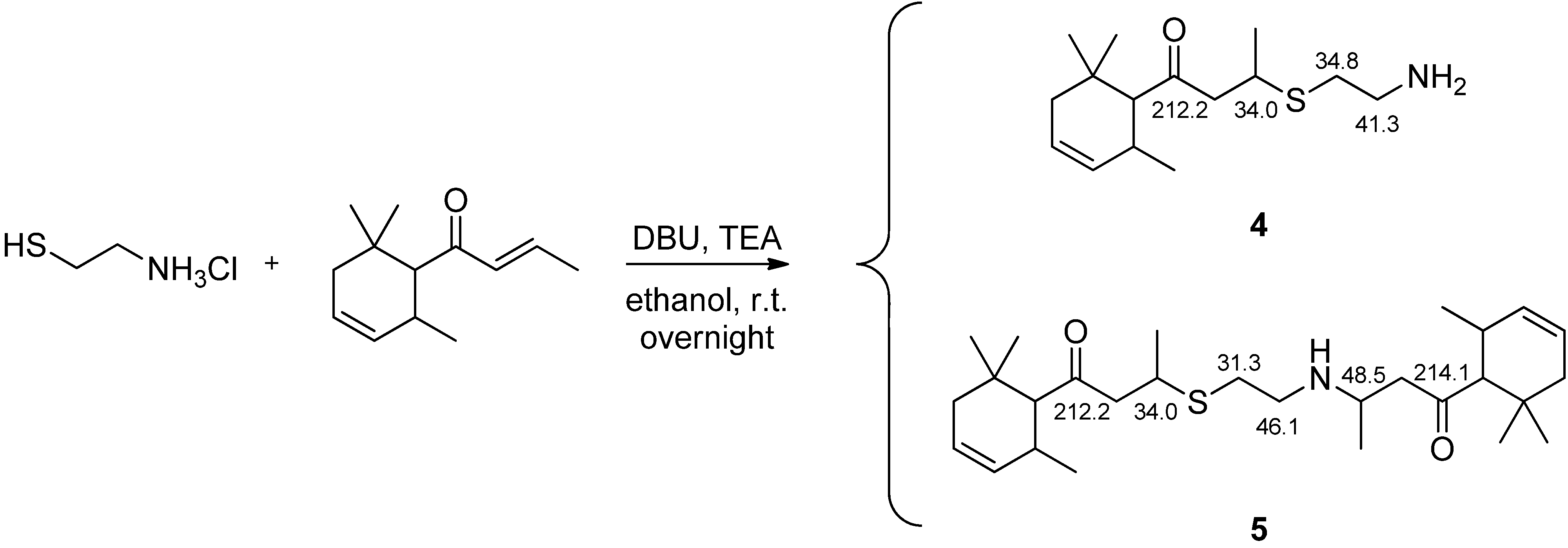
2.3. Preparation of 3-(2-Aminoethylthio)-1-(2,6,6-trimethyl-cyclohex-3-enyl)-butan-1-one (4) and 3-((2-((4-oxo-4-(2,6,6-Trimethylcyclohex-3-en-1-yl)butan-2-yl)amino)ethyl)thio)-1-(2,6,6-trimethylcyclohex-3-en-1-yl)butan-1-one (5)
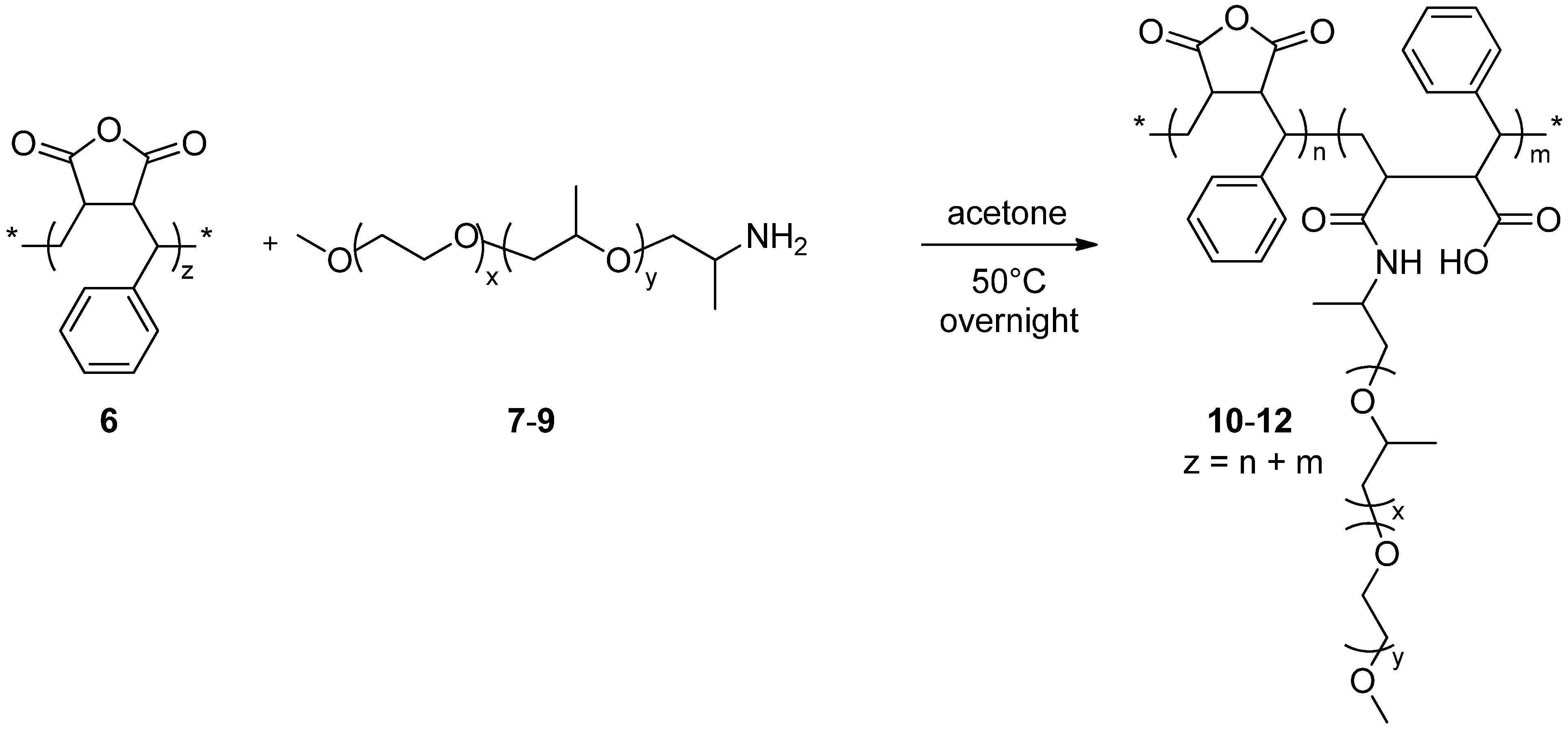
2.4. General Procedure to Prepare Copolymers of Styrene, Maleic Anhydride and 4-oxo-4-({ω-(2-Methoxyethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid
2.4.1. Copolymer of Styrene, Maleic Anhydride and 4-oxo-4-({ω-(2-Methoxyethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid (10) (Obtained by Grafting of Copolymer 7 with EO/PO = 0.11)
2.4.2. Copolymer of Styrene, Maleic Anhydride and 4-oxo-4-({ω-(2-Methoxyethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid (11) (Obtained by Grafting of Copolymer 8 with EO/PO = 0.14)
2.4.3. Copolymer of Styrene, Maleic Anhydride and 4-oxo-4-({ω-(2-Methoxyethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid (12) (Obtained by Grafting of Copolymer 9 with EO/PO = 3.60)
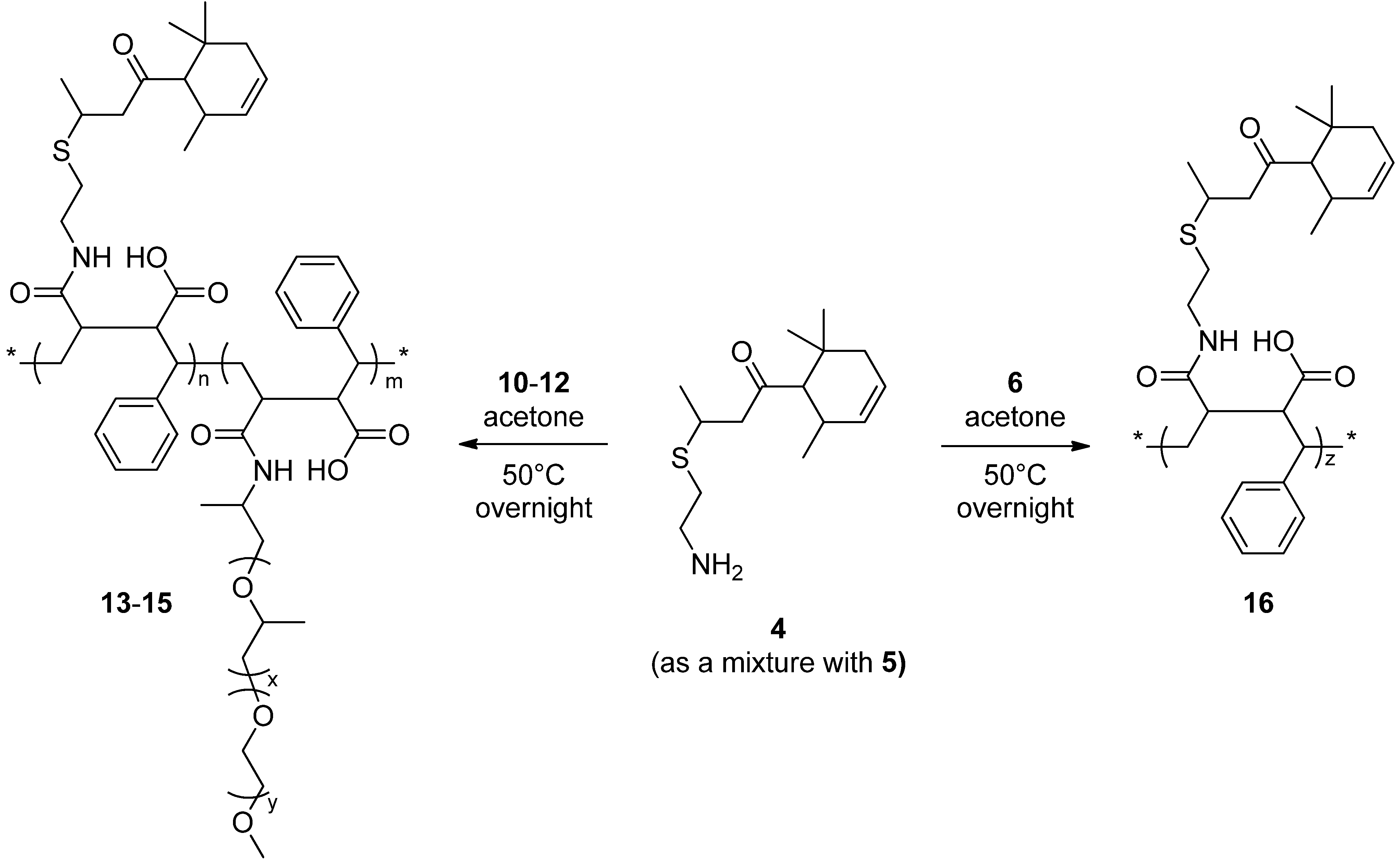
2.5. General Procedure to Prepare Copolymers of Styrene, 4-oxo-4-({ω-(2-Methoxy ethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid and 4-oxo-4-[{2-([4-oxo-4-(2,6,6-trimethylcyclohex-3-en-1-yl)butan-2-yl]thio)ethyl}amino]but-2-enoic Acid
2.5.1. Copolymer of Styrene, 4-oxo-4-({ω-(2-Methoxy ethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid (EO/PO = 0.11) and 4-oxo-4-[{2-([4-oxo-4-(2,6,6-Trimethylcyclohex-3-en-1-yl)butan-2-yl]thio)ethyl}amino]but-2-enoic Acid (13)
2.5.2. Copolymer of Styrene, 4-oxo-4-({ω-(2-Methoxy ethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid (EO/PO = 0.14) and 4-oxo-4-[{2-([4-oxo-4-(2,6,6-Trimethylcyclohex-3-en-1-yl)butan-2-yl]thio)ethyl}amino]but-2-enoic Acid (14)
2.5.3. Copolymer of Styrene, 4-oxo-4-({ω-(2-Methoxy ethyl)[poly(ethylene oxide)-co-poly(propylene oxide)]ethyl}amino)but-2-enoic Acid (EO/PO = 3.60) and 4-oxo-4-[{2-([4-oxo-4-(2,6,6-Trimethylcyclohex-3-en-1-yl)butan-2-yl]thio)ethyl}amino]but-2-enoic Acid (15)
2.5.4. Copolymer of Styrene and 4-oxo-4-[{2-([4-oxo-4-(2,6,6-Trimethylcyclohex-3-en-1-yl)butan-2-yl]thio)ethyl}amino]but-2-enoic Acid (16)
2.6. Preparation of Aqueous Surfactant Emulsions
2.7. Procedure for Polymer Deposition on Cotton and Dynamic Headspace Analysis [27]
3. Results and Discussion
3.1. Preparation of the δ-Damascone-Releasing Unit
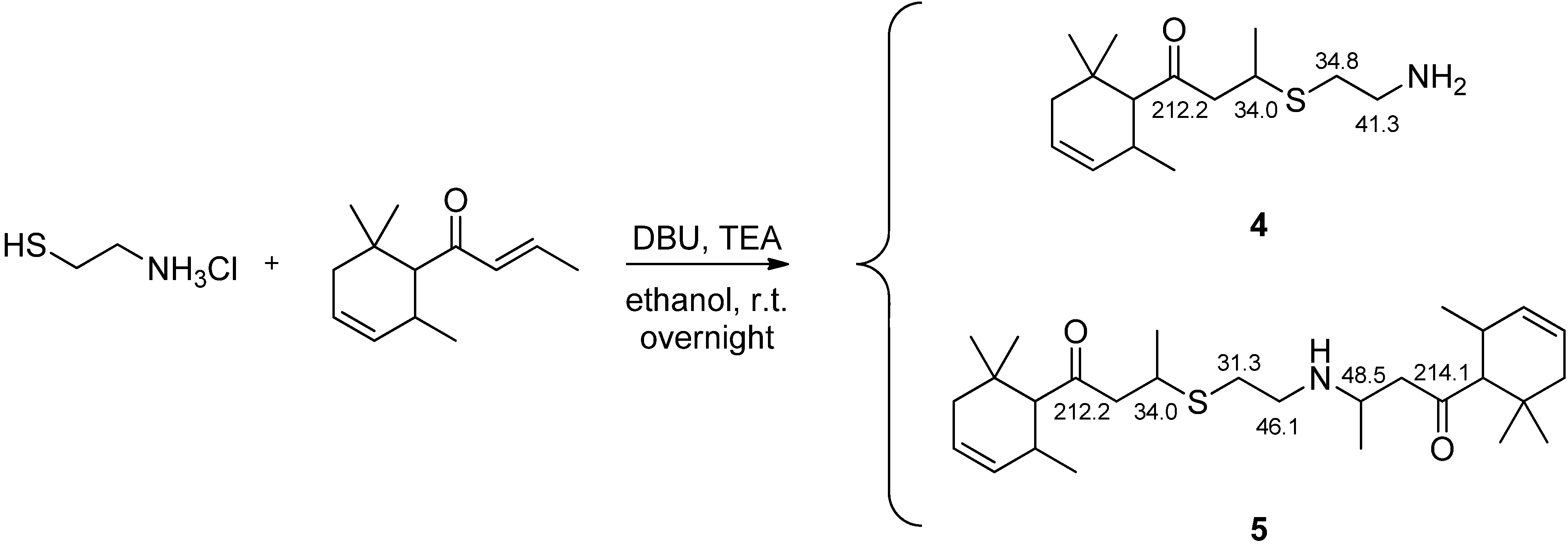
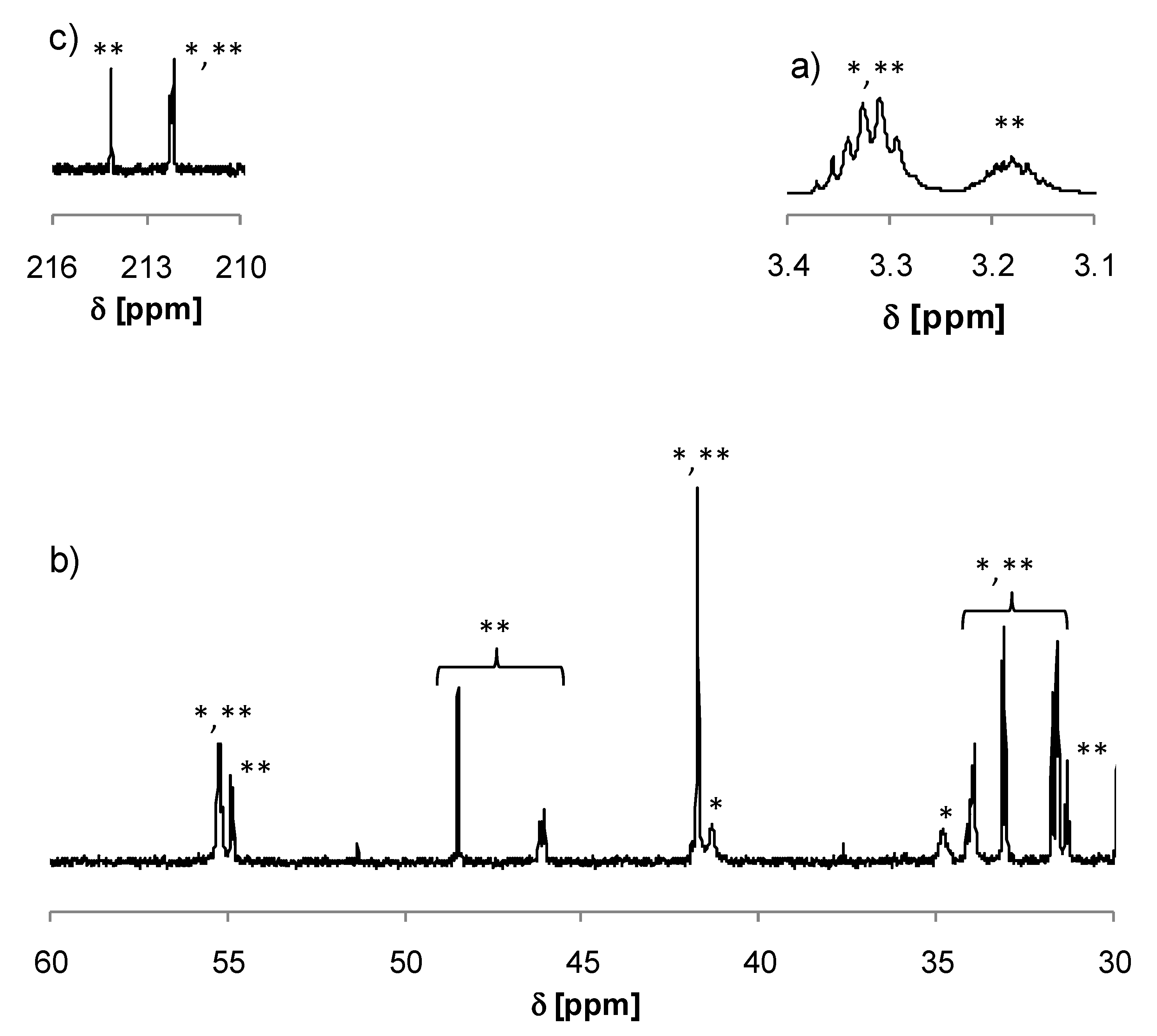
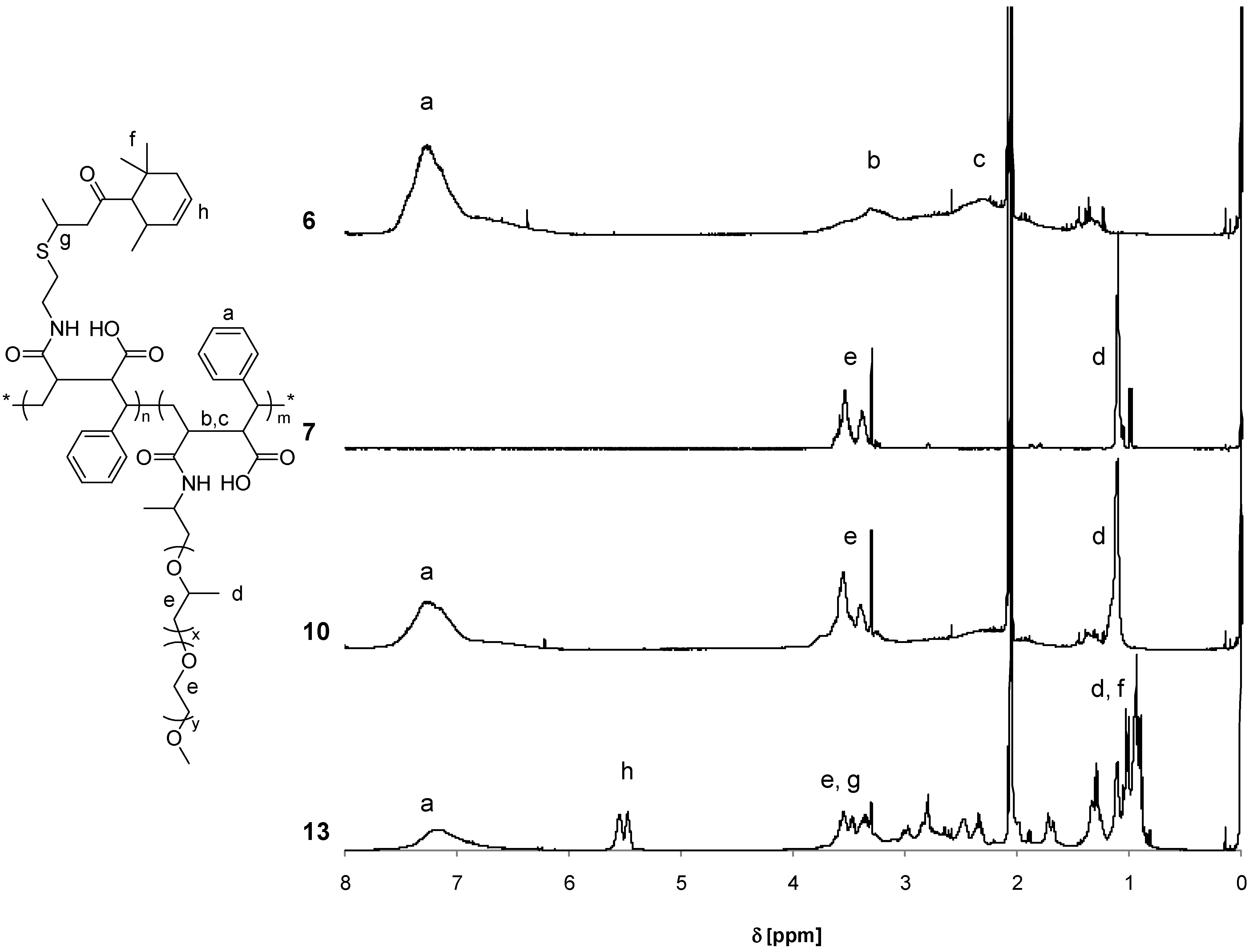
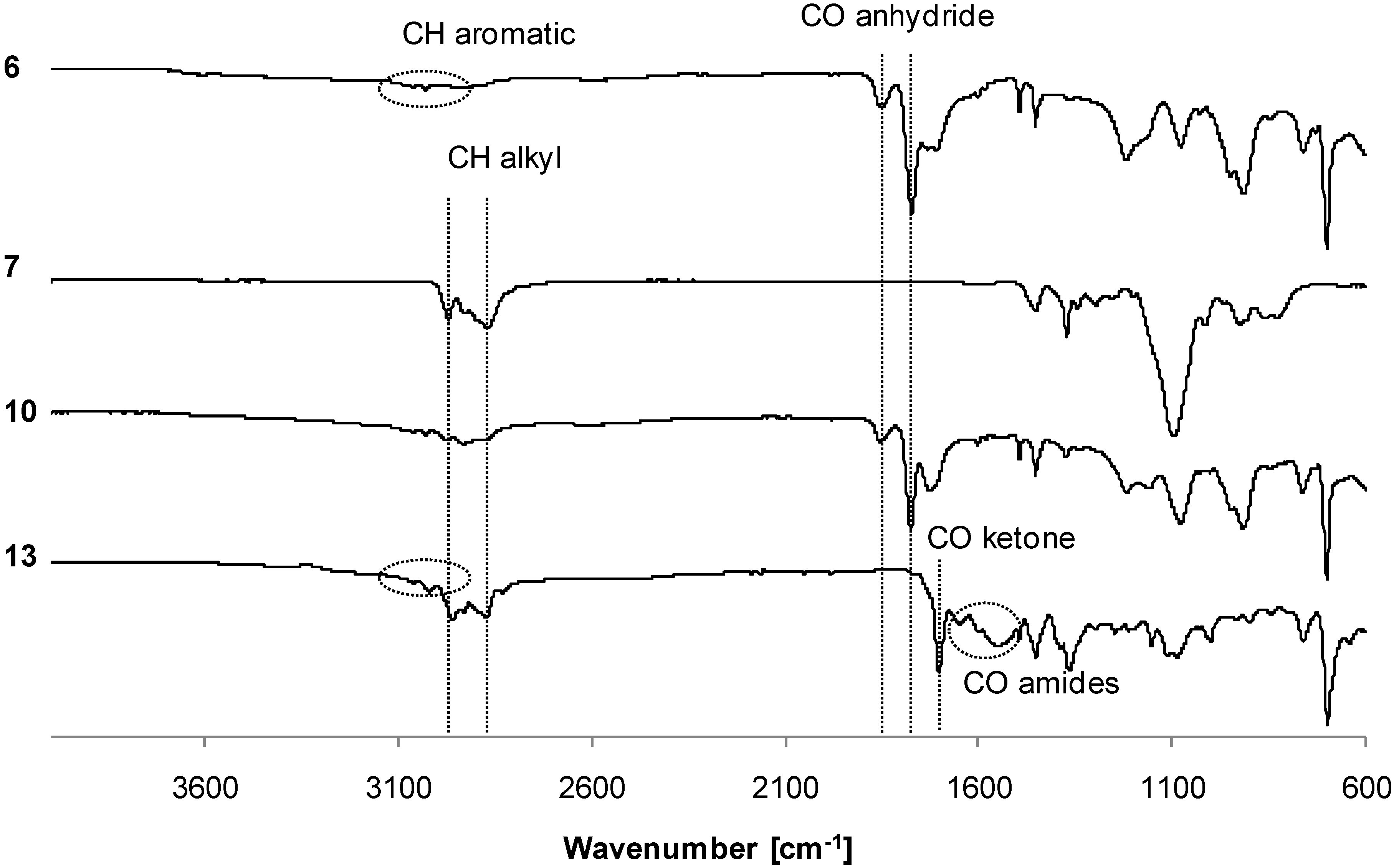
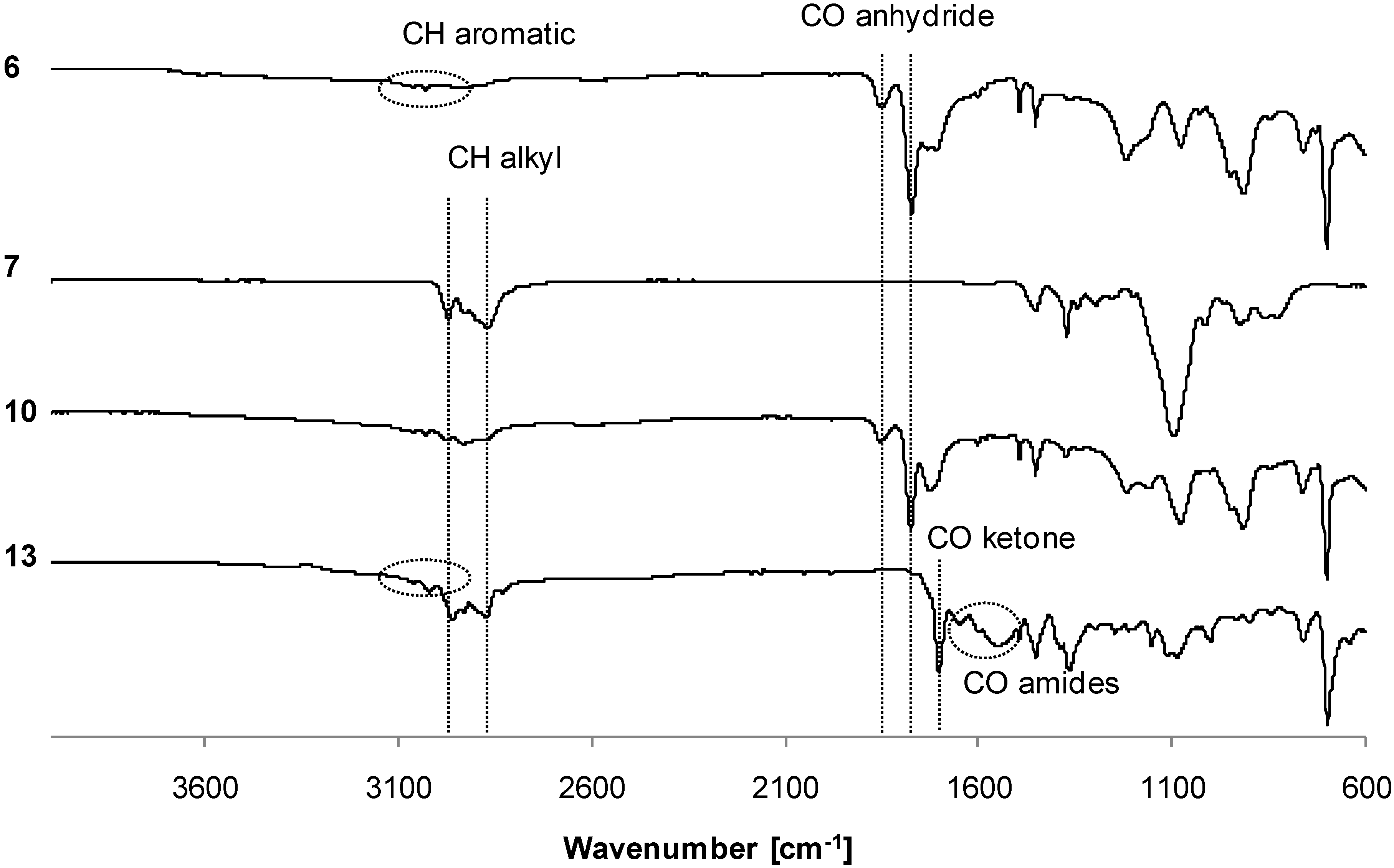
3.2. Synthesis of Copolymers
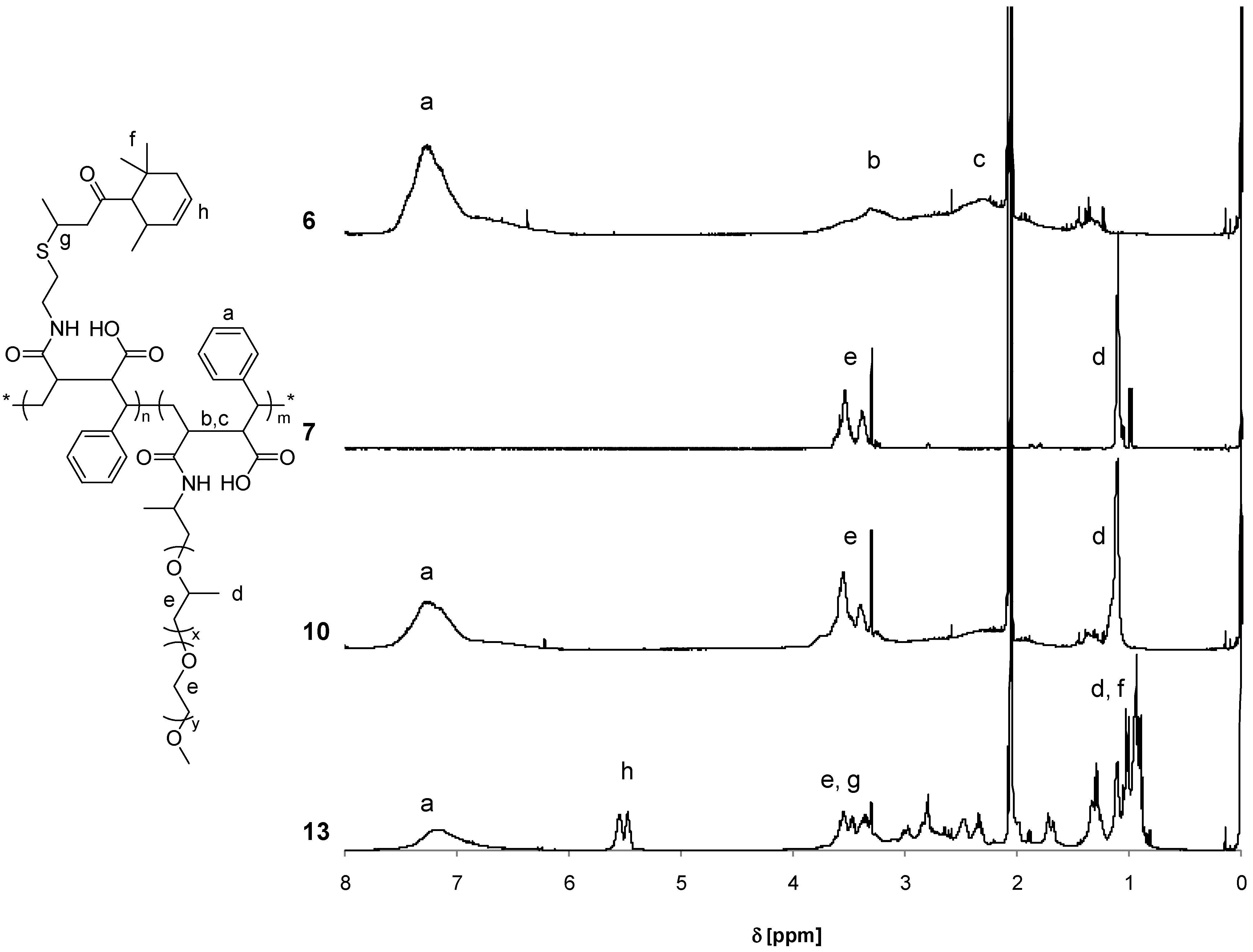

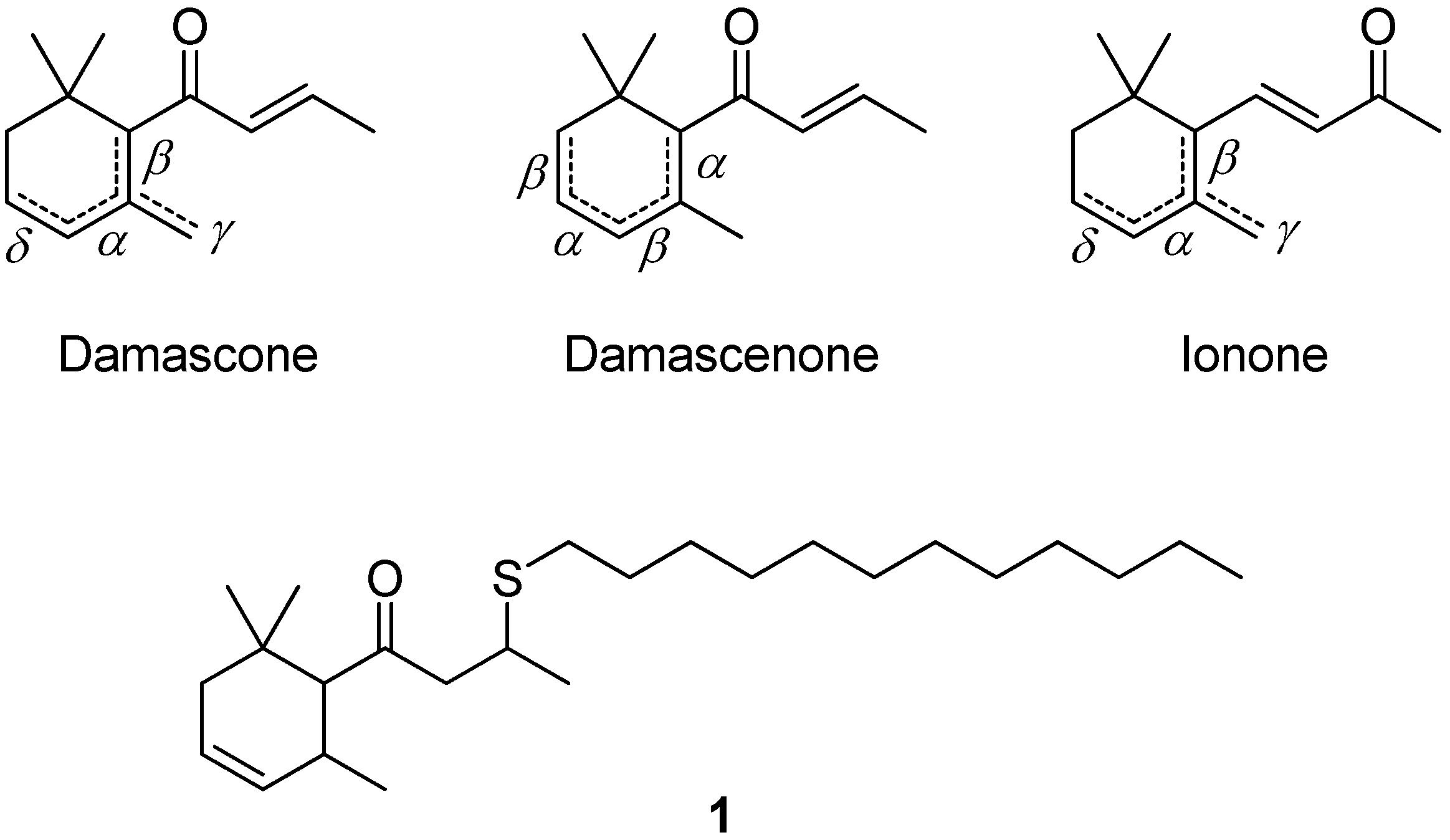{kind=link}
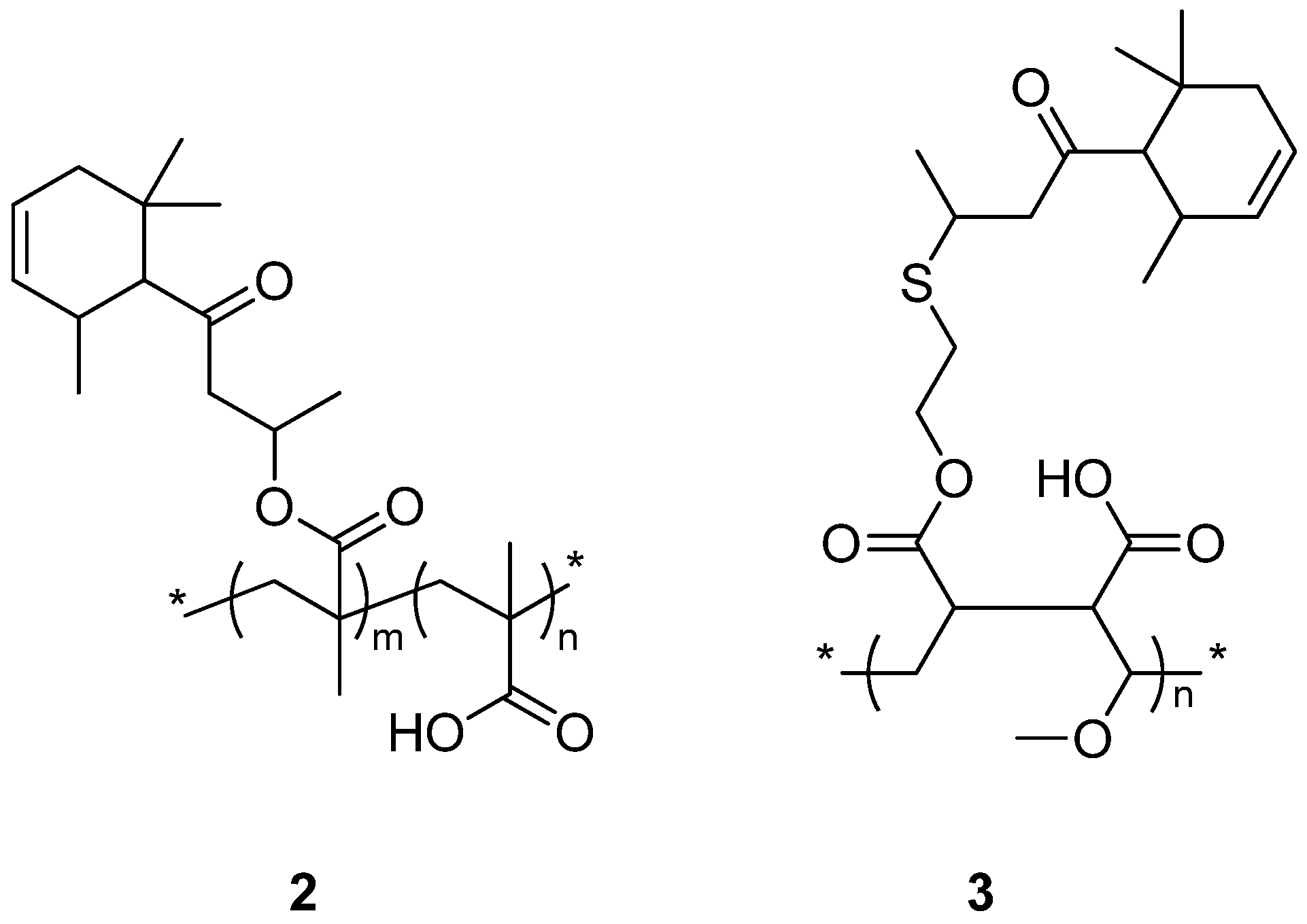{kind=link}
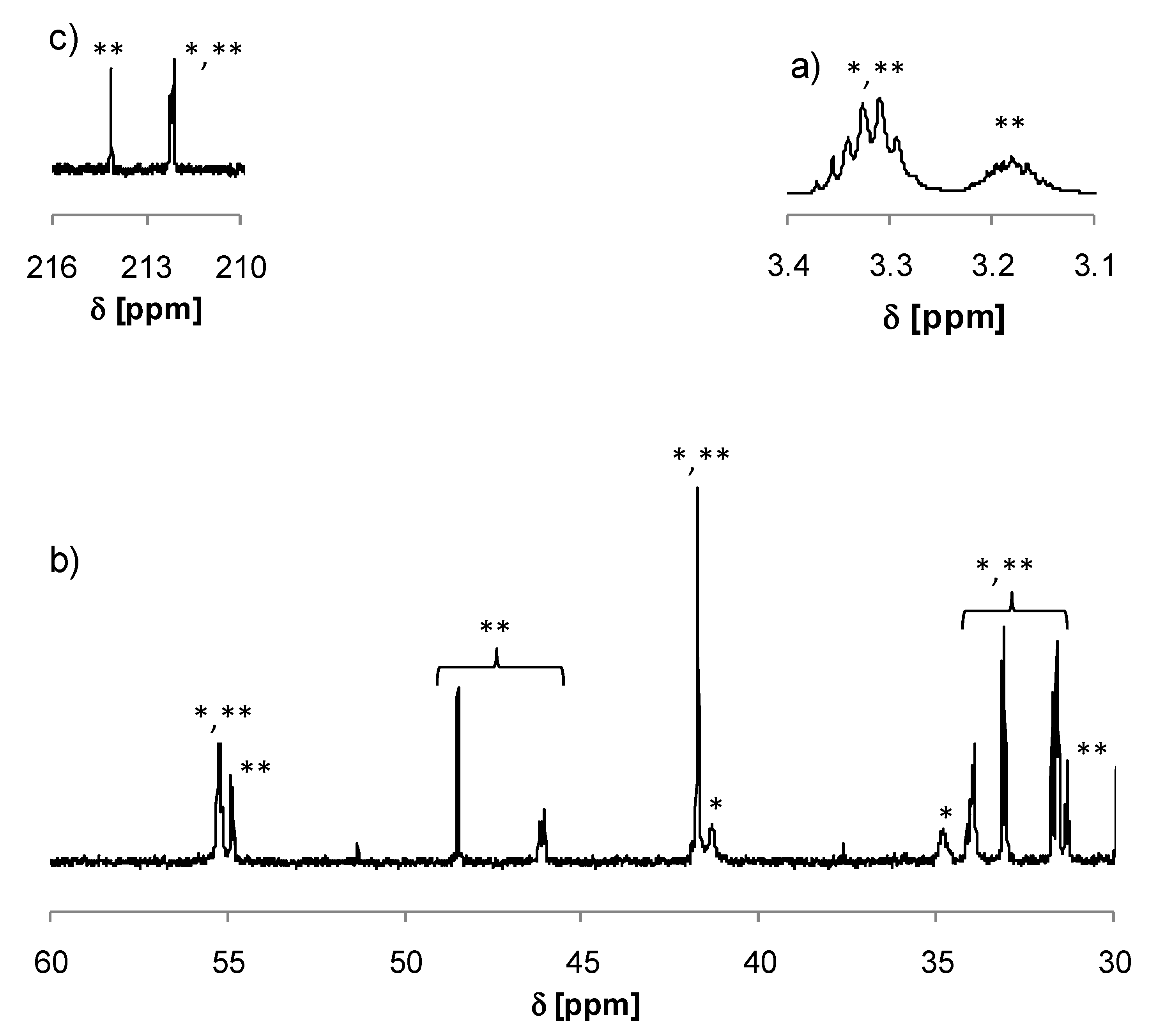{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Jeffamine® | From supplier | Measured by NMR | ||||
|---|---|---|---|---|---|---|---|
| n EO | n PO | EO/PO | n EO | n PO | EO/PO | ||
| 7 | M600 | 1 | 9 | 0.11 | 1 | 9 | 0.11 |
| 8 | M2005 | 6 | 29 | 0.21 | 6 | 42 | 0.14 |
| 9 | M2070 | 31 | 10 | 3.10 | 36 | 10 | 3.60 |

| Copolymer | Mn | Mw | PDI | Copolymer | Mn | Mw | PDI |
|---|---|---|---|---|---|---|---|
| 6 | 2100 | 2800 | 1.33 | 13 | 3100 | 5800 | 1.87 |
| 10 | 2600 | 4800 | 1.85 | 14 | 3900 | 9000 | 2.31 |
| 11 | 3600 | 7600 | 2.11 | 15 | 3500 | 7000 | 2.00 |
| 12 | 3560 | 6300 | 1.77 | 16 | 3200 | 7100 | 2.22 |
| Copolymer | wt % δ-damascone |
|---|---|
| 13 | 32.3 |
| 14 | 25.6 |
| 15 | 25.6 |
| 16 | 38.3 |
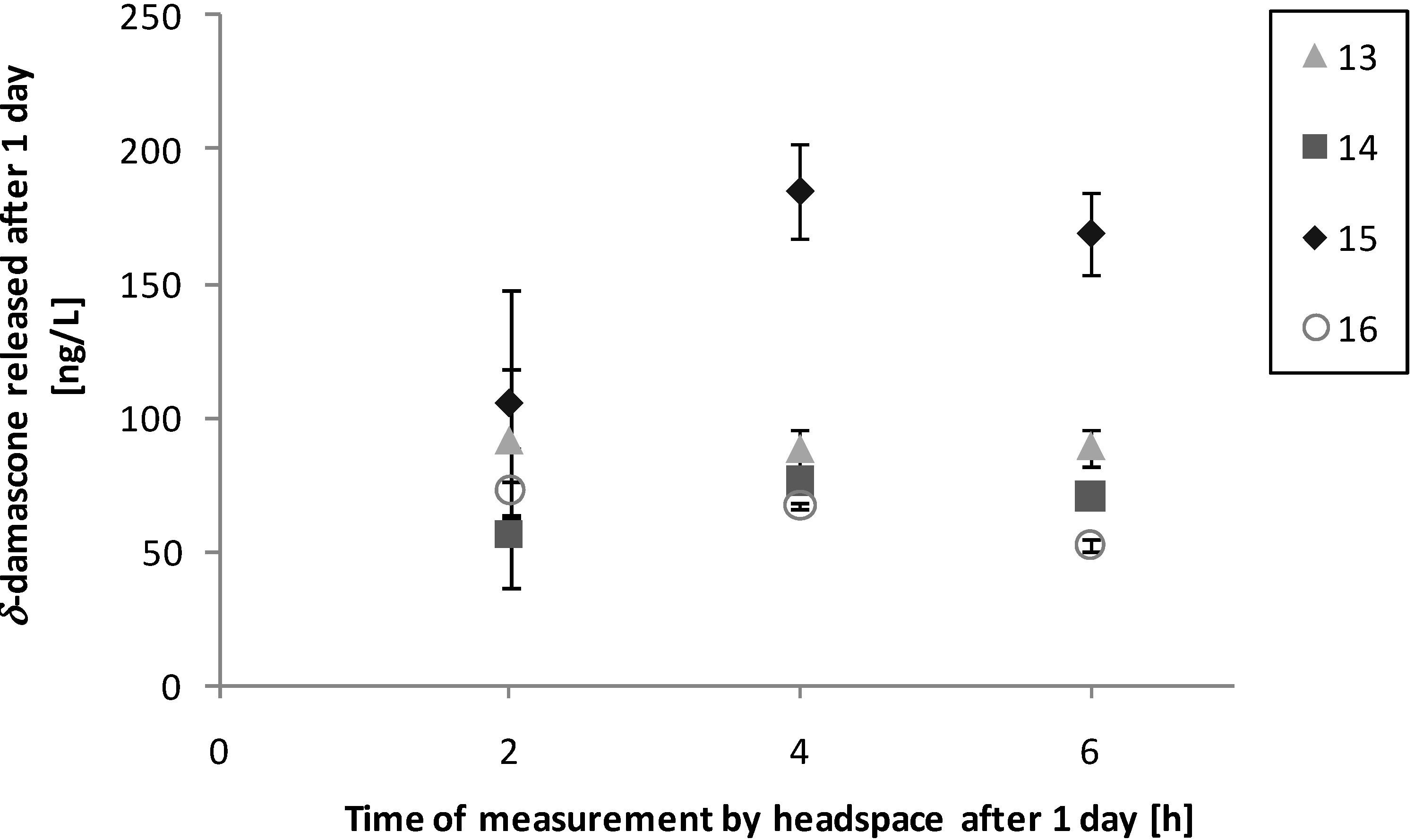
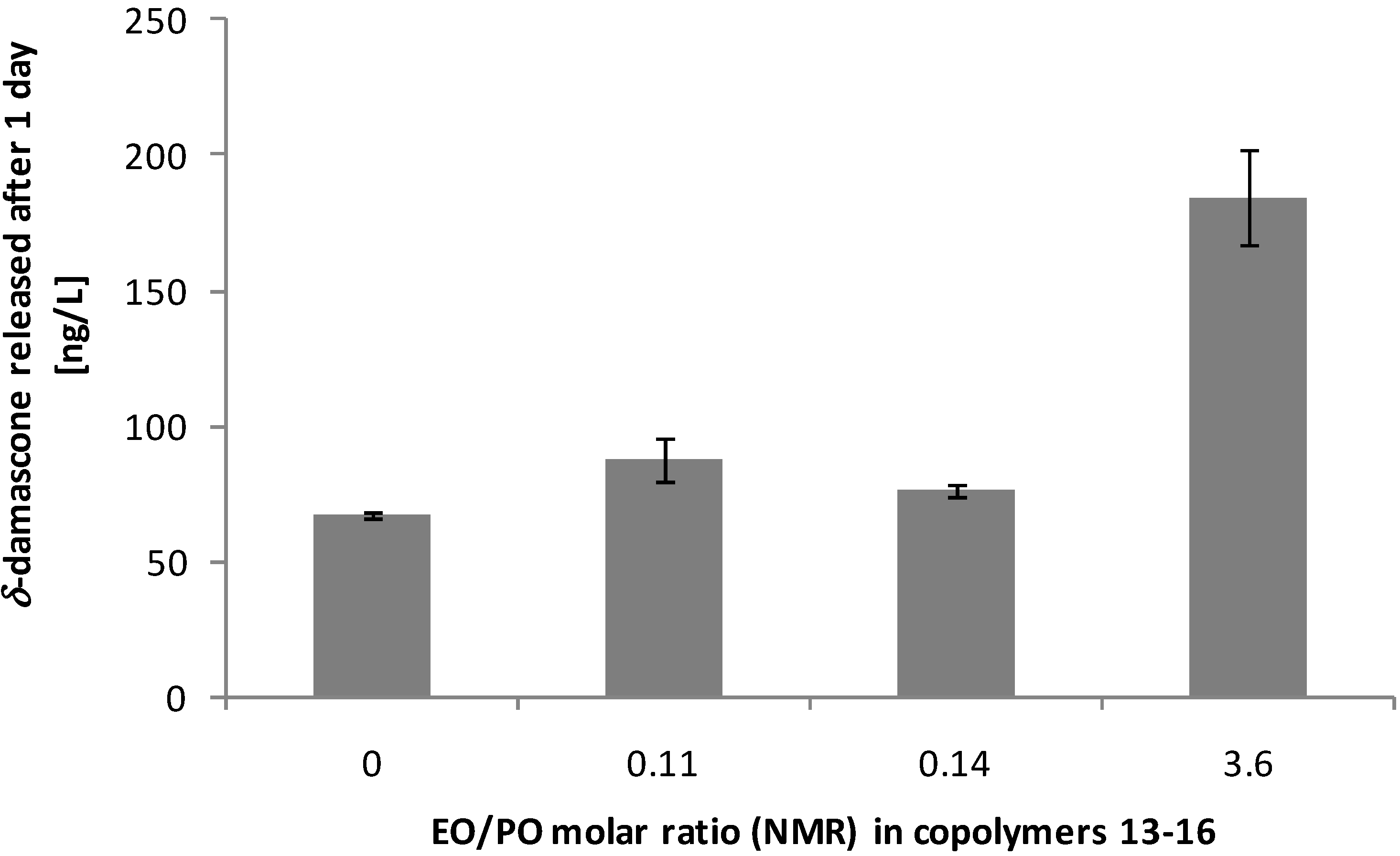
3.3. Performance on Fabric
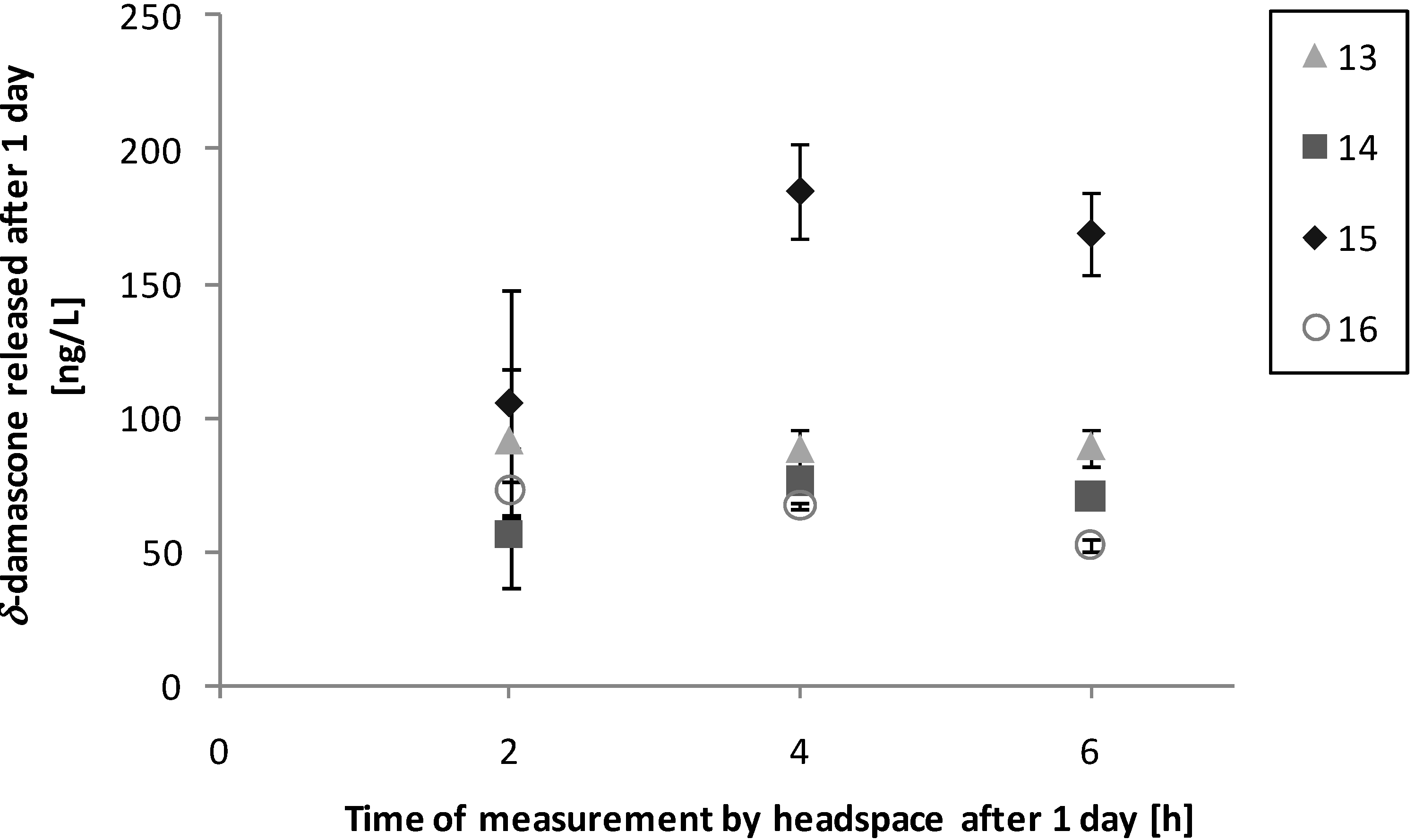

4. Conclusions
References
- Ohloff, G.; Pickenhagen, W.; Kraft, P. Scent and Chemistry: The Molecular World of Odors; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Herrmann, A. The Chemistry and Biology of Volatiles; John Wiley & Sons: Chichester, UK, 2010. [Google Scholar]
- Berger, R.G. Flavours and Fragrances—Chemistry, Bioprocessing and Sustainability; Springer-Verlag: Berlin, Germany, 2007. [Google Scholar]
- Sell, C. The Chemistry of Fragrances—From Perfumer to Consumer, 2nd ed; Royal Society of Chemistry: Cambridge, UK, 2006. [Google Scholar]
- Surburg, H.; Panten, J. Common Fragrance and Flavor Materials: Preparation, Properties and Uses, 5th ed; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Friberg, S.E. Fragrance compounds and amphiphilic association structures. Adv. Colloid Interface Sci. 1998, 75, 181–214. [Google Scholar] [CrossRef]
- Zhang, Z.; Friberg, S.E.; Aikens, P.A. Change of amphiphilic association structures during evaporation from emulsions in surfactant-fragrance-water systems. Int. J. Cosmet. Sci. 2000, 22, 181–199. [Google Scholar] [CrossRef]
- Milotic, D. The impact of fragrance on consumer choice. J. Consum. Behav. 2003, 3, 179–191. [Google Scholar] [CrossRef]
- Herrmann, A. Controlled release of volatiles under mild reaction conditions: From nature to everyday products. Angew. Chem. Int. Ed. 2007, 46, 5836–5863. [Google Scholar] [CrossRef]
- Herrmann, A. Profragrances and properfumes. In The Chemistry and Biology of Volatiles; John Wiley & Sons: Chichester, UK, 2010; pp. 333–362. [Google Scholar]
- Perry, R.J. “Pro-fragrant” silicone delivery polymers. In Delivery System Handbook for Personal Care and Cosmetic Products: Technology, Applications, and Formulations; Rosen, M.R., Ed.; William Andrew Publishing: Norwich, UK, 2005; pp. 667–682. [Google Scholar]
- Herrmann, A. Using photolabile protecting groups for the controlled release of bioactive volatiles. Photochem. Photobiol. Sci. 2012, 11, 446–459. [Google Scholar] [CrossRef]
- Derrer, S.; Flachsmann, F.; Plessis, C.; Stang, M. Applied photochemistry—Light controlled perfume release. Chimia 2007, 61, 665–669. [Google Scholar] [CrossRef]
- Schilling, B.; Kaiser, R.; Natsch, A.; Gautschi, M. Investigation of odors in the fragrance industry. Chemoecology 2010, 20, 135–147. [Google Scholar] [CrossRef]
- Rataj, V.; Ruyffelaere, F.; Aubry, J.-M. Libération contrôlée de molécules de parfum à partir de précurseurs. Cah. Formul. 2005, 12, 82–96. [Google Scholar]
- Kamogawa, H.; Mukai, H.; Nakajima, Y.; Nanasawa, M. Chemical release control—Schiff bases of perfume aldehydes and aminostyrenes. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 3121–3129. [Google Scholar] [CrossRef]
- Kamogawa, H.; Haramoto, Y.; Misaka, Y.; Asada, Y.; Ohno, Y.; Nanasawa, M. Chemical release control: sulfonate esters from perfume and herbicide alcohols and p-styrenesulfonyl chloride. J. Polym. Sci. Polym. Chem. Ed. 1985, 23, 1517–1526. [Google Scholar] [CrossRef]
- Galioğlu, O.; Akar, A. Perfume alcohols supported to sulfochlorinated poly(styrene-co-divinyl benzene) beads. J. Polym. Sci. A Polym. Chem. 1988, 26, 2355–2357. [Google Scholar] [CrossRef]
- Kamogawa, H.; Kohno, H.; Kitagawa, R. Chemical release control: carbamates of 3-vinylphenyl and 2-methacryloyloxyethyl isocyanates and perfume and herbicide alcohols. J. Polym. Sci. A Polym. Chem. 1989, 27, 487–495. [Google Scholar] [CrossRef]
- Berthier, D.; Trachsel, A.; Fehr, C.; Ouali, L.; Herrmann, A. Amphiphilic polymethacrylate- and polystyrene-based chemical delivery systems for damascones. Helv. Chim. Acta 2005, 88, 3089–3108. [Google Scholar] [CrossRef]
- Trachsel, A.; de Saint Laumer, J.-Y.; Haefliger, O.P.; Herrmann, A. Parameters influencing the release of tertiary alcohols from the surface of “spherical” dendrimers and “linear” stylomers by neighbouring-group-assisted hydrolysis of 2-carbamoylbenzoates. Chem. Eur. J. 2009, 15, 2846–2860. [Google Scholar]
- Aulenta, F.; Drew, M.G.B.; Foster, A.; Hayes, W.; Rannard, S.; Thornwaite, D.W.; Youngs, T.G.A. Fragrance release from the surface of branched poly(amide)s. Molecules 2005, 10, 81–97. [Google Scholar] [CrossRef]
- Morinaga, H.; Morikawa, H.; Wang, Y.; Sudo, A.; Endo, T. Amphiphilic copolymer having acid-labile acetal in the side chain as a hydrotrope: controlled release of aldehyde by thermoresponsive aggregation-dissociation of polymer micelles. Macromolecules 2009, 42, 2229–2235. [Google Scholar]
- Morinaga, H.; Morikawa, H.; Sudo, A.; Endo, T. Design of controlled releasing system: synthesis of an amphiphilic copolymer endowed with acid-labile side chains based on quaternarization of amine-containing prepolymer with benzyl halide having acetal moiety. J. Polym. Sci. A Polym. Chem. 2009, 47, 3241–3247. [Google Scholar] [CrossRef]
- Wang, Y.; Morinaga, H.; Sudo, A.; Endo, T. Synthesis of amphiphilic polyacetal by polycondensation of aldehyde and polyethylene glycol as an acid-labile polymer for controlled release of aldehyde. J. Polym. Sci. A Polym. Chem. 2011, 49, 596–602. [Google Scholar] [CrossRef]
- Wang, Y.; Morinaga, H.; Sudo, A.; Endo, T. Synthesis of amphiphilic copolymer having acid-labile bicyclo bisoxazolidine in the side-chain for controlled release of fragrance aldehyde. J. Polym. Sci. A Polym. Chem. 2011, 49, 1881–1886. [Google Scholar]
- Berthier, D.; Paret, N.; Trachsel, A.; Herrmann, A. Influence of the backbone structure on the release of bioactive volatiles from maleic acid-based polymer conjugates. Bioconjug. Chem. 2010, 21, 2000–2012. [Google Scholar] [CrossRef]
- Tree-udom, T.; Wanichwecharungruang, S.P.; Seemork, J.; Arayachukeat, S. Fragrant chitosan nanospheres: Controlled release systems with physical and chemical barriers. Carbohydr. Polym. 2011, 86, 1602–1609. [Google Scholar] [CrossRef]
- Fehr, C.; Galindo, J. Aldols by Michael addition: Application of the retro-Michael addition to the slow release of enones. Helv. Chim. Acta 2005, 88, 3128–3136. [Google Scholar] [CrossRef]
- Kastner, D. 25 Jahre Rosenketone—Thema mit Variationen. Parfuem. Kosmet. 1994, 75, 170–181. [Google Scholar]
- Williams, A. Rose ketones: Celebrating 30 years of success. Perfum. Flavorist 2002, 27, 18–32. [Google Scholar]
- Saito, Y.; Miura, K.; Tokuoka, Y.; Kondo, Y.; Abe, M.; Sato, T. Volatility and solubilization of synthetic fragrances by Pluronic® P-85. J. Dispers. Sci. Technol. 1996, 17, 567–576. [Google Scholar] [CrossRef]
- Suzuki, K.; Saito, Y.; Tokuoka, Y.; Abe, M.; Sato, T. Poly(ethylene oxide)/poly(propylene oxide)/poly(ethylene oxide) triblock copolymer as a sustained-release carrier for perfume compounds. J. Am. Oil Chem. Soc. 1997, 74, 55–59. [Google Scholar]
- Vauthey, S.; Leser, M.E.; Garti, N.; Watzke, H.J. Solubilization of hydrophilic compounds in copolymer aggregates. J. Colloid Interface Sci. 2000, 225, 16–24. [Google Scholar] [CrossRef]
- Kayali, I. Solubilization of fragrance compounds in block copolymer/water system. Jordan. J. Appl. Sci. 2003, 5, 42–49. [Google Scholar]
- Berthier, D.L.; Schmidt, I.; Fieber, W.; Schatz, C.; Furrer, A.; Wong, K.; Lecommandoux, S. Controlled release of volatile fragrance molecules from PEO-b-PPO-b-PEO block copolymer micelles in ethanol-water mixtures. Langmuir 2010, 26, 7953–7961. [Google Scholar]
- Berthier, D.; Herrmann, A. Polymer conjugates for a controlled release of active molecules. WO Patent 2008/044178, 17 April 2008. [Google Scholar]
- Rouseff, R.L.; Cadwallader, K.R. Headspace Analysis of Foods and Flavors: Theory and Practice; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2001. [Google Scholar]
- Rubiolo, P.; Sgorbini, B.; Liberto, E.; Cordero, C.; Bicchi, C. Analysis of the plant volatile fraction. In The Chemistry and Biology of Volatiles; Herrmann, A., Ed.; John Wiley & Sons: Chichester, UK, 2010; pp. 49–93. [Google Scholar]
- Levinson, M.I. Rinse-added fabric softener technology at the close of the twentieth century. J. Surfactants Deterg. 1999, 2, 223–235. [Google Scholar] [CrossRef]
- Mishra, S.; Tyagi, V.K. Ester quats: The novel class of cationic fabric softeners. J. Oleo Sci. 2007, 56, 269–276. [Google Scholar] [CrossRef]
- Levrand, B.; Fieber, W.; Lehn, J.-M.; Herrmann, A. Controlled release of volatile aldehydes and ketones from dynamic mixtures generated by reversible hydrazone formation. Helv. Chim. Acta 2007, 90, 2281–2314. [Google Scholar] [CrossRef]
- Buchs, B.; Godin, G.; Trachsel, A.; de Saint-Laumer, J.-Y.; Lehn, J.-M.; Herrmann, A. Reversible aminal formation: Controlling the evaporation of bioactive volatiles by dynamic combinatorial/covalent chemistry. Eur. J. Org. Chem. 2011, 681–695. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berthier, D.L.; Paret, N.; Trachsel, A.; Fieber, W.; Herrmann, A. Controlled Release of Damascone from Poly(styrene-co-maleic anhydride)-based Bioconjugates in Functional Perfumery. Polymers 2013, 5, 234-253. https://doi.org/10.3390/polym5010234
Berthier DL, Paret N, Trachsel A, Fieber W, Herrmann A. Controlled Release of Damascone from Poly(styrene-co-maleic anhydride)-based Bioconjugates in Functional Perfumery. Polymers. 2013; 5(1):234-253. https://doi.org/10.3390/polym5010234
Chicago/Turabian StyleBerthier, Damien L., Nicolas Paret, Alain Trachsel, Wolfgang Fieber, and Andreas Herrmann. 2013. "Controlled Release of Damascone from Poly(styrene-co-maleic anhydride)-based Bioconjugates in Functional Perfumery" Polymers 5, no. 1: 234-253. https://doi.org/10.3390/polym5010234