Thermal Properties of Aliphatic Polypeptoids

1
Professur für Makromolekulare Chemie, Department Chemie, Technische Universität Dresden, Zellescher Weg 19, 01062 Dresden, Germany
2
Functional Polymer Materials, Chair of Chemical Technology of Materials Synthesis, University Würzburg, Röntgenring 11, 97070 Würzburg, Germany
*
Author to whom correspondence should be addressed.
Polymers 2013, 5(1), 112-127; https://doi.org/10.3390/polym5010112
Submission received: 19 December 2012
/
Revised: 18 January 2013
/
Accepted: 21 January 2013
/
Published: 29 January 2013
Abstract
:A series of polypeptoid homopolymers bearing short (C1–C5) side chains of degrees of polymerization of 10–100 are studied with respect to thermal stability, glass transition and melting points. Thermogravimetric analysis of polypeptoids suggests stability to >200 °C. The study of the glass transition temperatures by differential scanning calorimetry revealed two dependencies. On the one hand an extension of the side chain by constant degree of polymerization decrease the glass transition temperatures (Tg) and on the other hand a raise of the degree of polymerization by constant side chain length leads to an increase of the Tg to a constant value. Melting points were observed for polypeptoids with a side chain comprising not less than three methyl carbon atoms. X-ray diffraction of polysarcosine and poly(N-ethylglycine) corroborates the observed lack of melting points and thus, their amorphous nature. Diffractograms of the other investigated polypeptoids imply that crystalline domains exist in the polymer powder.
1. Introduction
At the beginning of the 20th century Leuchs [1,2,3] discovered by chance a synthesis of α-amino acid N-carboxy anhydrides (NCAs), also known as Leuchs anhydrides. Subsequently, this class of molecules would be applied for the ring-opening polymerization to obtain synthetic polypeptides. Since the early 1950s many publications have dealt with mechanistic studies on the ring-opening polymerization of NCAs [4,5,6,7,8,9,10,11]. In the course of this, the class of N-substituted glycine N-carboxy anhydrides (NNCAs), represented solely by sarcosine-NCA (N-methylglycine-NCA), served predominantly to investigate the influence of amide proton on the polymerization. Other members of the resulting class of polymer (polypeptoids) were virtually unknown to polymer chemists until very recently. Sisido et al. only briefly mentioned poly(n-propylglycine) and poly(ethylglycine), without providing any characterization [12,13,14]. This limited interest in this polymer family may be attributed to an early report by Wessely et al., in which it was suggested that NNCAs are exceedingly sensitive to hydrolysis [5].
In the meantime, Sisido et al. [15], Okada et al. [16] and Kricheldorf et al. [17] demonstrated that the polymerization of sarcosine-NCA could be controlled more readily as compared to regular amino acid NCAs. More recently, Zhang and co-workers found that N-substituted NCAs could be polymerized yielding cyclic polypeptoids with excellent yield in a well-controlled manner [18,19,20,21].
The living character of the nucleophilic ring-opening polymerization (NuLROP) of NNCAs other than Sar-NCA was shown by Luxenhofer and co-workers [22]. In particular, the successful preparation of a pentablock quinquiespolymer highlighted the unique character of the NNCA NuLROP [23]. Moreover, efficient polymer analogue modification [24,25] and thermo-responsiveness [26,27] of polypeptoids was described.
The fact that polypeptoids can be prepared to yield highly defined linear or cyclic oligomer/polymers via step-wise solid phase synthesis or living ring-opening polymerization makes this class of polymer rather unique, as outlined in a recent review by Zhang et al. [28].
As this polymer class is rather new, little is known about the materials properties. Interestingly, Segalman studied a series of short sequence-specific polypeptoids (obtained by solid phase synthesis) in their crystallization and melting behavior [29]. As expected, melting points are lower than those of polypeptides (attributed to the lack of intermolecular H-bonding) and the crystallization behavior is strongly affected by the polymer side chain.
Here, we present a systematic study of the thermal stability, glass transition and melting points of polypeptoids bearing short (C1–C5) side chains of degrees of polymerization of 10–100. X-Ray diffraction studies confirm that polysarcosine and poly(N-ethylglycine) are amorphous solids while longer side chain yield semicrystalline materials.
2. Experimental Section
2.1. Materials
All the solvents and substances for the preparation of monomers and polymers were purchased from Aldrich or Acros and were used as received unless otherwise stated. Benzonitrile (BN) was dried by refluxing over P2O5, benzylamine over BaO and petrolether over CaH2 under dry argon atmosphere and subsequent distillation prior to use. Water levels were determined using a C20 compact coulometer (Mettler-Toledo, Giessen, Germany). In general, solvents were used at water levels <30 ppm. The monomers were handled preferably in a glovebox (UNIlab, MBraun, Garching, Germany).
2.2. Methods
Gel permeation chromatography (GPC) was performed on a PL-GPC-120 (Polymer Laboratories) running under WinGPC software (PSS, Mainz, Germany) with two consecutive Gram columns (100 and 1000 Å) with N,N-dimethylacetaminde (DMAc) (5 g/L LiBr, 70 °C, 1 mL/min) as eluent and calibrated against PMMA standards (PSS, Mainz, Germany).
NMR spectra were recorded on a Bruker DRX 500 at room temperature (295 K). The spectra were calibrated using the solvent signals (D2O 4.79 ppm, MeOD-d3 3.31 ppm, DMSO-d6 2.50, MeCN-d3 1.94 ppm).
Matrix assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectra were recorded on a Bruker Biflex IV (Bruker Daltonics, Bremen, Germany) using a N2 laser (λ = 337 nm). All spectra were recorded in positive reflector mode. The ions were accelerated using a potential of 19 kV and reflected using a voltage of 20 kV. Detection was typically set from 350 m/z to 10000 m/z with a matrix suppression of typically 350–1000 m/z. After parameter optimization for each measurement, the instrument was calibrated with Peptide Calibration Standard II and/or Oligonucleotide Calibration Standard (Bruker), depending on the m/z range of the individual sample. Samples were prepared with sinapinic acid (3,5-dimethoxy-4-hydroxycinnamic acid, SA) as matrix using the dried–droplet spotting technique (0.5–1 µL). Exemplarily, samples (1 g/L) were dissolved in methanol (supplemented with 1% (v/v) trifluoroacetic acid (TFA)). The solution was mixed 1:1 (v/v) with 20 g/L SA MeOH/1% TFA. Laser power was set slightly above the threshold, typically at 50%. Gauss distributions were calculated using Equation (1), where µ is the degree of polymerization (DP) obtained from the signal with highest intensity (MP). The obtained probabilities are plotted against the calculated m/z at the respective DP and overlaid with the experimental MALDI-ToF mass spectra to estimate the molar mass (Mn, Mw) and the dispersity of the polymers (ÐM).
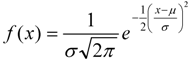
Thermogravimetric analysis was realized with a TG 50 modular unit and a TC 15 TA controller (Mettler-Toledo, Giessen, Germany).
DSC studies of the polypeptoids were conducted using a DSC 4 calorimeter (Perkin Elmer) under nitrogen. Powder samples sealed into the aluminum pans were first heated from 0 °C (except poly(N-butylglycine)25 and poly(N-pentylglycine)25 which started by −25 °C) to 250 °C at 5 K min−1, depending on the decomposition temperature of the sample, cooled to 0 °C by use of air cooling (~24 K min−1). Some additional measurements were conducted using a DSC 204 F1 Phoenix (Netzsch). Experimental data of glass transition temperature against the DP were fitted using Flory-Fox Equation (2).
Tg = Tg∞ – A/Mn
With Mn as derived from 1H-NMR and parameters listed in Table 1.
X-ray diffraction measurements were performed on the powder diffractometer Stadi P from STOE using the Cu-Kα radiation wavelength (λ = 1.54 Å). Measuring range comprised 10 to 80 2θ.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | Tg | |
|---|---|---|
| PSar | 71,000 K·g/mol | 428 K |
| P(N-EtGly) | 52,000 K·g/mol | 396 K |
| P(N-PrGly) | 69,000 K·g/mol | 373 K |
| P(N-PenGly) | 35,000 K·g/mol | 283 K |
2.3. Synthetic Procedures
2.3.1. Monomer Synthesis
Sarcosine-NCA:
Sarcosine-N-carboxyanhydride was synthesized in tetrahydrofuran (THF) in the presence of triphosgene as described previously [22].
N-Ethylglycine-, N-n-propylglycine-, N-n-butylglycine- and N-n-pentylglycine-NCA:
2.3.2. Preparation of Polymers
Poly(sarcosine)25 P2.
Sar-NCA (0.265 g, 2.3 mmol) was weighed into reaction vessel dissolved in 2.3 mL of dry benzonitrile. After complete dissolution, the initiator benzylamine (10.0 µL, 0.09 mmol) was added ([M]0/[I]0 = 25). Outside of the glovebox the reaction mixture was stirred at room temperature under constant pressure (20 mbar) for 2 h. Afterwards, reaction mixture was precipitated into 40 mL diethyl ether and isolated polysarcosine was dried under reduced pressure. After two precipitation steps polysarcosine was dissolved in water and subsequently freeze-dried (0.135 g, 83%).
GPC (DMAc): Mn = 1.3 kg/mol (ÐM = Mw/Mn = 1.31).
1H NMR (500 MHz; D2O): δ = 2.97 (66 H, br, CH3–), 4.29 (46 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.35 ppm (5 H, br, C6H5–).
All other polymers were synthesized accordingly.
Poly(sarcosine)10 P1.
1H NMR (500 MHz; D2O): δ = 2.99 (23 H, br, CH3–), 4.28 (17 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.33 ppm (5 H, br, C6H5–).
Poly(sarcosine)50 P3.
GPC (DMAc): Mn = 4.0 kg/mol (ÐM = Mw/Mn = 1.09).
1H NMR (500 MHz; D2O): δ = 2.96 (135 H, br, CH3–), 4.28 (90 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.32 ppm (5 H, br, C6H5–).
Poly(sarcosine)100 P4.
GPC (DMAc): Mn = 8.9 kg/mol (ÐM = Mw/Mn = 1.05).
1H NMR (500 MHz; D2O): δ = 2.95 (219 H, br, CH3–), 4.28 (146 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.33 ppm (5 H, br, C6H5–).
Poly(N-ethylglycine)10 P5.
1H NMR (500 MHz; D2O): δ = 1.14 (32 H, br, CH3–), 3.37 (19 H, br, CH3–CH2–), 4.25 (23 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.32 ppm (5 H, br, C6H5–).
Poly(N-ethylglycine)25 P6.
GPC (DMAc): Mn = 1.0 kg/mol (ÐM = Mw/Mn = 1.65).
1H NMR (500 MHz; D2O): δ = 1.14 (70 H, br, CH3–), 3.38 (45 H, br, CH3–CH2–), 4.26 (48 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.32 ppm (5 H, br, C6H5–).
Poly(N-ethylglycine)50 P7.
GPC (DMAc): Mn = 3.3 kg/mol (ÐM = Mw/Mn = 1.27).
1H NMR (500 MHz; D2O): δ = 1.14 (116 H, br, CH3–), 3.38 (73 H, br, CH3–CH2–), 4.22 (74 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.32 ppm (5 H, br, C6H5–).
Poly(N-ethylglycine)100 P8.
GPC (DMAc): Mn = 6.8 kg/mol (ÐM = Mw/Mn = 1.22).
1H NMR (500 MHz; D2O): δ = 1.14 (252 H, br, CH3–), 3.38 (164 H, br, CH3–CH2–), 4.25 (170 H, br, –CH2-CO- and C6H5–CH2–NH–), 7.32 ppm (5 H, br, C6H5–).
Poly(N-npropylglycine)10 P9.
GPC (DMAc): Mn = 0.6 kg/mol (ÐM = Mw/Mn = 1.58).
1H NMR (500 MHz; MeOD-d3): δ = 0.92 (38 H, br, CH3–), 1.60 (25 H, br, CH3–CH2–), 3.33 (n.d., br, CH3–CH2–CH2–), 4.25 (25 H, br, –CH2–CO– and C6H5–CH–NH–), 7.25 ppm (5 H, br, C6H5–).
Poly(N-npropylglycine)25 P10.
GPC (DMAc): Mn = 1.6 kg/mol (ÐM = Mw/Mn = 1.32).
1H NMR (500 MHz; MeOD-d3): δ = 0.92 (66 H, br, CH3–), 1.60 (45 H, br, CH3–CH2–), 3.32 (n.d., br, CH3–CH2–CH2–), 4.25 (43 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.25 ppm (5 H, br, C6H5–).
Poly(N-npropylglycine)50 P11.
GPC (DMAc): Mn = 5.0 kg/mol (ÐM = Mw/Mn = 1.22).
1H NMR (500 MHz; MeCN-d3): δ = 0.86 (50 H, br, CH3–), 1.52 (37 H, br, CH3–CH2–), 3.23 (32 H, br, CH3–CH2–CH2–), 4.10 (39 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.28 ppm (5 H, br, C6H5–).
Poly(N-npropylglycine)100 P12.
GPC (DMAc): Mn = 7.5 kg/mol (ÐM = Mw/Mn = 1.15).
1H NMR (500 MHz; MeOD-d3): δ = 0.92 (222 H, br, CH3–), 1.62 (149 H, br, CH3–CH2–), 3.33 (n.d., br, CH3–CH2–CH2–), 4.27 (146 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.26 ppm (5 H, br, C6H5–).
Poly(N-nbutylglycine)10 P13.
GPC (DMAc): Mn = 0.9 kg/mol (ÐM = Mw/Mn = 1.55).
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.87 (33 H, br, CH3–), 1.30 (23 H, br, CH3–CH2–), 1.61 (22 H, br, CH3–CH2–CH2–), 3.33 (22 H, br, CH3–(CH2)2–CH2–), 4.38 (25 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.17 ppm (5 H, br, C6H5–).
Poly(N-nbutylglycine)25 P14.
GPC (DMAc): Mn = 1.9 kg/mol (ÐM = Mw/Mn = 1.27).
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.86 (60 H, br, CH3–), 1.43 (82 H, br, CH3–CH2–CH2–), 3.34 (40 H, br, CH3–(CH2)2–CH2–), 4.32 (39 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
Poly(N-nbutylglycine)50 P15.
GPC (DMAc): Mn = 3.2 kg/mol (ÐM = Mw/Mn = 1.40).
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.87 (113 H, br, CH3–), 1.31 (77 H, br, CH3–CH2–), 1.62 (75 H, br, CH3–CH2–CH2–), 3.36 (76 H, br, CH3–(CH2)2– CH2–), 4.39 (80 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
Poly(N-nbutylglycine)100 P16.
GPC (DMAc): Mn = 4.2 kg/mol (ÐM = Mw/Mn = 1.16).
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.87 (172 H, br, CH3–), 1.30 (122 H, br, CH3–CH2–), 1.62 (115 H, br, CH3–CH2–CH2–), 3.36 (117 H, br, CH3–(CH2)2–CH2–), 4.34 (125 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
Poly(N-npentylglycine)10 P17.
GPC (DMAc): Mn = 0.9 kg/mol (ÐM = Mw/Mn = 1.38).
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.82 (30 H, br, CH3–), 1.27 (41 H, br, CH3–CH2–CH2–), 1.64 H (20 H, br, CH3–(CH2)2–CH2–), 3.35 (20 H, br, CH3–(CH2)3–CH2–), 4.39 (25 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
Poly(N-npentylglycine)25 P18.
GPC (DMAc): Mn = 2.1 kg/mol (ÐM = Mw/Mn = 1.20).
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.83 (51 H, br, CH3–), 1.27 (67 H, br, CH3–CH2–CH2–), 1.64 (34 H, br, CH3–(CH2)2–CH2–), 3.33 (34 H, br, CH3–(CH2)3–CH2–), 4.45 (36 H, br, –CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
Poly(N-npentylglycine)50 P19.
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.82 (111 H, br, CH3–), 1.27 (149 H, br, CH3–CH2–CH2–), 1.65 (73 H, br, CH3–(CH2)2–CH2–), 3.36 (75 H, br, CH3–(CH2)3–CH2–), 4.45 (79 H, br, CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
Poly(N-npentylglycine)100 P20.
1H NMR (500 MHz; TFA (DMSO-d6)): δ = 0.82 (190 H, br, CH3–), 1.27 (258 H, br, CH3–CH2–CH2–), 1.65 (127 H, br, CH3–(CH2)2–CH2–), 3.36 (130 H, br, CH3–(CH2)3–CH2–), 4.45 (140 H, br, CH2–CO– and C6H5–CH2–NH–), 7.16 ppm (5 H, br, C6H5–).
3. Results and Discussion
Polypeptoids with different degrees of polymerization (DP) and varying side-chain length comprising 1 to 5 carbon atoms were successfully synthesized (Scheme 1).
Scheme 1.
Schematic representation of synthesis of homo polypeptoids prepared in this work.

Characterization of the obtained polypeptoids was performed using 1H NMR spectroscopy, GPC and MALDI-ToF mass spectrometry (Table 2). The polymers P19 and P20 (poly(N-pentylglycine)) were insoluble in the used solvents (DMAc, MeOH), therefore the characterization by GPC and MALDI-ToF MS was not possible. Furthermore, the molar mass of P1 and P5 was too small for the GPC measurement under the given conditions.
| Polymer a | Mn b [kg/mol] | ÐM b | Mn c [kg/mol] | ÐM c | Mn d [kg/mol] | DP d | yield [%] | |
|---|---|---|---|---|---|---|---|---|
| P1 | PSar10 | - | - | 0.7 | 1.06 | 0.6 e | 7 e | 65 |
| P2 | PSar25 | 1.3 | 1.31 | 1.6 | 1.08 | 1.7 e | 22 e | 83 |
| P3 | PSar50 | 4.0 | 1.09 | 3.5 | 1.01 | 3.8 e | 52 e | 95 |
| P4 | PSar100 | 8.9 | 1.05 | 7.2 | 1.01 | 5.3 e | 73 e | >99 i |
| P5 | P(N-EtGly)10 | - | - | 1.0 | 1.06 | 1.0 e | 10.5 e | - |
| P6 | P(N-EtGly)25 | 1.0 | 1.65 | 2.0 | 1.05 | 2.1 e | 23 e | 62 |
| P7 | P(N-EtGly)50 | 3.3 | 1.25 | 3.3 | 1.06 | 3.9 e | 45 e | 92 |
| P8 | P(N-EtGly)100 | 6.8 | 1.22 | 6.6 | 1.01 | 7.2 e | 83 e | >99 i |
| P9 | P(N-PrGly)10 | 0.6 | 1.58 | 1.4 | 1.03 | 1.4 f | 13 f | 28 |
| P10 | P(N-PrGly)25 | 1.6 | 1.32 | 2.0 | 1.04 | 2.3 f | 22 f | 68 |
| P11 | P(N-PrGly)50 | 5.0 | 1.22 | 4.2 | 1.04 | 4.1 g | 40 g | 83 |
| P12 | P(N-PrGly)100 | 7.5 | 1.15 | 6.0 | 1.03 | 7.4 f | 74 f | 64 |
| P13 | P(N-BuGly)10 | 0.9 | 1.55 | 1.4 | 1.06 | 1.4 h | 11 h | 89 |
| P14 | P(N-BuGly)25 | 1.9 | 1.27 | 1.9 | 1.16 | 2.2 h | 18.5 h | 93 |
| P15 | P(N-BuGly)50 | 3.2 | 1.40 | 4.4 | 1.04 | 4.4 h | 38 h | 98 |
| P16 | P(N-BuGly)100 | 4.2 | 1.16 | - | - | 6.8 h | 59 h | >99 i |
| P17 | P(N-PenGly)10 | 0.9 | 1.38 | 1.4 | 1.03 | 1.5 h | 11 h | 64 |
| P18 | P(N-PenGly)25 | 2.1 | 1.2 | 1.9 | 1.16 | 2.3 h | 17 h | - |
| P19 | P(N-PenGly)50 | - | - | - | - | 4.9 h | 38 h | >99 i |
| P20 | P(N-PenGly)100 | - | - | - | - | 8.4 h | 65 h | >99 i |
a As determined from [M]0/[I]0; b As determined by gel permeation chromatography; c As calculated from Gauss distribution fitted to MALDI-ToF mass spectra; d As determined by end-group analysis from 1H NMR spectroscopy (signal intensity of aromatic protons of benzylamine-initiator vs main-chain and side-chain signal intensity); e Determined in D2O; f Determined in MeOD; g Determined in MeCN; h Determined in TFA-d1 with DMSO-d6 as external lock; i Traces of solvents could be contained in the polymers.
A comparison of the obtained molar masses with the theoretical masses calculated from [M]0/[I]0 using the example of P(N-PrGly) with different degrees of polymerization shows an increasing discrepancy with an increasing DP (Figure 1). This has been reported before by others and us [17,22,25]. To date, we lack a satisfactory explanation. Nevertheless, the experimentally determined degrees of polymerization generally follow the expected trend.
The thermal properties of the different polypeptoids have been investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Table 3).
Figure 1.
Correlation of the determined molar mass (by GPC, MALDI-ToF and 1H NMR spectroscopy, respectively) of different poly(N-propylglycine) (P(N-PrGly)) with the theoretical molar mass.
Figure 1.
Correlation of the determined molar mass (by GPC, MALDI-ToF and 1H NMR spectroscopy, respectively) of different poly(N-propylglycine) (P(N-PrGly)) with the theoretical molar mass.

| polymera | Tg [°C] | Tm [°C] | Tc [°C]b | Td [°C]c | |
|---|---|---|---|---|---|
| P1 | PSar10 | 28 | - | - | 175 |
| P2 | PSar25 | 127 | - | - | 210 |
| P3 | PSar50 | 136 | - | - | 215 |
| P4 | PSar100 | 143 | - | - | 250 |
| P5 | P(N-EtGly)10 | 77 | - | - | n.d. |
| P6 | P(N-EtGly)25 | 93 | - | - | 220 |
| P7 | P(N-EtGly)50 | 107 | - | - | 215 |
| P8 | P(N-EtGly)100 | 114 | - | - | 240 |
| P9 | P(N-PrGly)10 | 34 | - | - | 205 |
| P10 | P(N-PrGly)25 | 66 | 163 | - | 210 |
| P11 | P(N-PrGly)50 | 88 | 190 | 150 | 205 |
| P12 | P(N-PrGly)100 | 93 | 198 | 166 | 233 |
| P13 | P(N-BuGly)10 | - | 153 | - | 210 |
| P14 | P(N-BuGly)25 | 4 | 173 | - | n.d. |
| P15 | P(N-BuGly)50 | - | 215 (2nd Tm = 63 °C) | - | 215 |
| P16 | P(N-BuGly)100 | - | 225 (2nd Tm = 70 °C) | - | 213 |
| P17 | P(N-PenGly)10 | - | 145 (2nd Tm = 56 °C) | - | 205 |
| P18 | P(N-PenGly)25 | −3 | 176 (2nd Tm = 49 °C) | - | n.d. |
| P19 | P(N-PenGly)50 | 1 | 190 (2nd Tm = 70 °C) | - | 220 |
| P20 | P(N-PenGly)100 | 5 | 207 (2nd Tm = 67 °C) | - | 220 |
a As determined from [M]0/[I]0; b crystallization temperature; c decomposition temperature.
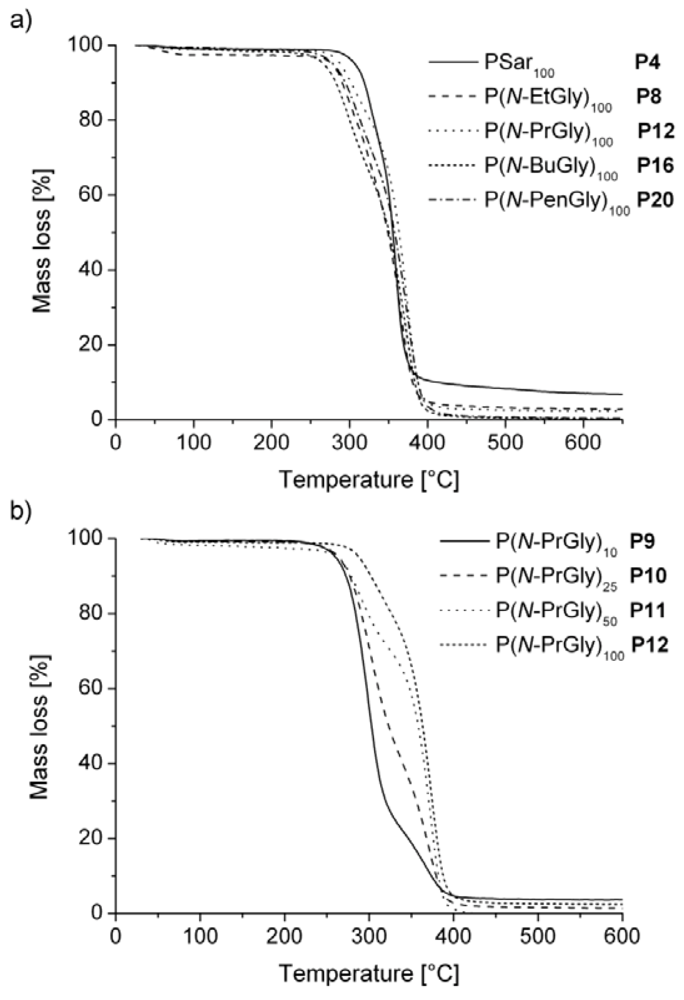
The TGA thermograms of all polypeptoid samples, except P1, reveal decomposition temperature thresholds over 200 °C, which is in accordance with earlier reports for several other polypeptoids with long-chain substituents [29]. In Figure 2, an assortment of TGA thermograms is displayed. On the one hand, polypeptoid samples with a increasing side chain length were compared (Figure 2a), on the other hand the influence of the DP based on different P(N-PrGly) samples is depicted (Figure 2b). The influence of the substituent on the decomposition temperature is relatively little; in contrast, with an increasing DP, a shift to higher decomposition temperatures is noticeable.
Figure 2.
Comparison of TGA thermograms of different polypeptoid samples with (a) an increase in side chain length and (b) different degrees of polymerization.
Figure 2.
Comparison of TGA thermograms of different polypeptoid samples with (a) an increase in side chain length and (b) different degrees of polymerization.
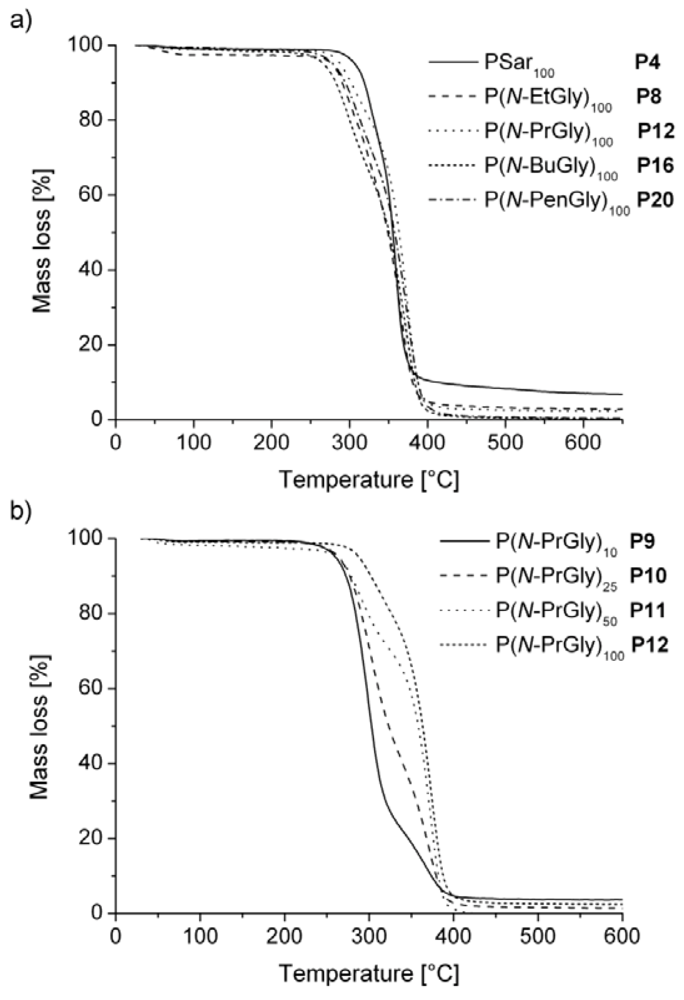
The decomposition temperature of polysarcosine is comparable with the sarcosine decomposition temperature of 212.5 °C. To the best of our knowledge, no decomposition temperatures have been published for the other corresponding N-substituted glycines.
Rosales et al. reported that poly(N-isopentylglycine), poly(N-hexylglycine) and other polypeptoids with longer side chains were stable up to 260 °C. In our case, the on-set temperatures were significantly lower than 260 °C. Unfortunately, a more detailed comparison with literature is not possible as no detailed decomposition values or thermograms were given [29].
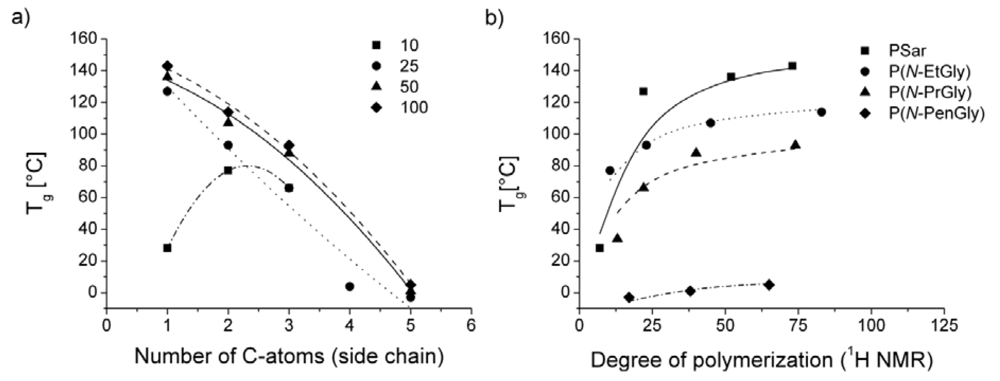
DSC studies were carried out below the decomposition temperatures to determine glass transition temperature (Tg) and melting points (Tm). The glass transition temperature as a function of the number of C-atoms in the side chain and of different degrees of polymerization was investigated (Figure 3). Different polypeptoid samples with the same DP, but increasing number of C-atoms in the side chain possess, as expected, decreasing glass transition temperatures (Figure 3a). Comparison of the Tg’s of P4 (Tg = 143 °C) and P20 (Tg = 5 °C) exhibit a difference of 138 °C. In the literature a Tg = 117 °C for polysarcosine with a DP of 103 can be found [30].
Figure 3.
Depiction of the glass transition temperature (Tg) as a function (a) of number of C-atoms in the side chain at different degrees of polymerization and (b) of the DP. Please note, lines in (a) represent only a guide for the eye. Lines in (b) are derived from Flory-Fox equation (Tg = Tg∞ – A/Mn) as outlined in experimental part.
Figure 3.
Depiction of the glass transition temperature (Tg) as a function (a) of number of C-atoms in the side chain at different degrees of polymerization and (b) of the DP. Please note, lines in (a) represent only a guide for the eye. Lines in (b) are derived from Flory-Fox equation (Tg = Tg∞ – A/Mn) as outlined in experimental part.

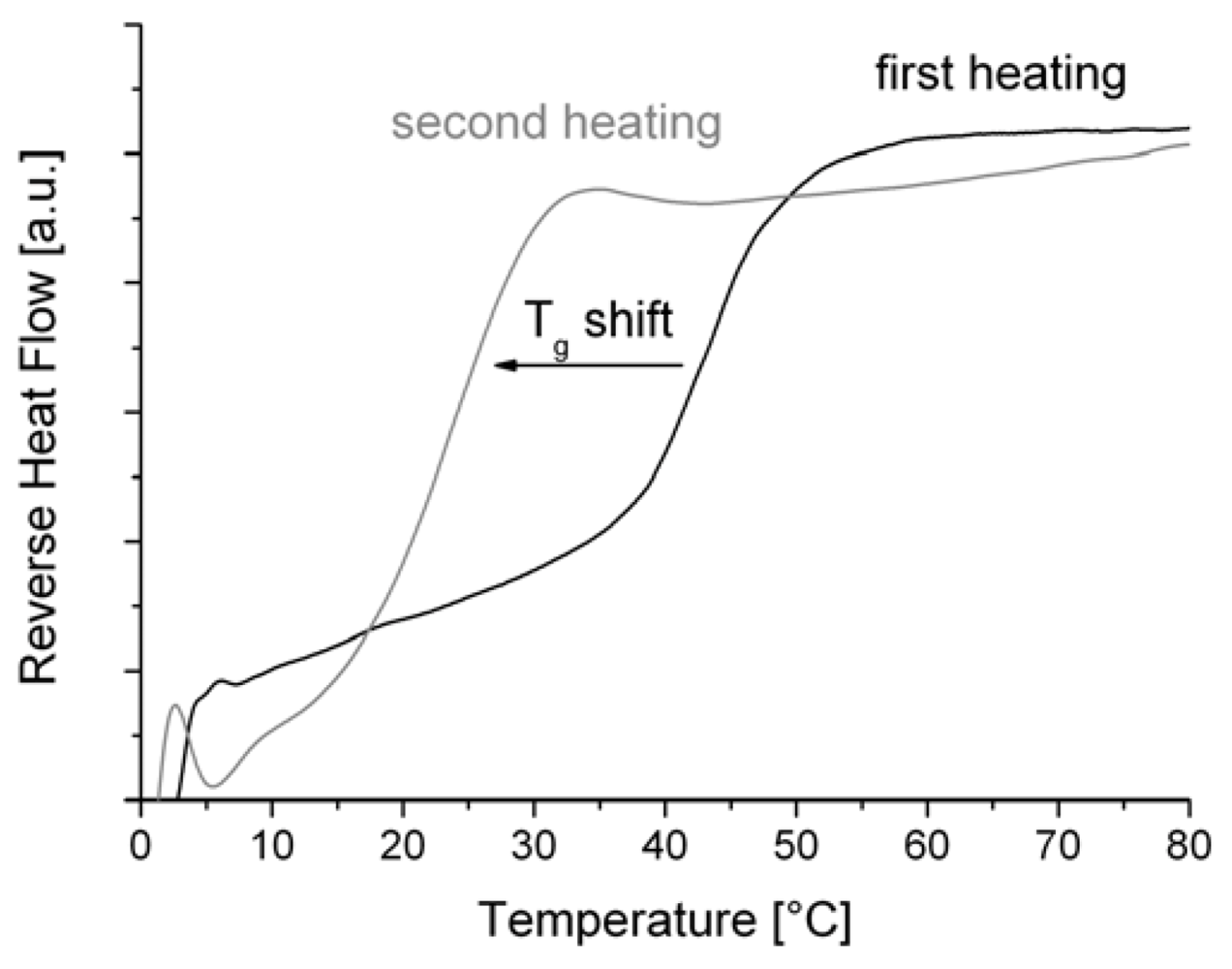
Notably, the Tg value of P1 does not fit in the observed trend. This may be attributed in part to the low decomposition on-set temperature. Possibly, the decomposition of the polymer starts in the first heating cycle, the consequence being that the glass transition was drastically decreased (Figure 4). However, the determined Tg from the first heating cycle is already very low.
Figure 4.
Shift of the glass transition temperature of P1 after the first heating cycle.

The decrease of the Tg by increasing side chain length is associated with extended flexibility of the backbone. Such decrease is expected and has also been studied in some detail for poly(2-oxazoline)s, constitutional isomers of polypeptoids [31]. Moreover, another trend is recognizable in Figure 3b. As is well known, an increase of the DP leads to an increase of the Tg [32,33]. An increase of the chain length decreases the flexibility of the polymer chains, the consequence being that the Tg increase to a constant value. In addition, the chain ends may act as plasticizers [34]. As the relative amount of polymer chain ends decreases with increase in chain length, the Tg reaches a plateau. For PSar, P(N-EtGly) and P(N-PrGly) this plateau appears to be reached at approx. DP = 50.
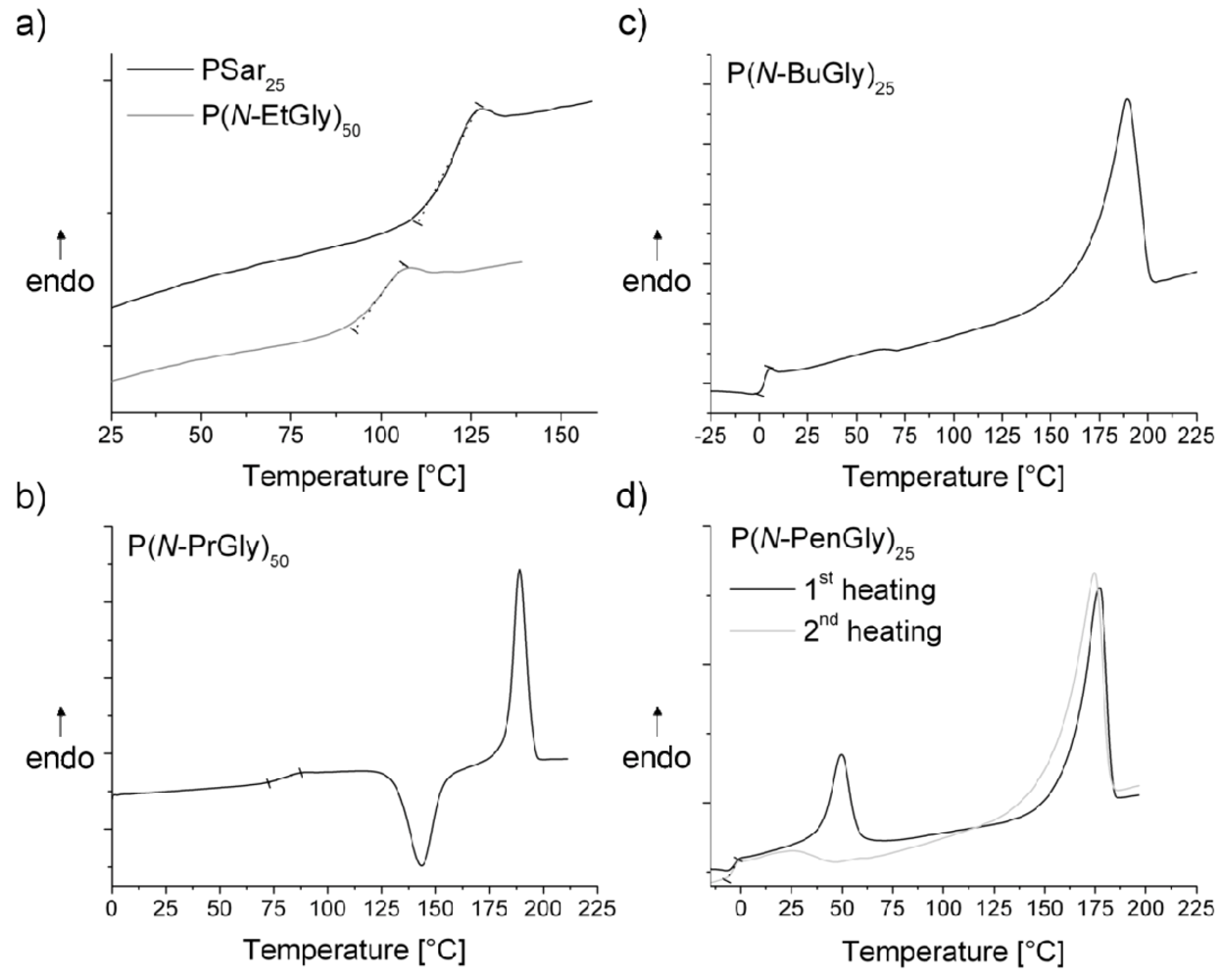
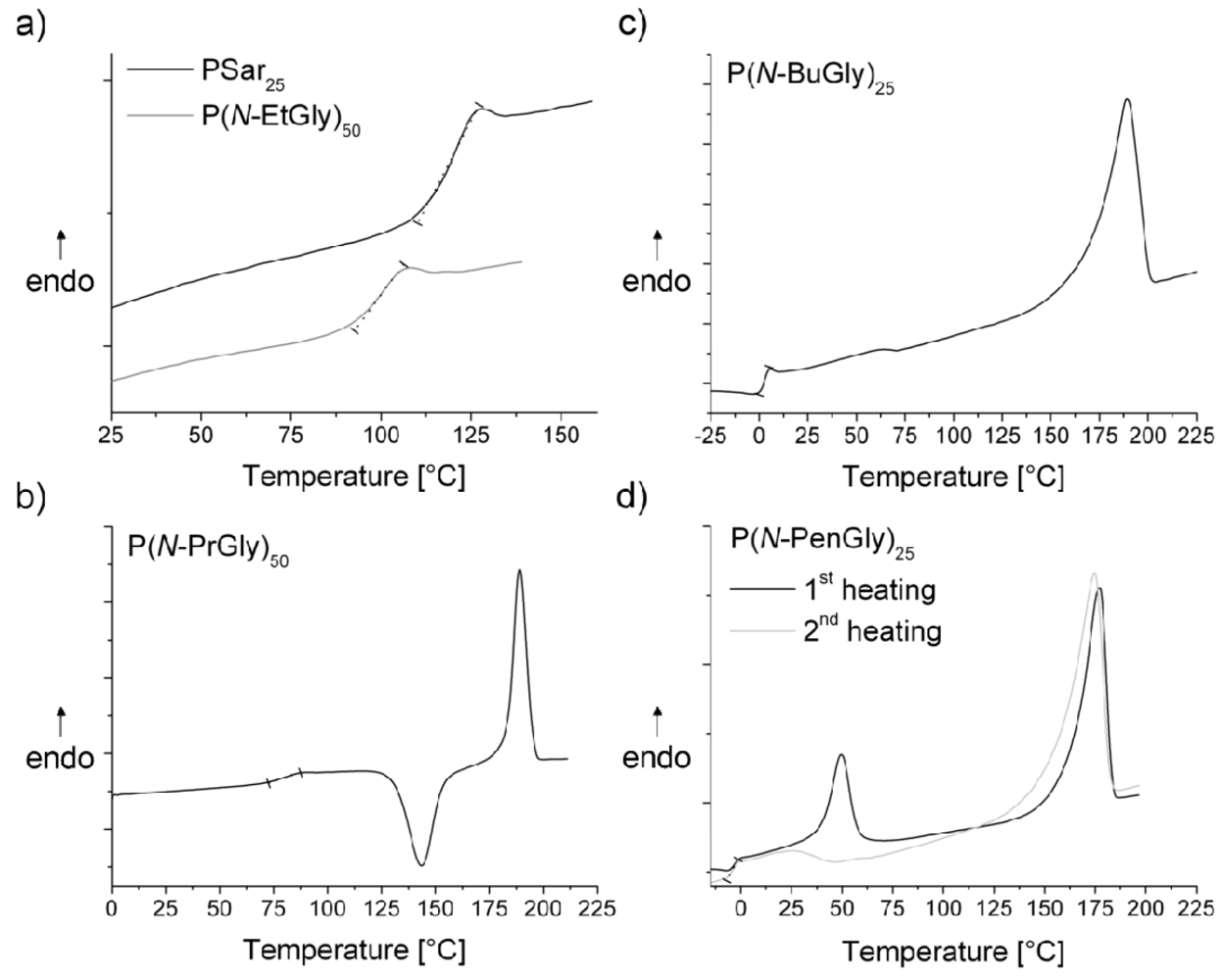
It should be noted that for unknown reasons P(N-BuGly) did not yield values for Tg for DPs other than 25 in this study. We are currently investigating this issue in more detail, as it may be that small residual of solvent is responsible for this. Some DSC response curves are depicted in Figure 5. We observed melting points for polypeptoids with a side chain comprising not less than three methyl carbon atoms. That implies that PSar and P(N-EtGly) are amorphous solids. Our data corroborates earlier reports by Okada et al. [30]. However, P(N-PrGly), P(N-BuGly) P(N-BuGly) and exhibit crystalline domains in the solid structure.
Figure 5.
(a), (b), (c) Second heating DSC thermograms of some synthesized polypeptoids and (d) comparison of the first and second heating DSC thermogram of P(N-PenGly)25 (P18). Glass transition temperature ranges were marked with a dotted line in each thermogram.
Figure 5.
(a), (b), (c) Second heating DSC thermograms of some synthesized polypeptoids and (d) comparison of the first and second heating DSC thermogram of P(N-PenGly)25 (P18). Glass transition temperature ranges were marked with a dotted line in each thermogram.
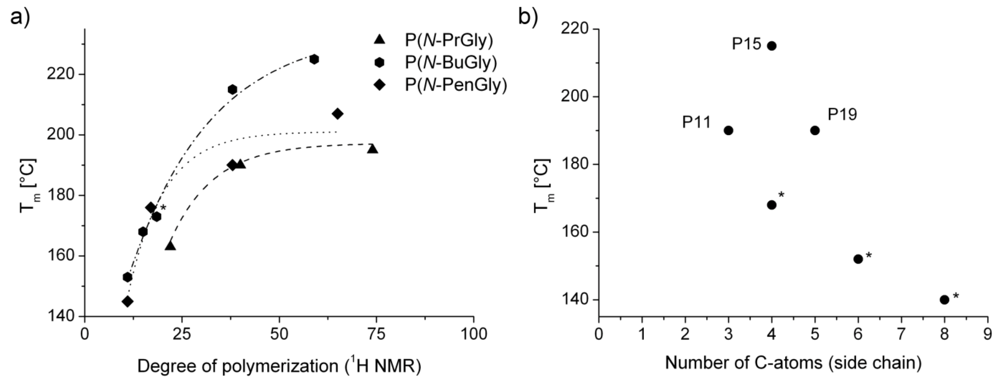
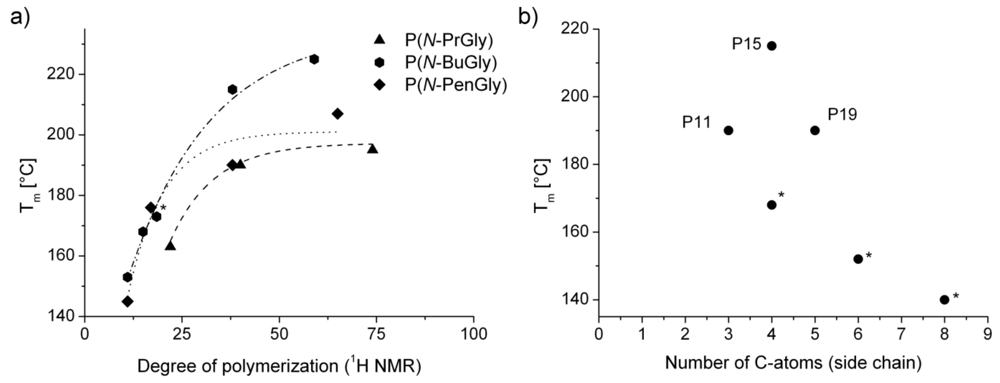
The obtained values for Tm of the polypeptoids follow the same trend as the glass transition temperature, i.e., with increasing chain length the Tm increases towards a plateau (Figure 6a). Interestingly, polymer Tm values at a fixed DP are highest for the P(N-BuGly). The value we report here for P(N-BuGly) again corroborate and supplement values reported by Rosales et al. Moreover, Tm values for P(N-PenGly) align roughly with literature values for poly(N-hexylglycine) and poly(N-octylglycine), although it must be noted that for those samples, a the smaller DP of 15 was reported (Figure 6b) [29]. In addition, these samples were monodispers.
Figure 6.
Comparison of polymer melting temperatures (Tm) as a function of (a) the degree of polymerization and (b) the polymer side chain length. Please note, values marked with asterisks were obtained by graphical extraction from ref. [29] and represent polymers with a degree of polymerization of 15. Lines in (a) only serve as guide for the eyes.
Figure 6.
Comparison of polymer melting temperatures (Tm) as a function of (a) the degree of polymerization and (b) the polymer side chain length. Please note, values marked with asterisks were obtained by graphical extraction from ref. [29] and represent polymers with a degree of polymerization of 15. Lines in (a) only serve as guide for the eyes.
The first and second heating DSC thermogram of P(N-PenGly)25 (P18) are shown in Figure 5d). The first heating thermogram exhibits two signals (in the range of 50 °C and 175 °C) which can be assigned to melting points. Interestingly, while for the other degrees of polymerization (P17, P19, P20) the lower temperature Tm persists in the second heating cycle no signal was detected at 50 °C therein for P18. In lieu thereof a buckle in the curve can be observed. Zhang et al. observed two melting points for the polypeptoid with a decyl substituent at the nitrogen and attributed this phenomenon to crystalline packing of the side chains and the main chains [35]. Accordingly, we also attribute the observed signals to crystalline packing of the side chains, but after the cooling cycle no melting point is observed in the second heating cycle. Repetition of the measurement revealed clearly smaller signals for the melting point in the range of 50 °C (data not shown). However, the melting point existed in the second heating curve as well.
The DSC thermogram of P(N-PrGly) shows an exothermic peak in the range of 143–155 °C, which can indicates a cold-crystallization process (Figure 5b). This peak describes the transition from amorphous solid to crystalline solid.
It should be noted that we observed a number of problems with repeatability of DSC measurements for some samples. In a number of cases, it appears that decomposition/discoloration started briefly after the glass transition temperature although TGA thermograms give no indication of a change in sample mass. The presence of flat and/or asymmetric peaks, decreasing Tg and Tm and discoloring of the samples after the measurement (yellow-brown), do hint towards changes in the polymer structure during these measurements. As may be expected, the problem with DSC measurement reproducibility was particularly pronounced for samples of low molar mass. We attribute this to more pronounced end-group effects for short polymer chains.
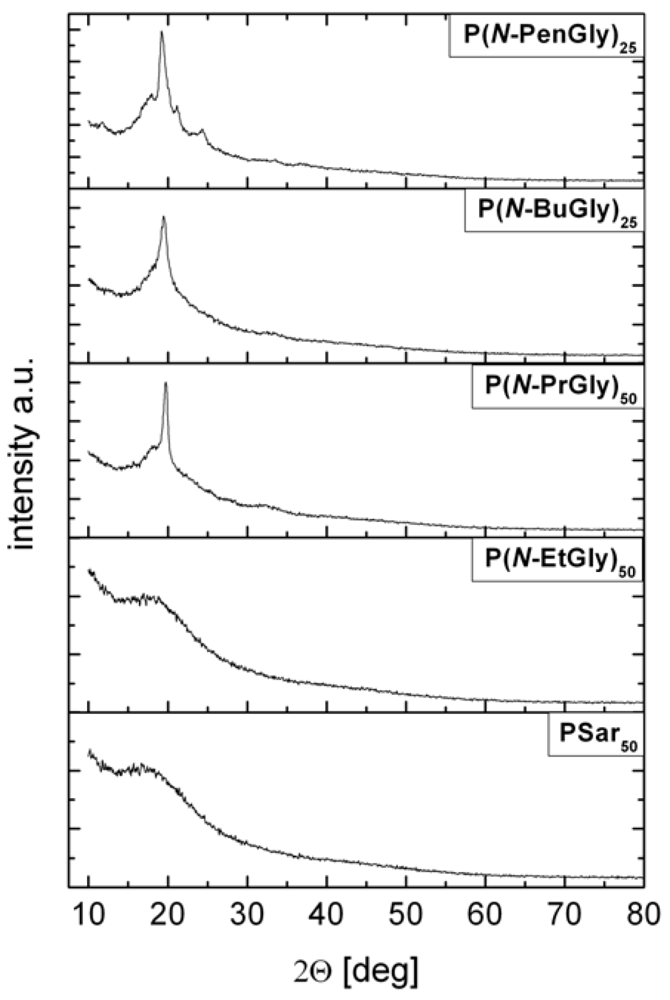
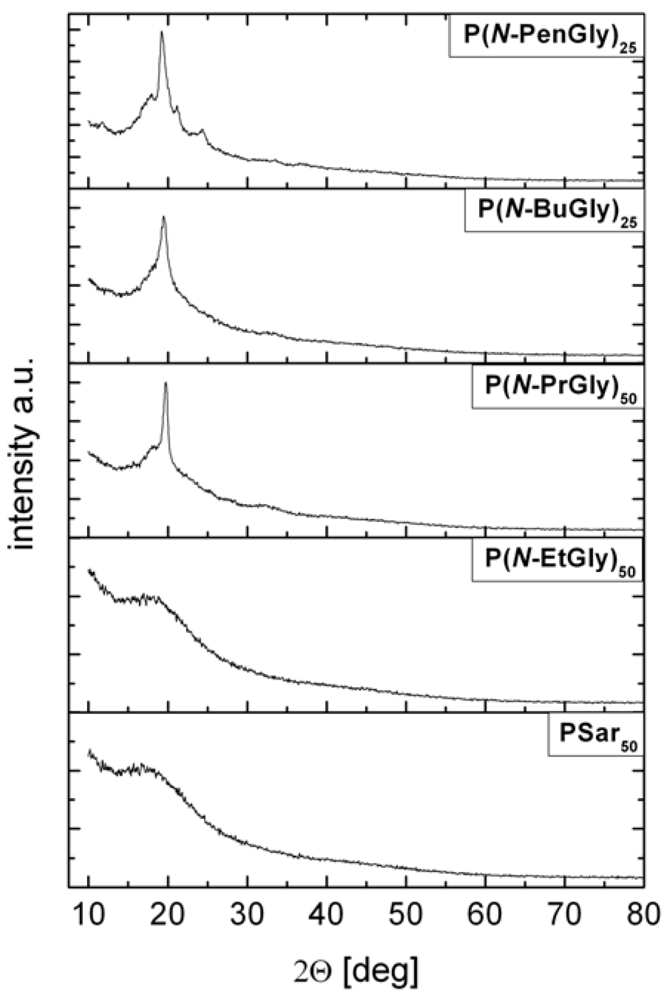
Crystallinity of polymer powders was also assessed using X-ray diffraction (Figure 7). Diffractograms of PSar and P(N-EtGly) show no sharp signals. This observation correspond amorphous solids and corroborates the observed lack of melting points by DSC. In contrast the diffractograms of the other three polymers (P(N-PrGly) (P11), P(N-BuGly) (P14) and P(N-PenGly) (P18) exhibit a sharp signal in a range of 20° 2θ, which is evidence for crystalline domains in the polymer powder. Rosales et al. detected a reflection in this range for all investigated peptoids and attributed this observation to the spacing between polymer chains (d = 4.5 Å) based on previously investigations [29,36]. Furthermore, P(N-PenGly) exhibits a reflection in the range of 25° 2θ. Zhang et al. investigated cyclic poly(N-decylglycine) with WAXS and attributed a reflection in the same range to the distance between adjacent repeating units (d = 3.6 Å) [35].
Figure 7.
Powder XRD diffractograms of some synthesized polypeptoids.
4. Conclusions
We have investigated a series of polypeptoid homopolymers bearing short (C1–C5) side chains of DP = 10–100 regarding thermal stability, glass transition and melting points. First, we synthesized polypeptoids by nucleophilic living ring-opening polymerization and characterized the obtained polymers with 1H NMR spectroscopy, MALDI-ToF mass spectrometry and gel permeation chromatography. Afterwards, thermal properties of the different polypeptoids have been investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry. TGA thermograms reveal decomposition temperature thresholds over 200 °C. Side chain length and DP affect the decomposition temperatures differently. The influence of the side chain is relatively little. However, an increasing DP leads to a shift to higher decomposition temperatures.
Furthermore, dependences of the glass transition temperature were investigated regarding side chain length and degree of polymerization. The obtained values confirm expected trends for Tg values and corroborate values reported earlier where available. In summary, an increasing side chain length leads to a decrease the Tg while an increase of the DP increases the Tg to a constant value. Similar observations occurred for the melting points, i.e., melting points increase to a constant value by increasing DP. However, three carbon atoms in the side chain are necessary to obtain semi-crystalline materials. Moreover, poly(N-butylglycine) and poly(N-pentylglycine) of degrees of polymerization of 50–100 exhibit two melting points which are attributed to crystalline packing of the side chains and the main chains.
X-ray diffraction measurements confirm crystalline domains in the polymer powder for polypeptoids with more than two carbon atoms in the side chain.
Acknowledgments
This publication is based on work supported by Award No. KUK-F1-029-32, made by King Abdullah University of Science and Technology (KAUST) and the Fonds der Chemischen Industrie (awarded to R.L.). We would like to acknowledge technical support by Matthias Kluge (TU Dresden) for TGA and DSC measurements. We kindly acknowledge technical support with XRD measurements by Anja Bensch (Prof. Michael Ruck, Inorganic Chemistry II at TU Dresden) The authors thank Julian Wagenhöfer for helpful discussions about XRD diffractograms. We also acknowledge Prof. Rainer Jordan (TU Dresden) for support and access to equipment. This publication was funded by the German Research Foundation (DFG) and the University of Wuerzburg in the funding programme Open Access Publishing.
References
- Leuchs, H.; Geiger, W. Über die Anhydride von α-Amino-N-Carbonsäuren und die von α-Aminosäuren. Chem. Ber. 1908, 41, 1721–1726. [Google Scholar] [CrossRef]
- Leuchs, H.; Manasse, W. Über die Isomerie der Carbäthoxyl-glycyl glycinester. Chem. Ber. 1907, 40, 3235–3249. [Google Scholar]
- Leuchs, H. Über die Glycin-Carbonsäure. Chem. Ber. 1906, 39, 857–861. [Google Scholar] [CrossRef]
- Sigmund, F.; Wessely, F. Untersuchungen über α-Amino-N-Carbonsäureanhydride. II. H-S Z. Physiol. Chem. 1926, 157, 91–105. [Google Scholar] [CrossRef]
- Wessely, F.; Riedl, K.; Tuppy, H. Untersuchungen über alpha-Amino-N-Carbonsäureanhydride VI. Monatsh. Chem. 1950, 81, 861–872. [Google Scholar] [CrossRef]
- Waley, S.G.; Watson, J. The Kinetics of the Polymerization of Sarcosine Carbonic Anhydride. P. Roy. Soc. Lond. A Mat. 1949, 199, 499–517. [Google Scholar] [CrossRef]
- Bamford, C.H.; Block, H.; Pugh, A.C. P. The polymerization of 3-substituted oxazolidine-2,5-diones. J. Chem. Soc. 1961, 1961, 2057–2063. [Google Scholar]
- Hanby, W.E.; Waley, S.G.; Watson, J. Synthetic Polypeptides. Part I. J. Chem. Soc. 1950, 3009–3013. [Google Scholar]
- Ballard, D.G.; Bamford, C.H. Kinetics of the Formation of Polypeptides from N-Carboxy-α-amino-acid Anhydrides. Nature 1953, 4365, 907–908. [Google Scholar]
- Ballard, D.G.H.; Bamford, C.H. Studies in polymerization. X. “The chain-effect”. P. Roy. Soc. Lond. A Mat. 1956, 236, 384–396. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of alpha-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar]
- Sisido, M.; Imanishi, Y.; Okamura, S. Polymerization of DL-beta-Phenylalanine N-Carboxyanhydride by Poly(N-n-Propylglycine) Diethylamide. Polym. J. 1970, 1, 198–203. [Google Scholar]
- Sisido, M.; Imanishi, Y.; Okamura, S. Polymerization of amino acid derivatives by polymer catalysts. V. Polymerization of DL-β-phenylalanine N-carboxyanhydride by several poly(N-alkylamino acid) diethylamides. Biopolymers 1970, 9, 791–797. [Google Scholar] [CrossRef]
- Sisido, M.; Imanishi, Y.; Okamura, S. Polymerization of amino acid derivatives by polymer catalysts. III. Chain effect polymerization induced by poly(N-ethylglycine) diethylamide. Biopolymers 1969, 7, 937–947. [Google Scholar] [CrossRef]
- Sisido, M.; Imanishi, Y.; Higashimura, T. Molecular weight distribution of polysarcosine obtained by NCA polymerization. Makromol. Chem. 1977, 178, 3107–3114. [Google Scholar] [CrossRef]
- Aoi, K.; Hatanaka, T.; Tsutsumiuchi, K.; Okada, M.; Imae, T. Synthesis of a novel star-shaped dendrimer by radial-growth polymerization of sarcosine N-carboxyanhydride initiated with poly (trimethyleneimine) dendrimer. Macromol. Rapid Commun. 1999, 20, 378–382. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; von Lossow, C.; Schwarz, G. Primary Amine-Initiated Polymerizations of Alanine-NCA and Sarcosine-NCA. Macromol. Chem. Phys. 2004, 205, 918–924. [Google Scholar] [CrossRef]
- Tanisaka, H.; Kizaka-Kondoh, S.; Makino, A.; Tanaka, S.; Hiraoka, M.; Kimura, S. Near-infrared fluorescent labeled peptosome for application to cancer imaging. Bioconjug. Chem. 2008, 19, 109–117. [Google Scholar] [CrossRef]
- Makino, A.; Kizaka-Kondoh, S.; Yamahara, R.; Hara, I.; Kanzaki, T.; Ozeki, E.; Hiraoka, M.; Kimura, S. Near-infrared fluorescence tumor imaging using nanocarrier composed of poly(L-lactic acid)-block-poly(sarcosine) amphiphilic polydepsipeptide. Biomaterials 2009, 30, 5156–5160. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, D. Cyclic poly(alpha-peptoid)s and their block copolymers from N-heterocyclic carbene-mediated ring-opening polymerizations of N-substituted N-carboxylanhydrides. J. Am. Chem. Soc. 2009, 131, 18072–18074. [Google Scholar]
- Guo, L.; Li, J.; Brown, Z.; Ghale, K.; Zhang, D. Synthesis and Characterization of Cyclic and Linear Helical Poly(alpha-peptoid)s by N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxyanhydrides. Biopolymers 2011, 96, 596–603. [Google Scholar] [CrossRef]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from N-Substituted Glycine N-Carboxyanhydrides: Hydrophilic, Hydrophobic, and Amphiphilic Polymers with Poisson Distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Fetsch, C.; Luxenhofer, R. Highly Defined Polypeptoids via Multiple Chain Extension and Macroinitiators. Macromol. Rapid. Commun. 2012, 33, 1708–1713. [Google Scholar] [CrossRef]
- Lahasky, S.H.; Serem, W.K.; Guo, L.; Garno, J.C.; Zhang, D. Synthesis and Characterization of Cyclic Brush-Like Polymers by N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Propargyl N-Carboxyanhydride and the Grafting-to Approach. Macromolecules 2011, 44, 9063–9074. [Google Scholar] [CrossRef]
- Robinson, J.W.; Schlaad, H. A versatile polypeptoid platform based on N-allyl glycine. Chem. Commun. 2012, 48, 7835–7837. [Google Scholar] [CrossRef]
- Lahasky, S.H.; Hu, X.; Zhang, D. Thermoresponsive Poly(α-peptoid)s: Tuning the Cloud Point Temperatures by Composition and Architecture. ACS Macro Lett. 2012, 1, 580–584. [Google Scholar] [CrossRef]
- Robinson, J.W.; Secker, C.; Weidner, S.; Schlaad, H. Thermo-responsive Poly(N-C3 glycine)s. Macromolecules 2013, 47. in print. [Google Scholar]
- Zhang, D.; Lahasky, S.H.; Guo, L.; Lee, C.-U.; Lavan, M. Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules 2012, 45, 5833–5841. [Google Scholar] [CrossRef]
- Rosales, A.M.; Murnen, H.K.; Zuckermann, R.N.; Segalman, R.A. Control of Crystallization and Melting Behavior in Sequence Specific Polypeptoids. Macromolecules 2010, 43, 5627–5636. [Google Scholar]
- Aoi, K.; Nakamura, R.; Okada, M. Polypeptide-synthetic polymer hybrids, 2. Miscibility of poly(vinyl alcohol) with polysarcosine. Macromol. Chem. Phys. 2000, 201, 1059–1066. [Google Scholar] [CrossRef]
- Rettler, E.F.J.; Kranenburg, J.M.; Lambermont-Thijs, H.M.L.; Hoogenboom, R.; Schubert, U.S. Thermal, Mechanical, and Surface Properties of Poly(2-N-alkyl-2-oxazoline)s. Macromol. Chem. Phys. 2010, 211, 2443–2448. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. The glass temperature and related properties of polystyrene. Influence of molecular weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]
- Claudy, P.; Létoffe, J.M.; Camberlain, Y.; Pascault, J.P. Glass transition of polystyrene versus molecular weight. Polym. Bull. 1983, 9, 208–215. [Google Scholar] [CrossRef]
- berreiter, K.; Kanig, G. Self-plasticization of polymers. J. Colloid Sci. 1952, 7, 569–583. [Google Scholar] [CrossRef]
- Lee, C.U.; Smart, T.P.; Guo, L.; Epps, T.H.; Zhang, D. Synthesis and Characterization of Amphiphilic Cyclic Diblock Copolypeptoids from N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-carboxyanhydride. Macromolecules 2011, 44, 9574–9585. [Google Scholar] [CrossRef]
- Nam, K.T.; Shelby, S.A.; Choi, P.H.; Marciel, A.B.; Chen, R.; Tan, L.; Chu, T.K.; Mesch, R.A.; Lee, B.C.; Connolly, M.D.; Kisielowski, C.; Zuckermann, R.N. Free-floating ultrathin two-dimensional crystals from sequence-specific peptoid polymers. Nat. Mater. 2010, 9, 454–460. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Fetsch, C.; Luxenhofer, R. Thermal Properties of Aliphatic Polypeptoids. Polymers 2013, 5, 112-127. https://doi.org/10.3390/polym5010112
AMA Style
Fetsch C, Luxenhofer R. Thermal Properties of Aliphatic Polypeptoids. Polymers. 2013; 5(1):112-127. https://doi.org/10.3390/polym5010112
Chicago/Turabian StyleFetsch, Corinna, and Robert Luxenhofer. 2013. "Thermal Properties of Aliphatic Polypeptoids" Polymers 5, no. 1: 112-127. https://doi.org/10.3390/polym5010112