Polyphosphazenes: Multifunctional, Biodegradable Vehicles for Drug and Gene Delivery


{kind=link}
{kind=link}
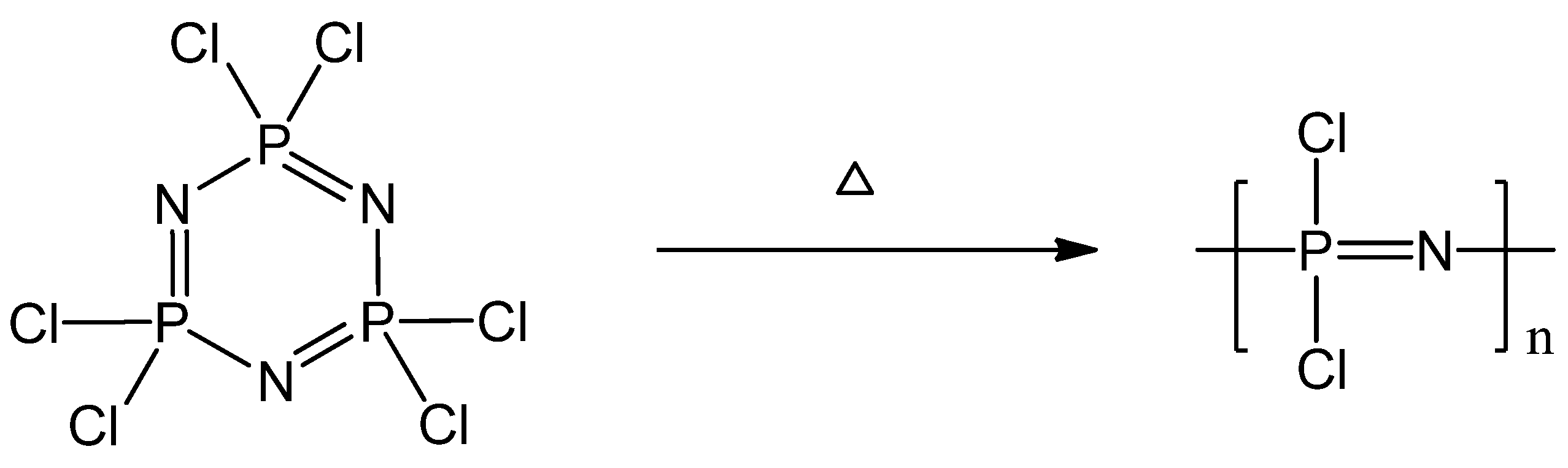{kind=link}
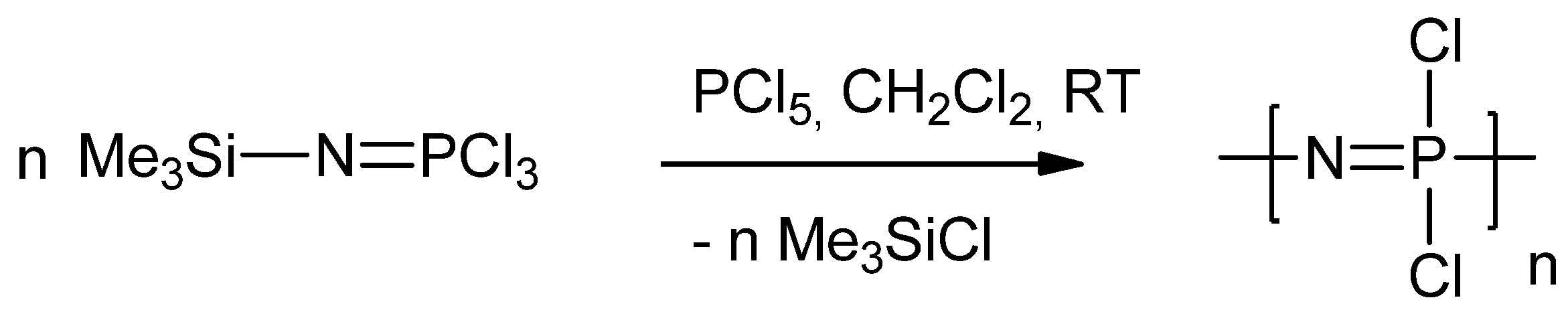{kind=link}
{kind=link}
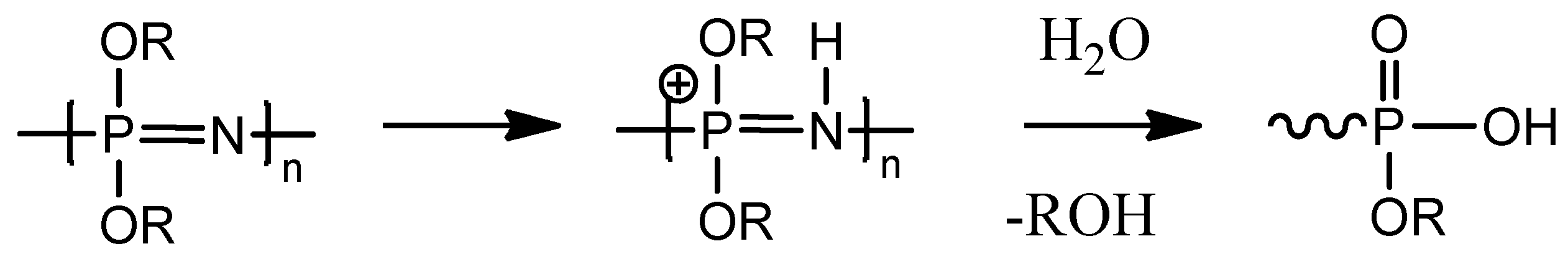{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Polymeric Carriers for Drug Delivery
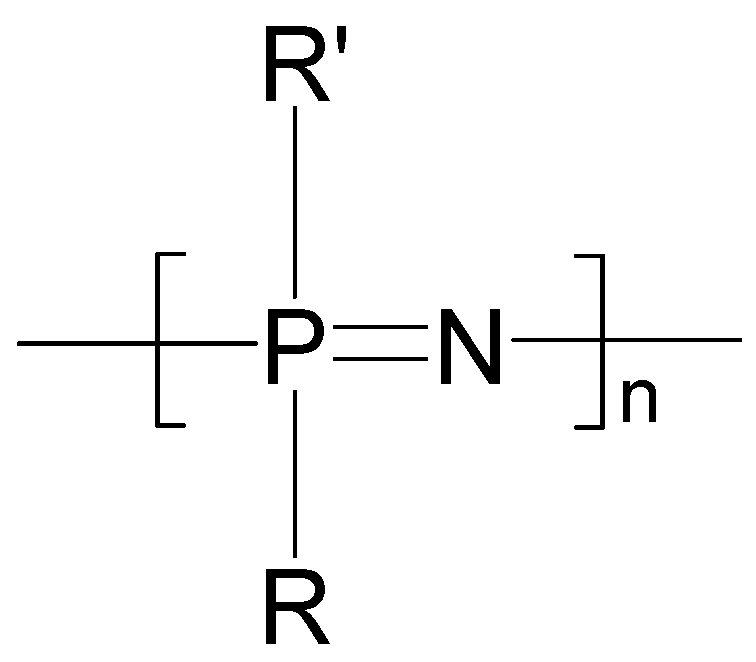
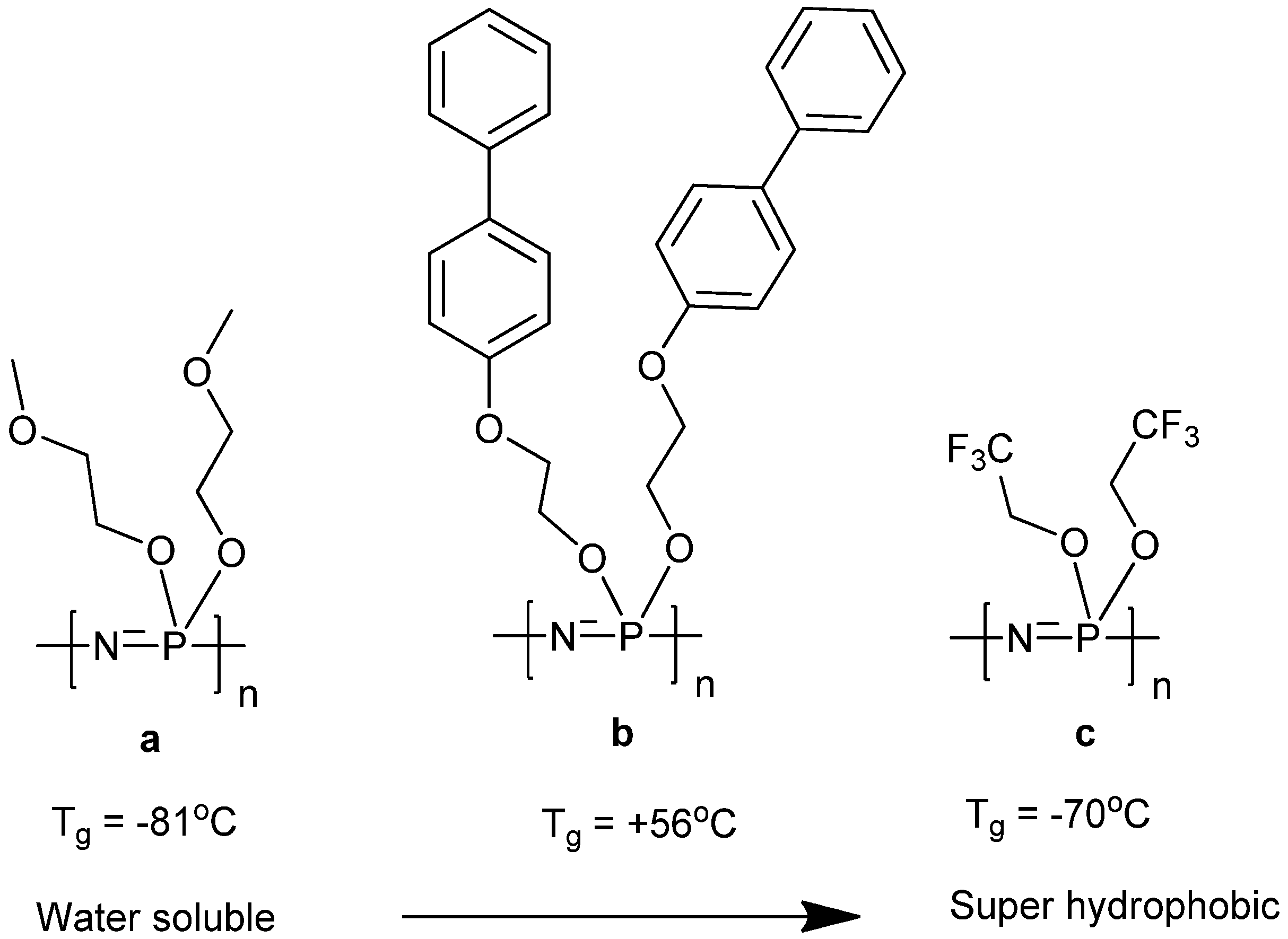
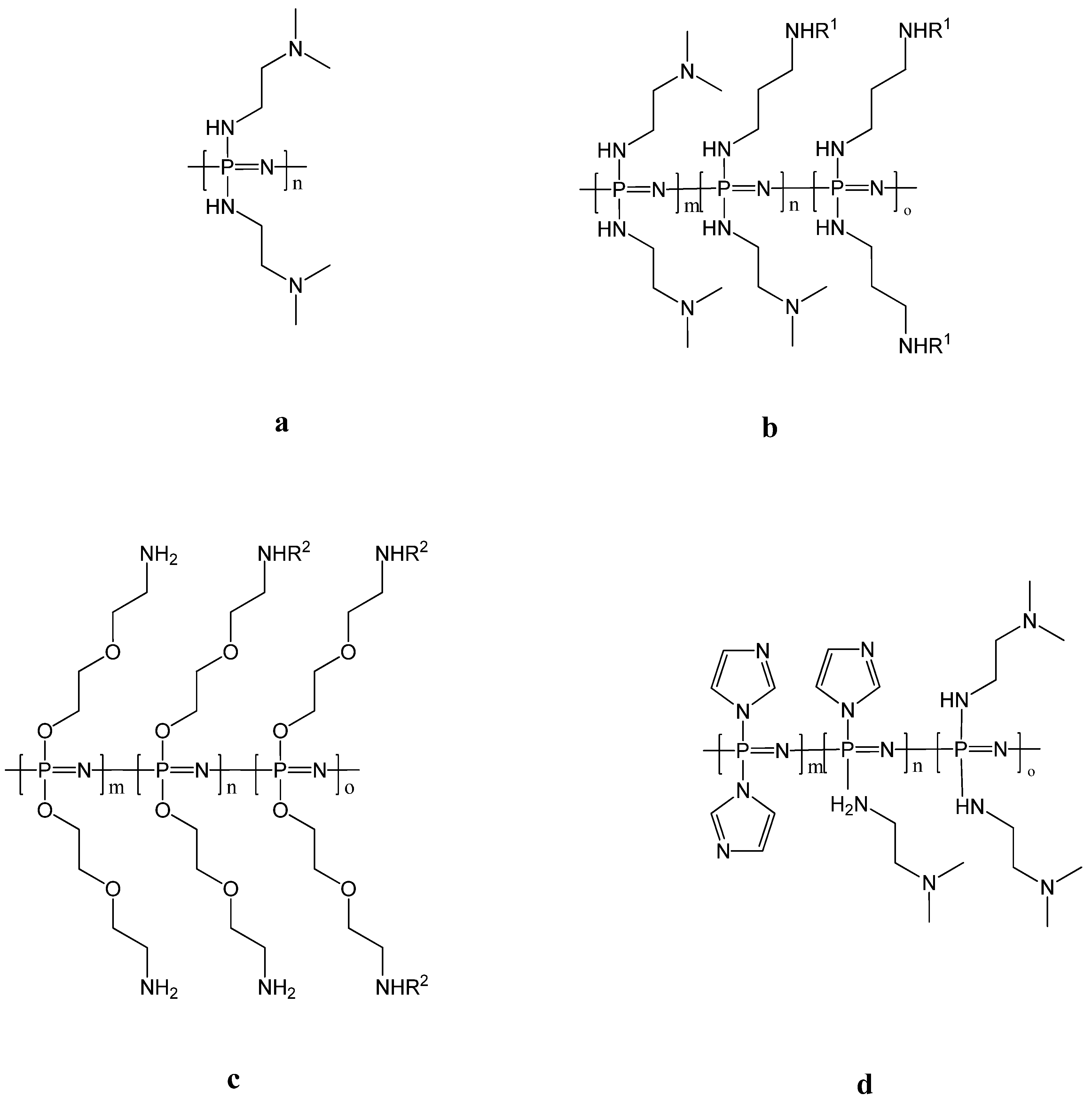
2. Poly[(organo)phosphazenes]
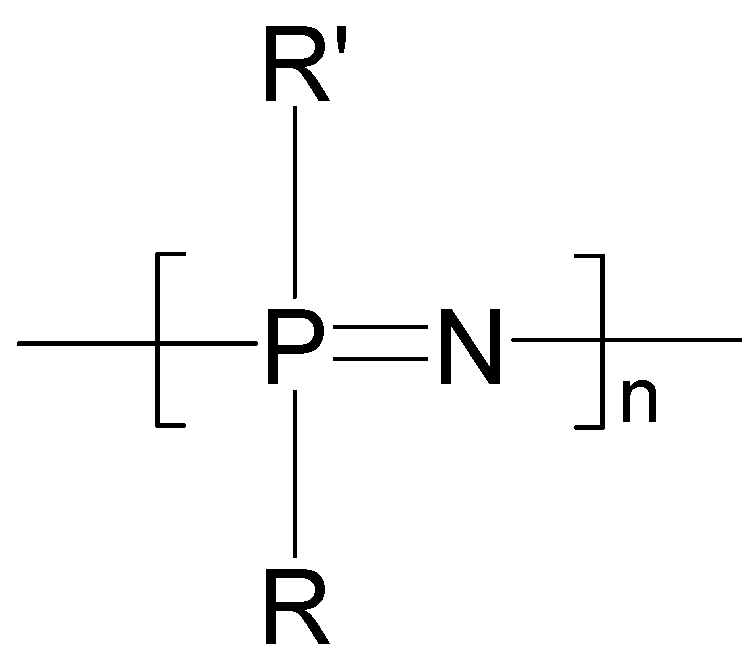
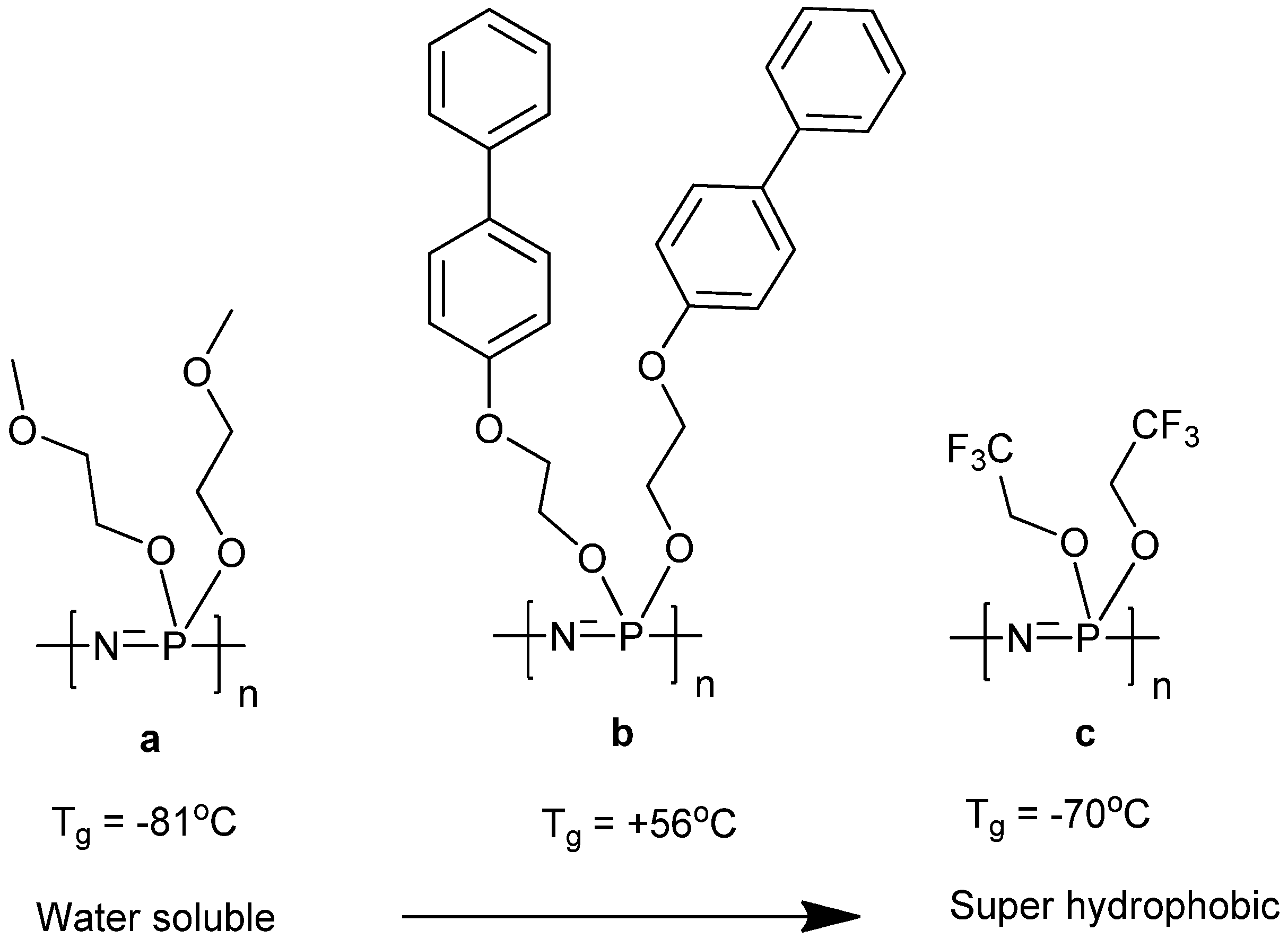
2.1. Synthetic Methods
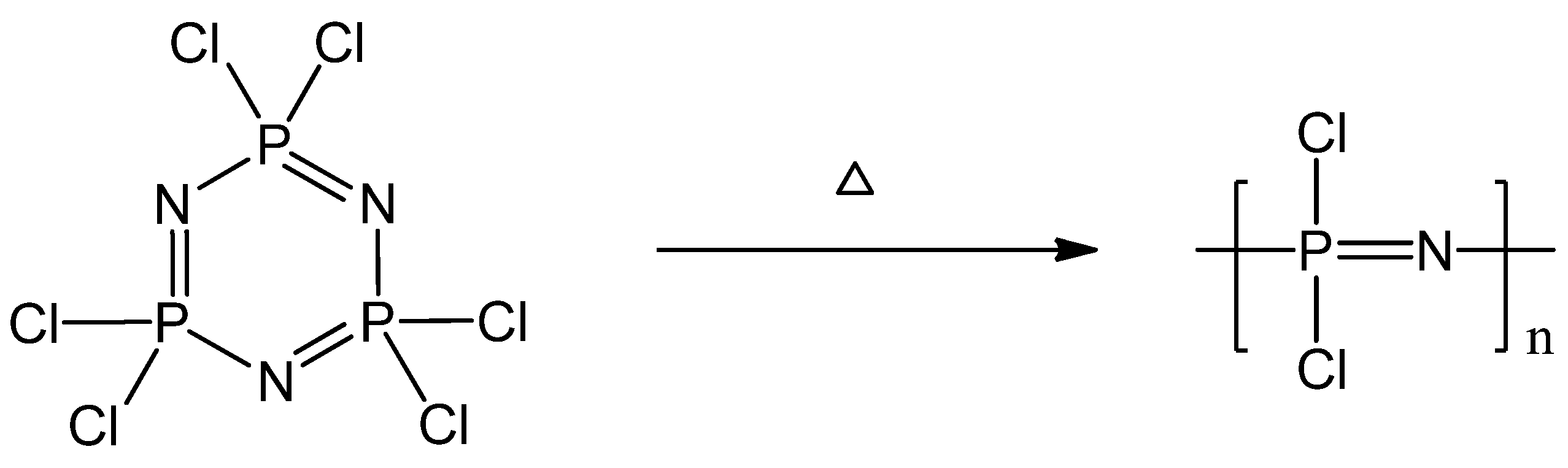
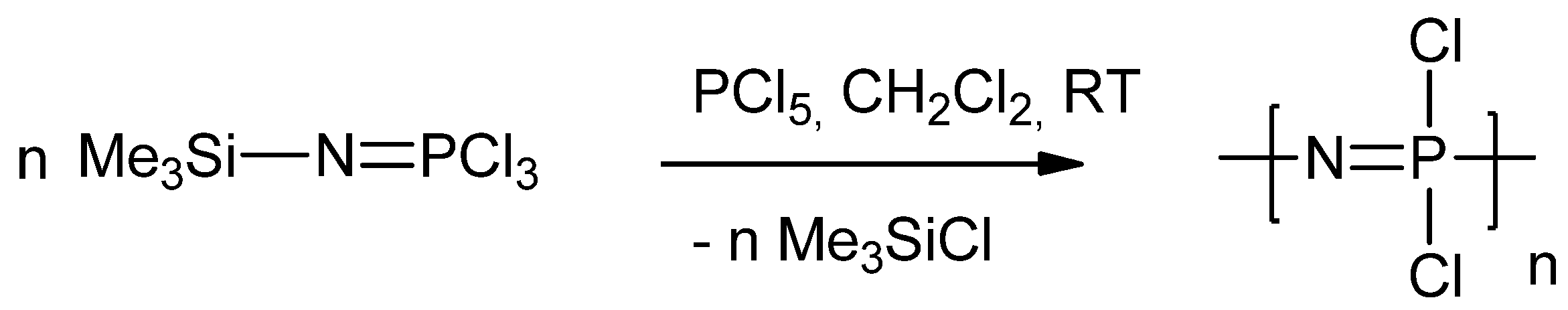
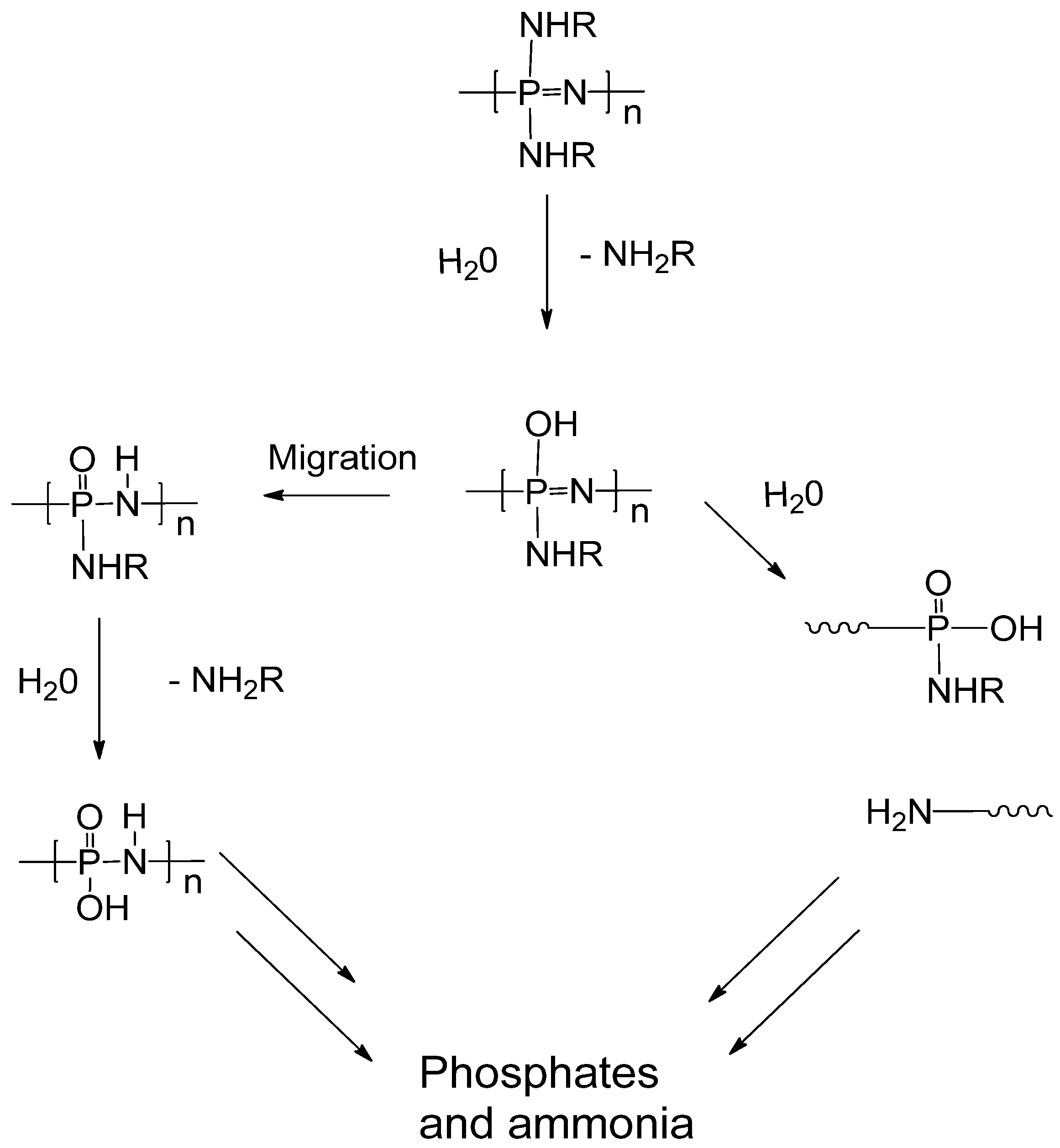
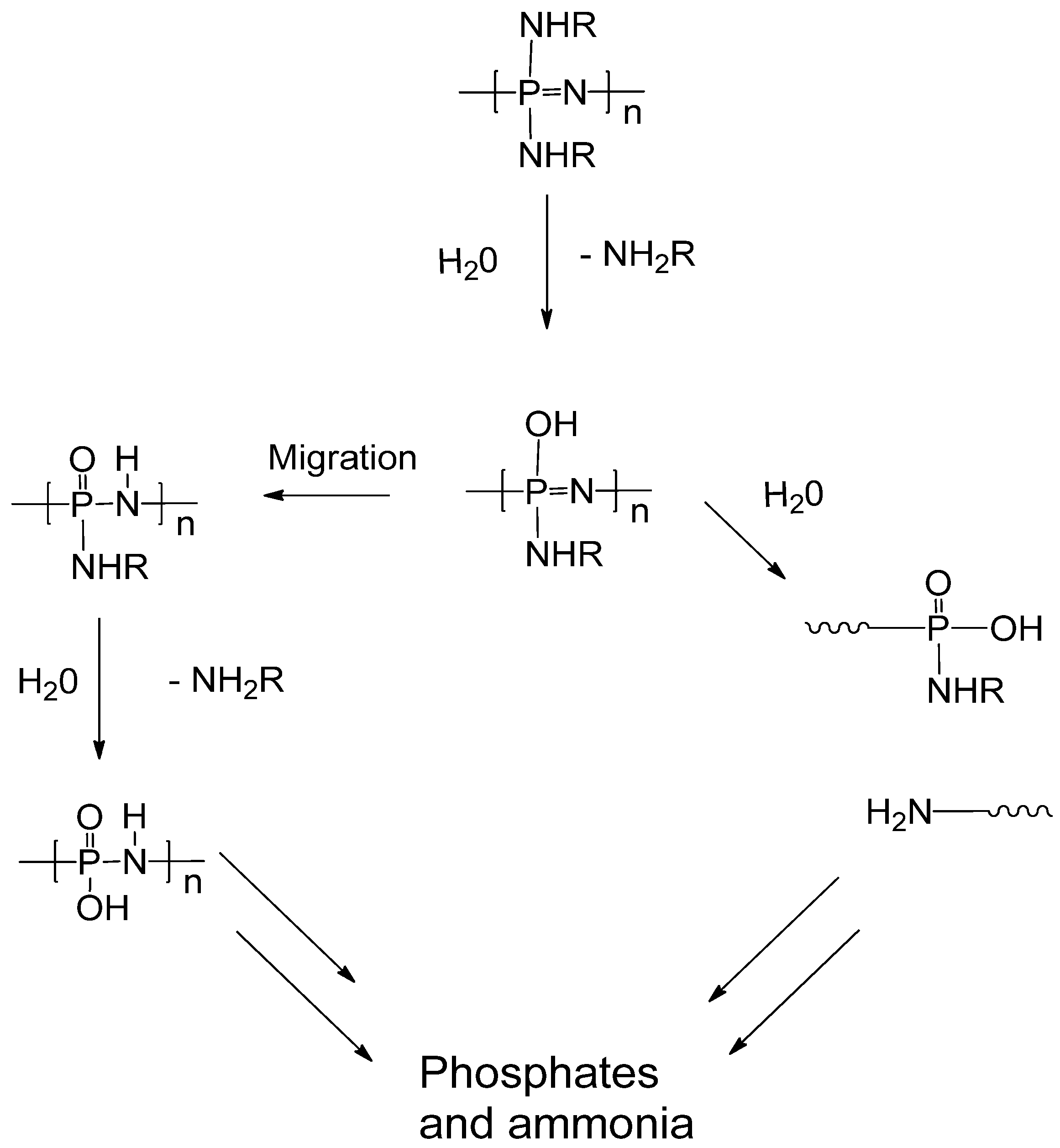
2.2. Biodegradation


3. Applications in Drug Delivery
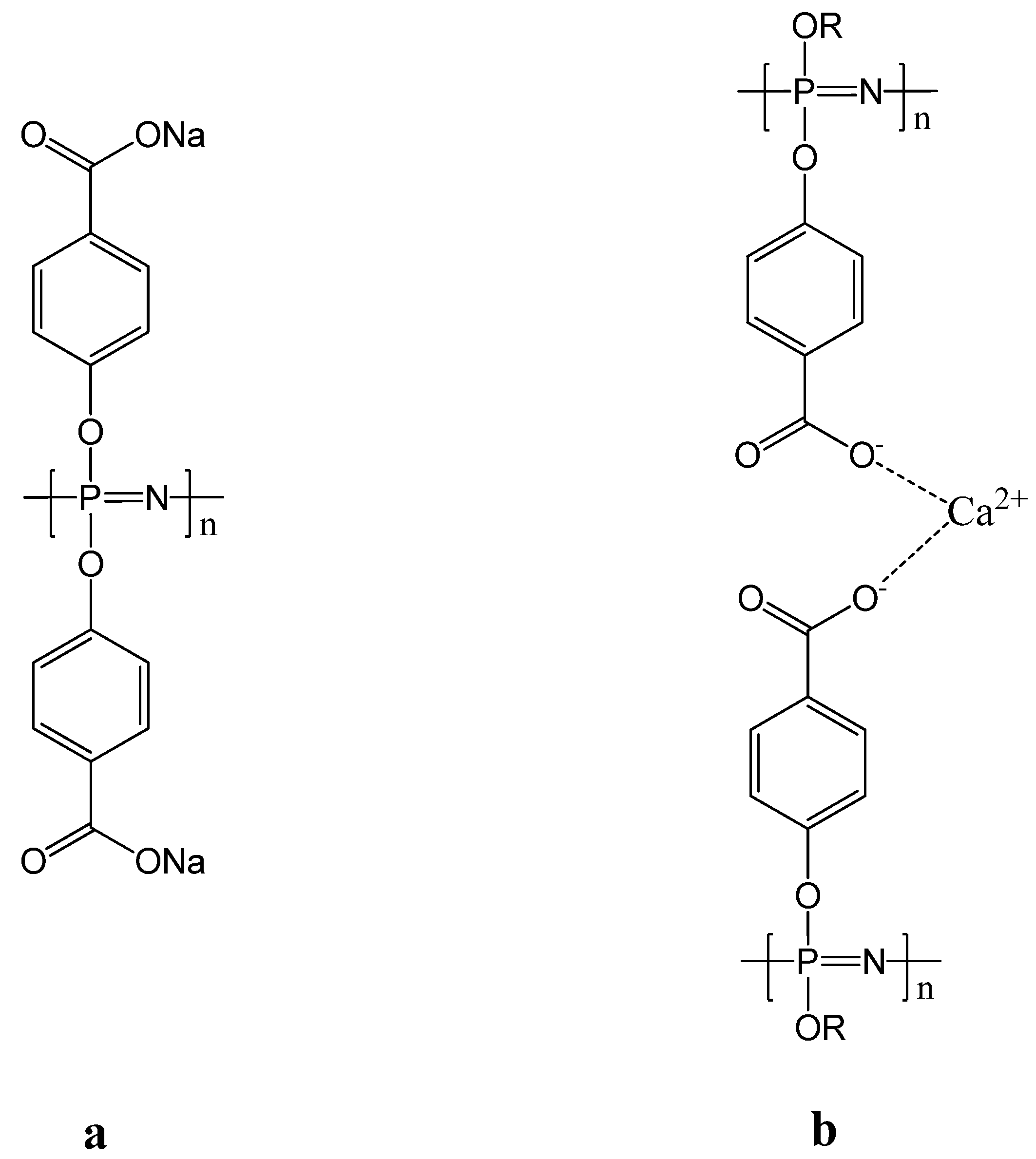
3.1. Matrix Encapsulation

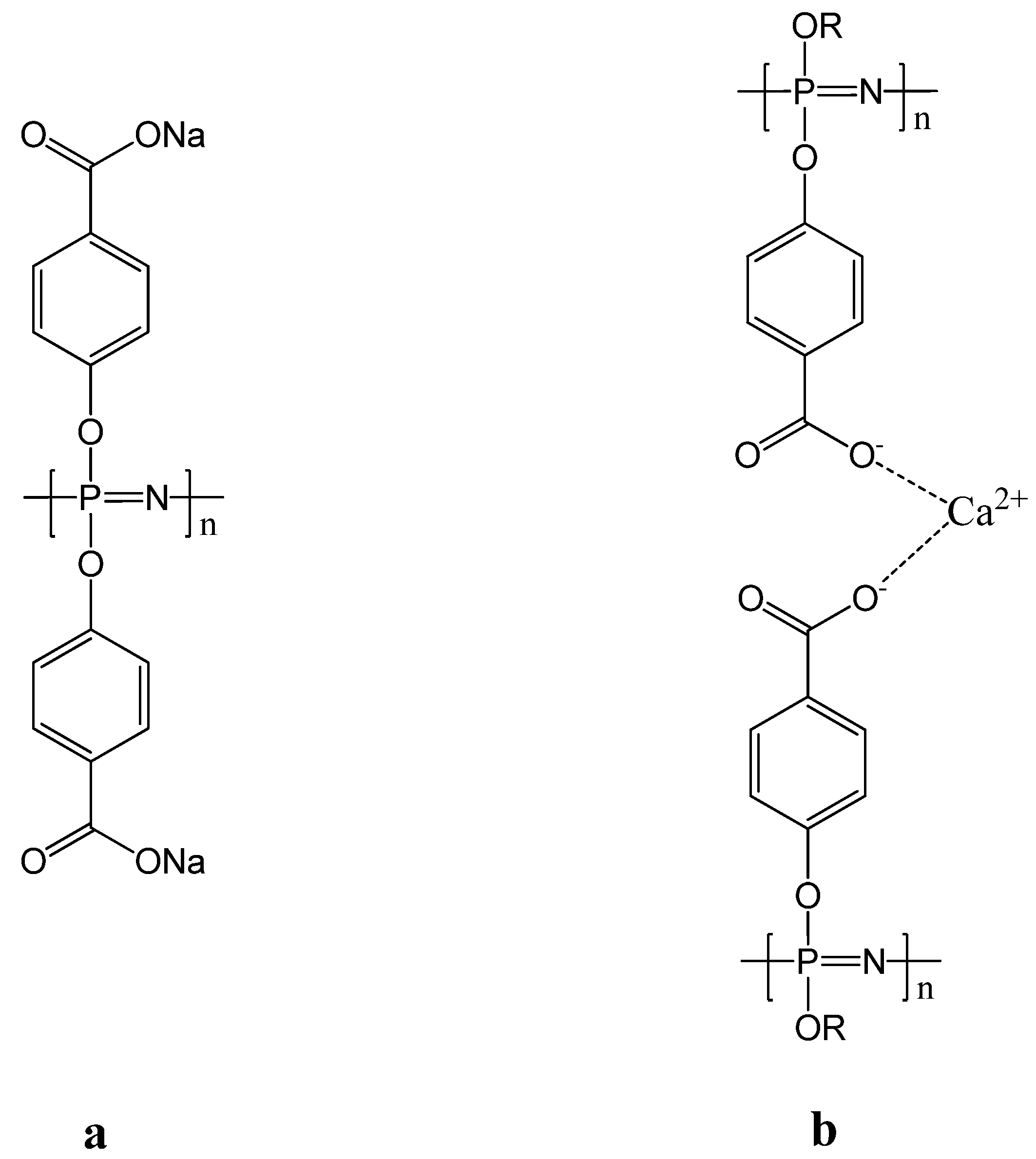
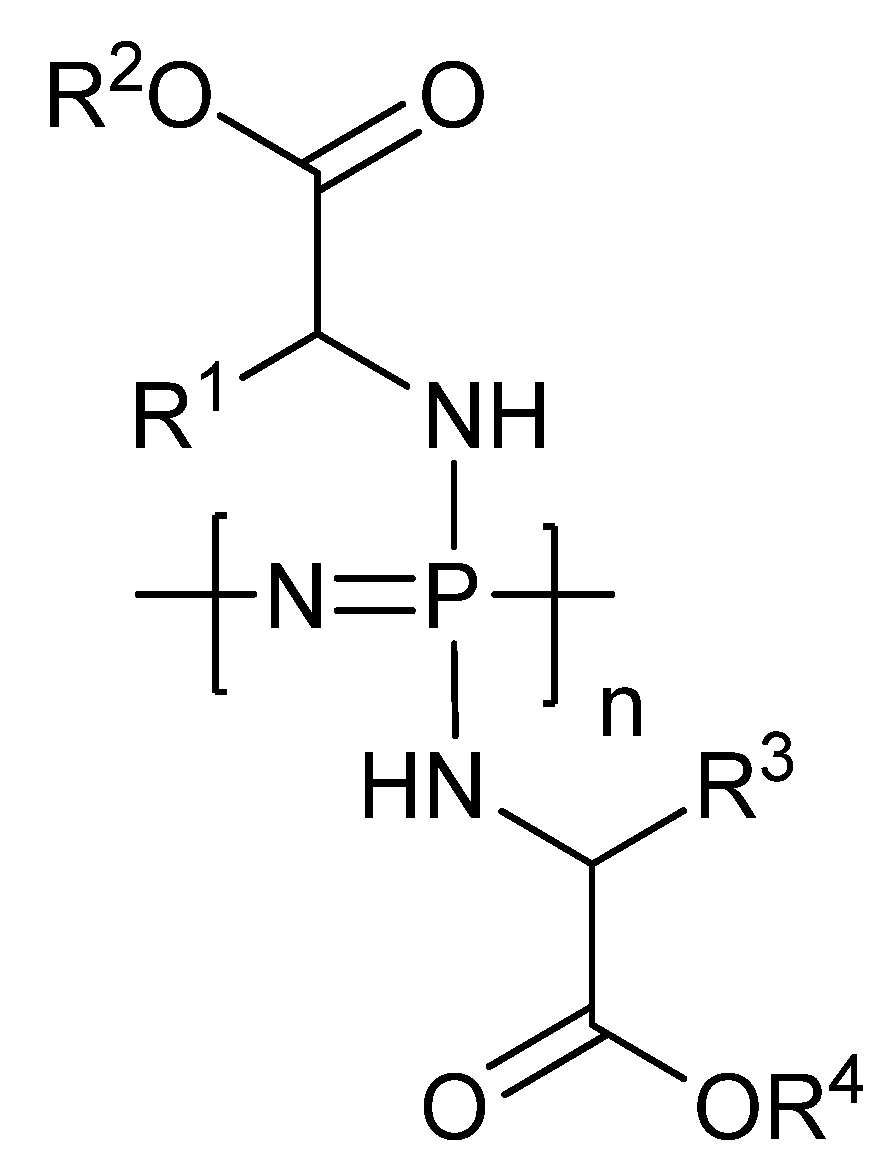
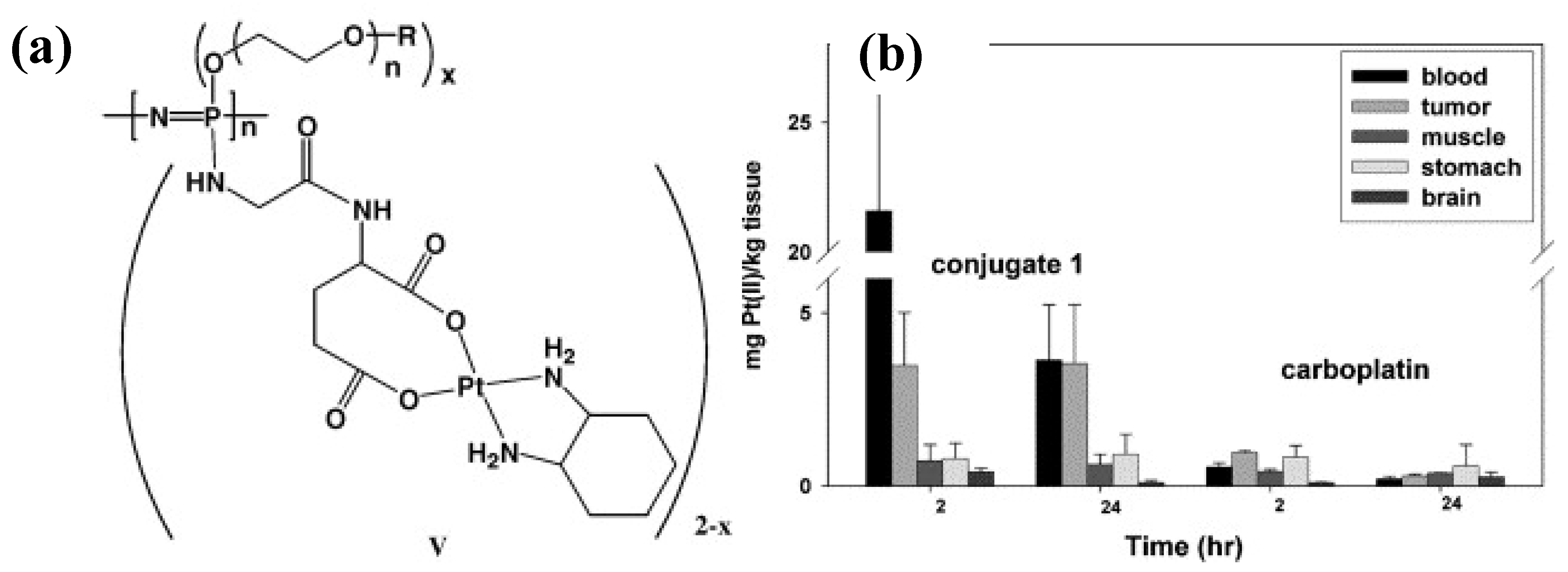
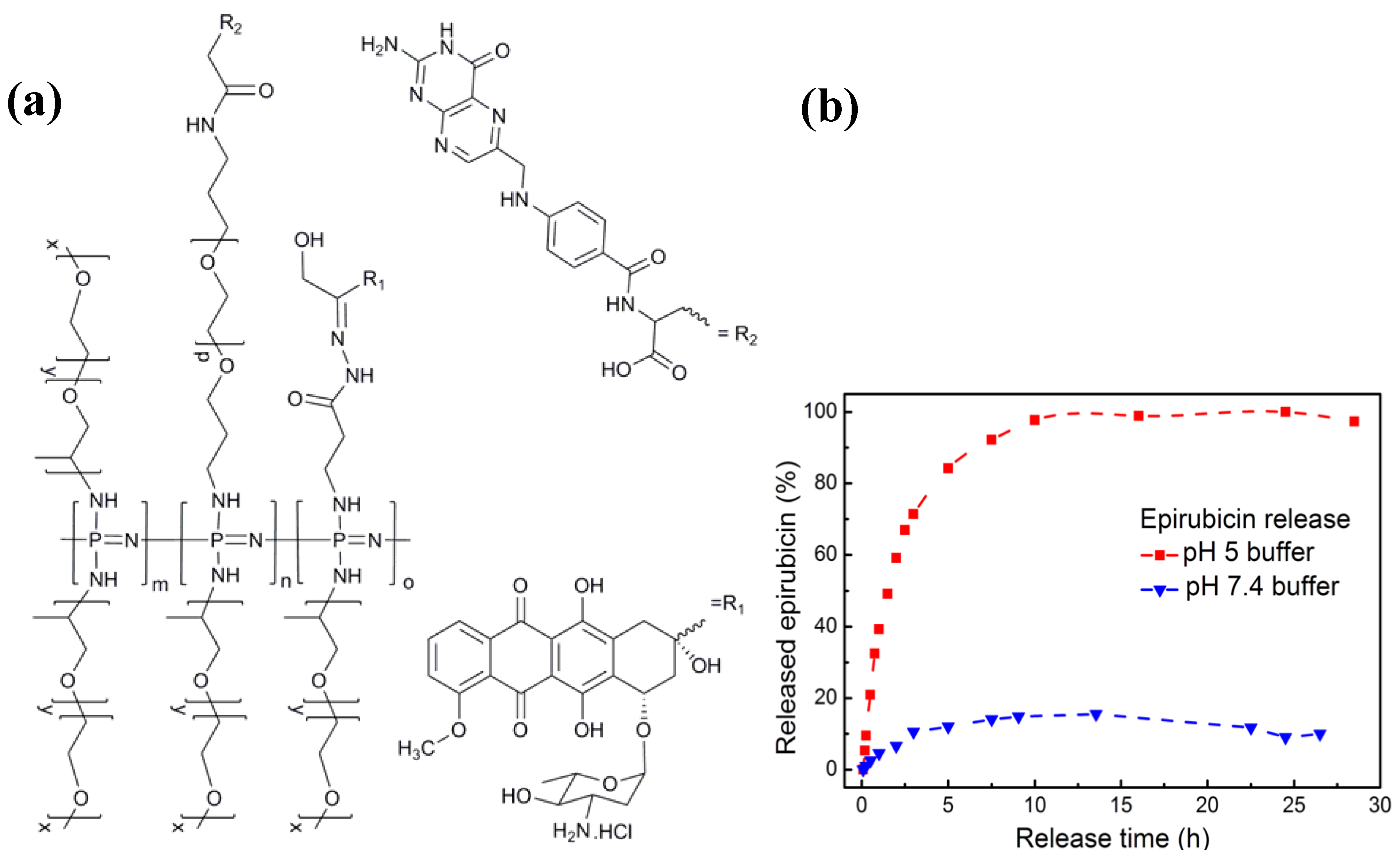
3.2. Anti-Cancer Drug-Conjugates
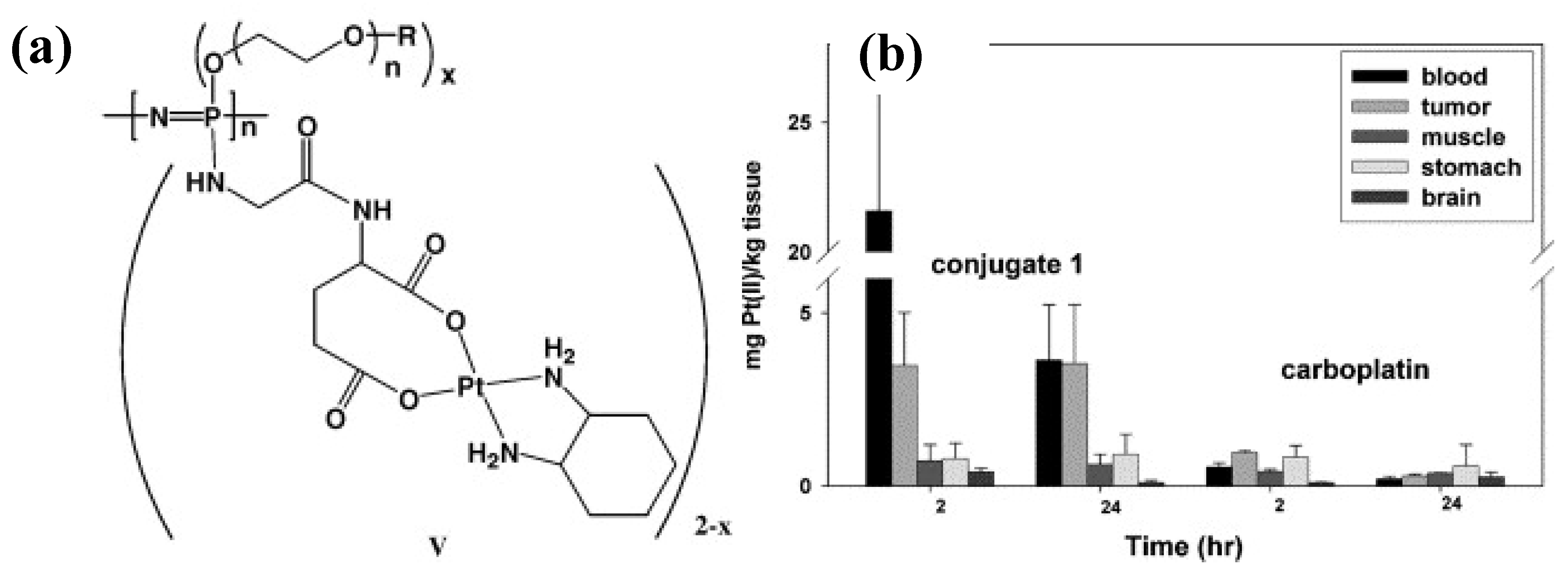
 ) and pH 7.4(
) and pH 7.4(  ). Originally published in [29] and reproduced by permission of The Royal Society of Chemistry.
) and pH 7.4( ). Originally published in [29] and reproduced by permission of The Royal Society of Chemistry.
). Originally published in [29] and reproduced by permission of The Royal Society of Chemistry.
) and pH 7.4( ). Originally published in [29] and reproduced by permission of The Royal Society of Chemistry.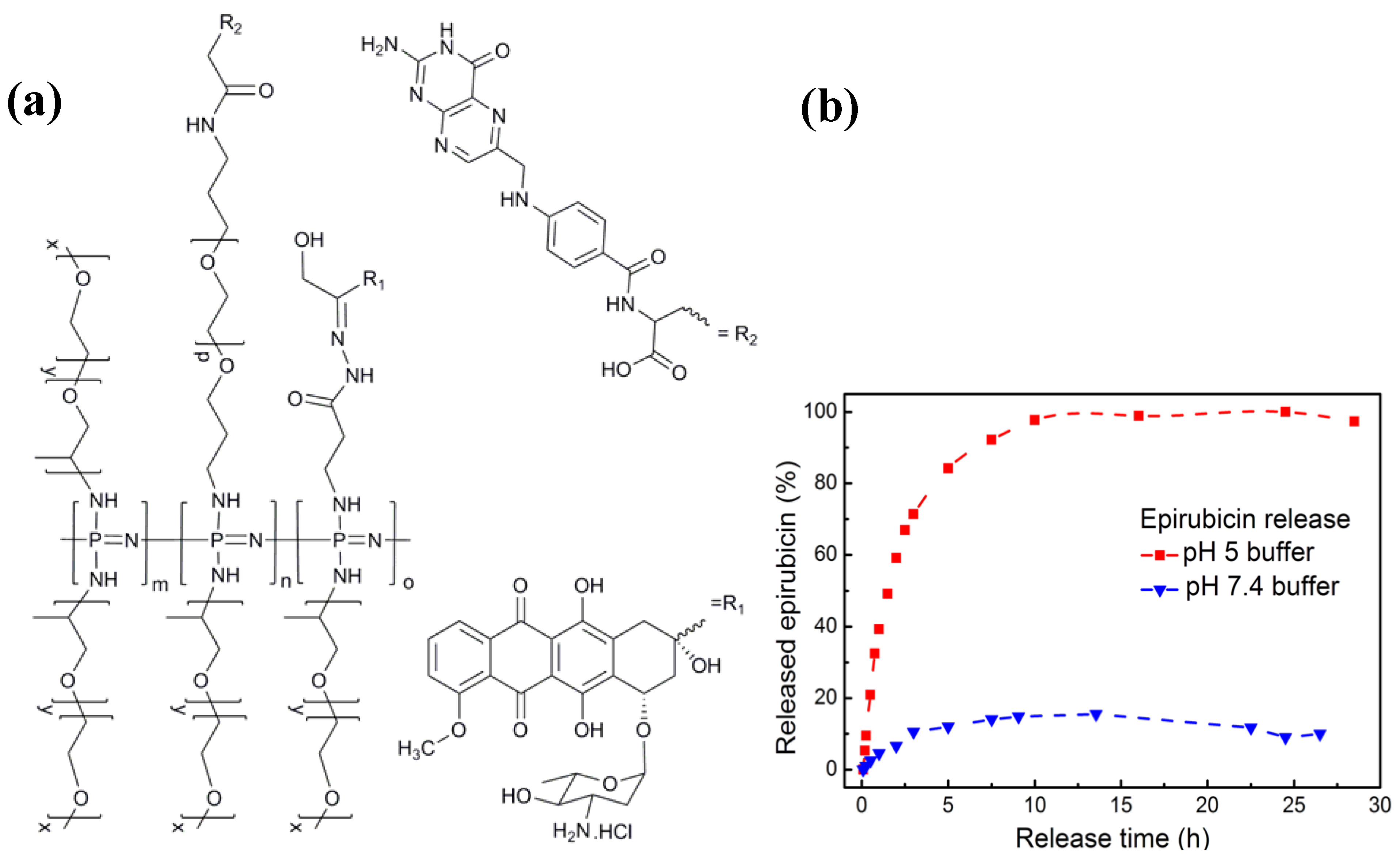
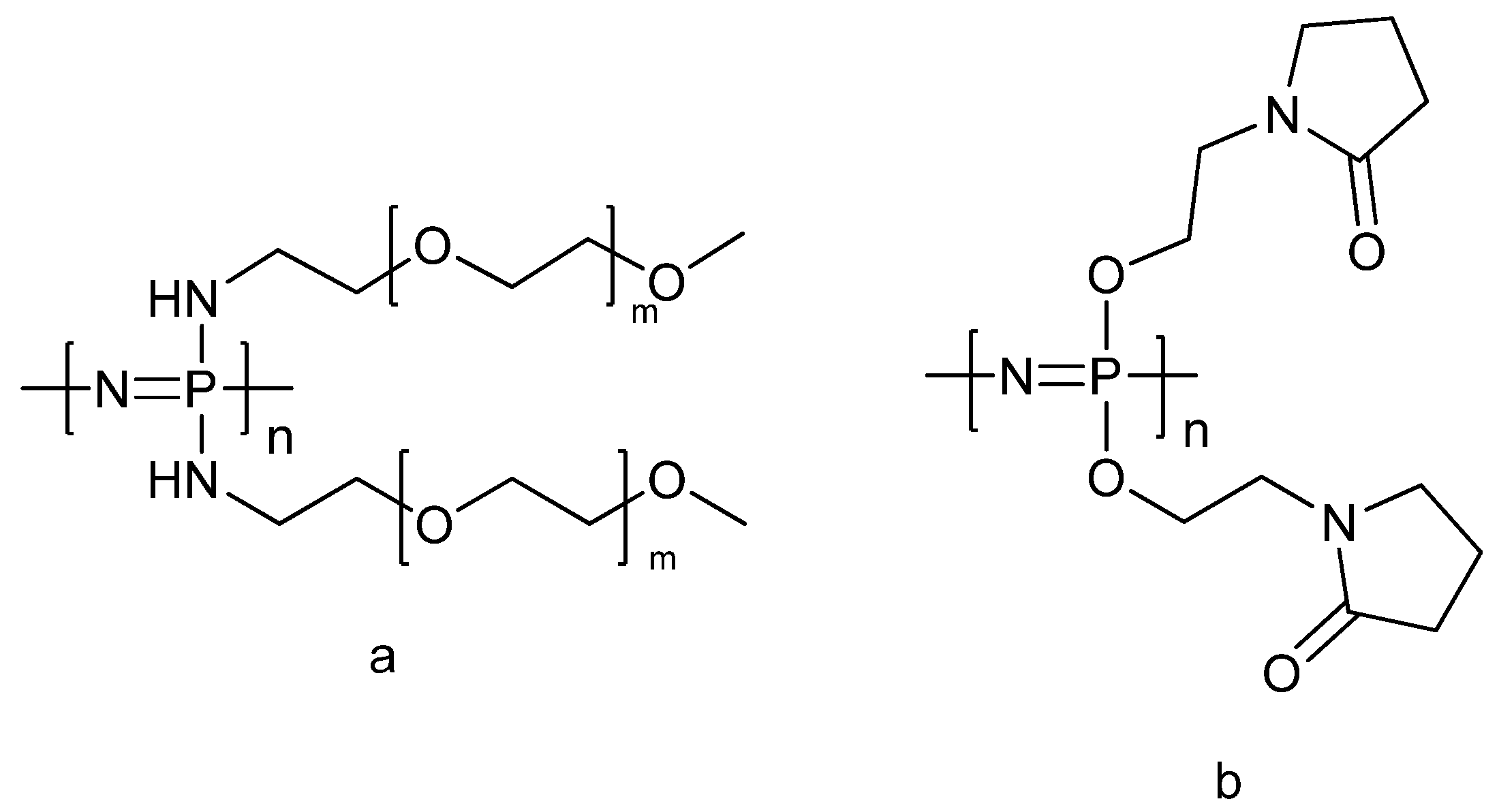
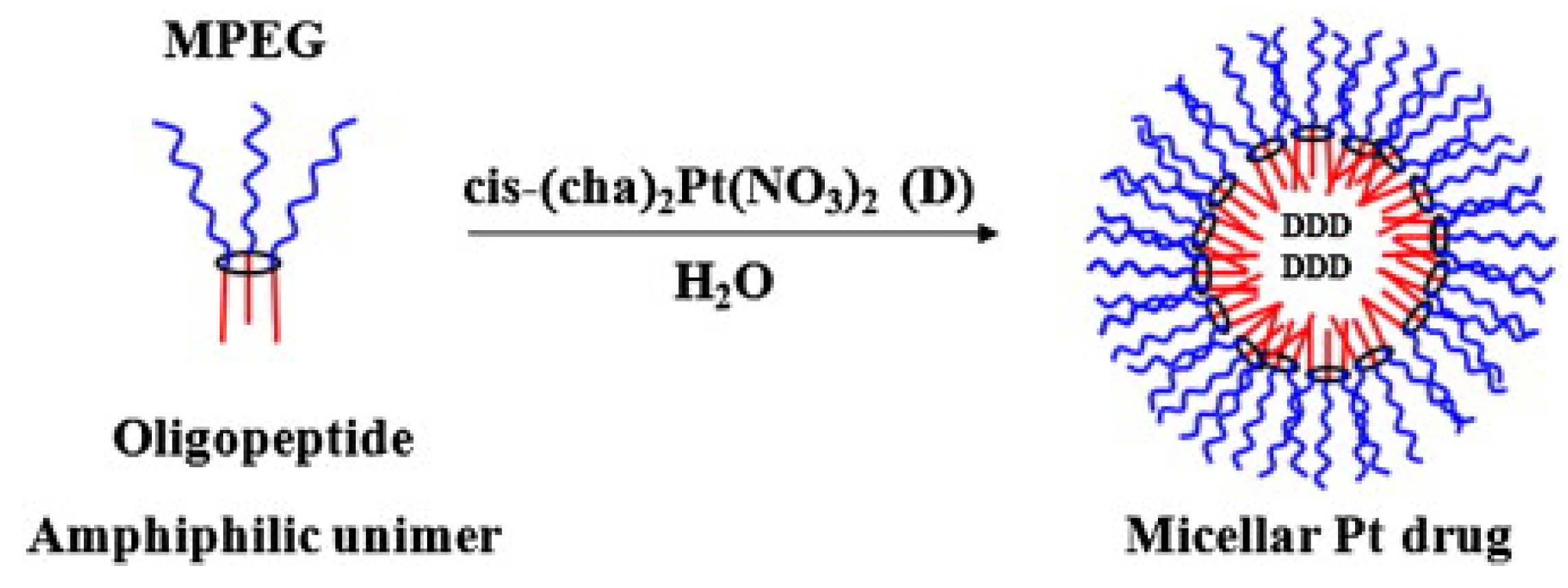
3.3. Self-Assembly
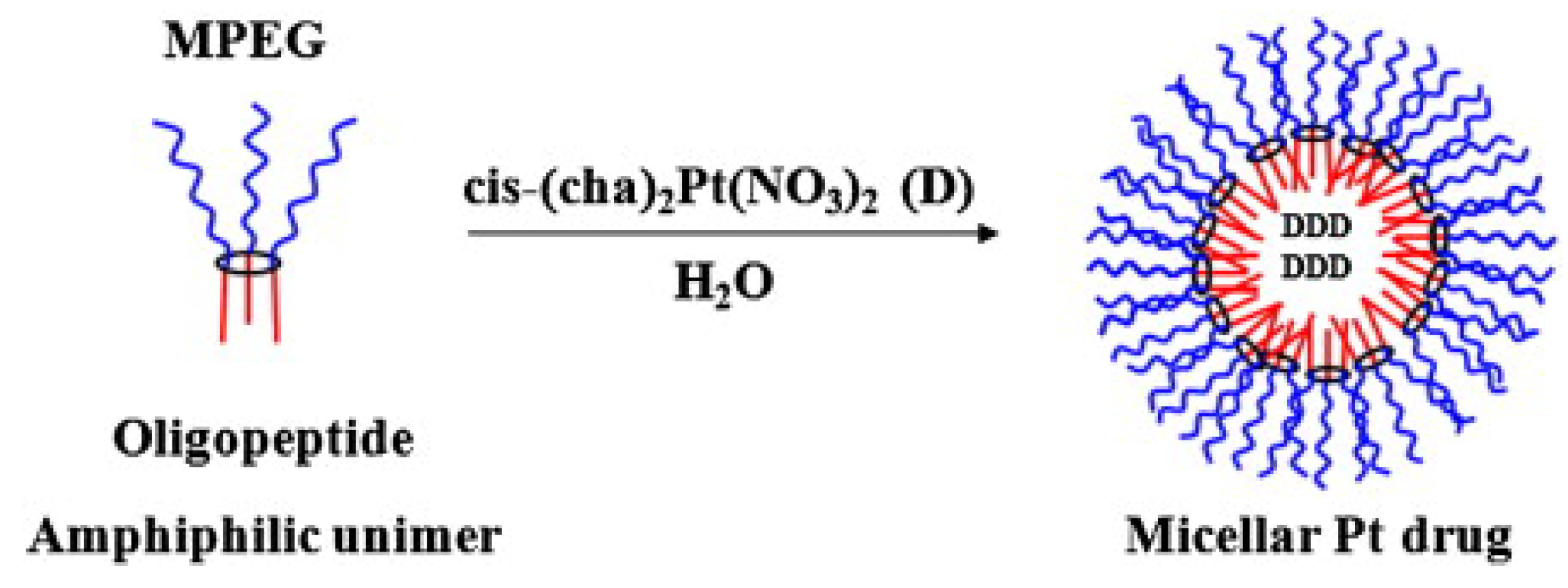
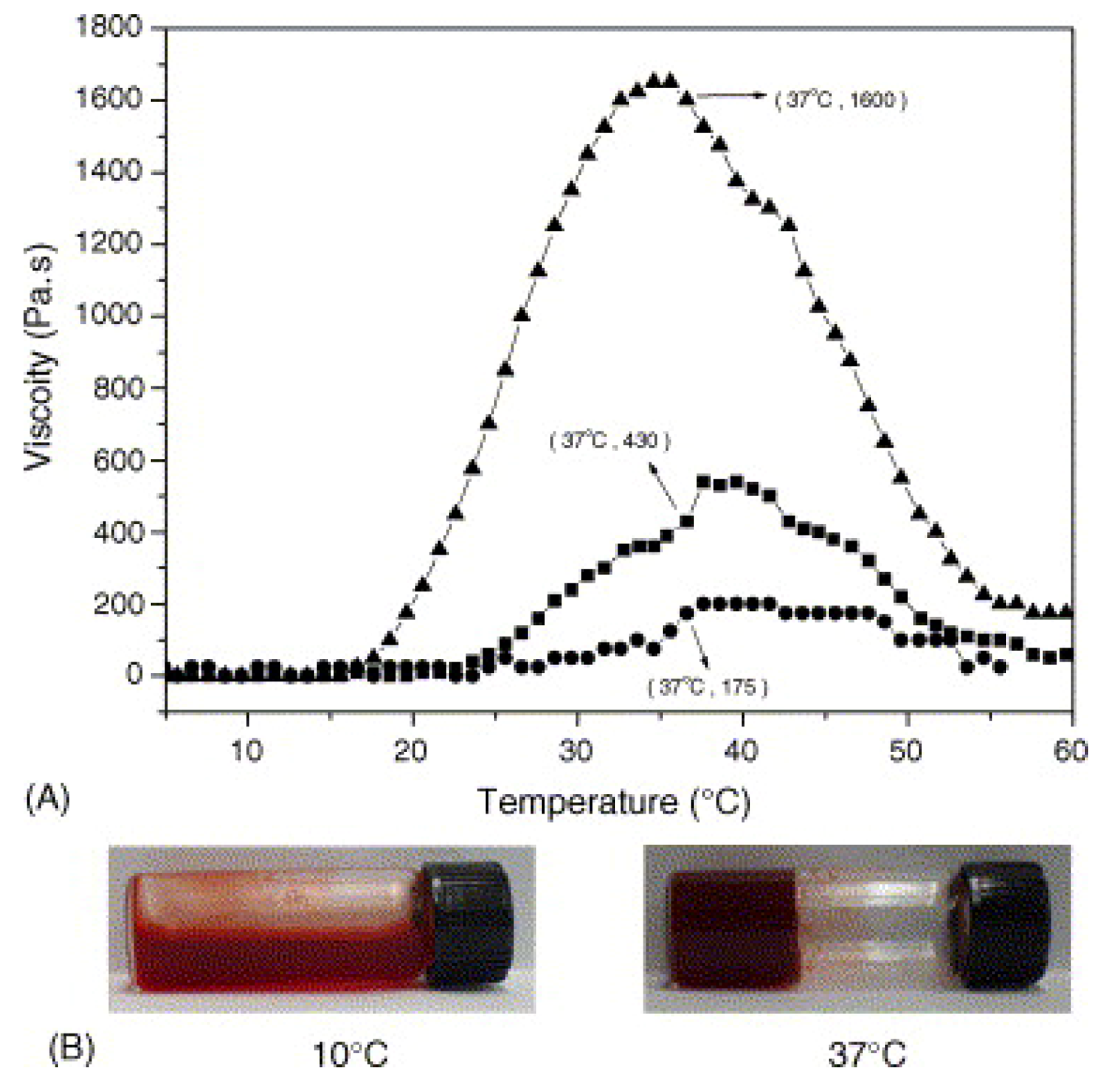
3.4. Thermosensitive Polyphosphazenes

3.5. Polyplexes for Gene Delivery
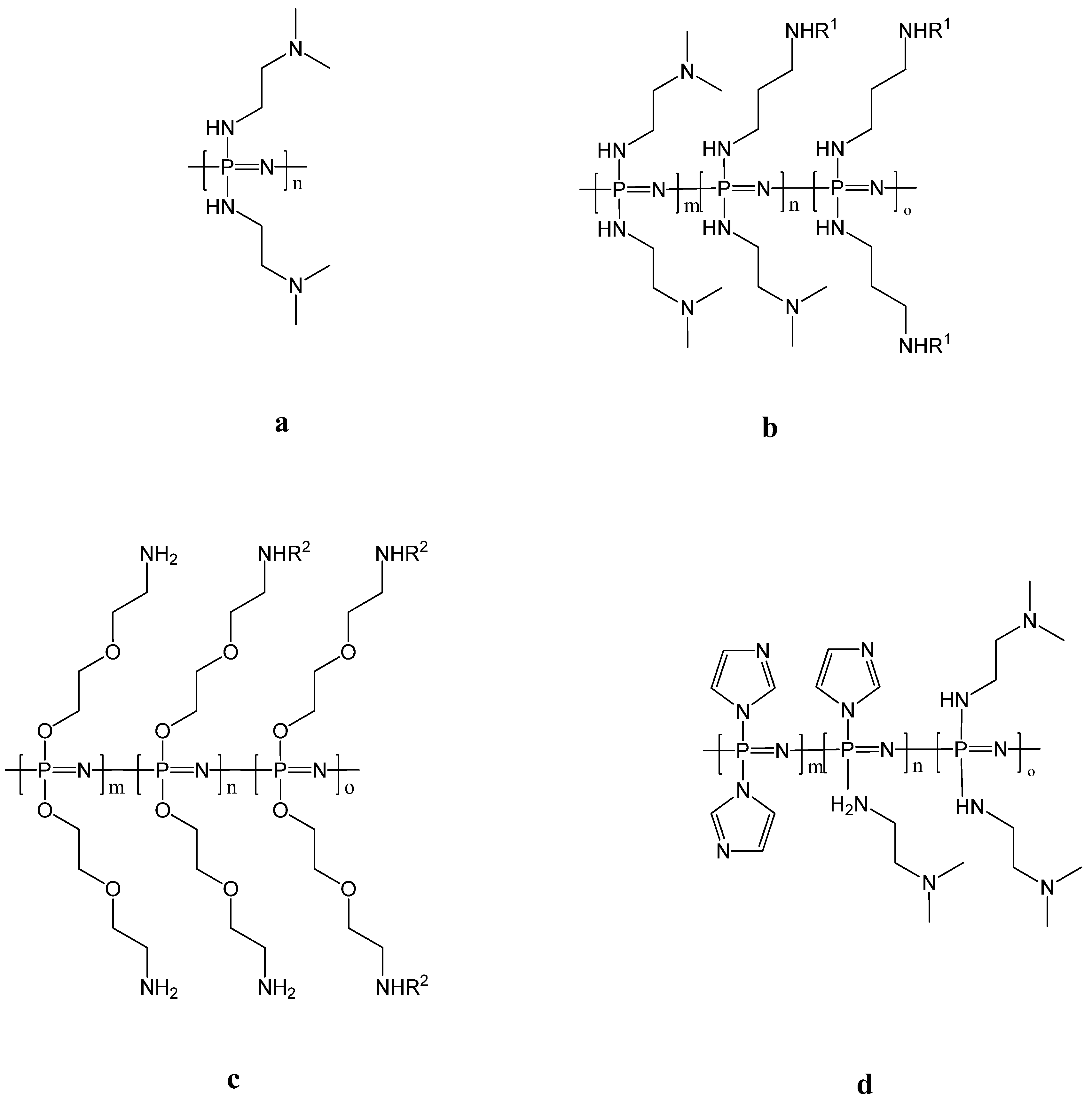
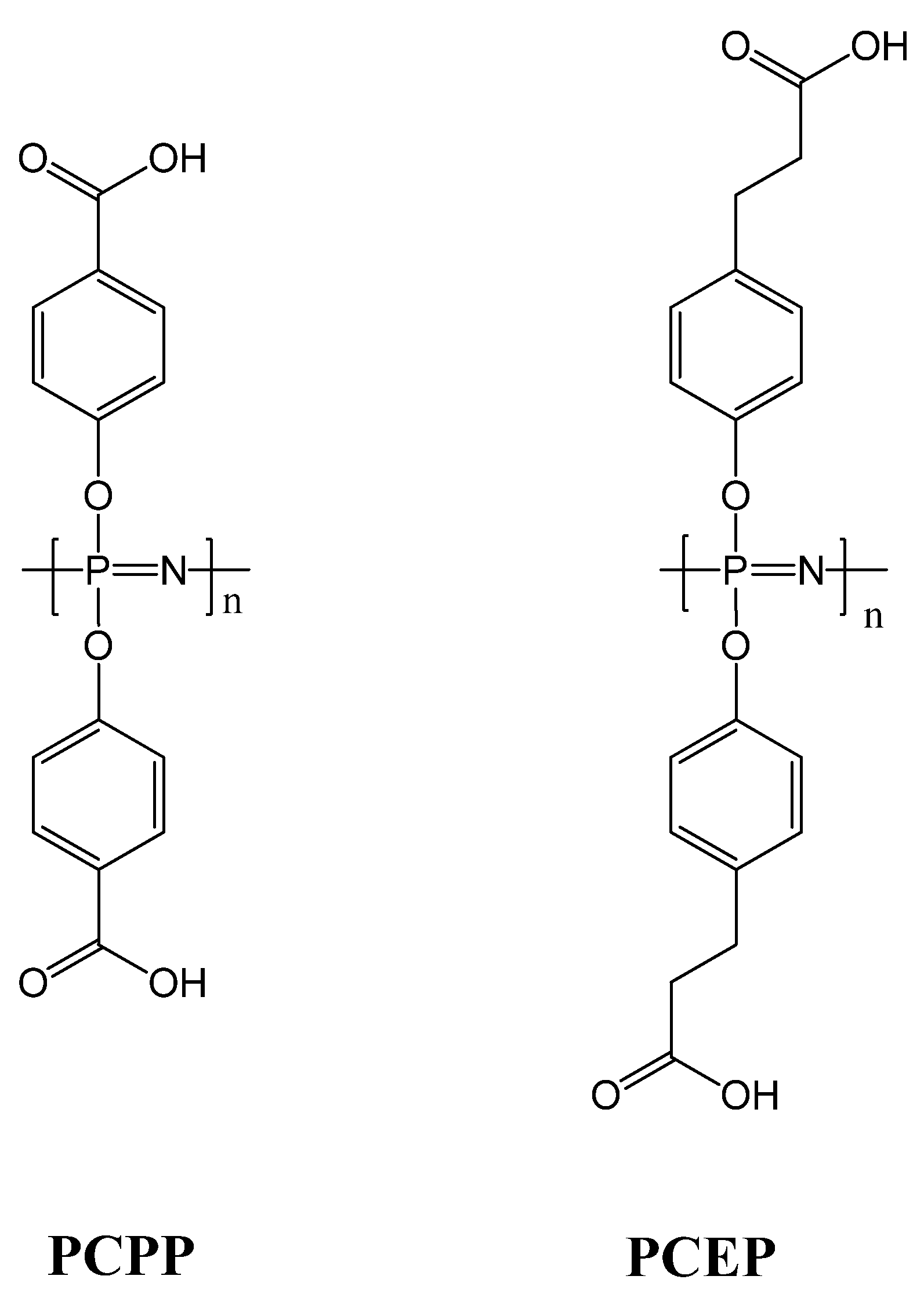
3.6. Vaccine Delivery and Immunomodulation
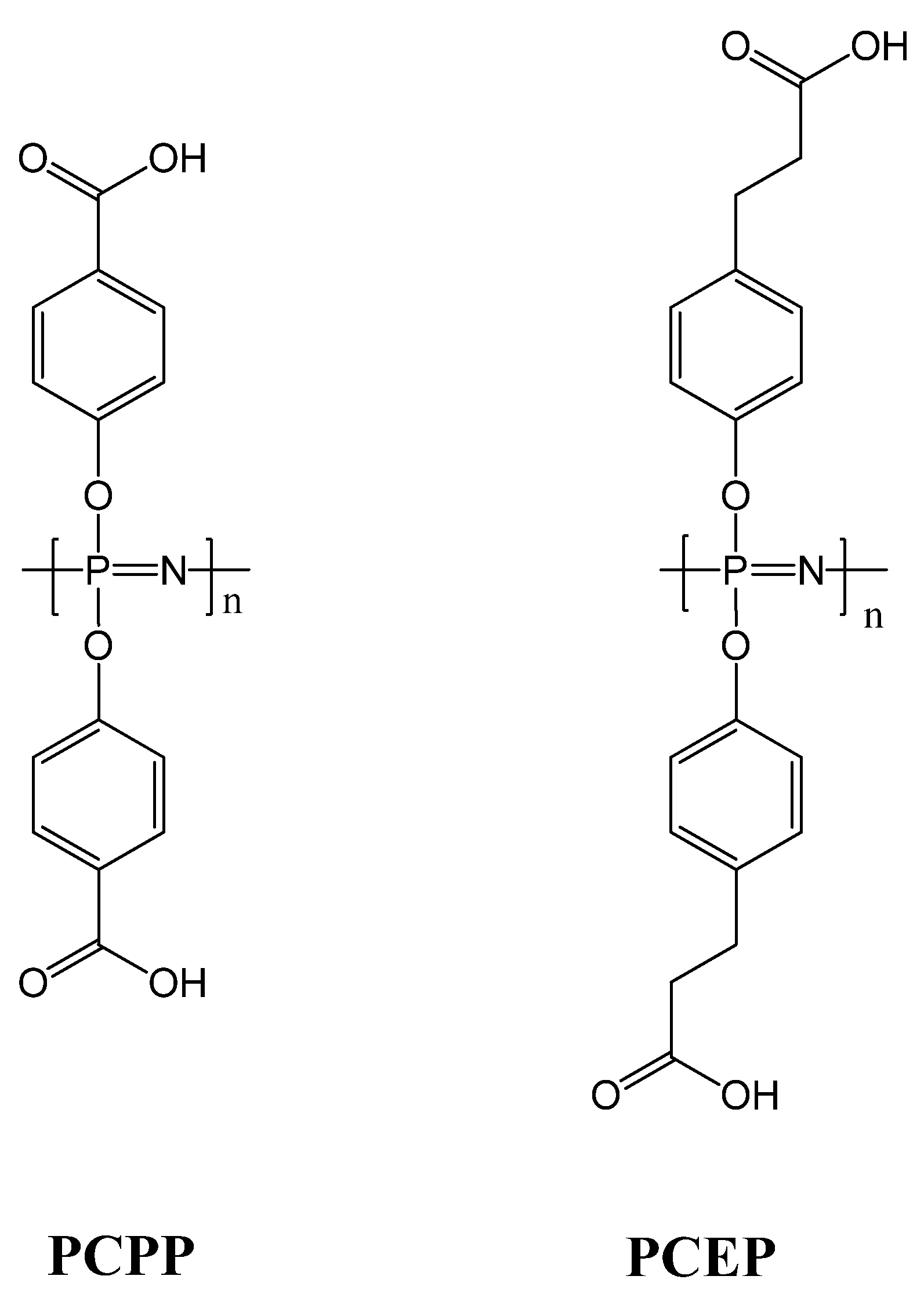
4. Conclusions
Acknowledgments
References
- Grund, S.; Bauer, M.; Fischer, D. Polymers in Drug Delivery—State of the Art and Future Trends. Adv. Eng. Mater. 2011, 13, B61–B87. [Google Scholar] [CrossRef]
- Lammers, T.; Subr, V.; Ulbrich, K.; Hennink, W.E.; Storm, G.; Kiessling, F. Polymeric nanomedicines for image-guided drug delivery and tumor-targeted combination therapy. Nano Today 2010, 5, 197–212. [Google Scholar] [CrossRef]
- Kopeček, J. Polymer–drug conjugates: Origins, progress to date and future directions. Adv. Drug Deliv. Rev. 2012, 65, 49–59. [Google Scholar]
- Haag, R.; Kratz, F. Polymer therapeutics: Concepts and applications. Angew. Chem. Int. Edit. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Ojima, I. Guided molecular missiles for tumor-targeting chemotherapy-case studies using the second-generation taxolds as warheads. Acc. Chem. Res. 2008, 41, 108–119. [Google Scholar] [CrossRef]
- Du, J.Z.; O'Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544–3561. [Google Scholar]
- Gillies, E.R.; Fréchet, J.M.J. Development of acid-sensitive copolymer micelles for drug delivery. Pure Appl. Chem. 2004, 76, 1295–1307. [Google Scholar] [CrossRef]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Nijsen, J.F.W.; Hennink, W.E. Polymeric Micelles in Anticancer Therapy: Targeting, Imaging and Triggered Release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Controlled Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Ruth, D. Polymer therapeutics as nanomedicines: new perspectives. Curr. Opin. Biotech. 2011, 22, 492–501. [Google Scholar]
- Duncan, R.; Vicent, M.J. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv. Drug Delivery Rev. 2010, 62, 272–282. [Google Scholar] [CrossRef]
- van der Poll, D.G.; Kieler-Ferguson, H.M.; Floyd, W.C.; Guillaudeu, S.J.; Jerger, K.; Szoka, F.C.; Fréchet, J.M. Design, synthesis, and biological evaluation of a robust, biodegradable dendrimer. Bioconjugate Chem. 2010, 21, 764–773. [Google Scholar] [CrossRef]
- Brinkhuis, R.P.; Stojanov, K.; Laverman, P.; Eilander, J.; Zuhorn, I.S.; Rutjes, F.P.J.T.; van Hest, J.C.M. Size dependent biodistribution and spect imaging of 111in-labeled polymersomes. Bioconjugate Chem. 2012, 23, 958–965. [Google Scholar]
- Maeda, H. Tumor-Selective Delivery of Macromolecular Drugs via the EPR Effect: Background and Future Prospects. Bioconjugate Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef]
- Reichert, S.; Welker, P.; Calderón, M.; Khandare, J.; Mangoldt, D.; Licha, K.; Kainthan, R.K.; Brooks, D.E.; Haag, R. Size-Dependant Cellular Uptake of Dendritic Polyglycerol. Small 2011, 7, 820–829. [Google Scholar] [CrossRef]
- Fox, M.E.; Szoka, F.C.; Fréchet, J.M.J. Soluble Polymer Carriers for the Treatment of Cancer: The Importance of Molecular Architecture. Acc. Chem. Res. 2009, 42, 1141–1151. [Google Scholar] [CrossRef]
- Qiu, L.Y.; Bae, Y.H. Polymer architecture and drug delivery. Pharm. Res. 2006, 23, 1–30. [Google Scholar] [CrossRef]
- Gillies, E.R.; Dy, E.; Frechet, J.M.J.; Szoka, F.C. Biological evaluation of polyester dendrimer: Poly(ethylene oxide) “Bow-Tie” hybrids with tunable molecular weight and architecture. Mol. Pharm. 2005, 2, 129–138. [Google Scholar]
- Uzgiris, E. The role of molecular conformation on tumor uptake of polymeric contrast agents. Invest. Radiol. 2004, 39, 131–137. [Google Scholar] [CrossRef]
- Edinger, D.; Wagner, E. Bioresponsive polymers for the delivery of therapeutic nucleic acids. WIREs Nanomed. Nanobiotechnol. 2011, 3, 33–46. [Google Scholar] [CrossRef]
- Hinrichs, W.L.J.; Schuurmans-Nieuwenbroek, N.M.E.; van de Wetering, P.; Hennink, W.E. Thermosensitive polymers as carriers for DNA delivery. J. Control. Release 1999, 60, 249–259. [Google Scholar]
- Putnam, D. Polymers for gene delivery across length scales. Nat. Mater. 2006, 5, 439–451. [Google Scholar] [CrossRef]
- Allcock, H.R. Chemistry and Applications of Polyphosphazenes; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Allcock, H.R. Recent developments in polyphosphazene materials science. Curr. Op. Solid St. M. 2006, 10, 231–240. [Google Scholar] [CrossRef]
- Luten, J.; van Steenbergen, M.J.; Lok, M.C.; de Graaff, A.M.; van Nostrum, C.F.; Talsma, H.; Hennink, W.E. Degradable PEG-folate coated poly(DMAEA-co-BA)phosphazene-based polyplexes exhibit receptor-specific gene expression. Eur. J. Pharm. Sci. 2008, 33, 241–251. [Google Scholar] [CrossRef]
- Allcock, H.R.; Steely, L.B.; Singh, A. Hydrophobic and superhydrophobic surfaces from polyphosphazenes. Polym. Int. 2006, 55, 621–625. [Google Scholar] [CrossRef]
- Teasdale, I.; Wilfert, S.; Nischang, I.; Bruggemann, O. Multifunctional and biodegradable polyphosphazenes for use as macromolecular anti-cancer drug carriers. Polym. Chem. 2011, 2, 828–834. [Google Scholar] [CrossRef]
- Zhang, J.X.; Qiu, L.Y.; Jin, Y.; Zhu, K.J. Thermally responsive polymeric micelles self-assembled by amphiphilic polyphosphazene with poly(N-isopropylacrylamide) and ethyl glycinate as side groups: Polymer synthesis, characterization, and in vitro drug release study. J. Biomed. Mater. Res. A 2006, 76A, 773–780. [Google Scholar] [CrossRef]
- Carriedo, G.A.; Crochet, P.; Alonso, F.J.G.; Gimeno, J.; Presa-Soto, A. Synthesis and catalytic activity of (eta(6)-p-cymene)(phosphane)ruthenium(II) complexes supported on poly(biphenoxyphosphazene) or chiral poly(binaphthoxyphosphazene) copolymers. Eur. J. Inorg. Chem. 2004, 18, 3668–3674. [Google Scholar]
- Sethuraman, S.; Nair, L.S.; El-Amin, S.; Nguyen, M.T.; Singh, A.; Krogman, N.; Greish, Y.E.; Allcock, H.R.; Brown, P.W.; Laurencin, C.T. Mechanical properties and osteocompatibility of novel biodegradable alanine based polyphosphazenes: Side group effects. Acta Biomater. 2010, 6, 1931–1937. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- El-Amin, S.F.; Kwon, M.S.; Starnes, T.; Allcock, H.R.; Laurencin, C.T. The biocompatibility of biodegradable glycine containing polyphosphazenes: A comparative study in bone. J. Inorg. Organomet. Polym. Mater. 2006, 16, 387–396. [Google Scholar]
- De Jaeger, R.; Gleria, M. Poly(organophosphazene)s and related compounds: Synthesis, properties and applications. Prog. Polym. Sci. 1998, 23, 179–276. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Chen, J.; LeGolvan, M.P. Poly(dichlorophosphazene) As a Precursor for Biologically Active Polyphosphazenes: Synthesis, Characterization, and Stabilization. Macromolecules 2003, 37, 414–420. [Google Scholar] [CrossRef]
- Carriedo, G.A.; Alonso, F.J.G.; Presa-Soto, A. High molecular weight phosphazene copolymers having chemical functions inside chiral pockets formed by (R)-(1,1'-binaphthyl-2,2'-dioxy)phosphazene units. Eur. J. Inorg. Chem. 2003, 24, 4341–4346. [Google Scholar]
- Mujumdar, A.N.; Young, S.G.; Merker, R.L.; Magill, J.H. A study of solution polymerization of polyphosphazenes. Macromolecules 1990, 23, 14–21. [Google Scholar] [CrossRef]
- Carriedo, G.A.; Alonso, F.L.G.; Gomez-Elipe, P.; Fidalgo, J.I.; Alvarez, J.L.G.; Presa-Soto, A. A simplified and convenient laboratory-scale preparation of N-14 or N-15 high molecular weight poly(dichlorophosphazene) directly from PCl5. Chem. Eur. J. 2003, 9, 3833–3836. [Google Scholar] [CrossRef]
- Sennett, M.S.; Hagnauer, G.L.; Singler, R.E.; Davies, G. Kinetics and mechanism of the boron-trichloride catalyzed thermal ring-opening polymerization of hexachlorocyclotriphosphazene in 1,2,4-trichlorobenzene solution. Macromolecules 1986, 19, 959–964. [Google Scholar] [CrossRef]
- Sohn, Y.S.; Cho, Y.H.; Baek, H.; Jung, O.-S. Synthesis and properties of low molecular weight polyphosphazenes. Macromolecules 1995, 28, 7566–7568. [Google Scholar] [CrossRef]
- Allcock, H.R.; Crane, C.A.; Morrissey, C.T.; Nelson, J.M.; Reeves, S.D.; Honeyman, C.H.; Manners, I. “Living” cationic polymerization of phosphoranimines as an ambient temperature route to polyphosphazenes with controlled molecular weights. Macromolecules 1996, 29, 7740–7747. [Google Scholar]
- Andrianov, A.K.; Chen, J.; LeGolvan, M.P. Poly(dichlorophosphazene) As a Precursor for Biologically Active Polyphosphazenes: Synthesis, Characterization, and Stabilization. Macromolecules 2004, 37, 414–420. [Google Scholar] [CrossRef]
- Blackstone, V.; Lough, A.J.; Murray, M.; Manners, I. Probing the Mechanism of the PCl5-Initiated Living Cationic Polymerization of the Phosphoranimine Cl3P=NSiMe3 using Model Compound Chemistry. JACS 2009, 131, 3658–3667. [Google Scholar] [CrossRef]
- Blackstone, V.; Pfirrmann, S.; Helten, H.; Staubitz, A.; Presa Soto, A.; Whittell, G.R.; Manners, I. A Cooperative Role for the Counteranion in the PCl5-Initiated Living, Cationic Chain Growth Polycondensation of the Phosphoranimine Cl3P=NSiMe3. JACS 2012, 134, 15293–15296. [Google Scholar]
- Allcock, H.R.; Reeves, S.D.; Nelson, J.M.; Crane, C.A.; Manners, I. Polyphosphazene block copolymers via the controlled cationic, ambient temperature polymerization of phosphoranimines. Macromolecules 1997, 30, 2213–2215. [Google Scholar] [CrossRef]
- Allcock, H.R.; Cho, S.Y.; Steely, L.B. New amphiphilic poly[bis(2,2,2-trifluoroethoxy)phosphazene]/poly(propylene glycol) triblock copolymers: Synthesis and micellar characteristics. Macromolecules 2006, 39, 8334–8338. [Google Scholar] [CrossRef]
- Nelson, J.M.; Allcock, H.R. Synthesis of triarmed-star polyphosphazenes via the “living” cationic polymerization of phosphoranimines at ambient temperatures. Macromolecules 1997, 30, 1854–1856. [Google Scholar] [CrossRef]
- Cho, S.Y.; Allcock, H.R. Dendrimers derived from polyphosphazene-poly(propyleneimine) systems: Encapsulation and triggered release of hydrophobic guest molecules. Macromolecules 2007, 40, 3115–3121. [Google Scholar] [CrossRef]
- Allcock, H.R.; Powell, E.S.; Chang, Y.Y.; Kim, C. Synthesis and micellar behavior of amphiphilic polystyrene-poly[bis(methoxyethoxyethoxy)phosphazene] block copolymers. Macromolecules 2004, 37, 7163–7167. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Prange, R.; Allcock, H.R.; Lee, S.C.; Kim, C. Amphiphilic poly[bis(trifluoroethoxy)phosphazene]-poly(ethylene oxide) block copolymers: Synthesis and micellar characteristics. Macromolecules 2002, 35, 8556–8559. [Google Scholar] [CrossRef]
- Cho, S.Y.; Allcock, H.R. Synthesis of Adamantyl Polyphosphazene-Polystyrene Block Copolymers, and beta-Cyclodextrin-Adamantyl Side Group Complexation. Macromolecules 2009, 42, 4484–4490. [Google Scholar] [CrossRef]
- Allcock, H.R.; Nelson, J.M.; Prange, R.; Crane, C.A.; de Denus, C.R. Synthesis of telechelic polyphosphazenes via the ambient temperature living cationic polymerization of amino phosphoranimines. Macromolecules 1999, 32, 5736–5743. [Google Scholar] [CrossRef]
- Allcock, H.R.; Powell, E.S.; Maher, A.E.; Prange, R.L.; de Denus, C.R. Telechelic polyphosphazenes: Reaction of living poly(dichlorophosphazene) chains with alkoxy and aryloxy phosphoranimines. Macromolecules 2004, 37, 3635–3641. [Google Scholar]
- Allcock, H.R.; de Denus, C.R.; Prange, R.; Laredo, W.R. Synthesis of norbornenyl telechelic polyphosphazenes and ring-opening metathesis polymerization reactions. Macromolecules 2001, 34, 2757–2765. [Google Scholar] [CrossRef]
- Montague, R.A.; Green, J.B.; Matyjaszewski, K. The Conversion of Phosphoranimines to Polyphosphazenes in the Presence of Electrophiles. J. Macromol. Sci. Pure Appl. Chem. 1995, A32, 1497–1519. [Google Scholar]
- Matyjaszewski, K.; Moore, M.K.; White, M.L. Synthesis of Polyphosphazene Block-Copolymers Bearing Alkoxyethoxy and Trifluoroethoxy Groups. Macromolecules 1993, 26, 6741–6748. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Miller, P.J.; Fossum, E.; Nakagawa, Y. Synthesis of block, graft and star polymers from inorganic macroinitiators. Appl. Organomet. Chem. 1998, 12, 667–673. [Google Scholar]
- Liu, X.; Tian, Z.; Chen, C.; Allcock, H.R. Synthesis and Characterization of Brush-Shaped Hybrid Inorganic/Organic Polymers Based on Polyphosphazenes. Macromolecules 2012, 45, 1417–1426. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Tian, Z.; Sen, A.; Allcock, H.R. Preparation of quaternized organic-inorganic hybrid brush polyphosphazene-co-poly[2-(dimethylamino)ethyl methacrylate] electrospun fibers and their antibacterial properties. Polym. Chem. 2012, 3, 2082–2091. [Google Scholar] [CrossRef]
- Soto, A.P.; Manners, I. Poly(ferrocenylsilane-b-polyphosphazene) (PFS-b-PP): A New Class of Organometallic-Inorganic Block Copolymers. Macromolecules 2009, 42, 40–42. [Google Scholar] [CrossRef]
- Soto, A.P.; Gilroy, J.B.; Winnik, M.A.; Manners, I. Pointed-Oval-Shaped Micelles from Crystalline-Coil Block Copolymers by Crystallization-Driven Living Self-Assembly. Angew. Chem. Int. Ed. 2010, 49, 8220–8223. [Google Scholar]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(Amino Acid Ester)Phosphazenes] as Substrates for the Controlled-Release of Small Molecules. Biomaterials 1994, 15, 563–569. [Google Scholar] [CrossRef]
- Allcock, H.R.; Fuller, T.J.; Mack, D.P.; Matsumura, K.; Smeltz, K.M. Synthesis of Poly[(Amino Acid Alkyl Ester)Phosphazenes]. Macromolecules 1977, 10, 824–830. [Google Scholar] [CrossRef]
- Ibim, S.E.M.; Ambrosio, A.M.A.; Kwon, M.S.; El-Amin, S.F.; Allcock, H.R.; Laurencin, C.T. Novel polyphosphazene/poly(lactide-co-glycolide) blends: miscibility and degradation studies. Biomaterials 1997, 18, 1565–1569. [Google Scholar]
- Allcock, H.R.; Morozowich, N.L. Bioerodible polyphosphazenes and their medical potential. Polym. Chem. 2012, 3, 578–590. [Google Scholar] [CrossRef]
- DeCollibus, D.P.; Marin, A.; Andrianov, A.K. Effect of Environmental Factors on Hydrolytic Degradation of Water-Soluble Polyphosphazene Polyelectrolyte in Aqueous Solutions. Biomacromolecules 2010, 11, 2033–2038. [Google Scholar] [CrossRef]
- Andrianov, A.K. Water-soluble polyphosphazenes for biomedical applications. J. Inorg. Organomet. Polym. Mater. 2006, 16, 397–406. [Google Scholar]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Synthesis of poly(orgnaophosphazenes) with glycolic acid ester and lactic acid ester side groups: Prototypes for new bioerodible polymers. Macromolecules 1994, 27, 1–4. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kwon, S. Glyceryl polyphosphazenes: Synthesis, properties, and hydrolysis. Macromolecules 1988, 21, 1980–1985. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R. Polyphosphazenes with glucosyl and methylamino, trifluoroethoxy, phenoxy, or (methoxyethoxy)ethoxy side groups. Macromolecules 1991, 24, 23–34. [Google Scholar] [CrossRef]
- Allcock, H.R.; Fuller, T.J.; Matsumura, K. Hydrolysis pathways for aminophosphazenes. Inorg. Chem. 1982, 21, 515–521. [Google Scholar] [CrossRef]
- Carriedo, G.A.; García Alonso, F.J.; García Álvarez, J.L.; Presa Soto, A.; Tarazona, M.P.; Laguna, M.T.R.; Marcelo, G.; Mendicuti, F.; Saiz, E. Experimental and Theoretical Study of the Acidic Degradation of Poly(2,2′-dioxy-1,1′-biphenyl)phosphazene. Macromolecules 2008, 41, 8483–8490. [Google Scholar]
- Luten, J.; van Steenis, J.H.; van Someren, R.; Kemmink, J.; Schuurmans-Nieuwenbroek, N.M.E.; Koning, G.A.; Crommelin, D.J.A.; van Nostrum, C.F.; Hennink, W.E. Water-soluble biodegradable cationic polyphosphazenes for gene delivery. J. Control. Release 2003, 89, 483–497. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(Amino-Acid-Ester)Phosphazenes]—Synthesis, Crystallinity, and Hydrolytic Sensitivity in Solution and the Solid-State. Macromolecules 1994, 27, 1071–1075. [Google Scholar] [CrossRef]
- Laurencin, C.T.; Norman, M.E.; Elgendy, H.M.; El-Amin, S.F.; Allcock, H.R.; Pucher, S.R.; Ambrosio, A.A. Use of polyphosphazenes for skeletal tissue regeneration. J. Biomed. Mater. Res. 1993, 27, 963–973. [Google Scholar] [CrossRef]
- Lakshmi, S.; Katti, D.S.; Laurencin, C.T. Biodegradable polyphosphazenes for drug delivery applications. Adv. Drug Deliv. Rev. 2003, 55, 467–482. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Peterson, P. Water-Soluble Biodegradable Polyphosphazenes Containing N-Ethylpyrrolidone Groups. Macromolecules 2005, 38, 7972–7976. [Google Scholar]
- Deng, M.; Nair, L.S.; Nukavarapu, S.P.; Jiang, T.; Kanner, W.A.; Li, X.; Kumbar, S.G.; Weikel, A.L.; Krogman, N.R.; Allcock, H.R.; Laurencin, C.T. Dipeptide-based polyphosphazene and polyester blends for bone tissue engineering. Biomaterials 2010, 31, 4898–4908. [Google Scholar]
- Nukavarapu, S.P.; Kumbar, S.G.; Allcock, H.R.; Laurencin, C.T. Biodegradable Polyphosphazene Scaffolds for Tissue engineering. In Polyphosphazenes for Biomedical Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 117–138. [Google Scholar]
- Deng, M.; Kumbar, S.G.; Wan, Y.; Toti, U.S.; Allcock, H.R.; Laurencin, C.T. Polyphosphazene polymers for tissue engineering: An analysis of material synthesis, characterization and applications. Soft Matter 2010, 6, 3119–3132. [Google Scholar] [CrossRef]
- Morozowich, N.L.; Nichol, J.L.; Mondschein, R.J.; Allcock, H.R. Design and examination of an antioxidant-containing polyphosphazene scaffold for tissue engineering. Polym. Chem. 2012, 3, 778–786. [Google Scholar] [CrossRef]
- Allcock, H.R.; Austin, P.E.; Neenan, T.X. Phosphazene high polymers with bioactive substituent groups: prospective anesthetic aminophosphazenes. Macromolecules 1982, 15, 689–693. [Google Scholar] [CrossRef]
- Laurencin, C.T.; Koh, H.J.; Neenan, T.X.; Allcock, H.R.; Langer, R. Controlled release using a new bioerodible polyphosphazene matrix system. J. Biomed. Mater. Res. 1987, 21, 1231–1246. [Google Scholar] [CrossRef]
- Ibim, S.M.; el-Amin, S.F.; Goad, M.E.; Ambrosio, A.M.; Allcock, H.R.; Laurencin, C.T. In vitro release of colchicine using poly(phosphazenes): The development of delivery systems for musculoskeletal use. Pharm. Dev. Technol. 1998, 3, 55–62. [Google Scholar] [CrossRef]
- Conforti, A.; Bertani, S.; Lussignoli, S.; Grigolini, L.; Terzi, M.; Lora, S.; Caliceti, P.; Marsilio, F.; Veronese, F.M. Anti-inflammatory activity of polyphosphazene-based naproxen slow-release systems. J. Pharm. Pharmacol. 1996, 48, 468–473. [Google Scholar] [CrossRef]
- Veronese, F.M.; Marsilio, F.; Caliceti, P.; de Filippis, P.; Giunchedi, P.; Lora, S. Polyorganophosphazene microspheres for drug release: Polymer synthesis, microsphere preparation, in vitro and in vivo naproxen release. J. Control. Release 1998, 52, 227–237. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Payne, L.G. Protein release from polyphosphazene matrices. Adv. Drug Deliv. Rev. 1998, 31, 185–196. [Google Scholar] [CrossRef]
- Ibim, S.M.; Ambrosio, A.A.; Larrier, D.; Allcock, H.R.; Laurencin, C.T. Controlled macromolecule release from poly(phosphazene) matrices. J. Control. Release 1996, 40, 31–39. [Google Scholar] [CrossRef]
- Caliceti, P.; Veronese, F.M.; Lora, S. Polyphosphazene microspheres for insulin delivery. Int. J. Pharm. 2000, 211, 57–65. [Google Scholar] [CrossRef]
- Song, S.C.; Lee, S.B.; Jin, J.I.; Sohn, Y.S. A new class of biodegradable thermosensitive polymers. I. Synthesis and characterization of poly(organophosphazenes) with methoxy-poly(ethylene glycol) and amino acid esters as side groups. Macromolecules 1999, 32, 2188–2193. [Google Scholar] [CrossRef]
- Cohen, S.; Bano, M.C.; Visscher, K.B.; Chow, M.; Allcock, H.R.; Langer, R. Ionically cross-linkable polyphosphazene—a novel polymer for microencapsulation. JACS 1990, 112, 7832–7833. [Google Scholar] [CrossRef]
- Bano, M.C.; Cohen, S.; Visscher, K.B.; Allcock, H.R.; Langer, R. A novel synthetic method for hybridoma cell encapsulation. Biotechnology 1991, 9, 468–471. [Google Scholar] [CrossRef]
- Cohen, S.; Bano, M.C.; Cima, L.G.; Allcock, H.R.; Vacanti, J.P.; Vacanti, C.A.; Langer, R. Design of synthetic polymeric structures for cell transplantation and tissue engineering. Clin. Mater 1993, 13, 3–10. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Cohen, S.; Visscher, K.B.; Payne, L.G.; Allcock, H.R.; Langer, R. Controlled-release using ionotropic polyphosphazene hydrogels. J. Control. Release 1993, 27, 69–77. [Google Scholar] [CrossRef]
- Goedemoed, J.H.; Degroot, K.; Claessen, A.M.E.; Scheper, R.J. development of implantable antitumor devices based on polyphosphazenes 2. J. Control. Release 1991, 17, 235–244. [Google Scholar] [CrossRef]
- Schacht, E.; Vandorpe, J.; Dejardin, S.; Lemmouchi, Y.; Seymour, L. Biomedical applications of degradable polyphosphazenes. Biotechnol. Bioeng. 1996, 52, 102–108. [Google Scholar]
- Goedemoed, J.H.; Mense, E.H.G.; Degroot, K.; Claessen, A.M.E.; Scheper, R.J. Development of injectable antitumor microspheres based on polyphosphazene. J. Control. Release 1991, 17, 245–257. [Google Scholar]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.J.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar]
- Yang, H.; Lopina, S.T.; DiPersio, L.P.; Schmidt, S.P. Stealth dendrimers for drug delivery: correlation between PEGylation, cytocompatibility, and drug payload. J. Mater. Sci. Mater. M. 2008, 19, 1991–1997. [Google Scholar] [CrossRef]
- Allcock, H.R.; Allen, R.W.; Obrien, J.P. Synthesis of Platinum Derivatives of Polymeric and Cyclic Phosphazenes. JACS 1977, 99, 3984–3987. [Google Scholar] [CrossRef]
- Allen, R.W.; Obrien, J.P.; Allcock, H.R. Crystal and Molecular-Structure of a Platinum-Cyclophosphazene Complex—Cis-Dichloro[Octa-(Methylamino)Cyclotetraphosphazene-N,N'']Platinum(Ii). JACS 1977, 99, 3987–3991. [Google Scholar] [CrossRef]
- Sohn, Y.S.; Baek, H.; Cho, Y.H.; Lee, Y.A.; Jung, O.S.; Lee, C.O.; Kim, Y.S. Synthesis and antitumor activity of novel polyphosphazene-(diamine)platinum(II) conjugates. Int. J. Pharm. 1997, 153, 79–91. [Google Scholar] [CrossRef]
- Lee, S.B.; Song, S.C.; Jin, J.I.; Sohn, Y.S. Synthesis and antitumor activity of polyphosphazene/methoxy-poly(ethylene glycol)/(diamine)platinum(II) conjugates. Polym. J. 1999, 31, 1247–1252. [Google Scholar] [CrossRef]
- Song, R.; Jun, Y.J.; Kim, J.I.; Jin, C.; Sohn, Y.S. Synthesis, characterization, and tumor selectivity of a polyphosphazene-platinum(II) conjugate. J. Control. Release 2005, 105, 142–150. [Google Scholar] [CrossRef]
- Jun, Y.J.; Kim, J.I.; Jun, M.J.; Sohn, Y.S. Selective tumor targeting by enhanced permeability and retention effect. Synthesis and antitumor activity of polyphosphazene-platinum (II) conjugates. J. Inorg. Biochem. 2005, 99, 1593–1601. [Google Scholar] [CrossRef]
- Schwach, G.; Horvarth, C.; Wilfert, S.; Schoefberger, W.; Brueggemann, O.; Pfragner, R.; Teasdale, I. An in vitro investigation of epirubicin-polyphosphazene polymer conjugates. 2013; unpublished work. [Google Scholar]
- Teasdale, I.; Waser, M.; Wilfert, S.; Falk, H.; Brueggemann, O. Photoreactive, water-soluble conjugates of hypericin with polyphosphazenes. Mon. Chem. 2012, 143, 355–360. [Google Scholar] [CrossRef]
- Allen, C.W. Regiochemical and stereochemical control in substitution-reactions of cyclophosphazenes. Chem. Rev. 1991, 91, 119–135. [Google Scholar]
- Cho, Y.W.; Lee, J.R.; Song, S.C. Novel thermosensitive 5-fluorouracil-cyclotriphosphazene conjugates: Synthesis, thermosensitivity, degradability, and in vitro antitumor activity. Bioconjugate Chem. 2005, 16, 1529–1535. [Google Scholar] [CrossRef]
- Song, S.C.; Lee, S.B.; Lee, B.H.; Ha, H.W.; Lee, K.T.; Sohn, Y.S. Synthesis and antitumor activity of novel thermosensitive platinum(II)-cyclotriphosphazene conjugates. J. Control. Release 2003, 90, 303–311. [Google Scholar] [CrossRef]
- Baek, H.; Cho, Y.; Lee, C.O.; Sohn, Y.S. Synthesis and antitumor activity of cyclotriphosphazene-(diamine)platinum(II) conjugates. Anti-Cancer Drugs 2000, 11, 715–725. [Google Scholar] [CrossRef]
- Jadhav, V.B.; Jun, Y.J.; Song, J.H.; Park, M.K.; Oh, J.H.; Chae, S.W.; Kim, I.S.; Choi, S.J.; Lee, H.J.; Sohn, Y.S. A novel micelle-encapsulated platinum(II) anticancer agent. J. Control. Release 2010, 147, 144–150. [Google Scholar] [CrossRef]
- Yu, J.Y.; Jun, Y.J.; Jang, S.H.; Lee, H.J.; Sohn, Y.S. Nanoparticulate platinum(II) anticancer drug: Synthesis and characterization of amphiphilic cyclotriphosphazene–platinum(II) conjugates. J. Inorg. Biochem. 2007, 101, 1931–1936. [Google Scholar]
- Jun, Y.J.; Jadhav, V.B.; Min, J.H.; Cui, J.X.; Chae, S.W.; Choi, J.M.; Kim, I.-S.; Choi, S.-J.; Lee, H.J.; Sohn, Y.S. Stable and efficient delivery of docetaxel by micelle-encapsulation using a tripodal cyclotriphosphazene amphiphile. Int. J. Pharm. 2012, 422, 374–380. [Google Scholar] [CrossRef]
- Li, X.; Li, B.; Li, Z.; Zhang, S. Self-assembly of nanoparticles from cyclotriphosphazenes grafted with hexa-[p-(carbonyl tryptophan ethyl ester) phenoxy)] group. RSC Adv. 2012, 2, 5997–6004. [Google Scholar]
- Chang, Y.; Lee, S.C.; Kim, K.T.; Kim, C.; Reeves, S.D.; Allcock, H.R. Synthesis and Micellar Characterization of an Amphiphilic Diblock Copolyphosphazene. Macromolecules 2000, 34, 269–274. [Google Scholar]
- Zhang, J.X.; Qiu, L.Y.; Zhu, K.J.; Jin, Y. Thermosensitive Micelles Self-Assembled by Novel N-Isopropylacrylamide Oligomer Grafted Polyphosphazene. Macromol. Rapid Commun. 2004, 25, 1563–1567. [Google Scholar] [CrossRef]
- Zhang, J.X.; Qiu, L.Y.; Wu, X.L.; Jin, Y.; Zu, K.J. Temperature-triggered nanosphere formation through self-assembly of amphiphilic polypho sphazene. Macromol. Chem. Phys. 2006, 207, 1289–1296. [Google Scholar] [CrossRef]
- Zhang, R.X.; Li, X.J.; Qiu, L.Y.; Li, X.H.; Yan, M.Q.; Jin, Y.; Zhu, K.J. Indomethacin-loaded polymeric nanocarriers based on amphiphilic polyphosphazenes with poly (N-isopropylacrylamide) and ethyl tryptophan as side groups: Preparation, in vitro and in vivo evaluation. J. Control. Release 2006, 116, 322–329. [Google Scholar] [CrossRef]
- Kang, G.D.; Cheon, S.H.; Song, S.C. Controlled release of doxorubicin from thermosensitive poly(organophosphazene) hydrogels. Int. J. Pharm. 2006, 319, 29–36. [Google Scholar] [CrossRef]
- Lee, S.M.; Chun, C.J.; Heo, J.Y.; Song, S.C. Injectable and Thermosensitive Poly(organophosphazene) Hydrogels for a 5-Fluorouracil Delivery. J. Appl. Polym. Sci. 2009, 113, 3831–3839. [Google Scholar] [CrossRef]
- Kang, G.D.; Heo, J.-Y.; Jung, S.B.; Song, S.-C. Controlling the Thermosensitive Gelation Properties of Poly(organophosphazenes) by Blending. Macromol. Rapid Commun. 2005, 26, 1615–1618. [Google Scholar] [CrossRef]
- Chun, C.; Lee, S.M.; Kim, C.W.; Hong, K.Y.; Kim, S.Y.; Yang, H.K.; Song, S.C. Doxorubicin-polyphosphazene conjugate hydrogels for locally controlled delivery of cancer therapeutics. Biomaterials 2009, 30, 4752–4762. [Google Scholar] [CrossRef]
- Cho, J.K.; Park, J.W.; Song, S.C. Injectable and Biodegradable Poly(organophosphazene) Gel Containing Silibinin: Its Physicochemical Properties and Anticancer Activity. J. Pharm. Sci. 2012, 101, 2382–2391. [Google Scholar] [CrossRef]
- Potta, T.; Chun, C.; Song, S.C. Chemically crosslinkable thermosensitive polyphosphazene gels as injectable materials for biomedical applications. Biomaterials 2009, 30, 6178–6192. [Google Scholar] [CrossRef]
- Park, M.-R.; Chun, C.; Ahn, S.-W.; Ki, M.-H.; Cho, C.-S.; Song, S.-C. Cationic and thermosensitive protamine conjugated gels for enhancing sustained human growth hormone delivery. Biomaterials 2010, 31, 1349–1359. [Google Scholar] [CrossRef]
- Schatzlein, A.G. Non-viral vectors in cancer gene therapy: principles and progress. Anti-cancer Drugs 2001, 12, 275–304. [Google Scholar] [CrossRef]
- de Wolf, H.K.; Luten, J.; Snel, C.J.; Oussoren, C.; Hennink, W.E.; Storm, G. In vivo tumor transfection mediated by polyplexes based on biodegradable poly(DMAEA)-phosphazene. J. Control. Release 2005, 109, 275–287. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, Z.; Jiang, J.; Gao, Y.; Gu, W.; Chen, L.; Tang, X.; Li, Y. Poly(imidazole/DMAEA)phosphazene/DNA self-assembled nanoparticles for gene delivery: Synthesis and in vitro transfection. J. Control. Release 2008, 127, 273–279. [Google Scholar] [CrossRef]
- Yang, Y.X.; Zhang, Z.W.; Chen, L.L.; Gu, W.W.; Li, Y.P. Galactosylated Poly(2-(2-aminoethyoxy)ethoxy)phosphazene/DNA Complex Nanoparticles: In Vitro and In Vivo Evaluation for Gene Delivery. Biomacromolecules 2010, 11, 927–933. [Google Scholar] [CrossRef]
- de Wolf, H.K.; de Raad, M.; Snel, C.; van Steenbergen, M.J.; Fens, M.; Storm, G.; Hennink, W.E. Biodegradable poly(2-dimethylamino ethylamino)phosphazene for in vivo gene delivery to tumor cells. Effect of polymer molecular weight. Pharm. Res. 2007, 24, 1572–1580. [Google Scholar] [CrossRef]
- Qiu, L.; Zheng, C.; Zhao, Q. Mechanisms of Drug Resistance Reversal in Dox-Resistant MCF-7 Cells by pH-Responsive Amphiphilic Polyphosphazene Containing Diisopropylamino Side Groups. Mol. Pharm. 2012, 9, 1109–1117. [Google Scholar]
- Schijns, V.; Lavelle, E.C. Trends in vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 539–550. [Google Scholar] [CrossRef]
- Eng, N.F.; Garlapati, S.; Gerdts, V.; Potter, A.; Babiuk, L.A.; Mutwiri, G.K. The potential of polyphosphazenes for delivery of vaccine antigens and immunotherapeutic agents. Curr. Drug Deliv. 2010, 7, 13–20. [Google Scholar] [CrossRef]
- Andrianov, A.K. Polyphosphazenes as Vaccine Adjuvants. In Vaccine Adjuvants and Delivery Systems; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 355–378. [Google Scholar]
- Awate, S.; Wilson, H.L.; Lai, K.; Babiuk, L.A.; Mutwiri, G. Activation of adjuvant core response genes by the novel adjuvant PCEP. Mol. Immunol. 2012, 51, 292–303. [Google Scholar] [CrossRef]
- Andrianov, A.K. Polyphosphazenes for Biomedical Applications; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Dar, A.; Lai, K.; Dent, D.; Potter, A.; Gerdts, V.; Babiuk, L.A.; Mutwiri, G.K. Administration of poly di(sodium carboxylatoethylphenoxy) phosphazene (PCEP) as adjuvant activated mixed Th1/Th2 immune responses in pigs. Veterinary Immunol. Immunop. 2012, 146, 289–295. [Google Scholar]
- Payne, L.G.; Jenkins, S.A.; Woods, A.L.; Grund, E.M.; Geribo, W.E.; Loebelenz, J.R.; Andrianov, A.K.; Roberts, B.E. Poly di(carboxylatophenoxy)phosphazene (PCPP) is a potent immunoadjuvant for an influenza vaccine. Vaccine 1998, 16, 92–98. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Teasdale, I.; Brüggemann, O. Polyphosphazenes: Multifunctional, Biodegradable Vehicles for Drug and Gene Delivery. Polymers 2013, 5, 161-187. https://doi.org/10.3390/polym5010161
Teasdale I, Brüggemann O. Polyphosphazenes: Multifunctional, Biodegradable Vehicles for Drug and Gene Delivery. Polymers. 2013; 5(1):161-187. https://doi.org/10.3390/polym5010161
Chicago/Turabian StyleTeasdale, Ian, and Oliver Brüggemann. 2013. "Polyphosphazenes: Multifunctional, Biodegradable Vehicles for Drug and Gene Delivery" Polymers 5, no. 1: 161-187. https://doi.org/10.3390/polym5010161