Effects of D-Lysine Substitutions on the Activity and Selectivity of Antimicrobial Peptide CM15

Abstract
: Despite their potent antimicrobial activity, the usefulness of antimicrobial peptides (AMPs) as antibiotics has been limited by their toxicity to eukaryotic cells and a lack of stability in vivo. In the present study we examined the effects of introducing D-lysine residues into a 15-residue hybrid AMP containing residues 1–7 of cecropin A and residues 2–9 of melittin (designated CM15). Diastereomeric analogs of CM15 containing between two and five D-lysine substitutions were evaluated for their antimicrobial activity, lysis of human erythrocytes, toxicity to murine macrophages, ability to disrupt cell membranes, and protease stability. All of the analogs caused rapid permeabilization of the Staphylococcus aureus cell envelope, as indicated by uptake of SYTOX green. Permeabilization of the plasma membrane of RAW264.7 macrophages was also observed for CM15, but this was substantially diminished for the D-lysine containing analogs. The introduction of D-lysine caused moderate decreases in antimicrobial activity for all analogs studied, with a much more pronounced reduction in toxicity to eukaryotic cells, leading to marked improvements in antimicrobial efficacy. Circular dichroism studies indicated a progressive loss of helical secondary structure upon introduction of D-lysine residues, with a good correspondence between helical content and eukaryotic cell cytotoxicity. Overall, these studies indicate that disruption of amphipathic secondary structure reduces both antimicrobial activity and eukaryotic cell toxicity, but that the reduction in eukaryotic cell cytotoxicity is more pronounced, leading to an overall gain in antimicrobial selectivity.1. Introduction
The extensive use of antibiotics over the past sixty years has led to an increased prevalence of antibiotic resistance in both hospital- and community-acquired infections [1-6], giving rise to a critical need for the development of new approaches for treatment of bacterial infections. In recent years there has been considerable interest in the development of antimicrobial peptides (AMPs) as novel antibiotics [7-11]. AMPs occur naturally within all kingdoms of life, from humans to prokaryotes, and constitute an important part of the innate immune response [7,8]. AMPs exhibit broad specificity against both Gram-positive and Gram-negative bacteria, including strains that are multi-drug resistant. AMPs are typically 12–50 amino acids in length, contain from 2–9 positively charged residues, and adopt amphipathic secondary structures upon binding to membranes [7-13]. Their positive charge allows AMPs to interact more strongly with the highly negatively-charged membranes of bacteria as opposed to the nearly neutral plasma membranes of eukaryotic cells [12,13], and their amphipathic secondary structure facilitates partitioning into the membrane bilayer [14]. For most AMPs, their mechanism of cell killing is believed to involve disruption of the membrane permeability barrier, either through the formation of discrete pores, detergent-like action, or perturbation of lipid packing [7-13,15]. Recent NMR studies have produced atomic-resolution structures for several important AMPs in membrane-mimetic environments, providing insights into their mechanism of action [16-19]. Although some AMPs do interact with intracellular targets [11], even these must cross the membrane in order to exert their biological effect.
Despite their potent antimicrobial activity, the usefulness of AMPs as antibiotics has been limited by their toxicity to eukaryotic cells and a lack of stability in vivo. Modifications to AMP structure designed to reduce cytotoxicity to eukaryotic cells and enhance stability include amino acid substitutions that disrupt amphipathic structure [20-22], the introduction of β- or D-amino acids [23-32], and manipulation of peptide charge and/or hydrophobicity [12,33-36]. In particular, the introduction of D-amino acids is an approach that allows disruption of secondary structure while conserving overall charge and hydrophobicity. Peptides containing a mixture of D- and L-amino acids have been shown to maintain antibacterial activity while having decreased toxicity to eukaryotic cells [12,23-28].
Of the many hundreds of known natural and designed AMPs [34,37], synthetic hybrids of the insect peptide cecropin and the bee venom peptide melittin are among the most potent and well-studied [38-50]. In particular, a hybrid containing residues 1–7 of cecropin and residues 2–9 of melittin (designated CM15) was found to maintain the antimicrobial activity of cecropin while decreasing the hemolytic effects of melittin [39]. CM15, in which five of the fifteen residues are lysine, forms α-helical secondary structure upon binding to membranes [39,43,44,46,48] and exhibits potent, broad-spectrum antimicrobial activity [36,38-40]. CM15 intercalates into lipid bilayers that mimic the bacterial plasma membrane and disrupts membrane structure [43,46,47,50]. Because of its small size, CM15 is readily amenable to manipulation of its structure and composition.
In these studies we examined the effects of modifying CM15 by substitution of D-lysine for L-lysine. It was anticipated that these modifications would disrupt the secondary structure of the peptide, in this case lowering α-helical content, without changing the overall charge or ratio of hydrophilic to hydrophobic amino acids. Analogs of CM15 containing between two and five D-lysine substitutions were evaluated for their antimicrobial activity, toxicity towards human erythrocytes and RAW264.7 macrophages, ability to permeabilize cell membranes, and protease stability. We find that changes in the biological activity of CM15 depend on both the number and position of D-lysine substitutions, and that such D-amino acid substitutions can dramatically lower toxicity to eukaryotic cells with only minimal decreases in antimicrobial activity. These studies support the growing consensus that disrupting the hydrophobic surface of amphipathic peptides can significantly diminish AMP toxicity to eukaryotic cells
2. Materials and Methods
2.1. Materials
Human serum albumin (HSA), Coomassie Blue G250, Triton X100, Sodium pyruvate, Penicillin/Streptomycin, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), trypsin, and chymotrypsin were obtained from Sigma-Aldrich (St. Louis, MO, USA). LB agar was obtained from Fischer Scientific (Pittsburgh, PA, USA). Tris/Tricine gels were obtained from BioRad. SYTOX green, fetal bovine serum (FBS), and DMEM (12100-046) were obtained from Invitrogen and Mueller-Hinton broth (MHB) was obtained from Difco.
2.2. Peptide Synthesis and Purification
Peptides with amidated C-termini and acetylated N-termini were obtained commercially (Biosynthesis Inc., Lewisville, TX, USA) or were synthesized on Rink amide MBHA resin using Fmoc (n-(9-fluorenyl)methoxycarbonyl) chemistry as previously described [43] on a CEM Discover microwave-assisted peptide synthesizer (CEM Biosciences, Matthews, NC, USA). Following precipitation of crude peptides with ice cold diethyl ether, the peptides were resuspended in water and purified by reverse phase HPLC on a 10-μm, 1.0 × 25 cm C8 column (Vydac, Hesperia, CA, USA) using a linear gradient of 20–80% acetonitrile in water/0.1% TFA over 40 min. Purified peptides were lyophilized and stored under vacuum until use. Peptide purity was assessed by analytical reverse-phase HPLC, and molecular mass was verified by MALDI-TOF mass spectrometry at the Protein and Nucleic Acid Shared Facility of the Medical College of Wisconsin. All peptides used in this study were >95% pure.
2.3. Liposome Preparation
Large unilameller vesicles (LUV) were prepared by extrusion using a mini-extrusion syringe device (Avanti Polar Lipids). Briefly, phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin (CL) (PE:PG:CL, molar ratio 70:25:5) were dried down under nitrogen and stored under vacuum overnight. Lipids were rehydrated in H2O, freeze-thawed using liquid nitrogen (5×), and extruded through a 0.1 micron filter (15×). Lipid concentration was determined by Stewart's assay [51].
2.4. Circular Dichroism
Circular dichroism (CD) spectra were obtained in buffer, in the presence of 50% trifluoroethanol (TFE), and in the presence of liposomes. For liposome samples, peptide and LUVs composed of PE:PG:CL were diluted in H2O (350 μL) to give a final concentration of 60 μM peptide at a lipid-to-peptide molar ratio of 100:1. Samples were equilibrated overnight at 4 °C and CD was performed at room temperature on a Jasco J-710 spectrapolarimeter (Tokyo, Japan) at a scan rate of 20 nm/min, 2 s response time, and 1 nm bandwidth. For liposome samples, spectra were truncated at 200 nm due to excessive light scattering at lower wavelengths. Helical content was estimated based on ellipticity at 208 nm as described by Greenfield and Fasman [52].
2.5. Antimicrobial Activity Assay
To assess antimicrobial activity, minimum inhibitory concentrations (MIC) were determined by broth-dilution assay [45]. Briefly, bacteria were grown at 37 °C overnight from frozen stock on LB or Pseudomonas-selective Vogel-Bonner minimal (VBM) agar plates with no antibiotic. Single colonies were inoculated into Mueller-Hinton broth (MHB), grown (37 °C, shaking at 150 rpm) until the OD at 600 nm was approximately 0.5–0.8, and then diluted in MHB to a working concentration of 2 × 105 cells/mL. In a clear bottom 96-well plate, we first added 100 μL MHB/0.1%HSA to each well. Peptide (100 μL, 256 μM in PBS) was then added to the first well in each row, followed by eight 2-fold serial dilutions. Finally, 100 μL of the bacterial working solution described above was added to each well, giving final concentrations of 64–0.5 μM peptide and 1 × 105 bacterial cells/mL. The plate was incubated overnight (37 °C, 150 rpm) and the OD600 values were measured using a SpectraMax M2 plate reader (Molecular Devices, Mountain View, CA, USA). MIC values are the lowest concentration of peptide at which there was no bacterial growth.
2.6. Hemolysis Assay
Outdated whole blood was obtained from the Blood Bank of Southeast Wisconsin. Human red blood cells (RBC) were separated by centrifugation (5,000g, 10 min, 4 °C), washed 3 times with cold PBS, and diluted to give a working stock of 10% (v/v) in PBS containing 5 mM glucose. Peptide samples (100 μL) in PBS were prepared in Eppendorf tubes by making 2-fold serial dilutions starting at 256 μM, followed by the addition of an equal volume of the RBC working stock solution to give a final concentration of 5% RBC by volume. Following a 1 h incubation (37 °C, with shaking at 150 rpm) samples were centrifuged (3 min, 1,000 rpm), the supernatants were transferred to a 96-well plate, and the OD at 543 nm was measured. Percent hemolysis was calculated relative to a negative control containing only PBS and a positive control containing 1% Triton X100.
2.7. Macrophage Toxicity by MTT Assay
RAW264.7 murine macrophages were cultured from frozen stock on tissue culture dishes (Sarstedt, 100 × 20 mm) in DMEM supplemented with sodium bicarbonate (44.05 mM), sodium pyruvate (1 mM), FBS (10%) and Penicillin/Streptomycin (1%) and maintained at 37 °C, 5% CO2 until confluent. Media was aspirated and cells were rinsed with PBS to remove dead cells and waste. Fresh media was added and cells were scraped, counted using a hemacytometer, and seeded into a 96-well plate at a concentration of 10,000 cells/well. Cells were allowed to attach overnight (37 °C, 5% CO2) and peptides (2 fold dilutions from 120 μM to 0.94 μM, final concentration) were added to each well. Plates were incubated for 1 hr (37 °C, 5% CO2) and MTT (20 μL of a 5 mg/mL stock solution in PBS) was added to each well. After a 4 h incubation (37 °C, 5% CO2) the media was removed and the plate was allowed to dry. The resulting formazan dye, formed by the active metabolism of MTT in viable cells, was resuspended in 100 μL of isopropanol and the OD at 570 nm was measured. Fractional survival was calculated relative to a control sample in PBS without peptide using the following equation:
2.8. Macrophage Toxicity by SYTOX Green Assay
Cultured RAW264.7 macrophages were grown and plated in a 96-well format as described above and incubated overnight (37 °C, 5% CO2). An appropriate concentration of peptide in 50 μL PBS was added to achieve a final concentration of four times the MIC value against S. aureus. Immediately following the addition of peptide, 100 μL of a working solution of SYTOX green (3 μL/5 mL DMEM, prepared according to the manufacturer's instructions) was added. Measurement of time course data was initiated immediately using a plate reader (Beckman Coulter DTX880) with excitation filter F485/20 and emission filter F535/25. Positive and negative controls without peptide contained 1% Triton X100 or PBS, respectively. To visualize SYTOX green uptake by fluorescence microscopy, cells were treated in the same manner as above and images were acquired every 15 min using a Nikon Eclipse TE200 microscope. Bright field and fluorescence images were obtained at 10× magnification.
2.9. S. aureus Membrane Permeability by Sytox Uptake
Bacterial cells were diluted to 2 × 107 cells per well in a 96 well plate (in 50 μL PBS). Peptide in PBS was added to give a final peptide concentration equal to the MIC value. 100 μL SYTOX green (2 μL/mL in PBS) was added to each well and a 2 h time course was obtained using a SpectraMax M2 plate reader (Molecular Devices, Mountain View, CA) with excitation at 488 nm, emission at 530 nm and cutoff of 495 nm. The fractional fluorescence intensity at 530 nm was quantitated relative to 100% live and dead cell suspensions prepared by a modified Live/Dead Baclight protocol (Invitrogen). Briefly, 30 mL cultures of S. aureus 6538p were grown in MHB to late log phase. Cells were pelleted via centrifugation (10,000g, 15 min), the supernatant discarded, and the pellet resuspended in sterile water (2 mL). Half of this preparation was added to 20 mL of sterile water while the other half was added to 20 mL of 70% isopropyl alcohol (IPA), incubated for 1 h with mixing every 15 min, pelleted via centrifugation (10,000g, 15 min), and washed with 20 mL sterile water.
2.10. Protease Susceptibility
Peptide (2.7 μg) and protease (10 ng, trypsin or chymotrypsin) were mixed in 10 μL of 10 mM Tris (pH 7.5 or pH 8, respectively). A proteolysis time course (1, 5, 10, 20, 30, and 60 min) was performed by removing aliquots at the designated times and quenching the protease with the addition of an equal volume of SDS PAGE sample buffer (200 mM Tris-HCl pH 6.8, 2% SDS, 40% glycerol, 0.04% Coomassie Blue). Samples were loaded into wells of a 16.5% Tris/Tricine gel (BioRad) and electrophoresed at room temperature (100 V, 75 min). Peptide bands were fixed in a solution of 40% methanol/10% acetic acid for 1 h, after which they were stained for 1 h (10% acetic acid/0.025% Coomassie Blue G-250). Gels were destained in 10% acetic acid to visualize the bands. Wells containing standard concentrations of intact peptide were included in each gel as controls. For quantitation, gels were imaged using an Alpha Imager (Alpha Innotech, Santa Clara, CA).
3. Results and Discussion
3.1. Peptide Design and Secondary Structure
To examine the effects of disrupting peptide secondary structure on the antimicrobial activity and cytotoxicity of CM15, we designed a set of peptides containing D-amino acid substitutions at lysine residues (Table 1). By consistently substituting D-lysine for L-lysine, these peptides retain the same charge and relative numbers of hydrophilic and hydrophobic amino acids as their parent peptide. Peptides D3,14, D3,7,14, and D3,6,7,13,14 were designed based on a lysine-enriched analog of CM15 (containing a Val14 to Lys substitution) that previously demonstrated improved selectivity [32]. In addition, we anticipated that D-lysine substitutions would increase resistance to digestion by trypsin-like proteases that cleave at cationic amino acid residues (i.e., lysine, arginine), and that placement of D-amino acids near the ends of the peptide would provide protection against carboxy- and aminopeptidases.
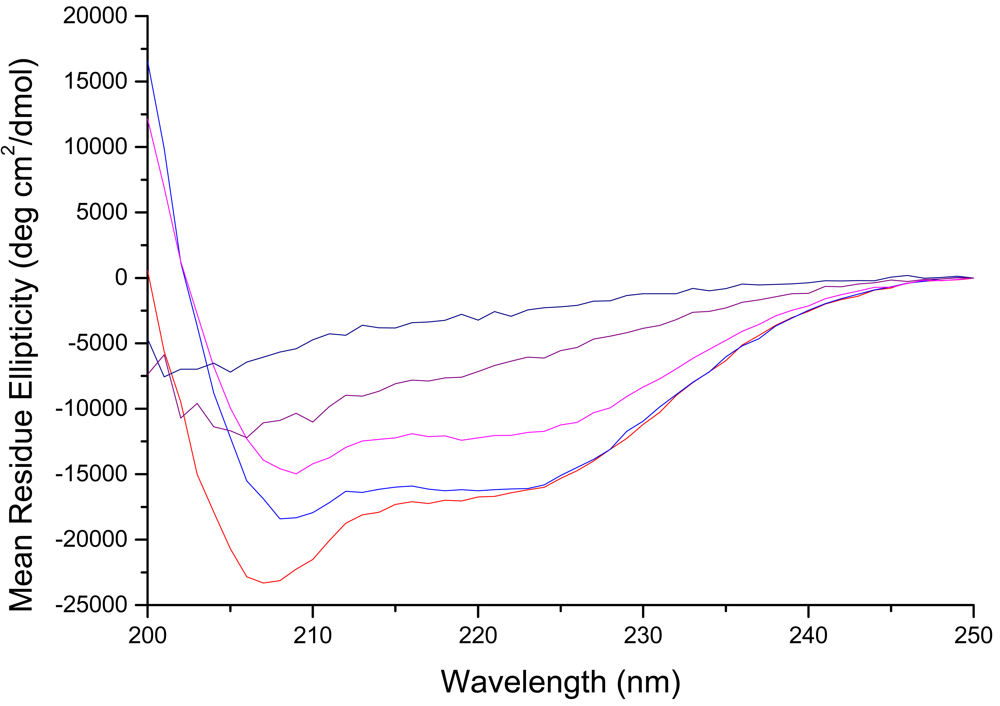
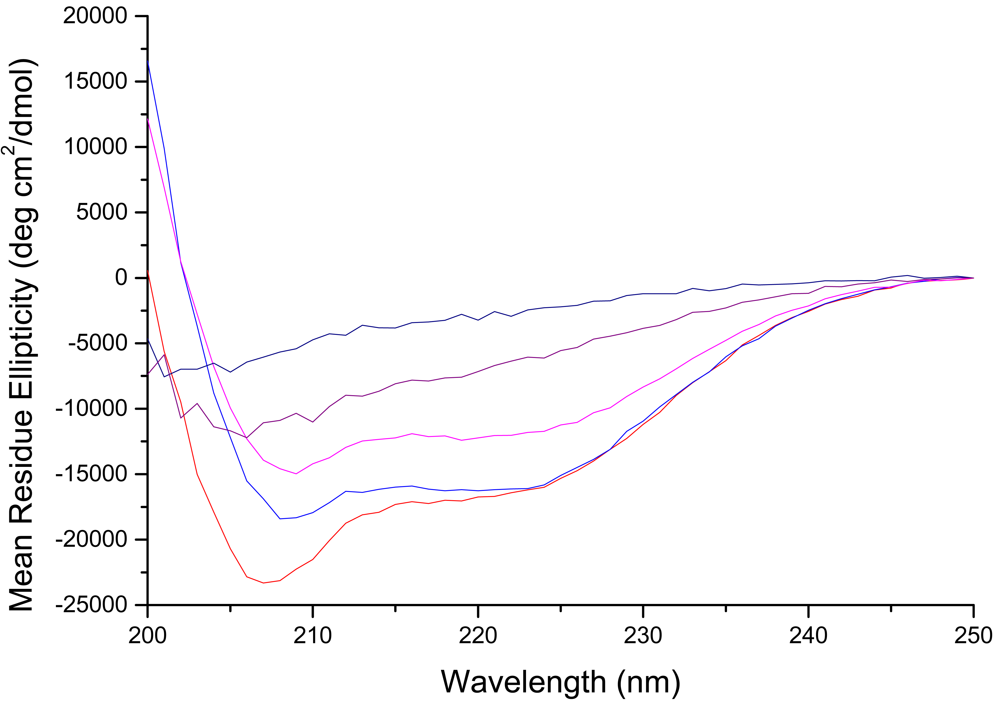
To evaluate changes in secondary structure of the D-lysine containing peptides, we examined their circular dichroism (CD) spectra. Previous studies of CM15 indicated α-helical contents of approximately 85% and 60% in the presence of 50% TFE and membranes, respectively [46], with a well-defined alpha helix extending from residues 4–14 in the membrane-bound form [43,48]. All of the peptides were unstructured in buffer (data not shown). In the presence of PE:PG:CL liposomes, mimicking the composition of the E. coli inner membrane, peptides CM15, D1,13, D3,13, and D3,14 all show prominent features at 208 and 222 nm, indicative of right-handed helical structure (Figure 1).
Of the D-lysine containing peptides, D1,13 retained the most helical content (55%, or ∼84% of that observed for CM15), followed by D3,13 and D3,14 which gave virtually identical CD spectra with ∼70% of the helical content observed for CM15. These results indicate that the α-helical structure in the central region of CM15 is largely preserved when D-amino acid substitutions are made near the ends of the peptide. In contrast, the CD spectrum of D3,7,13 bound to PE:PG:CL liposomes no longer exhibits well-defined minima at 208 and 222 nm, giving an estimated helical content of only 26%, and peptide D3,6,7,13,14 exhibits a CD spectrum consistent with an extended or random coil structure (Figure 1). Thus, these peptides give the desired progressive loss of helical secondary structure upon increasing D-amino acid substitution, and this effect is most pronounced when D-amino acids are introduced into the central region of the peptide.
3.2. Antimicrobial Activity
Broth dilution assays were used to compare antimicrobial activities of D-amino acid containing peptides to the parent peptide, CM15. Assays were performed against two Gram-positive bacterial species, Staphylococcus aureus and Staphylococcus epidermidis; and two Gram-negative species, Escherichia coli (strain BL21) and Pseudomonas aeruginosa (strain PAO1). Results are summarized in Table 2.
In agreement with previous studies e.g., [36,39,40], CM15 exhibited potent broad-spectrum antimicrobial activity against E. coli, S. aureus, and S. epidermidis, and good activity against P. aeruginosa (Table 2). Replacement of L-lysine with D-lysine at the first and thirteenth positions (D1,13) resulted in only a small decrease in antibacterial activity, with two to four-fold increases in MIC values against all strains tested. Similarly, an analog containing L- to D-lysine substitutions at the third and thirteenth positions (D3,13) also showed a slight increase in MIC values as compared to the parent peptide. However, both of these peptides retain good antimicrobial activity in general, particularly against E. coli and the two strains of Staphylococcus. We also tested an analog containing D-lysine substitutions at positions three and fourteen (D3,14), based on our previous studies indicating that an analog of CM15 containing a V14K substitution retained good antimicrobial activity and had significantly reduced hemolytic activity and toxicity to murine macrophages [36]. Surprisingly, this analog had substantially reduced activity against the E. coli and S. aureus test strains, although it retained good activity against S. epidermidis (Table 2). None of the D-lysine substituted peptides were particularly effective against P. aeruginosa (Table 2).
We also examined two peptides having a third D-lysine substitution near the center of the sequence (D3,7,13 and D3,7,14). Antimicrobial activities for D3,7,13 and D 3,7,14 decreased two-fold relative to D3,13 and D3,14, respectively. Nonetheless, MIC values for D3,7,13 remained at 8 μM or less for E. coli, S. aureus, and S. epidermidis. Finally, we examined a peptide containing five D-lysine residues, with D-lysine replacing all L-lysine residues except at the first position (which is N-acetylated). D3,6,7,13,14 had little or no antibacterial activity, even at high peptide concentrations (up to 128 μM), against any of the bacteria tested. Thus, there was an upper limit to the number of D-lysine substitutions that could be made while still retaining antimicrobial activity, and the gradual increase in MIC values with increasing D-amino acid substitution suggests that α-helical secondary structure does indeed play an important role in the potent antimicrobial activity of CM15.
3.3. Erythrocyte Lysis and Macrophage Toxicity
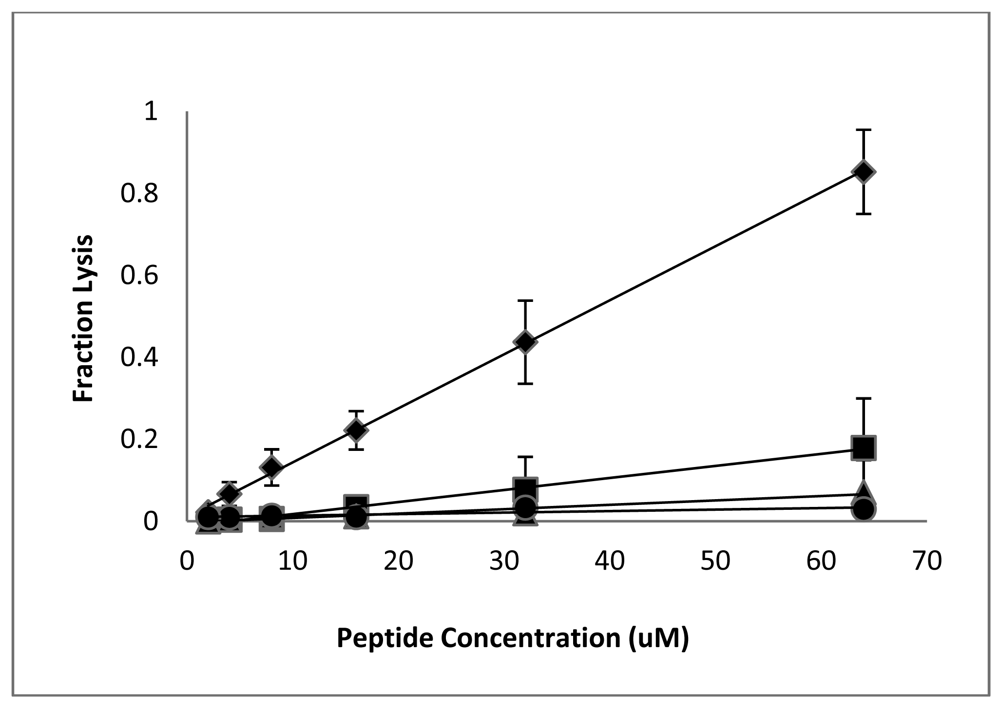
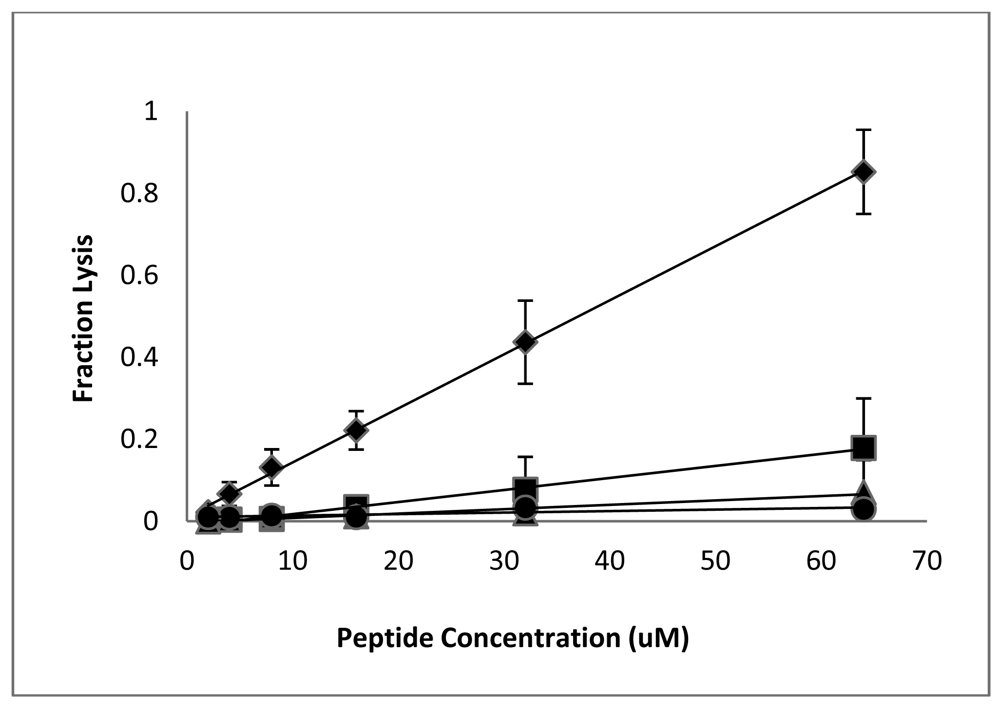
Having established that the D-lysine containing analogs of CM15 retain reasonably good antimicrobial activity (with the exception of D3,6,7,13,14), we next examined the effects of these peptides on eukaryotic cells. Lysis of red blood cells is commonly used to assess the cytotoxicity of antimicrobial peptides, and we previously observed significant hemolysis of human RBC by CM15 when tested at relatively high peptide concentrations [36]. The fractional hemolysis observed for a 5% suspension of human RBC following a 1 h incubation at 37 °C is shown as a function of peptide concentration in Figure 2. RBC lysis increased approximately linearly with peptide concentration. As observed previously [36], significant lysis was observed for CM15, with 64 μM peptide causing nearly 80% lysis (although it should be noted that this concentration is well above the MIC against any of the tested bacterial strains). D1,13 was the next most hemolytic, lysing approximately 16% of the red cell suspension at a peptide concentration of 64 μM. The remaining peptides were essentially non-hemolytic. D3,13 and D3,14 caused less than 6% hemolysis at 64 μM, while less than 1% hemolysis was observed for peptides D3,7,13, D3,7,14, and D3,6,7,13,14 (Figure 2). Thus, D-lysine substitution substantially reduces the hemolysis of human RBC by CM15 analogs, and does so in a manner that correlates with loss of secondary structure.
Although erythrocyte hemolysis is an important consideration and a convenient method for testing peptide toxicity to eukaryotic cells, human RBCs contain a relatively high cholesterol content, which may increase their resistance to AMP-induced lysis [13,53]. To further examine the toxicity of these peptides to eukaryotic cells we used murine RAW264.7 macrophages as a model eukaryotic cell. As with RBC hemolysis, CM15 significantly decreased macrophage viability even at relatively low concentrations, inhibiting mitochondrial respiration with a LD50 of 3.8 μM (Table 3). D1,13 was again the next most cytotoxic of the peptides tested, with an LD50 of 5.4 μM. The remainder of the peptides had substantially reduced toxicity to macrophages, with LD50 values ranging from 78 to 130 μM (Table 3). Notably, peptides D3,13 and D3,7,13, which retain strong antimicrobial activity, exhibited LD50 values of 78 μM and 98 μM, respectively. Thus, as with RBC lysis, D-lysine substitution dramatically lowered the cytotoxicity of these peptides to macrophages. Although low hemolytic activity can be misleading with regard to the general toxicity of AMPs to eukaryotic cells [12], in this study we observed a good correlation between hemolysis and toxicity to cultured macrophages.
3.4. Peptide-Induced Membrane Permeabilization
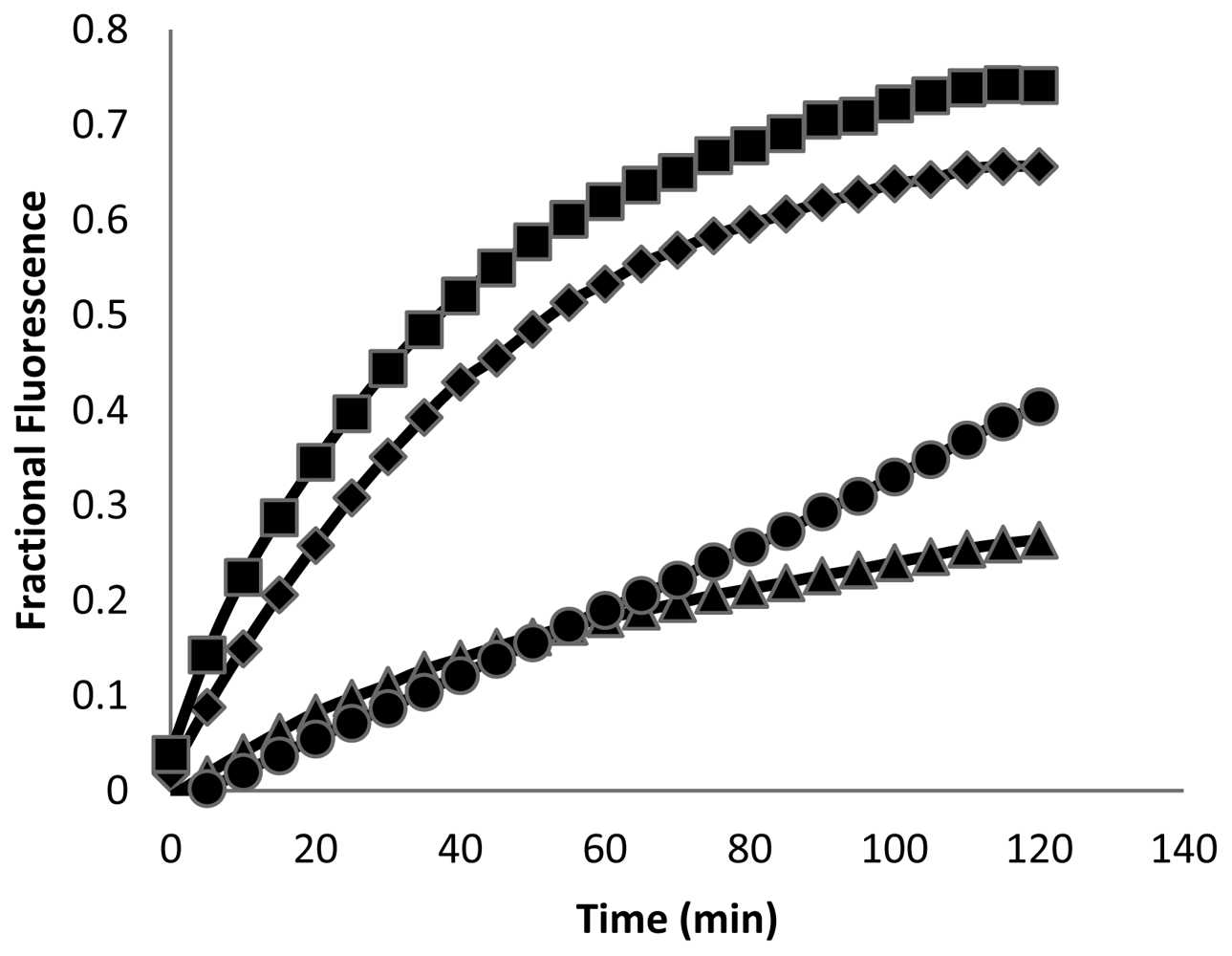
Membrane permeabilization is considered to be a key mechanism of bacterial cell killing by amphipathic α-helical AMPs [7-12,15]. To investigate the effects of these peptides on cell membrane integrity in both bacterial and eukaryotic cells, we examined their ability to facilitate uptake of the membrane-impermeable dye SYTOX green. Upon disruption of the membrane permeability barrier, SYTOX green diffuses into cells and binds DNA, producing a pronounced enhancement of its fluorescence [54]. As shown in Figure 3, CM15, D1,13, D3,13, and D3,7,13 all produced an immediate (<2 min from time of peptide addition) permeabilization of the S. aureus cell envelope to SYTOX green, consistent with membrane disruption as a prominent mechanism of bactericidal activity.
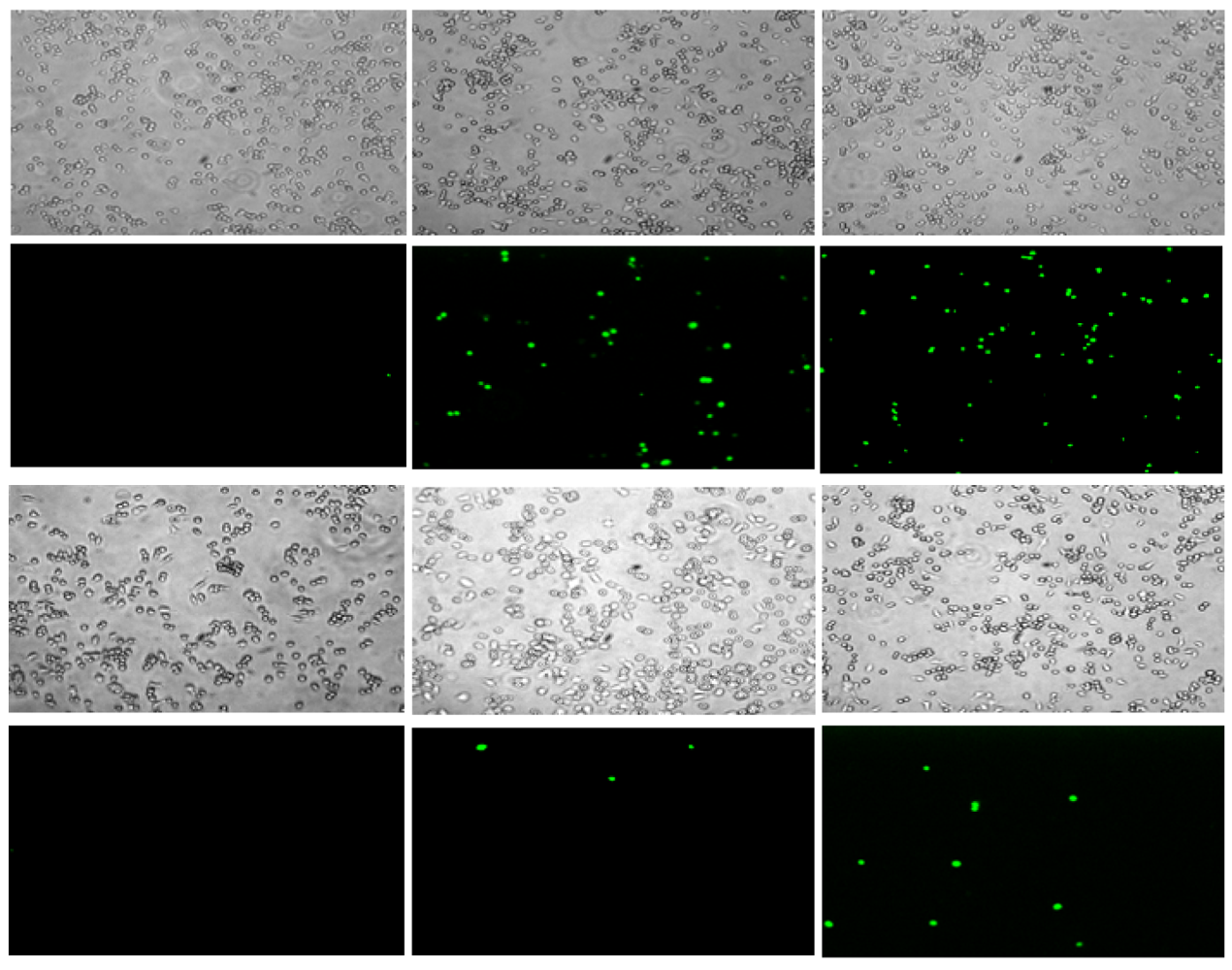
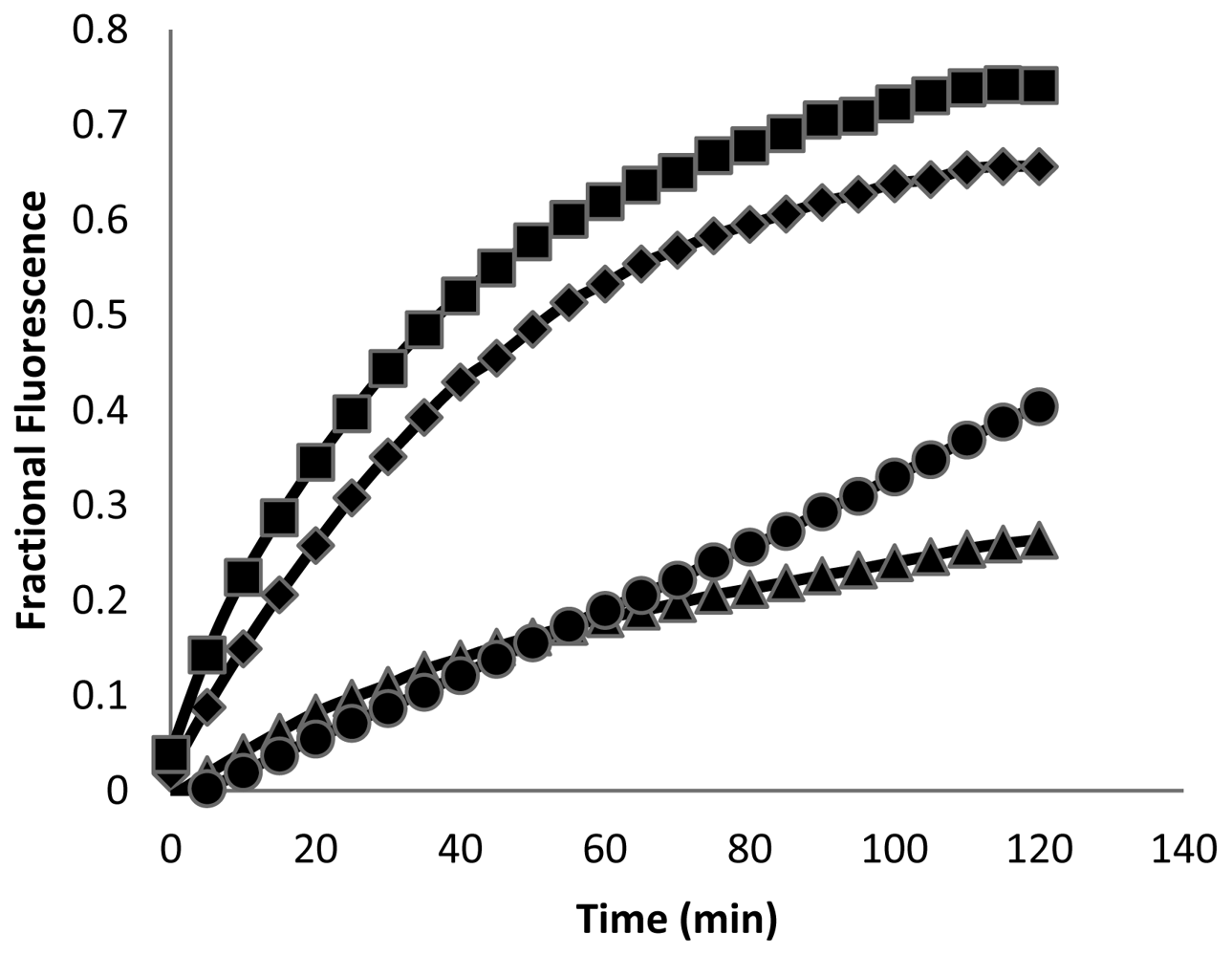
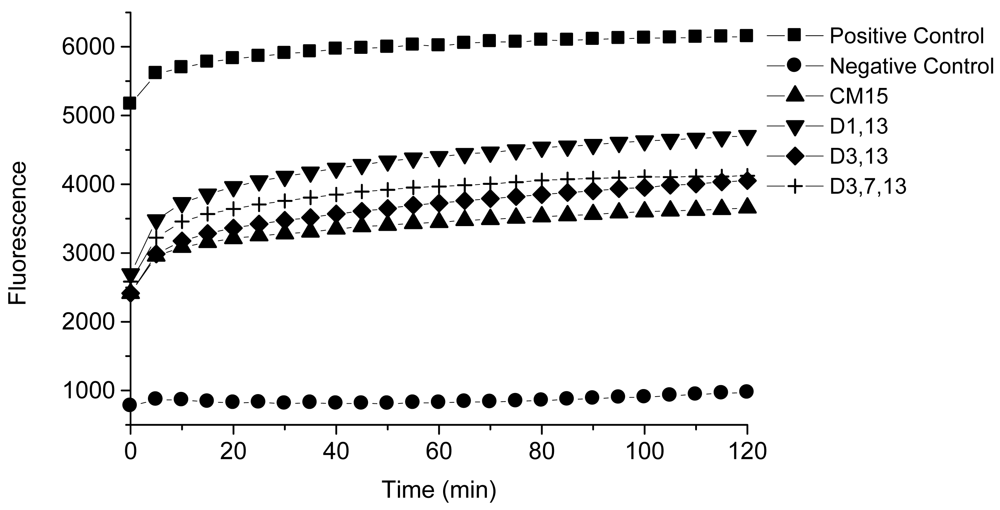
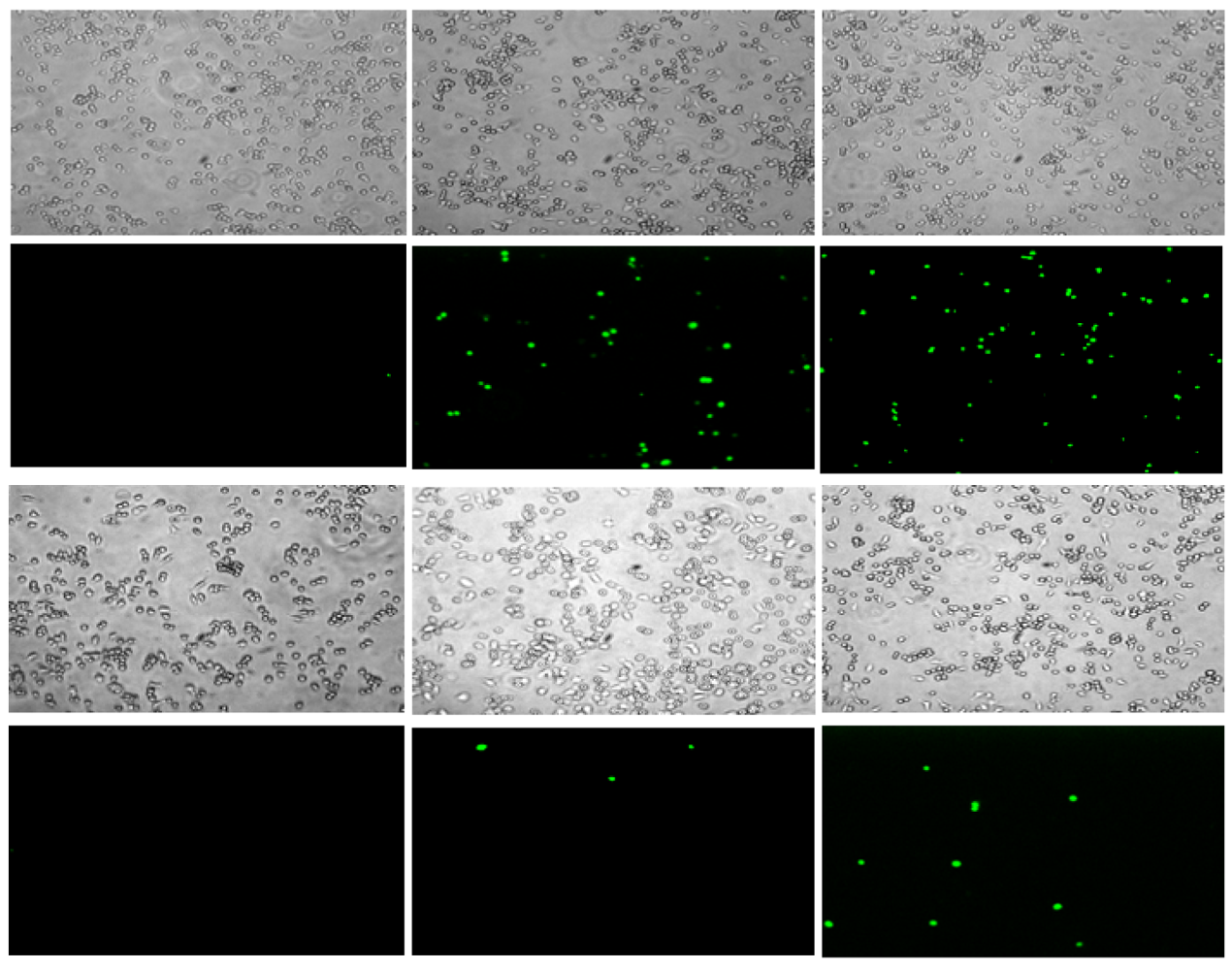
These peptides also caused permeabilization of the cell membrane in RAW264.7 macrophages, but at a slower rate and to a lesser degree than observed with S. aureus. Fluorescence microscopy images of macrophages exposed to CM15 or D-lysine containing analogs (e.g., D3,7,13, Figure 4) demonstrated a time-dependent increase in SYTOX-positive cells. As indicated by the representative data shown in Figure 4, little or no SYTOX fluorescence was observed immediately after peptide addition. Consistent with their relative effects on macrophage survival as assessed by MTT metabolism, the increase in SYTOX uptake was much greater for CM15 than for D3,7,13 (Figure 4) or for the other D-lysine containing peptides (data not shown). Time courses for the increase in SYTOX fluorescence produced by exposure of macrophages to CM15, D1,13, D3,13 and D3,7,13 are shown in Figure 5. As little or no SYTOX uptake was observed for peptides at their respective MIC values, each peptide was tested at a concentration 4 times its respective MIC value for S. aureus. High levels of SYTOX uptake (60–80% of the values obtained by complete membrane permeabilization with Triton X100) were observed for CM15 (2 μM) and D1,13 (4 μM), while substantially lower levels of SYTOX uptake were observed for D3,13 (16 μM) and D3,7,13 (32 μM) despite the much higher concentrations of the latter two peptides. Thus, these results parallel those based on MTT metabolism indicating the substantially reduced cytotoxicity of D3,13 and D3,7,13. These results are also consistent with damage to the plasma membrane as an important factor in the cytotoxicity of these peptides to eukaryotic cells.
3.5. Selectivity Indices
Antimicrobial efficacy is a combination of the ability to kill or inhibit growth of the pathogen and a lack of toxicity to host eukaryotic cells. As a comparative measure of in vitro efficacy, we calculated selectivity indices based on macrophage toxicity (Table 4) by taking the ratio of the macrophage LD50 to the MIC value for each peptide against each bacterial strain. Since lower toxicity to eukaryotic cells (reflected by an increased macrophage LD50) and greater antimicrobial activity (reflected by decreased MIC values) both increase this parameter, higher values of the selectivity index indicate a greater selectivity towards bacterial versus eukaryotic cells. Due to their substantially decreased macrophage toxicity, selectivity indices for peptides D3,13 and D3,7,13 were increased relative to CM15 against all of the tested bacteria (Table 4). Increases in selectivity for D3,13 relative to CM15 ranged from approximately 2.5 (for P. aeruginosa) to 5-fold (for S. epidermidis). Addition of a third D-lysine led to a generally decreased selectivity for D3,7,13 as compared to D3,13 (with the exception of P. aeruginosa PAO1, for which they were essentially equal), and for D3,7,14 as compared to D3,14. However, selectivity indices for D3,7,13 were uniformly greater than for D1,13, and were greater than for D3,14 against E. coli and S. aureus, indicating that efficacy was not determined solely by the number of D-amino acid substitutions. Overall, these results indicate that both the position and number of D-amino acid substitutions influence the relative selectivity of these diasteromeric CM15 analogs.
3.6. Protease Susceptibility
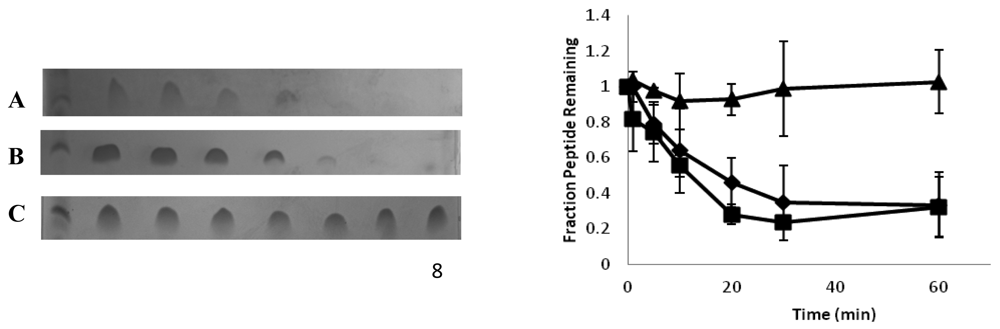
Introducing β- or D-amino acids into a peptide can increase its biological half-life by making it less susceptible to protease digestion. We used SDS PAGE to determine the relative susceptibility of CM15, D3,13 and D3,7,13 to trypsin and chymotrypsin. As seen in Figure 6, both CM15 and D3,13 were readily degraded, with almost all of the peptide being digested within the first 20 min of incubation. In contrast, D3,7,13 exhibited minimal digestion under our experimental conditions. As indicated by densitometric analysis, there was no significant difference in the amounts of CM15 and D3,13 remaining at any given time point during trypsin digestion, while the amount of intact D3,7,13 remained unchanged throughout the 60 min incubation time. Although neither of the aromatic residues (Trp2 or Phe5) were replaced with D-amino acids, a similar trend was observed with chymotrypsin (data not shown), suggesting that the presence of nearby D-lysine residues (i.e., at positions 3 and 7) impaired degradation by this protease.
3.7. Discussion
The ability to fold into amphipathic secondary structures in the a membrane environment is a unifying characteristic exhibited by a large majority of both synthetic and naturally-occurring AMPs [7-13]. Consequently, for many years it was assumed that the adoption of a well-defined amphipathic structure upon membrane binding was necessary for antimicrobial activity. Several studies have now called this assumption into question by indicating that interruption of the hydrophobic surface and/or disruption of amphipathic secondary structure primarily impacts the toxicity of AMPs to eukaryotic cells, while having more moderate effects on their antimicrobial activity e.g., [12,20-29]. Disruption of amphipathic secondary structure can be accomplished by the inclusion of polar or charged residues in the non-polar surface of helical AMPs [20-22] or by the introduction of D-amino [23-29] or β-amino acids [30-32]. The inclusion of β- or D-amino acids has the additional benefit of providing stability against protease digestion [24,25,28,31]. In the present study, we examined the impact of introducing D-amino acids into CM15, a well-established representative of the linear, amphipathic α-helical class of AMPs. Our results show that selective D-lysine substitution can indeed substantially decrease peptide toxicity to eukaryotic cells and prevent degradation by common proteases with the retention of good antimicrobial activity, resulting in an overall gain in selectivity for prokaryotic over eukaryotic cells.
As an experimental approach, the substitution of D-amino acids for their corresponding L-enantiomers allows one to alter secondary structure content without changing a peptide's charge or mean residue hydrophobicity. The use of diastereomers as less cytotoxic versions of all-L AMPs was first explored by Shai and co-workers [23-27]. Consistent with the present work, they found that D-amino acid substitutions had variable effects on antimicrobial activity depending on bacterial strain, but dramatically decreased or eliminated hemolytic activity concomitant with loss of α-helical secondary structure [24,25]. Peptides were either partially or fully protected against protease digestion depending on placement of the D-amino acid substitutions [26]. Additional studies of both α-helical [20,28] and β-sheet [29] peptides have also demonstrated substantially reduced hemolytic activity and decreased macrophage toxicity for diastereomeric AMPs. The use of diastereomers appears to be a more effective approach than simply using all-D enantiomers which, although they are protease resistant and retain full antimicrobial activity [42,55], may also retain the hemolytic properties of their parent all-L enantiomer [55]. Preliminary studies with all-D CM15 (data not shown) indicated an LD50 for RAW 264.7 macrophages similar to that observed for all-L CM15.
In the present work we have extended the study of peptide diastereomers to CM15. We found that the introduction of only two D-lysine residues into CM15 was sufficient to produce substantial decreases in RBC hemolysis and macrophage cytotoxicity. This is consistent with previous studies of small β-sheet peptides identified by high-throughput screening [29] and a 26-residue helical peptide [20] indicating that even a single D-amino acid substitution can significantly decrease hemolytic activity. The incorporation of D-lysine at the extreme ends of CM15 (in peptide D1,13), which decreased helical content only slightly, had the least impact on hemolysis and cytotoxicity. However, D-lysine substitution at positions 3 and 13 or at positions 3 and 14, either of which caused an approximately 30% reduction in helical content of the membrane-bound peptide, resulted in an ∼20-fold reduction in macrophage toxicity (i.e., a 20-fold increase in LD50) and similarly large reductions in hemolysis. The introduction of an additional D-lysine residue near the center of the peptide (at residue 7) further decreased helical content along with a concomitant decrease in both antimicrobial activity and cytotoxicity. In contrast to previous studies on 15-residue leucine-lysine model peptides [30,31] an analog of CM15 containing five D-amino acid substitutions (D3,6,7,13,14) retained little antimicrobial activity. Numerous studies have shown that, for most AMPs, there is a strong correlation between antimicrobial activity and the ability of a given peptide to disrupt the bacterial membrane permeability barrier. All of the peptides in this study permeabilized membranes to the cationic cyanine dye, SYTOX green. Permeabilization of S. aureus cell membranes occurred rapidly, with significant levels of SYTOX uptake observed immediately upon addition of peptide. This immediate disruption of the membrane permeability barrier is consistent with the very rapid bacterial cell killing (e.g., an approximately 5-log reduction in CFU within 15 min) observed for CM15 [45]. In contrast, permeabilization of the macrophage cell membrane to SYTOX occurred at a much slower rate, and the extent of SYTOX uptake into macrophages was markedly decreased for the relatively non-cytotoxic peptides D3,13 and D3,7,13. Thus, although there are many other potential mechanisms may that contribute to the effects of AMPs on cell viability [11], these studies suggest that disruption of the plasma membrane may play an important role in the eukaryotic cell cytotoxicity of CM15 and related AMPs.
In addition to reducing peptide toxicity to eukaryotic cells, D-amino acid substitutions can also protect against protease digestion [24,25,28], potentially extending biological half-life. Although D3,13 exhibited the greatest antimicrobial selectivity, the addition of two D-amino acid residues near the ends of CM15 did not substantially increase resistance to proteolytic digestion. In contrast, D3,7,13 was not degraded by either trypsin or chymotrypsin under the defined experimental conditions. Notably, the presence of D-lysine at the 3 and 7 positions was evidently sufficient to protect against trypsin digestion at Lys6, or against chymotrypsin digestion at Trp2 or Phe5.
4. Conclusions
This systematic study of CM15 analogs containing variable numbers of D-lysine substitutions demonstrates a strong correlation between helical content and toxicity to eukaryotic cells. These results support the emerging view that amphipathic secondary structure, which provides a continuous, hydrophobic surface that can facilitate perturbation of neutral membrane bilayers, plays a key role in the toxicity of AMPs to eukaryotic cells. In contrast, antimicrobial activity, which depends most prominently on electrostatic interactions, is less affected by the loss of amphipathic secondary structure. These studies led to the identification of two CM15 diastereomers, peptides D3,13 and D3,7,13, that were non-hemolytic and approximately 20-fold less cytotoxic to cultured murine macrophages than all-L CM15. In addition, D3,7,13 was resistant to digestion by both trypsin and chymotrypsin. Thus, the selective introduction of D-amino acid residues into linear, amphipathic AMPs is a viable approach for enhancing antimicrobial selectivity and increasing resistance to proteolytic degradation.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence * | % helix in PE:PG:CL |
|---|---|---|
| CM15 | KWKLFKKIGAVLKVL | 65 |
| D1,13 | kWKLFKKIGAVLkVL | 55 |
| D3,13 | KWkLFKKIGAVLkVL | 46 |
| D3,14 | KWkLFKKIGAVLKkL | 45 |
| D3,7,13 | KWkLFKkIGAVLkVL | 28 |
| D3,7,14 | KWkLFKkIGAVLKkL | 24 |
| D3,6,7,13,14 | KWkLFkkIGAVLkkL | 6 |
| Peptide | E. coli BL21 | Staph aureus (6835p) | Staph epidermidis | P. aeruginosa PA103 | P. aeruginosa PAO1 |
|---|---|---|---|---|---|
| CM15 | 0.5 | 0.5 | 0.5 | 8 | 4 |
| D1,13 | 1 | 2 | 1 | 16 | 16 |
| D3,13 | 4 | 4 | 2 | 16 | 32 |
| D3,14 | 16 | 32 | 2 | 8 | 16 |
| D3,7,13 | 8 | 8 | 4 | 32 | 32 |
| D3,7,14 | >128 | 64 | 4 | 16 | 32 |
| D3,6,7,13,14 | >128 | >128 | >128 | >128 | >128 |
| Peptide | LD50 (μM) |
|---|---|
| CM15 | 3.8 ± 0.3 |
| D1,13 | 5.4 ± 0.8 |
| D3,13 | 78 ± 13 |
| D3,14 | 84 ± 14 |
| D3,7,13 | 98 ± 8.4 |
| D3,7,14 | 126 ± 11 |
| D3,6,7,13,14 | 131 ± 15 |
| Peptide | E. coli BL21 | Staph. aureus 6835p | Staph. epidermidis | P. aeruginosa PA103 | P. aeruginosa PA01 |
|---|---|---|---|---|---|
| CM15 | 7.6 | 7.6 | 7.6 | 0.5 | 0.9 |
| D1,13 | 5.4 | 2.7 | 5.4 | 0.3 | 0.3 |
| D3,13 | 20 | 20 | 40 | 5 | 2.5 |
| D3,14 | 5 | 2.5 | 42 | 10 | 5 |
| D3,7,13 | 12 | 12 | 24 | 3 | 3 |
| D3,7,14 | --- | 2 | 32 | 8 | 4 |
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of General Medical Sciences grant GM068829.
References
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; Craig, A.S.; Zell, E.R.; Fosheim, G.E.; McDougal, L.K.; Carey, R.B.; Fridkin, S.K. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar]
- Martinez, J.L. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc. Biol. Sci. 2009, 276, 2521–2530. [Google Scholar]
- Furuya, E.Y.; Lowy, F.D. Antimicrobial-resistant bacteria in the community setting. Nat. Rev. Micro. 2006, 4, 36–45. [Google Scholar]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Micro. 2007, 5, 175–186. [Google Scholar]
- Hancock, R.E. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar]
- Fernandez-Vidal, M.; Jayasinghe, S.; Ladokhin, A.S.; White, S.H. Folding amphipathic helices into membranes: Amphiphilicity trumps hydrophobicity. J. Mol. Biol. 2007, 370, 459–470. [Google Scholar]
- Huang, H.W. Action of antimicrobial peptides: Two-state model. Biochemistry 2000, 39, 8347–8352. [Google Scholar]
- Domadia, P.; Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Structure, interactions, and antimicrobial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: Role of the helical hairpin conformation in outer membrane permeabilization. J. Am. Chem. Soc. 2010, 132, 18417–18428. [Google Scholar]
- Bhunia, A.; Domadia, P.; Torres, J.; Hallock, K.J.; Ramamoorthy, A.; Bhattacharjya, S. NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles. J. Biol. Chem. 2010, 285, 3883–3895. [Google Scholar]
- Porcelli, F.; Verardi, R.; Shi, L.; Henzler-Wildman, K.A.; Ramamoorthy, A.; Veglia, G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. [Google Scholar]
- Porcelli, F.; Buck-Koehntop, B.A.; Thennarasu, S.; Ramamoorthy, A.; Veglia, G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 2006, 45, 5793–5799. [Google Scholar]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar]
- Hawrani, A.; Howe, R.A.; Walsh, T.R.; Dempsey, C.E. Origin of low mammalian cell toxicity in a class of highly active antimicrobial amphipathic helical peptides. J. Biol. Chem. 2008, 283, 18636–18645. [Google Scholar]
- Dempsey, C.E.; Hawrani, A.; Howe, R.A.; Walsh, T.R. Amphipathic antimicrobial peptides— From biophysics to therapeutics? Protein Pept. Lett. 2010, 17, 1334–1344. [Google Scholar]
- Shai, Y.; Oren, Z. Diastereomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem. 1996, 271, 7305–7308. [Google Scholar]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar]
- Oren, Z.; Hong, J.; Shai, Y. A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J. Biol. Chem. 1997, 272, 14643–14649. [Google Scholar]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar]
- Braustein, A.; Papo, N.; Shai, Y. In vitro activity and potency of an intravenously injected antimicrobial peptide and its DL amino acid analog in mice infected with bacteria. Antimicrob. Agents Chemother. 2004, 48, 3127–3129. [Google Scholar]
- Wang, P.; Nan, Y.; Yang, S.-T.; Kang, S.; Kim, Y.; Park, I.-S.; Hahm, K.-S.; Shin, S. Cell selectivity and anti-inflammatory activity of a Leu/Lys-rich alpha-hleical model amtimicrobial peptide and its diastereomeric peptides. Peptides 2010, 31, 1251–1261. [Google Scholar]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar]
- Epand, R.F.; Schmitt, M.A.; Gellman, S.H.; Epand, R.M. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two alpha/beta-antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1343–1350. [Google Scholar]
- Schmitt, M.A.; Weisblum, B.; Gellman, S.H. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of alpha/beta-peptides. J. Am. Chem. Soc. 2007, 129, 417–428. [Google Scholar]
- Olsen, C.A.; Ziegler, H.L.; Nielsen, H.; Frimodt-Moller, N.; Jaroszewski, J.; Franzyk, H. Antimicrobial, hemolytic, and cytotoxic activities of beta-peptoid-peptide hybrid oligomers: Improved properties compared to natural AMPs. Chembiochem 2010, 11, 1356–1360. [Google Scholar]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar]
- Sato, H.; Feix, J.B. Lysine-enriched cecropin-mellitin antimicrobial peptides with enhanced selectivity. Antimicrob. Agents Chemother. 2008, 52, 4463–4465. [Google Scholar]
- Wang, Z.; Wang, G. APD: The antimicrobial peptide database. Nucleic Acids Res. 2004, 32, D590–D592. [Google Scholar]
- Boman, H.G.; Wade, D.; Boman, I.; Wahlin, B.; Merrifield, R.B. Antibacterial and antimalarial properties of peptides that are cecropin-melittin hybrids. FEBS Lett. 1989, 259, 103–106. [Google Scholar]
- Andreu, D.; Ubach, J.; Boman, A.; Wahlin, B.; Wade, D.; Merrifield, R.B.; Boman, H.G. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 1992, 296, 190–194. [Google Scholar]
- Wade, D.; Andreu, D.; Mitchell, S.A.; Silveira, A.M.; Boman, A.; Boman, H.G.; Merrifield, R.B. Antibacterial peptides designed as analogs or hybrids of cecropins and melittin. Int. J. Pept. Protein Res. 1992, 40, 429–436. [Google Scholar]
- Piers, K.L.; Hancock, R.E. The interaction of a recombinant cecropin/melittin hybrid peptide with the outer membrane of Pseudomonas aeruginosa. Mol. Microbiol. 1994, 12, 951–958. [Google Scholar]
- Merrifield, R.B.; Juvvadi, P.; Andreu, D.; Ubach, J.; Boman, A.; Boman, H.G. Retro and retroenantio analogs of cecropin-melittin hybrids. Proc. Natl. Acad. Sci. USA 1995, 92, 3449–3453. [Google Scholar]
- Bhargava, K.; Feix, J.B. Membrane binding, structure, and localization of cecropin-mellitin hybrid peptides: A site-directed spin-labeling study. Biophys. J. 2004, 86, 329–336. [Google Scholar]
- Sato, H.; Feix, J.B. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1245–1256. [Google Scholar]
- Sato, H.; Feix, J.B. Osmoprotection of bacterial cells from toxicity caused by antimicrobial hybrid peptide CM15. Biochemistry 2006, 45, 9997–10007. [Google Scholar]
- Pistolesi, S.; Pogni, R.; Feix, J.B. Membrane insertion and bilayer perturbation by antimicrobial peptide CM15. Biophys. J. 2007, 93, 1651–1660. [Google Scholar]
- Bastos, M.; Bai, G.; Gomes, P.; Andreu, D.; Goormaghtigh, E.; Prieto, M. Energetics and partition of two cecropin-melittin hybrid peptides to model membranes of different composition. Biophys. J. 2008, 94, 2128–2141. [Google Scholar]
- Respondek, M.; Madl, T.; Gobl, C.; Golser, R.; Zangger, K. Mapping the orientation of helices in micelle-bound peptides by paramagnetic relaxation waves. J. Am. Chem. Soc. 2007, 129, 5228–5234. [Google Scholar]
- Cao, Y.; Yu, R.Q.; Liu, Y.; Zhou, H.X.; Song, L.L.; Cao, Y.; Qiao, D.R. Design, recombinant expression, and antibacterial activity of the cecropins-melittin hybrid antimicrobial peptides. Curr. Microbiol. 2010, 61, 169–175. [Google Scholar]
- Fantner, G.E.; Barbero, R.J.; Gray, D.S.; Belcher, A.M. Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat. Nanotechnol. 2010, 5, 280–285. [Google Scholar]
- Stewart, J.C.M. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar]
- Greenfield, N.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar]
- Lebaron, P.; Catala, P.; Parthuisot, N. Effectiveness of SYTOX Green stain for bacterial viability assessment. Appl. Environ. Microbiol. 1998, 64, 2697–2700. [Google Scholar]
- Chen, Y.; Vasil, A.I.; Rehaume, L.; Mant, C.T.; Burns, J.L.; Vasil, M.L.; Hancock, R.E.; Hodges, R.S. Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem. Biol. Drug Des. 2006, 67, 162–173. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kaminski, H.M.; Feix, J.B. Effects of D-Lysine Substitutions on the Activity and Selectivity of Antimicrobial Peptide CM15 . Polymers 2011, 3, 2088-2106. https://doi.org/10.3390/polym3042088
Kaminski HM, Feix JB. Effects of D-Lysine Substitutions on the Activity and Selectivity of Antimicrobial Peptide CM15 . Polymers. 2011; 3(4):2088-2106. https://doi.org/10.3390/polym3042088
Chicago/Turabian StyleKaminski, Heather M., and Jimmy B. Feix. 2011. "Effects of D-Lysine Substitutions on the Activity and Selectivity of Antimicrobial Peptide CM15 " Polymers 3, no. 4: 2088-2106. https://doi.org/10.3390/polym3042088