Gas Dissolution Foaming as a Novel Approach for the Production of Lightweight Biocomposites of PHB/Natural Fibre Fabrics

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
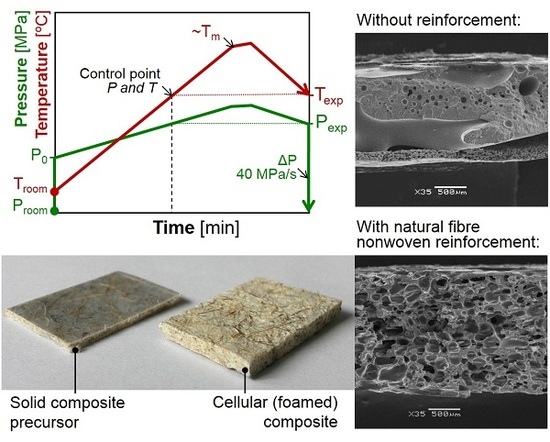
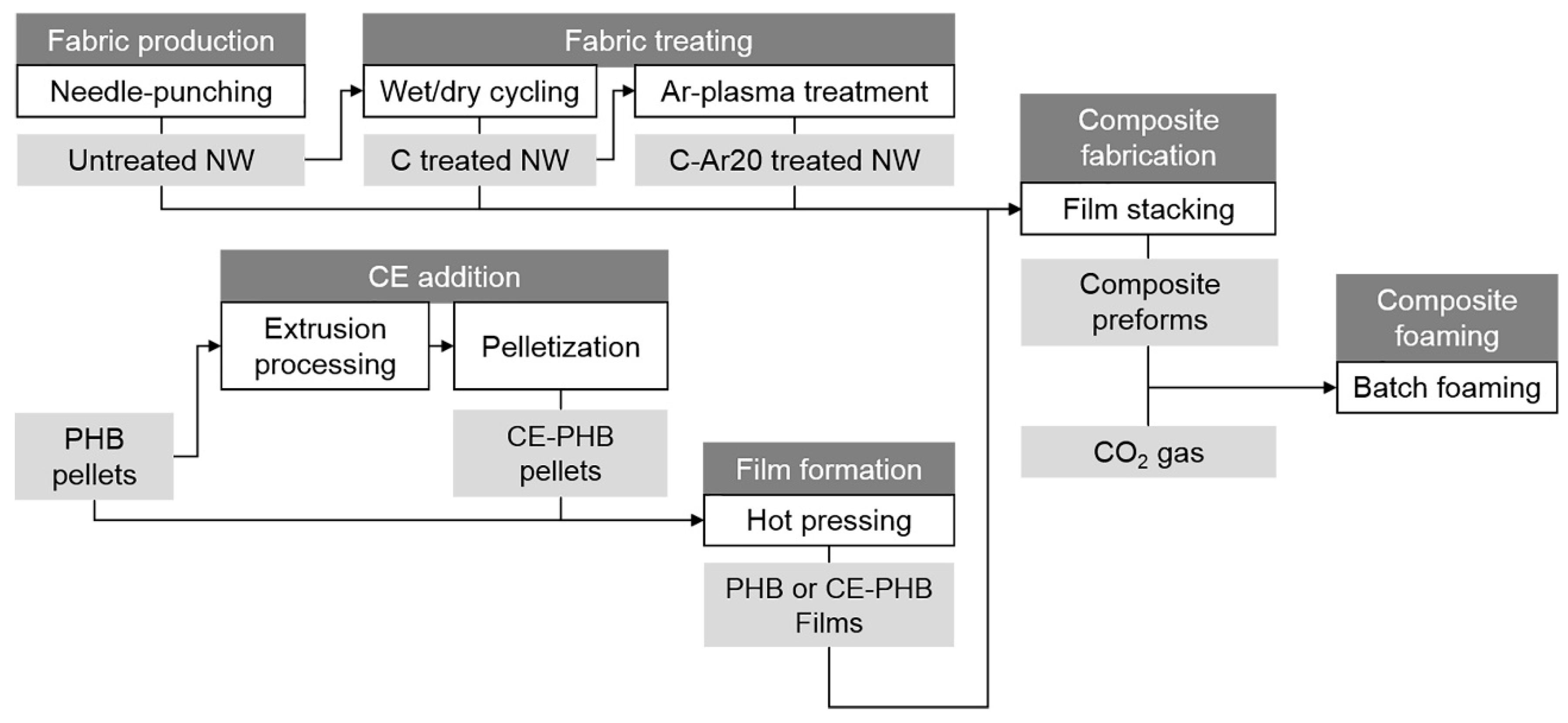
2.1. Foamed Composites Production
2.1.1. Production and Treatment of the Natural Fibre Nonwoven Fabrics
2.1.2. CE Addition to the Matrix
2.1.3. Fabrication of Solid Composite Precursors
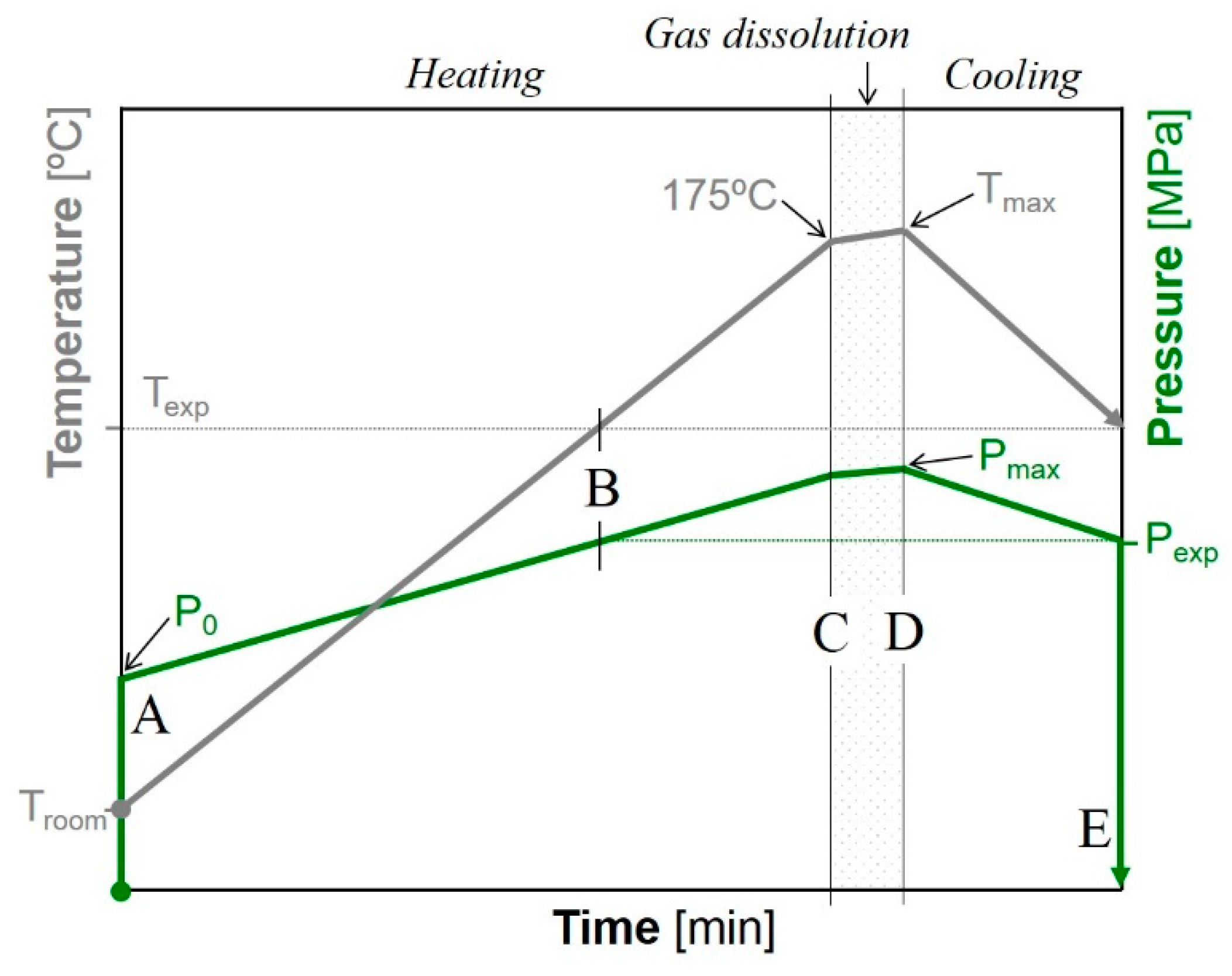
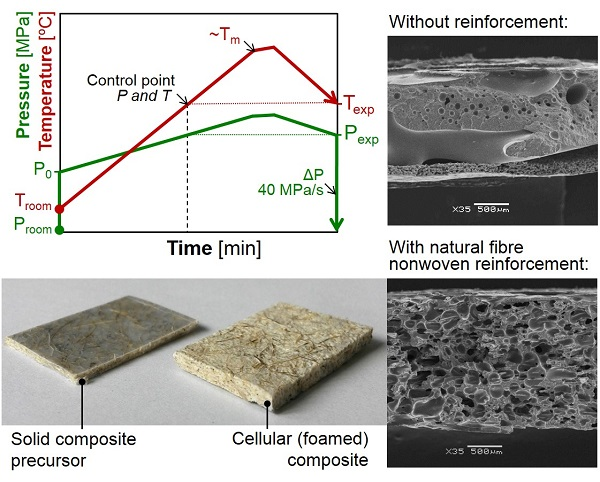
2.1.4. Foaming Process of the Solid Composite Precursors
2.2. Samples Characterisation
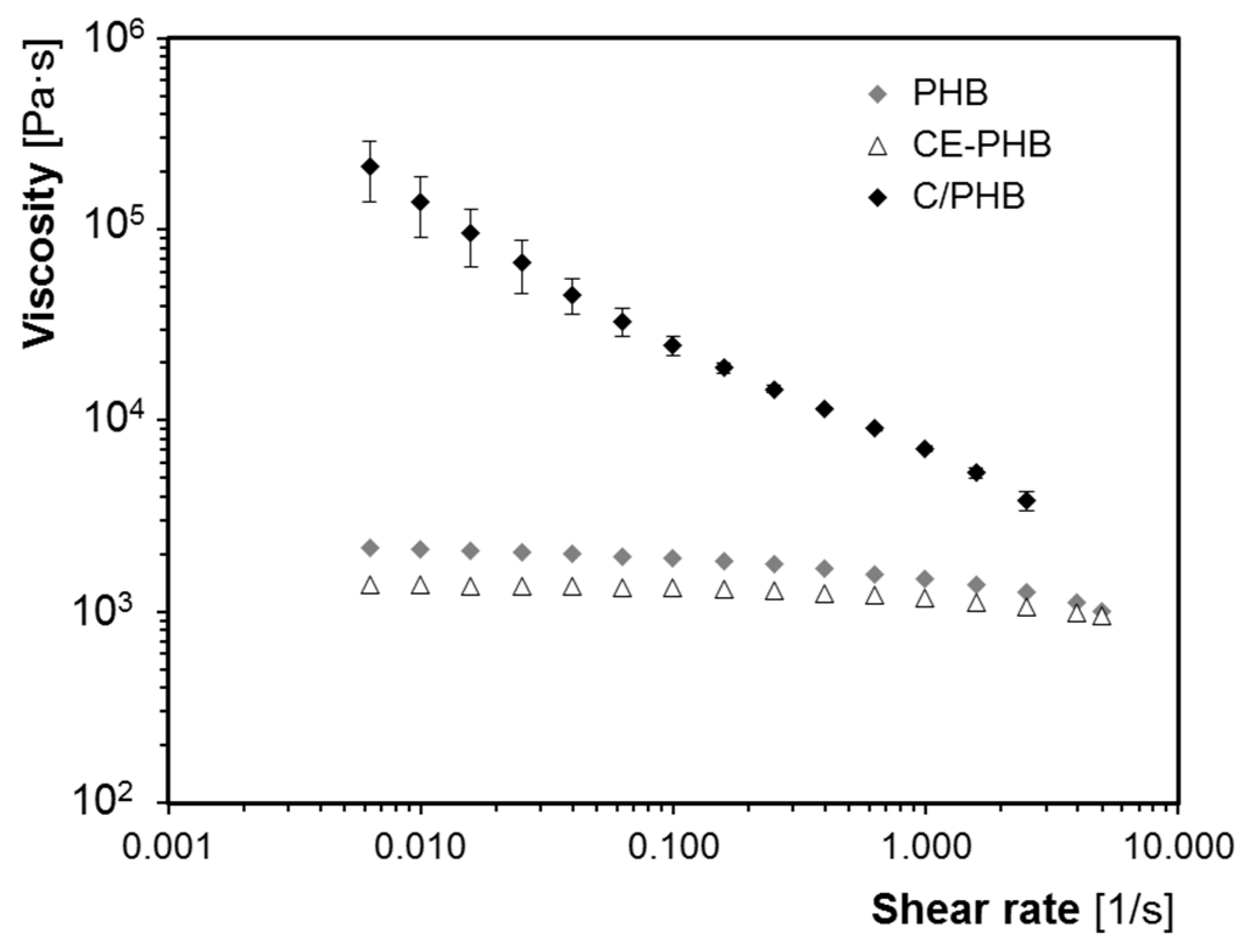
2.2.1. Rheology Measurements
2.2.2. Density
2.2.3. Cell Size and Cell Density
2.2.4. Mechanical Properties
3. Results and Discussion
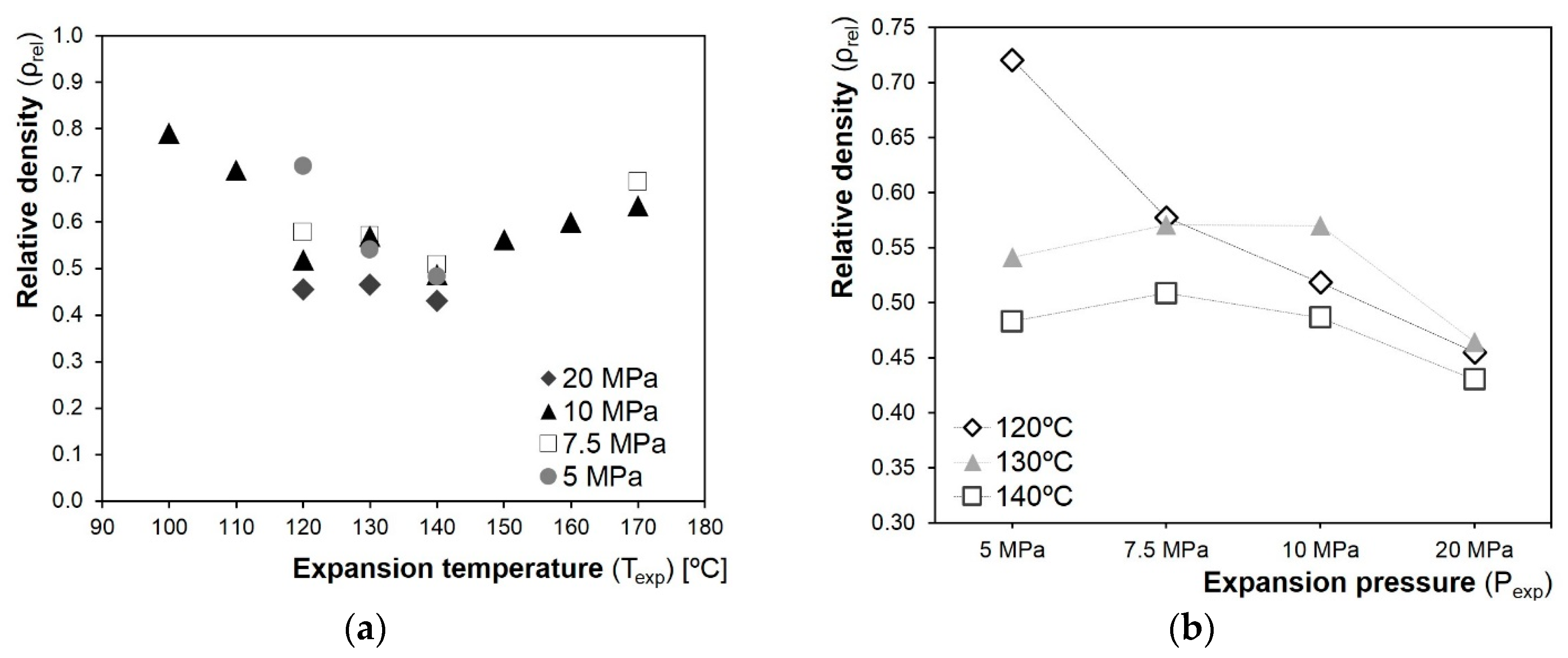
3.1. Preliminary Tests
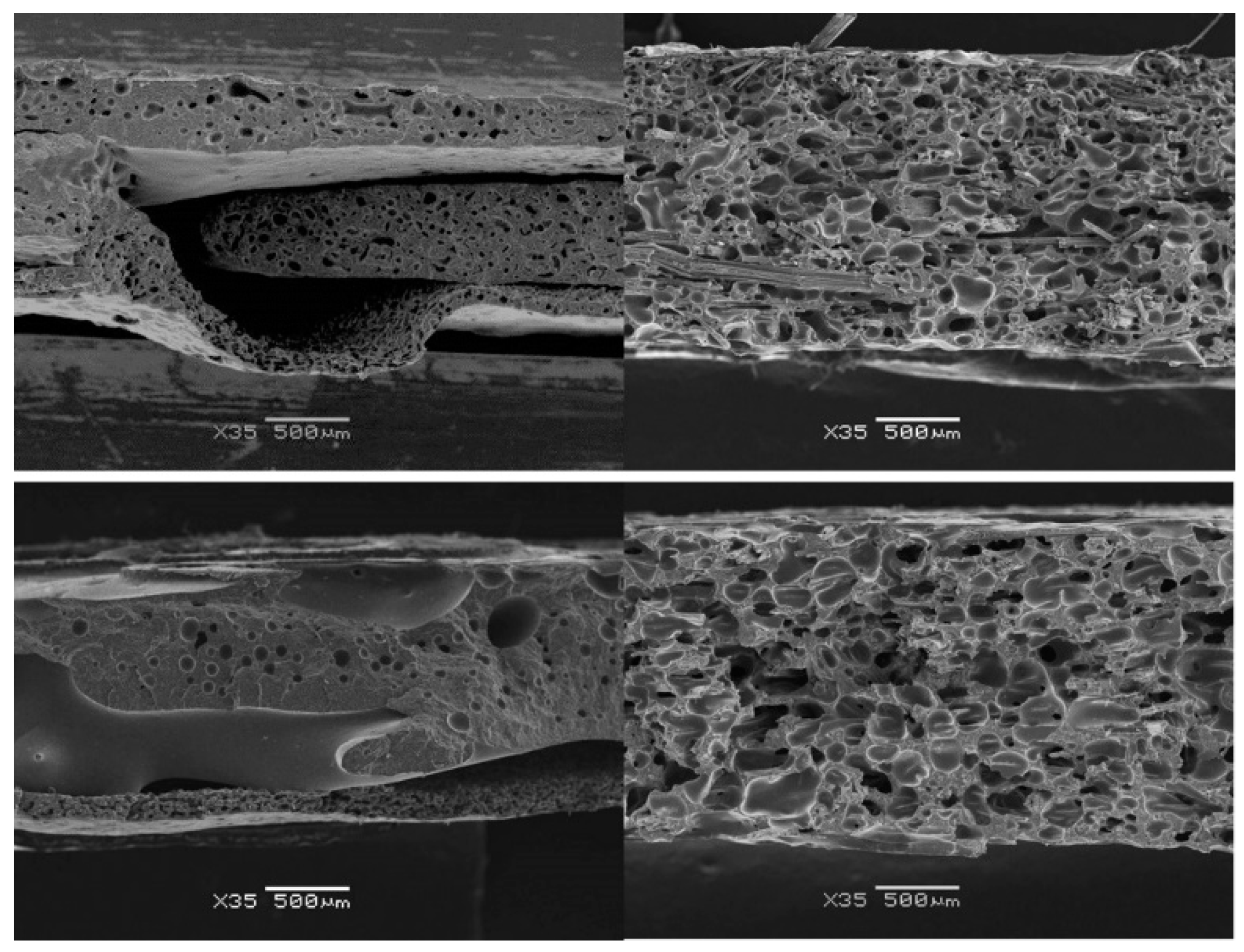
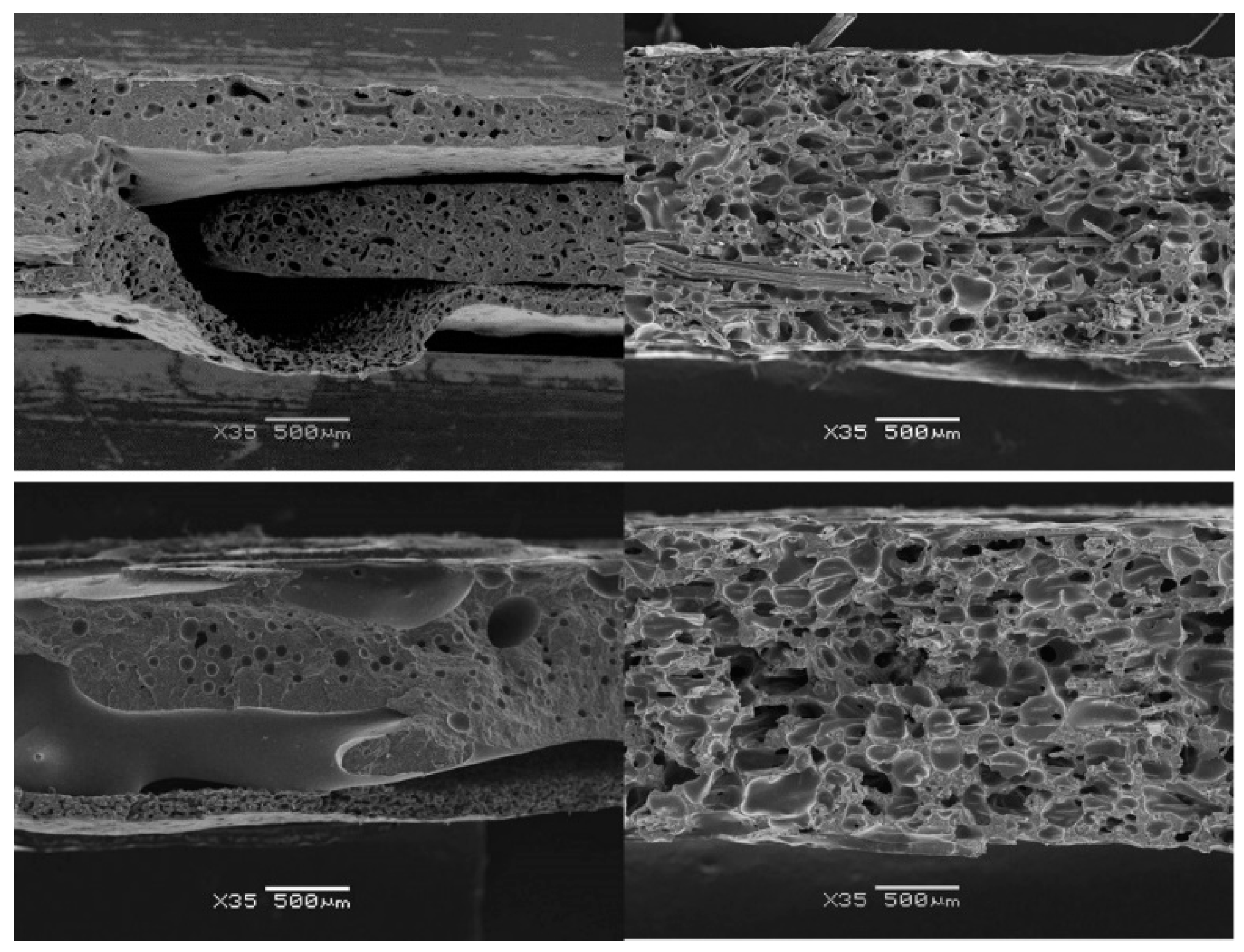
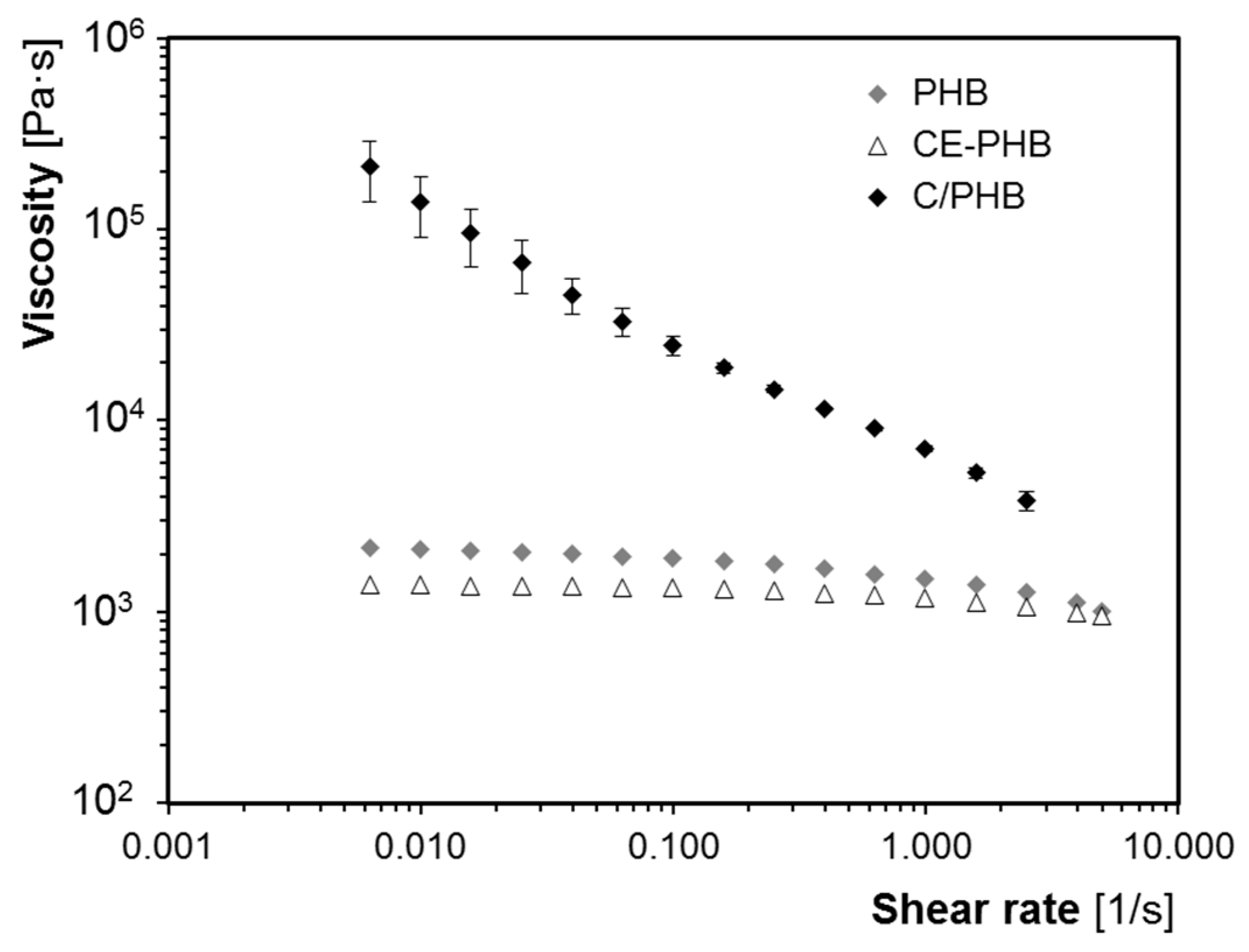
3.2. Influence of Fibres on the Foaming Process
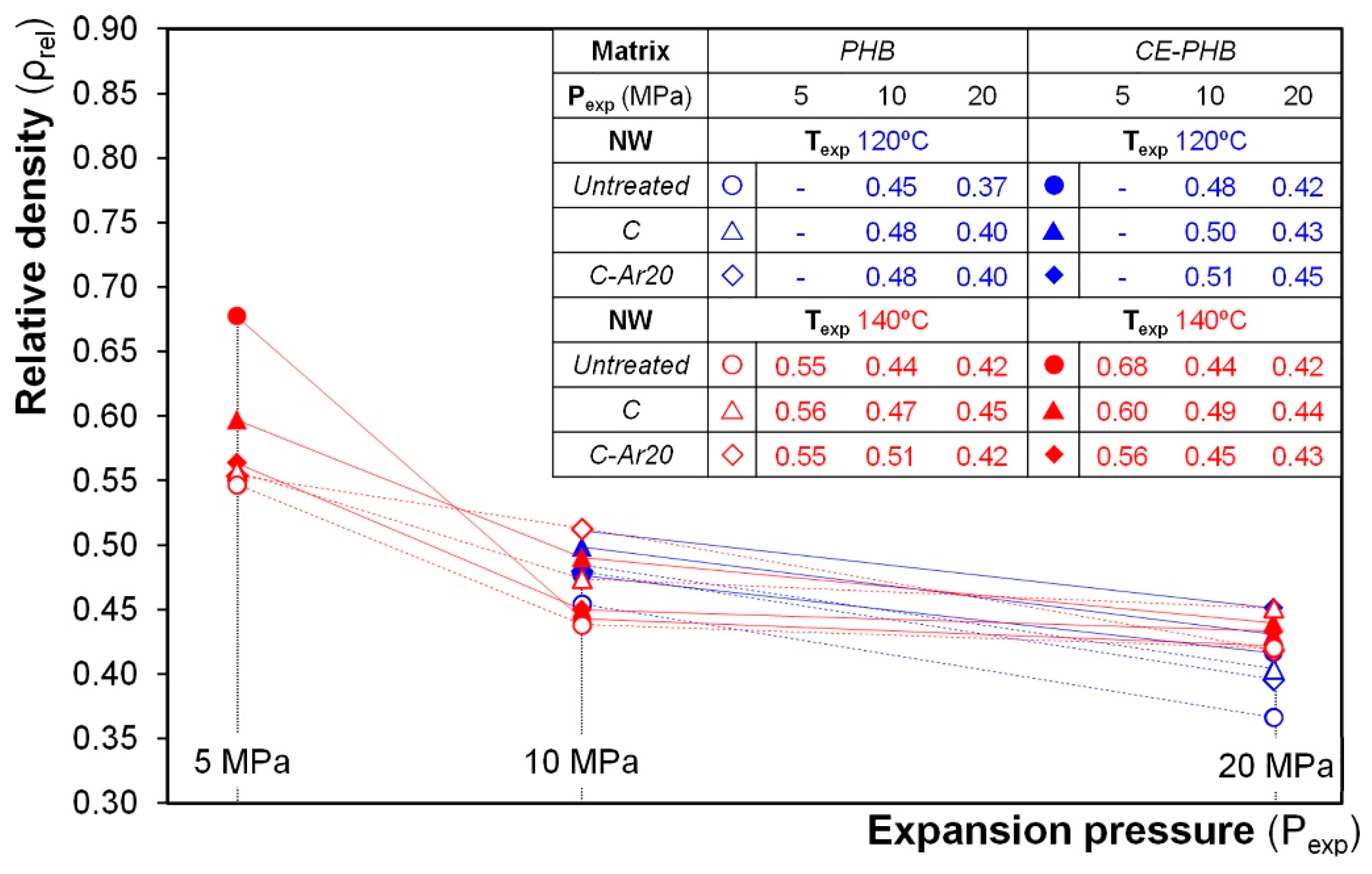
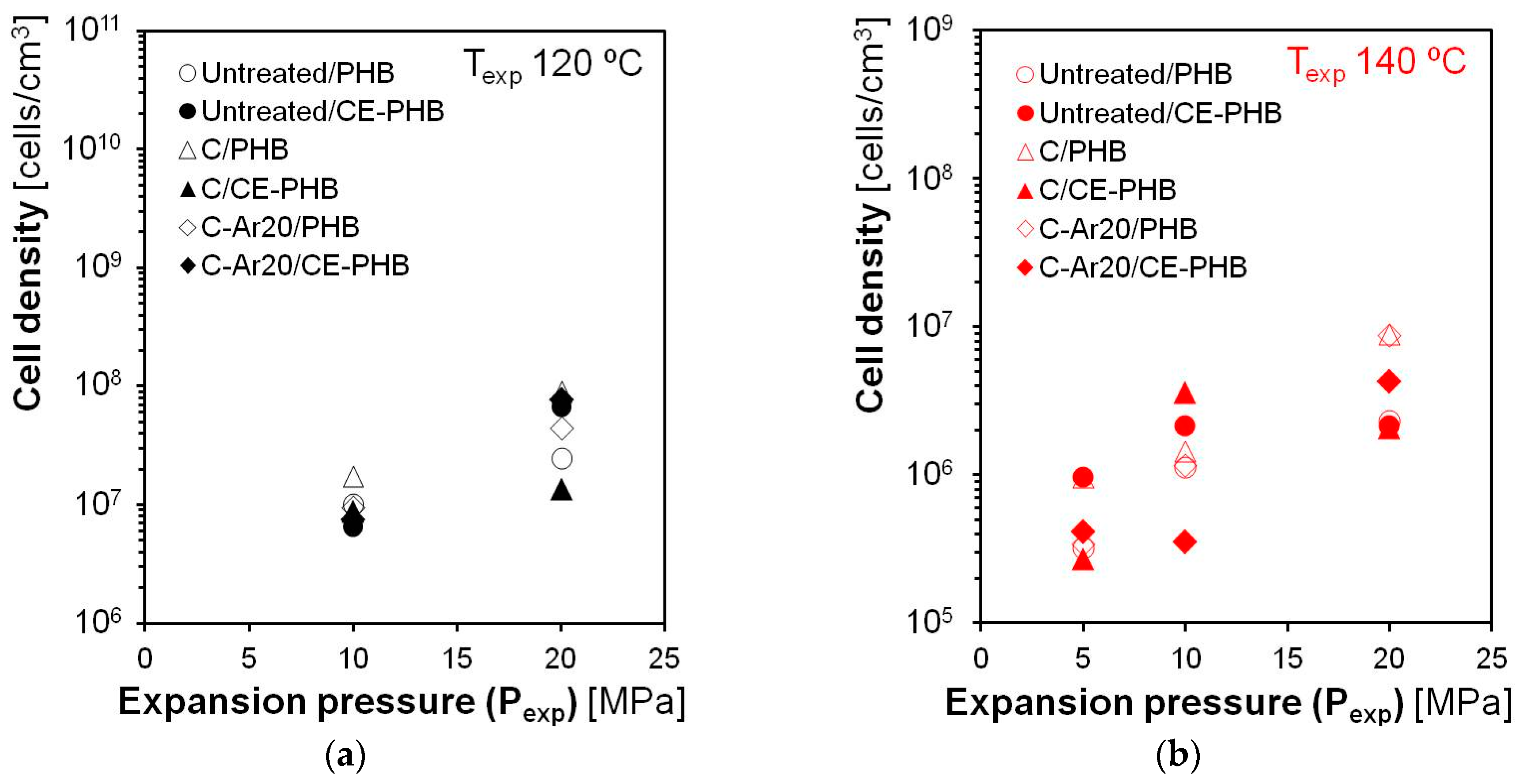
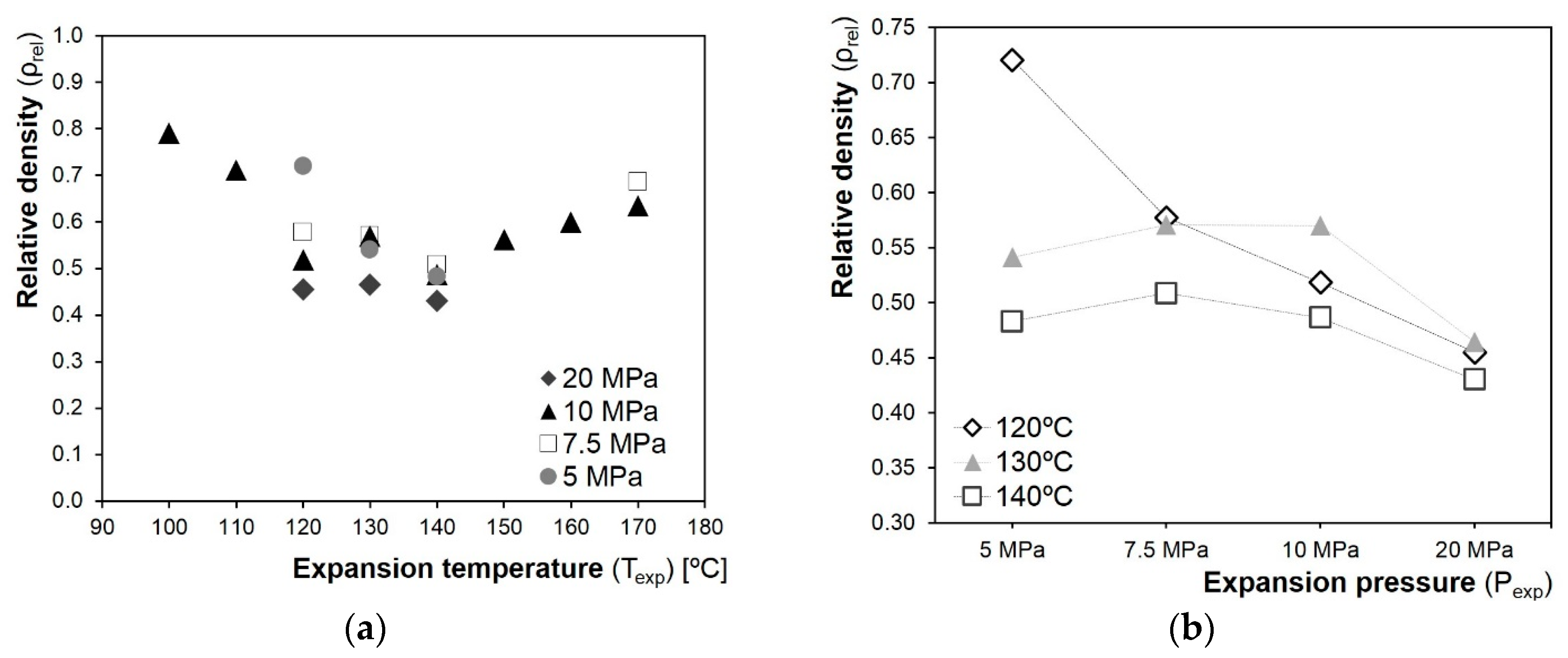
3.3. Influence of the Processing Conditions on the Cellular Composites’ Properties
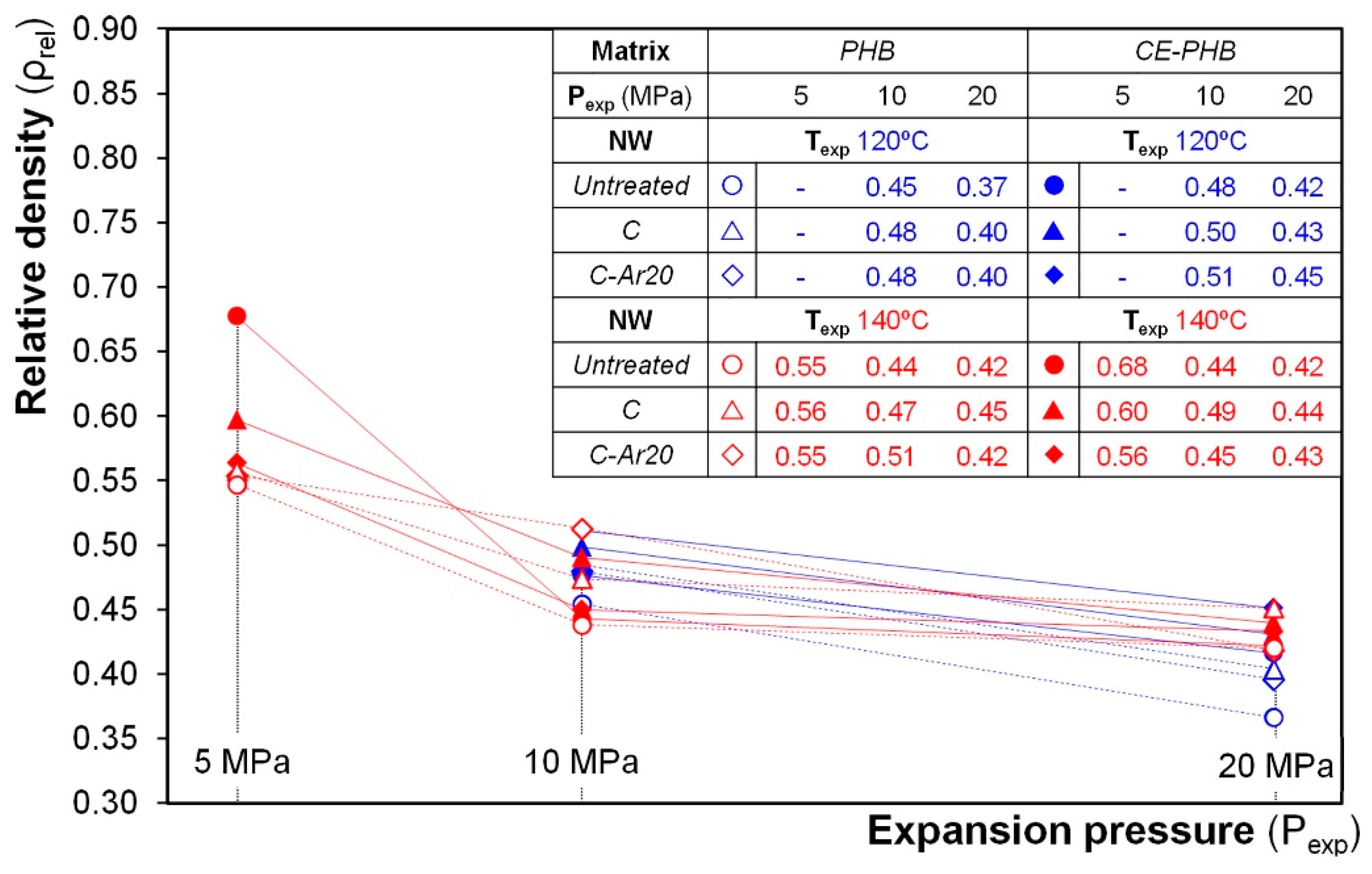
3.3.1. Density
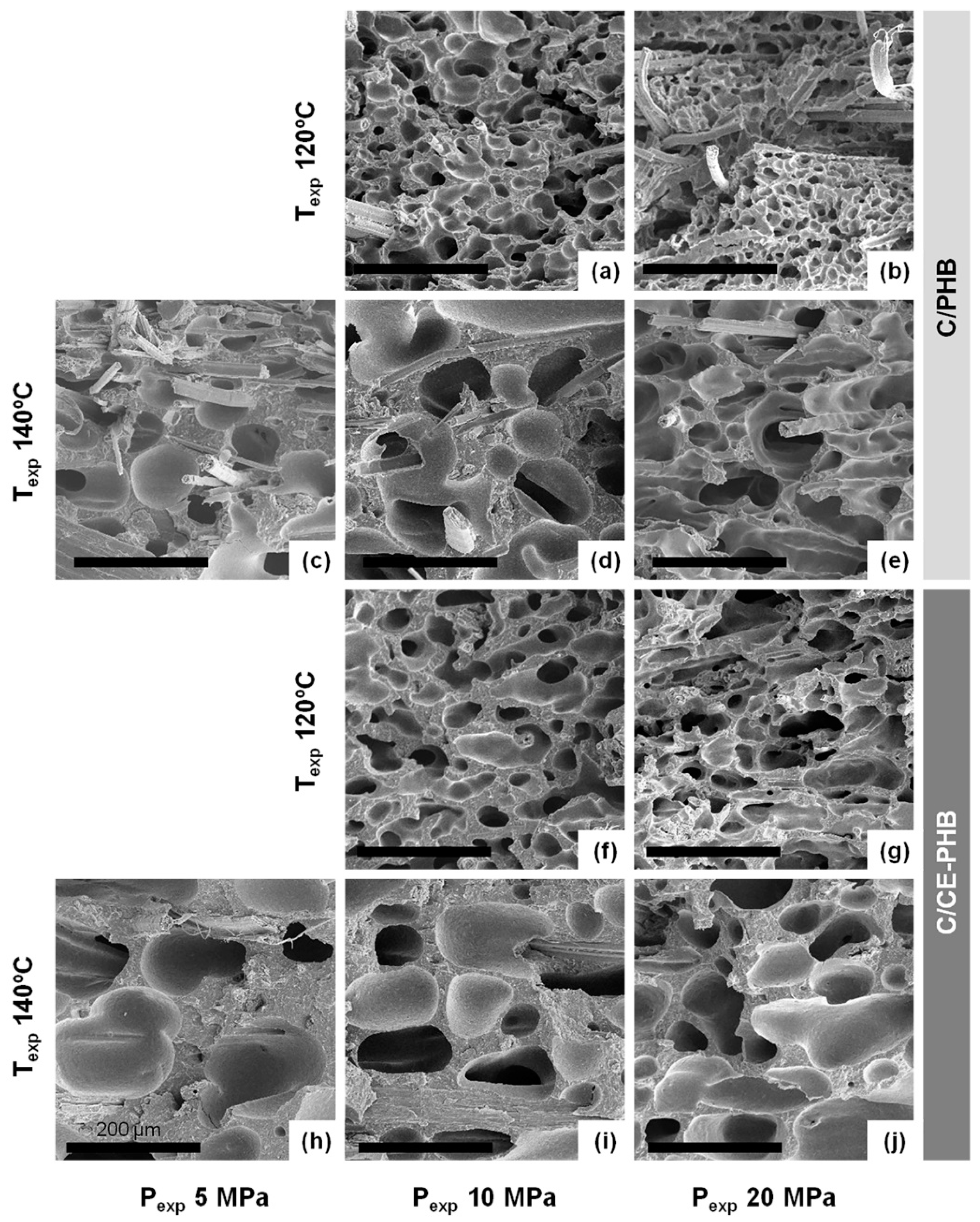
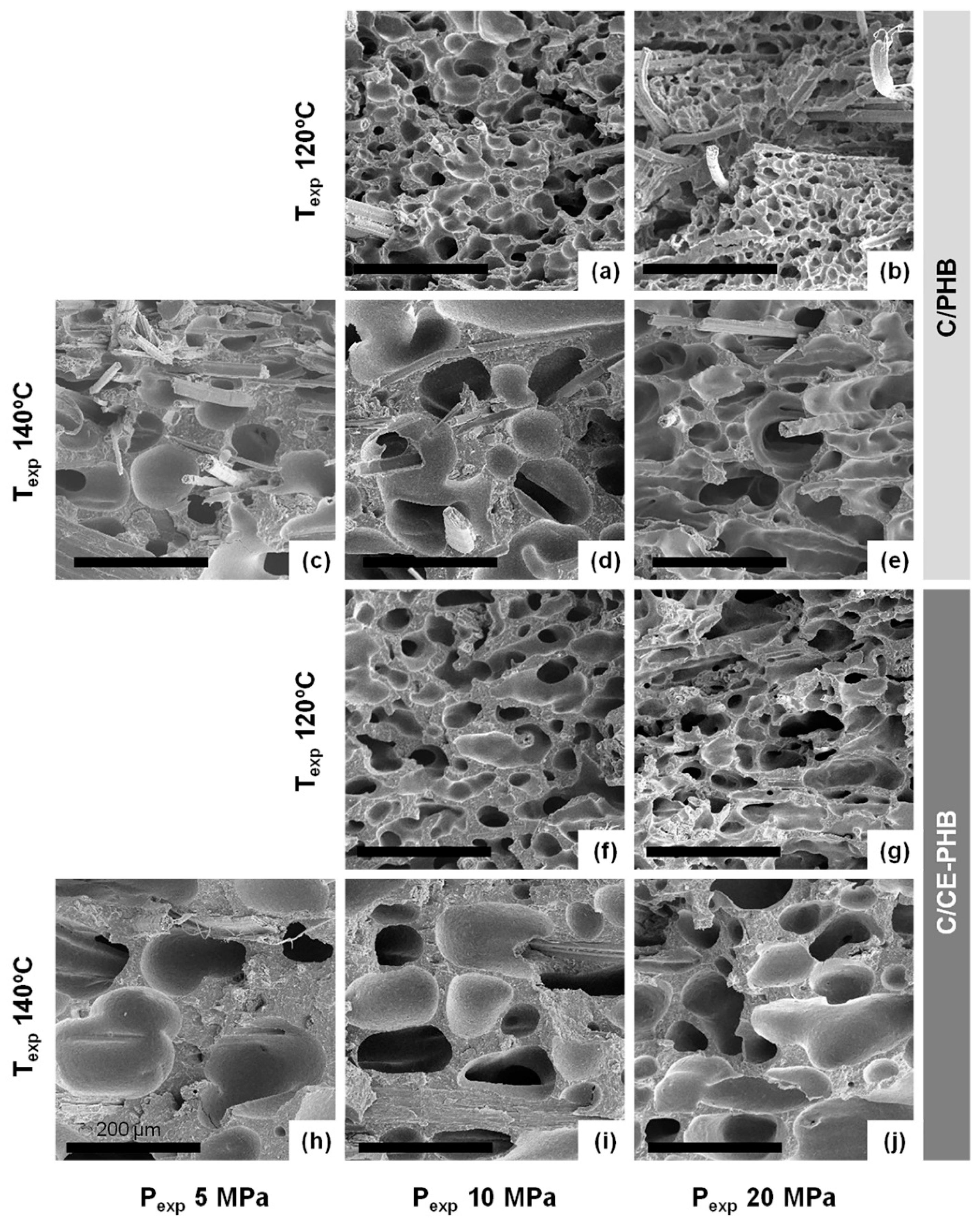
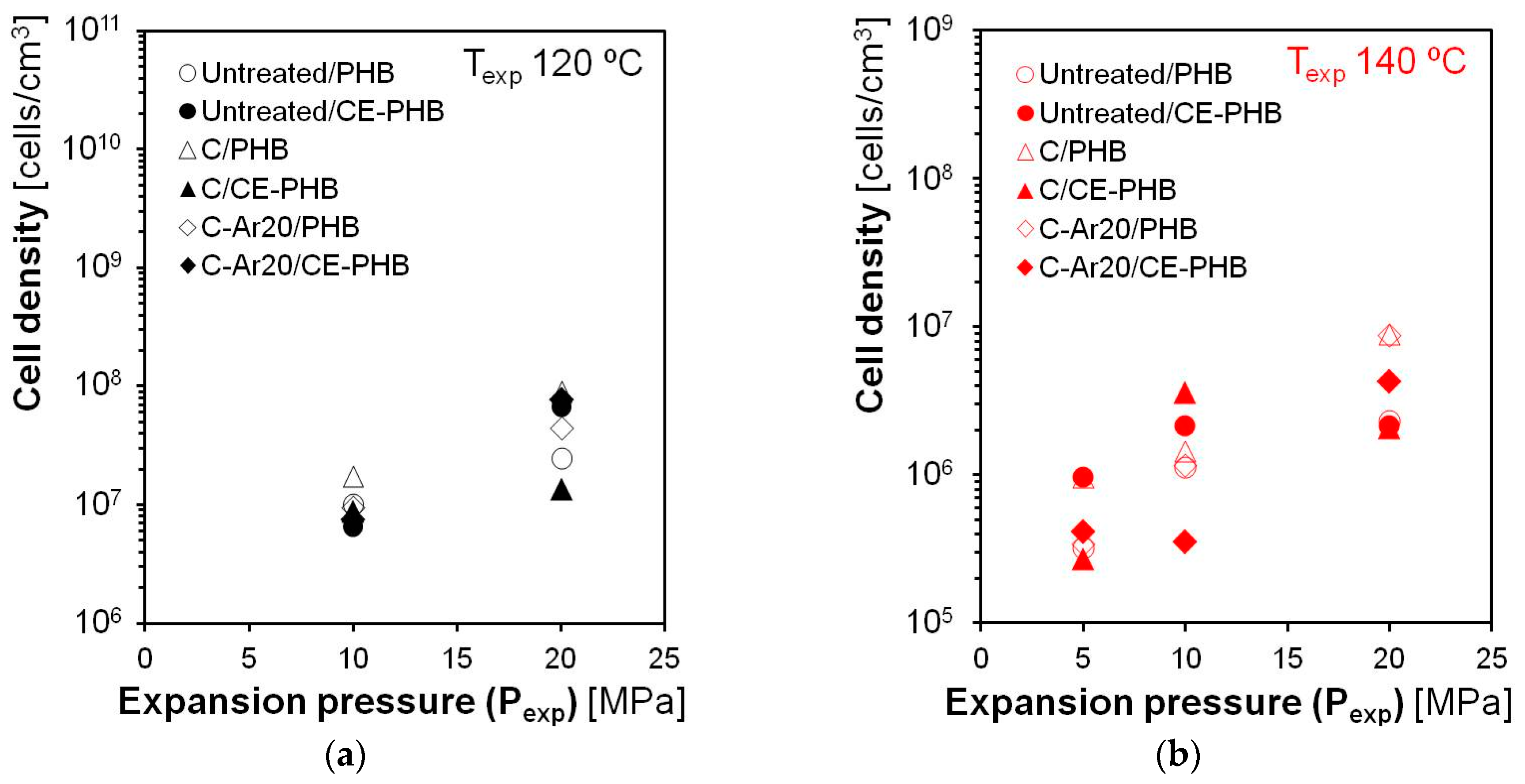
3.3.2. Cellular Structure
3.3.3. Mechanical Behaviour
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sorrentino, L.; Cafiero, L.; D’Auria, M.; Iannace, S. Cellular thermoplastic fibre reinforced composite (CellFRC): A new class of lightweight material with high impact properties. Compos. Part A Appl. Sci. Manuf. 2014, 64, 223–227. [Google Scholar] [CrossRef]
- Chanprateep, S. Current trends in biodegradable polyhydroxyalkanoates. J. Biosci. Bioeng. 2010, 110, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Netravali, A.N. Interfacial and mechanical properties of environment-friendly “green” composites made from pineapple fibers and poly(hydroxybutyrate-co-valerate) resin. J. Mater. Sci. 1999, 34, 3709–3719. [Google Scholar] [CrossRef]
- Graupner, N.; Müssig, J. A comparison of the mechanical characteristics of kenaf and lyocell fibre reinforced poly(lactic acid) (PLA) and poly(3-hydroxybutyrate) (PHB) composites. Compos. Part A Appl. Sci. Manuf. 2011, 42, 2010–2019. [Google Scholar] [CrossRef]
- Peterson, S.; Jayaraman, K.; Bhattacharyya, D. Forming performance and biodegradability of woodfibre–BiopolTM composites. Compos. Part A Appl. Sci. Manuf. 2002, 33, 1123–1134. [Google Scholar] [CrossRef]
- Barkoula, N.M.; Garkhail, S.K.; Peijs, T. Biodegradable composites based on flax/polyhydroxybutyrate and its copolymer with hydroxyvalerate. Ind. Crops Prod. 2010, 31, 34–42. [Google Scholar] [CrossRef]
- Bodros, E.; Pillin, I.; Montrelay, N.; Baley, C. Could biopolymers reinforced by randomly scattered flax fibre be used in structural applications? Compos. Sci. Technol. 2007, 67, 462–470. [Google Scholar] [CrossRef]
- Jeon, B.; Kim, H.K.; Cha, S.W.; Lee, S.J.; Han, M.-S.; Lee, K.S. Microcellular foam processing of biodegradable polymers—Review. Int. J. Precis. Eng. Manuf. 2013, 14, 679–690. [Google Scholar] [CrossRef]
- Ventura, H.; Laguna-Gutiérrez, E.; Rodriguez-Perez, M.A.; Ardanuy, M. Effect of chain extender and water-quenching on the properties of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) foams for its production by extrusion foaming. Eur. Polym. J. 2016, 85, 14–25. [Google Scholar] [CrossRef]
- Liao, Q.; Tsui, A.; Billington, S.; Frank, C.W. Extruded foams from microbial poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and its blends with cellulose acetate butyrate. Polym. Eng. Sci. 2012, 52, 1495–1508. [Google Scholar] [CrossRef]
- Szegda, D.; Daungphet, S.; Song, J.; Tarverdi, K. Extrusion foaming and rheology of PHBV. In Proceedings of the 8th International Conference on Foam Materials & Technology (Foams), Seattle, WA, USA, 28 September–1 October 2010. [Google Scholar]
- Wright, Z.C.; Frank, C.W. Increasing cell homogeneity of semicrystalline, biodegradable polymer foams with a narrow processing window via rapid quenching. Polym. Eng. Sci. 2014, 54, 2877–2886. [Google Scholar] [CrossRef]
- Pilla, S.; Kim, S.G.; Auer, G.K.; Gong, S.; Park, C.B. Microcellular extrusion-foaming of polylactide with chain-extender. Polym. Eng. Sci. 2009, 49, 1653–1660. [Google Scholar] [CrossRef]
- Ludwiczak, J.; Kozlowski, M. Foaming of polylactide in the presence of chain extender. J. Polym. Environ. 2015, 23, 137–142. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, W.; Zhang, H.; Park, C.B. Continuous processing of low-density, microcellular poly(lactic acid) foams with controlled cell morphology and crystallinity. Chem. Eng. Sci. 2012, 75, 390–399. [Google Scholar] [CrossRef]
- Duangphet, S.; Szegda, D.; Song, J.; Tarverdi, K. The effect of chain extender on poly(3-hydroxybutyrate-co-3-hydroxyvalerate): Thermal degradation, crystallization, and rheological behaviours. J. Polym. Environ. 2013, 22, 1–8. [Google Scholar] [CrossRef]
- Javadi, A.; Srithep, Y.; Lee, J.; Pilla, S.; Clemons, C.; Gong, S.; Turng, L.-S. Processing and characterization of solid and microcellular PHBV/PBAT blend and its RWF/nanoclay composites. Compos. Part A Appl. Sci. Manuf. 2010, 41, 982–990. [Google Scholar] [CrossRef]
- Javadi, A.; Srithep, Y.; Pilla, S.; Lee, J.; Gong, S.; Turng, L.-S. Processing and characterization of solid and microcellular PHBV/coir fiber composites. Mater. Sci. Eng. C 2010, 30, 749–757. [Google Scholar] [CrossRef]
- Boissard, C.I.R.; Bourban, P.-E.; Tingaut, P.; Zimmermann, T.; Manson, J.-A.E. Water of functionalized microfibrillated cellulose as foaming agent for the elaboration of poly(lactic acid) biocomposites. J. Reinf. Plast. Compos. 2011, 30, 709–719. [Google Scholar] [CrossRef]
- Le Moigne, N.; Sauceau, M.; Benyakhlef, M.; Jemai, R.; Benezet, J.-C.; Rodier, E.; Lopez-Cuesta, J.-M.; Fages, J. Foaming of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/organo-clays nano-biocomposites by a continuous supercritical CO2 assisted extrusion process. Eur. Polym. J. 2014, 61, 157–171. [Google Scholar] [CrossRef]
- Chauvet, M.; Sauceau, M.; Fages, J. Extrusion assisted by supercritical CO2: A review on its application to biopolymers. J. Supercrit. Fluids 2017, 120, 408–420. [Google Scholar] [CrossRef]
- Ventura, H.; Ardanuy, M.; Capdevila, X.; Cano, F.; Tornero, J.A. Effects of needling parameters on some structural and physico-mechanical properties of needle-punched nonwovens. J. Text. Inst. 2014, 105, 1065–1075. [Google Scholar] [CrossRef]
- Ventura, H.; Claramunt, J.; Navarro, A.; Rodriguez-Perez, M.; Ardanuy, M. Effects of wet/dry-cycling and plasma treatments on the properties of flax nonwovens intended for composite reinforcing. Materials 2016, 9, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, J.; Solorzano, E.; Rodriguez-Perez, M.A.; De Saja, J.A. Characterization of the cellular structure based on user-interactive image analysis procedures. J. Cell. Plast. 2013, 49, 555–575. [Google Scholar] [CrossRef]
- Colton, J.S. The nucleation of microcellular foams in semi-crystalline thermoplastics. Mater. Manuf. Process. 1989, 4, 253–262. [Google Scholar] [CrossRef]
- Takahashi, S.; Hassler, J.C.; Kiran, E. Melting behavior of biodegradable polyesters in carbon dioxide at high pressures. J. Supercrit. Fluids 2012, 72, 278–287. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Sample Type | Texp (°C) | Pexp (Mpa) | Specific Stiffness, E/ρ (GPa/g·cm−3) |
|---|---|---|---|---|
| PHB | Matrix | - | - | 1.11 |
| Untreated/PHB | Precursor | - | - | 1.78 |
| Foam | 120 | 10 | 1.40 | |
| 140 | 10 | 1.47 | ||
| 140 | 20 | 1.43 | ||
| C/PHB | Precursor | - | - | 1.75 |
| Foam | 120 | 10 | 1.81 | |
| 140 | 10 | 1.74 | ||
| 140 | 20 | 1.44 | ||
| C-Ar20/PHB | Precursor | - | - | 1.66 |
| Foam | 120 | 10 | 1.23 | |
| 140 | 10 | 1.50 | ||
| 140 | 20 | 1.43 | ||
| CE-PHB | Matrix | - | - | 1.41 |
| Untreated/CE-PHB | Precursor | - | - | 2.21 |
| Foam | 120 | 10 | 1.48 | |
| 140 | 10 | 1.58 | ||
| 140 | 20 | 1.37 | ||
| C/CE-PHB | Precursor | - | - | 2.42 |
| Foam | 120 | 10 | 1.62 | |
| 140 | 10 | 1.64 | ||
| 140 | 20 | 1.45 | ||
| C-Ar20/CE-PHB | Precursor | - | - | 2.04 |
| Foam | 120 | 10 | 1.70 | |
| 140 | 10 | 1.65 | ||
| 140 | 20 | 1.65 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventura, H.; Sorrentino, L.; Laguna-Gutierrez, E.; Rodriguez-Perez, M.A.; Ardanuy, M. Gas Dissolution Foaming as a Novel Approach for the Production of Lightweight Biocomposites of PHB/Natural Fibre Fabrics. Polymers 2018, 10, 249. https://doi.org/10.3390/polym10030249
Ventura H, Sorrentino L, Laguna-Gutierrez E, Rodriguez-Perez MA, Ardanuy M. Gas Dissolution Foaming as a Novel Approach for the Production of Lightweight Biocomposites of PHB/Natural Fibre Fabrics. Polymers. 2018; 10(3):249. https://doi.org/10.3390/polym10030249
Chicago/Turabian StyleVentura, Heura, Luigi Sorrentino, Ester Laguna-Gutierrez, Miguel Angel Rodriguez-Perez, and Monica Ardanuy. 2018. "Gas Dissolution Foaming as a Novel Approach for the Production of Lightweight Biocomposites of PHB/Natural Fibre Fabrics" Polymers 10, no. 3: 249. https://doi.org/10.3390/polym10030249