A Facile, One-Pot, Surfactant-Free Nanoprecipitation Method for the Preparation of Nanogels from Polyglycerol–Drug Conjugates that Can Be Freely Assembled for Combination Therapy Applications


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Instrumentation
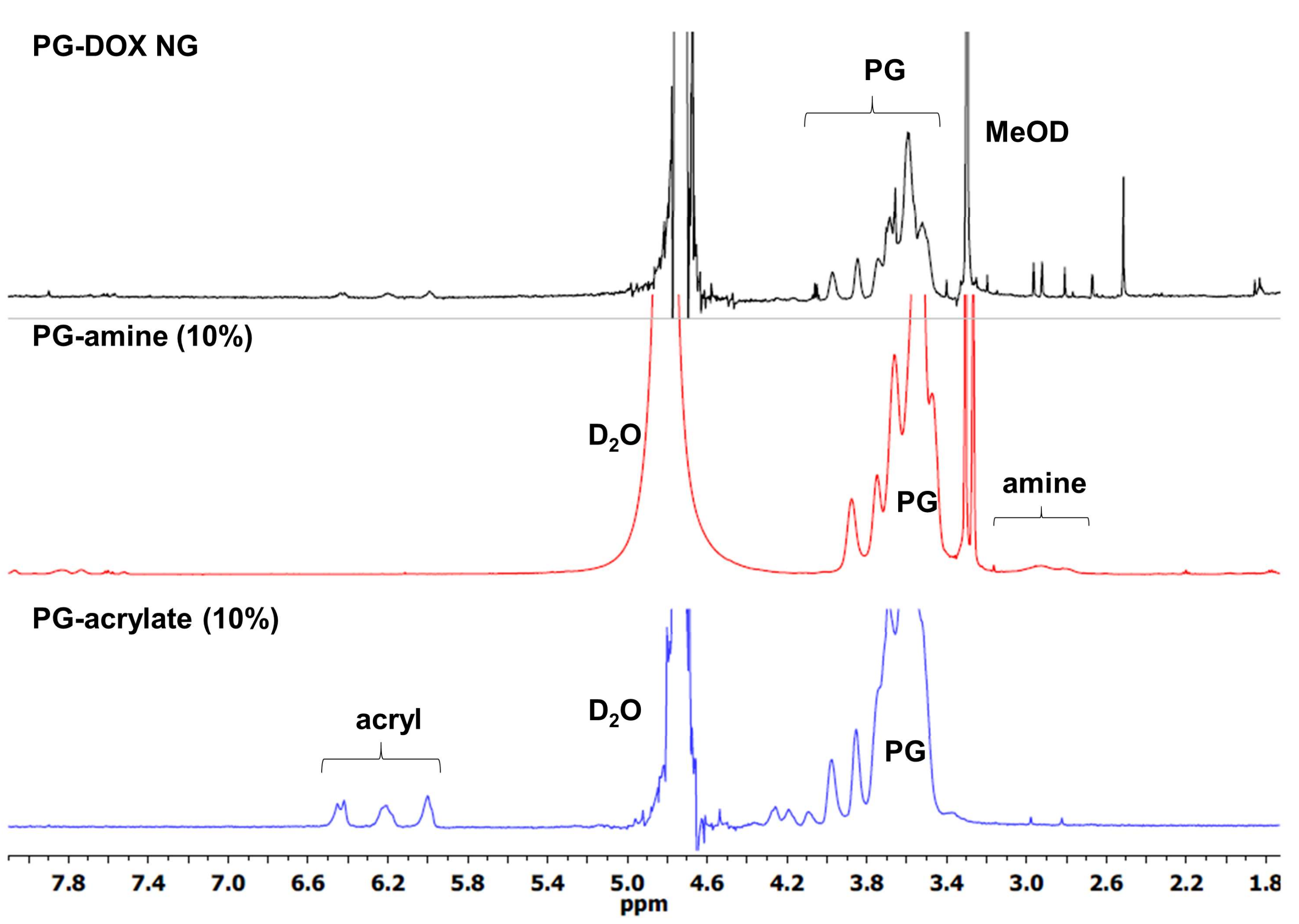
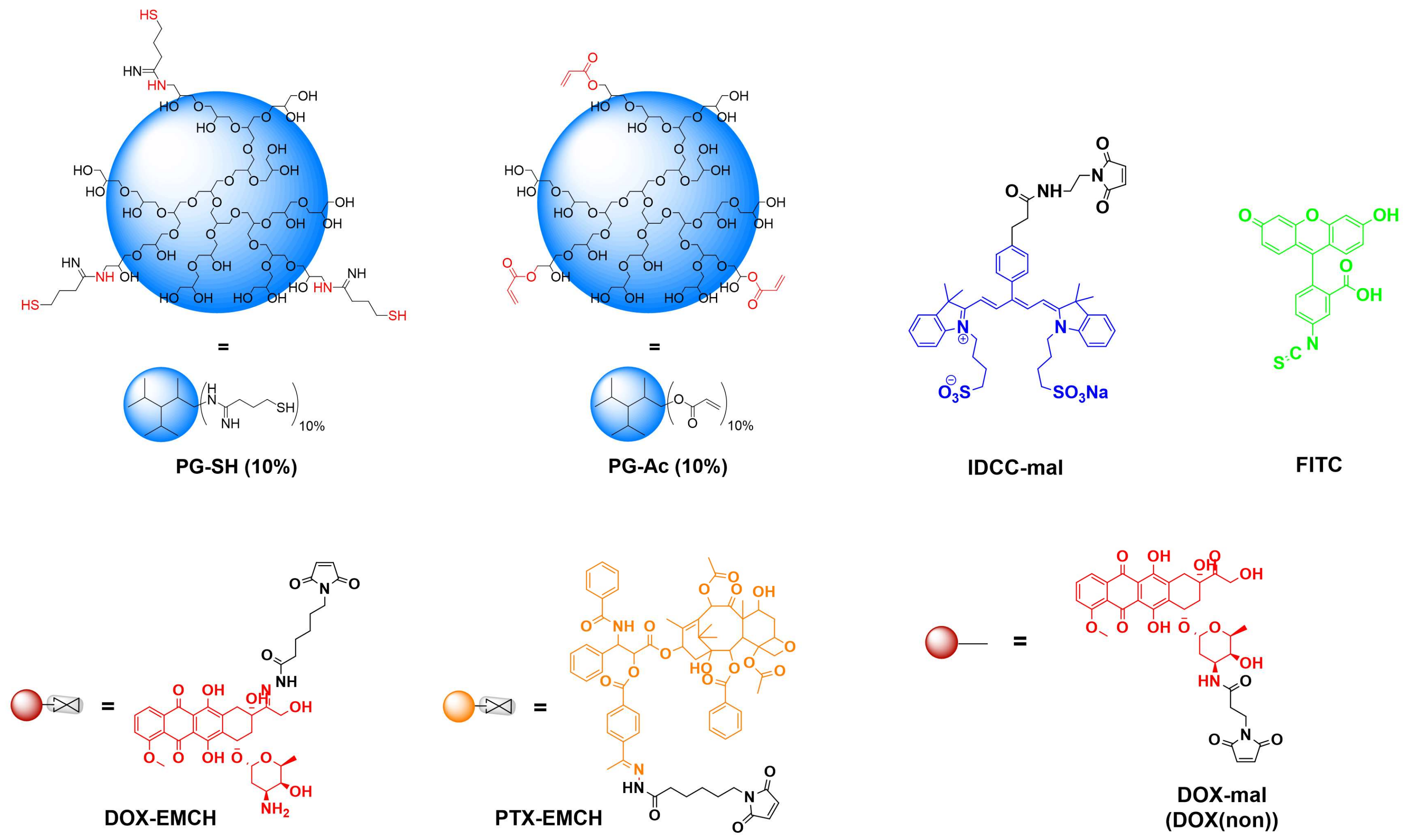
2.3. Synthesis of PG-Acrylate
2.4. Synthesis of PG-Amine
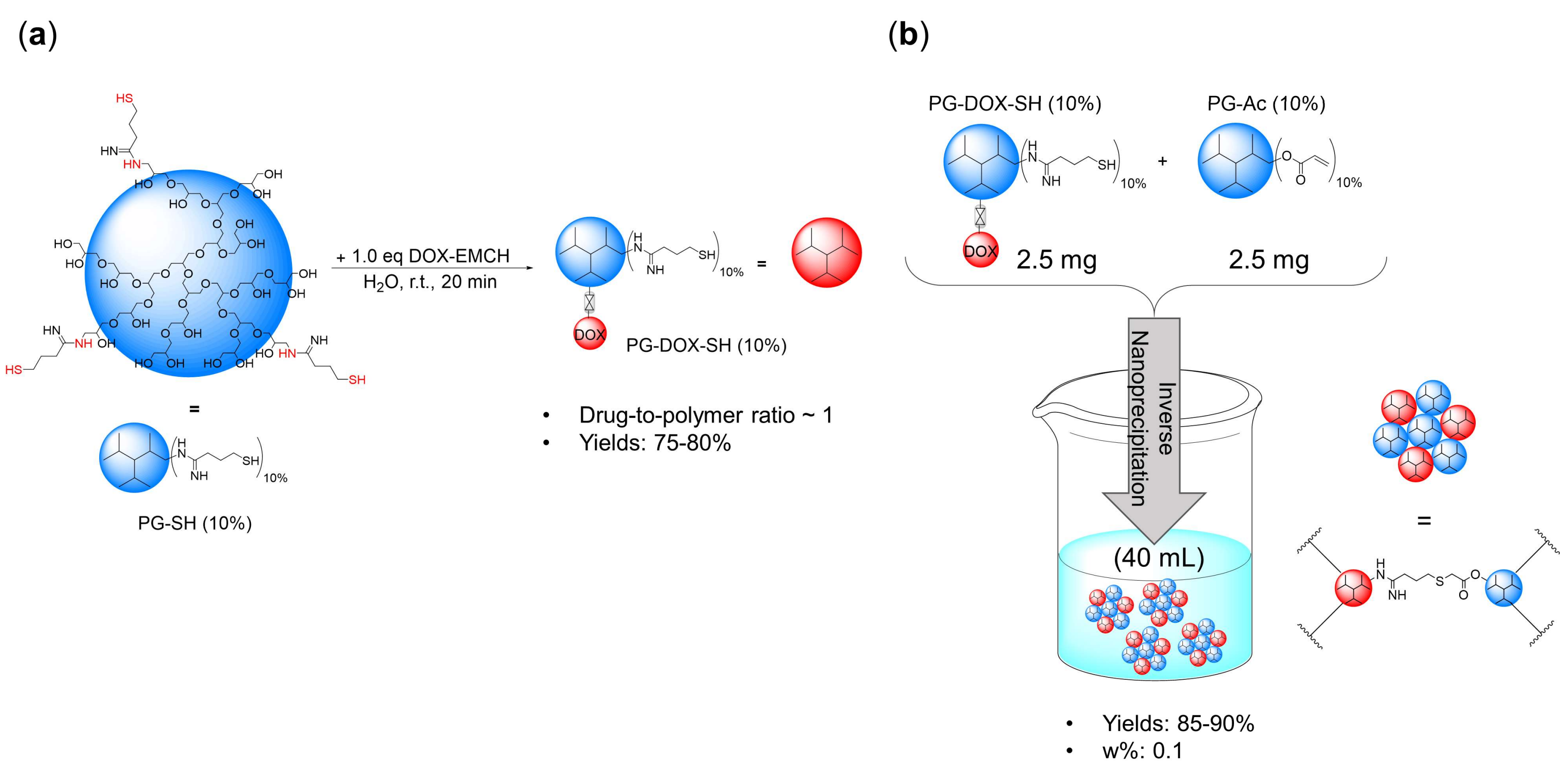
2.5. Synthesis of PG–Drug/Dye–SH Conjugates
2.6. General Procedure for the Preparation of PG-NGs
2.7. RP-HPLC Analysis of PTX Release
2.8. GPC Analysis of PG-DOX NG
2.9. Fluorescence Analysis of DOX Release
3. Results and Discussion
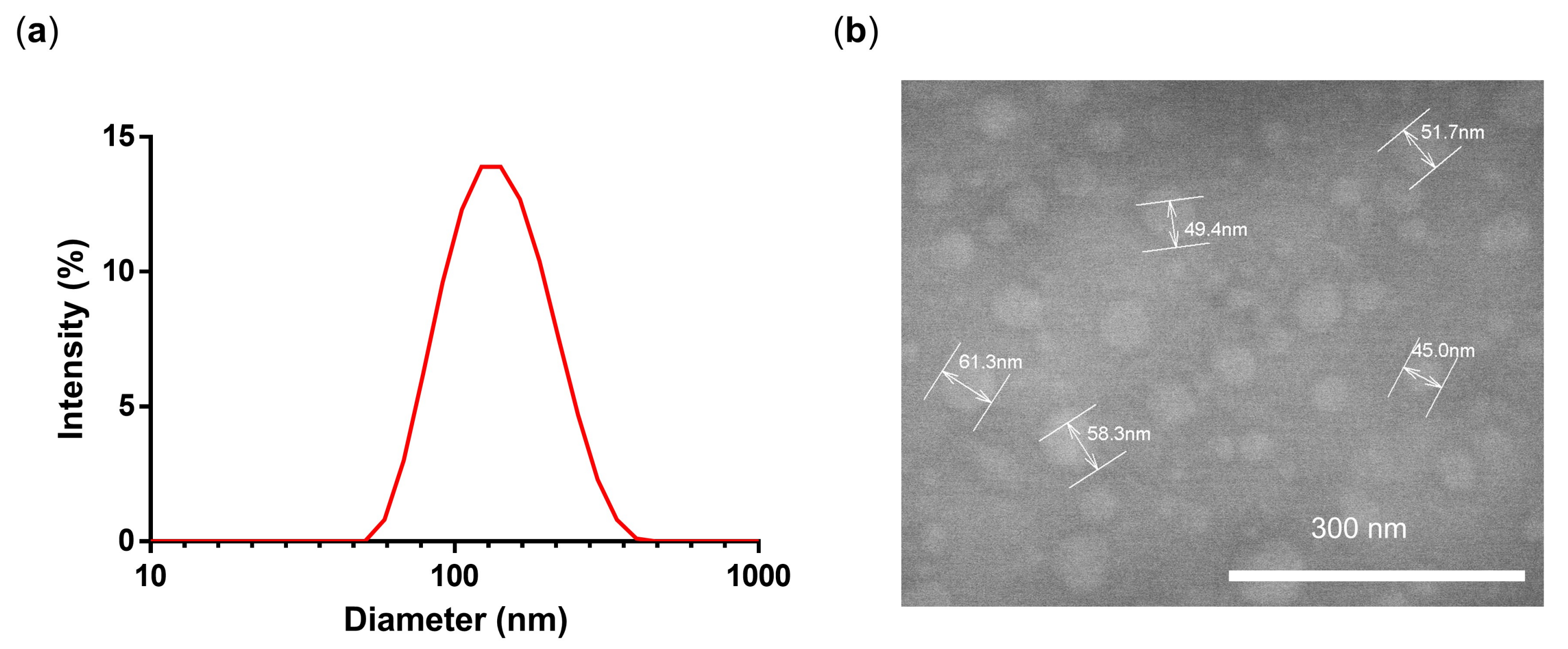
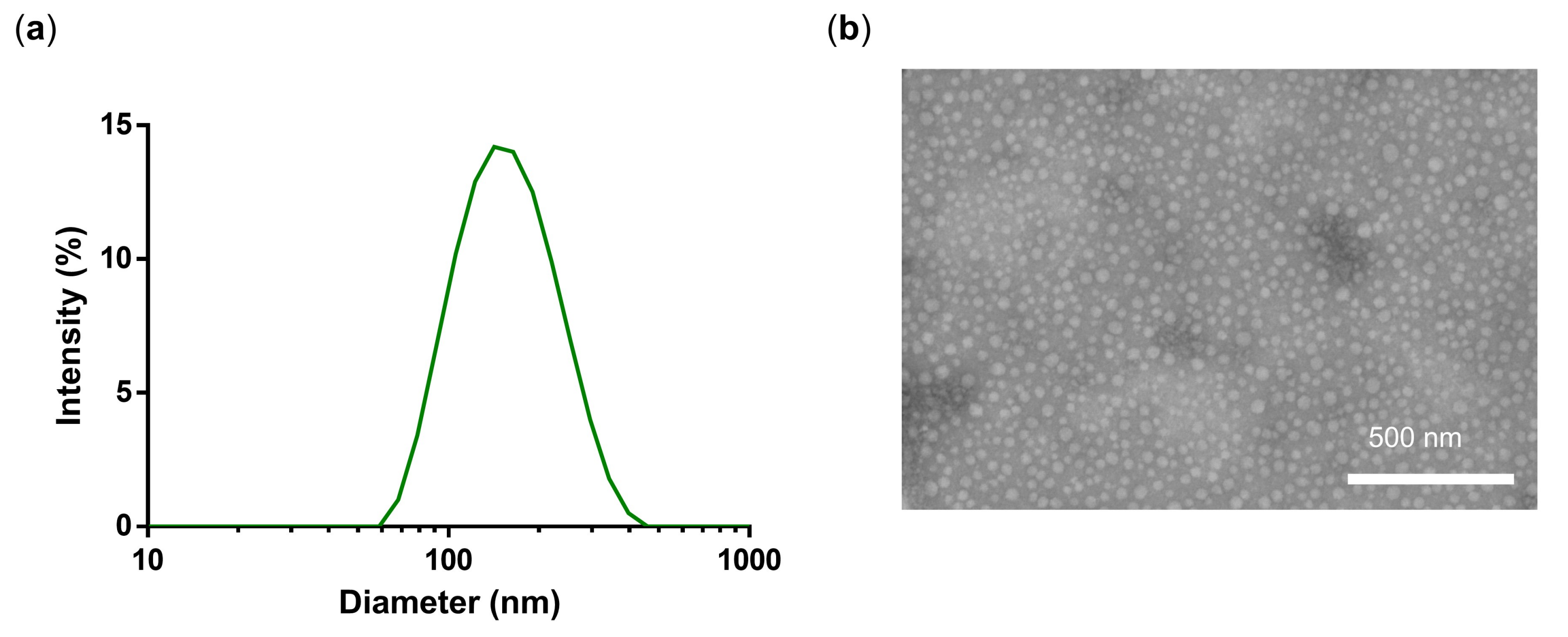
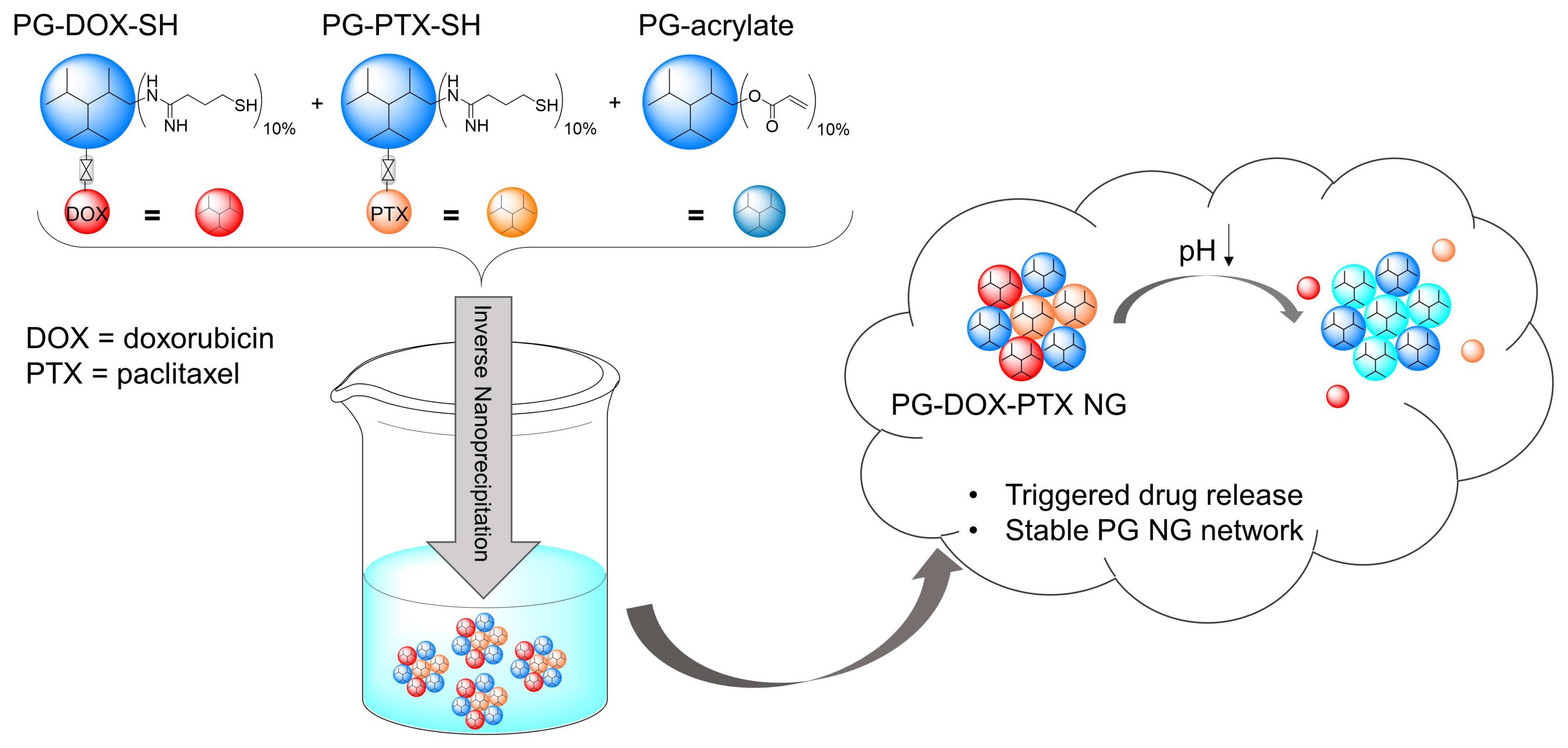
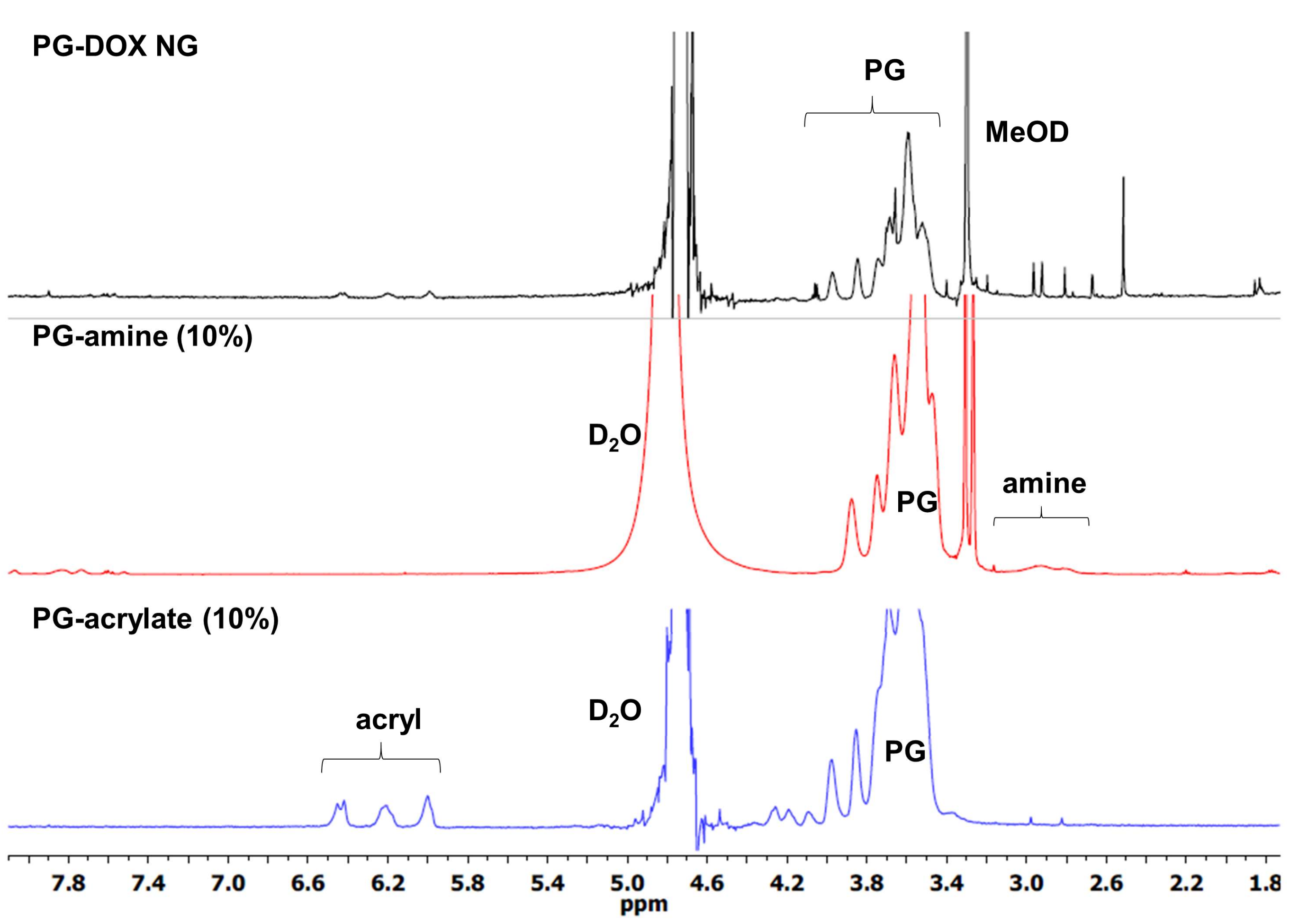
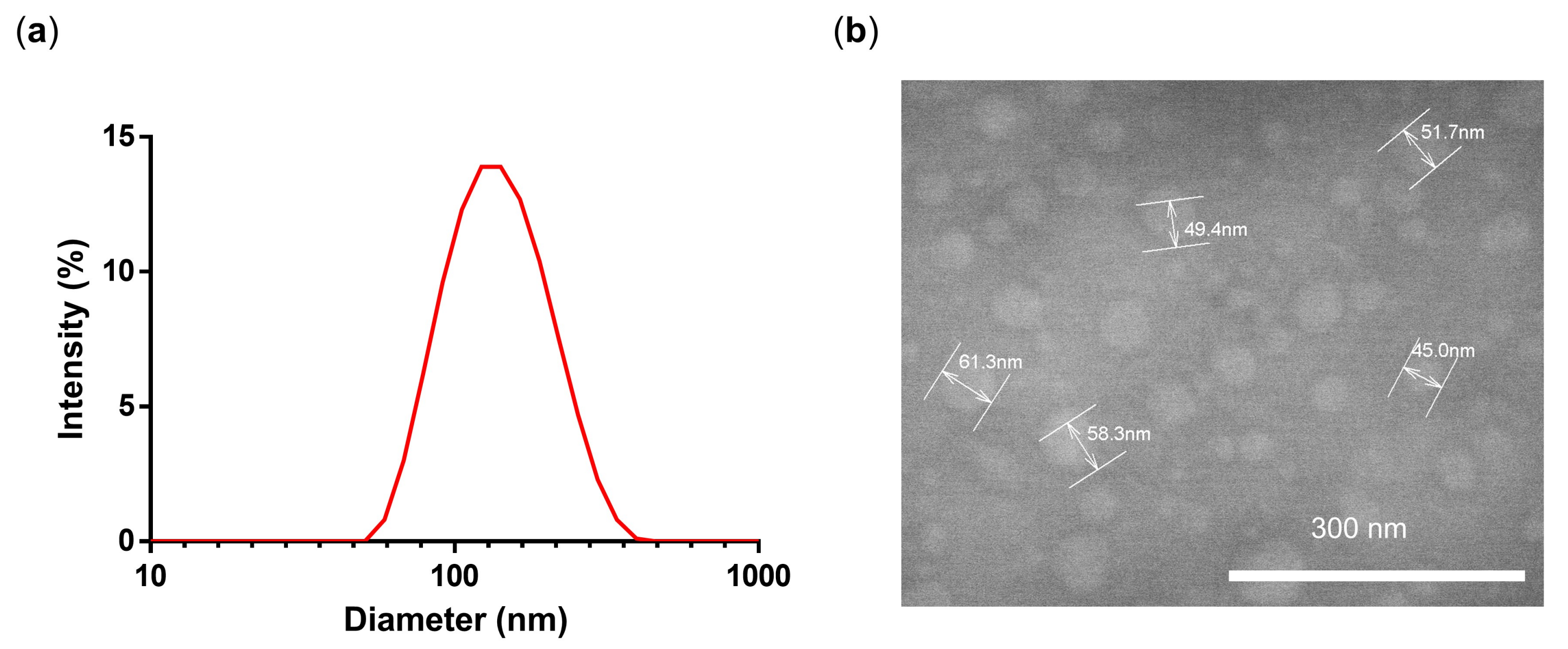
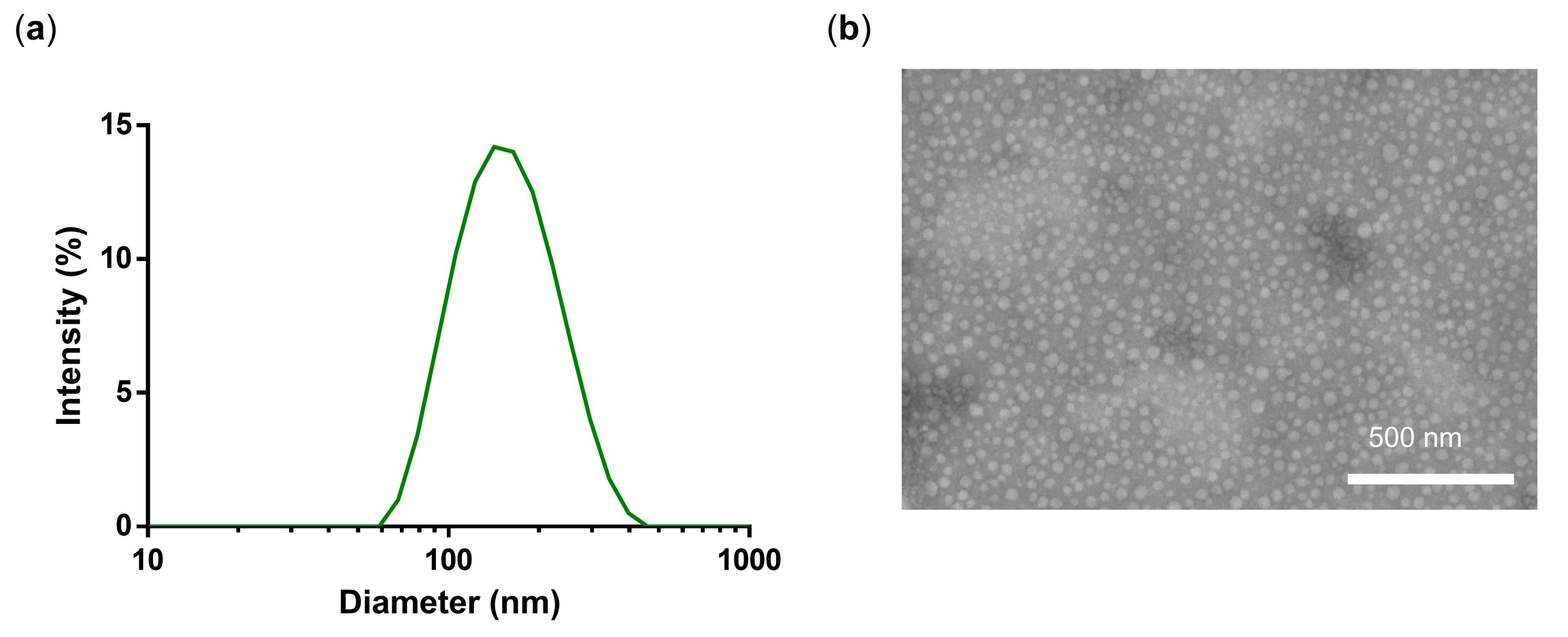
3.1. Synthesis and Characterization of PG-DOX-PTX NGs by an Inverse Nanoprecipitation Method
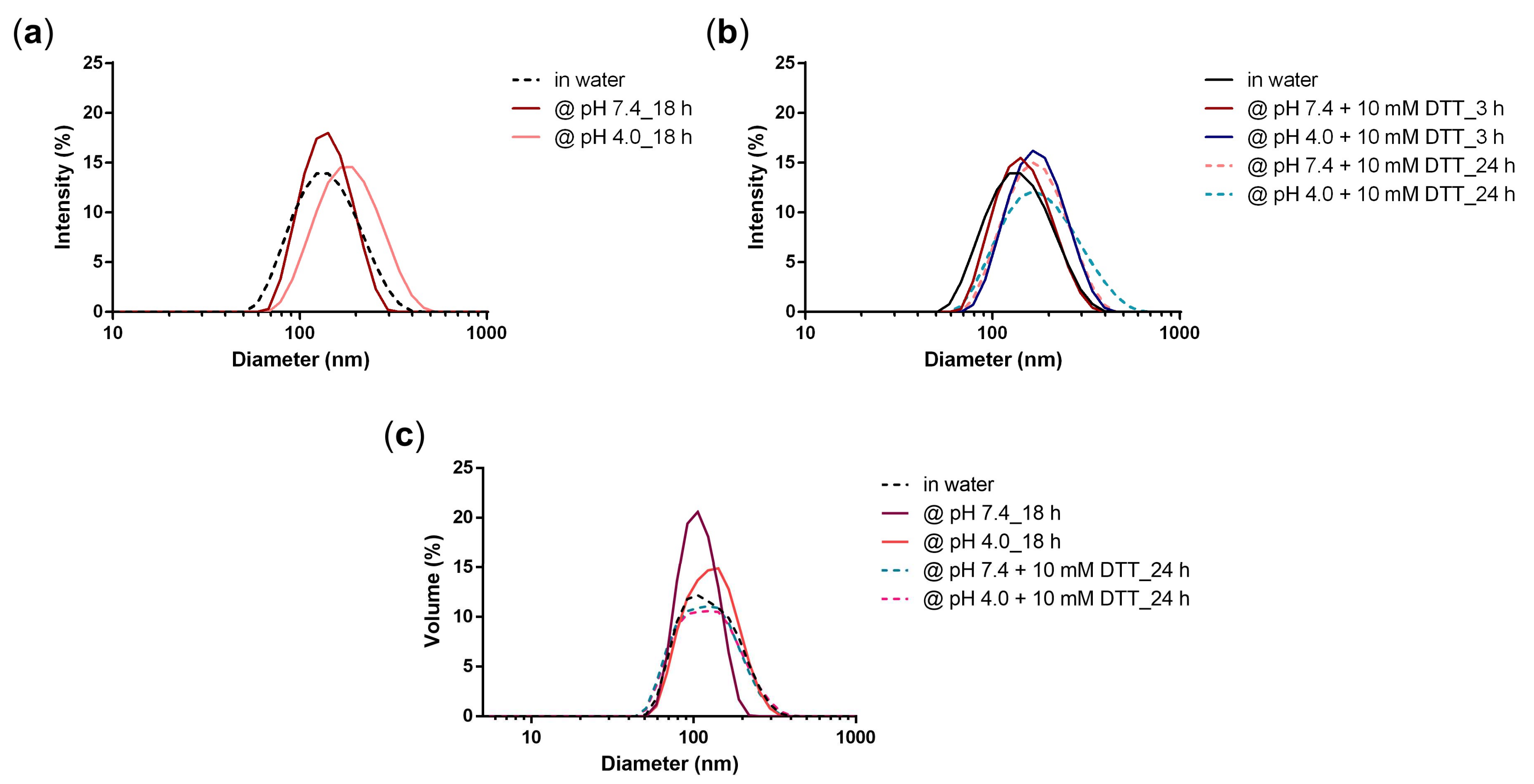
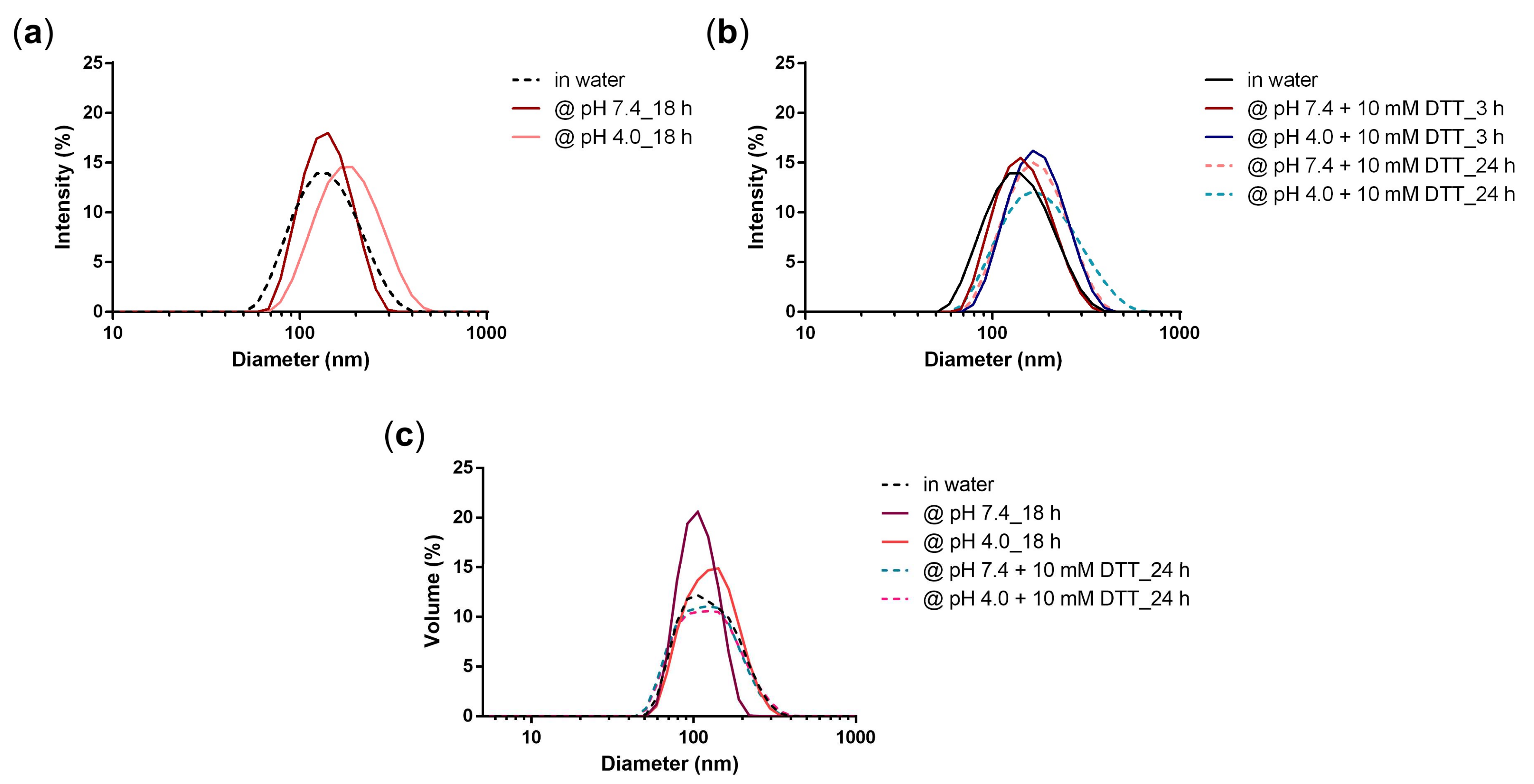
3.2. Stability of PG-DOX NG
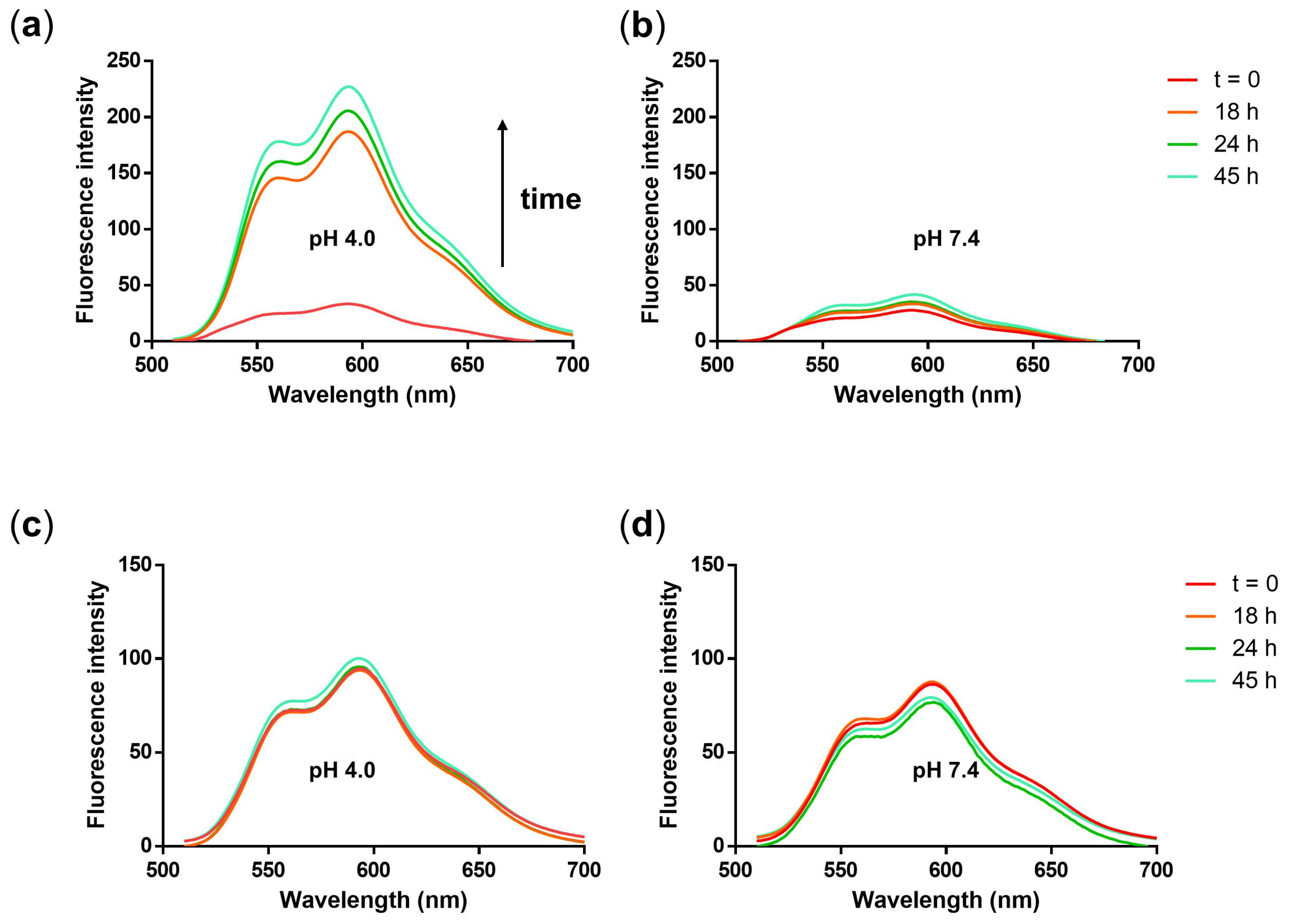
3.3. Triggered Release of DOX and PTX from the NG
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Logman, J.F.; Heeg, B.M.; Botteman, M.F.; Kaura, S.; van Hout, B.A. Economic evaluation of zoledronic acid for the prevention of osteoporotic fractures in postmenopausal women with early-stage breast cancer receiving aromatase inhibitors in the UK. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2010, 21, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Boock, A.; Polyak, D.; Scomparin, A.; Satchi-Fainaro, R. Nano-sized polymers and liposomes designed to deliver combination therapy for cancer. Curr. Opin. Biotechnol. 2013, 24, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Greco, F.; Vicent, M.J. Combination therapy: Opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv. Drug Deliv. Rev. 2009, 61, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Warnecke, A. Finding the optimal balance: Challenges of improving conventional cancer chemotherapy using suitable combinations with nano-sized drug delivery systems. J. Control. Release 2012, 164, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 2007, 19, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Calderón, M.; Quadir, M.A.; Sharma, S.K.; Haag, R. Dendritic polyglycerols for biomedical applications. Adv. Mater. 2010, 22, 190–218. [Google Scholar] [CrossRef] [PubMed]
- Khandare, J.; Calderón, M.; Dagia, N.M.; Haag, R. Multifunctional dendritic polymers in nanomedicine: Opportunities and challenges. Chem. Soc. Rev. 2012, 41, 2824–2848. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as pharmaceutical carriers: Finite networks of infinite capabilities. Angew. Chem. Int. Ed. Engl. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [PubMed]
- Asadian-Birjand, M.; Sousa-Herves, A.; Steinhilber, D.; Cuggino, J.C.; Calderon, M. Functional nanogels for biomedical applications. Curr. Med. Chem. 2012, 19, 5029–5043. [Google Scholar] [CrossRef] [PubMed]
- Landfester, K.; Willert, M.; Antonietti, M. Preparation of polymer particles in nonaqueous direct and inverse miniemulsions. Macromolecules 2000, 33, 2370–2376. [Google Scholar] [CrossRef]
- Steinhilber, D.; Seiffert, S.; Heyman, J.A.; Paulus, F.; Weitz, D.A.; Haag, R. Hyperbranched polyglycerols on the nanometer and micrometer scale. Biomaterials 2011, 32, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Sisson, A.L.; Steinhilber, D.; Rossow, T.; Welker, P.; Licha, K.; Haag, R. Biocompatible functionalized polyglycerol microgels with cell penetrating properties. Angew. Chem. Int. Ed. Engl. 2009, 48, 7540–7545. [Google Scholar] [CrossRef] [PubMed]
- Antonietti, M. Microgels—Polymers with a special molecular architecture. Angew. Chem. Int. Ed. Engl. 1988, 27, 1743–1747. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, J.; Deng, C.; Suuronen, E.J.; Zhong, Z. Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering. Biomaterials 2014, 35, 4969–4985. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Delaney, J.J.T.; Schubert, U.S. Nanoprecipitation and nanoformulation of polymers: From history to powerful possibilities beyond poly (lactic acid). Soft Matter 2011, 7, 1581–1588. [Google Scholar] [CrossRef]
- Zhang, X.; Achazi, K.; Steinhilber, D.; Kratz, F.; Dernedde, J.; Haag, R. A facile approach for dual-responsive prodrug nanogels based on dendritic polyglycerols with minimal leaching. J. Control. Release Soc. 2014, 174, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Witting, M.; Zhang, X.; Staegemann, M.; Paulus, F.; Friess, W.; Kuchler, S.; Haag, R. Surfactant free preparation of biodegradable dendritic polyglycerol nanogels by inverse nanoprecipitation for encapsulation and release of pharmaceutical biomacromolecules. J. Control. Release Soc. 2013, 169, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Giulbudagian, M.; Asadian-Birjand, M.; Steinhilber, D.; Achazi, K.; Molina, M.; Calderon, M. Fabrication of thermoresponsive nanogels by thermo-nanoprecipitation and in situ encapsulation of bioactives. Polym. Chem. 2014, 5, 6909–6913. [Google Scholar] [CrossRef]
- Dimde, M.; Sahle, F.F.; Wycisk, V.; Steinhilber, D.; Camacho, L.C.; Licha, K.; Lademann, J.; Haag, R. Synthesis and validation of functional nanogels as ph-sensors in the hair follicle. Macromol. Biosci. 2017, 17, 1600505. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Herves, A.; Wedepohl, S.; Calderon, M. One-pot synthesis of doxorubicin-loaded multiresponsive nanogels based on hyperbranched polyglycerol. Chem. Commun. 2015, 51, 5264–5267. [Google Scholar] [CrossRef] [PubMed]
- Shatsberg, Z.; Zhang, X.; Ofek, P.; Malhotra, S.; Krivitsky, A.; Scomparin, A.; Tiram, G.; Calderón, M.; Haag, R.; Satchi-Fainaro, R. Functionalized nanogels carrying an anticancer microrna for glioblastoma therapy. J. Control. Release 2016, 239, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Giulbudagian, M.; Honzke, S.; Bergueiro, J.; Isk, D.; Schumacher, F.; Saeidpour, S.; Lohan, S.B.; Meinke, M.C.; Teutloff, C.; Schafer-Korting, M.; et al. Enhanced topical delivery of dexamethasone by [small beta]-cyclodextrin decorated thermoresponsive nanogels. Nanoscale 2018, 10, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Giulbudagian, M.; Yealland, G.; Honzke, S.; Edlich, A.; Geisendorfer, B.; Kleuser, B.; Hedtrich, S.; Calderon, M. Breaking the barrier—Potent anti-inflammatory activity following efficient topical delivery of etanercept using thermoresponsive nanogels. Theranostics 2018, 8, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Baabur-Cohen, H.; Vossen, L.I.; Krüger, H.R.; Eldar-boock, A.; Yeini, E.; Landa-Rouben, N.; Tiram, G.; Wedepohl, S.; Markovsky, E.; Leor, J.; et al. In Vivo comparative study of distinct polymeric architectures bearing a combination of paclitaxel and doxorubicin at a synergistic ratio. J. Control. Release 2017, 257, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly (ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Trail, P.A.; Hofstead, S.J.; King, H.D.; Lasch, S.J.; Braslawsky, G.R.; Greenfield, R.S.; Kaneko, T.; Firestone, R.A. (6-maleimidocaproyl)hydrazone of doxorubicin. A new derivative for the preparation of immunoconjugates of doxorubicin. Bioconj. Chem. 1993, 4, 521–527. [Google Scholar] [CrossRef]
- Krüger, H.R.; Schütz, I.; Justies, A.; Licha, K.; Welker, P.; Haucke, V.; Calderón, M. Imaging of doxorubicin release from theranostic macromolecular prodrugs via fluorescence resonance energy transfer. J. Control. Release 2014, 194, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Scheuermann, K.; Stockmar, C.; Maier, G.; Fiebig, H.; Unger, C.; Mulhaupt, R.; Kratz, F. Synthesis and in vitro efficacy of acid-sensitive poly(ethylene glycol) paclitaxel conjugates. Bioorg. Med. Chem. Lett. 2003, 13, 355–360. [Google Scholar] [CrossRef]
- Roller, S.; Zhou, H.; Haag, R. High-loading polyglycerol supported reagents for mitsunobu- and acylation-reactions and other useful polyglycerol derivatives. Mol. Divers. 2005, 9, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Cuggino, J.C.; Alvarez, I.C.I.; Strumia, M.C.; Welker, P.; Licha, K.; Steinhilber, D.; Mutihac, R.-C.; Calderon, M. Thermosensitive nanogels based on dendritic polyglycerol and n-isopropylacrylamide for biomedical applications. Soft Matter 2011, 7, 11,259–11,266. [Google Scholar] [CrossRef]
- Sunder, A.; Mülhaupt, R.; Haag, R.; Frey, H. Hyperbranched polyether polyols: A modular approach to complex polymer architectures. Adv. Mater. 2000, 12, 235–239. [Google Scholar] [CrossRef]
- Riener, C.K.; Kada, G.; Gruber, H.J. Quick measurement of protein sulfhydryls with ellman’s reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 2002, 373, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Ferber, S.; Tiram, G.; Sousa-Herves, A.; Eldar-Boock, A.; Krivitsky, A.; Scomparin, A.; Yeini, E.; Ofek, P.; Ben-Shushan, D.; Vossen, L.I.; et al. Co-targeting the tumor endothelium and p-selectin-expressing glioblastoma cells leads to a remarkable therapeutic outcome. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Herves, A.; Wurfel, P.; Wegner, N.; Khandare, J.; Licha, K.; Haag, R.; Welker, P.; Calderon, M. Dendritic polyglycerol sulfate as a novel platform for paclitaxel delivery: Pitfalls of ester linkage. Nanoscale 2015, 7, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Calderón, M.; Welker, P.; Licha, K.; Graeser, R.; Kratz, F.; Haag, R. Development of efficient macromolecular prodrugs derived from dendritic polyglycerol. J. Control. Release 2010, 148, e24–e25. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.F.; Krüger, H.R.; Kampmeier, F.; Weissbach, T.; Licha, K.; Kratz, F.; Haag, R.; Calderon, M.; Barth, S. Targeted delivery of dendritic polyglycerol—Doxorubicin conjugates by scfv-snap fusion protein suppresses egfr + cancer cell growth. Biomacromolecules 2013, 14, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Corbo, C.; Dominguez-Asenjo, B.; Vossen, L.I.; Perez-Pertejo, Y.; Munoz-Fenandez, M.A.; Balana-Fouce, R.; Calderon, M.; Reguera, R.M. Pegylated dendritic polyglycerol conjugate delivers doxorubicin to the parasitophorous vacuole in leishmania infantum infections. Macromol. Biosci. 2017, 17, 1700098. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Dong, B.; Song, X.; Wang, C.; Zhang, N.; Lin, W. Dual turn-on fluorescence signal-based controlled release system for real-time monitoring of drug release dynamics in living cells and tumor tissues. Theranostics 2018, 8, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Aime, S.; Hennink, W.E.; Storm, G.; Kiessling, F. Theranostic nanomedicine. Acc. Chem. Res. 2011, 44, 1029–1038. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | c [Monomers] | Acetone (mL) | d [nm] Water | PDI |
|---|---|---|---|---|
| NG1 | 10 mg/1 mL | 40 | 187 | 0.16 |
| NG2 | 5 mg/1 mL | 20 | 210 | 0.49 |
| NG3 | 5 mg/2 mL | 40 | 133 | 0.14 |
| # | Name | d [nm] Water | PDI | w% DOX | w% PTX | w% FITC | w% IDCC |
|---|---|---|---|---|---|---|---|
| NG3 | PG-DOX NG | 133 | 0.14 | 1.0 | - | ||
| NG4 | PG-DOX1-PTX1 NG | 148 | 0.12 | 0.5 | 0.9 | ||
| NG5 | PG-PTX NG | 148 | 0.23 | - | 1.9 | ||
| NG6 | PG-DOX5-PTX1 NG | 110 | 0.21 | 0.8 | 0.3 | ||
| NG7 | PG-FITC NG | 125 | 0.18 | - | - | 0.3 | - |
| NG8 | PG-DOX-IDCC NG | 164 | 0.30 | 0.3 | - | - | 0.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vossen, L.I.; Wedepohl, S.; Calderón, M. A Facile, One-Pot, Surfactant-Free Nanoprecipitation Method for the Preparation of Nanogels from Polyglycerol–Drug Conjugates that Can Be Freely Assembled for Combination Therapy Applications. Polymers 2018, 10, 398. https://doi.org/10.3390/polym10040398
Vossen LI, Wedepohl S, Calderón M. A Facile, One-Pot, Surfactant-Free Nanoprecipitation Method for the Preparation of Nanogels from Polyglycerol–Drug Conjugates that Can Be Freely Assembled for Combination Therapy Applications. Polymers. 2018; 10(4):398. https://doi.org/10.3390/polym10040398
Chicago/Turabian StyleVossen, Laura I., Stefanie Wedepohl, and Marcelo Calderón. 2018. "A Facile, One-Pot, Surfactant-Free Nanoprecipitation Method for the Preparation of Nanogels from Polyglycerol–Drug Conjugates that Can Be Freely Assembled for Combination Therapy Applications" Polymers 10, no. 4: 398. https://doi.org/10.3390/polym10040398