Polysaccharides: The “Click” Chemistry Impact

 and
and
Abstract
: Polysaccharides are complex but essential compounds utilized in many areas such as biomaterials, drug delivery, cosmetics, food chemistry or renewable energy. Modifications and functionalizations of such polymers are often necessary to achieve molecular structures of interest. In this area, the emergence of the “click” chemistry concept, and particularly the copper-catalyzed version of the Huisgen 1,3-dipolar cycloaddition reaction between terminal acetylenes and azides, had an impact on the polysaccharides chemistry. The present review summarizes the contribution of “click” chemistry in the world of polysaccharides.1. Introduction
1.1. “Click Chemistry”: When Copper Salts Met Polysaccharides
The impact of the “click” chemistry concept [1] and, especially, the copper catalyzed azide/alkyne cycloaddition (CuAAC) [2-4] do not need to be re-demonstrated in this paper. Indeed several reviews have previously developed applications of the triazole chemistry in many areas [5-8]. Fundamental and mechanistic aspects of the CuAAC were fully described in [9]. Materials, polymer and dendrimer science were also approached in [10], as wasthe area of drug discovery [11], bioconjugations, peptidomimetics, chemical ligations, etc. However no review was designated to the role of the CuAAC on polysaccharides. Therefore, the present paper will try to describe how the ‘polyosides’ behaved towards the azide/alkyne-“click”-reaction, and assess if, also in this chemistry domain, “everything is as easy as a click” [12].
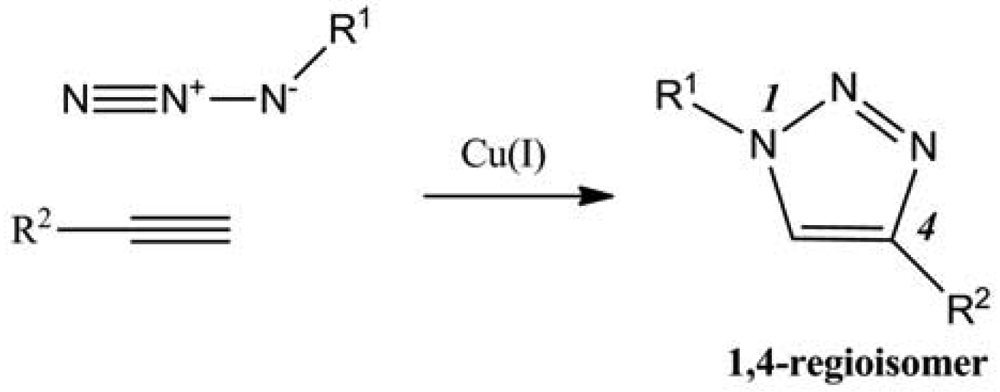
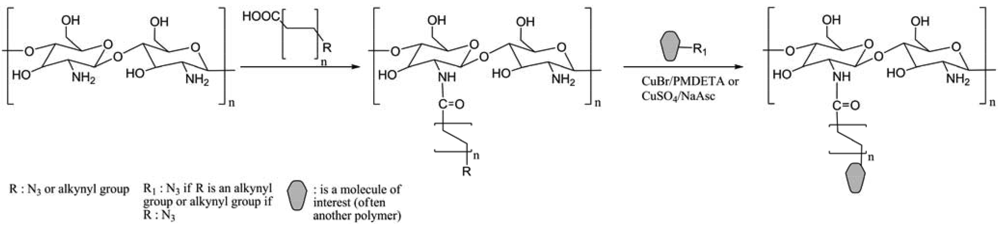
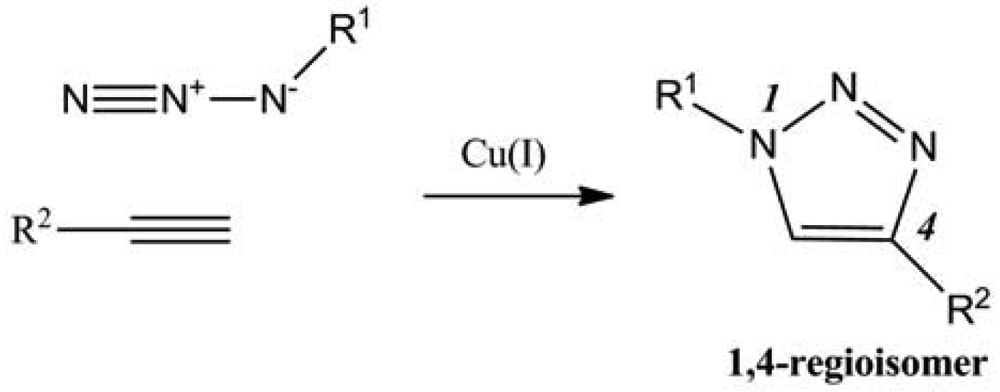
Among the multiple reactions that could be named “click”, the focus will be on the catalyzed version of Huigen 1,3-dipolar cycloaddition reaction between azides and terminal alkynes (Figure 1). Indeed polysaccharides were often modified using this reaction for different purposes such as creating new materials, new therapeutic or diagnostic agents, or merely to understand the reactivity of such complex structures in terms of regioselectivity, degree of substitution (DS), cross-linking, etc.
A non-catalytic variation of the 1,3-dipolar azide-alkyne cycloaddition was also utilized to functionalize polysaccharides, thanks notably to difluorocyclooctynes (DIFO), with interesting applications in the biochemical area, because of the removal of copper toxicity (Figure 2) [13,14]. The copper-free reaction was recently developed and proved suitable for many applications, notably in the glycans area. This reaction, achieved by increasing the reactivity of the alkyne by introducing ring strain, was named Strain Promoted Alkyne Azide Cycloaddition (SPAAC).
Through the reactions described above, different families of polysaccharides will be investigated in terms of their reactivity and their applications, in order to understand the influence of “click” chemistry on the polyosides.
1.2. The World of Polysaccharides: Various Entities Associated to Various Reactivities
The polysaccharides regroup lots of types of macromolecules. Among them, only eight categories were identified to have been impacted by the CuAAC or the SPAAC (Table 1), which are cellulose, dextran, chitosan, starch, guar, hyaluronan, (1→3)-(β-D-glucan, and glycans. The different approaches of modifications will be detailed in the paper. Indeed, even if the “click” reactions are by definition reactions with quantitative yields, high tolerance of functional groups, etc. [1], they require however particular starting reagents. Thus, the terminal alkyne or azide groups need to be “inserted” in the polymer. In order to control the final structure/composition of the macromolecules, regioselective reactions are often necessary.
2. A Need for Pre-“Click” Functionalization
Considering the hydroxyl/amine positions in the polymer structure (except for hyaluronan), a well-defined functionalization is a preliminary step towards the “click” modification. Several attempts were undertaken, with an asymptotic trend towards the regioselective functionalization. To understand the problem, three examples of modifications are developed below, for four different polysaccharides: starch, chitosan, cellulose and (1→3)-β-D-glucan.
2.1. Positions 2, 3 and 6 in Starch
The repeating units of starch present three potentially reactive sites that correspond to the hydroxyls in positions 2, 3 and 6 of the sugar. In this way, Hopf and co-workers described an etherification of potato starch using sodium hydroxide or Li dimsyl in dimethylsulfoxide, and propargyl bromide [49]. Thus, they established an order of reactivity, which was 2 > 6 ≫ 3. Then a further functionalization by CuAAC with benzyl azide was carried out and led to a N-benzyltriazole derivatized starch (Figure 3).
2.2. From Chitosan to Cellulose
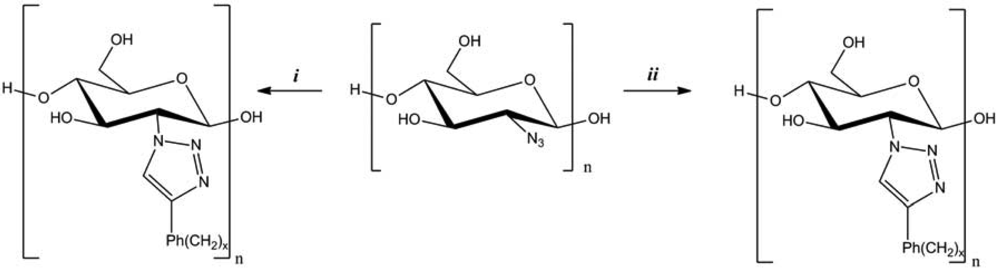
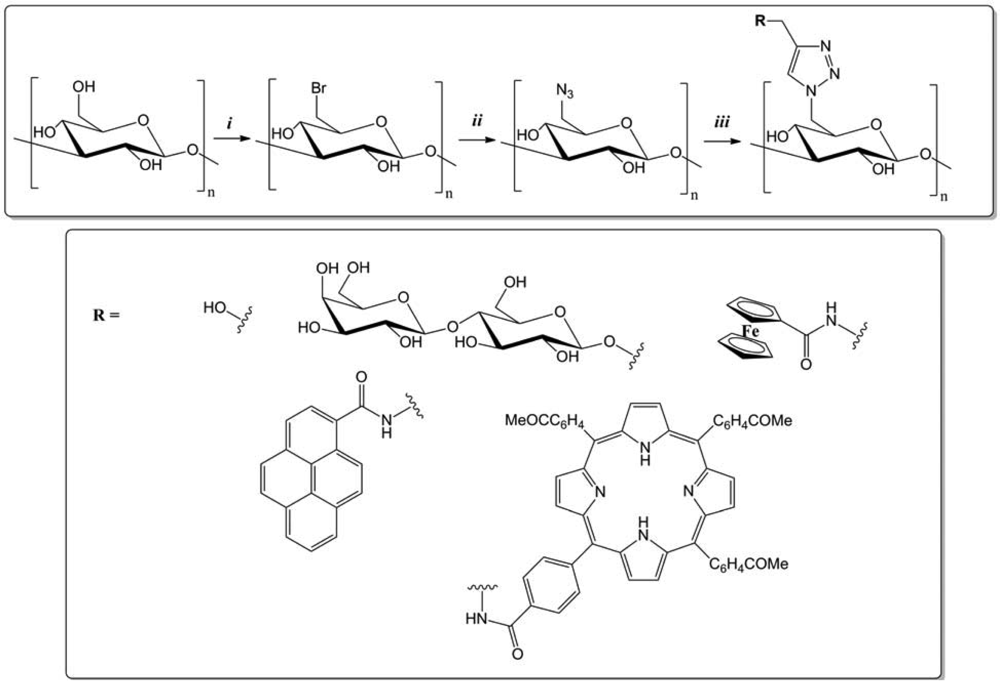
An original pathway was established by Vasella and co-workers to synthesize 2-azido-2-deoxycellulose from chitosan (Figure 4) [34]. The azido-functionalization was carried out using a diazo transfer from trifluoromethanesulfonyl azide (TfN3), among four to five reaction steps. Then the reactivity of 2-azido-2-deoxycellulose was evaluated by the CuAAC, in the presence of CuI, dimethylsulfoxide (DMSO), and phenyl-, (phenyl)alkyl-, and alkylacetylenes. New cellulose derivatives were thus obtained, in high yield.
2.3. Position 6: The Case of Cellulose and (1→3)-β-D-Glucan
According to the literature, the position 6 of polysaccharides seems to be more accessible in terms of reactivity. Indeed the azido-derivatives are easily synthesized, introducing first a good leaving group in the polysaccharide structure, then by a nucleophilic displacement using sodium azide. In this way, Shinkai and co-workers developed a convenient approach to elaborate (1→3)-(β-D-glucan with various functional appendages (Figure 5) [56].
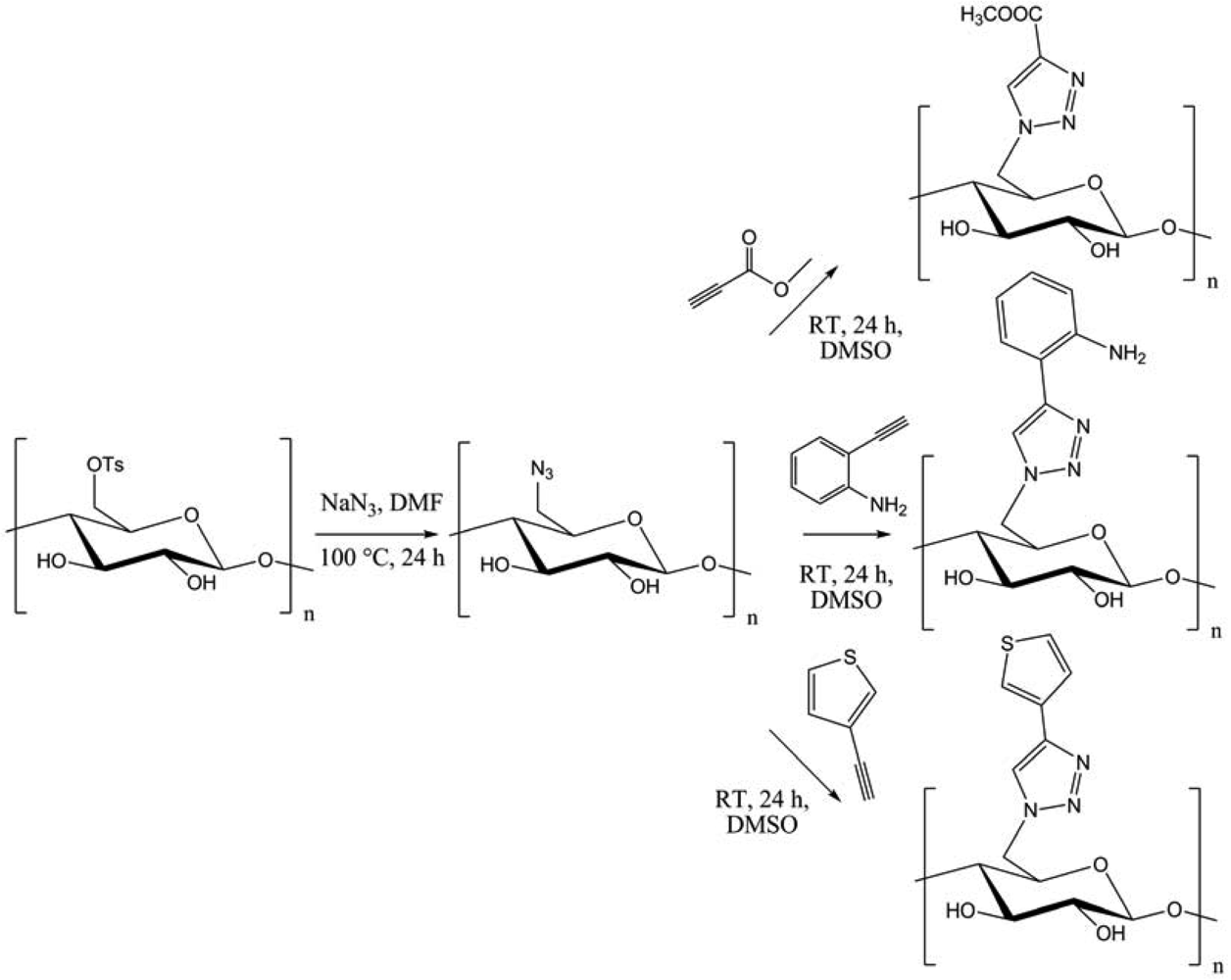
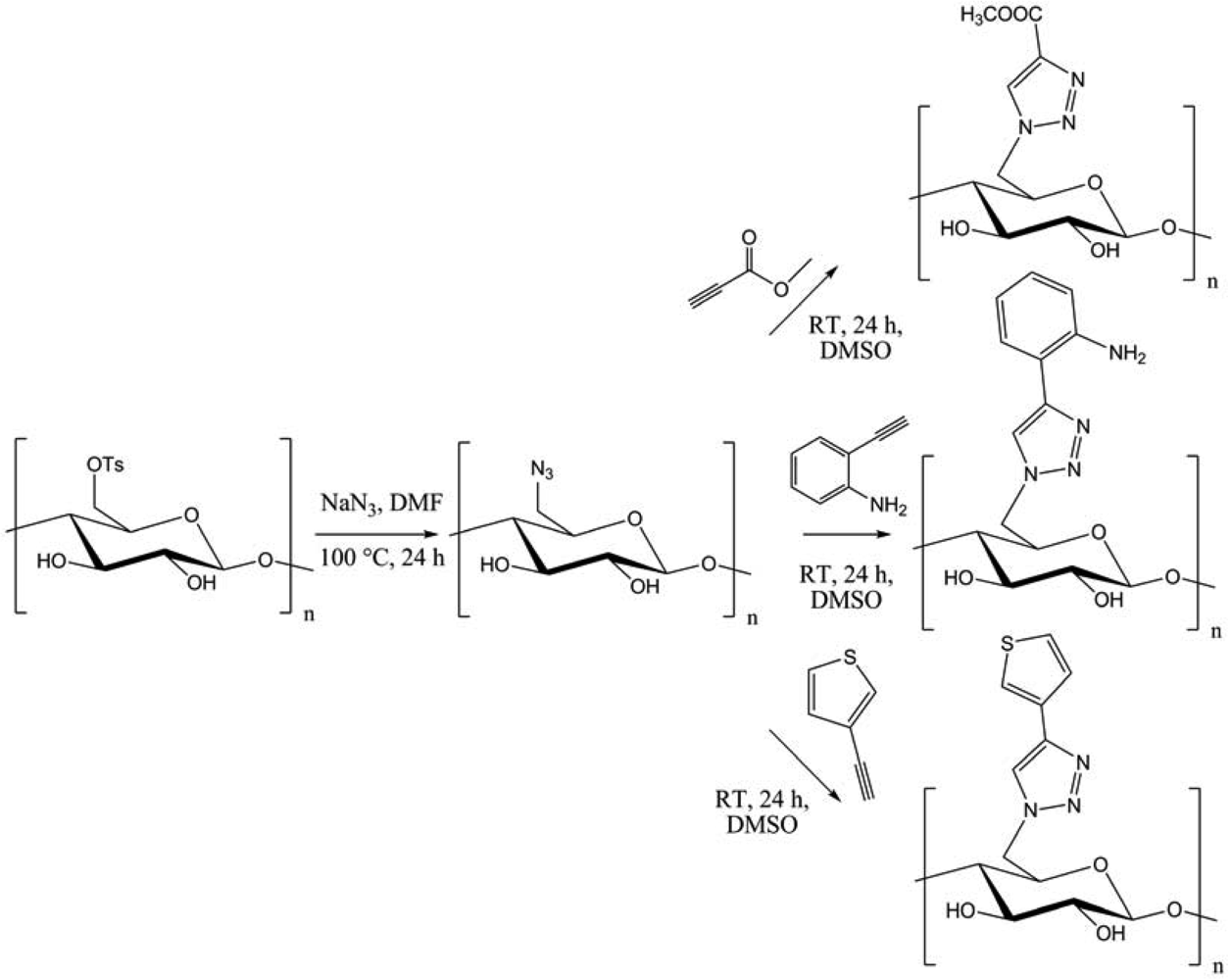
Bromination-azidation of curdlan afforded 6-azido-6-deoxycurdlan. The chemoselective [3 + 2] cycloaddition with several functional modules led to functionalized (1→3)-(β-D-glucan, with possible applications in gene carriers, redox-active or fluorescent “green” materials. In another context, Heinze et al. introduced azide group into cellulose using the tosyl derivative (Figure 6) [15].
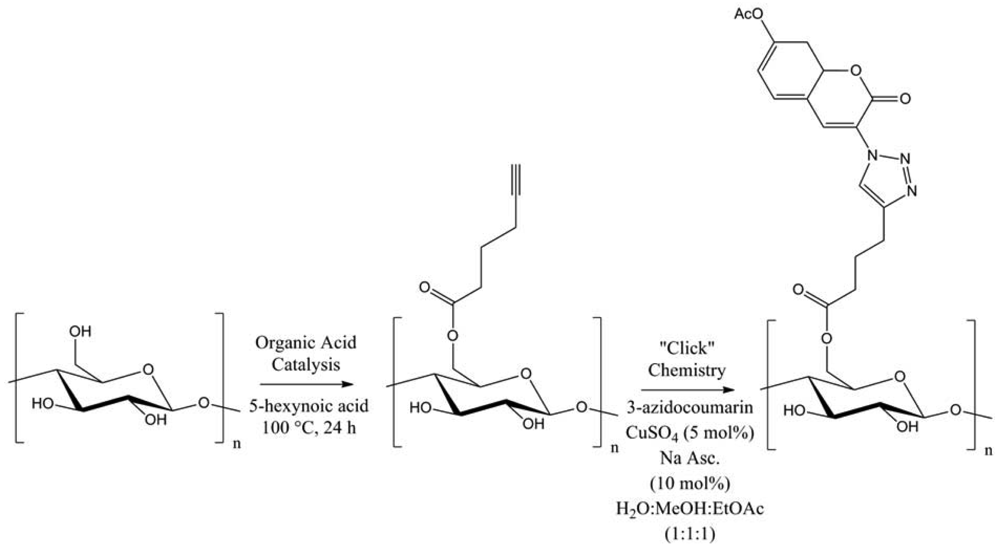
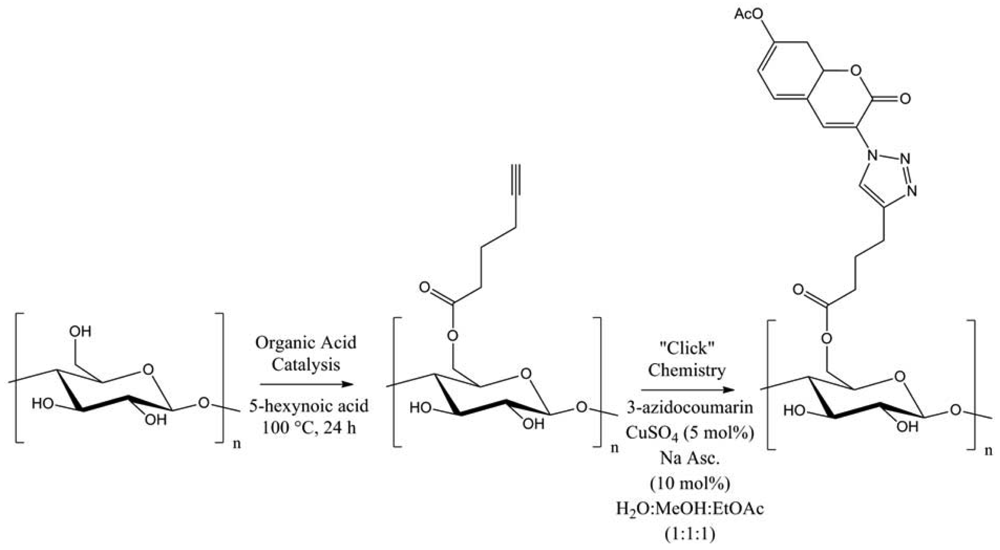
Several functional moieties were then “clicked”, such as methylcarboxylate, 2-aniline or 3-thiophene. Hydrolytically stable cellulose derivatives were so generated, with high conversions and DS up to 0.9. Always using cellulose, Hafrén and co-workers described a simple and original method, combining a first acid-catalyzed esterification followed by CuAAC, in order to attach fluorescent probes (Figure 7) [23].This was the first approach describing the modification of polysaccharides surfaces by a combination of organocatalysis and “click” chemistry under environmentally friendly reaction conditions.
3. Materials
Nowadays, oily fossil resource materials play an important role in the economy, particularly, in packaging, car industries, agriculture and furniture applications. Two major drawbacks are related to these commodity products: they are obtained from a non-renewable resource, and at the end of their life, there is an accumulation of this non-biodegradable waste (within a reasonable laps of time). Recently, the need to develop materials from renewable and biodegradable resources has known a growing interest [57-60], because bio-based materials and composites could be a promising solution both in terms of environmental and performance aspects [24]. In this context, the use of natural polysaccharides can constitute an interesting viable alternative to the formerly discussed problems. Thus, these macromolecular architectures can provide several advantages, namely: their low density, their bio-renewable character, their ubiquitous availability at low cost and their interesting mechanical properties. Therefore the attention they received from chemists has continued to grow in recent years.
3.1. Dendrimers
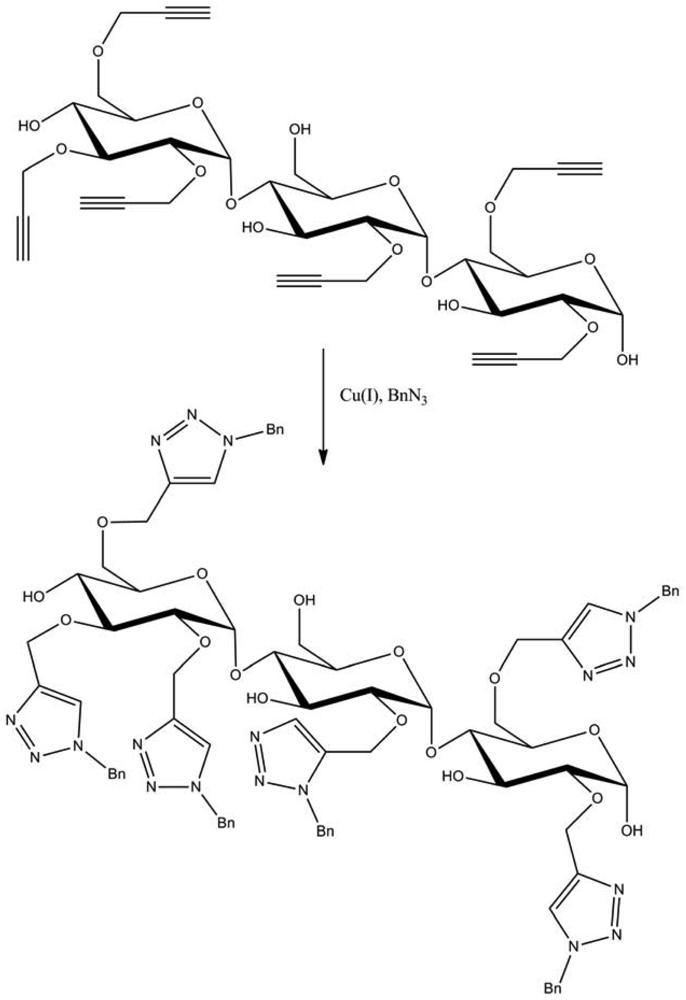
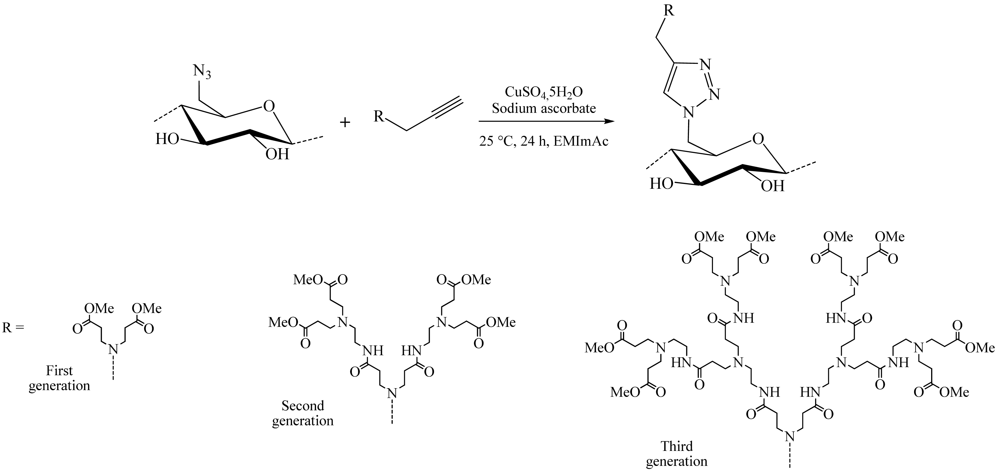
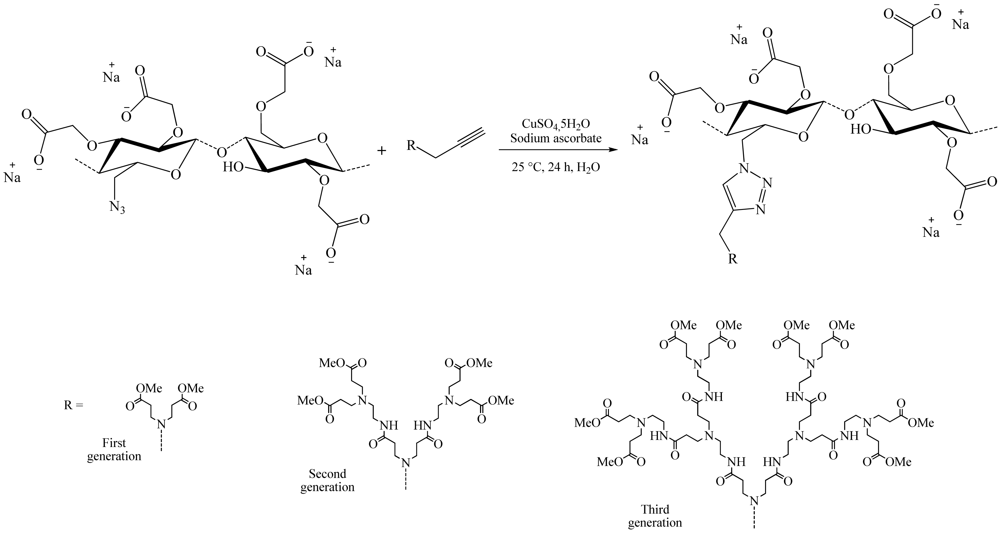
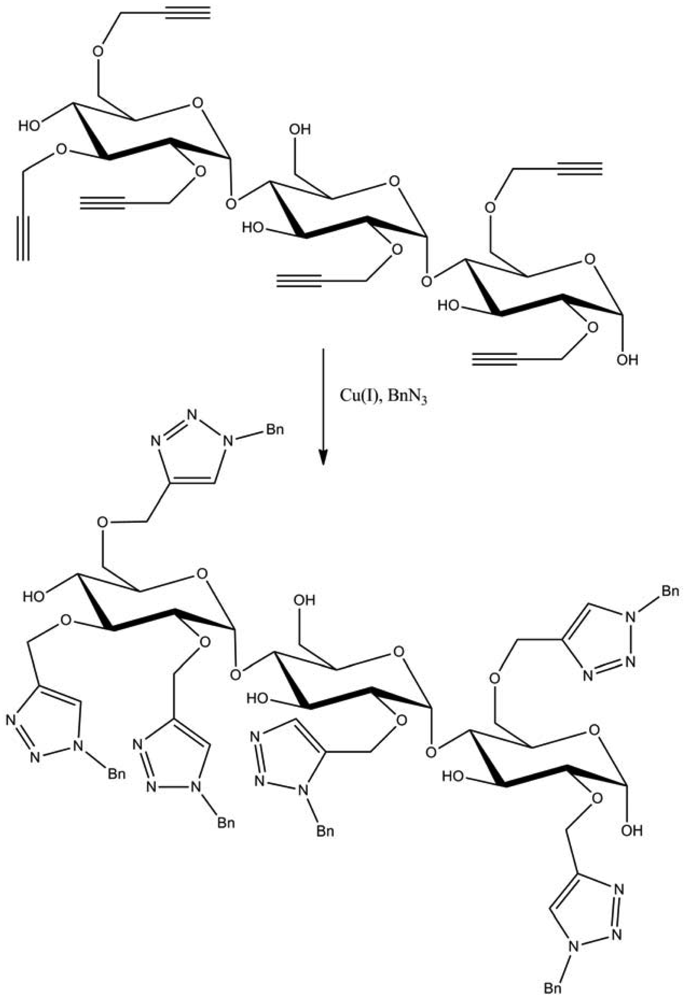
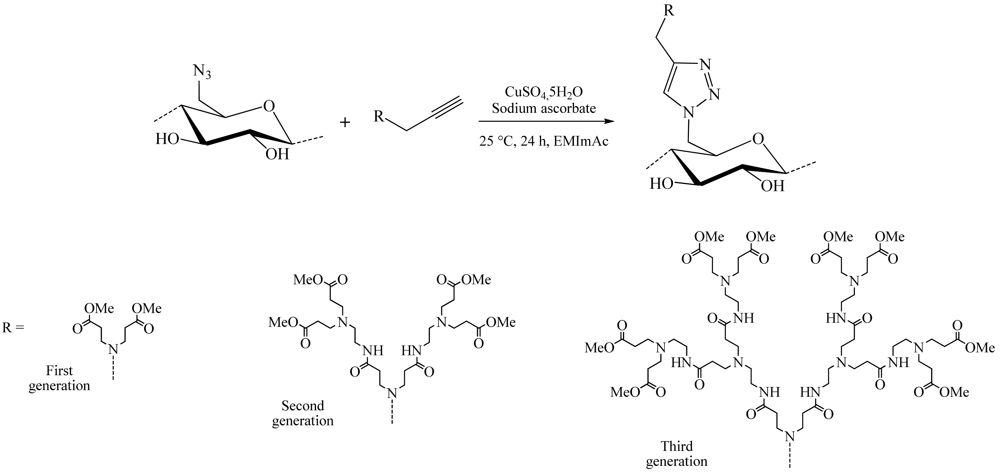
Among these large fields of application, one of the most studied subjects is the grafting of dendron molecules onto cellulose. This work was performed by Thomas Heinze and co-workers. The products so-obtained were used as biomaterials or even as liquid crystals. The first results were published in 2008 [16]. In this approach, the dendrons (propargyl-polyamidoamine) were grafted to the 6-azido-6-deoxycellulose (DS 0.75) using the CuAAC reaction in ionic liquid medium (1-ethyl-3-methylimidazolium acetate) to promote dissolution of the compounds (Figure 8). The reaction was catalyzed by copper sulfate reduced by an aqueous solution of sodium ascorbate and took place at RT for 24 h. 1-Ethyl-3-methylimidazolium acetate (EMImAc) dissolved very well 6-azido-6-deoxy cellulose with a DS of 0.75 and represented a suitable reaction medium for the homogenous conversion of 6-azido-6-deoxycellulose with propargyl-dendrons (PAMAM) via copper catalyzed Huisgen reaction. In this way, it was possible to prepare PAMAM-triazolo-cellulose derivatives from the first to the third generation with DS values of up to 0.60 using the CuAAC reaction.
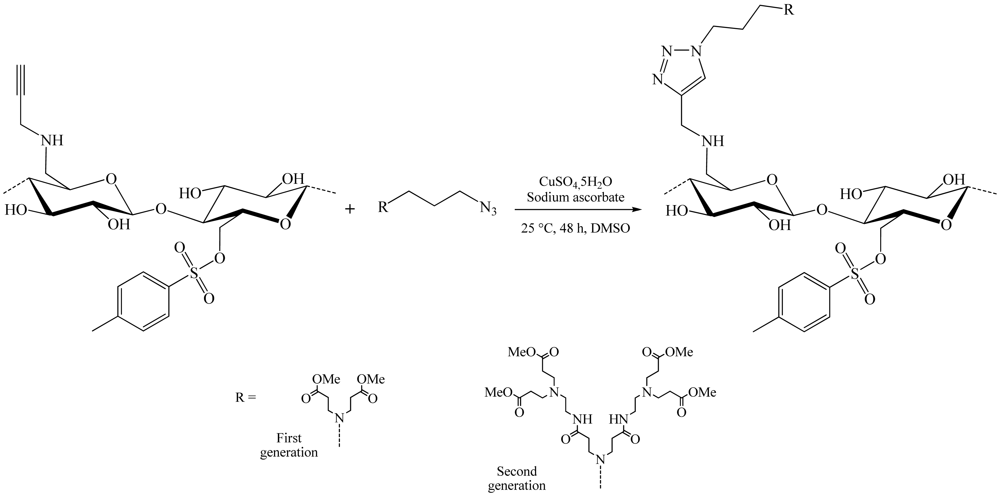
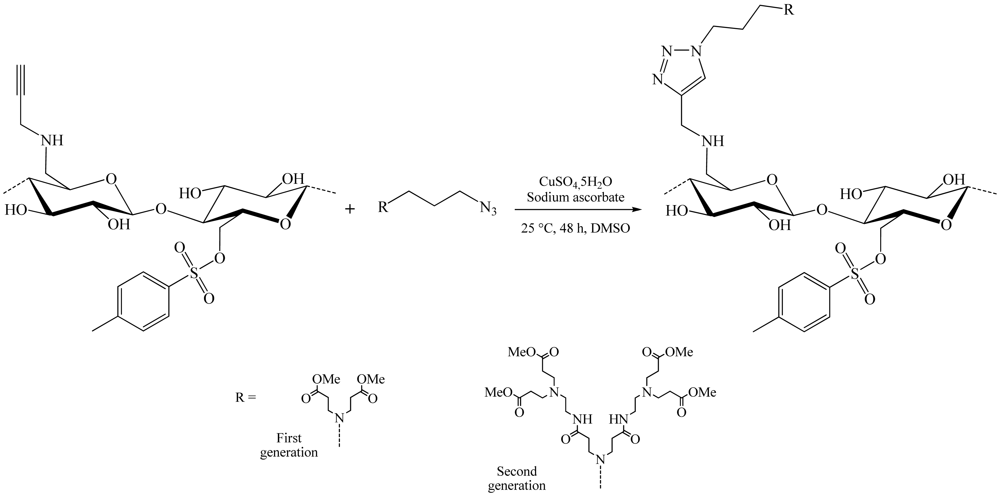
During the same year, a slightly different synthesis method of these dendrons cellulose was developed (Figure 9) [19]. This time, azidopropyl-dendrons and the 6-deoxy-6-cellulose aminopropargyl (DS 0.41) facilitated modified cellulose by nucleophilic displacement of the 6-tosylcellulose (DS 0.58), using propargyl amine. The copper-catalyzed Huisgen reaction in DMSO using CuSO4,5H2O/sodium ascorbate as catalytic system, yield 6-deoxy-6-amino-(4-methyl-[1,2,3-triazolo]-1-propyl- polyamido amine) cellulose derivatives of the first (DS 0.33) and the second (DS 0.25) generation.
To facilitate the solubilization of dendronized cellulose in water, Heinze and colleagues published works in 2009 showing new compounds obtained from the 6-deoxy-6-azidocellulose [17]. Water-soluble deoxy-azidocellulose derivatives were synthesized by heterogeneous carboxymethylation, applying 2-propanol/aqueous NaOH as slurry medium (Figure 10). The novel, carboxymethyl deoxy-azidocellulose provided a convenient starting material for the selective dendronization of cellulose via the copper-catalyzed Huisgen reaction, in water, yielding water-soluble carboxymethyl 6-deoxy-(1-N-[1,2,3-triazolo]-4-polyamidoamine) cellulose derivatives of first (DS 0.51), second (DS 0.44) and third generation (DS 0.39).
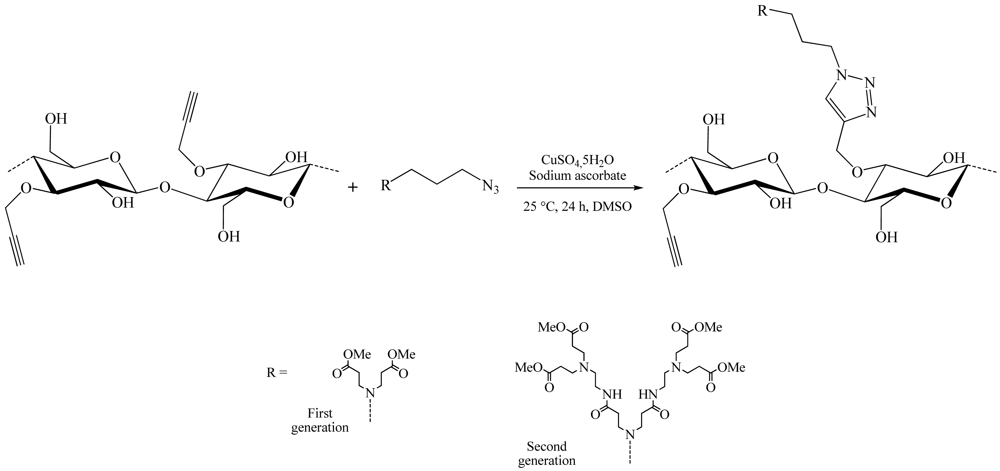
Over this year, the same team also proposed a different structure from previous ones [28]. This time, the position 3 of the anhydroglucose units was referred. This structure was obtained by reacting as shown before the azido-PAMAM and 3-O-propargyl cellulose using CuSO4,5H2O/sodium ascorbate as catalytic system in a water/DMSO mixture. The synthesis of 3-O-propargyl cellulose (DS 1) was realized using the thexyldimethylsilyl moiety as protecting group. The treatment of 3-O-propargyl-2,6-di-O-thexyldimethylsilyl cellulose with tetrabutylammonium fluoride trihydrate led to the complete removal of the silicon containing groups. A comprehensive analysis of the obtained products showed a conversion rate of the triple bonds of 25% and 13% respectively for the first and second generation dendrons (Figure 11).
3.2. Gels Preparation
Another area of application of “click” chemistry to polysaccharides concerns the formation of gel by crosslinking. In 2009, Zhang and co-workers published the synthesis of a composite gel having interesting properties of swelling and temperature resistance [18]. This material is an azido-cellulose coupled to a polyacrylamide based polymer. Azide-modified cellulose and the alkyne-modified P(NIPAAm-co-HEMA) were synthesized in two steps each. Then a series of novel hydrogels were prepared by mixing the solution of two components functionalized with alkyne and azide groups through Cu(I)-catalyzed Huisgen's 1,3-dipolar cycloaddition (Figure 12). The resulting hydrogels exhibited a porous network structure and favorable thermosensitivity in terms of temperature dependence of swelling ratio, deswelling kinetics and reswelling kinetics.
In 2010, three research groups published their work on the development of new gels from three different polysaccharides (cellulose, chitosan and guar gum) and a spacer molecule to cross-link these polysaccharides. Zampano and co-workers [35] used an aliphatic or an aromatic chain bearing two alkyne functions to cross-link the 6-deoxy-6-azido chitosan (Figure 13). The azido-functionalized products reacted efficiently in Cu(I)-catalyzed “click” cycloaddition at room temperature with a model reagent phenylacetylene, the di-alkynes 1,7-octadiyne and 1,4-diethynylbenzene used for cross-linking. All alkynes reacted efficiently both in heterogeneous and homogeneous phases. Thus the “click” functionalized/cross-linked derivatives were easily deprotected with hydrazine in order to restore the amino groups. The cross-linked NH2-deprotected products showed a pH-dependent swellability in water.
Argyropoulos et al. described the formation of nanoplatelet gel-like nanomaterials formed using cellulose nanocrystals (CNC) as starting materials (Figure 14) [27]. Initially, the primary hydroxyl groups on the surfaces of the CNCs were selectively activated to carboxylic acids by using TEMPO-mediated hypohalite oxidation. In the next step, 11-azido-3,6,9-trioxaundecan-1-amine was grafted onto the activated oxidized surface CNCs via an amidation reaction. The grafted amine compounds contained terminal alkyne or azide functionalities and as such two sets of precursors were prepared. The alkyne and azide, surface-functionalized CNC precursors, were then brought together using “click” chemistry, creating for the first time unique nanoplatelet gels.
Fleury and co-workers [50] designed new thermosensitive guar-based hydrogels by CuAAC cross-linking between alkyne-functionalized guar chains and α,ω-diazido poly[(ethylene glycol)-co-(propylene glycol)] (PEG-co-PPG) (Figure 15). The reaction was successfully performed in water under mild conditions with gelation times relatively shorter than other described guar-based hydrogels [61]. They demonstrated that both mechanical and swelling properties can be efficiently tuned by varying experimental parameters, such as the CuAAC coupling temperature, the guar molecular weight and the mass fraction of guar/cross-linker chains. It appears that for sufficient cross-linker content, the resulting hydrogel shrinks while heating. Preliminary data concerning the capability of these networks to act as capsule for a temperature controlled drug delivery are given, owing to the thermo-dependant microstructure of hydrogels.
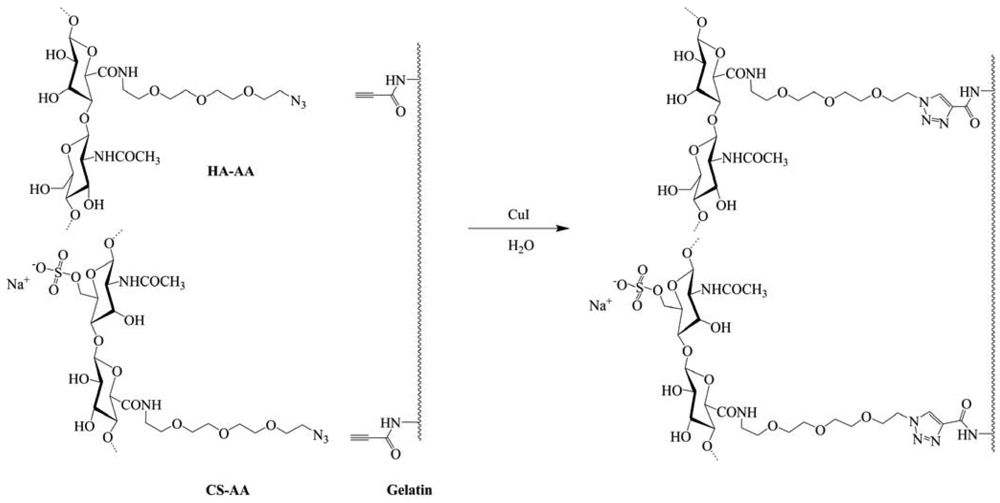
Very recently, two works on this area were published. The first comes from Gao et al., [51] they synthesized a biological hydrogel from the biopolymers hyaluronic acid (HA), chondroitin sulfate (CS) and gelatin via “click” chemistry, in order to mimic the natural cartilage extracellular matrix, which is composed of core proteins and glycosaminoglycans. HA and CS were modified with 11-azido-3,6,9-trioxaundecan-1-amine (AA) and gelatin was modified with propiolic acid (PA). The biological hydrogel was readily formed at room temperature within 5 min after HA–AA, CS–AA and G–PA were mixed together in the presence of Cu(I) as catalyst at a concentration of 0.95 mg mL−1 (Figure 16). The hydrogel obtained was in a highly swollen state and showed the characteristics of an elastomer. Incubation in phosphate-buffered saline for 4 weeks resulted in a weight loss of up to 45%. Moreover, about 20% gelatin and 10% CS were released from the hydrogel in 2 weeks. In vitro cell culture confirmed that chondrocytes could adhere and proliferate on the hydrogel. Thus the molecular modification endowed the biopolymers with good reactivity to form a biological hydrogel under very mild conditions by “click” chemistry, which has also shown the potential for biomedical applications.
The second comes from Heinze et al. [26]. They synthesized a novel cellulose-based hydrogel. Cellulose derivatives bearing azide and alkyne moieties were prepared by conversion of cellulose p-toluenesulphonic acid ester with sodium azide, on the one hand, and propargylamine, on the other. The products obtained were carboxymethylated to yield water soluble multifunctional cellulose derivatives. The CuAAC was applied for the cross-linking. Gel formation occurred within 55 and 1,600 s after mixing the aqueous solutions with both compounds and CuSO4/Ascorbic acid. The gelation time was found to depend on the degree of functionalization and the amount of copper (I) catalyst. The gels contain up to 98.4% water. Freeze-drying led to spongy materials with a porous structure (Figure 17).
3.3. Miscellaneous Materials
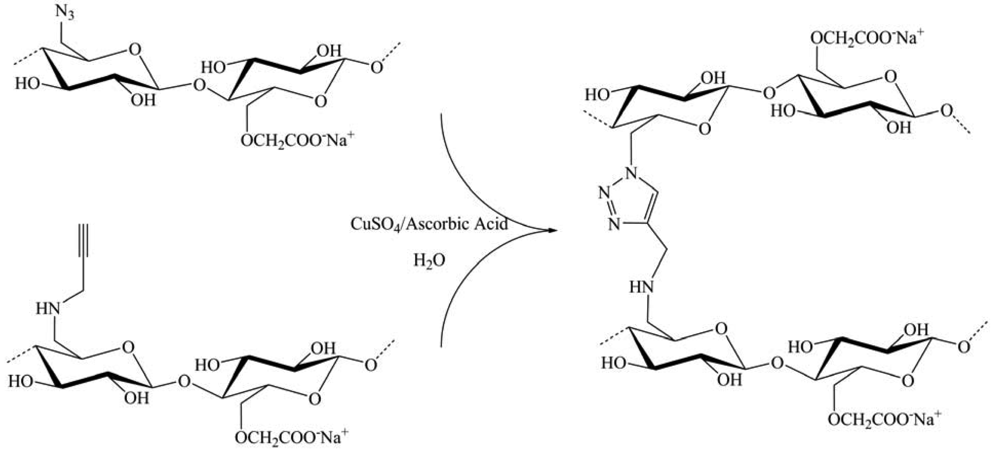
Cross-linking through the “click” chemistry did not develop only gels. Indeed, during 2008, De Geest and colleagues published work on the preparation of biodegradable dextran-based multilayer films or capsules. On the other hand, Bras and co-workers showed the feasibility of developing new materials from cellulose using a heterogeneous CuAAC reaction. The biodegradable [29] film consists in alternating layers of dextran modified with azide and alkyne groups. The dextran was modified in such a way that the azide and alkyne moieties were connected to the dextran backbone by a hydrolyzable carbonate ester function (Figure 18). Therefore, the multilayered structures obtained after successive CuAAC reactions can be degraded by simple hydrolysis in alkaline medium. This type of multilayer could be interesting for the fields of drug delivery and tissue engineering.
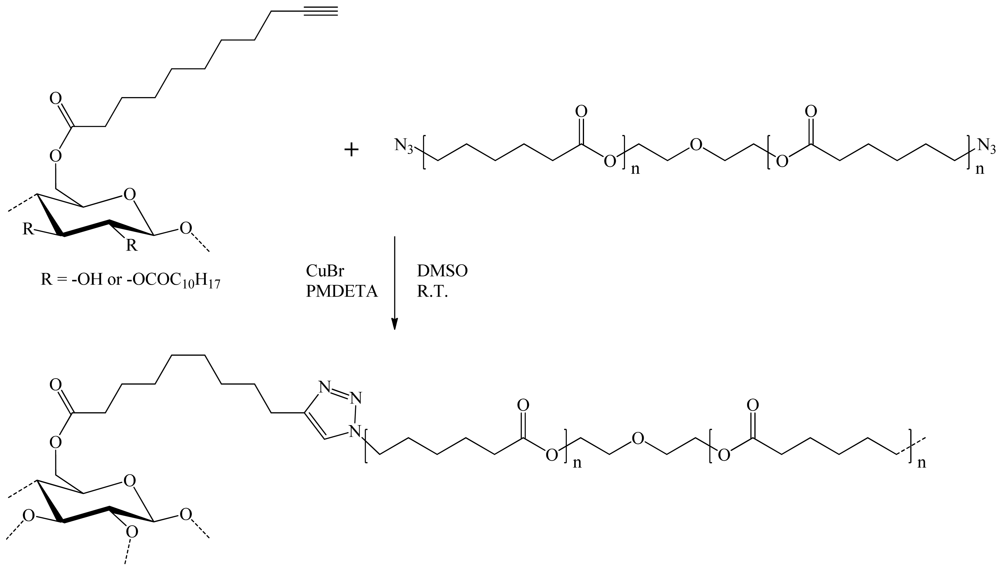
Bras et al. [25] used cellulose ester bearing a terminal alkyne function and a diazido-polycaprolactone (N3-PCL-N3) for the preparation of their material. Esterification of cellulose was realized in a moderately swelling solvent mixture to limit the modification to the superficial surface hydroxyl groups only (DS 0.1). Convertion of commercial polycaprolactone-diol (PCL-diol) into diazido-polycaprolactone was then achieved in two steps through a nucleophilic displacement reaction between di-p-toluenesulfonyl-polycaprolactone (Ts-PCL-Ts) and NaN3. The heterogeneous copper (I)-catalyzed reaction between azido-polycaprolactone and undecynoate cellulose in tetrahydrofuran (THF) led to a weight gain of 20% (Figure 19). This procedure was further expected to produce highly grafted cellulose particles, which can be considered as thermoformables.
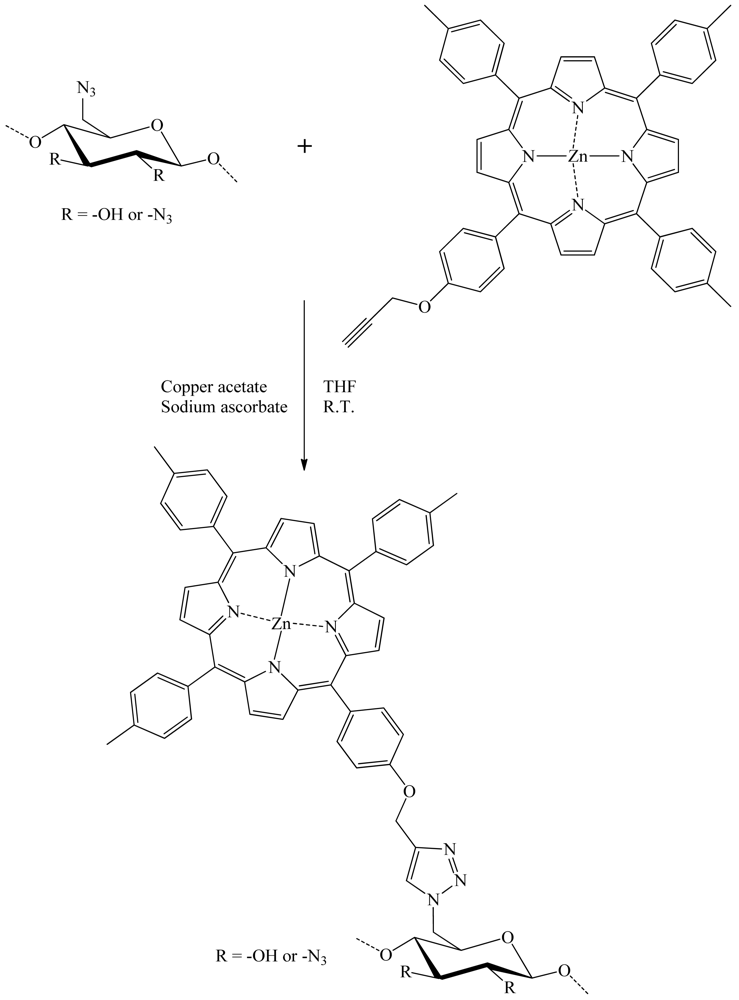
Sol and colleagues published in 2009 [22] the results of their work on the development of a new photobactericidal tissue. They were elaborated by grafting meso-arylporphyrin on cotton fabric by the means of direct cellulose azidation, followed by “click” chemistry reaction in THF and water with acetylenic porphyrin (Figure 20). Under irradiation with visible light, this material displayed an antibacterial activity against representative strains of Escherichia Coli and Staphylococcus Aureus. This new photobactericidal textile showed potential for industrial, medical, and household applications.
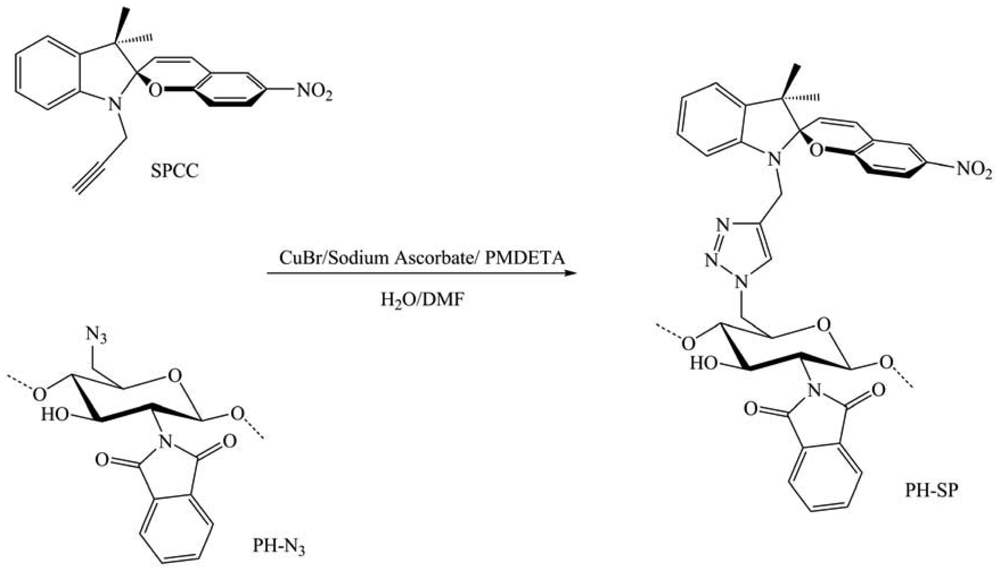
In 2011, Nazzi et al. published their work on the creation of new materials by combining chromophores and polysaccharides [36]. They questioned the effect of the polysaccharide backbone on their properties. A new light responsive polysaccharide based on a precisely defined N-phthaloyl chitosan with a covalent bound photochromic spiropyran moiety in C-6 was synthesized through CuAAC reaction between 6-azido-6-deoxy, N-phthaloyl chitosan and a new spiropyran derivative containing an alkynyl group (SPCC) (Figure 21). Various analyses were performed on the product cast as a film or in solution to evaluate these reactions. The new chitosan-based material with covalently bound spiropyran groups showed an interesting photochromic response. Indeed, after UV light irradiation, the colorless material became intensely colored either in diethyl ether suspension or solid film, due to the conversion of the spiropyran in the merocyanine form. The photo-excited merocyanine form lived more than 24 h as suspension in diethyl ether and more than two months as dry film at room temperature. By contrast, the unbound spiropyran in ethanol or diethyl ether bleached in few minutes. These findings indicate a very well controlled route to provide a polysaccharide, in particular chitosan, with a specific photoresponse to light irradiation, which is affected and modified to large extent by chromophore-biomacromolecule nonbonded interaction.
3.4. Development of Chemical Tools
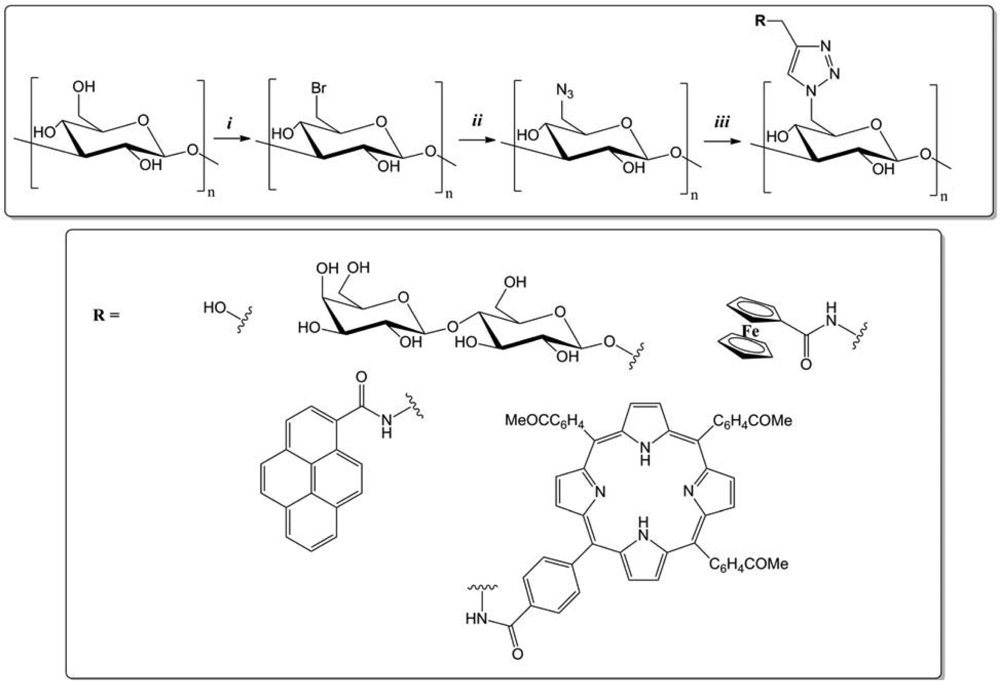
CuAAC applied to polysaccharides also helped to develop tools for synthesis, preparation or purification of compounds. Regarding the latter application area, three groups presented their work in 2010 and 2011, covering both the purification of proteins, or water. Ye and co-workers immobilized boronic acid ligands on Sepharose for an effective separation of glycoproteins [62]. A new “clickable” boronic acid ligand was synthesized by introducing a terminal acetylene group onto commercially available 3-aminophenyl boronic acid. The “clickable” ligand, 3-(prop-2-ynyloxycarbonylamino)phenylboronic acid, was easily coupled to azide-functionalized Sepharose using CuAAC reaction in aqueous methanol (Figure 22). Compared to other boronic acid affinity gels, the new affinity gel displayed superior effectiveness in separating model glycoproteins from closely related bovine serum albumin and RNase A in the presence of Escherichia Coli proteins.
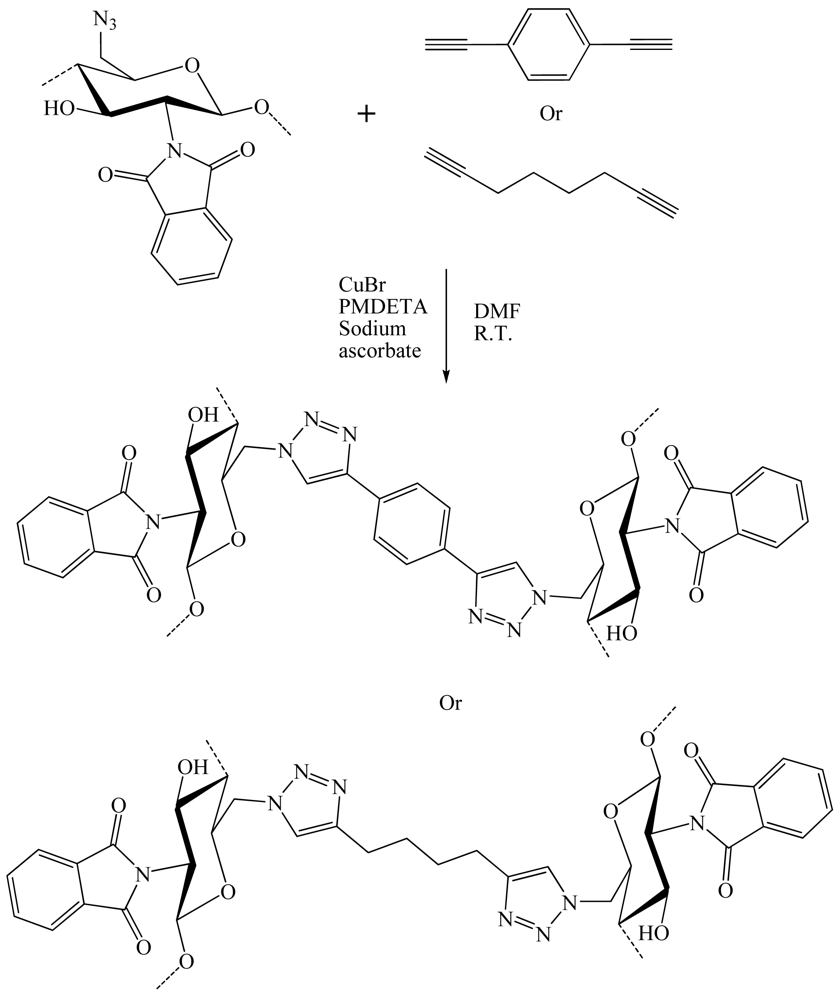
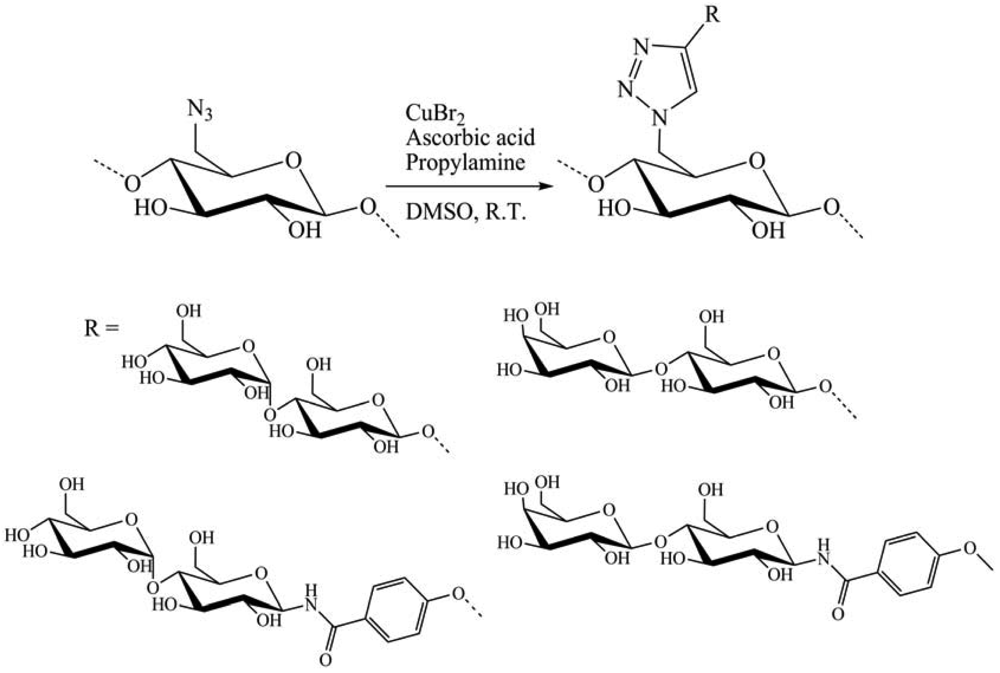
In 2011, Hasegawa et al. [20] presented the synthesis of cellulose glycoclusters having an affinity for proteins, lectin and more. Oligosaccharide appendages (O-/N-linked β-maltoside and O-/N-linked β-lactoside) with a terminal alkyne function were grafted onto C-6 positions of all UAG of 6-azido-6-deoxycellulose prepared from cellulose through a regioselective and quantitative bromination/azidation. Oligosaccharides carrying terminal alkyne were prepared through two different synthetic routes. Per-acetylated maltose (AcMal) and lactose (AcLac) were coupled with propargyl alcohol by treating with boron trifluoride and then deacetylated. They could otherwise be brominated first with hydrogen bromide, azidated with sodium azide, and then hydrogenated to give the corresponding oligosaccharide derivatives having amino functionalities at their anomeric positions. The resultant oligosaccharides were coupled to a benzoic acid derivative having terminal alkyne and then, deprotected to give N-linked β-maltoside and β-lactoside having terminal alkyne. CuAAC reaction was then realized in DMSO with CuBr, ascorbic acid and propylamine, at R.T. during 12 h (Figure 23). The resultant cellulose derivatives showed improved water solubility in comparison to native cellulose. However, they bound to carbohydrate-binding proteins in a rather non-specific manner. Molecular dynamics calculations revealed that these properties were attributable to rigid sheet-like structures of the cellulose derivatives and the subsequent exposure of their hydrophobic moieties to solvents.
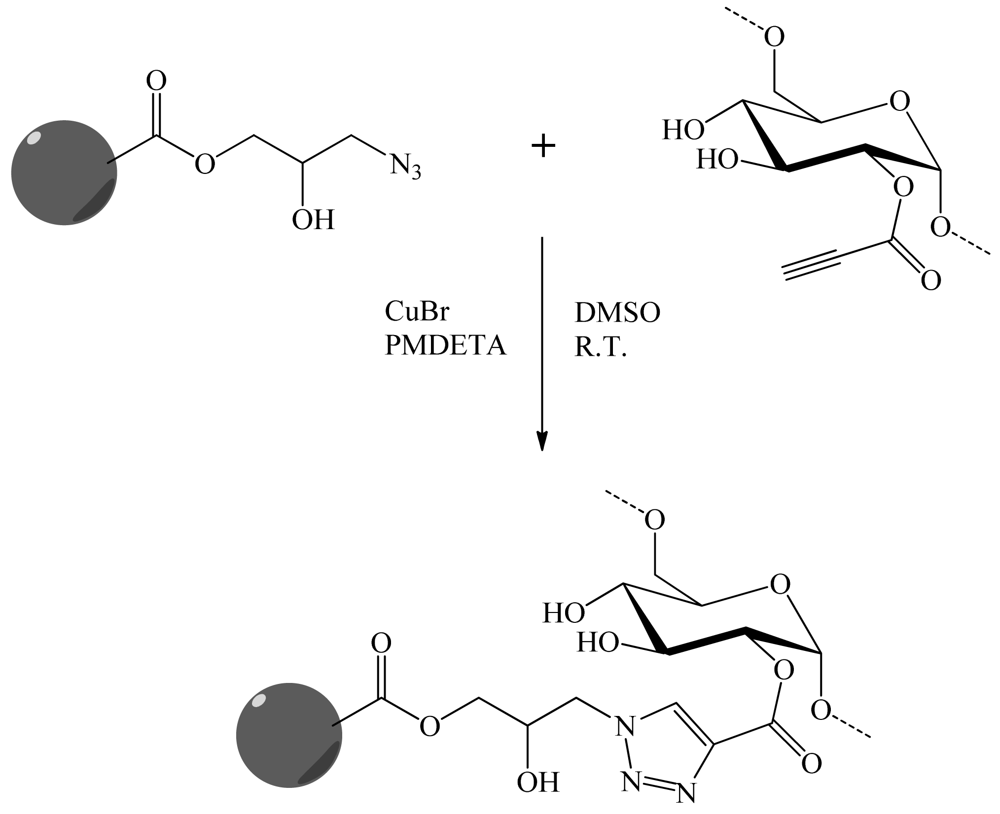
To solve problems of water contamination by boron, Tuncel and co-workers [30] have developed porous particles supporting dextran molecular brushes for boron removal. The monodisperse porous poly(glycidyl methacrylate-co-ethylene dimethacrylate) (poly(GMA-co-EDM)) particles 6 μm in size, were obtained by “modified seeded polymerization”. They were then derivatized. The azide groups were obtained by the reaction between NaN3 and the epoxypropyl functionality. In parallel, an alkyne carrying ligand, propiolic acid was covalently linked to the dextran via activation with a water soluble carbodiimide. Dextran was finally grafted onto the particles via “click” chemistry (Figure 24). A second sorbent was synthesized by the direct covalent attachment of dextran onto the poly(GMA-co-EDM), particles. Boron sorption capacities of both sorbents were investigated using model boron solutions and compared with the commercial resins. Monodisperse-porous particles with dextran-based molecular brushes were an effective sorbent for boron-removal from water.
Save and colleagues [31] applied “click” chemistry to dextran in order to produce an emulsion stabilizer for the polymerization ab initio of poly(vinyl acetate) (PVAc). The dextran end-functionalized by a xanthate moiety was synthesized by Huisgen's 1,3-dipolar cycloaddition catalyzed by copper. It was applied as a macromolecular “reversible addition fragmentation chain transfer” agent in surfactant-free emulsion polymerization of vinyl acetate to form in situ an amphiphilic block copolymer able to efficiently stabilize the latex particles. The reducing end of dextran was propargyled using propargylamine. In parallel, α-azide-functionalized chain transfer agent (CTA) is prepared by alkylation of O-ethyl xanthic acid potassium salt by the 2-bromopropionic acid 3-azido-propyl ester. CuAAC reaction was then realized at R.T. in a DMSO/H2O solvent system with CuSO4,5 H2O/sodium ascorbate as catalytic system. Finally this method of polymerization afforded the preparation of high solids content lattices coated by dextran.
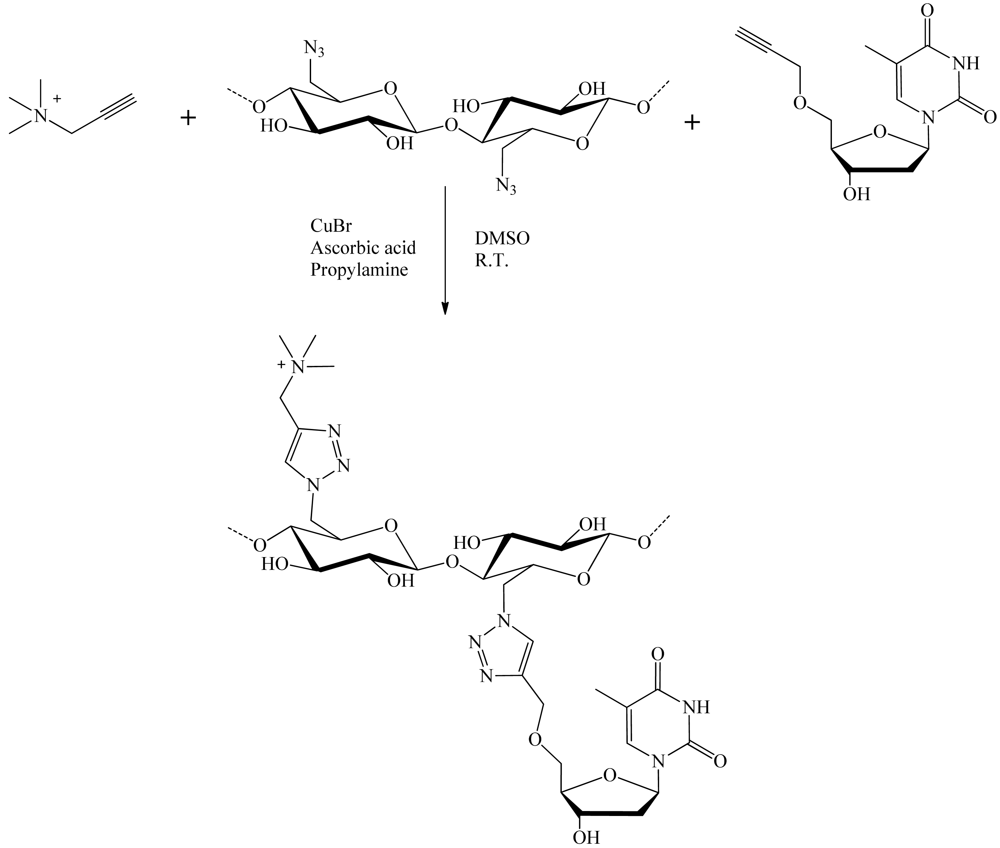
Hasegawa et al. [21] have shown that nucleosides grafted onto cellulose chains could be used to facilitate the dispersion of carbon nanotubes in water (Figure 25). Cellulose derivatives having thymidine and/or trimethylammonium appendages exclusively at C-6 position can be prepared in a convenient manner through C-6 direct azidation on cellulose to afford 6-azido-6-deoxycellulose, followed by a CuAAC reaction in DMSO with CuBr2/ascorbic acid and propylamine as a catalytic system. These cellulose derivatives took unique sheet-like structures and functions as “wrapping papers” to effectively disperse single walled carbon nanotubes in water. Although the resultant nanocomposites finally aggregated to form black precipitates after several weeks mainly due to the intrinsic nature of cellulose main chain to induce strong interstranded interactions.
4. Therapeutics
In therapeutic domains, polysaccharides are often used to deliver drugs with micelles, vesicles and polymersomes (similar structure to viral capsids). They can also be useful in hiding peptide in a cell, to target cancer related protein receptors or DNA and to enhance efficiency of lytic peptide for cancer treatment. One modification most commonly found of polysaccharides is the formation of drug delivery agent using chitosan (CS) and dextran. Indeed CS is a typical natural polyaminosaccharide with good biocompatibility, biodegradability, nontoxicity, and bioactivity. Therefore, it presents wide applications in the biomedical field.
4.1. Modification of Position 6
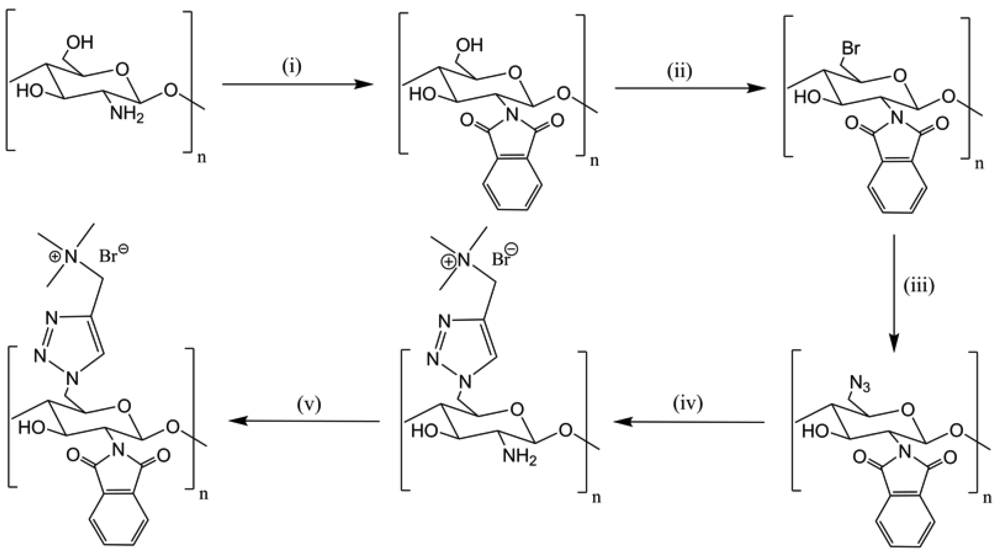
“Click” reactions were done using CuSO4/NaAsc according to Li et al. [37] The modification on C-6 of CS is the selective introduction of a trimethylammonium cationic group which led to the 6-N,N,N-trimethyltriazole-CS (TCs) (Figure 26). This product showed a good solubility in water, a strong DNA binding ability and a high protection of DNA against nuclease degradation. These results suggested that TCs could be an efficient and safe material for gene delivery by transfection.
The graft modification has been explored as a convenient method to overcome the insolubility of CS in common solvents and to develop multifunctional CS copolymers. These copolymers owned self-assembling properties to form polymersomes and micelles. Two positions of CS are commonly modified, NH2 on position 2 and OH on position 6. For the modification of position 6, the first step is the protection of NH2 (Figure 27). Then a second step concerns the linking of a carboxylic arm with a propargyl or azido group for “click” reaction. Finally the molecule of interest is grafted by “click” chemistry on modified CS.
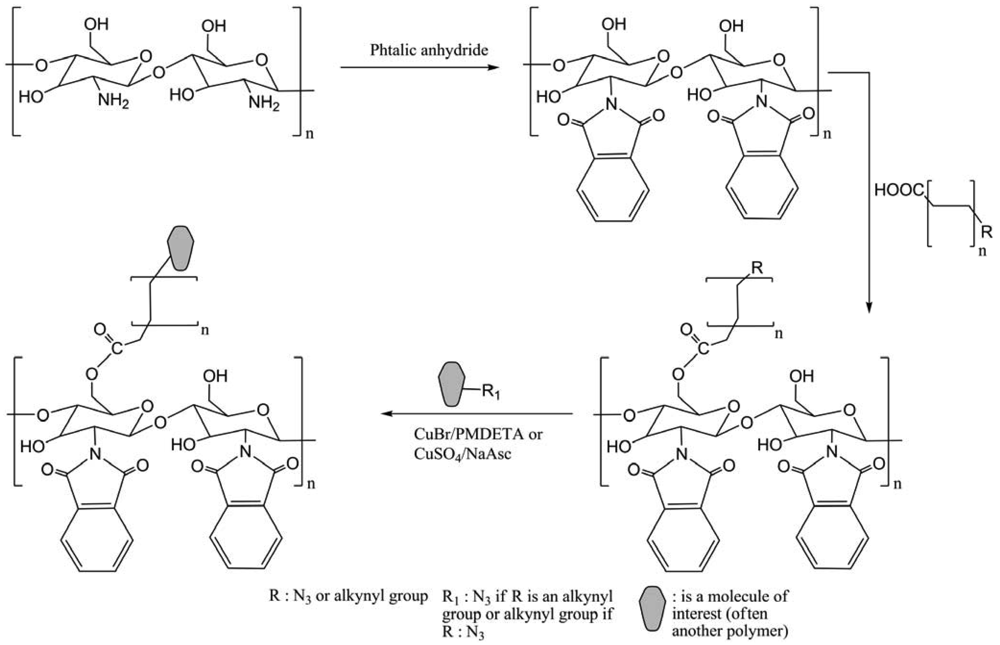
Ifuku et al. developed a highly regioselective 6-substitution of chitosan derivatives using “click” chemistry, and Cu(SO4)/NaAsc as catalytic system [39]. Generally, reactions were performed using CuBr/PMDETA (N,N,N′,N′,N-pentamethyldiethylenetriamine) to form CS copolymers, which all have self-assembling abilities in water to form polymersomes and micelles. Amphiphilic chitosan-graft-poly(2-(N,N-diethylamino)ethyl methacrylate)-block-poly(ethylene oxide) (CS-g-(PDEAEMA-b-PEO)) copolymer was synthesized by the combination of atom transfer radical polymerization (ATRP) and “click” chemistry (Figure 28) [40]. The micelle sizes were adjusted through the alteration of the pH values of solutions. The presence of PEO segments improved hydrophilicity of copolymer and avoided excessive aggregation of the micelles. CS-g-(PDEAEMA-b-PEO) copolymer presented both the unique properties of PDEAEMA-b-PEO and CS.
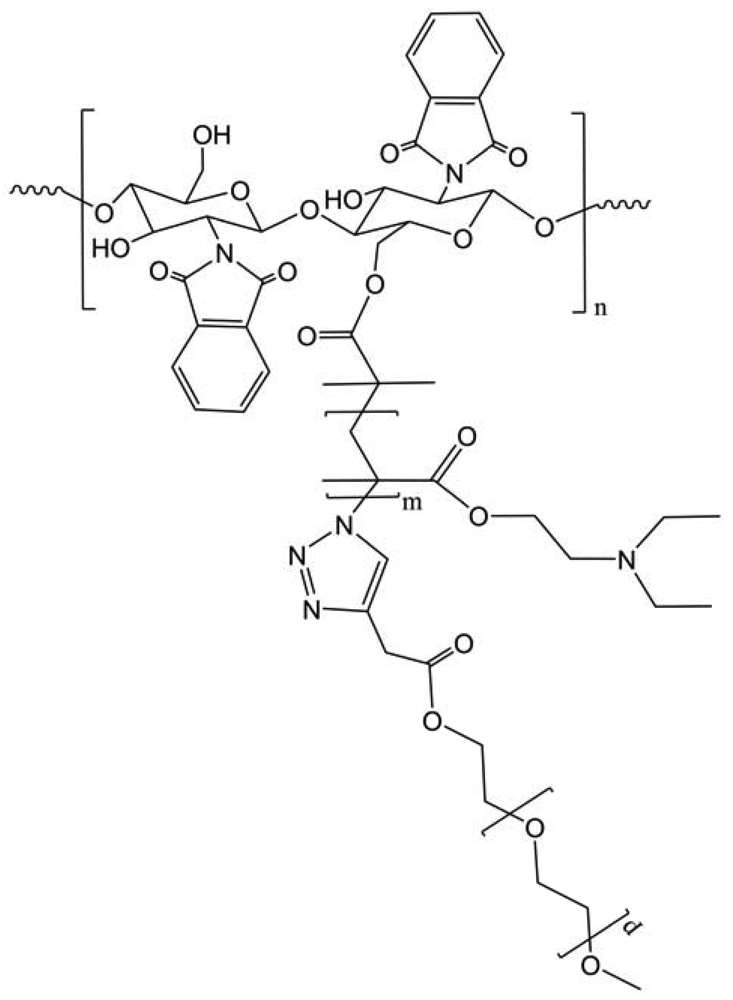
Amphiphilic chitosan-graft-P(2-(2-methoxyethoxy)ethylmethacrylate-co-oligo(ethylene glycol) methacrylate) (CS-g-P(MEO2MA-co-OEGMA)) copolymers have a self-assembling behavior and tunable thermosensitive properties (Figure 29) [41]. The lower critical solution temperature (LCST) values of CS copolymer micelle solutions were tuned by altering the molar ratio of MEO2MA and OEGMA in copolymers. Furthermore, the copolymer micelles can reversibly swell and shrink in response to external temperature. The obtained tunable thermosensitive amphiphilic CS copolymers have both the unique properties of P(MEO2MA-co-OEGMA) and chitosan.
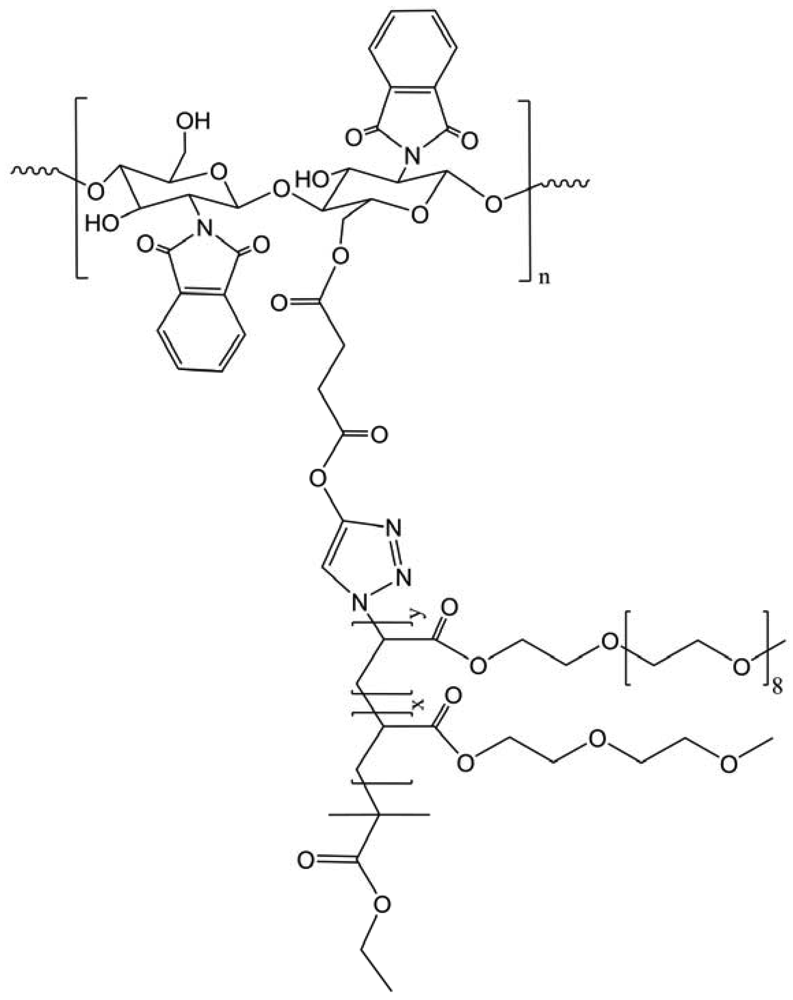
Another amphiphilic chitosan-g-poly(ε-caprolactone)-(g-poly(2-(2-methoxyethoxy)ethyl methacrylate)-co-oligo(ethylene glycol) methacrylate) (CS-g-PCL(-g-P(MEO2MA-co-OEGMA))) copolymers with double side chains of PCL and P(MEO2MA-co-OEGMA) were synthesized via combination of ring-opening polymerization (ROP), ATRP and “click” chemistry (Figure 30) [42]. As the precedent product, the micelles can reversibly swell and shrink in response to temperature, impacting the fluorescent intensity. The model drug release from the micelles indicated that the release rate of drug could be effectively controlled by altering temperature.
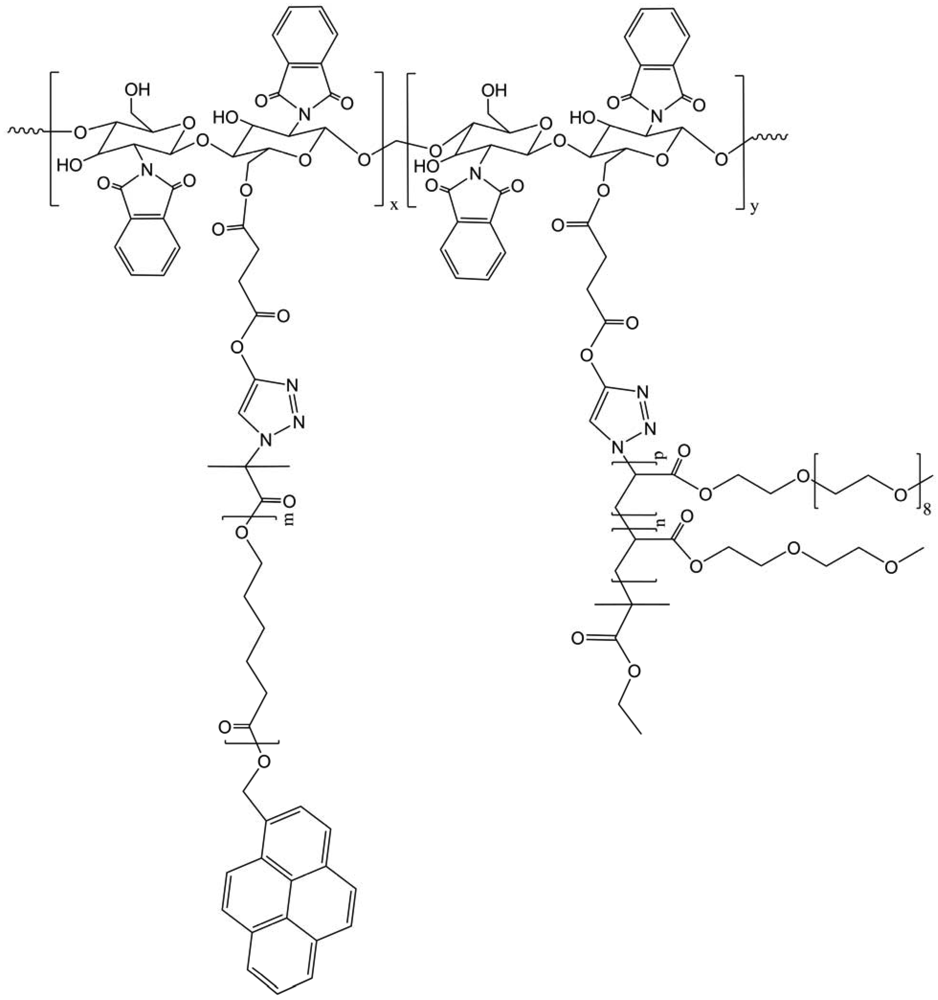
Melting, crystallization behaviors and crystalline morphology of the chistosan-g-poly(ε-caprolactone) and poly(ethylene oxide) (CS-g-PCL/PEO) copolymers were adjusted by alteration of the feed ratio of PCL and PEO segments. CS-g-PCL/PEO copolymers revealed crystalline morphology different from that of linear alkynyl PCL and alkynyl PEO due to the influence of brush structure of copolymers and the mutual influence of PCL and PEO segments (Figure 31) [43]. Hydrophilicity of the CS copolymers was improved and adjusted by the alteration of the composition of PCL and PEO segments. Investigation showed that the size of CS copolymer micelles increased with the increase of the content of hydrophobic PCL segments in copolymers, which indicated that the micellar behavior of the copolymers can be controlled by the adjustment of the ratio of PCL and PEO segments in copolymer.
Starch is another substrate to create micelles. Recently, Xiao et al. synthesized a starch grafted poly(vinyl acetate) (PVAc) using CuAAC [48]. Authors controlled the structure and the composition of their final compound, which may expand field of exploitation of starch/PVAc biohybrid.
Furthermore, in the same spirit, Huerta-Angeles et al. N-acetylated the hyaluronan and modified on position 6 [52]. The obtained amide derivatives were applied as precursor for crosslinking reactions. Their modifications led to hydrogels, which might have applications in tissue engineering.
4.2. Modification of Position 2
For the modification of the position 2, most of the works have a common strategy of modification (Figure 32). The first step is to graft an azido or a propargyl arm on CS with a reaction of amidation between the amino group of CS and the carboxylic functional group (COOH) of the arm. The second step is to graft by “clicking” the molecule of interest on modified CS using CuSO4, H2O/NaAsc or SPAAC.
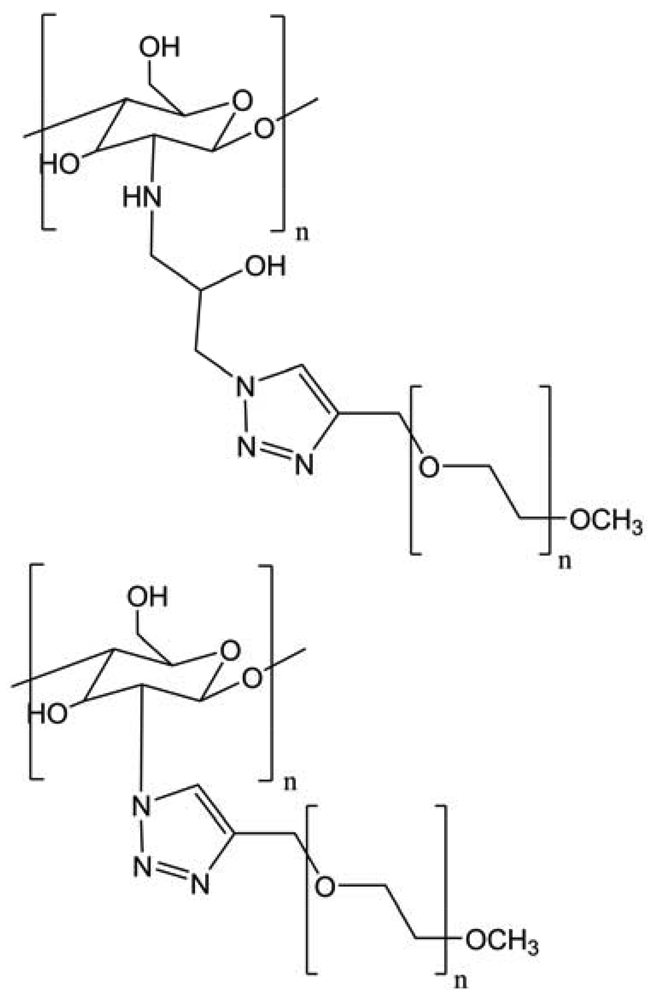
The publication of Kulbokaite and colleagues, in 2009, compared the azidation of position 2 using four different ways with azidated epichlorohydrin, sodium azide, sodium nitrite, trifluoromethane sulfonyl azide or imidazole-1-sulfonyl azide hydrochloride (Figure 33) [38]. The two last reagents are the most suitable for N-azidation of chitosan giving the degree of azidation (DA) of chitosan up to 40% and 65%, respectively. N-azidated chitosans with DA of about 60% are insoluble in aqueous and common organic solvents, but dissolve in 5% LiCl solution in N-methyl-2-pyrrolidone, which is one of the very few solvents for chitin. Chitosan-methoxy poly(ethylene glycol) derivatives containing triazolyl moiety (chitosan-N-TMPEG comb copolymers) were prepared for the first time by coupling via 1,3-dipolar cycloaddition between pendant azide and end alkyne groups of chitosan and MPEG, respectively. Comb copolymers chitosan-N-TMPEG with DS equal to DA were synthesized at a certain excess of MPEG alkyne proving “click” chemistry to be useful in exact predicting composition of the graft copolymers. “Clicking” of MPEG alkyne onto soluble azidated chitosan was successful in binary mixture of water and methylene chloride but failed in 5% LiCl solution in N-methyl-2-pyrrolidone. Significant breakdown of chitosan backbone took place under “clicking” of MPEG in the presence of Cu(II)/ascorbate catalyst resulting in graft copolymers with bimodal molecular weight distribution. Chitosan-N-TMPEG copolymers contained a certain residual amount of Cu and were soluble in acetate buffer (pH 3.7).
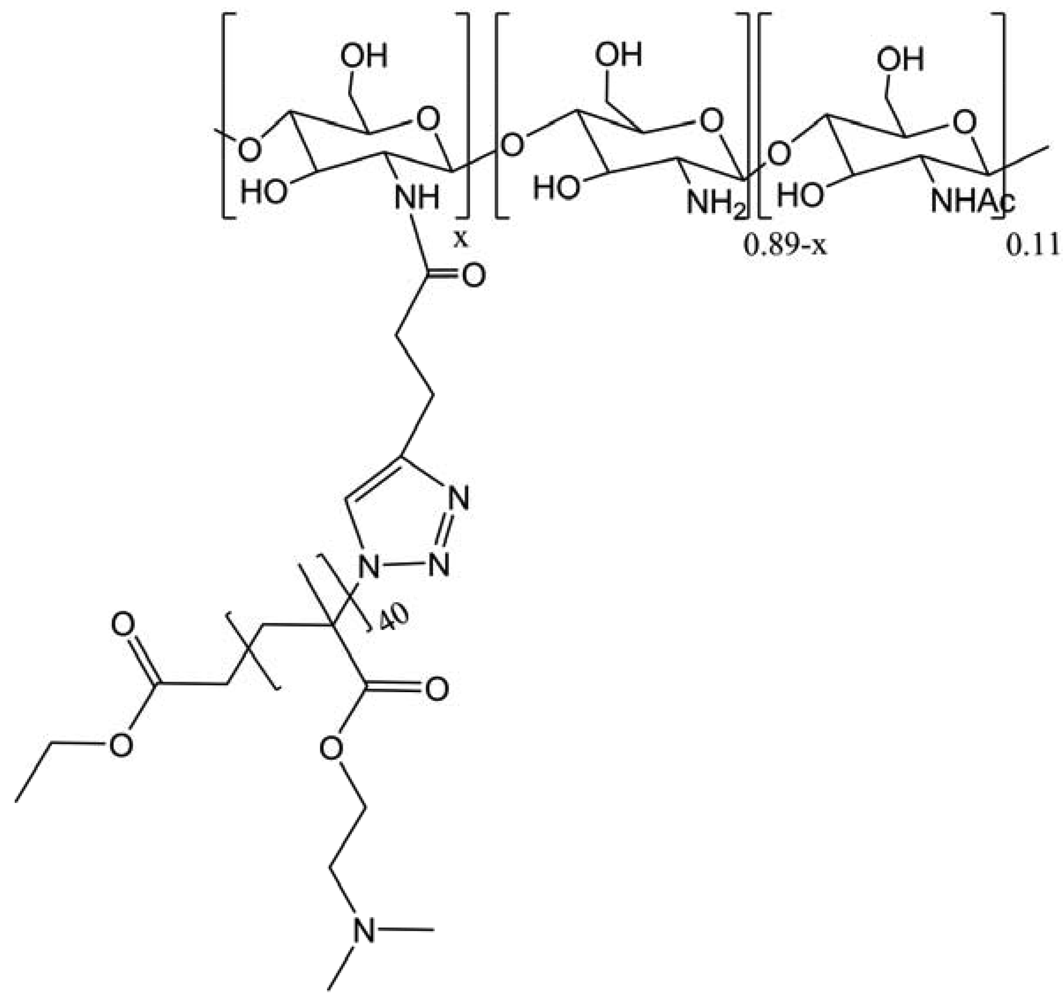
In this study, a well-defined and dually stimuli-responsive graft copolymer, CS-g-(poly(N-isopropylacrylamide) or PNIPAM, was successfully synthesized via the combination of ATRP and “click” reaction techniques (Figure 34) [44]. The thermo-induced self-assembling behavior of the copolymer in dilute solutions was investigated in order to determine that at elevated temperatures (>32 °C), the core-shell structured micelles with PNIPAM as a core and ionized CS as a shell were formed in an acidic environment (pH ∼ 4). By increasing pH at 40 °C, the uniform micelles turned into larger aggregates gradually and precipitated at pH 7. Furthermore, the cosolute effects on the low critical solution temperature (LCST) phase transition of the concentrated CS-g-PNIPAM solutions were also studied by micro differential scanning calorimetry (DSC) and UV turbidimetry.
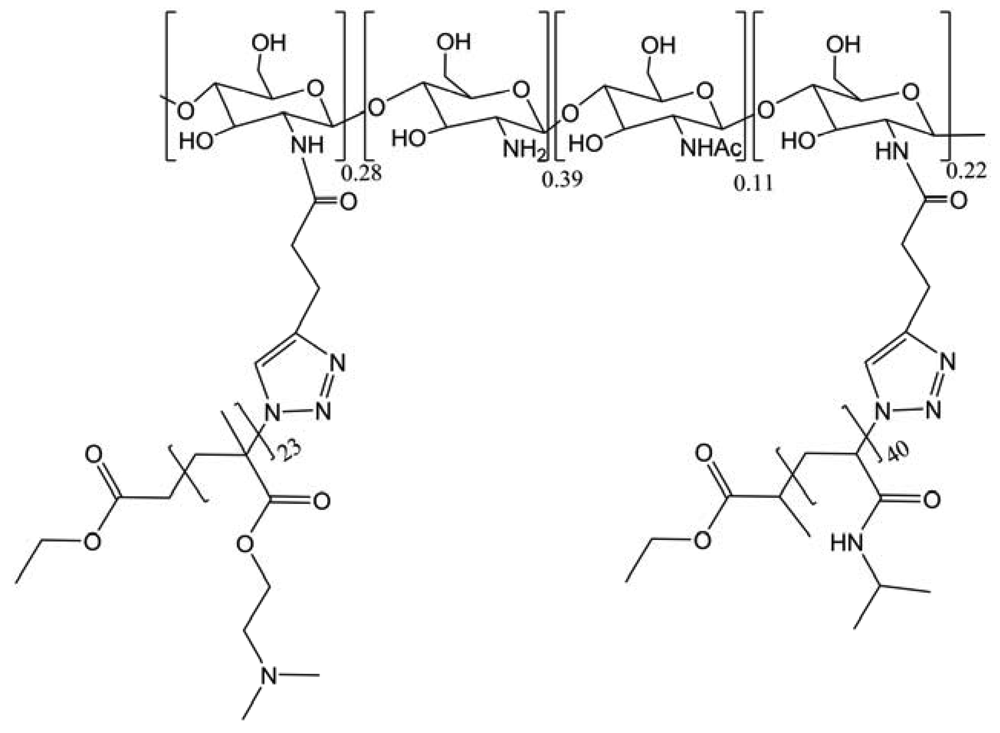
In the same class of modification, a novel dually stimuli-responsive graft terpolymer, CS(-g-(poly((2-dimethylamino)ethyl methacrylate)) or PDMAEMA)-g-(poly(N-isopropylacrylamide) or PNIPAM), based on the CS derivative, was synthesized using the combination of ATRP and “click” reaction (Figure 35) [45]. The use of ATRP not only allowed to modulate the side chains flexibly but also guaranteed their narrow molecular weight distribution, which further brought on a low polydispersity of the terpolymer. The “click” reaction taking place in the mild and homogeneous solution was proved to be an efficient coupling method to obtain a high degree of grafting on CS. The grafted terpolymer could aggregate into three-layer “onion-like” micelles at 25 °C when pH is above 7. Moreover, spherical micelles with PNIPAM cores were obtained around 40 °C in acidic solution (pH < 4). Most importantly, switching between different types of aggregates and unimers can be reversibly achieved by properly tuning pH and temperature of the aqueous solutions. This work represents the first example of nonlinear dual hydrophilic CS copolymer showing the micellization behavior.
Some other work used the SPAAC in order to avoid copper salts that have two major side effects to polysaccharides: depolymerization and contamination [46]. So in order to perform selective functionalization of polysaccharides based system, the use of SPAAC appears as an efficient alternative that eliminates the requirement of Cu(I). In this context, the application of SPAAC for the efficient decoration of CS-g-PEG nanostructures with an IgG under physiological conditions represents a step forward in the development of environmentally friendly bioconjugation technologies for the preparation of immunonanoparticles. The very recent enhancement of the SPAAC rate to values comparable to that of CuAAC, by appending propargylic fluorine atoms or fused phenyl rings to the cyclooctyne probe, suggests a rich future for SPAAC in nanotechnology and materials science.
Recently, Zhang et al. used CS to form microcapsules composed of a multilayer CS. These layers were obtained by “click” reaction using CaCO3. They obtained a versatile platform for designing novel vehicles with applications in targeted drug delivery field, particularly in liver [47].
4.3. Modification of Position 1
In the same field of application, but with another polysaccharide, dextran is here modified on the first position in this work to form a polysaccharide block-polypeptide (Figure 36) [32]. Dextran is a water-soluble microbial polysaccharide composed of D-glucopyranose units linked by α(1-6) glycosidic bonds with a low percentage of (1-3)-linked side chains. The polypeptide used as a model is a poly(γ-benzyl L-glutamate). This kind of compound has a real ability to self-assemble into small vesicles, which could be used to construct a new generation of drug and gene-delivery systems with high affinities for the surface glycoproteins of living cells. It has also been noted previously that the structure and properties of polymersomes are similar to those of viral capsids.
Another polysaccharide was used and consisted of block copolymers composed of a polypeptide segment and hyaluronan (Figure 36) [53]. This polysaccharide with its hydrophilic stabilizing block, combined with its specific ligand properties to CD44 glycoprotein receptors that are overexpressed in many cancers, provides a means to obtain a synergy between structure and biofunctionality within a single material.
Another polysaccharides modification on position 1 was performed using glycans, commonly used to hide from the defense mechanism of cells' medical active polypeptide [54].
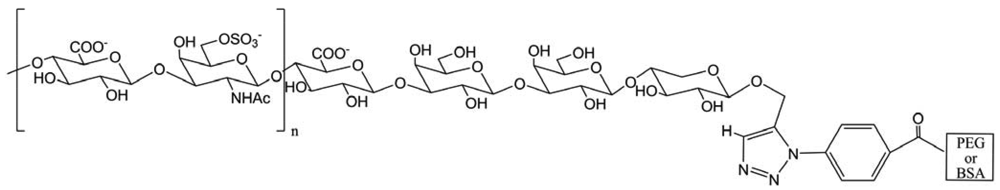
Here, Yamaguchi and co-workers performed the preparation of an alkyne containing chondroitin 6 sulfate (ChS) using an enzymatic method and synthesized an azido group containing polyethylene glycol or bovine serum albumin (Figure 37). These compounds were used for “click” chemistry to form neoproteoglycans. Biological assays employing these neo-proteoglycans are now under investigation.
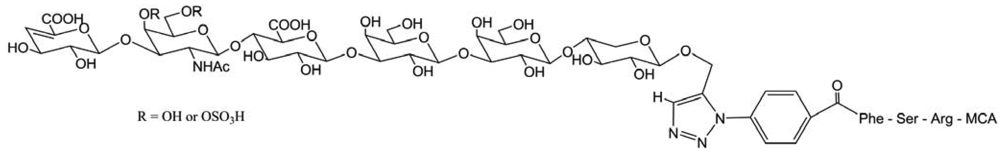
Another ChS-conjugated therapeutic peptides and proteins can be made to acquire resistance to proteolysis, which would improve their performance as medicines from two viewpoints (Figure 38) [55]. First, their resistance to proteolysis would extend their biological halflife, and second, their binding to specific tissues would enable them to deliver drugs precisely to a target site. Thus, addition of ChS would improve the biopharmaceutical properties of drugs. The approach proposed here will be broadly used for utilization of ChS.
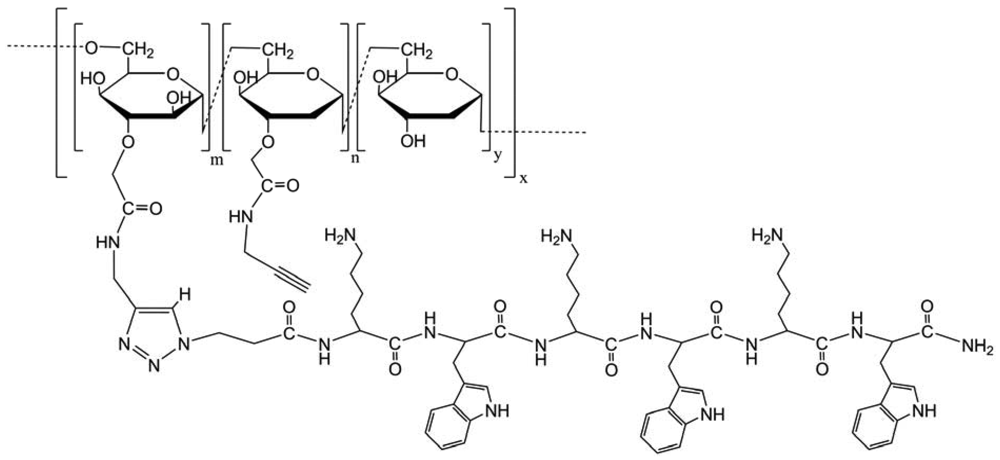
After the modification of the positions 2, 6 and 1, Zhong et al. modified the position 3 on dextran. This modification aims to synthesize a polyvalent lytic peptide-polymer conjugate Dex-(KW)3 (Figure 39) [33]. The new conjugate showed significantly enhanced activity against cancer cells compared to monomeric peptides, and the anticancer activity was not affected by the acquisition of multidrug resistance in cancer cells. The detrimental effect on erythrocytes was negligible. The encouraging in vitro data showed the potential of this new conjugate for systemic treatment against drug-resistant cancer. For the first time, the mechanism of polyvalency-mediated activity enhancement of lytic peptides was investigated by quantitative thermodynamic study. At the same bulk concentration, the polyvalent conjugate caused a stronger perturbation of membrane structure, which should be attributed to the increased local concentration of peptides in a polyvalent conjugate.
5. Diagnostic Agents
In biological systems, a wide range of polysaccharides plays an important role. Usually known as glycans, these macromolecules are present in various complex structures: independent polysaccharides, glycoproteins, glycolipids and some glycoconjugates with small molecules. These glycans are involved in many physiological events (fertilization, development, immunity, inflammation, etc.) and in pathophysiological phenomena (i.e., viral entry, bacteria-host interactions and tumor progression) [63]. The importance in biological systems relies on the structural heterogeneity of glycans. Indeed, this diversity is the result of chemical properties of saccharides, which compose the glycans (composition amongst nine different hexoses, anomeric linkage, regio-linkage, branching) and of auxiliary chemical modifications (sulfation, phosphorylation, acetylation, methylation and epimerization) [63]. All of the biosynthetic processes are non-templated and glycans are often bound to lipid or protein scaffolds, render small quantities, difficult to isolate. These factors make the comprehension of glycans synthesis and roles very difficult. In order to avoid these difficulties, glycans need to be directly visualized in vivo, by biocompatible and fast processes.
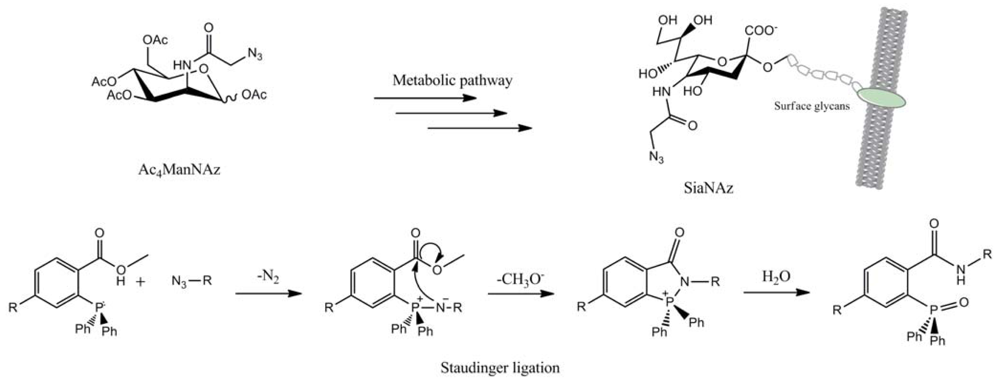
In this regard, the works on Staudinger ligation made by Bertozzi and co-workers are prominent [64]. A unnatural azido-sugar, the peracetylatedazidoacetylmannosamine (Ac4ManNAz), was metabolically modified into the corresponding N-azidoacetylsialic acid (SiaNAz) and incorporated into the cell's glycans. Thus, these cells were incubated with a biotinylatedtriphenylphosphine and the Staudinger ligation led to an amide bond between the marked surface glycans and the phosphine (Figure 40). The introduction of fluorescein isothiocyanate (FITC)–avidin allowed the visualization of surface glycans by fluorescence.
This process does not affect the cell viability and the ligation can be realized within three hours. This progress in in vivo engineering, even applied on living animals [65], has some drawbacks as this is a pH-dependent reaction and the triphenylphosphines are binding to use (air oxidation, poor water solubility and limited reaction rate).
In parallel, “click” chemistry was developed, especially the CuAAC, and was widely used throughout numerous fields of chemistry. The CuAAC was applied to biological systems such as virus particles [65] and Escherichia coli cells [66-69] quite early on. Considering that the harmless introduction of modified sugars in glycans had already been studied, [70,71] the “click” chemistry was naturally employed for its mild conditions and its short reaction time. At this time, two ways were offered to “click” glycans in biological systems: the CuAAC or the SPAAC.
5.1. The CuAAC Guide to Label Glycans
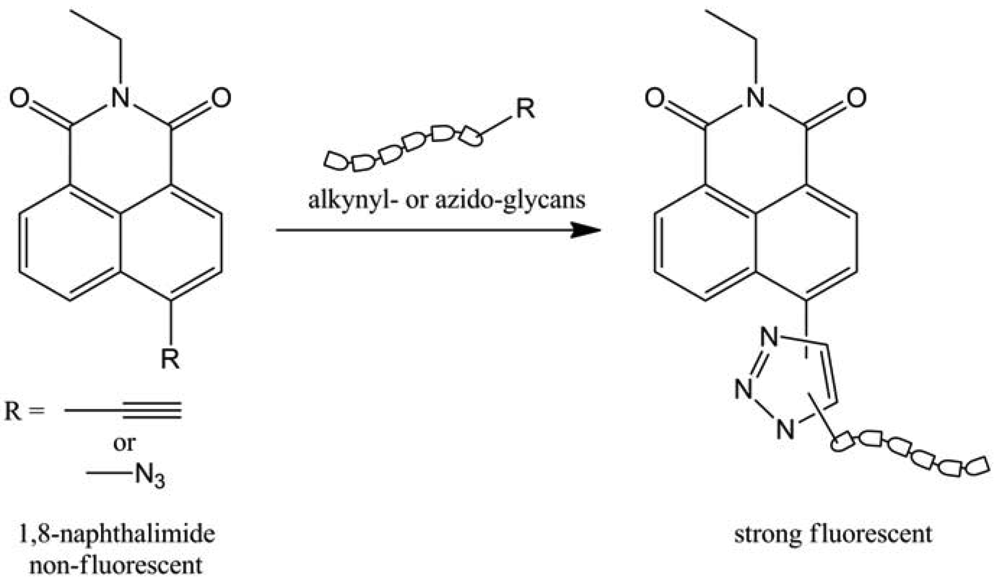
In order to label glycans, CuAAC was first used by Wong and colleagues [72] by developing a “click”-activated fluorescent labeling technique. They tried to investigate and to visualize the fucosylation in Jurkat cancer cells' glycans. They used the triazole formation generated by CuAAC to activate the fluorescence of 1,8-naphtalimide derivatives (Figure 41).
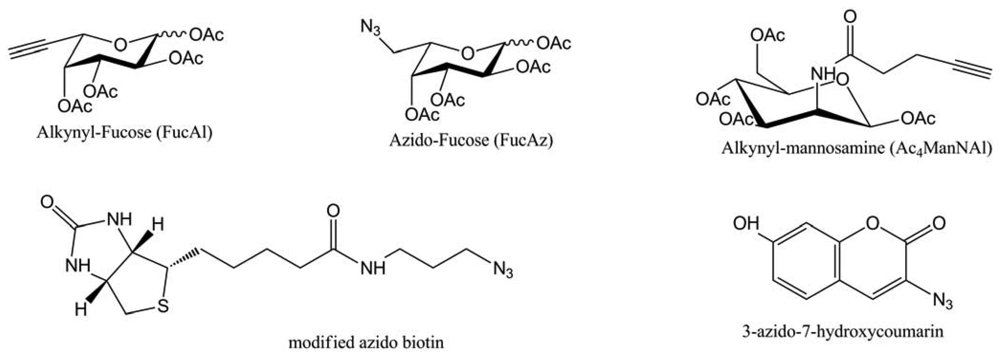
The well-known toxicity of copper was decreased (but not avoided) by the introduction of a tris-(triazoleamine) ligand and the copper sources were CuBr, or CuSO4 reduced by sodium ascorbate. Thanks to this process, they managed to visualize fucosylated glycans at the cell surface and inside the cell by fluorescence, with a two-steps method: introduction of azido-fucose and in-situ “click” chemistry. In the line of this work, this team extended this method to other modified sugars, probes (Figure 42) and on two more cancer cell lines: Hep3B and MCF-7 [73].
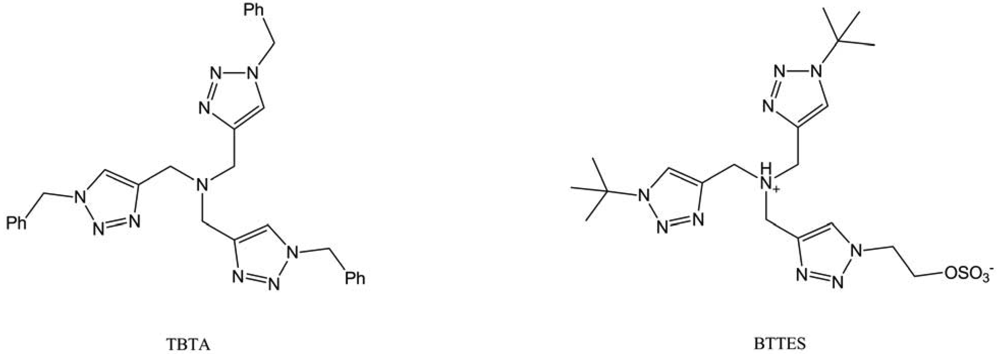
This time, they compared the “click”-generated fluorescence of 3-azido-7-hydroxycoumarine with the immunomarked fluorescence of azido-biotin. The two systems led to equivalent signals, proving the interest of designing “click”-activated probes, which requires fewer steps (one less incubation and no washing step). Nevertheless, the major issue in this method is the use of cytotoxic copper, even in the presence of ligand like the tris-[(1-benzyl-1H-1,2,3-triazol-4-yl) (TBTA). That's why Wu and co-workers tried to improve these tris-triazoleamine ligands by screening their water solubility and their reactivity [74]. The best ligand, called BTTES (Figure 43), allowed avoiding cell death due to copper presence and did not seem to affect cells proliferation rate. This was tested on four cancer cell lines (Jurkat, CHO, HEK and LNCaP) and on Zebrafish embryos.
Even in CuAAC conditions, this ligand greatly decreased copper toxicity (development abnormalities in Zebrafish embryos are only observed four days after “click” reaction) and led to very fast reaction (within minutes).
To sum up, the CuAAC has proven its efficiency in biological systems, labeling glycans in a short time. The strength of this “CuAAC way” is the wide range of possibilities in markers synthesis and the “click”-generated fluorescence, conducted by small bio-compatible molecules, limits the steps necessary to visualize glycans. Thus, progress in copper ligation nearly avoided the cytotoxic effect of catalyst. All of these parameters still need to be improved and further applications in biological systems, like living animals, are not yet demonstrated.
5.2. The Impact of SPAAC
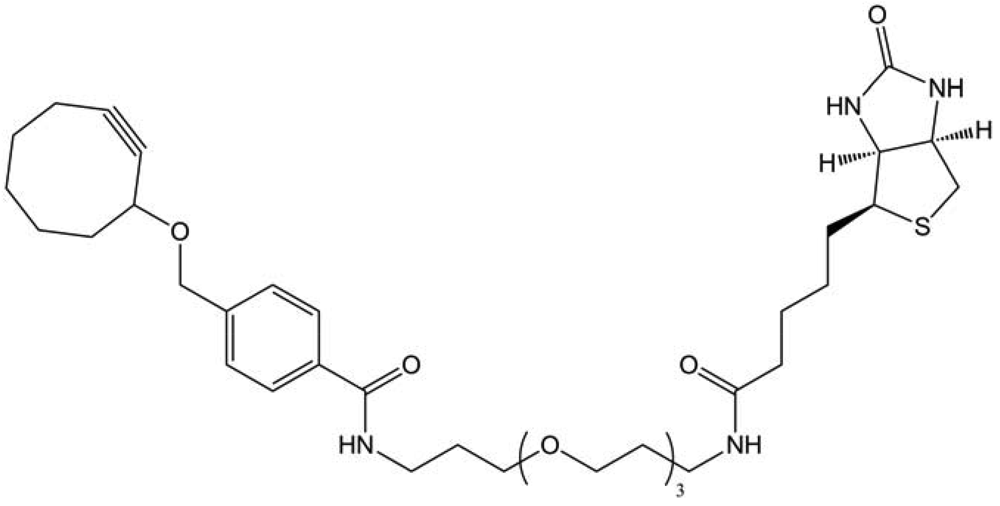
The first attempt to “click” glycans in living systems was made by Bertozzi and co-workers [13], by using the strain-promoted azide-alkyne cycloaddition. They introduced SiaNAz in Jurkat cells glycans, as described previously in Staudinger ligation, and made the cycloaddition thanks to a biotinylated cyclooctyne (Figure 44).
This method seemed to be faster than the Staudinger ligation one (within 1.5 h), but the mean fluorescence is half as efficient. Cells viability did not seem to be affected as there was no use of toxic copper catalyst. These encouraging results, led Bertozzi's team to develop new cyclooctyne markers, including fluorescent moieties [75]. They employed modified DIFOs, because of their high reaction rate, due to the electron-withdrawing effect of the difluoromethylene group (Figure 45).
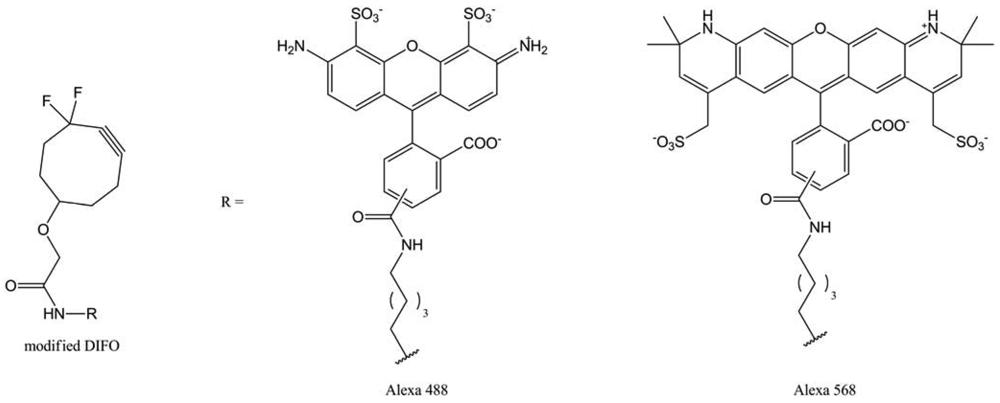
This time, the SPAAC-labeling, using these DIFOs, was about 20-times more efficient than the Staudinger ligation. This short reaction time opened the way to dynamic labeling. Bertozzi et al. synthesized two new DIFO derivatives with different fluorescent moieties (Alexa Fluor 488 and Alexa Fluor 568). Then, they proceeded to a two-step labeling: they labeled cells bearing SiaNAz residues with DIFO-488, reincubated it with Ac4ManNAz and realized a second labeling with DIFO-568. With this method, they were able to visualize the glycan trafficking from the cells internal compartments to the lysosomes, suggesting this labeling protocol did not disturb this biological phenomenon. However, the major limit of this method was the DIFO synthesis, which is conducted in 12 steps with an overall yield of 1%.
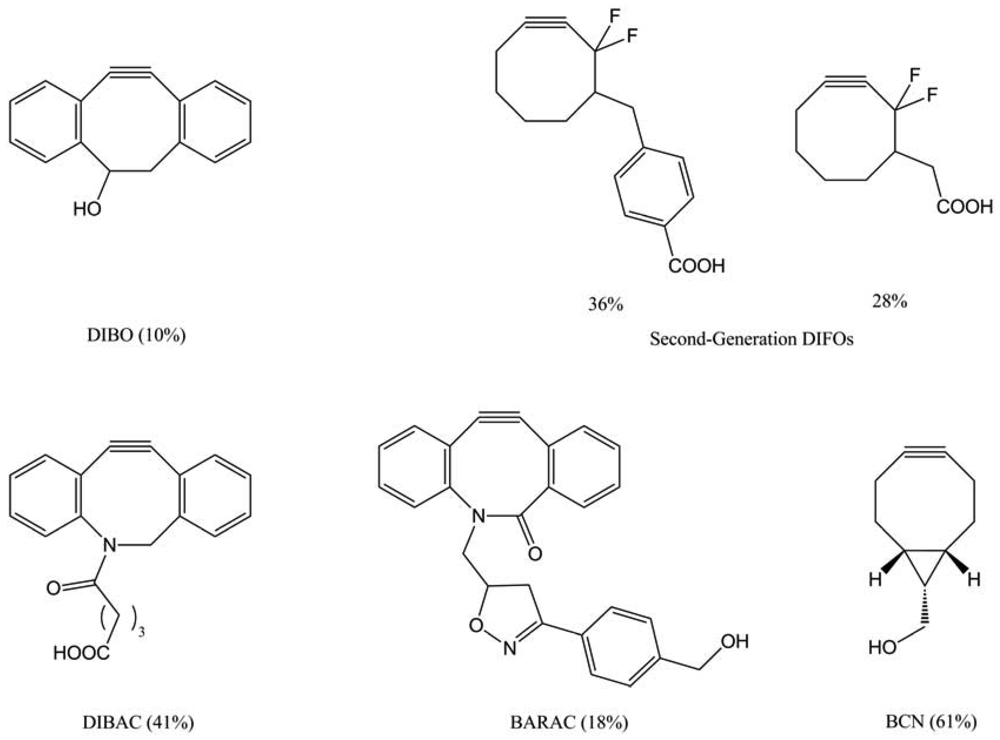
Boons and co-workers proposed another type of cyclooctynes: dibenzocyclooctynes (DIBO) [76]. In that case, a four-step synthesis yielded the desired compound in 10%, but the DIBO-labeling conduced to a less important mean fluorescence than DIFO. In parallel, Bertozzi and colleagues developed another generation of DIFO, which possessed a C-C bond to a linker substituent at C-4, rather than a C-O bond at C6 (Figure 46). They synthesized two of these second-generation DIFOs in 28% and 36% overall yield for six steps. These novel reagents can selectively tag azide-labeled glycans with efficiencies approaching that of the first generation DIFOs. We can also report the synthesis of the dibenzoazacyclooctyne (DIBAC) [77], obtained in an overall yield of 41% over 9 steps, and the synthesis of biarylazacyclooctynone (BARAC) [78] conducted in 6 steps to a lesser overall yield of 18%. More recently, Van Delft and co-workers [79] described the synthesis of a promising bicyclononyne (BCN) issued by a short synthetic route (4 steps) with only two purifications in an overall yield of 61%. The BCN-labeling showed interesting results, with a higher reactivity than the other cyclooctynes and a very good sensitivity.
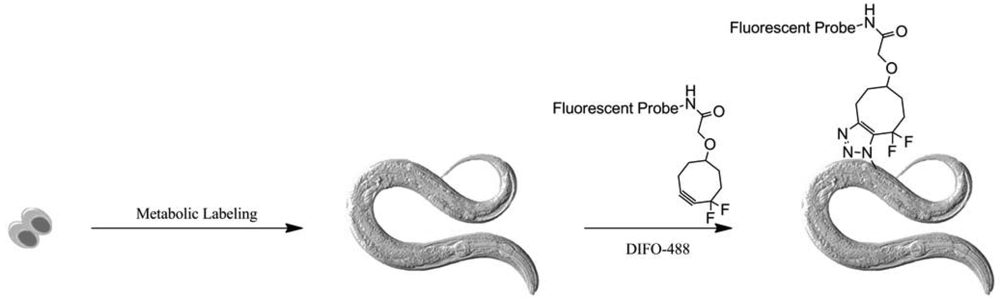
In the meantime, Bertozzi and co-workers pursued the extension of her method to other biological systems. First, they used Alexa Fluor combined first-generation DIFO (DIFO-488 and -568) to visualize glycans in C. Elegans (Figure 47) [80]. As we saw earlier, they applied the two-color labeling strategy to observe the glycans biosynthesis from larval stage to adulthood.
No toxicity was observed during this process and labeling showed some good results at the larval stages. However, in adult stage, only the organs accessible to solutions are readily labeled by DIFO, highlighting their limited penetrance into the organism.
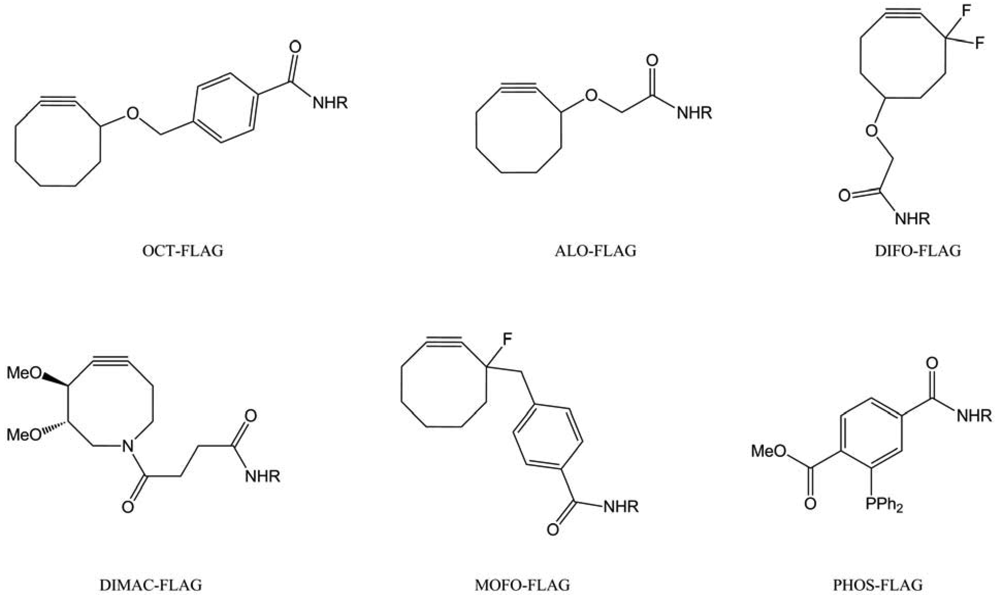
Finally, the SPAAC was investigated on more complex living system: mice [82]. In this study, the “chemical reporters” were the unnatural azido-sugar Ac4ManNAz and several cyclooctynes (Figure 48) have been used, conjugated to a small peptide (FLAG). This FLAG peptide allowed further analysis thanks to anti-FLAG antibody.
Attempts with OCT-FLAG and MOFO-FLAG were not conductive to any labeling, showing its poor water solubility and lesser reactivity. The ALO-FLAG only weakly labeled intestines; meanwhile DIMAC- and DIFO-FLAG tagged all of the harvested organs (intestines, liver and heart). In comparison, PHOS-FLAG appears to access the same organs. The DIFO-FLAG proved to be the best cyclooctyne to label modified glycans, due to its high reactivity, but it also had a high affinity to serum albumin, which compromised its bioavailability.
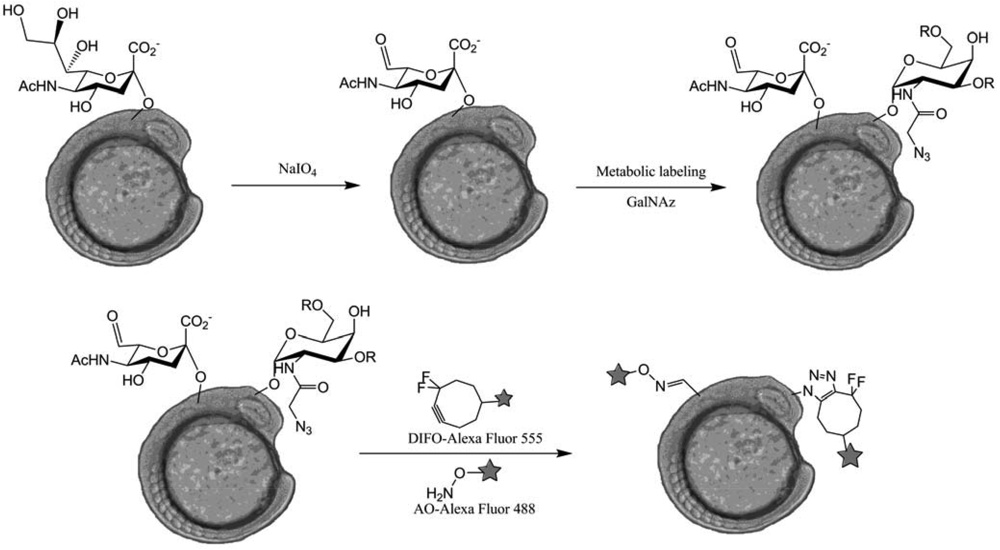
The most advanced work on SPAAC labeling was conducted by Bertozzi et al. [82] as they managed to directly visualize glycans in membrane cells of zebrafish embryos during their development. Injection of Ac4GalNAz combined with a DIFO-fluorophore conjugates labeling led to very interesting observations about O-glycans distribution in zebrafish embryos. They were able to proceed to a simultaneous visualization of two types of glycans using two independent labeling: the O-glycans labeling, using DIFO, and the sialic acid labeling, using NaIO4 and aminooxy fluorophores (AO) (Figure 49).
Their experiments validate the ability of using two independent chemistries to visualize different classes of glycan in the same organism. Moreover, a time-lapse monitoring of mitotic cells showed a very fast reorganization of membrane O-glycans to the cleavage furrow. In order to visualize the glycans trafficking during this phenomenon, they tried to apply the two-color labeling on that case but the observed reorganization took place on a faster scale than that of the labeling protocol.
In conclusion, SPAAC used in glycans imaging is now at an advanced stage and has been applied to various biological models: cultured cells, zebrafish embryos, nematodes or mice. Despite all of these progresses, the SPAAC labeling suffers from the difficult cyclooctynes synthesis and design (reactivity and poor water solubility). The real stake of this labeling way relies on organic chemistry and one of these challenges will be the design of “click”-generated fluorescent cyclooctynes.
6. Conclusions
Over the past ten years, polysaccharides have been clearly impacted by “click” chemistry, and particularly by the CuAAC and SPAAC. Among the early work on polyoses, few different scientific areas were involved, such as materials, therapeutics, or diagnostic agents. In several cases, a functionalization approach was considered, often leading to the issue of regioselectivity in polymer chemistry; leading to the interrogation whether to “click” or not to “click” on polysaccharides. The answer might well be yes, but not forgetting that the click concept should be ideally applicable to all the reaction steps, and not only to the cycloaddition step. Finally the first studies of the use of “simple” reactions on polysaccharides are just emerging, and the combination of new “click” reactions to specific polysaccharides could open a wide field of discovery in future research.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Polysaccharides | Modifications pre-“click” (positions) | Regio-selectivity | Reference |
|---|---|---|---|---|
| 1 | 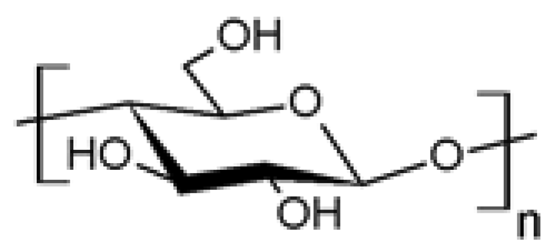 Cellulose Cellulose | Azidation (C6) | Yes | [15-21] |
| Azidation | No | [22] | ||
| Esterification (C6) | Yes | [23-25] | ||
| Amination (C6) | Yes | [19,26] | ||
| Amidation (C6) | Yes | [27] | ||
| Propargylation (C3) | Yes | [28] | ||
| 2 | 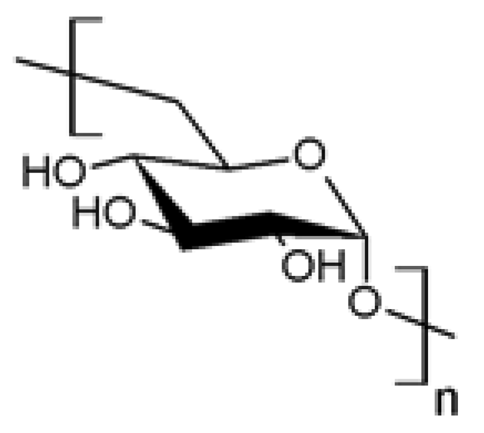 Dextran Dextran | Esterification (C3) | Yes | [29] |
| Esterification (C2) | Yes | [30] | ||
| Reductive amination(reductive end) | Yes | [31,32] | ||
| Etherification (C3) | Yes | [33] | ||
| 3 | 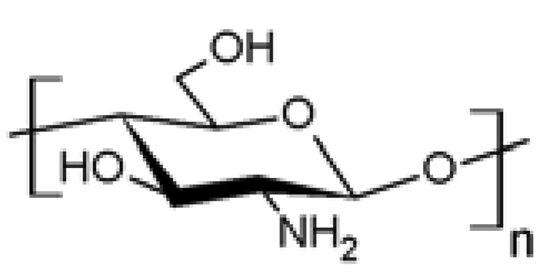 Chitosan Chitosan | Azidation (C6, C2) | Yes | [34-38] |
| Esterification (C6) | Yes | [39-43] | ||
| Amidation (C2) | Yes | [44-47] | ||
| 4 | 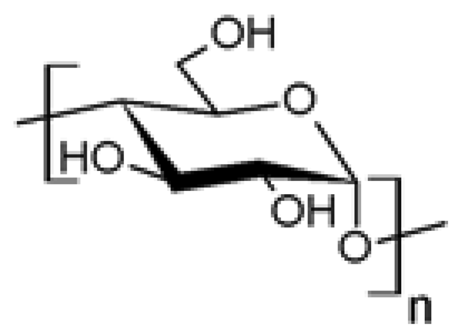 Starch Starch | Azidation | No | [48] |
| Propargylation | No | [49] | ||
| 5 | 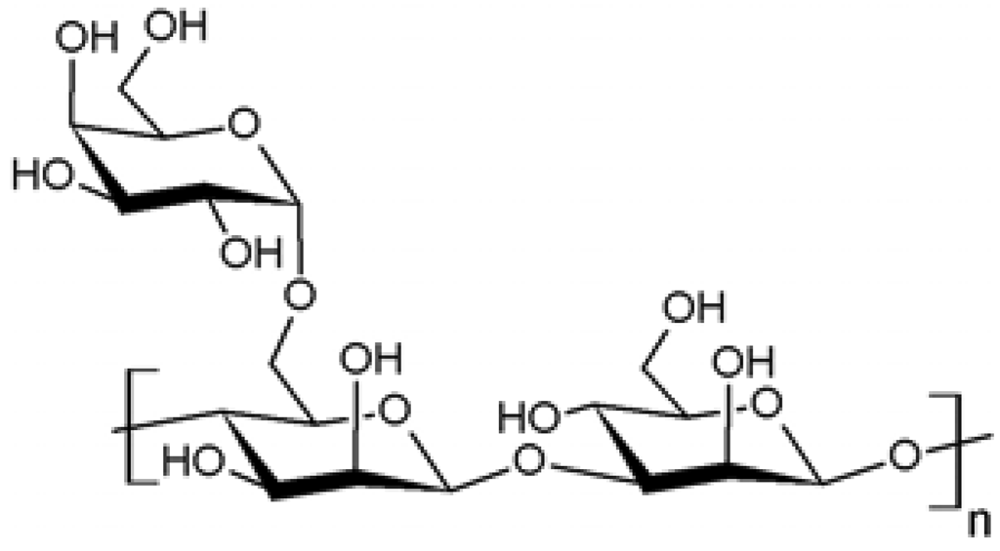 Guar Guar | Propargylation | No | [50] |
| 6 | 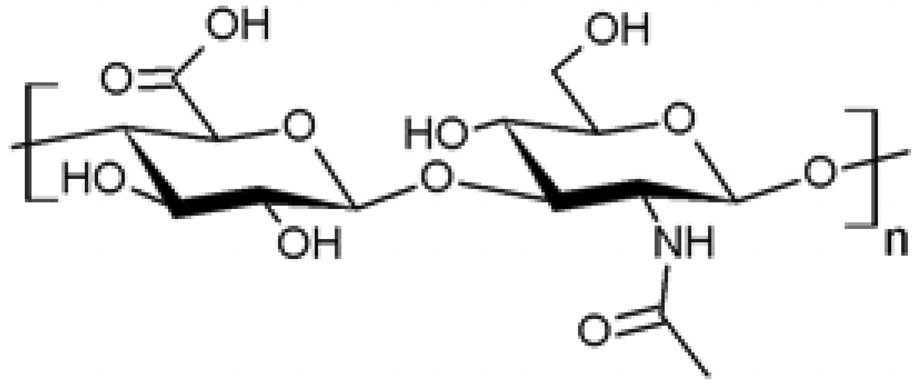 Hyaluronan Hyaluronan | Amidation (C6) | Yes | [51,52] |
| Reductive amination(reductive end) | Yes | [53] | ||
| 7 | Glycans | Propargylation (reductive end) | Yes | [54,55] |
| 8 | 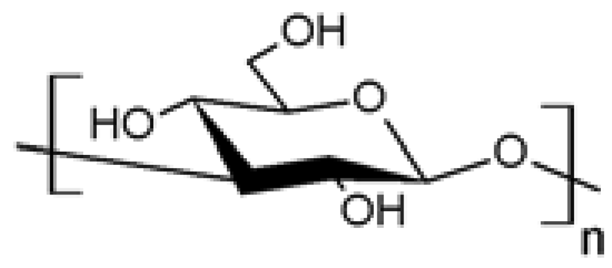 (1→3)̃β̃-D-glucan (1→3)̃β̃-D-glucan | Azidation (C6) | Yes | [56] |
Acknowledgments
The authors thank Jessica Roper for her help in writing the manuscript.
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar]
- Tornøe, C.W.; Meldal, M. Peptidotriazoles: Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions on Solid-Phase. In Peptides 2001, Proc. Am. Pept. Symp.; Lebl, M., Houghten, R.A., Eds.; American Peptide Society and Kluwer Academic Publishers: San Diego, CA, USA, 2001; pp. 263–264. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar]
- Bock, V.D.; Hiemstra, H.; Maarseveen, J.H. CuI-catalyzed alkyne-azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur. J. Org. Chem. 2006, 2006, 51–68. [Google Scholar]
- Dondoni, A. Triazole: The Keystone in glycosylated molecular architectures constructed by a click reaction. Chem. Asian J. 2007, 2, 700–708. [Google Scholar]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2007, 28, 278–308. [Google Scholar]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar]
- Binder, W.H.; Sachsenhofer, R. “Click” chemistry in polymer and materials science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 24, 1128–1137. [Google Scholar]
- Lucas, R.; Zerrouki, R.; Krausz, P. And if everything was as easy as a “click”. Cu(I)-catalyzed 1,3-dipolar cycloaddition between terminal alkynes and azides. L'actualité Chim. 2009, 335, 5–9. [Google Scholar]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar]
- Codelli, J.A.; Baskin, J.M.; Agard, N.J.; Bertozzi, C.R. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J. Am. Chem. Soc. 2008, 130, 11486–11493. [Google Scholar]
- Liebert, T.; Hänsch, C.; Heinze, T. Click chemistry with polysaccharides. Macromol. Rapid Commun. 2006, 27, 208–213. [Google Scholar]
- Heinze, T.; Schöbitz, M.; Pohl, M.; Meister, F. Interactions of ionic liquids with polysaccharides. IV. dendronization of 6-azido-6-deoxy cellulose. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 3853–3859. [Google Scholar]
- Pohl, M.; Morris, G.A.; Harding, S.E.; Heinze, T. Studies on the molecular flexibility of novel dendronized carboxymethyl cellulose derivatives. Eur. Polym. J. 2009, 45, 1098–1110. [Google Scholar]
- Zhang, J.; Xu, X.-D.; Wu, D.-Q.; Zhang, X.-Z.; Zhuo, R.-X. Synthesis of thermosensitive p(NIPAAm-co-HEMA)/cellulose hydrogels via “click” chemistry. Carbohyd. Polym. 2009, 77, 583–589. [Google Scholar]
- Pohl, M.; Heinze, T. Novel biopolymer structures synthesized by dendronization of 6-deoxy-6-aminopropargyl cellulose. Macromol. Rapid Commun. 2008, 29, 1739–1745. [Google Scholar]
- Negishi, K.; Mashiko, Y.; Yamashita, E.; Otsuka, A.; Hasegawa, T. Cellulose chemistry meets click chemistry: Syntheses and properties of cellulose-based glycoclusters with high structural homogeneity. Polymer 2011, 3, 489–508. [Google Scholar]
- Kawagoe, N.; Kasori, Y.; Hasegawa, T. Highly C6-selective and quantitative modification of cellulose: Nucleoside-appended celluloses to solubilize single walled carbon nanotubes. Cellulose 2011, 18, 83–93. [Google Scholar]
- Ringot, C.; Sol, V.; Granet, R.; Krausz, P. Porphyrin-grafted cellulose fabric: New photobactericidal material obtained by “click-chemistry” reaction. Mater. Lett. 2009, 63, 1889–1891. [Google Scholar]
- Hafrén, J.; Zou, W.; Córdova, A. Heterogeneous ‘organoclick’ derivatization of polysaccharides. Macromol. Rapid Comm. 2006, 27, 1362–1366. [Google Scholar]
- Hafren, J.; Cordova, A. Direct organocatalytic polymerization from cellulose fibers. Macromol. Rapid Commun. 2005, 26, 82–86. [Google Scholar]
- Krouit, M.; Bras, J.; Belgacem, M.N. Cellulose surface grafting with polycaprolactone by heterogeneous click-chemistry. Eur. Polym. J. 2008, 44, 4074–4081. [Google Scholar]
- Koschella, A.; Hartlieb, M.; Heinze, T. A “click-chemistry” approach to cellulose-based hydrogels. Carbohydr. Polym. 2011, 86, 154–161. [Google Scholar]
- Filpponen, I.; Argyropoulos, D.S. Regular linking of cellulose nanocrystals via click chemistry: Synthesis and formation of cellulose nanoplatelet gels. Biomacromolecules 2010, 11, 1060–1066. [Google Scholar]
- Fenn, D.; Pohl, M.; Heinze, T. Novel 3-O-propargyl cellulose as a precursor for regioselective functionalization of cellulose. React. Funct. Polym. 2009, 69, 347–352. [Google Scholar]
- De Geest, B.G.; van Camp, W.; Du Prez, F.E.; De Smedt, S.C.; Demeester, J.; Hennink, W.E. Degradable multilayer films and hollow capsules via a ‘click’ strategy. Macromol. Rapid Commun. 2008, 29, 1111–1118. [Google Scholar]
- Samatya, S.; Orhan, E.; Kabay, N.; Tuncel, A. Comparative boron removal performance of monodisperse-porous particles with molecular brushes via “click chemistry” and direct coupling. Colloid. Surface. A: Physicochem. Eng. Asp. 2010, 372, 102–106. [Google Scholar]
- Bernard, J.; Save, M.; Arathoon, B.; Charleux, B. Preparation of a xanthate-terminated dextran by click chemistry: Application to the synthesis of polysaccharide-coated nanoparticles via surfactant-free ab initio emulsion polymerization of vinyl acetate. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 2845–2857. [Google Scholar]
- Schatz, C.; Louguet, S.; Le Meins, J.-F.; Lecommandoux, S. Polysaccharide-block-polypeptide copolymer vesicles: Towards synthetic viral capsids. Angew. Chem. Int. Ed. 2009, 48, 2572–2575. [Google Scholar]
- Zhong, J.; Chau, Y. Synthesis, characterization, and thermodynamic study of a polyvalent lytic peptide-polymer conjugate as novel anticancer agent. Bioconjugate Chem. 2010, 21, 2055–2064. [Google Scholar]
- Zhang, F.; Bernet, B.; Bonnet, V.; Dangles, O.; Sarabia, F.; Vasella, A. 2-Azido-2-deoxycellulose: Synthesis and 1,3-dipolar Cycloaddition. Helv. Chim. Acta 2008, 91, 608–617. [Google Scholar]
- Zampano, G.; Bertoldo, M.; Ciardelli, F. Defined chitosan-based networks by C-6-Azide-alkyne “click” reaction. React. Funct. Polym. 2010, 70, 272–281. [Google Scholar]
- Bertoldo, M.; Nazzi, S.; Zampano, G.; Ciardelli, F. Synthesis and photochromic response of a new precisely functionalized chitosan with “clicked” spiropyran. Carbohydr. Polym. 2011, 85, 401–407. [Google Scholar]
- Gao, Y.; Zhang, Z.; Chen, L.; Gu, W.; Li, Y. Synthesis of 6-N,N,N-trimethyltriazole chitosan via “click chemistry” and evaluation for gene delivery. Biomacromolecules 2009, 10, 2175–2182. [Google Scholar]
- Kulbokaite, R.; Ciuta, G.; Netopilik, M.; Makuska, R. N-PEG'ylation of chitosan via “click chemistry” reactions. React. Funct. Polym. 2009, 69, 771–778. [Google Scholar]
- Ifuku, S.; Wada, M.; Morimoto, M.; Saimoto, H. Preparation of highly regioselective chitosan derivatives via “click chemistry”. Carbohydr. Polym. 2011, 85, 653–657. [Google Scholar]
- Yuan, W.; Zhao, Z.; Gu, S.; Ren, T.; Ren, J. Synthesis and self-assembly of pH-responsive chitosan graft copolymer by the combination of atom transfer radical polymerization and click chemistry. Mater. Lett. 2011, 65, 793–796. [Google Scholar]
- Li, X.; Yuan, W.; Gu, S.; Ren, J. Synthesis and self-assembly of tunable thermosensitive chitosan amphiphilic copolymers by click chemistry. Mater. Lett. 2010, 64, 2663–2666. [Google Scholar]
- Yuan, W.; Li, X.; Gu, S.; Cao, A.; Ren, J. Amphiphilic chitosan graft copolymer via combination of ROP, ATRP, and click chemistry: Synthesis, self-assembly, thermosensitivity, fluorescence, and controlled drug release. Polymer 2010, 52, 658–666. [Google Scholar]
- Yuan, W.; Zhao, Z.; Gu, S.; Ren, J. Synthesis, characterization, and properties of amphiphilic chitosan copolymers with mixed side chains by click chemistry. J. Polym. Sci. Part A: Polym. Chem. 2010, 48, 3476–3486. [Google Scholar]
- Bao, H.; Li, L.; Leong, W.C.; Gan, L.H. Thermo-responsive association of chitosan-graft-poly(N-isopropylacrylamide) in aqueous solutions hongqian. J. Phys. Chem. B. 2010, 114, 10666–10673. [Google Scholar]
- Bao, H.; Li, L.; Gan, L.H.; Ping, Y.; Li, J.; Ravi, P. Thermo- and pH-responsive association behavior of dual hydrophilic graft chitosan terpolymer synthesized via ATRP and click chemistry. Macromolecules 2010, 43, 5679–5687. [Google Scholar]
- Lallana, E.; Fernandez-Megia, E.; Riguera, R. Surpassing the use of copper in the click functionalization of polymeric nanostructures: A strain-promoted approach. J. Amer. Chem. Soc. 2009, 131, 5748–5750. [Google Scholar]
- Zhang, J.; Li, C.; Xue, Z.-Y.; Cheng, H.-W.; Huang, F.-W.; Zhuo, R.-X.; Zhang, X.-Z. Fabrication of lactobionic-loaded chitosan microcapsules as potential drug carriers targeting the liver. Acta Biomater. 2011, 7, 1665–1673. [Google Scholar]
- Xiao, C.; Lu, D.; Xu, S.; Huang, L. Tunable synthesis of starch-poly(vinyl acetate) bioconjugate. Starch 2011, 63, 209–216. [Google Scholar]
- Tankam, P.F.; Müller, R.; Mischnick, P.; Hopf, H. Alkynyl polysaccharides: Synthesis of propargyl potato starch followed by subsequent derivatizations. Carbohydr. Res. 2007, 342, 2049–2060. [Google Scholar]
- Tizzotti, M.; Labeau, M.-P.; Hamaide, T.; Drockenmuller, E.; Charlot, A.; Fleury, E. Synthesis of thermosensitive guar-based hydrogels with tunable physico-chemical properties by click chemistry. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2733–2742. [Google Scholar]
- Hu, X.; Li, D.; Zhou, F.; Gao, C. Biological hydrogel synthesized from hyaluronic acid, gelatin and chondroitin sulfate by click chemistry. Acta Biomat. 2011, 7, 1618–1626. [Google Scholar]
- Huerta-Angeles, G.; Šmejkalová, D.; Chládková, D.; Ehlová, T.; Buffa, R.; Velebný, V. Synthesis of highly substituted amide hyaluronan derivatives with tailored degree of substitution and their crosslinking via click chemistry. Carbohydr. Polym. 2011, 84, 1293–1300. [Google Scholar]
- Upadhyay, K.K.; Le Meins, J.-F.; Misra, A.; Voisin, P.; Bouchaud, V.; Ibarboure, E.; Schatz, C.; Lecommandoux, S. Biomimetic Doxorubicin loaded polymersomes from hyaluoran-block-poly(γ-benzyl glucamate) copolymers. Biomacromolecules 2009, 10, 2802–2808. [Google Scholar]
- Yamaguchi, M.; Kojima, K.; Hayashi, N.; Kakizaki, I.; Kon, A.; Takagaki, K. Efficient and widely applicable method of constructing neo-proteoglycan utilizing copper(I) catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 7455–7458. [Google Scholar]
- Yamaguchi, M.; Takagaki, K.; Kojima, K.; Hayashi, N.; Chen, F.; Kakizaki, I.; Kon, A.; Endo, M. Novel proteoglycan glycotechnology: Chemoenzymatic synthesis of chondroitin sulfate-containing molecules and its application. Glycoconjugate J. 2010, 27, 189–198. [Google Scholar]
- Hasegawa, T.; Umeda, M.; Numata, M.; Li, C.; Bae, A.-H.; Fujisawa, T.; Haraguchi, S.; Sakurai, K.; Shinkai, S. ‘Click Chemistry’ on polysaccharides: A convenient, general, and monitorable approach to develop (1->3)-β-D-glucans with various functional appendages. Carbohydr. Res. 2006, 341, 35–40. [Google Scholar]
- Belgacem, M.N.; Gandini, A. Monomers, Polymers and Composites from Renewable Resources; Elsevier: Amesterdam, The Netherlands, 2008. [Google Scholar]
- Thomas, S.; Pothan, L. Cellulose Fibre Reinforced Polymer Composites; Old City Publishing: Philadelphia, PA, USA, 2008. [Google Scholar]
- Wool, R.P.; Sun, X.S. Bio-Based Polymers and Composites; Elsevier: Amesterdam, The Netherlands, 2005. [Google Scholar]
- Eichhorn, S.J.; Baillie, C.A.; Zafeiropoulos, N.; Mwaikambo, L.Y.; Ansell, M.P.; Dufresne, A.; Entwistle, K.M.; Herrera-Franco, P.J.; Escamilla, G.C.; Groom, L. Current international research into cellulosic fibres and composites. J. Mater. Sci. 2001, 36, 2107–2131. [Google Scholar]
- Li, X.; Wu, W.; Liu, W. Synthesis and properties of thermo-responsive guar gum/poly(N-isopropylacrylamide) interpenetrating polymer network hydrogels. Carbohydr. Polym. 2008, 71, 394–402. [Google Scholar]
- Suksrichavalit, T.; Yoshimatsu, K.; Prachayasittikul, V.; Bülow, L.; Ye, L. “Clickable” affinity ligands for effective separation of glycoproteins. J. Chromatogr. A 2010, 1217, 3635–3641. [Google Scholar]
- Hanson, S.R.; Greenberg, W.A.; Wong, C.-H. Probing glycans with the copper(I)-catalyzed [3 + 2] azide-alkyne cycloaddition. QSAR Comb. Sci. 2007, 11-12, 1243–1252. [Google Scholar]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified Staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar]
- Prescher, J.A.; Dube, D.H.; Bertozzi, C.R. Chemical remodelling of cell surfaces in living animals. Nature 2004, 430, 873–877. [Google Scholar]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar]
- Link, A.J.; Tirrell, D.A. Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 11164–11165. [Google Scholar]
- Link, A.J.; Vink, M.K.S.; Tirrell, D.A. Presentation and detection of azide functionality in bacterial cell surface proteins. J. Am. Chem. Soc. 2004, 126, 10598–10602. [Google Scholar]
- Beatty, K.E.; Xie, F.; Wang, Q.; Tirrell, D.A. Selective dye-labeling of newly synthesized proteins in bacterial cells. J. Am. Chem. Soc. 2005, 127, 14150–14151. [Google Scholar]
- Keppler, O.T.; Horstkorte, R.; Pawlita, M.; Schmidt, C.; Reutter, W. Biochemical engineering of the N-acyl side chain of sialic acid: Biological implications. Glycobiology 2001, 11, 11R–18R. [Google Scholar]
- Hang, H.C.; Yu, C.; Kato, D.L.; Bertozzi, C.R. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. PNAS 2003, 100, 14846–14851. [Google Scholar]
- Sawa, M.; Hsu, T.-L.; Itoh, T.; Sugiyama, M.; Hanson, S.R.; Vogt, P.K.; Wong, C.-H. Glycoproteomic probes for fluorescent imaging of fucosylated glycans in vivo. PNAS 2006, 103, 12371–12376. [Google Scholar]
- Hsu, T.-L.; Hanson, S.R.; Kishikawa, K.; Wang, S.-K.; Sawa, M.; Wong, C.-H. Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells. PNAS 2007, 104, 2614–2619. [Google Scholar]
- Soriano Del Amo, D.; Wang, W.; Jiang, H.; Besanceney, C.; Yan, A.C.; Levy, M.; Liu, Y.; Marlow, F.L.; Wu, P. Biocompatible copper(I) catalysts for in vivo imaging of glycans. J. Am. Chem. Soc. 2010, 132, 16893–16899. [Google Scholar]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-free click chemistry for dynamic in vivo imaging. PNAS 2007, 104, 16793–16797. [Google Scholar]
- Ning, X.; Guo, J.; Wolfert, M.A.; Boons, G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. 2008, 47, 2253–2255. [Google Scholar]
- Debets, M.F.; van Berkel, S.S.; Schoffelen, S.; Rutjes, F.P.J.T.; van Hest, J.C.M.; van Delft, F.L. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 2010, 46, 97–99. [Google Scholar]
- Jewett, J.C.; Sletten, E.M.; Bertozzi, C.R. Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 2010, 132, 3688–3690. [Google Scholar]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L.J.A.; Rutjes, F.P.J.T.; van Hest, J.C.M.; Lefeber, D.J.; Friedl, P.; van Delft, F.L. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. 2010, 49, 9422–9425. [Google Scholar]
- Laughlin, S.T.; Bertozzi, C.R. In vivo imaging of Caenorhabditis elegans glycans. ACS Chem. Biol. 2009, 4, 1068–1072. [Google Scholar]
- Changa, P.V.; Preschera, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-free click chemistry in living animals. PNAS 2010, 107, 1821–1826. [Google Scholar]
- Baskin, J.M.; Dehnert, K.W.; Laughlin, S.T.; Amacher, S.L.; Bertozzi, C.R. Visualizing enveloping layer glycans during zebrafish early embryogenesis. PNAS 2010, 107, 10360–10365. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Elchinger, P.-H.; Faugeras, P.-A.; Boëns, B.; Brouillette, F.; Montplaisir, D.; Zerrouki, R.; Lucas, R. Polysaccharides: The “Click” Chemistry Impact. Polymers 2011, 3, 1607-1651. https://doi.org/10.3390/polym3041607
Elchinger P-H, Faugeras P-A, Boëns B, Brouillette F, Montplaisir D, Zerrouki R, Lucas R. Polysaccharides: The “Click” Chemistry Impact. Polymers. 2011; 3(4):1607-1651. https://doi.org/10.3390/polym3041607
Chicago/Turabian StyleElchinger, Pierre-Henri, Pierre-Antoine Faugeras, Benjamin Boëns, François Brouillette, Daniel Montplaisir, Rachida Zerrouki, and Romain Lucas. 2011. "Polysaccharides: The “Click” Chemistry Impact" Polymers 3, no. 4: 1607-1651. https://doi.org/10.3390/polym3041607