Diasteromeric Effect on the Homolysis of the C–ON Bond in Alkoxyamines: A DFT Investigation of 1,3-Diphenylbutyl-TEMPO

Abstract
:1. Introduction
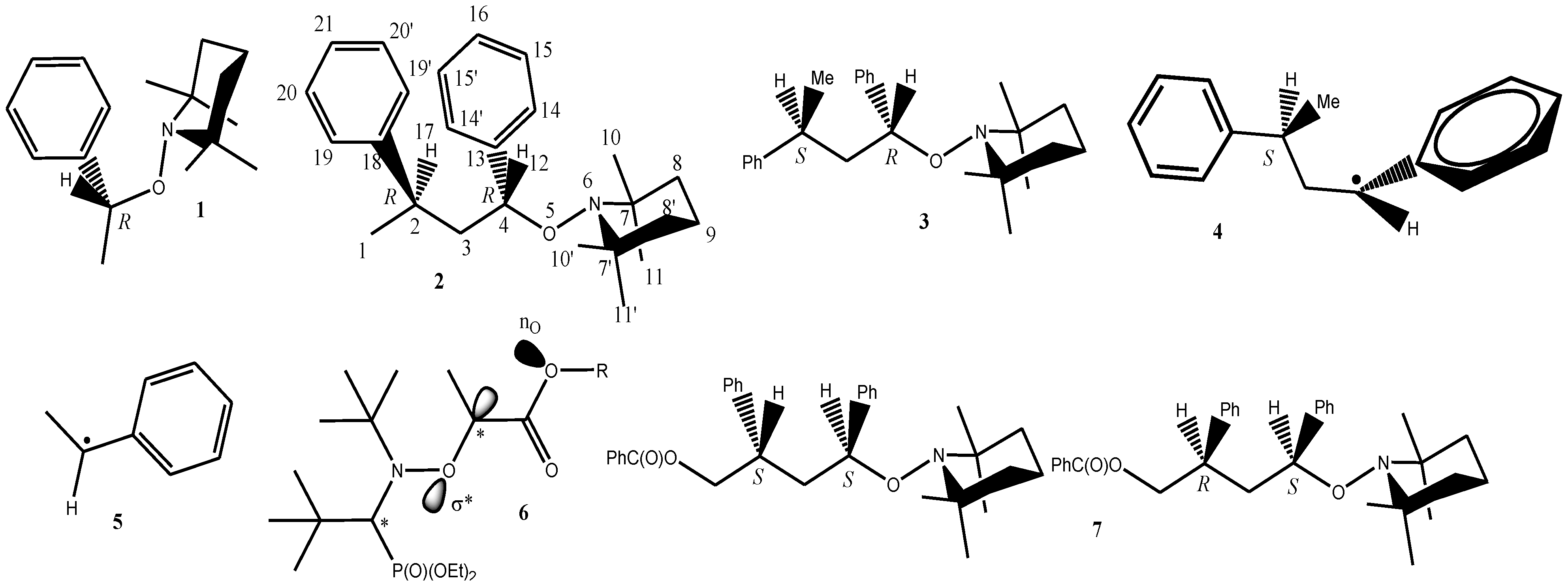
2. Computational Method
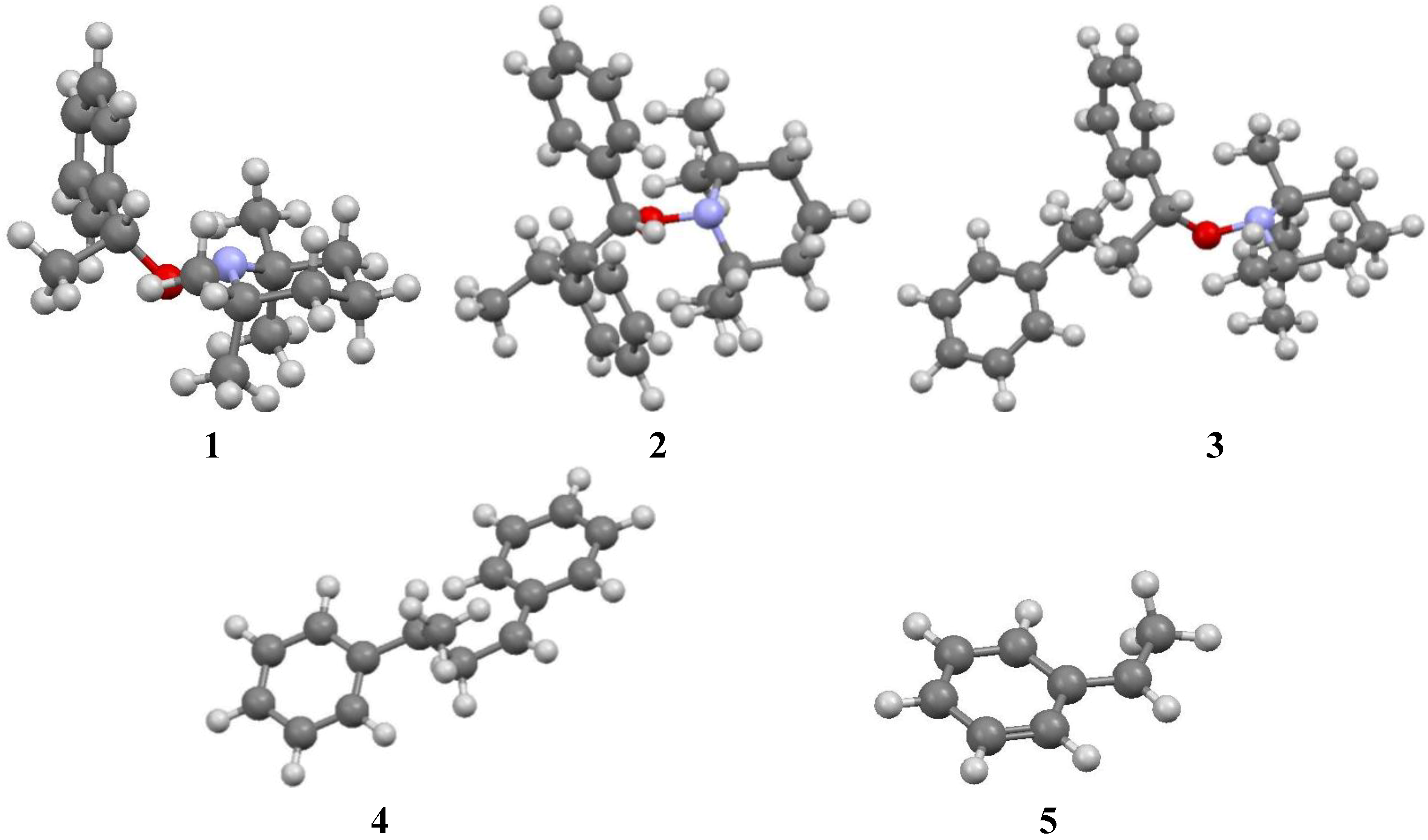
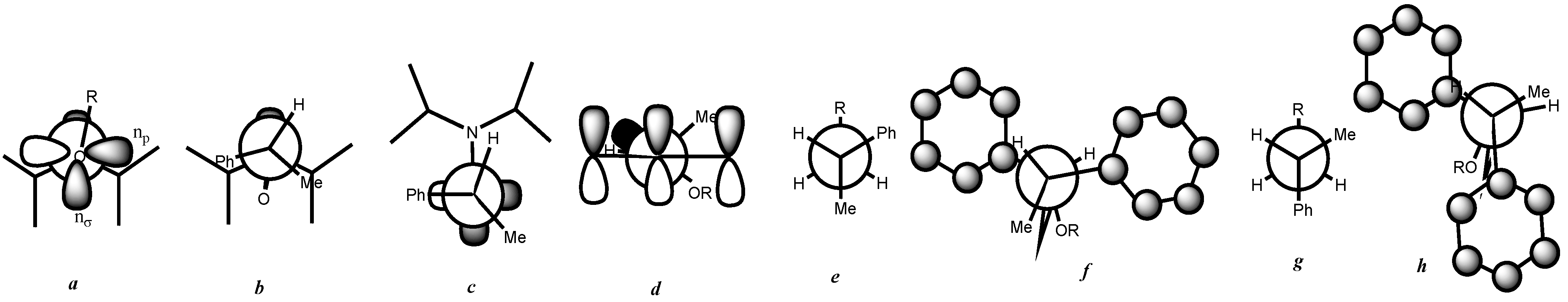
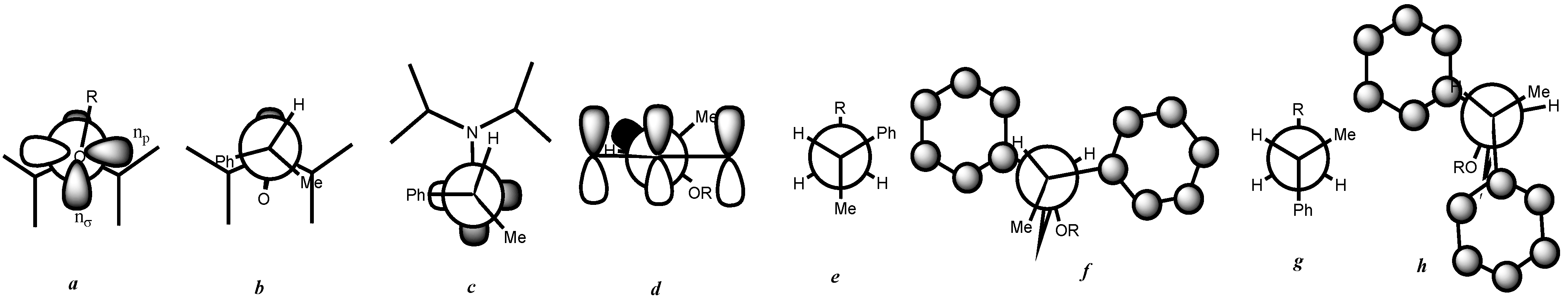
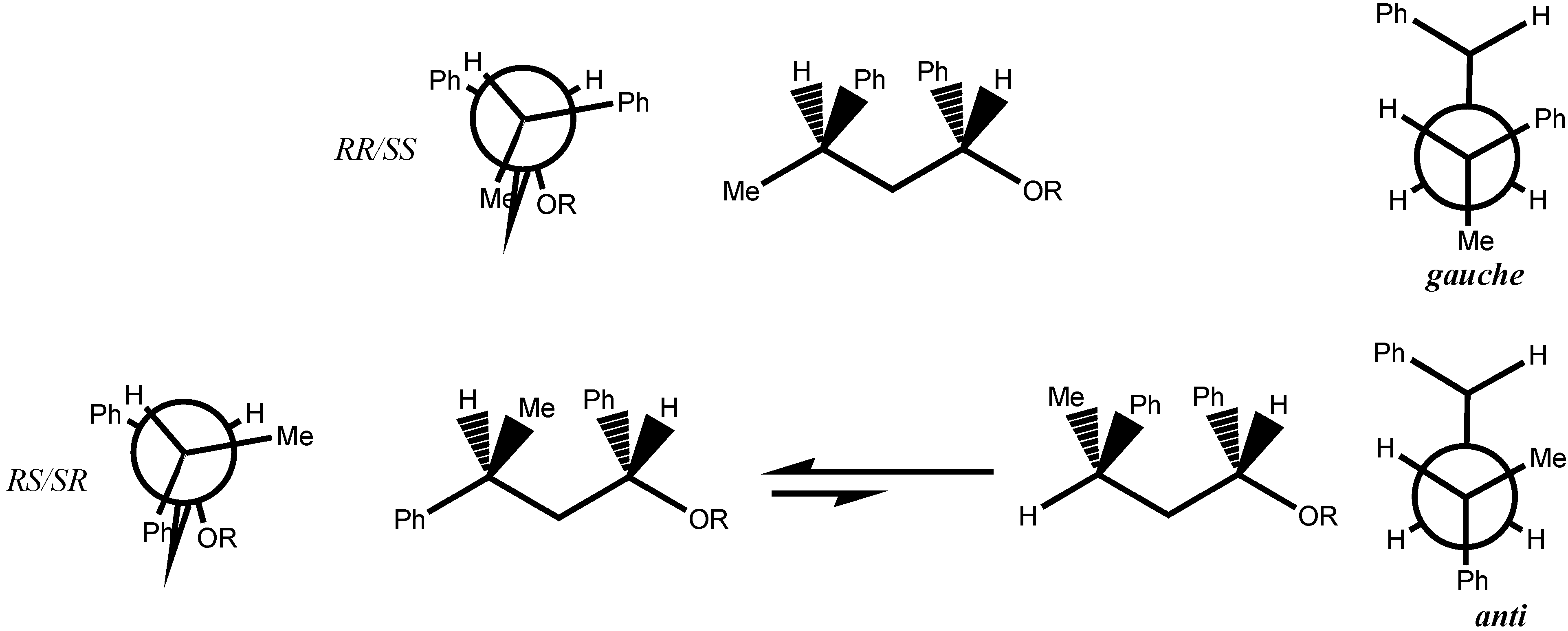
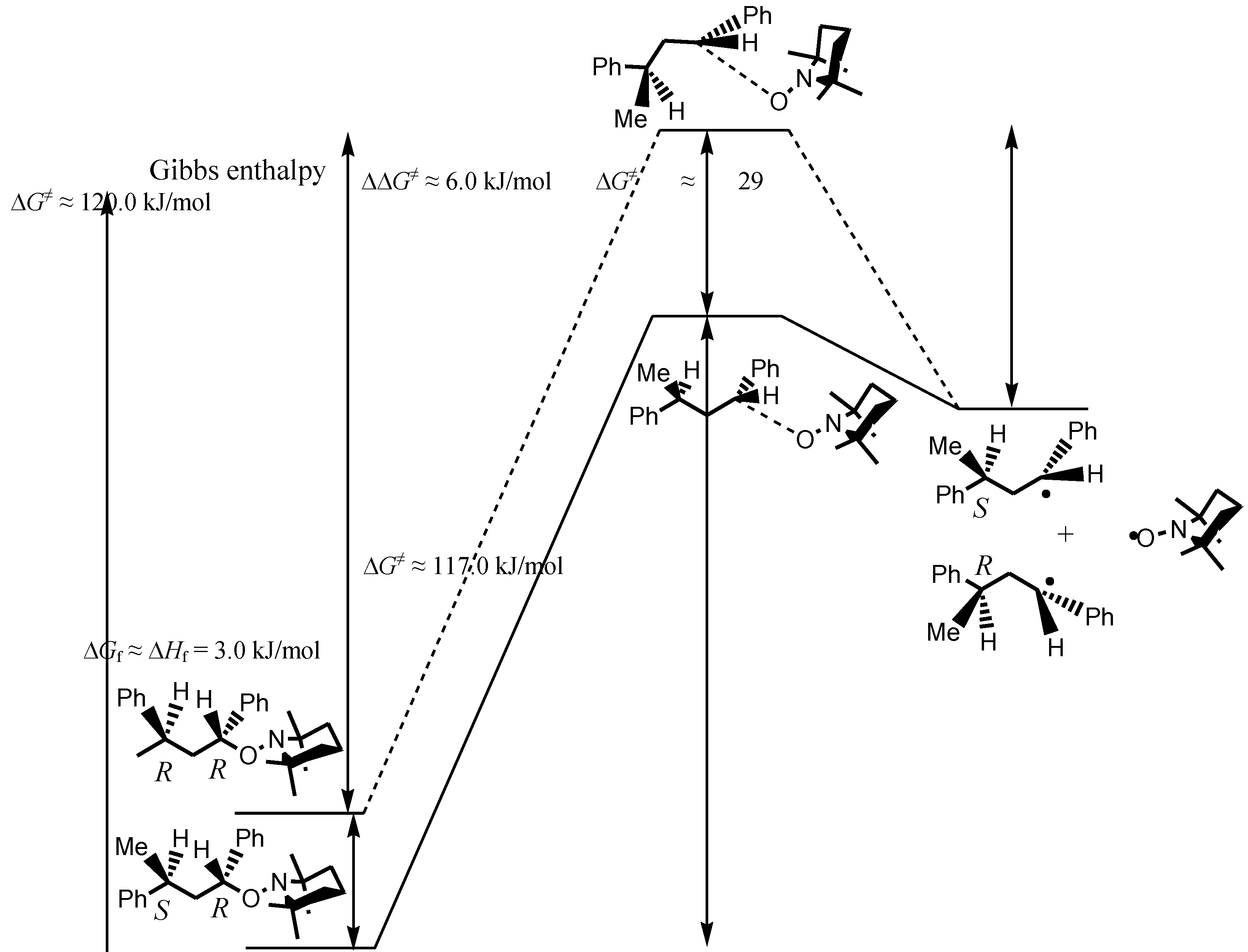
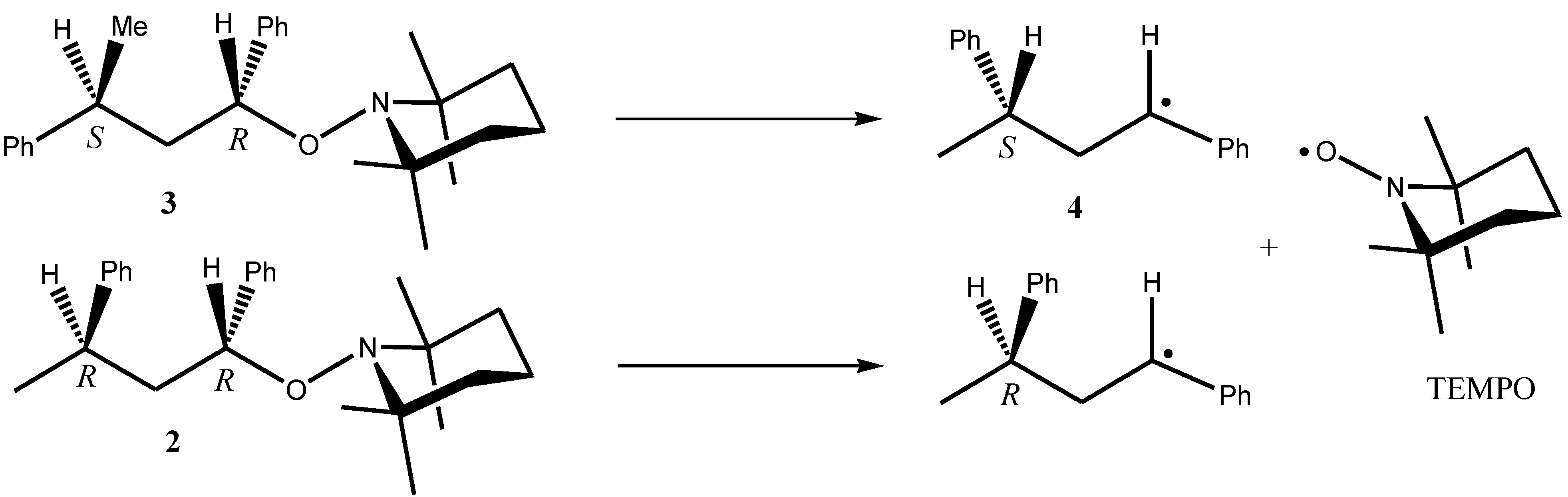
3. Results and Discussion

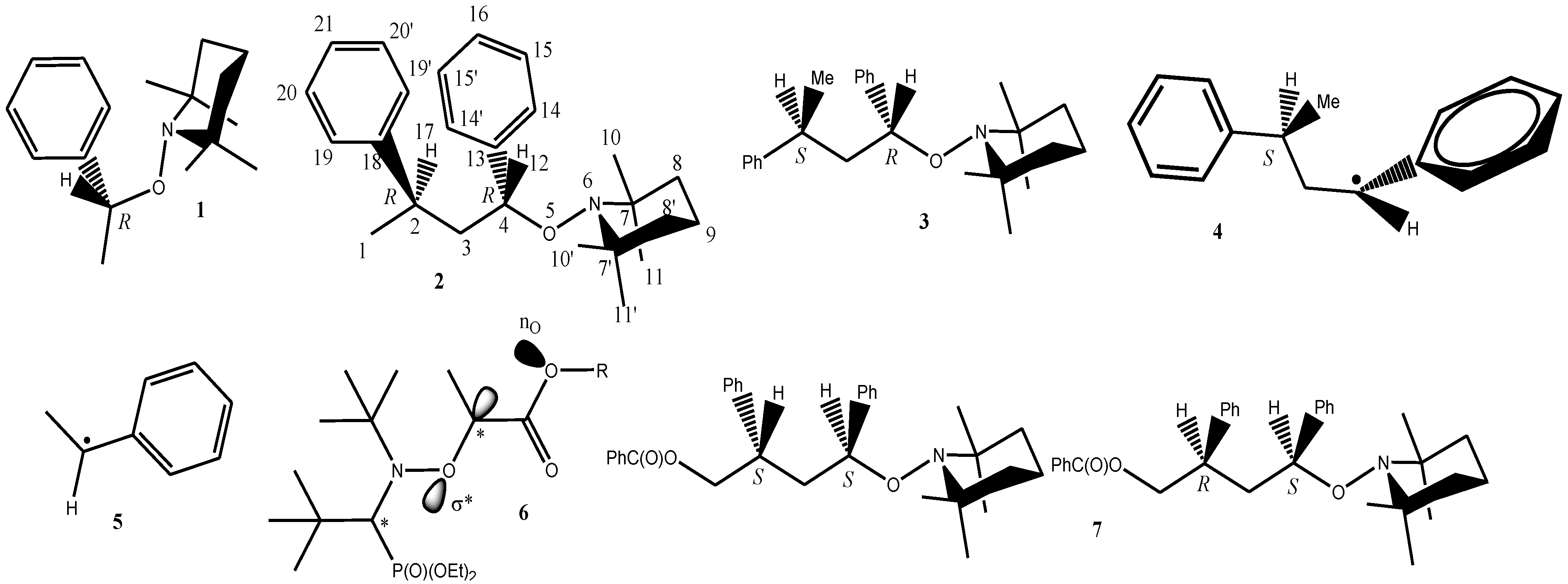
| l (Å) | 1 (X-ray) | 1(R) | 2(R4R2) | 3(R4S2) | 4(S2) | 5 |
|---|---|---|---|---|---|---|
| O5–C4 | 1.452 | 1.447 | 1.450 | 1.449 | – | – |
| N6–O5 | 1.458 | 1.453 | 1.453 | 1.453 | – | – |
| C4–C13 | 1.505 | 1.521 | 1.521 | 1.520 | 1.416 | 1.416 |
| C3–C4 | 1.521 | 1.533 | 1.539 | 1.541 | 1.498 | 1.497 |
| C2–C3 | – | – | 1.546 | 1.547 | 1.566 | – |
| d (Å) | ||||||
| C4···N6 | 2.420 | 2.427 | 2.432 | 2.430 | – | – |
| H12···C10' | 2.707 | 2.708 | 2.637 | 2.686 | – | – |
| C13···C3 | 2.509 | 2.508 | 2.540 | 2.536 | 2.594 | 2.582 |
| H12···C18/1 | – | – | 2.862 | 2.886 | 3.184 | – |
| C13···H17 | – | – | 2.706 | 2.699 | 3.200 | – |
| H12···H17 | – | – | 3.183 | 3.197 | 3.508 | – |
| C13···C2 | – | – | 3.100 | 3.097 | 3.461 | – |
| C2···C4 | – | – | 2.611 | 2.618 | 2.573 | – |
| α (°) | ||||||
| <N6O5C4> | 112.5 | 113.7 | 113.9 | 113.7 | – | – |
| <C3C4O5> | 105.0 | 106.1 | 104.7 | 104.9 | – | – |
| <C13C4O5> | 112.3 | 113.5 | 113.7 | 113.1 | – | – |
| θ (°) | ||||||
| <O5C4C13C14> | 61.6 | 54.6 | 60.3 | 58.5 | – | – |
| <C4O5N6σN6>b | −12.0 | −13.7 | −15.4 | −14.0 | – | – |
| <N6O5C4H12> | −29.6 | −23.5 | −28.2 | −25.2 | – | – |
| <O5N6C7C11> | 50.3 | 49.8 | 50.0 | 50.4 | – | – |
| <C19C18C2C3> | – | – | −61.0 | 67.5 | 67.0 | – |
| <C1C2C3C4> | – | – | 172.7 | −62.8 | 60.6 | – |
| <C2C3C4C13> | – | – | −58.2 | −58.2 | 86.5 | – |
| Interaction energies (kJ/mol) | ||||||
| πβ,C18→σ∗β,C3–C4 | – | – | – | – | 6.0 | – |
| σβ,C3–C4→β-LUMO | – | – | – | – | 11.0 | 14.0c |
| α-SOMO→σ*α,C3–C4 | – | – | – | – | 22.0 | 15.0d |
| σα,C3–C4→π*α,C18 | – | – | – | – | 5.0 | – |
| α-SOMO→π*α,C13 | – | – | – | – | 165.0 | 162.0 |
| nσ,O→σ*C3–C4 | – | 0.0e | 2.0 | 2.0 | – | – |
| σC3–C4→π*C18 | – | – | 7.0 | 9.0 | – | – |
| πC13→σ*C–O | – | 19.0 | 22.0 | 21.0 | – | – |
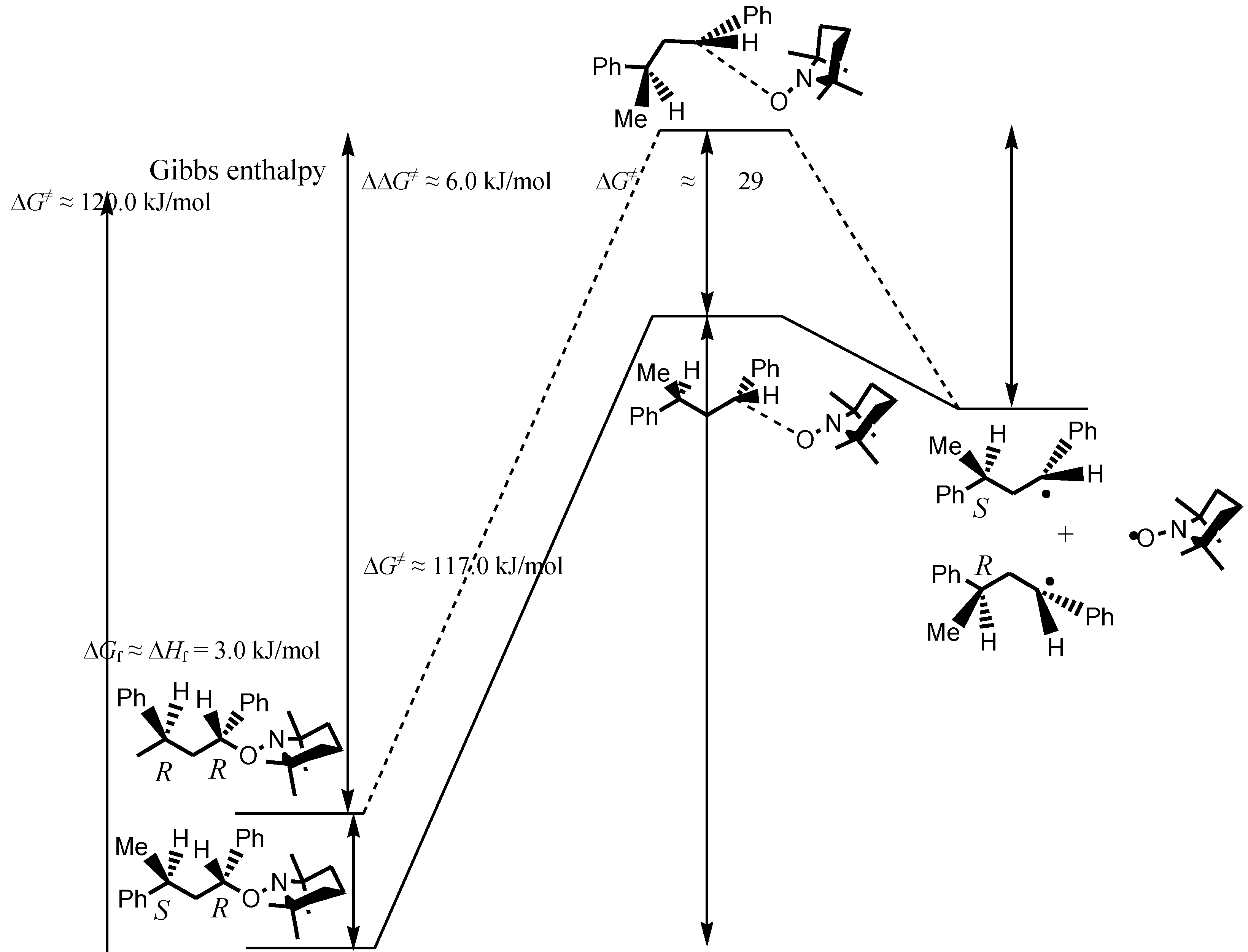
| ΔHf (kJ/mol) | – | – | – | −3.0f | – | – |


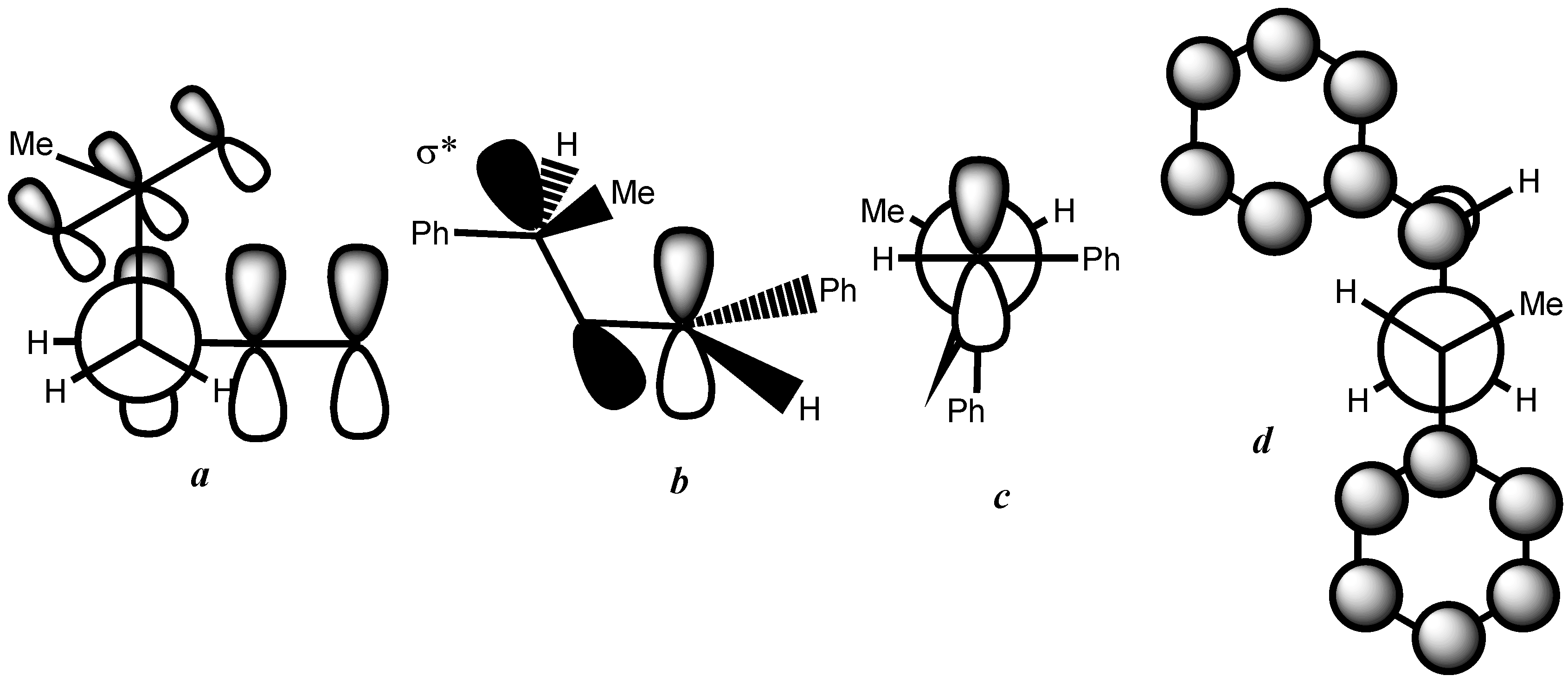
{kind=link}
{kind=link}
{kind=link}
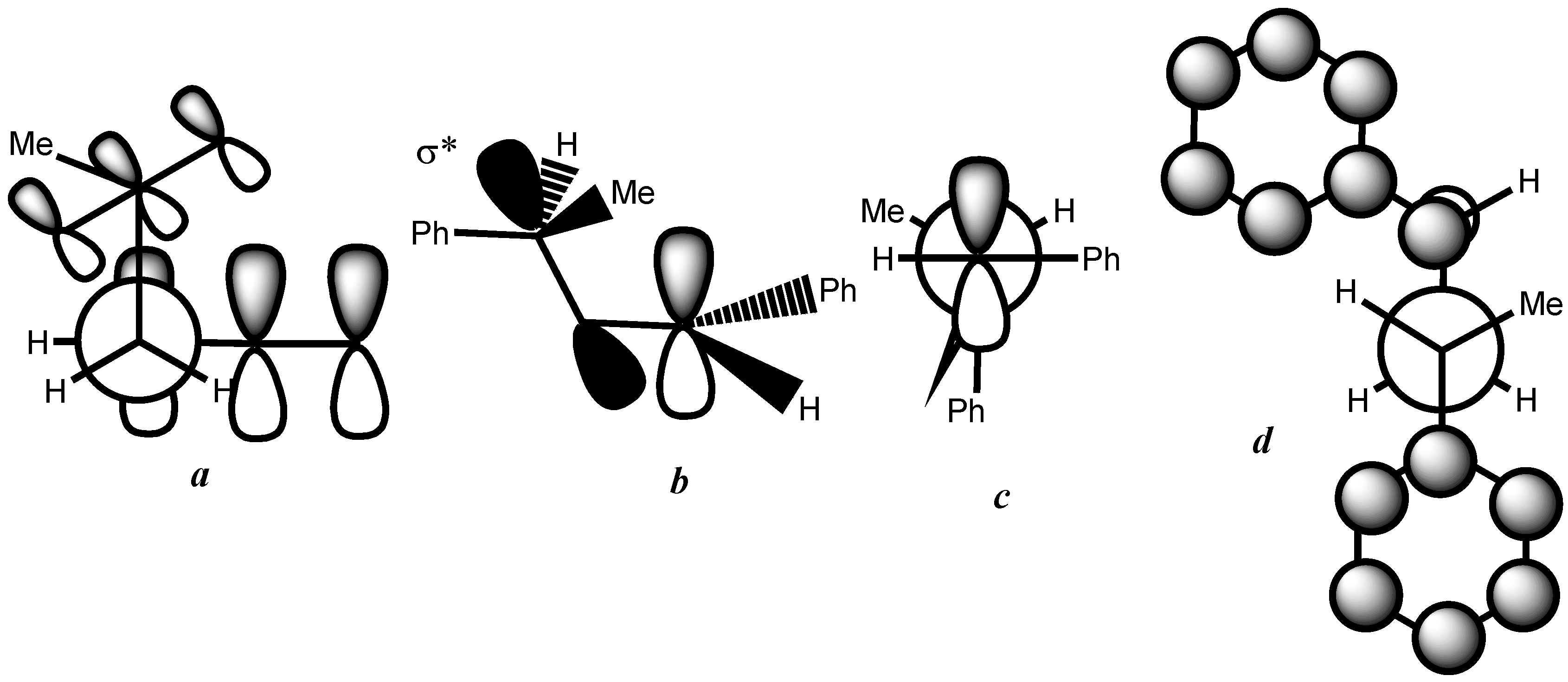{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusion
References and Notes
- Solomon, D.H.; Rizzardo, E; Cacioli, P. Polymerization process and polymers produced thereby. U.S. Patent 4,581,429, 11 July 1984. [Google Scholar]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar]
- Goto, A.; Fukuda, T. Kinetics of living radical polymerization. Progr. Polym. Sci. 2004, 29, 329, and references cited therein. [Google Scholar]
- Fischer, H. The persistent radical effect: A principle for selective radical reactions and living radical polymerizations. Chem. Rev. 2001, 101, 3581, and references cited therein. [Google Scholar]
- Fischer, H.; Souaille, M. The persistent radical effect in living radical polymerizatio—Borderline cases and side reactions. Chimia 2001, 55, 109–113. [Google Scholar]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Bertin, D.; Gigmes, D.; Marque, S.R.A. Trialkylhydroxylamines (Alkoxyamines) in Radical Chemistry: Preparation, Stability and Applications. Recent Res. Devel. Org. Chem. 2006, 10, 63–121. [Google Scholar]
- Chauvin, F.; Dufils, P.E.; Gigmes, D.; Guillaneuf, Y.; Marque, S.R.A.; Tordo, P.; Bertin, D. Nitroxide-mediated polymerization: The pivotal role of the k(d) value of the initiating alkoxyamine and the importance of the experimental conditions. Macromolecules 2006, 39, 5238–5250. [Google Scholar] [CrossRef]
- Bagryanskaya, E.; Bertin, D.; Gigmes, D.; Kirilyuk, I.; Marque, S.R.A.; Reznikov, V.; Roshchupkina, G.; Zhurko, I.; Zubenko, D. Can the first addition of alkyl radicals play a role in the fate of NMP? Macromol. Chem. Phys. 2008, 209, 1345–1357. [Google Scholar] [CrossRef]
- Bertin, D.; Dufils, P.-E.; Durand, I.; Gigmes, D.; Giovanetti, B.; Guillaneuf, Y.; Marque, S.R.A.; Phan, T.; Tordo, P. Effect of the penultimate unit on the C–ON bond homolysis in SGi-based alkoxyamines. Macromol. Chem. Phys. 2008, 209, 220–224. [Google Scholar] [CrossRef]
- Nicolas, J.; Dire, C.; Mueller, L.; Belleney, J.; Charleux, B.; Marque, S.R.A.; Bertin, D.; Magnet, S.; Couvreur, L. Living character of polymer chains prepared via nitroxide-mediated controlled free-radical polymerization of methyl methacrylate in the presence of a small amount of styrene at low temperature. Macromolecules 2006, 39, 8274–8282. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Gigmes, D.; Marque, S.R.A.; Tordo, P.; Bertin, D. Nitroxide-mediated polymerization of methyl methacrylate using an SG1-based alkoxyamine: How the penultimate effect could lead to uncontrolled and unliving polymerization. Macromol. Chem. Phys. 2006, 207, 1278–1288. [Google Scholar] [CrossRef]
- Li, L.C.; Hamer, G.K.; Georges, M.K. A quantitative H-1 NMR method for the determination of alkoxyamine dissociation rate constants in stable free radical polymerization. Application to styrene dimer alkoxyamines. Macromolecules 2006, 39, 9201–9207. [Google Scholar] [CrossRef]
- Configuration and conformation of the S,S diastereoisomer was determined by X-ray analysis. See Reference 13.
- Bon, S.A.F.; Chambard, G.; German, A.L. Nitroxide-mediated living radical polymerization: Determination of the rate coefficient for alkoxyamine C–O bond homolysis by quantitative ESR. Macromolecules 1999, 32, 8269. [Google Scholar] [CrossRef]
- Goto, A.; Fukuda, T. Kinetic study on nitroxide-mediated free radical polymerization of tert-butyl acrylate. Macromolecules 1999, 32, 618–623. [Google Scholar] [CrossRef]
- Guillaneuf, Y.; Gigmes, D.; Marque, S.R.A.; Tordo, P.; Bertin, D. First effective nitroxide-mediated polymerization of methyl methacrylate. Macromolecules 2007, 40, 3108–3114. [Google Scholar] [CrossRef]
- Edeleva, M.V.; Kirilyuk, I.A.; Zubenko, D.P.; Zhurko, I.F.; Marque, S.R.A.; Gigmes, D.; Guillaneuf, Y.; Bagryanskaya, E.G. Kinetic Study of H-atom Transfers in Imidazoline-, Imidazolidine-, and Pyrrolidine-Based Alkoxyamines: Consequences for Nitroxide Mediated Polymerization. J. Polym. Sci. Polym. Chem. A 2009, 47, 6579–6595. [Google Scholar] [CrossRef]
- Beaudoin, E.; Bertin, D.; Gigmes, D.; Marque, S.R.A.; Siri, D.; Tordo, P. Alkoxyamine C–ON bond homolysis: Stereoelectronic effects. Eur. J. Org. Chem. 2006, 7, 1755–1768. [Google Scholar] [CrossRef]
- Bertin, D.; Gigmes, D.; Marque, S.R.A.; Siri, D.; Tordo, P.; Trappo, G. Effect of the carboxylate salt on the C–ON bond homolysis of SG1-based alkoxyamines. Chem. Phys. Chem. 2008, 9, 272–281. [Google Scholar] [PubMed]
- Marque, S.R.A.; Siri, D. Is Experimental Evidence Sufficient Enough To Account for the Stabilization Effect of Bisnitroxide on the Fate of NMP Experiments? Macromolecules 2009, 42, 1404–1406. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Sofware for calculations: Gaussian 03; Revision C.02. Gaussian, Inc.: Wallingford, CT, USA, 2004. Available online: http:/www.gaussian.com (accessed on 21 September 2010).
- It is well known that geometry optimization performed by DFT with the B3LYP method provides geometries as reliable as those obtained with the ab initio MP2 methods. See: Koch, W.; Holthausen, M.C. A Chemist's Guide to DFT, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2001; Chapter 8; pp. 119–136. [Google Scholar]
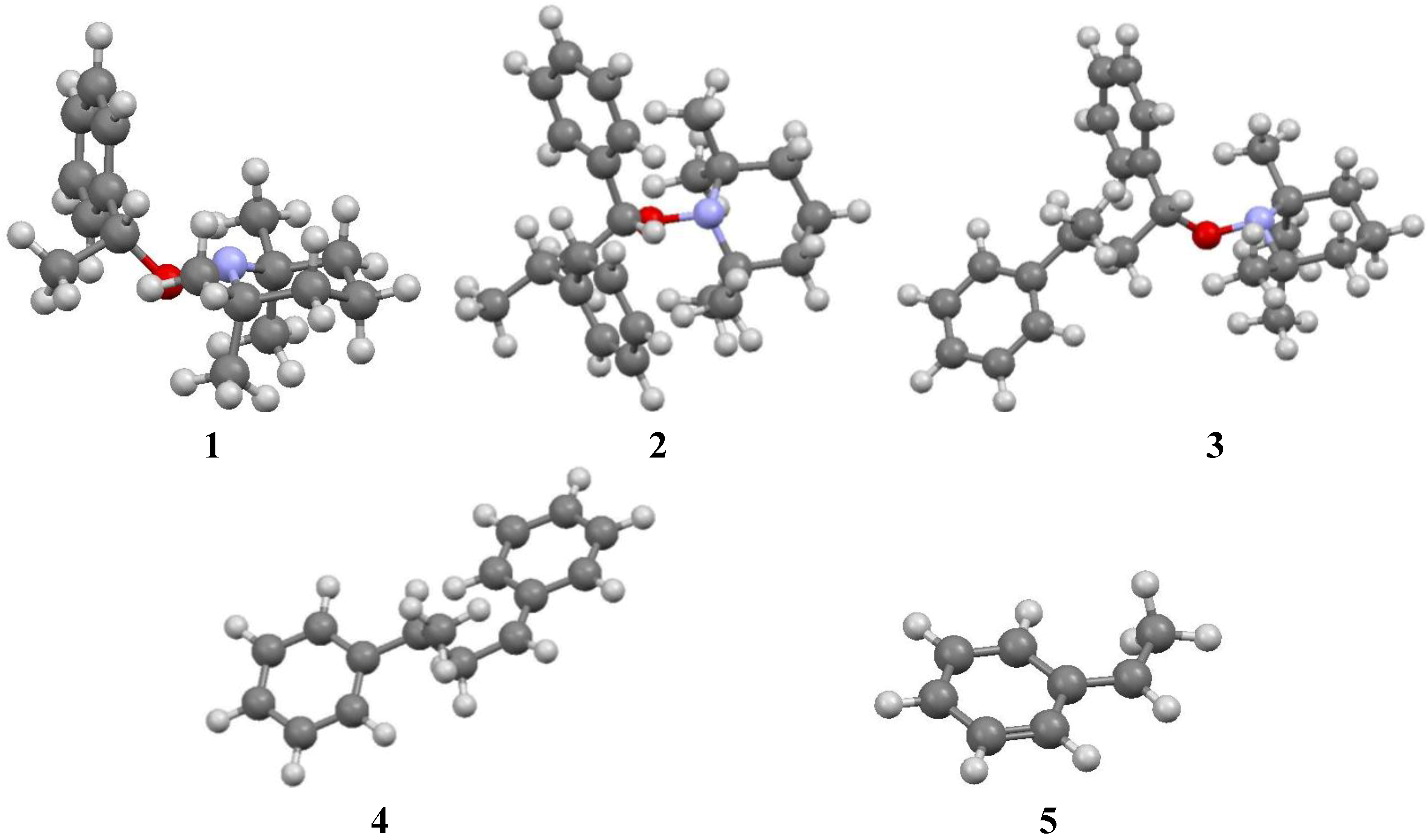
- Calculations were started from conformations different from those displayed in Figure 1 and Figure 2. In each case, conformations displayed in Figure 1 and Figure 2 appeared as the most stable.
- The B3P86 method was chosen to calculate energies because it is known to provide the most accurate values. See: Feng, Y.; Liu, L.; Wang, J.-T.; Huang, H.; Guo, Q.-X. Assessment of Experimental Bond Dissociation Energies Using Composite ab Initio Methods and Evaluation of the Performances of Density Functional Methods in the Calculations of Bond Dissociation Energies. J. Chem. Inf. Comput. Sci. 2003, 4, 2005–2013. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F.A. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E. Alkoxyamine-initiated living radical polymerization: Factors affecting alkoxyamine homolysis rates. Macromolecules 1995, 28, 8722–8728. [Google Scholar] [CrossRef]
- Lagrille, O.; Cameron, N.R.; Lovell, P.A.; Blanchard, R.; Goeta, A.E.; Koch, R. Novel acyclic nitroxides for nitroxide-mediated polymerization: Kinetic, electron paramagnetic resonance spectroscopic, X-ray diffraction, and molecular modeling investigations. J. Polym. Sci. Polym. Chem. 2006, 44, 1926–1940. [Google Scholar] [CrossRef]
- TS involving large molecules and conformational changes are time-consuming and often difficult to determine, especially when the difference between TS is expected to be small. Thus, we preferred to discuss our results applying the Hammond postulate.
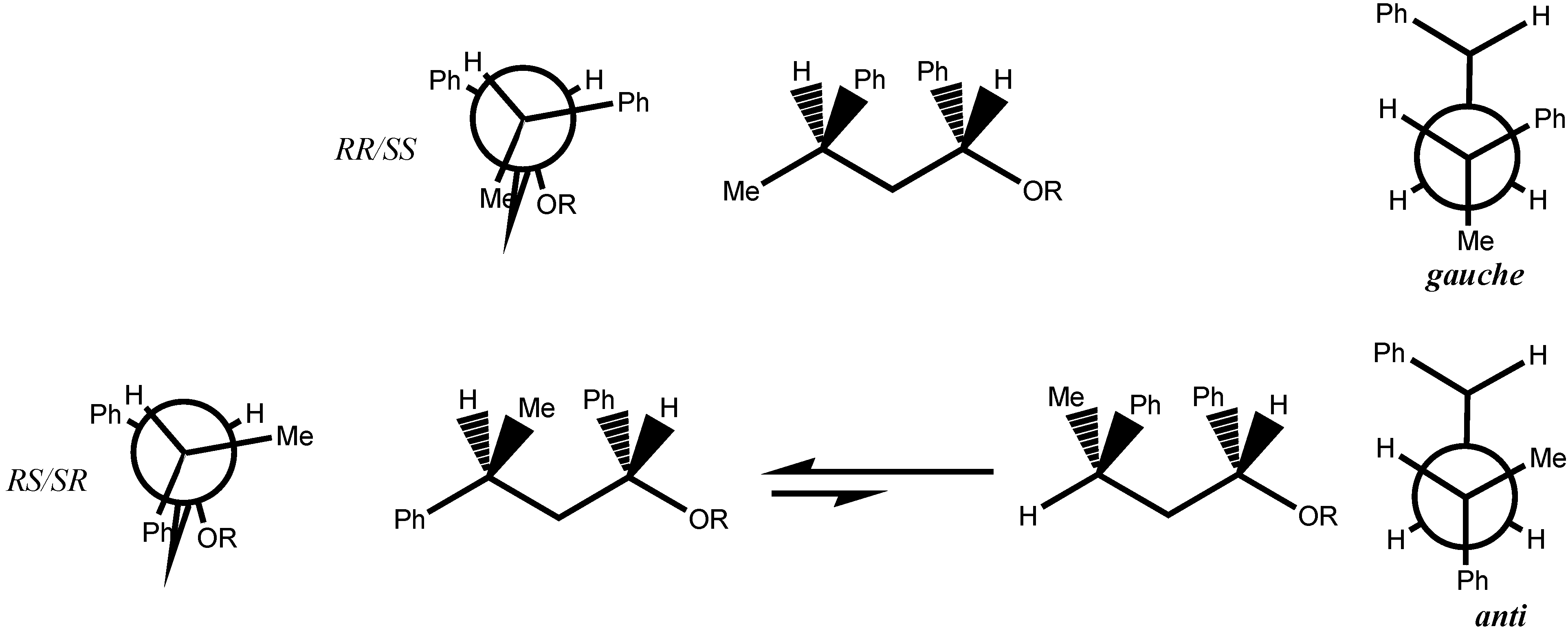
- At first glance, it was not obvious that the phenyl group was bulkier than the methyl group because it is known as a Janus group whose size can be much smaller or much larger than that of the methyl group.
- The conformation calculated for the enantiomer RR of 2 correspond to the conformation reported for the SS enantiomer of 7 [13]. It was assumed that the conformation given by the X-ray data was the most stable.
- As TS were not calculated [29], the activation barriers ΔG≠ were estimated with the Arrhenius parameters reported in [13]. As the cross-coupling rate constants to yield the unimeric (kc = 2.2 × 108 l·mol−1·s−1) and polymeric (kc = 1.8 × 108 l·mol−1·s−1) species at 20 °C were very close ( Guillaneuf, Y.; Bertin, D.; Castignolles, P.; Charleux, B. New experimental procedure to determine the recombination rate constants between nitroxides and macroradicals. Macromolecules 2005, 38, 4638–4646. [Google Scholar]), ΔG≠ for 2 and 3 to reach TS from the products were likely very close to the one reported for the formation of 1 (ΔG≠ = 28.6 kJ·mol−1, A = 4.0 × 1011 l·mol−1·s−1, Ea = 15.5 kJ·mol−1, Sobek, J.; Martschke, R.; Fischer, H. Entropy control of the cross-reaction between carbon-centered and nitroxide radicals. J. Am. Chem. Soc. 2001, 123, 2849–2857. [Google Scholar] ).
- It was assumed that the contribution of the entropic term to the total energy of the molecule was negligible to the enthalpic term, and thus, ΔGf ≈ ΔHf.
- Although PhCOO group is larger than H atom it should not generate larger strain than the methyl group of the model 2/3 due to its conformation in the same plane than the backbone as given by the X-ray structure (see Reference 13). The effect of the polarity of PhCOO group (σI,AcOCH2 = 0.15) on the reactive center is buffered by the sequence CH2CPhHCH2.
- Marque, S.; Le Mercier, C.; Tordo, P.; Fischer, H. Factors influencing the C–O– bond homolysis of trialkylhydroxylamines. Macromolecules 2000, 33, 4403–4410. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Blachon, A.; Marque, S.R.A.; Roubaud, V.; Siri, D. Diasteromeric Effect on the Homolysis of the C–ON Bond in Alkoxyamines: A DFT Investigation of 1,3-Diphenylbutyl-TEMPO. Polymers 2010, 2, 353-363. https://doi.org/10.3390/polym2030353
Blachon A, Marque SRA, Roubaud V, Siri D. Diasteromeric Effect on the Homolysis of the C–ON Bond in Alkoxyamines: A DFT Investigation of 1,3-Diphenylbutyl-TEMPO. Polymers. 2010; 2(3):353-363. https://doi.org/10.3390/polym2030353
Chicago/Turabian StyleBlachon, Alexandra, Sylvain R. A. Marque, Valérie Roubaud, and Didier Siri. 2010. "Diasteromeric Effect on the Homolysis of the C–ON Bond in Alkoxyamines: A DFT Investigation of 1,3-Diphenylbutyl-TEMPO" Polymers 2, no. 3: 353-363. https://doi.org/10.3390/polym2030353