3.1. Polymer Properties of Plasticized Polyhydroxyalkanoates (PHA) Pellets
The pre-plasticized PHB pellet showed poor processability because of stiffness and low solubility. Copolymerization with the biodegradable polyesters, PBS and PLA, improved the mechanical properties of PHA. According to the catalog specification values, copolymerization with PBS increased elongation and Izod impact strength, while copolymerization with PLA increased melt flow rate and flexural modulus. Actually, when installing a sample into the solid-state NMR rotor, the pre-plasticized PHB was difficult to cut and/or crush, even though the plasticized PHA copolymers were easily deformable to prepare a solid-state NMR sample.
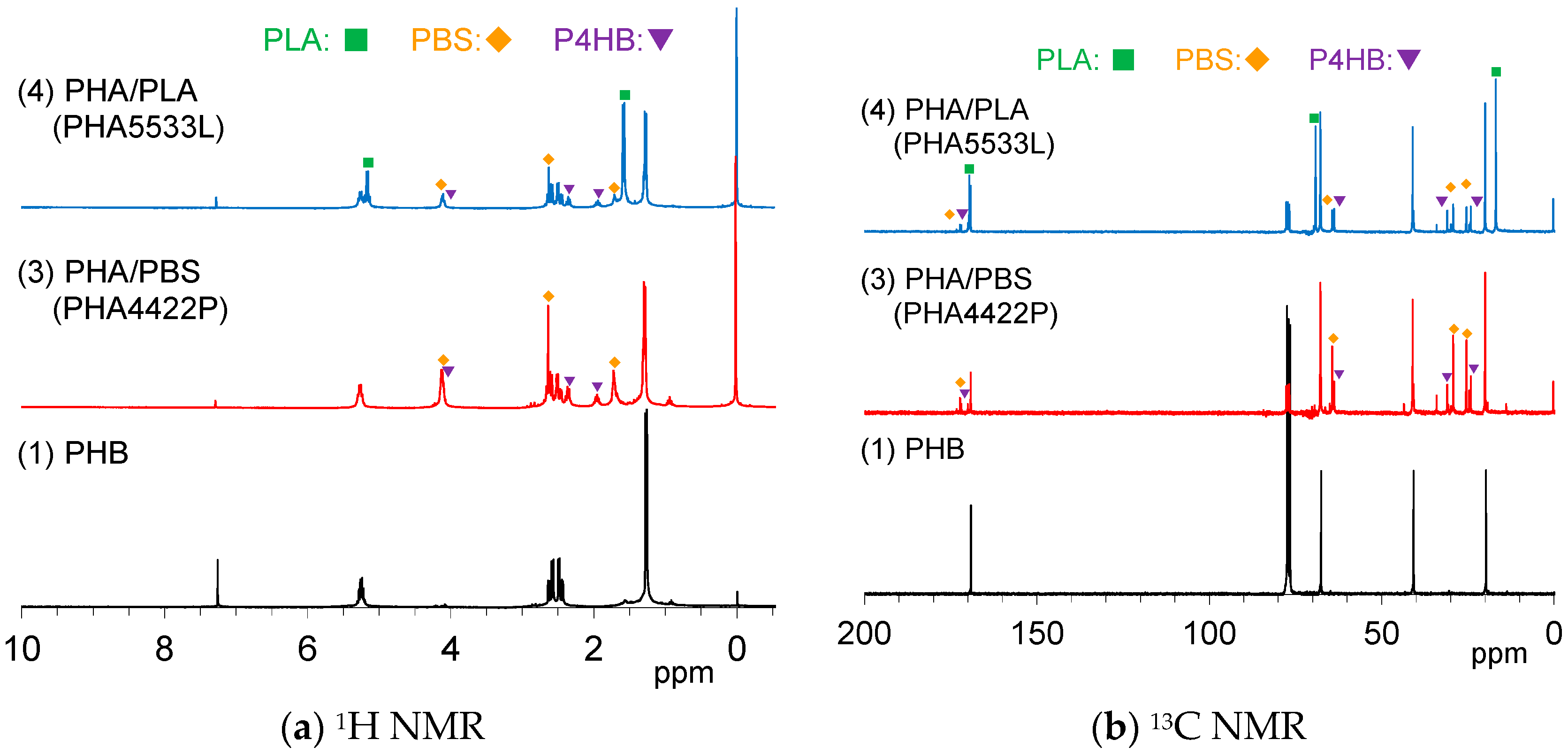
In order to determine in detail the constituents of the plasticized PHA pellets connected with the mechanical properties, solution NMR spectra were measured in CDCl
3.
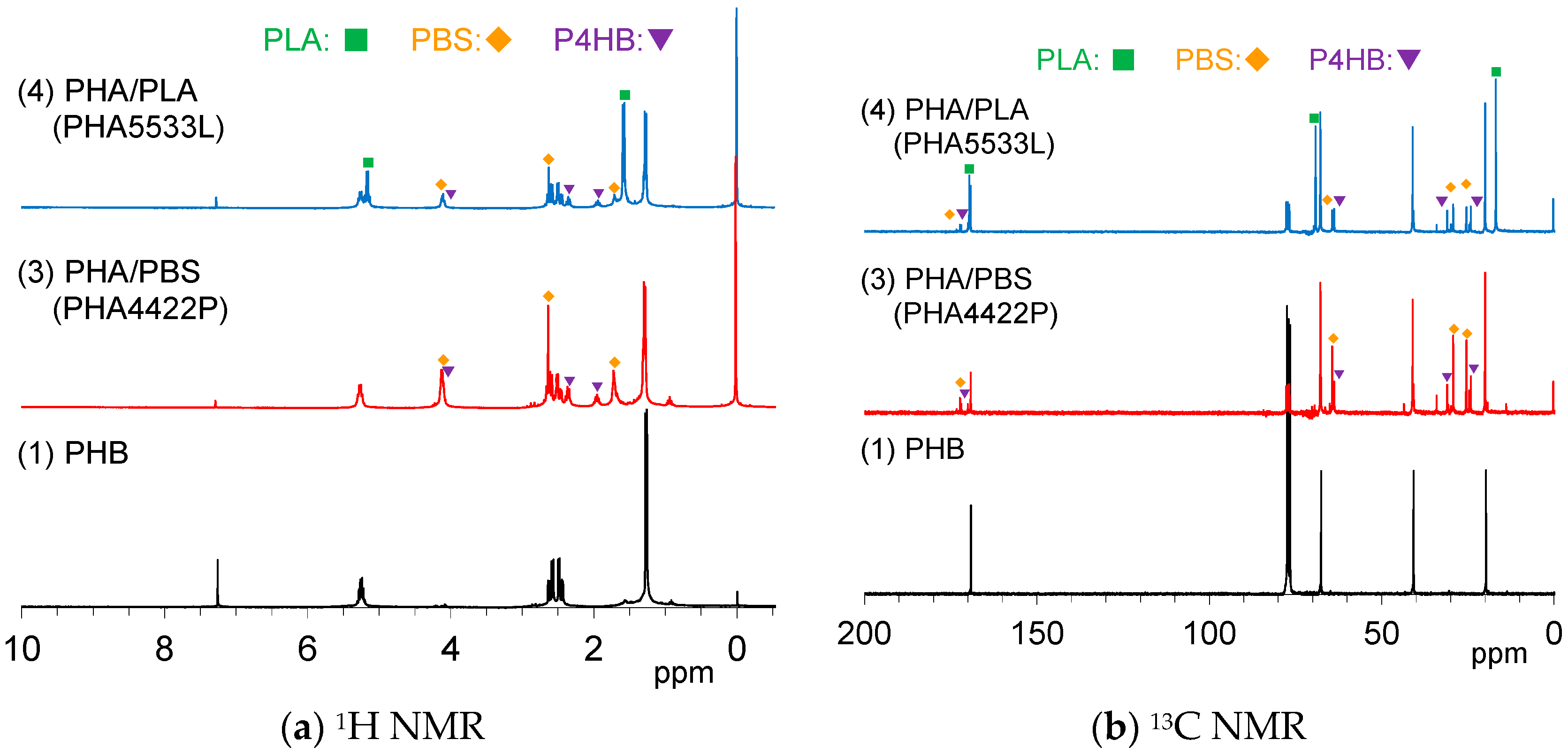
Figure 1a shows
1H NMR spectra of the pre-plasticized PHB and the plasticized PHA pellets. The PHA4422P pellet (3) showed three PHB signals, doublet (CH
3, 1.28 ppm), quartet-doublet (CH
2, 5.24 ppm), and quartet (CH, 5.26 ppm), which can be also observed for the original PHB (1). PHA4422P also had small amount of poly(4-hydrobutyrate) (P4HB) [
29], and the added PBS for the copolymerization. One can see a trace peak at 0.98 ppm, in which region a CH
3 group of PHA having long alkyl groups might appear; however, the actual structure could not be assigned at this time. Meanwhile,
1H NMR spectra also indicated that the PHA5533L pellet (4) consisted of PHB, the added PLA, and a small amount of PBS and P4HB. Minor constituents in each pellet could be confirmed by
13C NMR spectra (
Figure 1b). The signals of P4HB in both PHA4422P (3) and PHA5533L (4) also appeared not only in the aliphatic region but also in the carbonyl region.
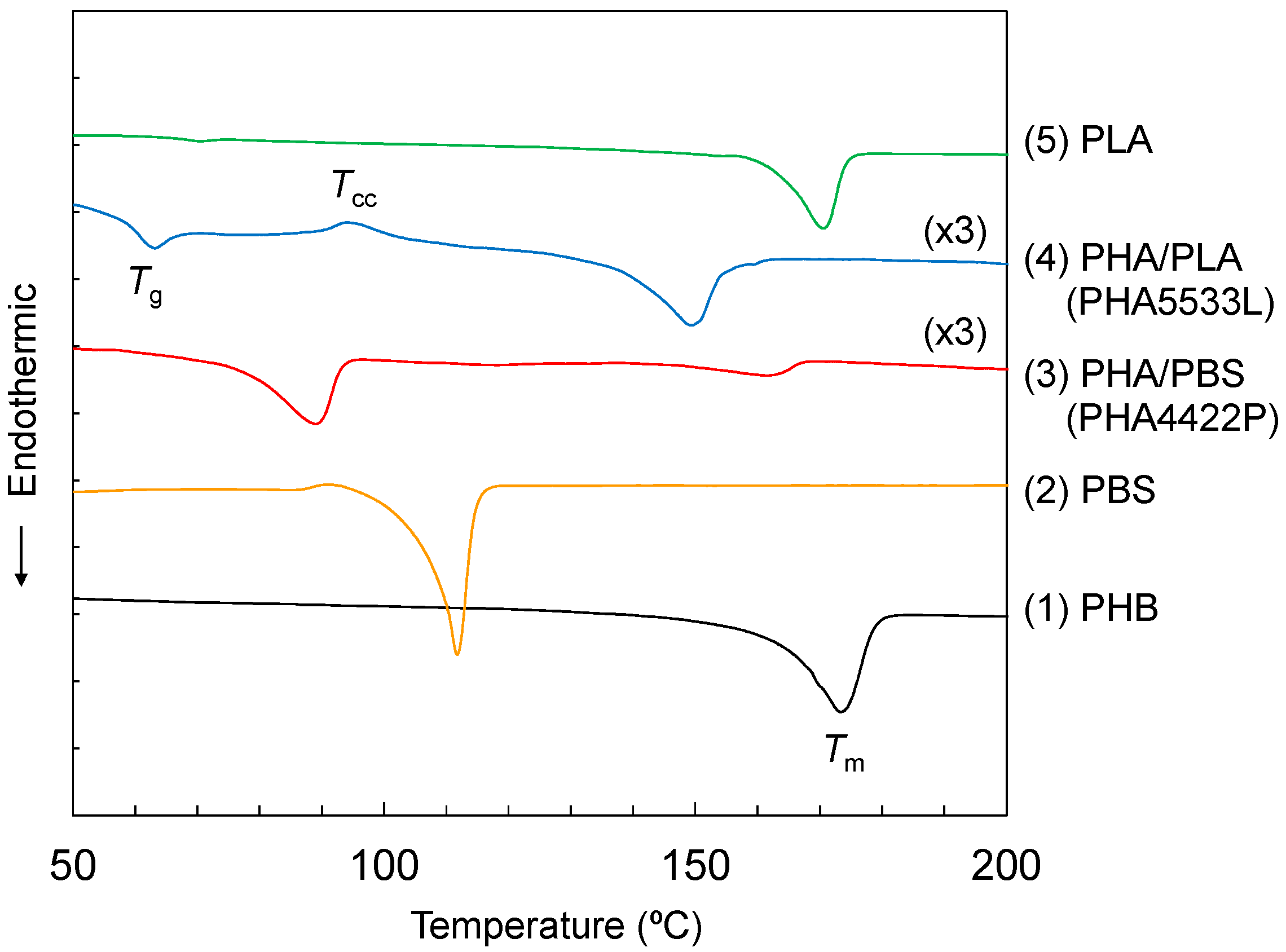
The improvement of mechanical properties due to the copolymerization with the biodegradable polyesters could be explained by a change of thermal characteristics.
Figure 2 shows the DSC curves for the modified PHA pellets and the reference polymers. The melting peak (
Tm) of the original PHB (1) appeared at a relatively high temperature (174 °C) with no other endothermic and exothermic peaks. Of the constituent polyesters used for the copolymerization, PBS (2) had a lower
Tm (112 °C) while PLA (5) also had a high
Tm (171 °C). Interestingly, the copolymerization resulted in a decrease of
Tm to a value lower than the initial
Tm of the constituent polyesters, although the heat capacities of both plasticized PHA pellets were smaller than the original PHB. The PHA4422P pellet (3) had a lower
Tm (89 °C) with higher temperature small endothermic peaks. In contrast, the PHA5533L pellet (4) had a glass transition peak with relaxation (
Tg: 64 °C) and cold crystallization (
Tcc: 95 °C), and a quite high
Tm (150 °C). Considering the DSC results together with the solution NMR, small amounts of P4HB and PBS were extremely effective for changing the thermal properties of the PHA5533L pellet. Moreover, the DSC results showed that the plasticization of PHA improved processability because of the decrease of
Tm (PBS) or
Tg (PLA).
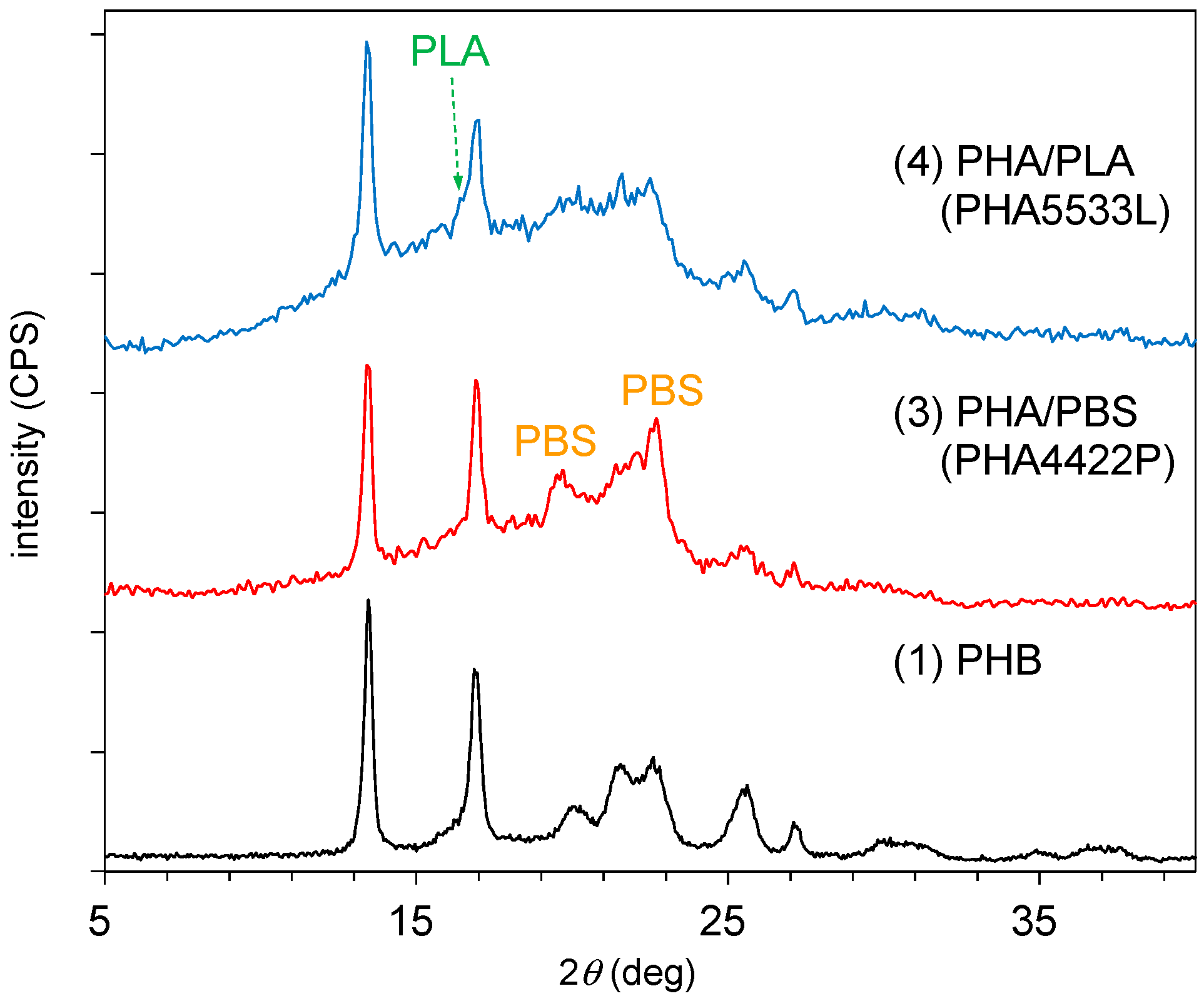
The copolymerization with the biodegradable polyesters also changed the crystallinity of the PHA.
Figure 3 shows changes in the XRD patterns of PHA by the addition of other polyesters. The original PHB (1) had moderate crystallinity, showing orthorhombic crystal planes with major sharp peaks at 2θ = 13.5° (020) and 16.9° (110). After the copolymerization of PHA with PBS [PHA4422P (3)], new peaks that originated from PBS appeared at 2θ = 19.6° (020) and 22.6° (110) overlapped with the (021) and (111) peaks of PBS [
30]. At the same time, the amorphous peak of PHA clearly increased. The copolymerization of PHA with PLA [PHA5533L (4)] increased further the amorphous peak of PHA; only a small peak of PLA was observed at 2θ = 16.4° (100) [
24]. In both cases of PHA4422P and PHA5533L, characteristic XRD peaks from P4HB could not be assigned. The XRD results showed that the copolymerization of both biodegradable polymers decreased the crystallinity of PHA and that the decrease was more pronounced for the copolymerization with PLA than with PBS.
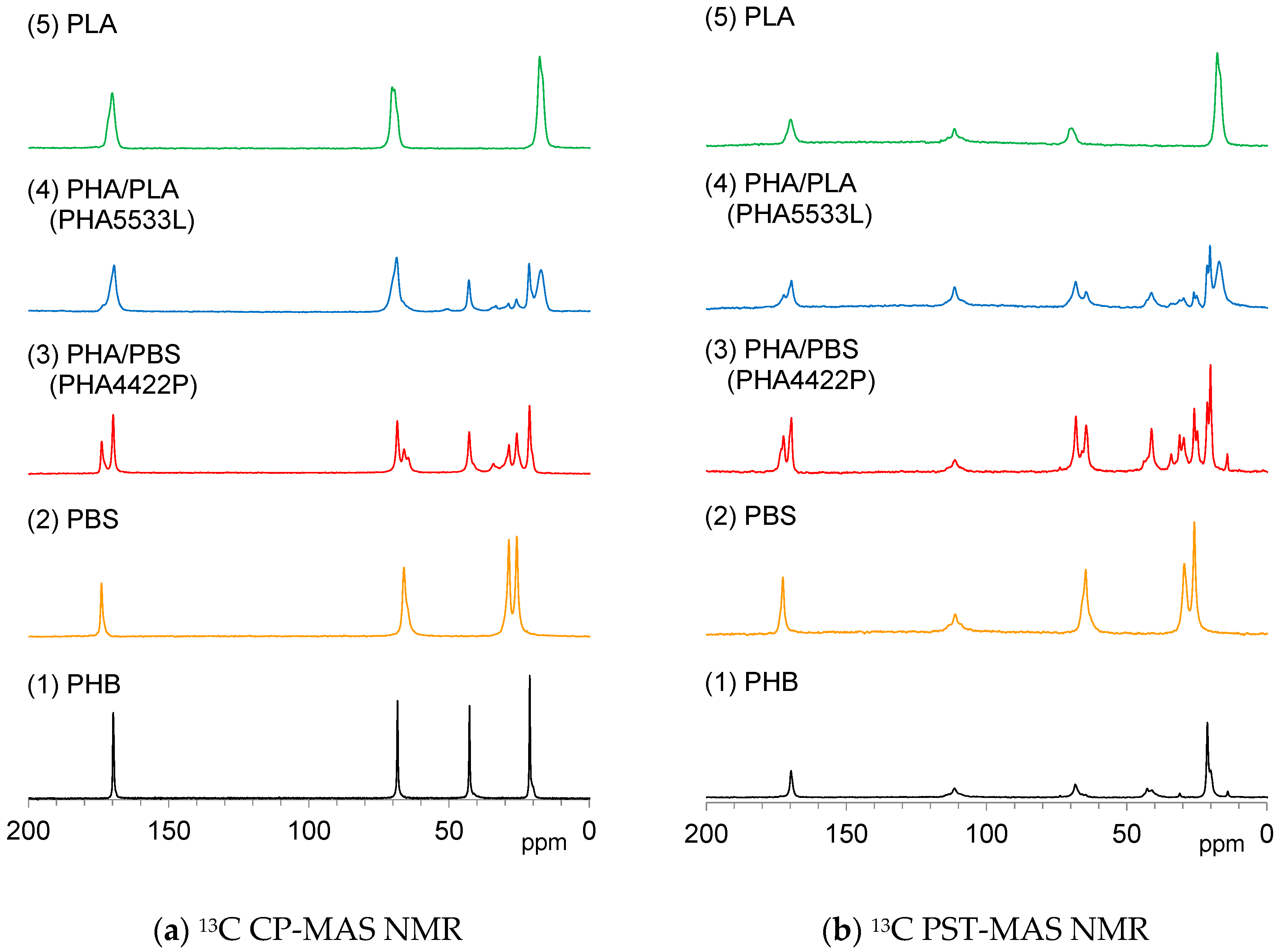
3.2. 13C Magic-Angle Spinning (MAS) NMR of Plasticized PHA Copolymers and Constituent Polymers
Solid-state
13C NMR is a powerful tool for analyzing the crystallinity and mobility of polymer chains. The
13C CP-MAS NMR variant is commonly used in polymer science; it utilizes the magnetization transfer from
1H nuclei to
13C nuclei, enhancing the signal of a rigid substituent.
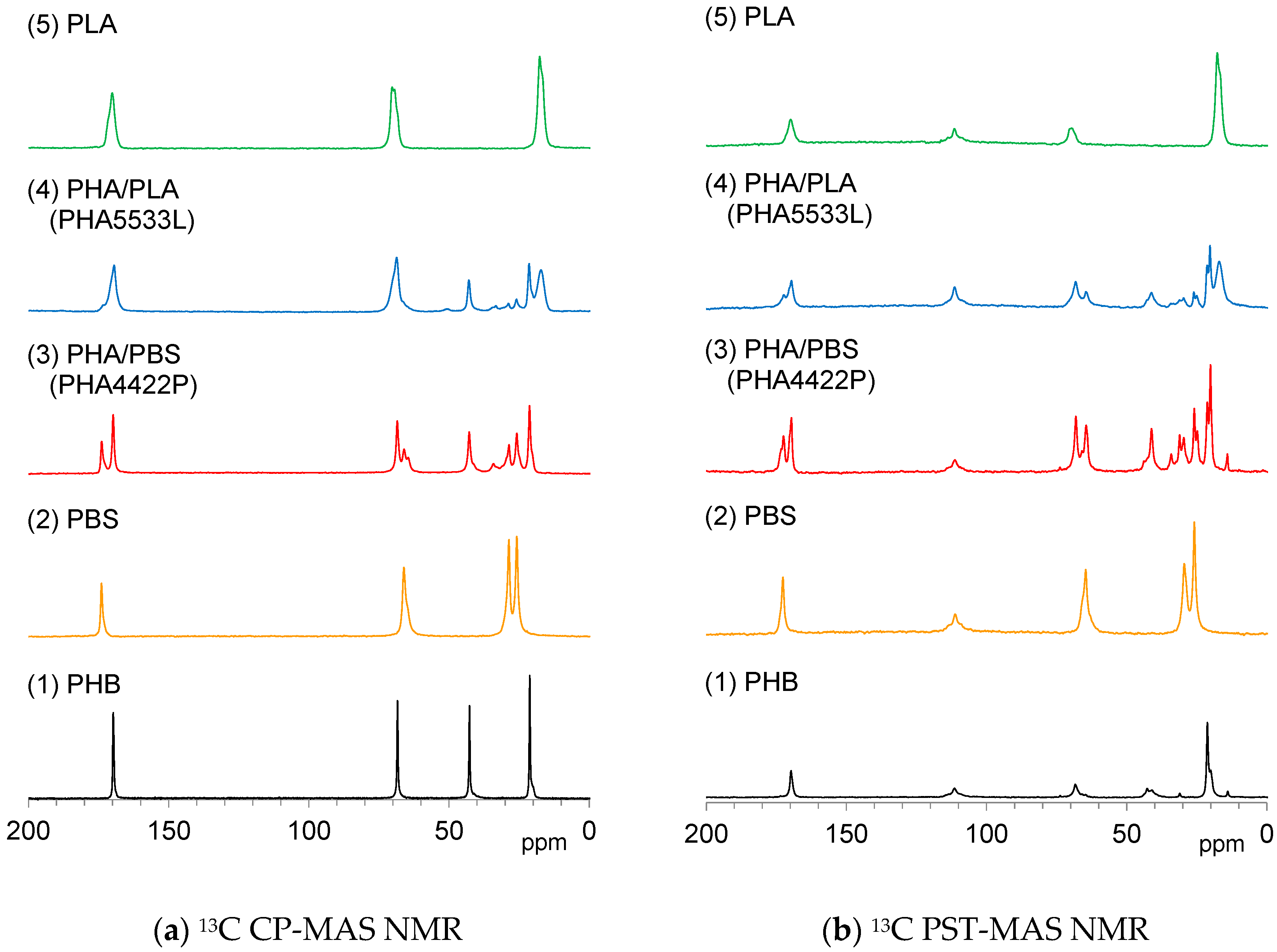
Figure 4a shows
13C CP-MAS NMR spectra of plasticized PHA copolymers and constituent polymers. Each signal for the polymer constituents, PHB (1), PBS (2) and PLA (5) could be effectively detected in the
13C CP-MAS NMR spectra regardless of the flexibility and the rigidity of each polymer. The
13C CP-MAS signals of the plasticized PHA4422P (3) could be assigned as PHB and PBS, while those of the plasticized PHA5533L (4) as PHB, PLA, and PBS. The isomeric polymer P4HB could be also detected in both PHA4422P (3) and PHA5533L (4).
Meanwhile,
13C PST-MAS NMR is a method utilizing the nuclear Overhauser effect, resulting in the enhancement of a signal of a substituent having high mobility near a hydrogen atom [
27].
Figure 4b shows
13C PST-MAS NMR spectra of plasticized PHA copolymers and constituent polymers. Actually, the CH
3 group of PLA (5) and the OCH
2CH
2 group of PBS (2) showed larger intensities than other substituents because the amplitudes of their molecular motions were higher. For the pre-plasticized PHB (1), the CH
3 group had a high signal intensity compared to the other signals. Interestingly, the copolymerization of PHA with PBS [PHA4422P (3)] increased the signal from CH
2 and CH
3 groups accompanied by the appearance of signals from the minor isomeric polymer P4HB. In both PHA (1) and PHA4422P (3), trace signals appeared at the higher field near the CH
3 groups’ signal. These small signals originated from trace amounts of PHA having long alkyl groups. In contrast, the copolymerization of PHA with PLA [PHA5533L (4)] did not increase the CH
2 and CH
3 groups’ signals but only gave the sum of the signals of PHA, P4HB, PLA, and PBS. The results of the
13C PST-MAS NMR spectra confirmed that PBS enhanced molecular mobility of the whole copolymer system. Next, we consider the different trends in molecular mobility between the plasticization with PBS [PHA4422P (3)] and PLA [PHA5533L (4)] revealed by their spin-lattice relaxation times.
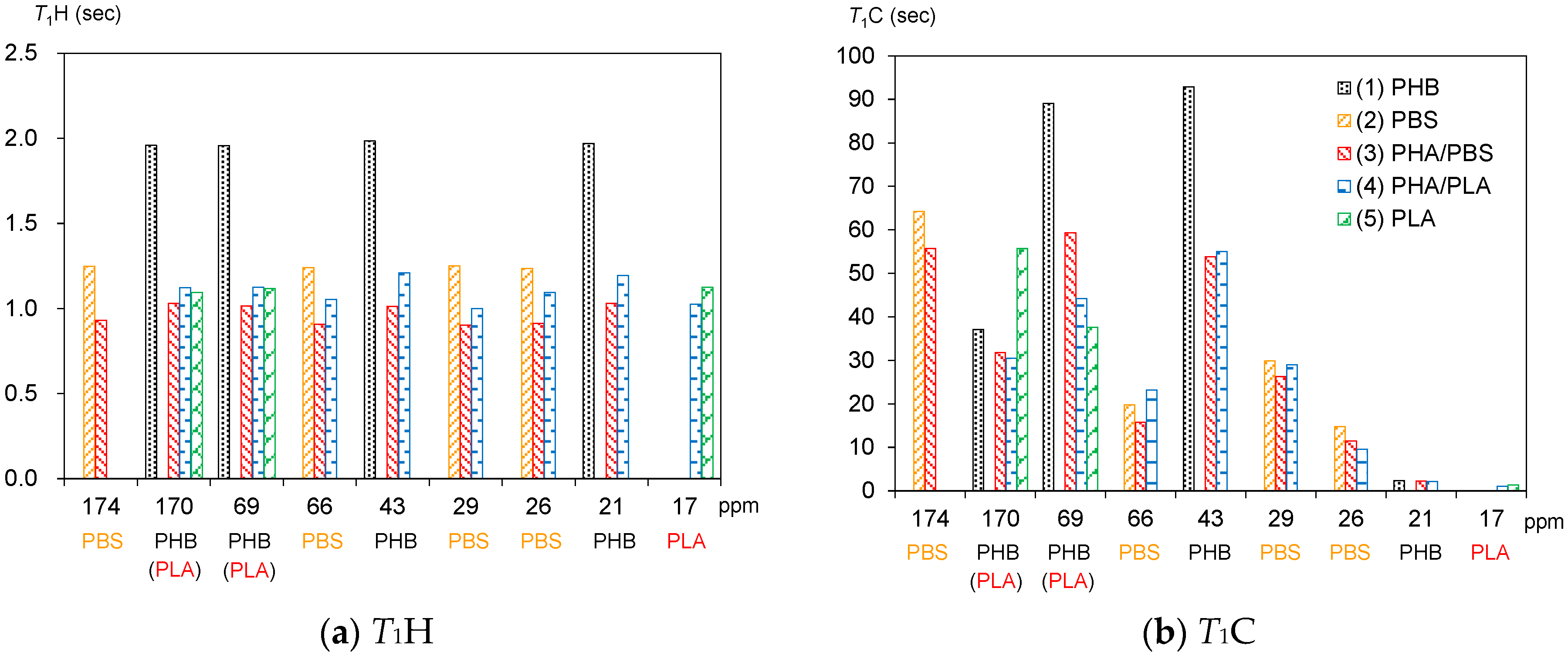
3.3. Spin-Lattice Relaxation Times in the Laboratory Frame of Plasticized PHA Copolymers and Constituent Polymers
The 1H spin-lattice relaxation time in the laboratory frame (T1H) is reduced by rapid molecular motions matched to the Larmor frequency. For example, the 1H spin-lattice relaxation in the laboratory frame (T1H relaxation) of PLA is promoted by the rotation of a CH3 group around its C3 axis. Moreover, the T1H value of each substituent in the same polymer chain adopts an averaged value because of 1H spin diffusion.
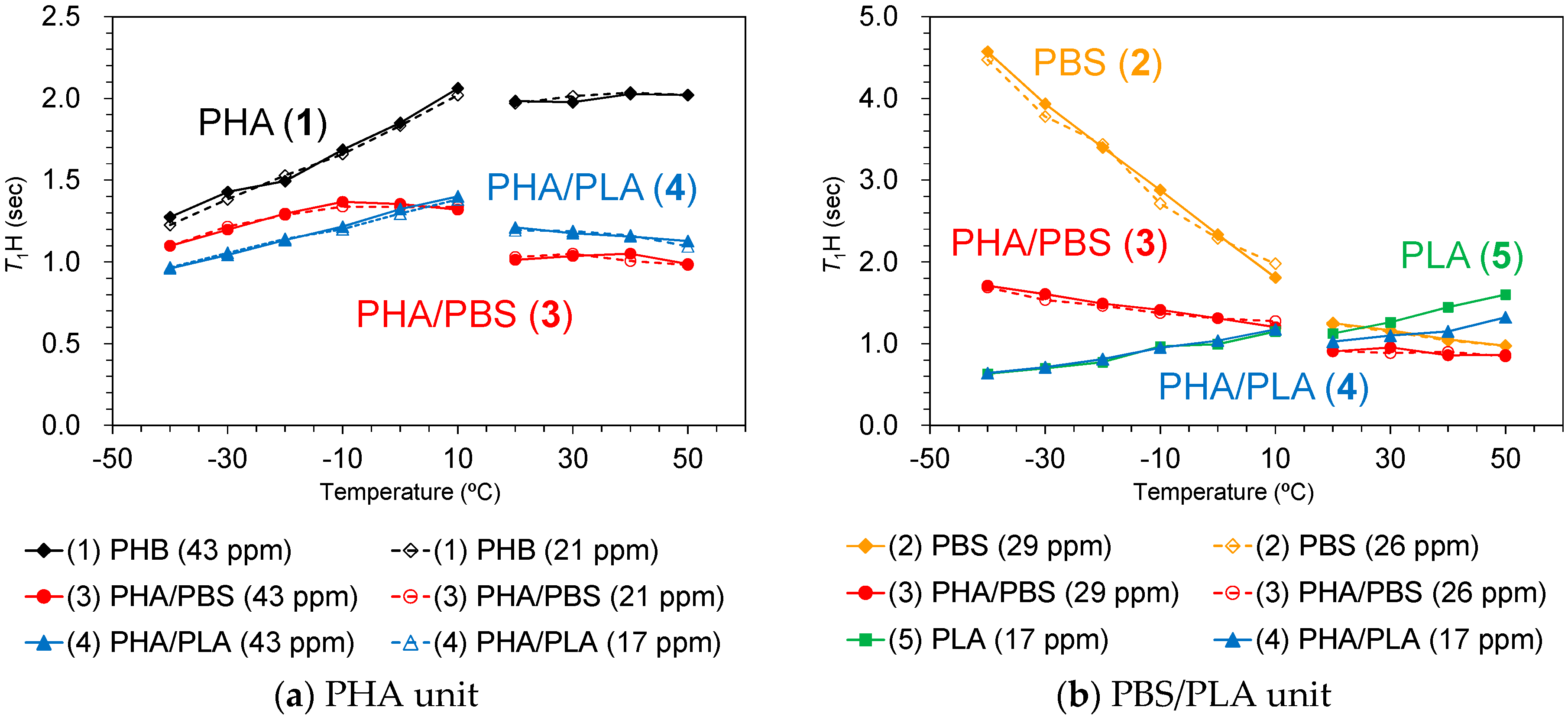
Figure 5a shows
T1H values of plasticized PHA copolymers and constituent polymers. As described in the above paragraph, the
T1H values of PLA (5) were averaged by the spin diffusion to have almost the same values (1.1 s) due to the
T1H relaxation via the rotation of the CH
3 group. Although the
T1H values of PBS (2) also showed averaged values (1.2 s) similar to PLA, the
T1H relaxation of PBS cannot be explained directly by molecular motions of a specific substituent, such as a CH
3 group. Moreover, the
T1H values of PHB (1) had longer averaged values (2.0 s) even though the
T1H relaxation occurred via the motion of CH
3 group. The different
T1H values of PLA (5) and PHB (1) having the same
T1H relaxation route were due to whether the frequency of CH
3 motion matched the Larmor frequency, or not.
Copolymerization with the biodegradable polyesters significantly decreased the
T1H values of PHB. For the plasticized PHA4422P (3) in particular, the addition of PBS decreased not only the
T1H values of the PHB unit (1.0 s) but also the
T1H values of the PBS unit (0.9 s). Meanwhile, for the plasticized PHA5533L (4), the addition of PLA also decreased the
T1H values of the PHB unit (1.2 s), although the
T1H values of the PLA unit kept the original values of the PLA pellet (1.05 s). At the same time, the
T1H values of trace amounts of the PBS unit slightly decreased (1.0 s) in PHA5533L. These
T1H reductions were caused by the enhancement of the
T1H relaxation due to the heterogeneity that arose from the amorphous moiety, as we have previously described for PLA copolymer fibers [
24].
The
13C spin-lattice relaxation time in the laboratory frame (
T1C) can provide information on molecular motions for each substituent more directly because it excludes the effect of
1H spin diffusion.
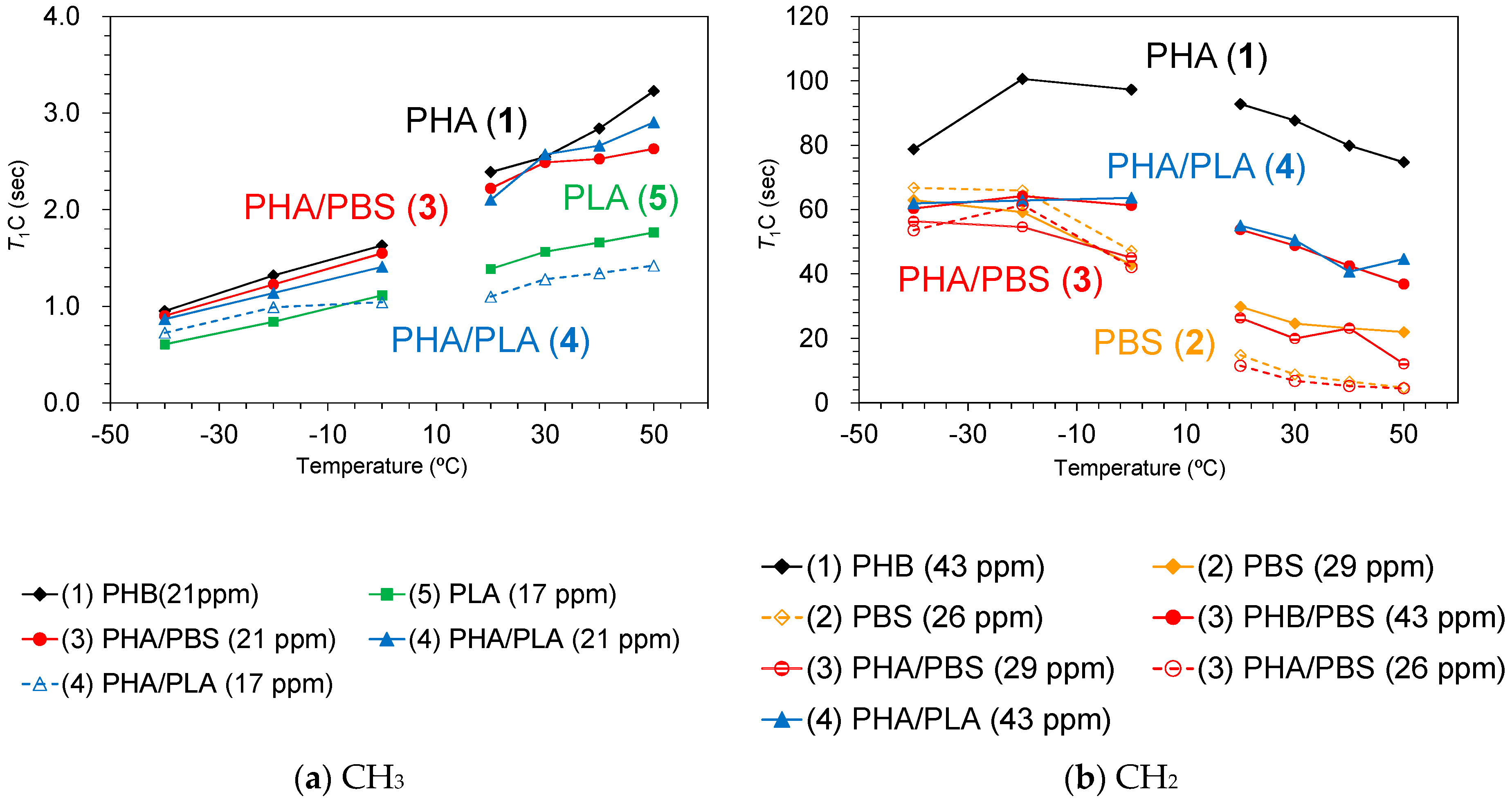
Figure 5b shows
T1C values of plasticized PHA copolymers and constituent polymers. One can see a different
T1C value for each substituent in the pre-plasticized PHB (1): C=O, 37 s; CH, 89 s; CH
2, 92 s; CH
3, 2.4 s. The
T1C value of CH
3 nearly equaled the
T1H (2.0 s) value because the
13C spin-lattice relaxation (
T1C relaxation) was also promoted by the CH
3 rotation, the same as for the
T1H relaxation. This closeness between the
T1C relaxation of CH
3 and the
T1H due to the CH
3 rotation also arose in PLA (5) (
T1C, 1.4 s;
T1H, 1.1); however, the C=O group (56 s) showed a longer value than the CH group (38 s) in PLA, unlike the pre-plasticized PHB. One can also see the longer
T1C value of the C=O group (64 s) and moderate
T1C values of CH
2 groups (15–30 s) in PBS (2). Although PBS had high flexibility, the molecular motion of each substituent did not match with the Larmor frequency of the
13C nuclei.
The reduction of
T1C values due to the copolymerization was also observed not only in PHA4422P (3) but also in PHA5533L (4), similarly to the
T1H values. The
T1C reduction of the PHB unit was significant for C(=O) CH
2 [original 89 s; 4422(PBS), 59 s; 5533(PLA), 44 s] and OCH
2 [original, 93 s; 4422(PBS), 54 s; 5533(PLA), 55 s]. In contrast, the
T1C value of the C=O of the PBS unit changed less [original, 37 s; 4422(PBS), 30 s; 5533(PLA), 32 s] as well as the
T1C of CH
3 [original, 2.4 s; 4422(PBS), 2.2 s; 5533(PLA), 2.1 s]. Moreover, the
T1C values of the PBS unit in PHA4422P (3) and the
T1C values of the PLA unit CH
3 in PHA5533L (4) were slightly reduced by the copolymerization, compared with the original
T1C values. Since the copolymerization with either PBS or PLA decreased the crystallinity of the PHB unit (
Figure 3), the reduction of the
T1C values of the PHB unit could be also explained by a change of
T1C relaxation due to the heterogeneity arising from the amorphous moiety. We will discuss the relationship of the heterogeneity with the compatibility of the copolymer components and molecular mobility later.
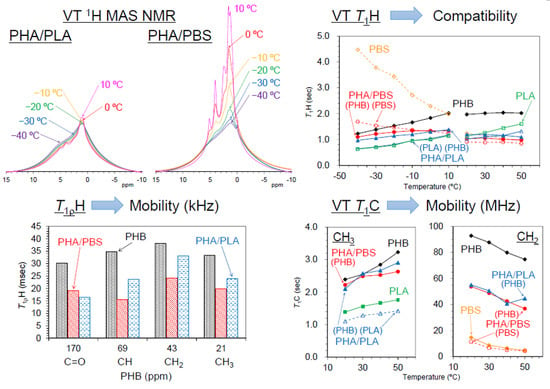
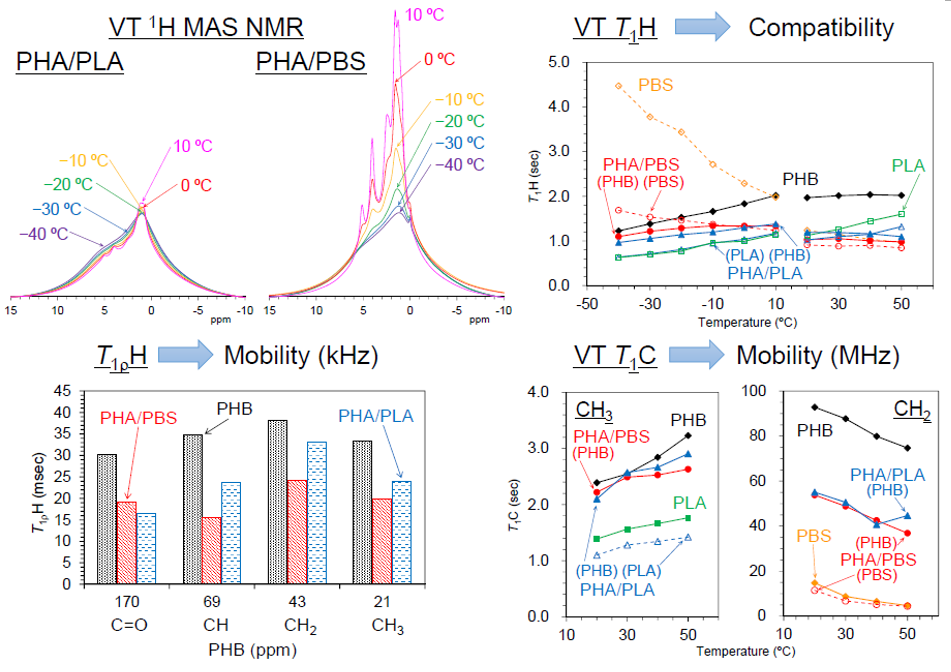
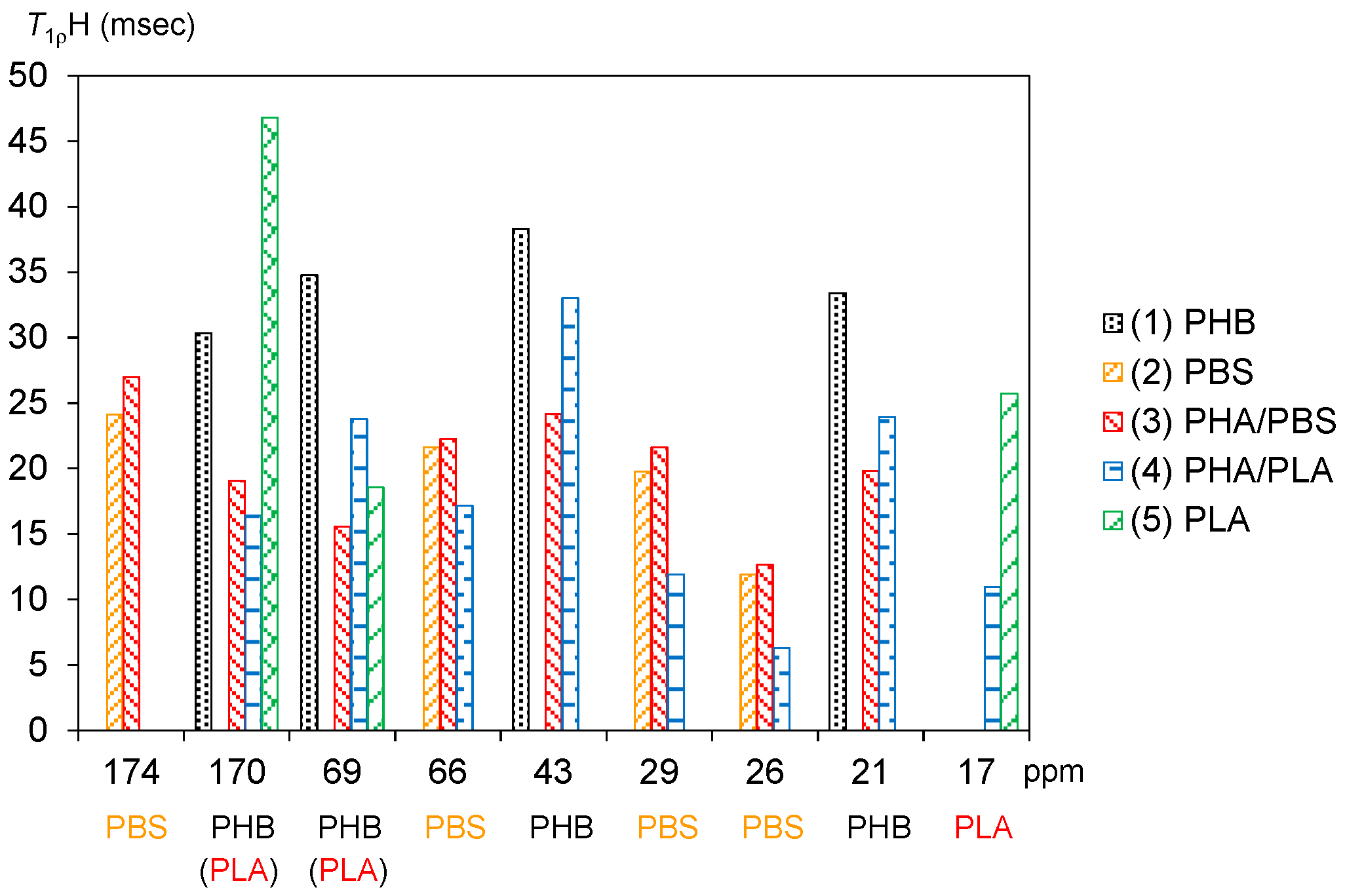
3.4. Spin-Lattice Relaxation Time in the Rotating Frame of Plasticized PHA Copolymers and Constituent Polymers
The 1H spin-lattice relaxation in the rotating frame (T1ρH relaxation) is promoted by molecular motions near the spin-locking frequency, which is much less than the Larmor frequency. That is, the 1H spin-lattice relaxation time in the rotating frame (T1ρH) is affected more than the T1H value by slower molecular motions, such as the main chain motion of polymers.
Figure 6 shows
T1ρH values of plasticized PHA copolymers and constituent polymers. The significant decreases of the
T1H values for PHB due to the copolymerization with PBS and PLA were also observed for the
T1ρH values. The original PHB (1) showed comparable
T1ρH values for each substituent as follows: C=O, 30 ms; CH, 35 ms; CH
2, 38 ms; CH
3, 33 ms. The
T1ρH values of the PHB unit were obviously reduced by the copolymerization with the biodegradable polyesters. Except for the C=O group, the reduction ratios of
T1ρH values were larger in the copolymer with PBS [PHA4422P (3): 16–24 ms] than in the copolymer with PLA [PHA5533L (4): 24–33 ms]. Meanwhile, PBS (2) took lower
T1ρH values, such as C=O (24 ms), OCH
2 (22 ms), C(=O) CH
2 (20 ms), CH
2 (12 ms). The
T1ρH values of PBS unit stayed almost unchanged for PHA4422P (3) (13–27 ms) while they decreased in PHA5533L (4) (6–17 ms). The
T1ρH values of CH
3 in PLA (5) also decreased with the copolymerization (PLA, 26 ms; PHA5533L, 11 ms).
As shown above, since the spin-lattice relaxation times in the laboratory frame (
T1H and
T1C) showed a similar trend between PHA4422P (PBS) and PHA5533L (PLA), rapid molecular motions (MHz) matched with the Larmor frequency in both plasticized PHA copolymers was enhanced by the plasticization. On the other hand, the spin-lattice relaxation time in the rotating frame (
T1ρH) decreased more in PHA4422P. Therefore, the plasticization with the flexible PBS enhanced slow molecular motions (kHz) corresponding to the spin-locking frequency more than did PLA. We previously discussed the correlation of increase of the PST-MAS signals and
T1ρH values for phenol formaldehyde resin-impregnated soft wood [
25]. Similar to this previous result, the increases of the
13C PST-MAS NMR signals here could be explained by the enhancement of slow molecular motions (kHz) due to the plasticization with PBS. We will discuss the increase of
1H MAS NMR signals related to the slow molecular motion in the next subsection.
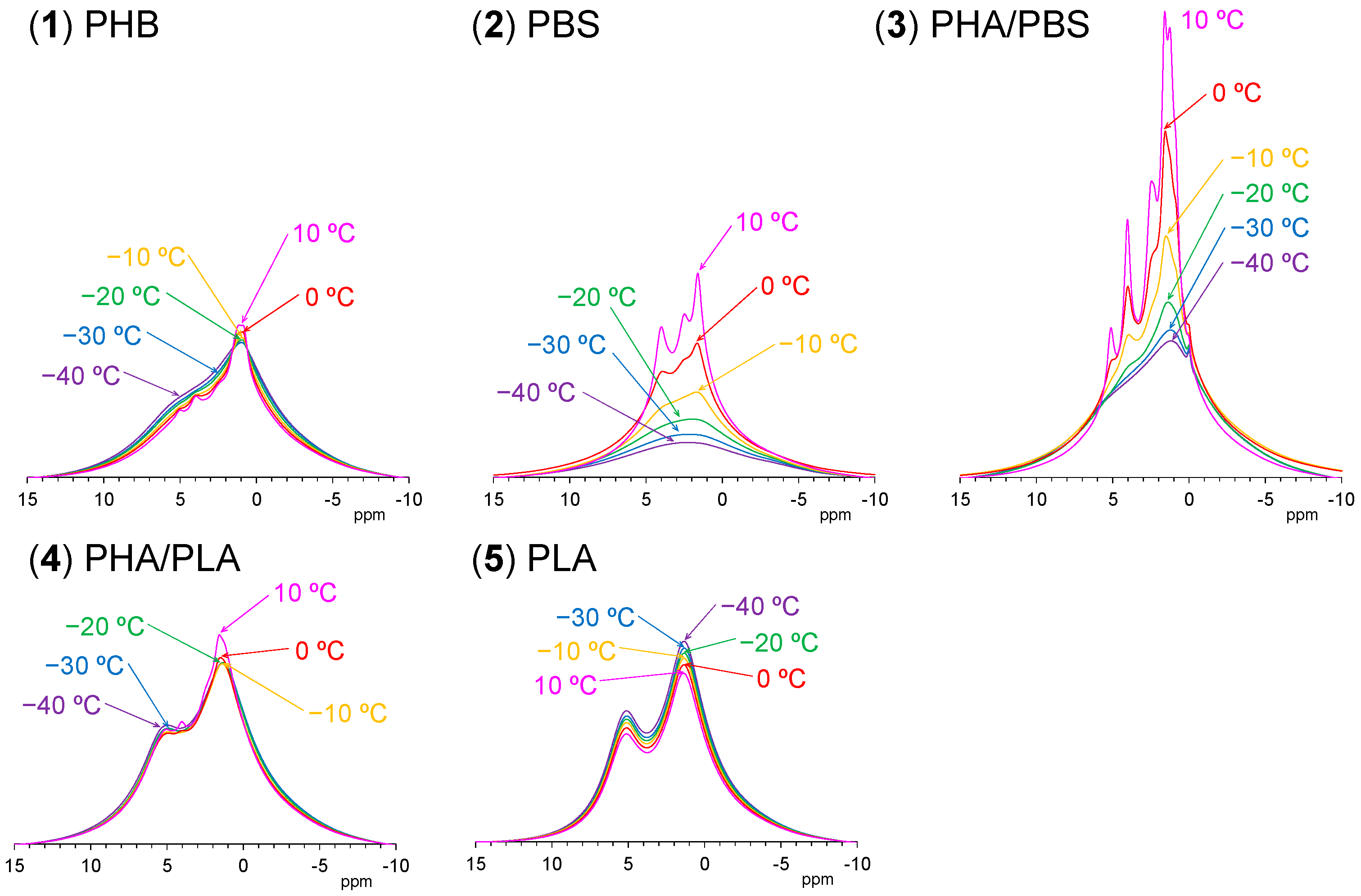
3.5. Changes of 1H MAS NMR with Rising Temperature
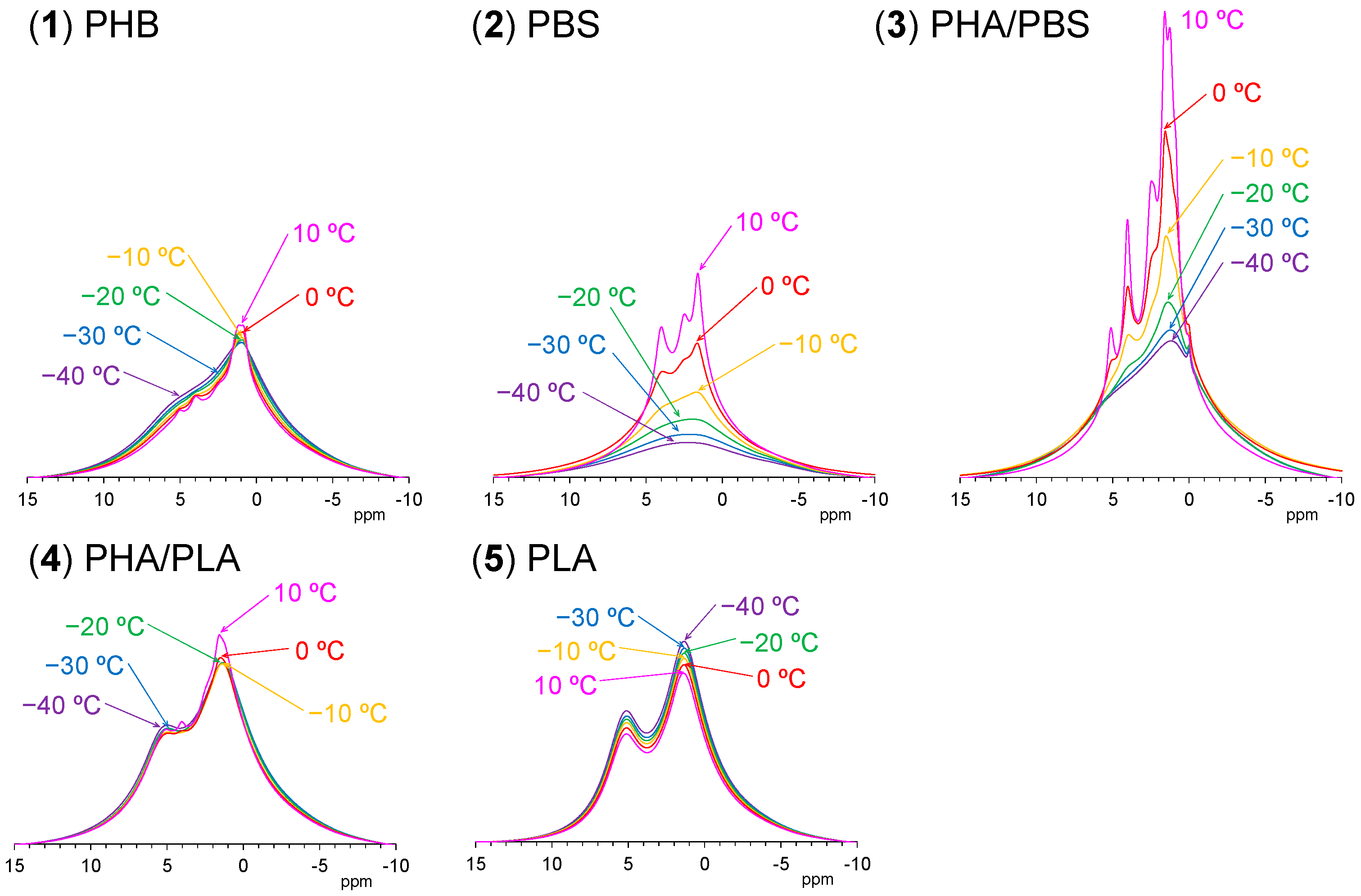
Figure 7 shows variable temperature
1H MAS NMR spectra of plasticized PHA copolymers and constituent polymers. Because of strong dipolar–dipolar interactions of
1H nuclei with each other, the
1H signals in the solid-state NMR appear as broad overlapped signals. The original PHB (1) showed only such signals, which changed little with increasing temperature; the CH
3 signal sharpened and increased and the broader peaks in the CH and CH
2 regions decreased at the same time. This showed that the molecular mobility of PHB (1) alone remained almost unchanged by the temperature rise. For the flexible PBS (2), however, every signal significantly sharpened and increased with increasing temperature, even though only very broad and weak peaks appeared at low temperatures (−40 °C). That is, the mobility of flexible PBS (2) was greatly affected by the temperature rise, resulting in a sharpened and larger
1H MAS signals. Interestingly, the copolymerization of PHA with PBS [PHA4422P (3)] enlarged and sharpened the signals of not only the PBS unit but also the PHB unit. The rate of increase of the
1H signals for both PHA and PBS units in PHA4422P (3) was greater than for PBS (2) alone; thus, the copolymerization of PHA with PBS increased the mobility of not only the PHA unit but also the PBS unit compared with the original polymers. Meanwhile, the
1H signal intensity of rigid PLA (5) decreased with increasing temperature as seen for woody materials [
26,
27]. These
1H signal decreases may occur due to sensitivity change of the detecting coil in the probe; now, we are still searching other reasons related to the magnetic relaxation. The plasticized PHA5533L (4) showed only the overlapped
1H signal of PHB and PLA units, which simply changed in the same manner as each polymer alone, with no increase in signal or sharpness. According to the variable
1H MAS NMR results, the flexibility of the polymer significantly enhanced slow molecular motions (kHz) corresponding to the
T1ρH relaxation, resulting in sharp and large
1H signals after plasticization with PBS. The slow molecular motion (kHz order) corresponded to the motion of polymer chains, which was enhanced at temperatures over −10 °C.
3.6. Changes of Spin-Lattice Relaxation Times in the Laboratory Frame with Rising Temperature
In order to investigate the rapid molecular motions (MHz) that participated in the spin-lattice relaxation in the laboratory frame, rising temperature
T1H and
T1C measurements were performed in two temperature regions. The
T1H and
T1C changes due to the temperature rises are summarized in
Figure 8 and
Figure 9, respectively.
The change in trends of
T1H and
T1C values was closely related to the existence of CH
3 groups. The
T1H values of PHB (1) (
Figure 8a, black line) and PLA (5) (
Figure 8b, green line), both of which had CH
3 groups, increased with increasing temperature except for the high temperature region of PHB (1). In contrast, PBS (2) (
Figure 8b, orange line), which had no CH
3 group, decreased with increasing temperature; the rate of decrease was significantly faster in the low-temperature region. The different change trend that correlates with the existence of CH
3 groups was also observed in the
T1C values. The
T1C values of CH
3 groups increased monotonically with increasing temperature in both PHB (1) (black line) and PLA (5) (green line) up to 50 °C (
Figure 9a). Conversely, a decrease with rising temperature was observed for the
T1C value of each substituent in PBS (
Figure 9b, orange line). The CH
2 group in PHB (43 ppm) has a very long
T1C value (over 80 s), which increased in the low temperature and decreased in the high temperature regions (
Figure 9b, black line). These trends of
T1 values showed that the rotation of CH
3 in PHB and PLA had a lower correlation time (τ
c) than molecular motions of CH
2 in PHB and PBS.
Although the
T1H value of the PHA unit in PHA4422P (3) increased to −10 °C, it started to decrease over −10 °C (
Figure 8a, red line). The
T1H value of PBS unit in PHA4422P (3) conversely decreased and the rate of decrease became smaller in the high-temperature region (
Figure 8b, red line). As a result, the
T1H values of PHA and PBS units in PHA4422P (3) approached each other over −10 °C and these approached the
T1H values of PHA and PBS units which maintained their relationship in the higher temperature region. The approached
T1H values are caused by the
1H spin diffusion via the interface between PHB and PBS, which increases with improving polymer compatibility of PHA and PBS units above −10 °C. This interfacial
1H spin diffusion possibly enhanced the T
1H relaxation in the whole system of PHB/PBS copolymer. The improved compatibility for PHA4422P (3) also made
1H MAS NMR signals sharper and larger at temperatures over −10 °C (
Figure 7). Although a
T1H reduction of the PHA unit was also observed in PHA5533L (4) (
Figure 8a, blue line), no inflection points of the
T1H curve were observed in the low-temperature region and the
1H MAS NMR spectra changed only slightly in PHA5533L (4). These results indicated that the polymer compatibility between PHA and PLA was less improved in PHA5533L (4) although the
T1H relaxation was enhanced by the heterogeneity.
The T1C value of the CH2 groups in the plasticized PHA copolymers gave information about rapid molecular motions (MHz order) of the polymer chain. In all temperature ranges, the PHB unit of the plasticized PHA copolymers has a low T1C value, compared with the pre-plasticized PHB, although the PBS units had similar T1C values regardless of the plasticization. The heterogeneity that had arisen from the amorphous moiety also increased the molecular mobility of the PHB chain, which enhanced the T1C relaxation to produce the lowered T1C value of the PHB unit. Meanwhile, the T1C value of CH3 groups in the plasticized PHA copolymers provides information on the local motion of CH3 closely related to the T1H relaxation. The reduction of the T1C value of CH3 groups due to copolymerization was limited, unlike the T1H value. Thus, the change of T1H was not caused by the molecular motion of CH3 groups, but by the interaction between constituent polymers.
In conclusion, T1H values can act as an index of polymer compatibility due to plasticization, while T1C values of CH2 groups can provide an index of rapid molecular motions of constituent polymers. Moreover, the T1ρH values can index slow molecular motions of the polymer main chain, along with the 1H and 13C PST-MAS NMR spectra. Since copolymerization of PHA with PBS improved elongation and impact properties, the increase in slow molecular motions and compatibility of the PHA/PBS copolymer is possibly related to such polymer properties. Next, we are planning to study other biomass-based polymers to relate polymer properties to the indexes in solid-state NMR analytical methods proposed in the present study.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}