
Structural Aspects of Porphyrins for Functional Materials Applications


1
Department of Materials Science and Engineering, The Catholic University of America, Washington, DC 20064, USA
2
Department of Chemistry, The Catholic University of America, Washington, DC 20064, USA
3
Materials Measurement Science Division, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA
*
Author to whom correspondence should be addressed.
Crystals 2017, 7(7), 223; https://doi.org/10.3390/cryst7070223
Submission received: 19 April 2017
/
Revised: 22 June 2017
/
Accepted: 12 July 2017
/
Published: 15 July 2017
(This article belongs to the Special Issue Crystallography of Functional Materials)
Abstract
:Porphyrinic compounds comprise a diverse group of materials which have in common the presence of one or more cyclic tetrapyrroles known as porphyrins in their molecular structures. The resulting aromaticity gives rise to the semiconducting properties that make these compounds of interest for a broad range of applications, including artificial photosynthesis, catalysis, molecular electronics, sensors, non-linear optics, and solar cells. In this brief review, the crystallographic attributes of porphyrins are emphasized. Examples are given showing how the structural orientations of the porphyrin macrocycle, and the inter-porphyrin covalent bonding present in multiporphyrins influence the semiconducting properties. Beginning with porphine, the simplest porphyrin, we discuss how the more complex structures that have been reported are described by adding peripheral substituents and internal metalation to the macrocycles. We illustrate how the conjugation of the π-bonding, and the presence of electron donor/acceptor pairs, which are the basis for the semiconducting properties, are affected by the crystallographic topology.
1. Introduction
Porphyrins are defined as a group of heterocyclic macrocycle organic compounds, composed of four modified pyrrole subunits interconnected at their carbon atoms via methine (=CH–) bridges [1]. Since the porphyrin macrocyclic structure was first proposed by Küster over a century ago [2], research in the field has grown dramatically, resulting in a vast body of literature, which is continuing to expand at a rapid pace. As an indication, the current series of the “Handbook of Porphyrin Science,” initiated in 2010, at present encompasses 44 volumes containing a total of 214 chapters [3]. In addition to purely scientific importance, as evidenced by the award of several Nobel prizes for porphyrin-related research through the years [4], much of the recent growth has been driven by the potential for applications in materials science and engineering [5,6]. A large part of the progress to date has been facilitated by the application of X-ray crystallography (including synchrotron) and neutron crystallography to the determination of porphyrin crystal structures. Currently, more than 4000 porphyrin crystal structures are reported in the Cambridge Structural Database (CSD) [7]. It is the objective of the present paper to provide a brief outline of what is known of porphyrin solid state structures from a crystallographic perspective, in the hope that it may be useful to materials scientists as they become involved with porphyrin research. To ensure adequate focus in this review, in general we do not include porphyrin derivatives and related compounds such as corrins, chlorins, phthalocyanines, metal-organic frameworks (MOFs) and covalent organic frameworks (COFs).
2. Porphine, the Parent Compound
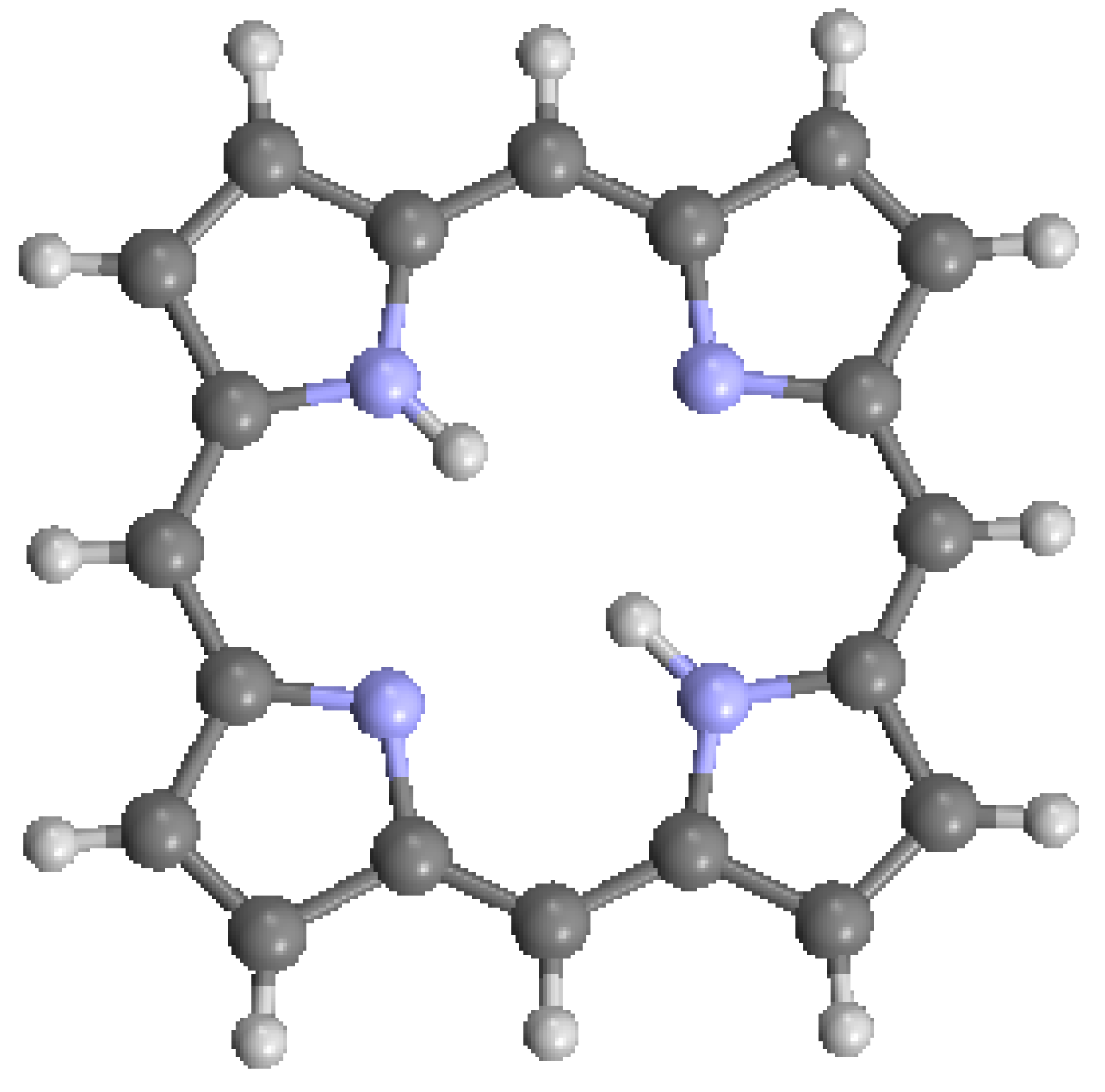
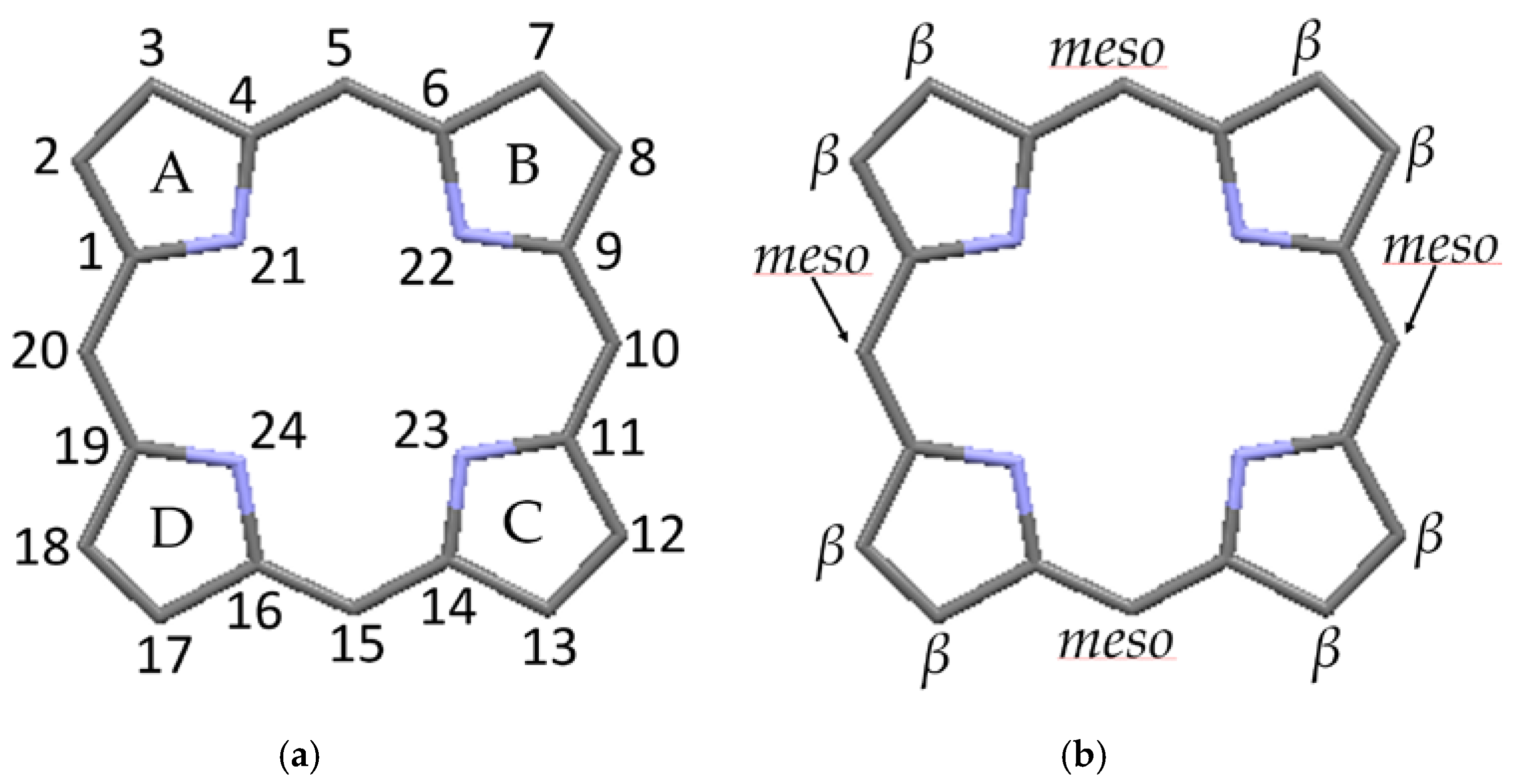
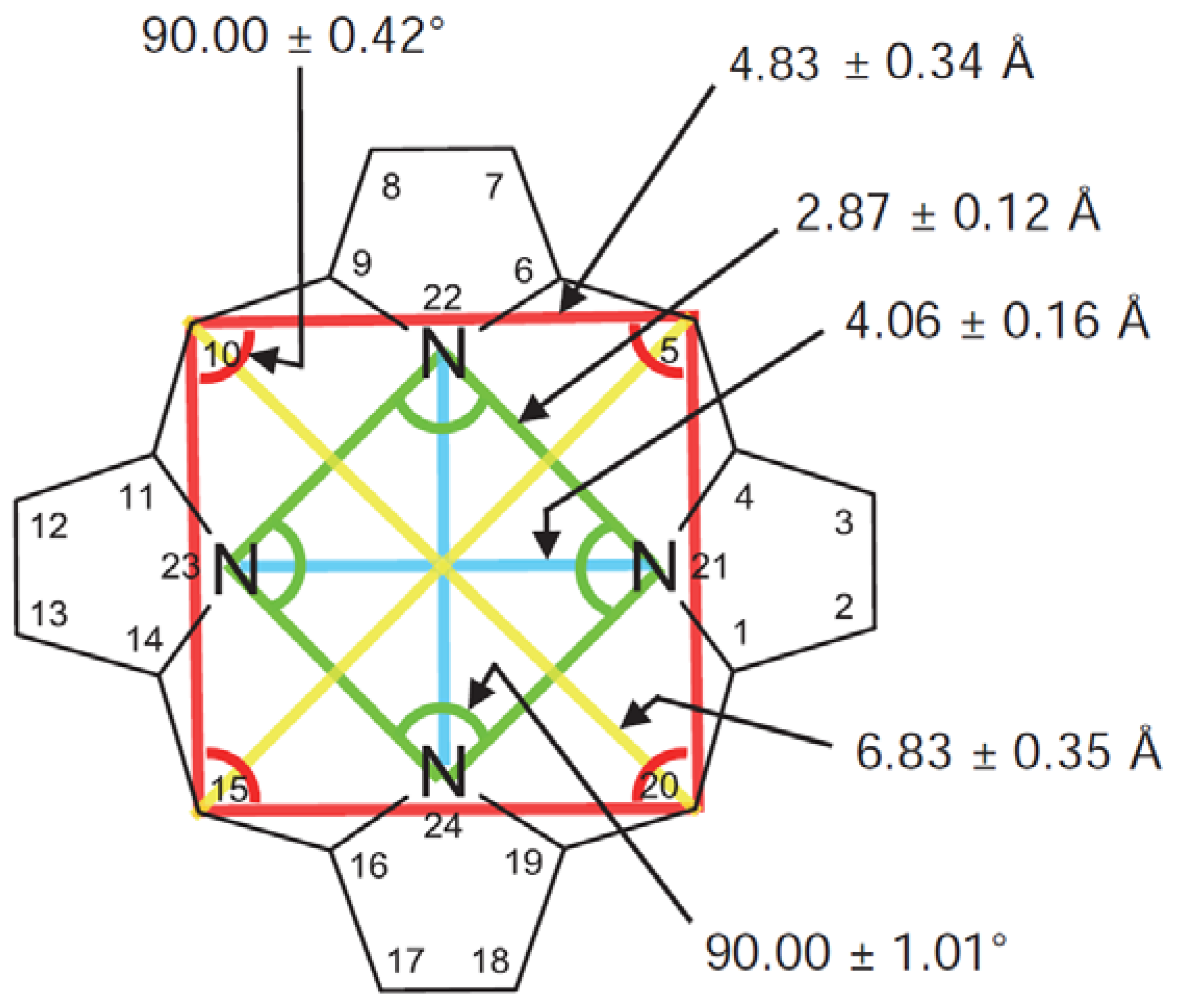
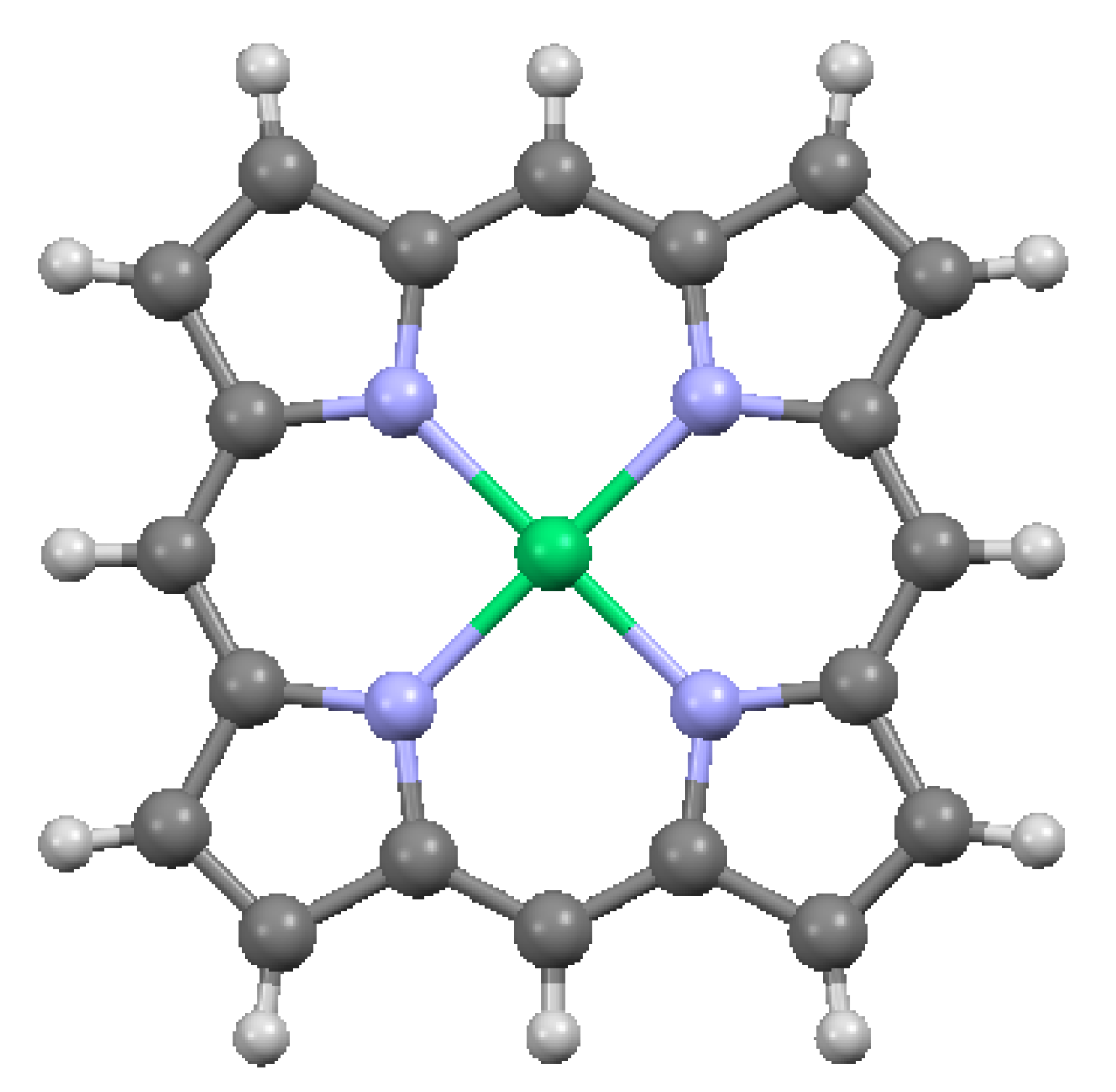
The structure of the porphyrin parent compound, porphine (C20H14N4), which forms a stable solid at room temperature, is shown in Figure 1. The various carbon and nitrogen positions have been indicated in Figure 2 according to the nomenclature suggested by IUAPC [8]. The carbons of the macrocycle are numbered from 1 to 20, and the four interior nitrogens are at positions 21–24 in Figure 2a. The nitrogens at positions 21 and 23 are bonded to hydrogens, which project toward the interior of the macrocycle. Numerous sidearm substituents are possible at the β and meso sites shown in Figure 2b, but in the basic porphine structure, these substitutions are lacking. Mean bond angles and distances for porphine macrocycles are indicated in Figure 3. These macrocycle dimensions are relatively constant for all porphyrins, including complex multiporphyrins, as well as the simpler porphine derivatives.
The crystallographic parameters of porphine are given in Table 1. The structure is monoclinic, with space group P 21/c. The unit cell structure (not shown) consists of four stacked, but offset macrocycles with an interlayer spacing of 3.355 Å. The macrocycles themselves are planar, coplanar with each other, and stacked in a rotated, offset manner. In more complex porphyrins, the planarity of the individual macrocycles may undergo distortions of up to a few tenths of Ångstroms, termed “ruffling” or “doming”.
3. Peripheral Substituents
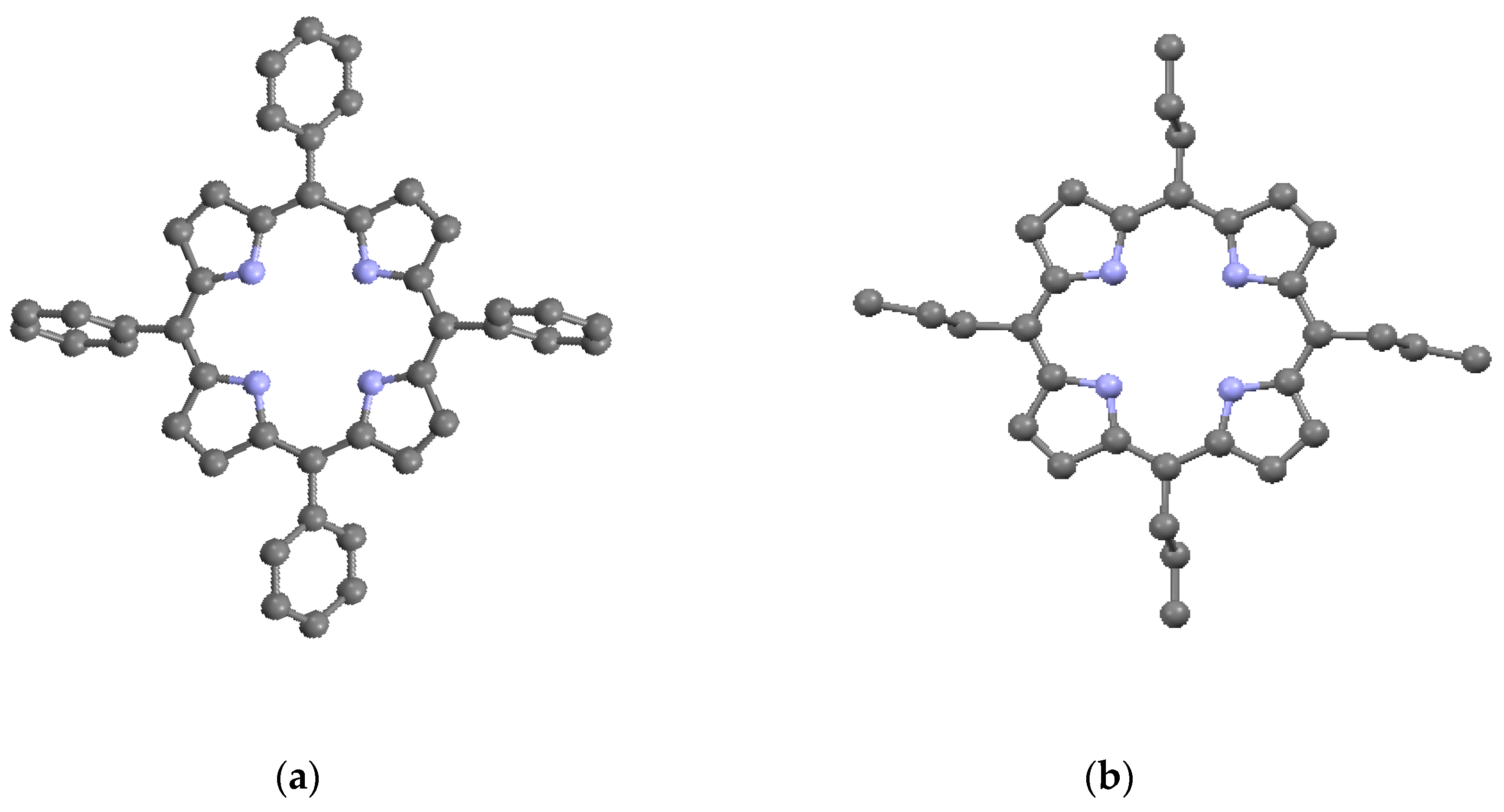
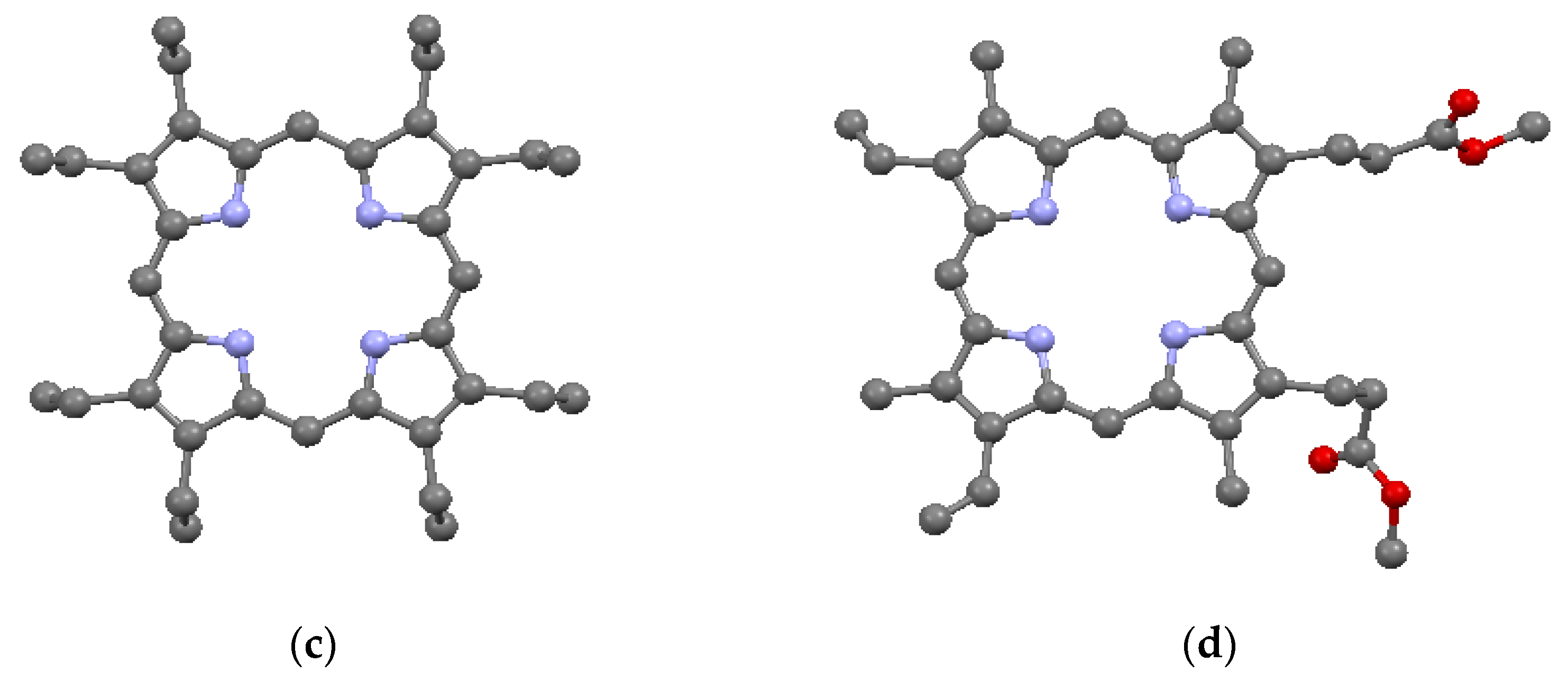
The porphyrin macrocycle is capable of accommodating several different types of substituents by bonding to the external carbons in the β- and the meso-positions on the ring. Simple substitutions based on the porphine structure lead to the common porphyrins given in Table 2 and shown in Figure 4a–c; these are, respectively, tetraphenyl porphyrin (TPP) (C44H30N4), tetrapropyl porphyrin (TPrP) (C32H38N4), and octaethyl porphyrin (OEP) (C36H46N4). TPP and TPrP have phenyl (–C6H5) and propyl (-CH2CH2CH3) substitutions, respectively, on the four meso-positions, while OEP has ethyl (-CH2CH3) substitutions on the eight β-positions. Somewhat more complicated substitutions on the β-positions lead to protoporphyrin IX (PP9) (C36H38N4O4), as described in Table 2 and Figure 4d. PP9 is a well-known compound closely related to hemin [11]. The β substituents on PP9 are not symmetrical, with four methyl (–CH3) groups on β-positions 3, 8, 12, and 18, two vinyl (–CH=CH2) groups on β-positions 2 and 7, and two propionic acid (-CH2CH2COOH) groups on β-positions 13 and 17. Among the substituents, two of the TPP phenyls and the two PP9 propionic acids have the greatest steric deviations from the macrocycle plane. In Table 2, it can be seen that the substituents have lowered the densities and most of the crystal symmetries relative to the basic porphine of Table 1. However, in general, the presence of simple substituents does not substantially affect the crystallographic parameters of the basic macrocycle itself. Most instances of macrocycle distortion involve metalation and/or the presence of complex substituents requiring non-planar inter-porphyrin bonding arrangements.
4. Metalation of the Macrocycle Core
Another structural variation possible involves deprotonating the interior nitrogens of the macrocycle at positions 21 and 23 and inserting a metal ion, which is bonded to all four nitrogens. In some cases, to satisfy charge balance, metalation may be accompanied by axial bonding of the metal to a cation such as a halide. In other situations, coordinate covalent bonding between the metal and a neutral ligand may occur. More than 50 metals and semi-metals have been inserted in various porphyrins.
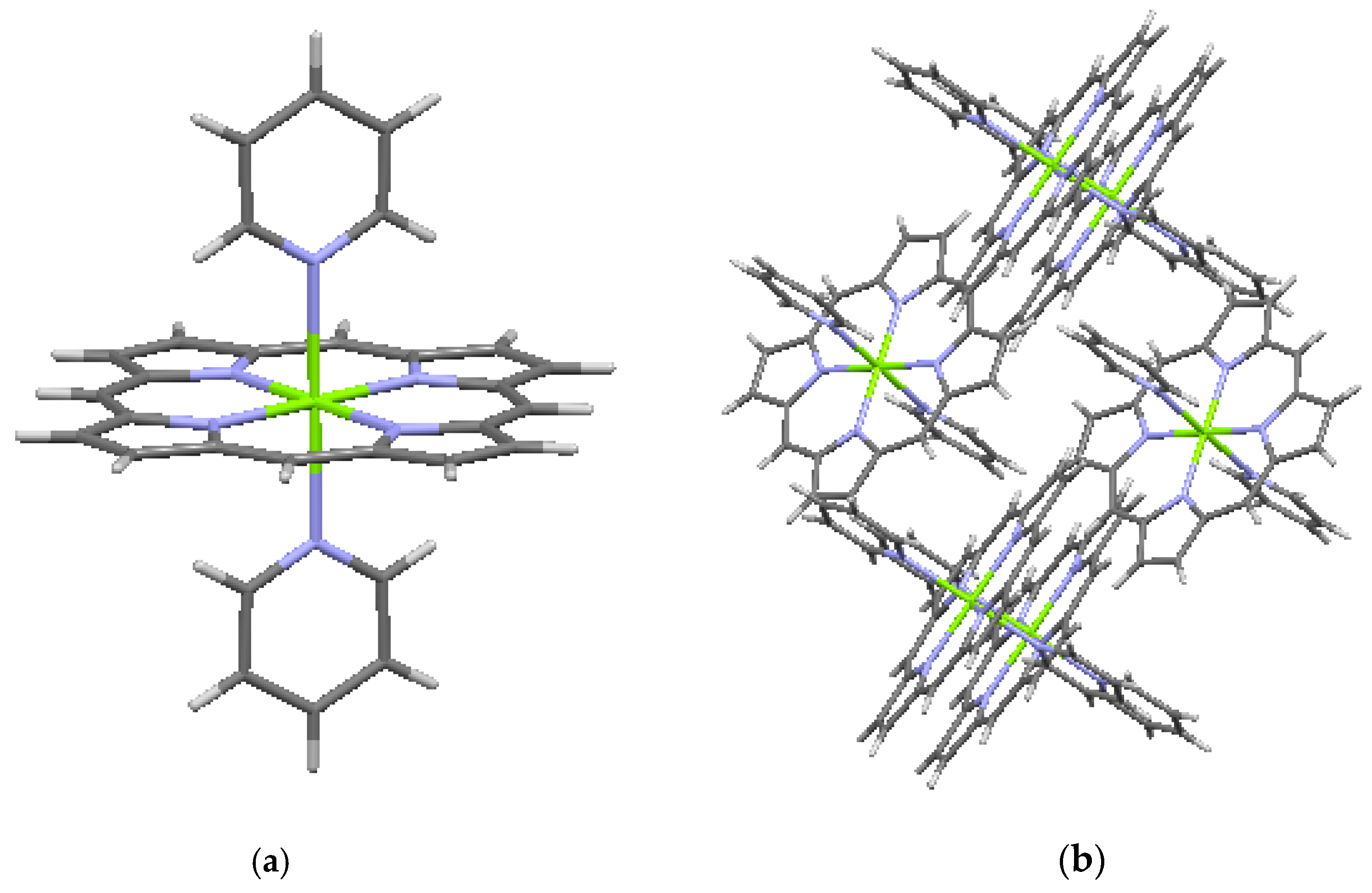
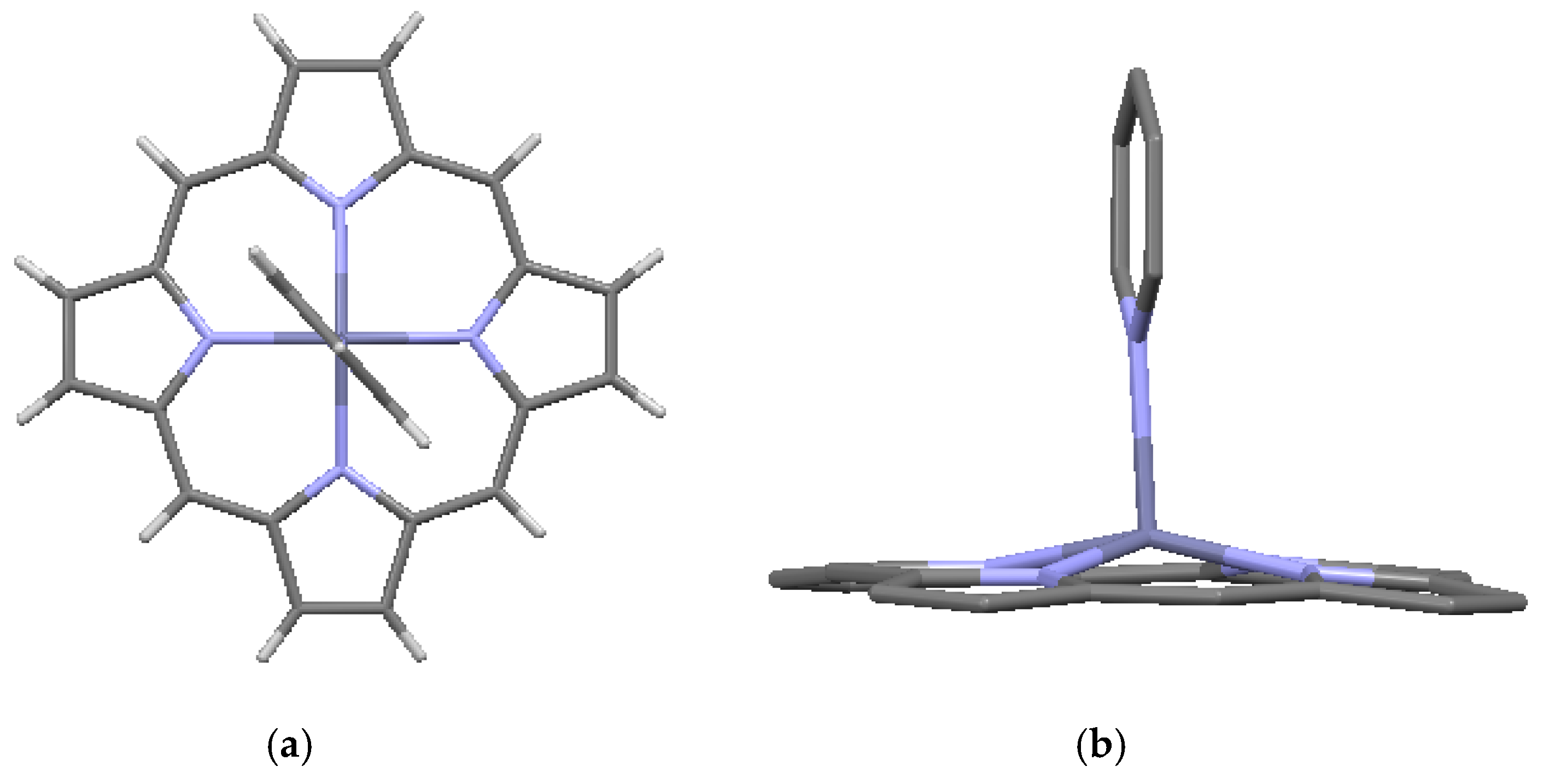
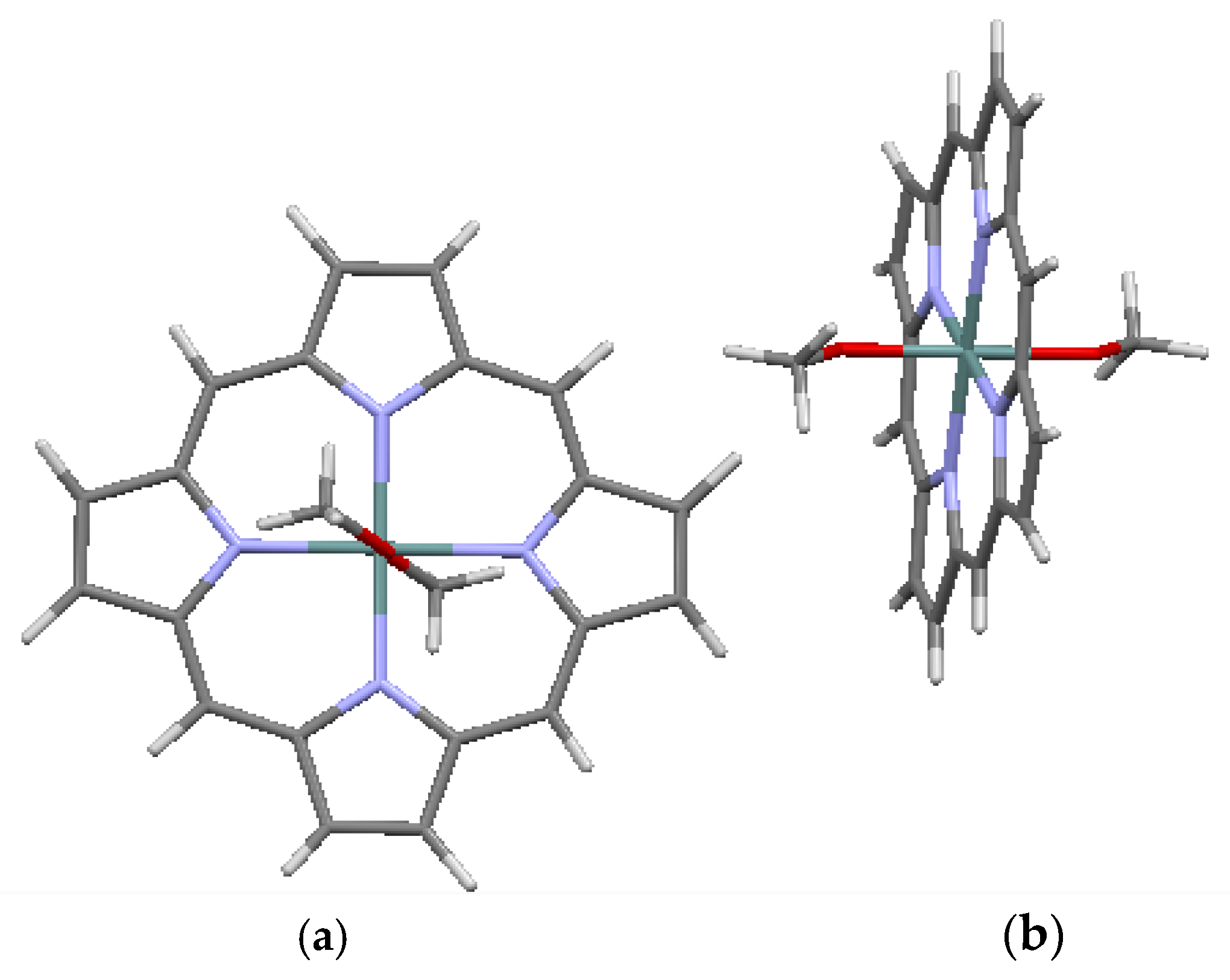
Crystallographic data are available for several metalated porphines, of which data for four compounds are included in Table 3. Included in the table are a Ni-metalated variant, a Mg-metalated variant, a Zn-metalated variant, and a Ge-metalated variant. All are monoclinic with space group P 21/c, with the exception of the Mg variant, which is monoclinic with space group C 2/c. In the Ni-metalated variant, the Ni is coplanar with the macrocycle (Figure 5) and the structure is otherwise similar to that of porphine, with an interlayer distance of 3.347 Å. In the Mg-metalated variant (Figure 6a) the Mg is also coplanar with the macrocycle, but is axially bonded to pyridine groups (C5H5N) above and below the plane of the macrocycle. This results in a more complicated stacking arrangement in which the macrocycles of successive layers are not all coplanar, but the pyridine extensions are nearly parallel and coplanar with one another (Figure 6b). The distance between the canted macrocycles, which are closest at the edges, is 3.315 Å, less than the interplanar stacking distance in porphine. In the Zn-metalated variant, the Zn is positioned slightly above the plane of the macrocycle and is axially bonded to the nitrogen of a single pyridine (Figure 7a,b). The plane of the macrocycle is puckered upward slightly (domed) toward the Zn (Figure 7b). In the Ge-variant, which contains two axially bonded methoxide (–CH3O) units on opposite sides of the macrocycle, the Ge is in the plane of the macrocycle (Figure 8). As listed in Table 3, the root-mean-square displacement of atoms from the mean macrocycle plane is as follows: Ni-porphine, 0.0191 Å; Mg-porphine, 0.0242 Å; Zn-porphine, 0.0873 Å; and Ge-porphine, 0.0157 Å [10]. The Zn-porphine atoms have the highest RMS departure from the mean plane due to lifting of the Zn by coordination with a single pyridine. The compounds are all monoclinic; three have the same space group as the basic porphine (Table 1). As expected, all have higher densities than the porphine, although the Mg-variant has substantially lower density than the other three, presumably because the lower atomic mass of Mg is supplemented by the less efficient crystal packing of the bulkier molecules.
Devillers et al. [10] have calculated average bond lengths and bond angles for the pyrrole units among a sampling of eight porphine macrocycles (including both non-metalated and metalated, but with no substituents). The results do not deviate significantly from the mean values. Thus, based on this small sample, the effect of metalation by itself on the crystallographic dimensions of the macrocycle appears to be small. However, the presence of ligands axially bonded to the metals may sterically require differences in the overall stacking mode, which can affect the overall crystal properties.
As noted by Fleischer [19], the coordination of the metal in metalated porphyrins, depending on the metal, may be described as: four-coordinated (square-planar), five-coordinated (square pyramidal), six-coordinated (distorted octahedral), or eight-coordinated (square-antiprismatic, as when sandwiched in a dimer). The type of coordination is influenced by the size of the metal ion. Divalent metals that can readily fit into the plane of the macrocycle core can have square-planar coordination, as shown for the nickel in Figure 5. High-spin ferric iron has five-coordination, with a halogen at the apex, and the four nitrogens at the base of the coordination pyramid. The germanium in Figure 8 can be regarded as octahedrally coordinated. Seven-coordination is also possible when the metal is out of the macrocycle plane. The metal coordination can have a substantial effect on the optical absorption spectrum and the electrical and magnetic properties.
5. Role of Covalent Inter-Molecular Bonding
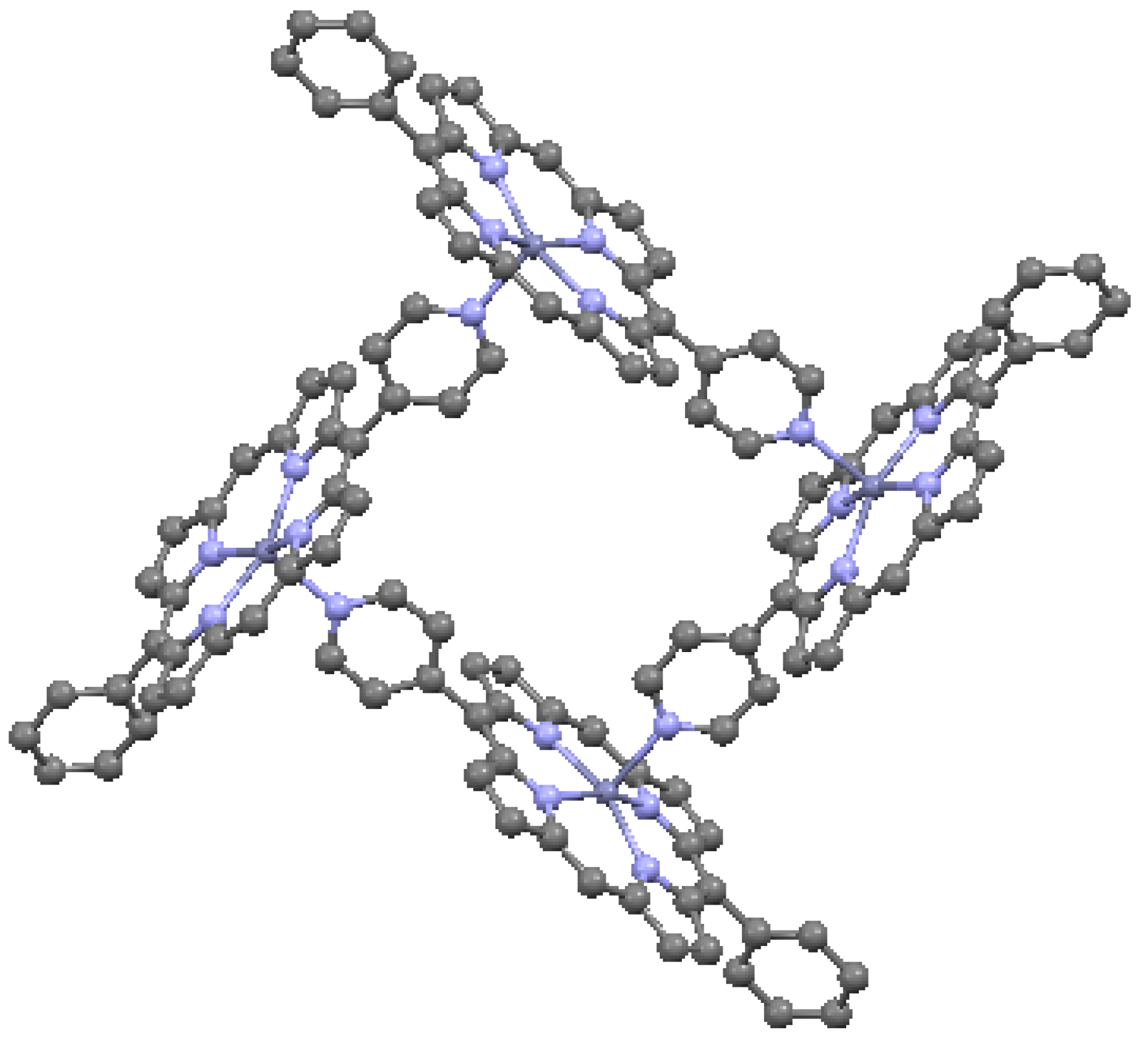
Many porphyrins crystallize as molecular compounds. Intermolecular interactions are important in determining the overall crystallographic properties and include hydrogen bonding, van der Waals bonding, π–π stacking, covalent bonding, coordinate covalent bonding, and electrostatic interactions. As a result of the first two types of interaction, discrete porphyrin molecules are attached to one another in the crystal structure by hydrogen- or van der Waals-bonds. For example, we have described the presence of hydrogen bonding in hemin, an Fe-porphyrin [11]. Coordinate covalent bonding can be important for the configuration of neutral axial ligands. Electrostatic interactions may play a role in the self-assembly of porphyrin molecules. As noted throughout this review, π–π conjugation plays an essential role in determining electronic properties. Covalent intermolecular bonding in porphyrins, by virtue of the geometrical control exerted, has a direct effect on π–π stacking. Relative to hydrogen and van der Waals bonding, covalent bonding results in the linking of molecules by stronger bridges. Such bridges include C2H2, C2H4, –N=N–, =C=C=, –S–S–, –C(OH)–, etc., which link porphyrins to form crystalline dimers, trimers or other oligomers [20]. Covalency can occur as meso-meso, β-β, or meso-β links. The geometries of the linkages are controlled by the bond angles present in the bridges. The C2H2 bridge allows for some flexibility in crystal packing as it can fold to allow more compact crystal packing. In linkages involving C2H4, rotational flexibility of the linear –C–C– single bond also allows for some flexibility in molecular packing. Table 4 gives parameters for two examples of covalent dimer bonding, as illustrated in Figure 9 and Figure 10. In Figure 9, the –N=N– meso-meso bridge linking the TPP dimer has a zig-zag geometry that results in a displacement of the macrocycles horizontally and vertically relative to one another, but which nonetheless allows them to remain nearly parallel with one another. In Figure 10, the OEP dimer is β-β linked at a high angle due to the angularity of the –C(OH) – bridge. Thus, there are many possibilities for the formation of 3-D molecular structures based on the packing of covalently-bonded oligomeric macrocycles; structures are influenced and limited by the nature of the linkages, as well as by the steric chemistry of the substituents on the linked macrocycles. Structural possibilities are further extended by the fusion or direct linkage of macrocycles.
6. Electronic Properties as Related to Structure
The semiconducting nature of porphyrins is of great interest to materials scientists for applications ranging from artificial photosynthesis to catalysis, molecular electronics, sensors, non-linear optics, and charge separation in solar cell materials. The semiconducting properties arise from the aromaticity of the macrocycle, which leads to a significant electron mobility. In particular, stacking of the macrocycles in crystal structures such that there is close proximity of the π-bonding regions results in enhanced capability for electron transfer. Additionally, π-bonding conjugation may occur in planes parallel to the macrocycle through covalent linkages, as outlined above. Furthermore, in certain structures, porphyrins act as electron donors when bonded to electron acceptors, such as fullerenes. By varying the linkages in these and other dyads, the kinetics of electron–hole recombination can be tuned. Factors important in optimizing porphyrin semiconducting properties include the planarity of the macrocycle, and its parallelism with others in the structure. When discussing electronic properties, it is important to differentiate among different structural regimes, i.e., whether molecular, single-crystal, thin-film, or bulk-polycrystalline environments are being considered. Thin-film materials may have the added complexity of epitaxy or texturing, which unless fully characterized, can make precise interpretation of data difficult, as electrical conductivity is a second-rank tensor property.
6.1. Properties at the Molecular Level
Porphyrins, including fused or aromatically linked linear architectures, have a number of properties at the molecular level which make them suitable for electronic applications [24]. They can be readily oxidized to π radical cations, or reduced to π radical anions; the HOMO/LUMO energy gap is generally only about 2 eV; metal chelation at the core allows control over the redox properties; selection of outer ring substituents gives additional control over solubilities and functionalities. Solubilities in various solvents allow for flexibility in the use of chemical deposition methods. Porphyrins are robust; many are stable under ambient conditions, as well as having thermal stability up to 300 °C or 400 °C [25]. Jurow et al. [26] have reviewed the potential applications of porphyrins in functional molecular electronic devices such as diodes, rectifiers, wires, capacitors, and electronic or magnetic memory. Many molecular applications involve self-assembled monolayers (SAMs) on electronically active substrates, which can then be patterned and assembled into nano- or micro-scale devices. Surface effects are of prime importance; the fabrication and performance of 2-D porphyrin structures have been recently reviewed by [27]. However we are concerned here mainly with the crystallographic controls on molecular electronic properties.
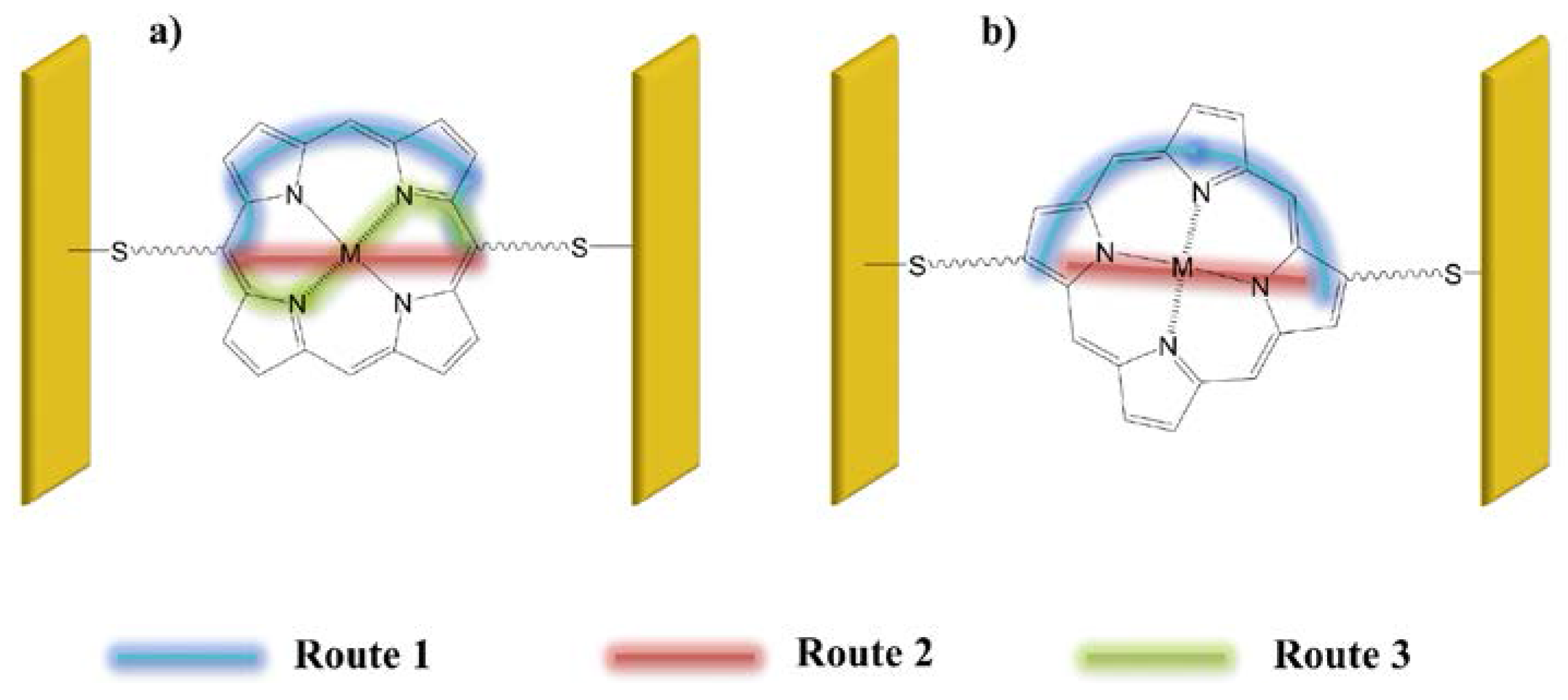
It is known that in molecules connected to electrodes with tunneling barriers, electron currents are determined by relative HOMO and LUMO spacing, spin, and vibrational modes [28,29]. Figure 11 shows three possible transport paths within a porphyrin macrocycle, depending on whether connection is made at meso sites or at β sites. Route (1) involves transport through the conjugated bonds of the macrocycle and avoids the metal; route (2) takes the electrons straight across the macrocycle, through the metal; and route (3) involves conjugated bonds as well as the metal. Theoretical calculations [30] predict that if the porphyrin is connected via opposite meso sites, transport is most likely to occur via the conjugated bonds. On the other hand, if linkages are made via β sites, the transport will be controlled by the central metal.
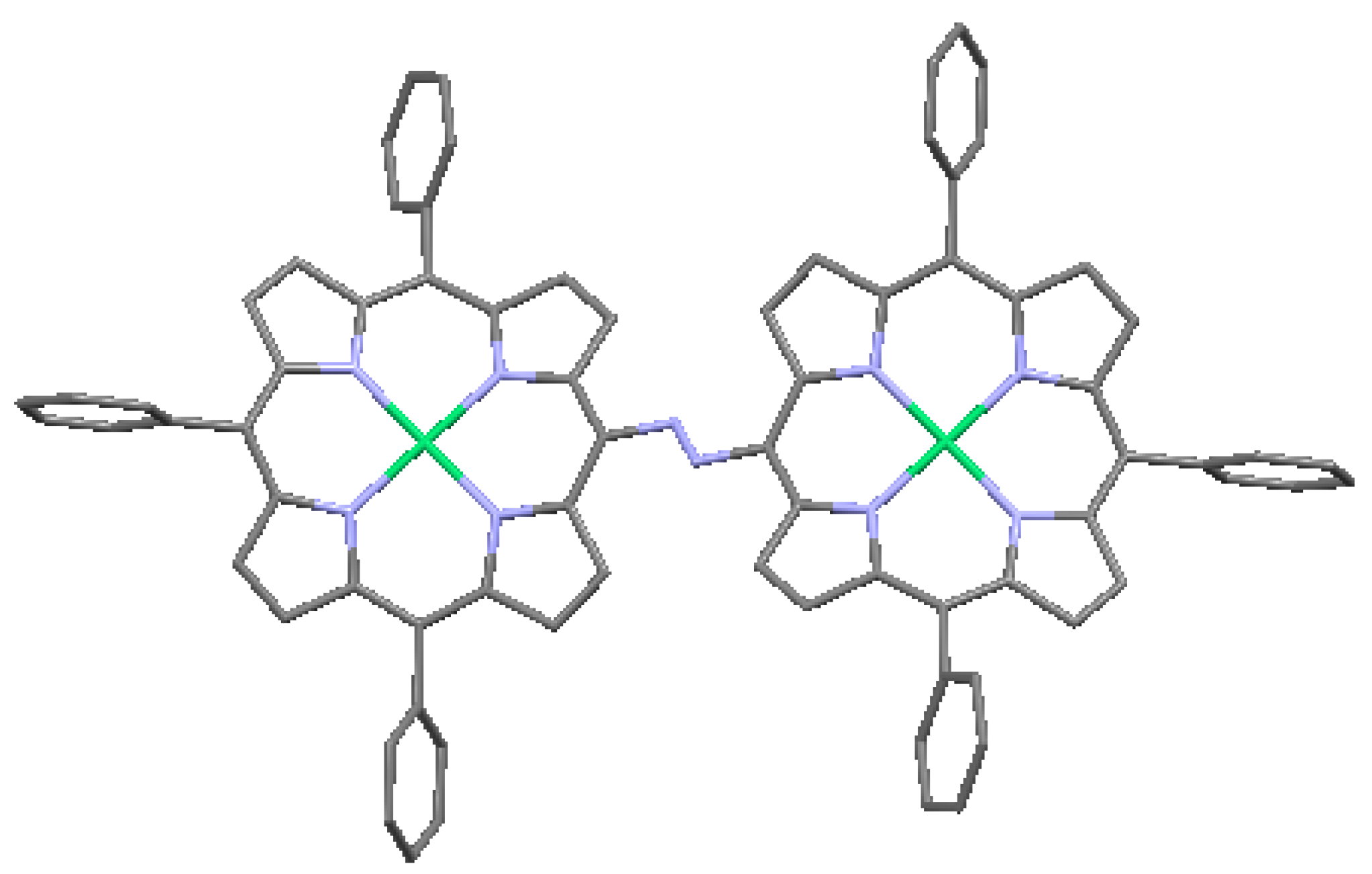
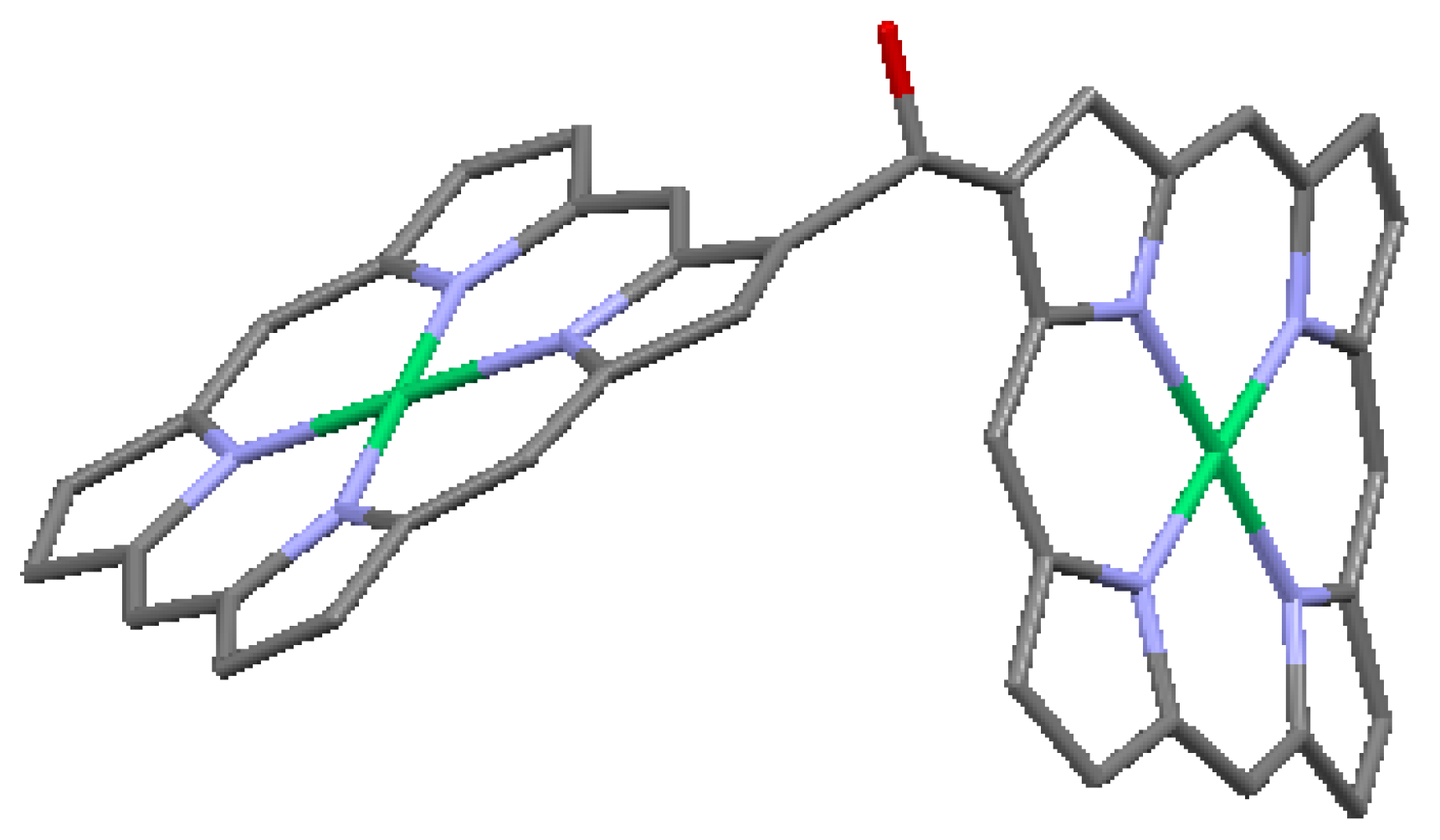
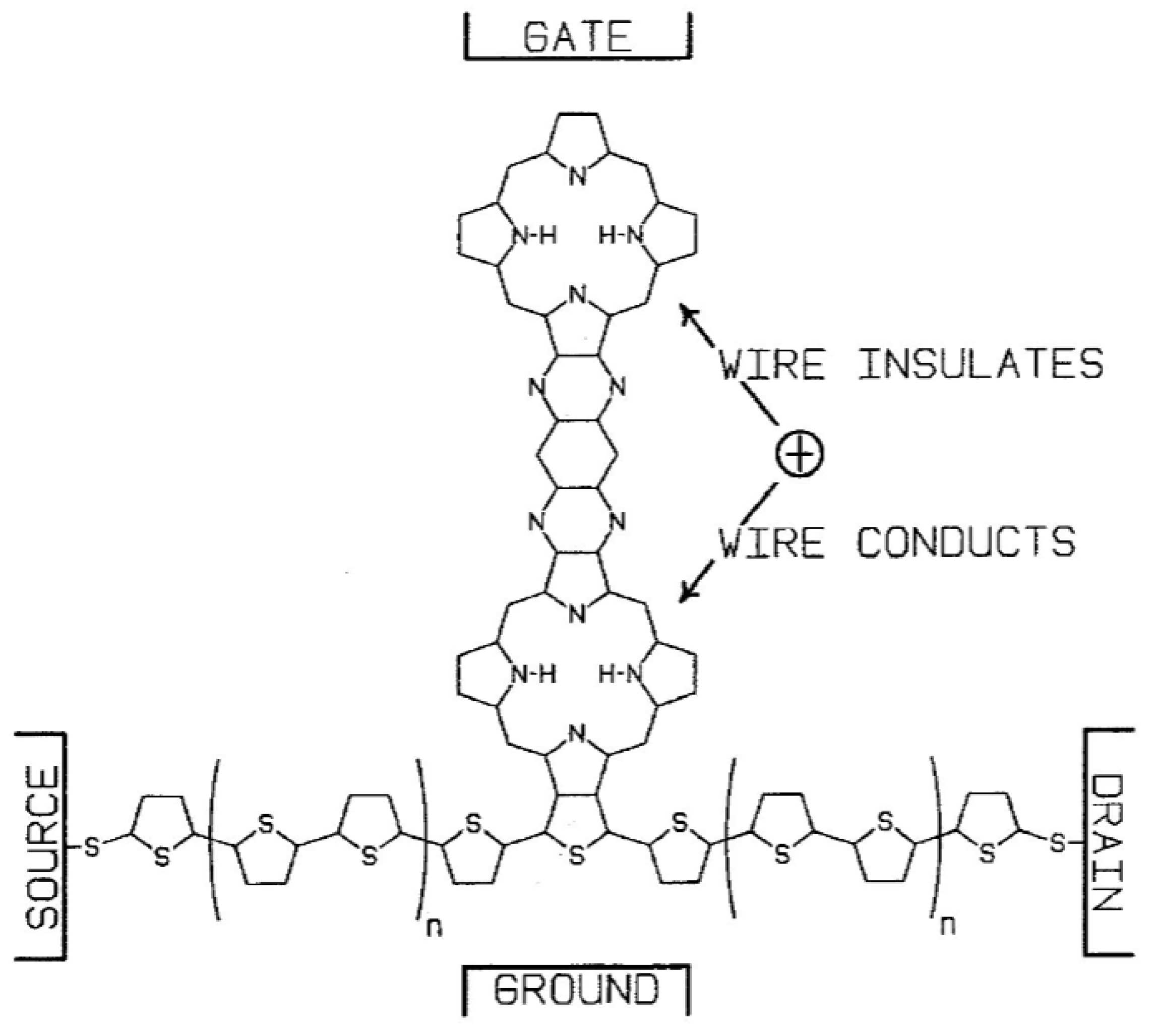
Crossley and Burn [24] synthesized molecular wires comprised of a linearly conjugated tetrakisporphyrin, as shown schematically in Figure 12. Based on spectroscopic studies, the HOMO/LUMO gap was reduced by about 0.8 eV, compared with the same gap in simple porphyrins. This effect is largely due to the presence of conjugated bridges of 1,4,5,8-tetra-azaanthracene. The presence of the meta-di-t-butylphenyl substituents on the macrocyles enhances solubility. Reimers et al. [32] performed a quantum-chemical analysis of this type of oligomer and discussed in detail the effect of oligomer size and geometry on electron- and hole-conduction. Based on their analysis, they suggest a design for a molecular switch, a modification of the design of Aviram [33], in which an oligoporphyrin is linked to a polythiophene wire as a side chain, as shown in Figure 13. The conduction between the source and drain electrodes can be switched on or off depending on the location of an excess electron hole on the porphyrin side chain, as controlled by the electric field at the gate electrode.
Yoon et al. [34] measured electrical conduction through two types of porphyrin wires using nanoelectrodes. One type consisted of directly meso-meso-linked Zn(II) porphyrin arrays with 48 Zn(II) porphyrin moieties. These arrays have an orthogonal configuration such that adjacent porphyrins are oriented at right angles to one another along the chain. A second type consisted of flat tape-like arrays with eight porphyrin units in which the porphyrin macrocycles are triply linked at meso-meso, β-β, and β-β positions, resulting in a linear fused array. I–V curves for both types were measured and compared. The orthogonal type showed hysteresis, which suggests the possibility of application to memory devices, whereas the fused tape-like type showed no hysteresis. The hysteresis is interpreted as arising from the rotation of the meso-meso carbon-carbon bond in the orthogonal configuration, whereas rotation of the meso-meso bonds in the fused array is hindered by the triple linkage. The room-temperature resistance of the orthogonal array was estimated as 125–670 MΩ, whereas that for the fused array was estimated as 50 MΩ, which suggests a higher conductivity for the latter, as would be expected from the co-planar orientation of the fused macrocycles. Porphyrin-based molecular wires were investigated by Sedghi et al. [35] using STM methods. Three architectures were prepared: (A) meso-butadiene-linked porphyrin chains; (B) twisted meso-meso directly linked chains; and (C) multiply-fused (at both β and meso positions) chains. Allowing for the length of the structures, the fused versions (C) had the greatest molecular conductance, and the twisted versions (B) had the lowest conductance. This is consistent with the higher macrocycle co-planarity in (C) and the resulting increase in conjugation.
Wang et al. [36] demonstrated self-assembled synthesis of porphyrin nanotubes based on Sn(IV)-porphyrin with meso-substituted pyridines. The Sn(IV) was axially bonded above and below the macrocycle plane to Cl– or OH–. The nanotubes were micrometers in length, with 50–70 nm diameters and ~20 nm-thick walls and showed moderate crystallinity. Electrostatic forces between the porphyrin building blocks are thought to contribute to the self-assembly process, in addition to van der Waals, hydrogen bonding, and axial coordination interactions. The nanotubes were found to be photocatalytic, and are stable for extended periods (months) when stored in the dark. They are mechanically responsive to light, i.e., when illuminated with strong light, the nanotubes become rods, in a reversible manner. This response may be due either to a softening of the tube walls and a collapse due to local heating, or to the result of photoinitiated intermolecular electron transfer that disrupts the charge balance and the rigidity of the ionic structure.
6.2 Conductivity in Single Crystals
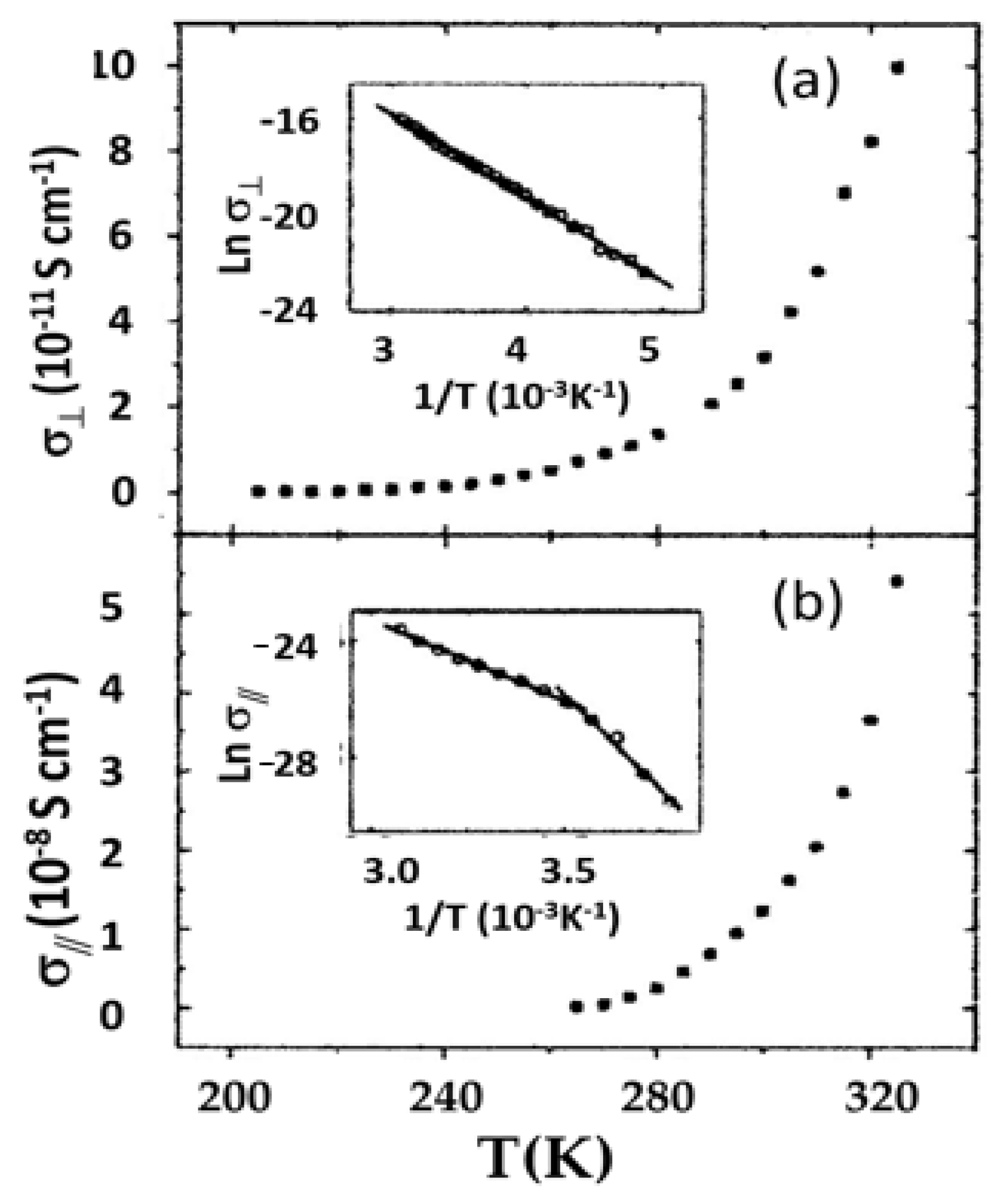
Tetragonal porphyrin single crystals with parallel open channels and interconnected columns of macrocycles were synthesized by Li et al. [37]. These features allowed simultaneous ionic (lithium) conductivity and electrical conductivity. Electrical conductivities of 10−9 S cm−1 were reported; by insertion of conducting aniline into the channels, the conductivity was increased to 10−3 S cm−1. Single crystal conductivity data was reported by Chen et al. [38] for a porphyrin salt based on 5,10,15,20-tetrakis (4-pyridyl) porphyrin, or H2TPyP. This triclinic salt, H2TEPyP4+·4I−, contains four ethyl groups and four iodines, with macrocycle stacking in the a crystallographic direction. Conductivity was measured by a four-probe method. Data as a function of temperature are shown in Figure 14 for orientations parallel and perpendicular to the stacking direction. The room temperature conductivity was found to be ~3 orders of magnitude higher along the stacking axis relative to measurements perpendicular to it (3.2 × 10−8 S cm−1 vs. 1.2 × 10−11 S cm−1). The nature of the structure results in higher π-bond conjugation, and thus conductivity, in the direction of stacking.
Although phthalocyanines are not directly considered in this review, it is instructive to include an example of single crystal conductivity measurements on a phthalocyanine. The phthalocyanine macrocycle (C32H18N8) is closely related to porphyrin in that it is based on four pyrrole-like moieties which are linked together to form a ring much like the porphyrin macrocycle, and thus it has some similar aromatic properties. The differences are that nitrogens are present instead of carbons at the meso positions (refer to Figure 2b), and a benzene is fused to each pyrrole at the β positions, such that the four pyrroles become isoindoles (C8H7N). Schramm et al. [39] measured electrical conductivity of single crystals of nickel phthalocyanine iodide, a compound which is composed of stacks of planar nickel phthalocyanine molecules in a one-dimensional configuration parallel with adjacent chains of I3− molecules. A very high conductivity (250 to 650 S cm−1 at room temperature) was observed in the stacking direction (c-axis), and the temperature dependence confirmed the conductivity as metallic. The carrier mean free path of the most highly conductive crystals was estimated to be ~60 Å, indicating the effectiveness of the stacked macrocyclic electron donor/linear iodine acceptor configuration in promoting the metallic conductivity along the stacking direction.
6.3. Thin-Film Conductivities
In early work, Livshits and Blyumenfel’d [40] measured the dark electrical conductivity of PP9 thin-films, and reported a value of ~10−11 S cm−1 at 20 °C. In an attempt to improve conductivity, Kobayashi et al. [41] explored increasing the size of the porphyrin ring complex by extending the length of the substituents, thereby enlarging the conjugation system, and in principle lowering the activation energy for conduction. They prepared 50 nm thin films of four compounds with progressively larger ring complexes: zinc tetraphenylporphyrin, zinc tetrabenzoporphyrin, zinc tetranaphthaloporphyrin, and zinc tetraanthraporphyrin. The measured room temperature conductivities were, respectively, <10−11 S cm−1, 4 × 10−10 S cm−1, 3 × 10−4 S cm−1, and 1 × 10−7 S cm−1, suggesting a ring size limit above which the conductivity no longer increases, but rather, decreases. HOMO/LUMO gaps decreased as the size of the π-conjugation system increased. Since the films were prepared in air, the conduction mechanism is thought to involve oxygen doping and electron transfer from the porphyrin ring to an oxygen acceptor molecule. Jones et al. [42] investigated dark conductivity in multilayer Langmuir–Blodgett films and showed that an Ag-porphyrin similar in structure to metalated PP9 had relatively high conductivity (2.5 × 10−3 S cm−1 at 18 °C) in the plane of the film, opening up possibilities for device applications. The conduction was observed to be anisotropic, and two models were suggested for the conduction: (1) structural defects in the films; and (2) a semiconductor-type depletion layer at the interface. Analogs with Cu(II), Ni(II), Cd(II), and Au(III) as the metal were substantially less conductive than the Ag version.
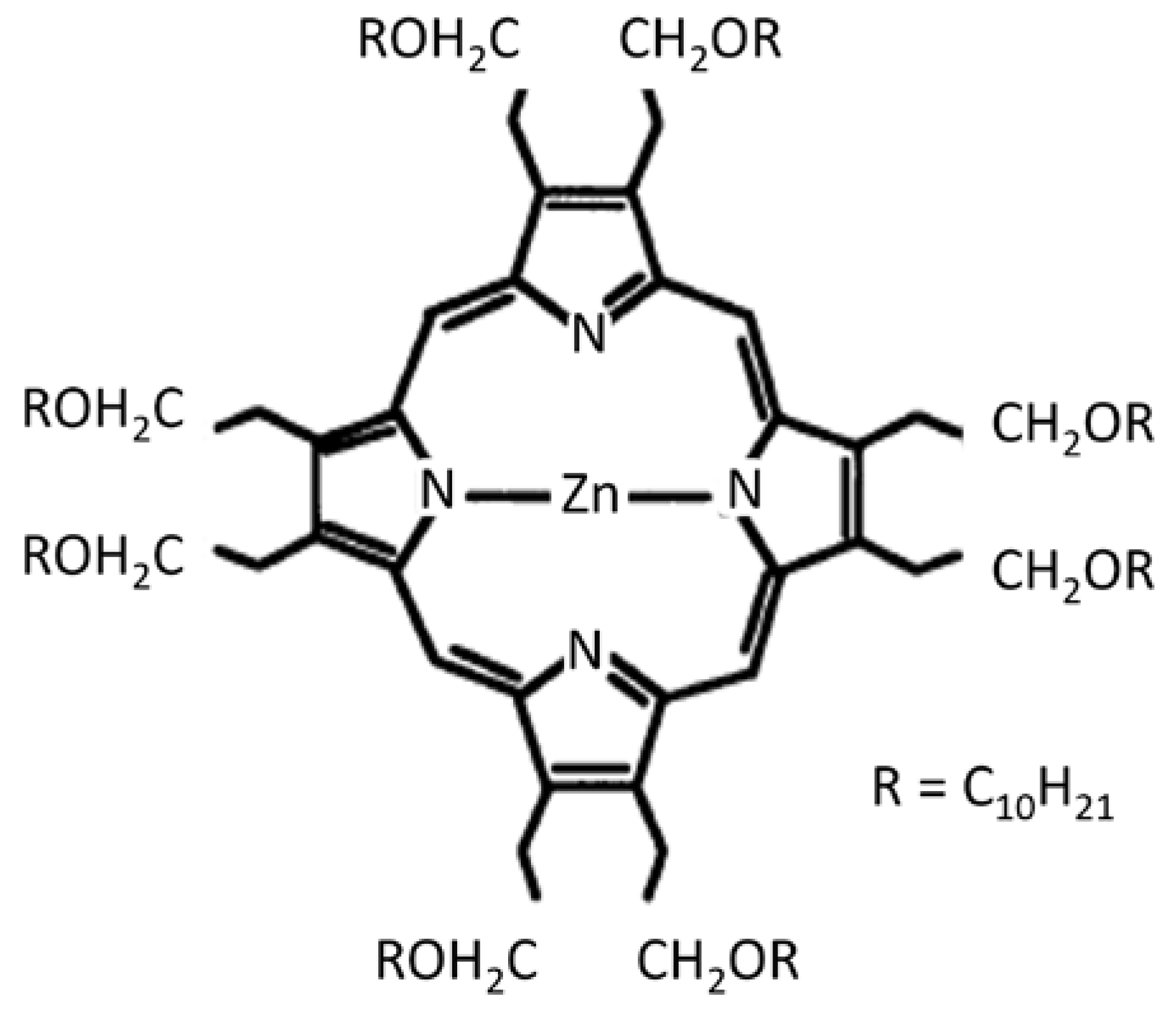
Liu et al. [43] observed an electrooptical memory effect in thin films of the photoconductor zinc-octakis(β-decoxyethyl) porphyrin (ZnODEP). Information, as trapped charge, can be written, read, and erased by simultaneous application of an electric field and a light pulse. During irradiation and the simultaneous application of an electric field, electron–hole pairs are generated. These become “frozen” within the films when irradiation is interrupted. Subsequent re-irradiation releases charge and generates a transient photocurrent. The structure of ZnODEP, a liquid crystal, is shown schematically in Figure 15.
6.4. Conductivity in Bulk-Polycrystalline Porphyrins
We use the combined term bulk-polycrystalline to clearly differentiate these materials from thin film polycrystalline or single crystal materials. Wood et al. [44] studied pressed powders of eight different porphyrins under pressures up to 90 kbar. These compounds included: (a) protoporphyrin (PP9), and five related compounds, i.e., hematoporphyrin free base, hematoporphyrin dihydrochloride, copper protoporphyrin, hemin, and hematin; and (b) etioporphyrin and vanadyl etioporphyrin (relative to PP9, etioporphyrins have a methyl and an ethyl group at the β positions of each pyrrole). At low pressure, the compounds in Group (a) had higher conductivities than Group (b), which the authors attribute to overlap of the carbonyls in Group (a). Conductivities for the hemin, hematin, and vanadyl etioporphyrin show marked increases with pressure, whereas the others do not. For example, it is reported that the room temperature conductivity of hemin increases from ~10−10 S cm−1 at ambient pressure to ~10−6 S cm−1 at 90 kbar. On the other hand, with increasing pressure for PP9, hematoporphyrin free base, hematoporphyrin dihydrochloride, and copper protoporphyrin, conductivities remained relatively constant at 10−11 to10−10 S cm−1. A full explanation of these differences was not given, as the crystal structures were not adequately known. A possibility could involve oxidation/reduction of the iron and vanadium, as the experiments were not sealed.
Collman et al. [45,46] described a strategy in which conductive metalloporphyrins with a linear so-called “shish-kebab” structure were designed by combining three variables: the nature of the macrocycle, the bridging group between macrocycles, and the transition metal in the macrocycle core. Accordingly, they synthesized Fe(II/III)-, Ru(II/III)-, and Os(II/III)-octaethylporphyrins bridged with pyrazine, 4,4′-bipyridine, or 1,4-diazabicyclo[2,2,2]octane, and found that pressed powders of the partially oxidized ruthenium and osmium versions were highly conductive. Conductivity increased in polymers relative to monomers. The conductivity was found to found to be affected by molecular weight according to Os > Ru > Fe, possibly due to a progressive decrease in the size of the band gaps with increasing molecular weight. It was found that doping with oxidants, namely ferricenium hexafluorophosphate, nitrosyl hexafluorophosphate, or iodine, resulted in a substantial increase in conductivity. ESR and electrochemistry were used to investigate the conduction path, which was determined to be influenced primarily by the metal/ligand backbone of the structure, rather than the macrocycle. Room temperature conductivities ranged from 10−11 S cm−1 for undoped pyrazine Fe monomers, to 10−2 S cm−1 for doped pyrazine-linked Os polymers.
6.5. Ferroelectric Porphyrins
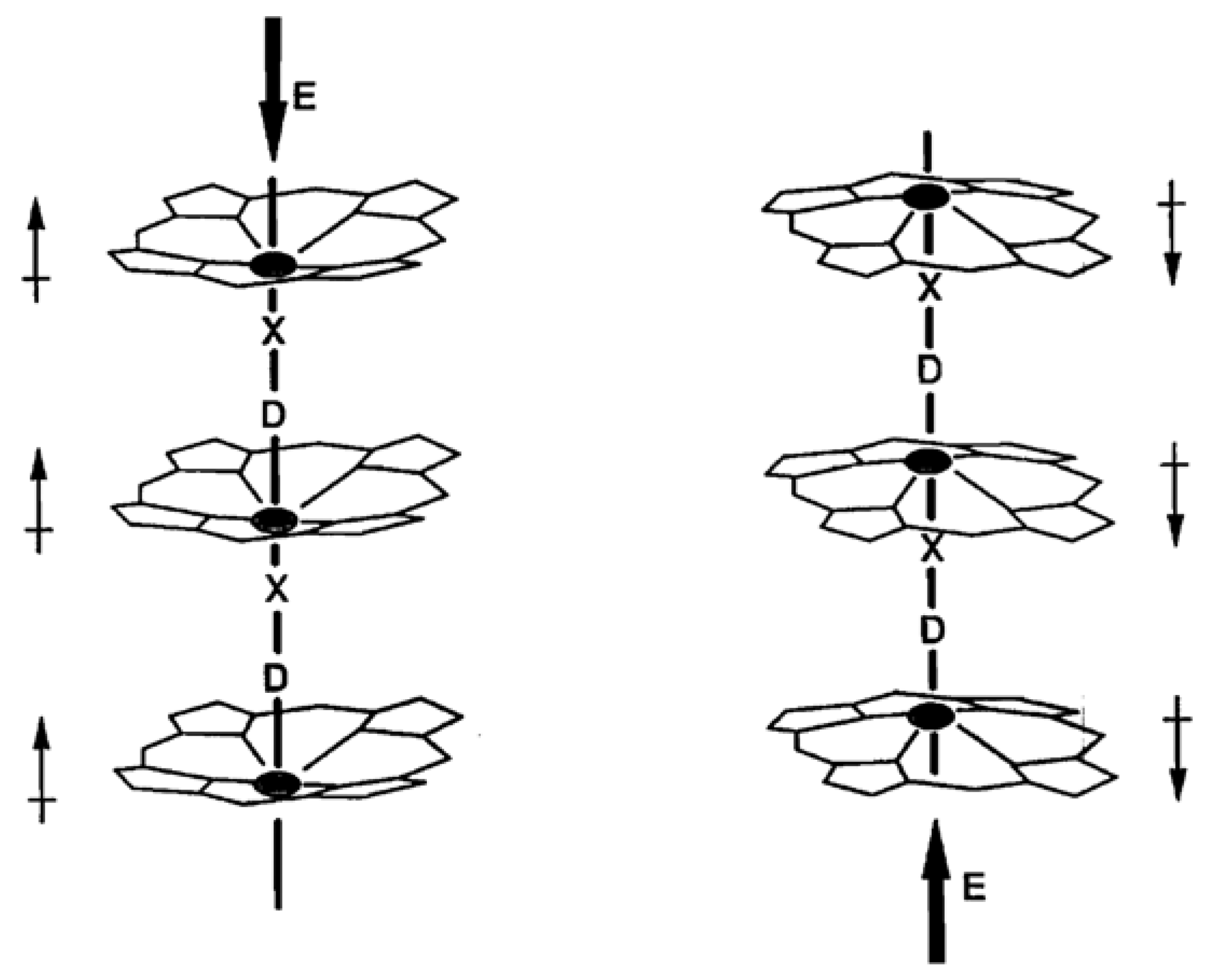
Suslick and Chen [47] and Chen and Suslick [48] have described work on linear “shish-kebab” porphyrins in which compounds with possible ferroelectric properties were synthesized. Of particular interest is the polymeric compound [FeIII(TPP)(ImPhO)]∞, where Im = imidazole and Ph = phenyl, and ImPhO is the composite axial ligand linking the macrocycles. This compound, which crystallizes in the non-centrosymmetric space group Pna21, is expected to have a permanent dipole moment parallel with the c-axis. In this compound the iron is pulled out of the macrocycle plane by bonding to the axial ligand. A small doming effect is the result. An applied electric field could be expected to have the effect illustrated in Figure 16, in which the direction of the dipole can be reversed by reversing the electric field. In principle, such materials can be fabricated into electronic oscillators, high-frequency filters, converters, and non-linear capacitors.
7. Structural Examples of Complex Multiporphyrins
The long-term technological potential of porphyrins stems from the large number of ways individual molecules can be linked together to form larger molecular structures, or arrays. In the following discussion, we are guided by the work of Aratani and Osuka [49], who categorized porphyrinic arrays based on the type of linkage, both in terms of chemistry and structure. We do not include porphyrinic MOFs and COFs, which are primarily of interest for their sorption properties, and have been reviewed elsewhere (e.g., [6,50]). The compounds in Table 5 provide examples of some of the multiporphyrin structures that have been determined by X-ray diffraction and illustrate the structural features that influence porphyrin molecular semiconducting properties. In Table 5, the compounds are referenced to the CSD database and to the figures following the tables. The figures have been prepared using the Mercury crystal structure software suite [7]. It is noteworthy that the majority of the compounds in Table 5 are triclinic or monoclinic, with the highest symmetry belonging to the sole orthorhombic compound in the table. This is most likely due to the complexity of the crystal packing required for the bulky, asymmetric molecules constituting these compounds.
7.1. Ethyne-Bridged Multiporphyrins
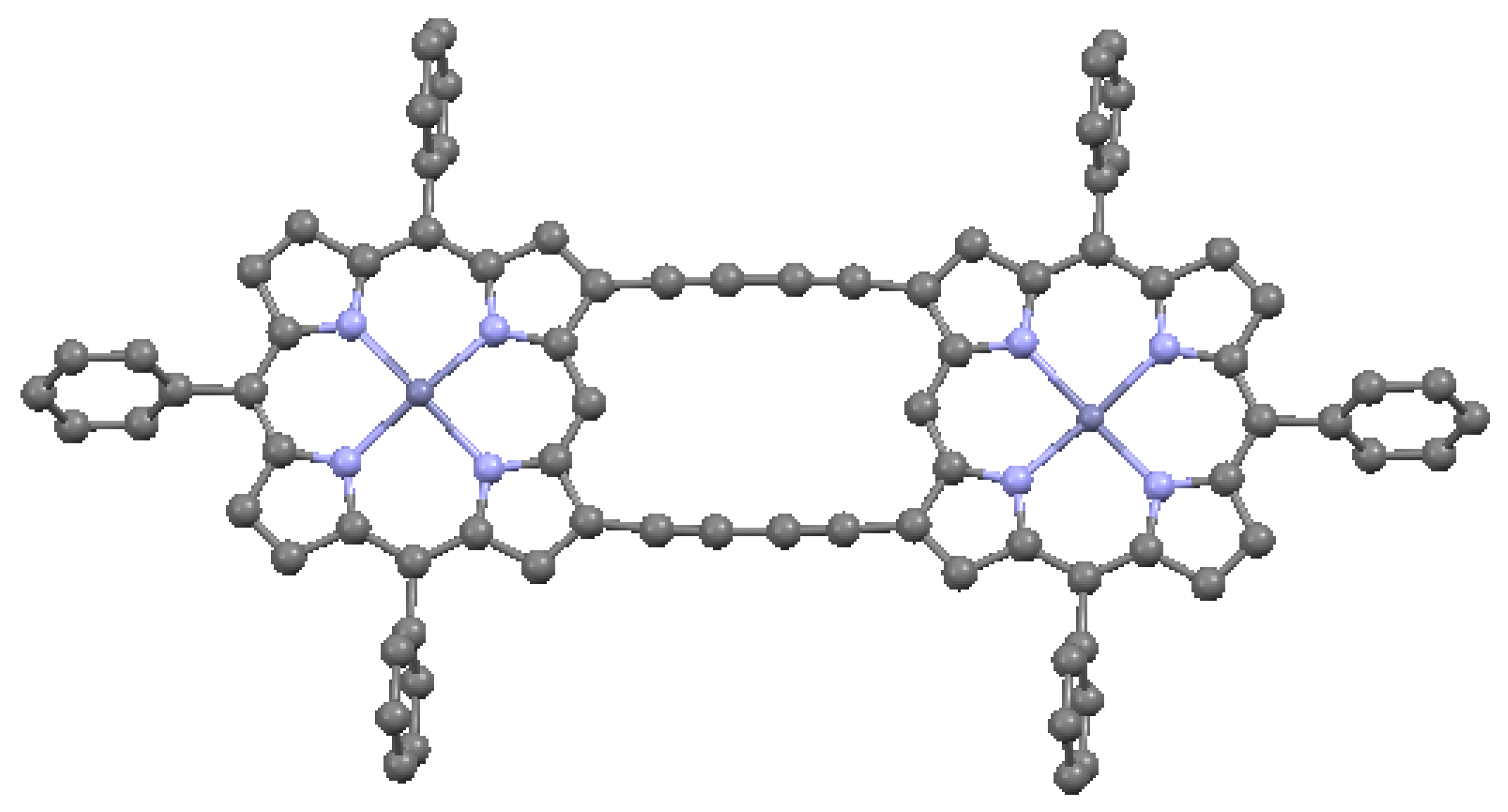
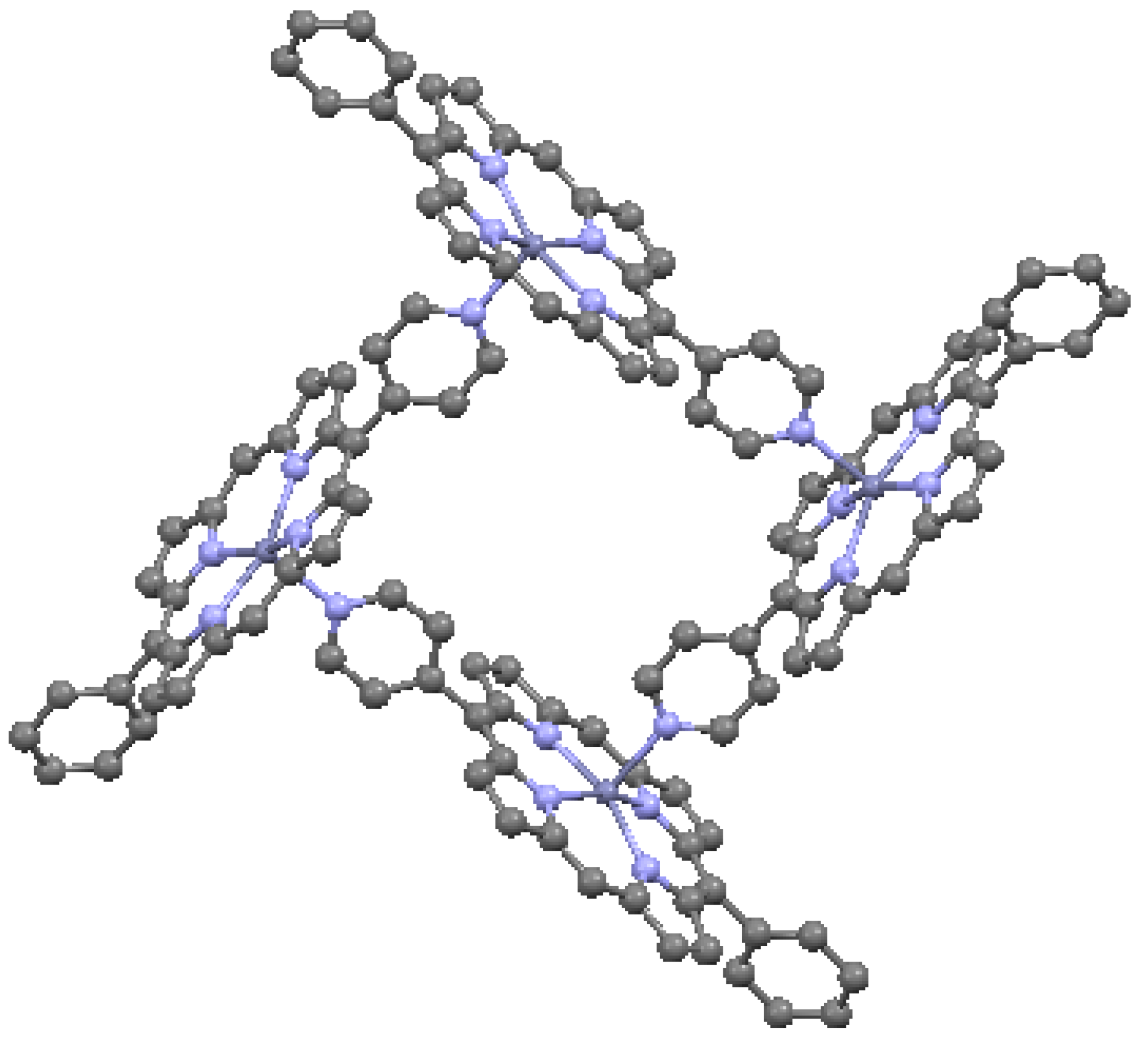
When the aromatic macrocycles are linked by appropriate ligands, conjugation occurs, resulting in extension of the π–π bonding [24]. As noted, this influences the electronic properties. Ethyne-bridges (based on the acetylene bond) can have an important modulating effect on electronic coupling when attached to either the meso- or the β- sites of the macrocycle. Typically, ethyne-bridged porphyrins show strong exitonic coupling, and large optical absorption bands in the visible and near-IR regions [49]. The selection of bonding sites can be important. For example, a structure with vinylene-meso-bridged porphyrin dimers exhibits a large electronic interaction [51], in contrast to β-bridged analogs, which allow twisting of the macrocycles out of conjugation. The vinylene-meso-bridged structure also shows a substantial non-linear optical effect. A longer linkage comprised of double β-to-β butadiyne (H–C≡C–C≡C–H) linkages is shown in Figure 17. This structure is relatively planar, with only 0.053 Å deviation from the average plane formed by the porphyrin cores and the bridges. The planarity results in significant enhancement of two-photon absorption properties. A cyclic ethyne-meso-bridged mixed-metal OEP porphyrin tetramer is shown in Figure 18. This structure contains Rh atoms at the center of the two laterally-positioned macrocycles which are bonded axially in both directions to a phosphine group and thence to ethyne bridges connected to zinc porphyrins at the bottom and top of the cyclic tetramer. The zincs of the upper and lower macrocycles are bonded across the interior of the tetramer to each other with pyridyl groups. Mixed metal porphyrins such as this can conceivably allow fine-tuning of redox and other useful properties by varying the identity of the metals.
7.2. Cofacial Dimers
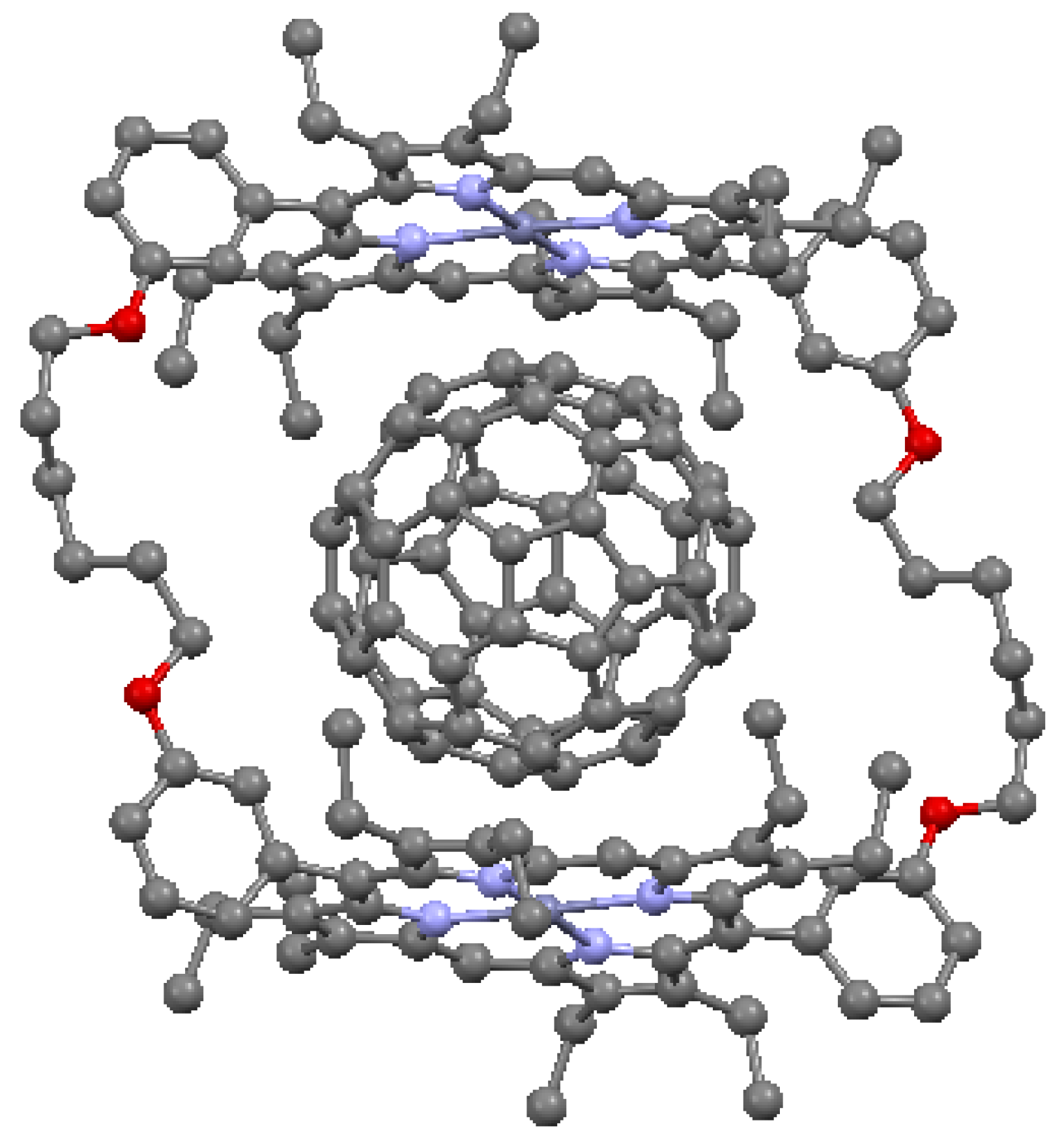
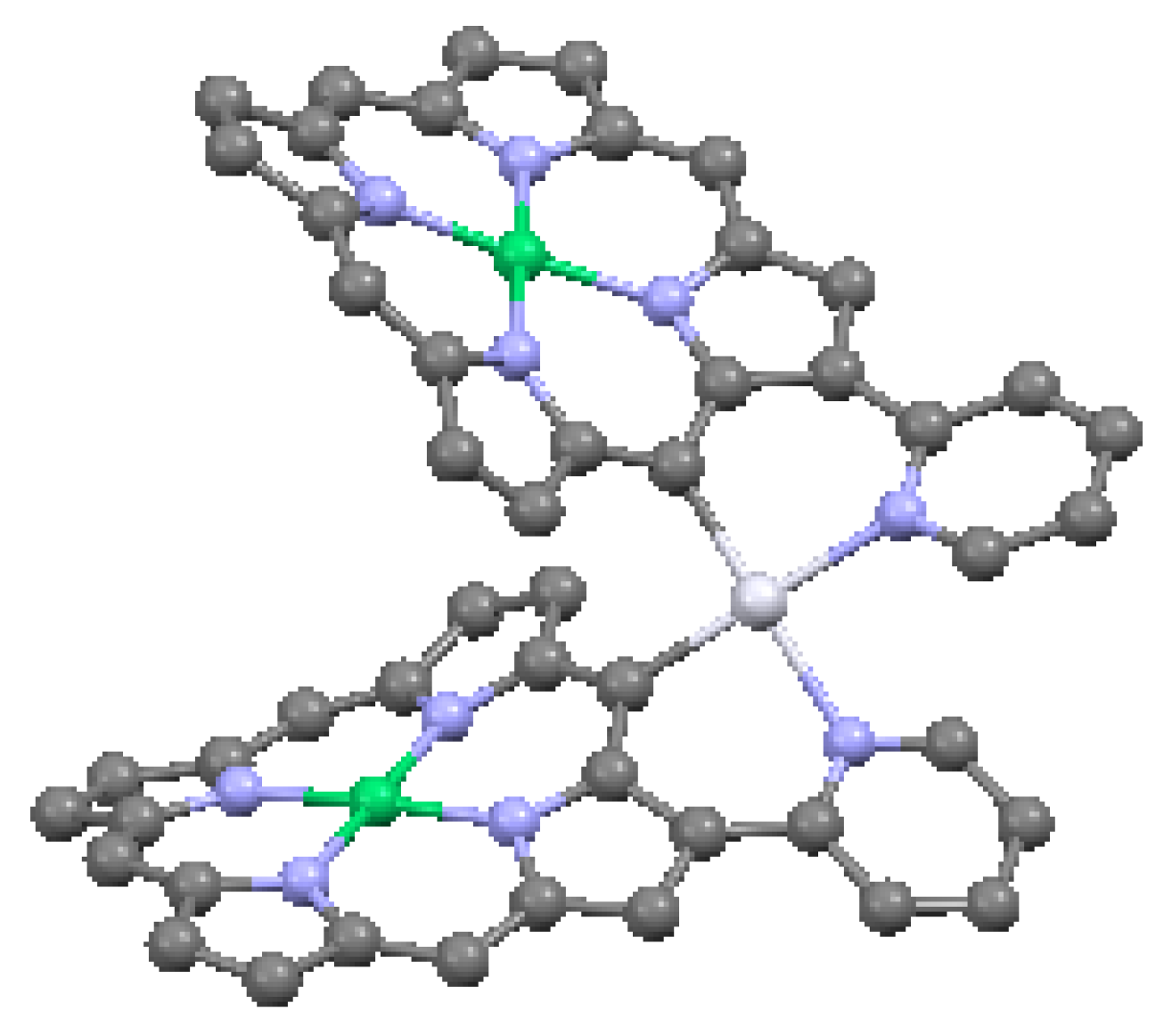
An important attribute of cofacial porphyrin dimers is the ability to capture other structural groups internally, in particular, fullerenes. These combinations can result in enhanced electron and energy transfer [52]. The ability to capture fullerenes depends on the metal atoms of the macrocycle. In Figure 19, a structure is illustrated with cofacial zinc porphyrins bound to an internal C60 molecule. The distance between the pyrrole carbon atoms of the porphyrins and the nearest carbons of the fullerene is shorter than the typical interlayer distance of graphite (3.354 Å), suggesting that a π interaction is present between the two moieties. A cofacial pillared zinc bisporphyrin is shown in Figure 20. This type of architecture, known to have a “Pac-Man” effect, is characterized by the ability under chemical stimulation to open and close its binding pocket by over 4 Å longitudinally. The ruffled porphyrin moieties at the top and bottom are not equivalent; the rotation of the porphyrins to maximize π interactions is prevented by the inflexibility of the dibenzofuran (C12H8O) spacer with regard to twisting in this direction.
7.3. Directly-Linked Porphyrins.
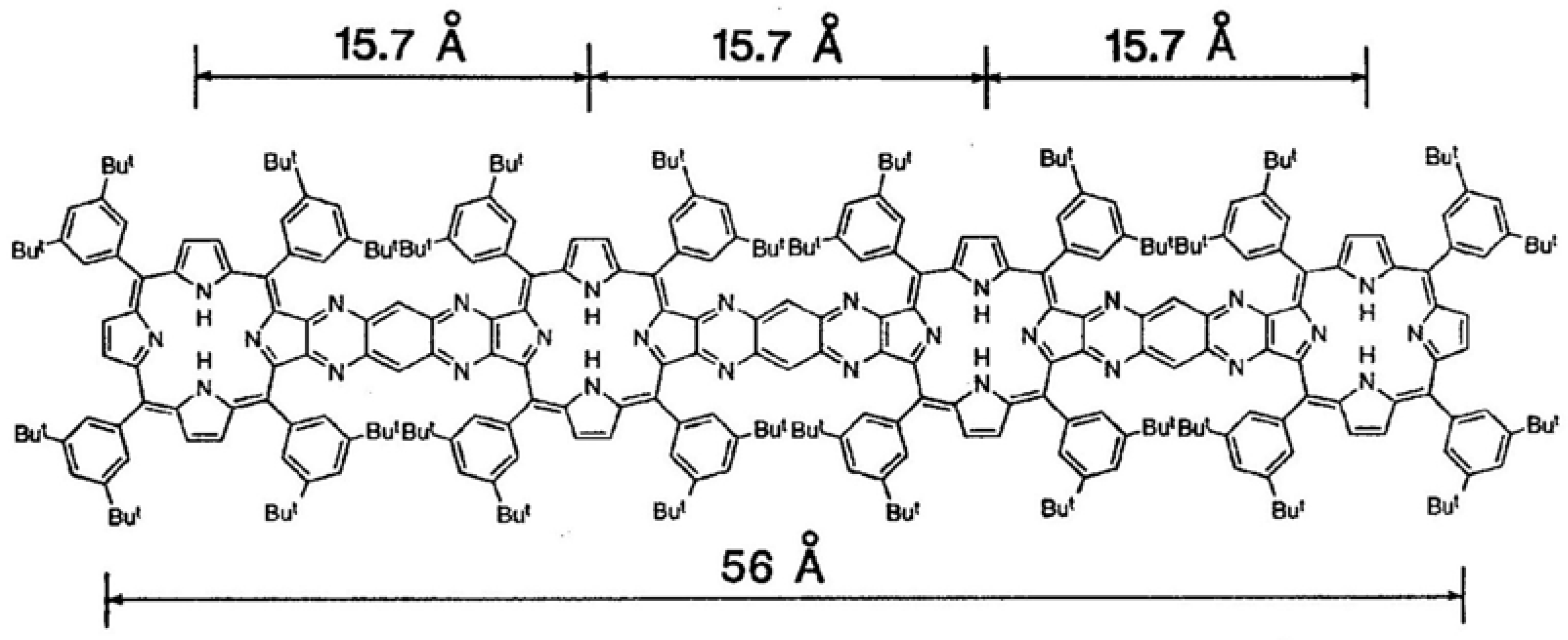
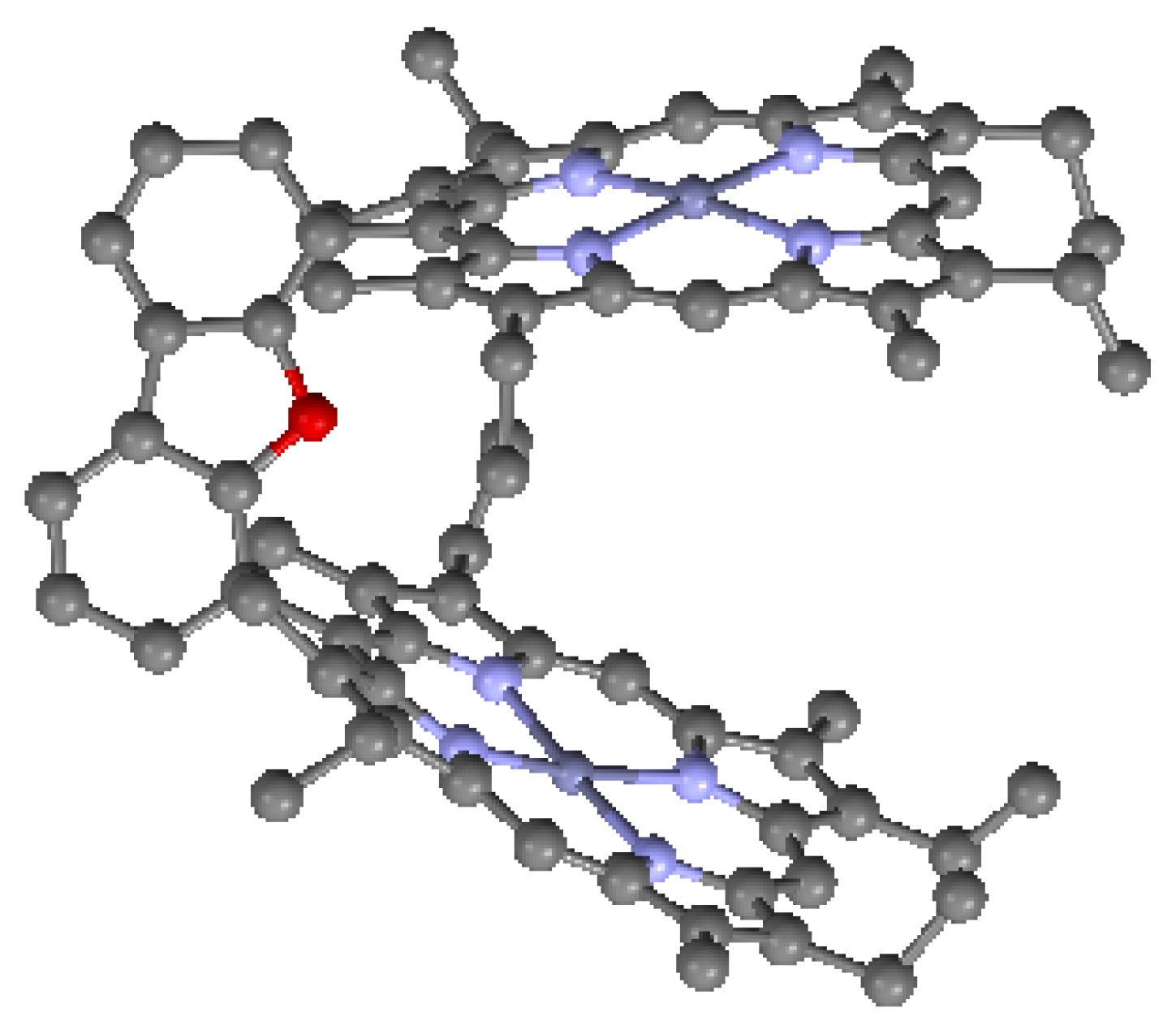
In many directly-linked architectures, the neighboring porphyrins are linked together by meso-meso bonds. This arrangement results in higher molecular symmetry and larger, more regular electronic interactions, due to the continuous porphyrinic π-electronic network. Such architectures can lead to 3-D arrays useful in light harvesting [61]. Furthermore, individual directly-linked subunits can be combined by linking with spacers to form larger units. Large porphyrin wheels have been prepared by combining directly-linked porphyrin subunits via meso-meso-linked phenylene spacers [62]. In addition to direct linkage by meso-meso bonding, linkages based on β-β, and meso-β combinations have been reported. An example of the latter with two porphyrins is shown in Figure 21. The mean planes of the macrocycles are almost orthogonal due to the steric requirements of the bonding, and therefore it is expected that the inter-porphyrin conjugation is limited for this particular architecture.
7.4. Multiply-Linked Structures
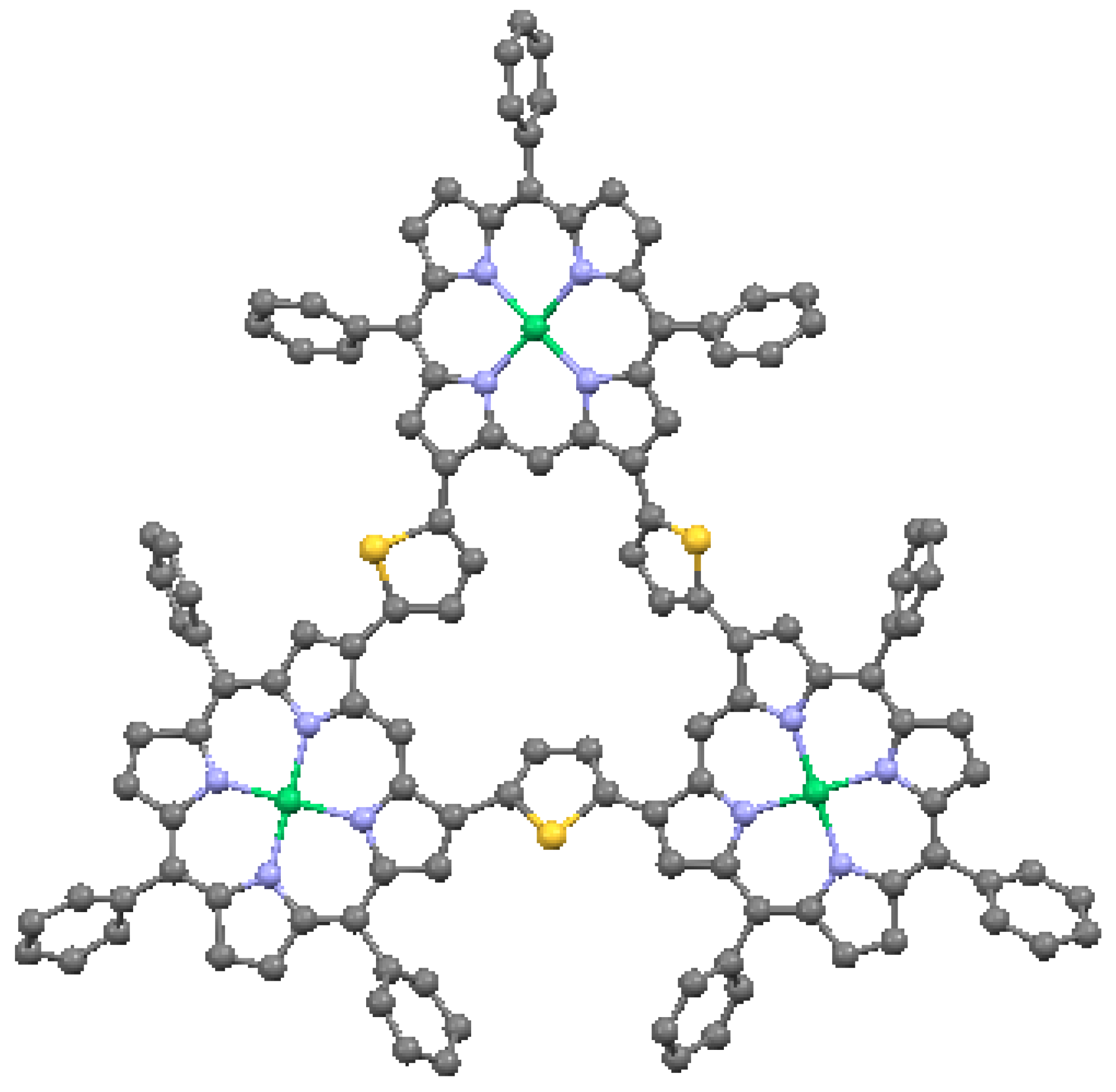
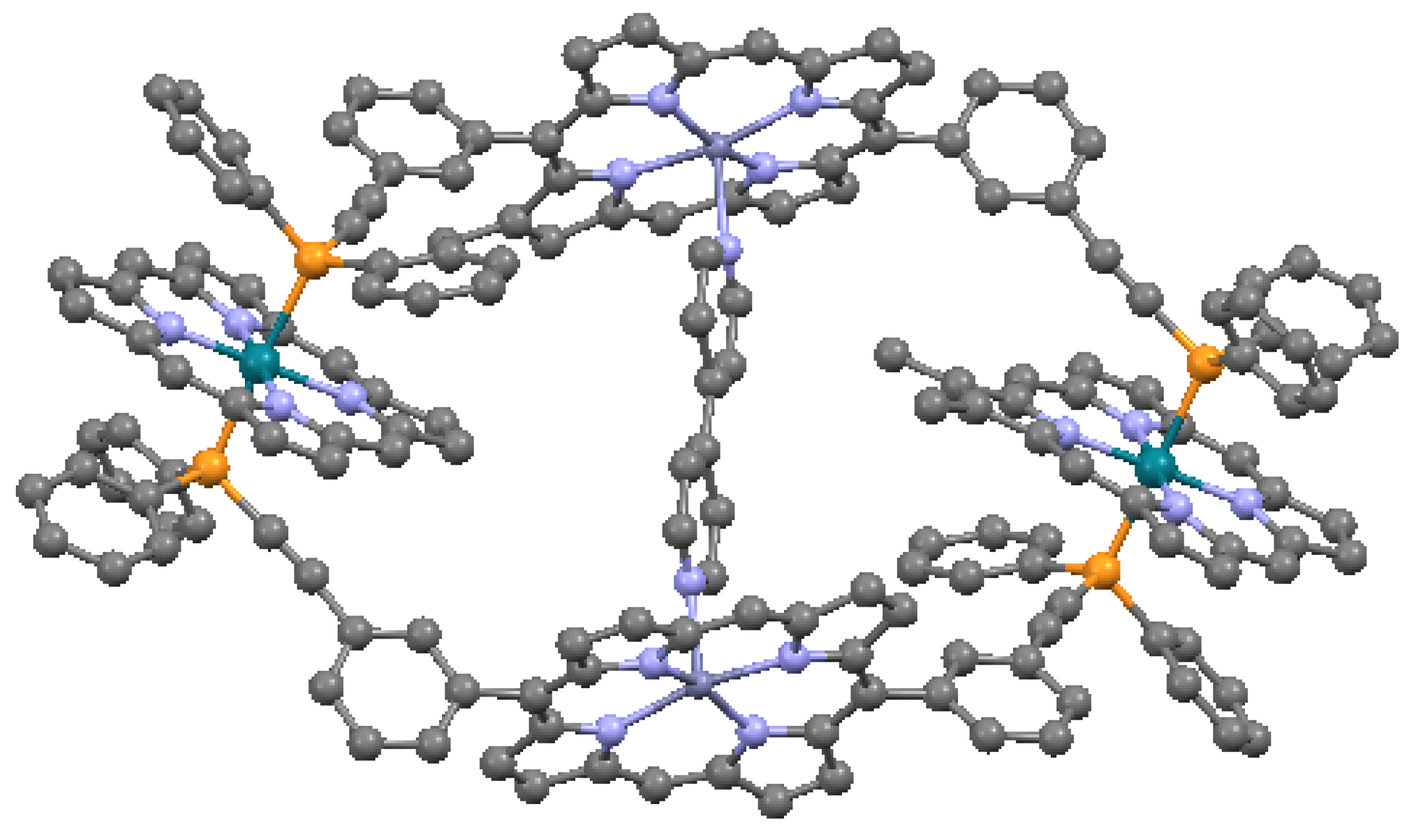
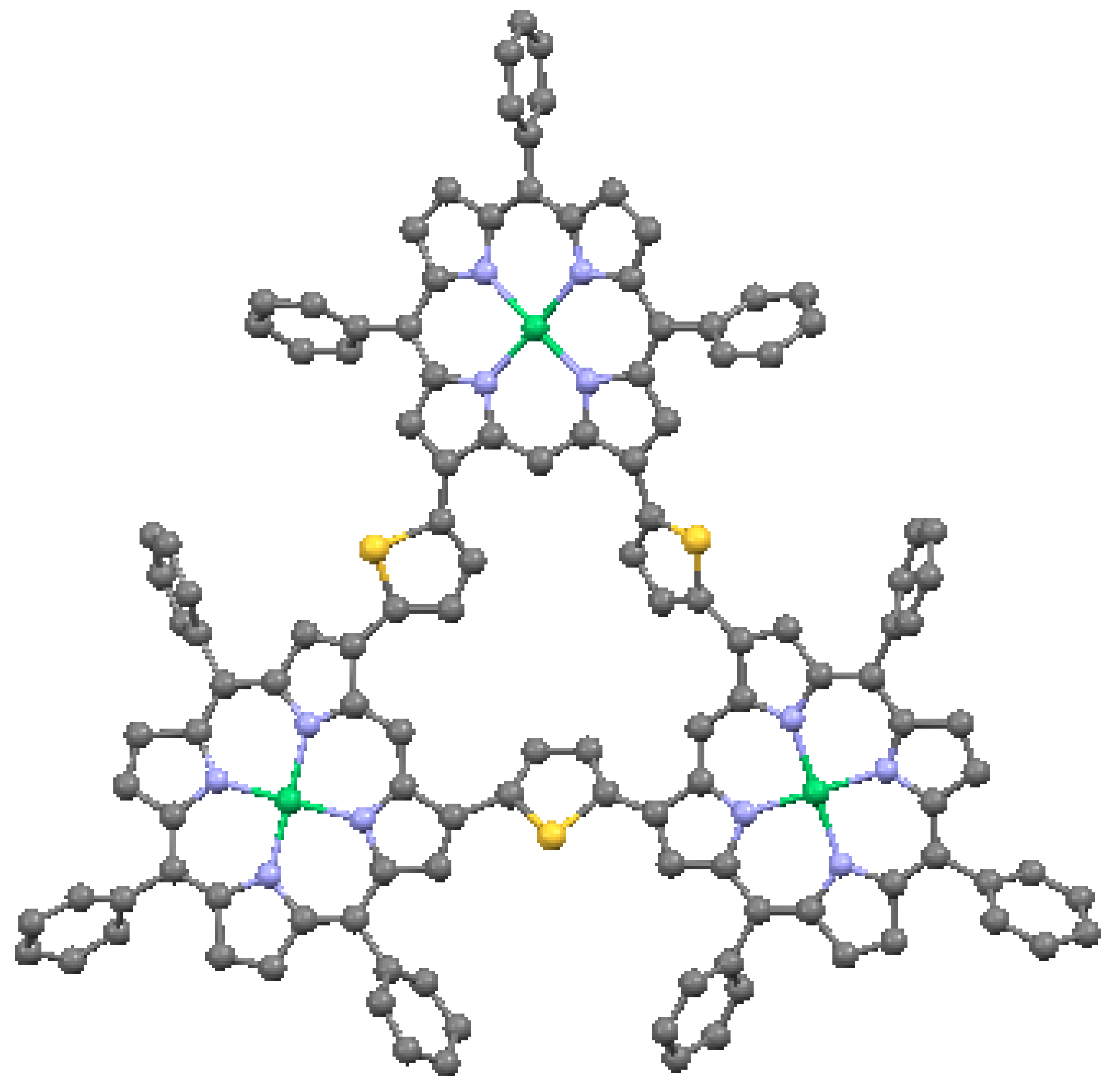
An important benefit of multiple linkages is the enhanced π-conjugation that can be achieved [35]. In general, this results from the co-planarity enforced by multiple bonding of the macrocycles, or in some cases enforced by direct fusion of the pyrroles. Effects such as narrowing of the HOMO-LUMO gaps have been observed, as well as high two-photon absorptions. An example of a multiply-linked structure is the β-thienylene-bridged nickel triporphyrin in Figure 22. In this triangular porphyrin trimer, the porphyrin cores are ruffled, with a maximum carbon displacement from the mean plane of 0.915 Å. The thiophenes (C4H4S) have tilts of 26.6° to 36.1° relative to the adjacent pyrroles. The structural distortion caused by the double-bridging of the macrocycles is reflected by the perturbation of the optical absorption spectrum.
7.5. Self-Assembled Arrays
For efficient synthesis of more complicated porphyrins, self-sorting methods that utilize mixtures of components with desired structural attributes that combine spontaneously have proved beneficial. Tsuda et al. [59] used this approach to synthesize 3-D porphyrin boxes. The structure of 5-p-pyridyl-15-(3,5-di-tert-butylphenyl) Zn(II) porphyrin is shown in Figure 23. The inter-porphyrin electronic interactions in this structure are decidedly weak by comparison with other meso-meso linked diporphyrins, due to the perpendicular orientations of the macrocycles.
7.6. Metal-Bridged Arrays
The linking of porphyrins by metal bridges adds another dimension to their structural diversity. A Pt(II)-bridged cofacial nickel diporphyrin is shown in Figure 24. The Pt (II) is square-planar-coordinated. The macrocycles have a ruffled configuration, with mean plane deviations of 0.323 Å for the nitrogens. The compound has an inherent helical chirality. Yoshida et al. [63] have explored the reactivity of related Pt (II) and Pt (IV) porphyrinic compounds, which show interesting redox properties. Potentially, there are applications in catalysis.
8. Conclusions
Many of the promising materials applications for porphyrins will rely on optimization of solid state semiconducting properties, which in turn depend on the underlying crystallography. Of foremost importance are the planarity of the macrocycle and the conjugation of π-bonding through the proximity of adjacent macrocycles or the interconnection of macrocycles via appropriate covalently-bonded ligands. For some structures, conductivity also depends on the presence of electron donor/acceptor pairs. We have described for selected examples how semiconducting properties originate at the molecular level. Starting with the basic porphine parent molecule, we have shown how structures can be modified by addition of substituents and by metalation of the macrocycle core. Complex 3-D multiporphyrins have been illustrated which show interesting and varied topologies. A major need is further understanding of the relation between semiconducting properties at the molecular level and at the macroscopic level (single crystals, and polycrystalline materials). Most of the porphines, porphyrins, and multiporphyrins discussed have relatively low symmetry (triclinic or monoclinic), due to the complexity of the crystal packing required for the porphyrin moieties. Given the large number of possible metal insertions, substituent attachments, and linkages, the number of unstudied crystallographic combinations appears to be very large; obviously, not all will have useful properties. Devising efficient synthesis methods for processing of sufficient quantities to measure properties and evaluate potential applications continues to be a challenge, but one with major potential overall payoff.
Author Contributions
L.P.C. researched the literature, organized the results, and was the principal author of the paper. G.B. provided oversight on the structural organic chemistry and assisted with writing the paper. W.W.-N. assisted with writing the paper and prepared the majority of the illustrations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Porphyrin. Available online: https://en.wikipedia.org/w/index.php?title=Porphyrin&oldid=781762373 (accessed on 10 June 2017).
- Küster, W. Beitrage zur Kenntnis des Bilirubins und Hämins. Hoppe Seyler’s Z. Physiol. Chem. 1912, 82, 463–483. [Google Scholar] [CrossRef]
- Kadish, W.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010–2016; Volume 44. [Google Scholar]
- Moore, M.W. An historical introduction to porphyrin and chlorophyll synthesis. In Tetrapyrroles: Birth, Life, and Death; Warren, M., Smith, A., Eds.; Landes Bioscience and Springer Science+Business Media: Berlin, Germany, 2009; pp. 1–28. [Google Scholar]
- Suslick, K.S.; Rakow, N.A.; Kosal, M.E.; Chou, J.H. The materials chemistry of porphyrins and metalloporphyrins. J. Porphyr. Phthalocyanines 2000, 4, 407–413. [Google Scholar] [CrossRef]
- Cook, L.P.; Wong-Ng, W.; Brewer, G. Porphyrin-based chemistry for carbon capture and sequestration. In Advances in Materials Science for Environmental and Energy Technologies V; Ohji, T., Kanakhala, R., Matyáš, J., Marijooran, N., Pickrell, G., Wong-Ng, W., Eds.; Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 201–221. [Google Scholar]
- Cambridge Crystallographic Data Center. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 31 March 2017).
- International Union of Pure and Applied Chemistry. Available online: http://www.chem.qmul.ac.uk/iupac/tetrapyrrole/ (accessed on 31 March 2017).
- Saltsman, I.; Goldberg, I.; Balasz, Y.; Gross, Z. Porphine and pyrrole-substituted porphyrin from cyclocondensation of tripyrrane with mono-substituted pyrolles. Tetrahedron Lett. 2007, 48, 239–244. [Google Scholar] [CrossRef]
- Devillers, C.H.; Fleurat-Lessard, P.; Lucas, D. “Porphine,” the fully unsubstituted porphyrin: A comprehensive overview. In Handbook of Porphyrin Science; Kadish, W., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Singapore, 2014; Volume 37, pp. 75–231. [Google Scholar]
- Kaduk, J.A.; Wong-Ng, W.; Cook, L.P.; Chakraborty, B.; Lapidus, S.H.; Ribaud, L.; Brewer, G. Synchrotron X-ray investigation of α-chlorohemin, C34H32ClFeN4O4, an Fe-porphyrin. Solid State Sci. 2016, 53, 63–70. [Google Scholar] [CrossRef]
- Silvers, S.J.; Tulinsky, A. The crystal and molecular structure of triclinic tetraphenyl porphyrin. J. Am. Chem. Soc. 1967, 89, 3331–3337. [Google Scholar] [CrossRef] [PubMed]
- Codding, P.W.; Tulinsky, A. Structure of tetra-n-propylporphine. Average structure for the free base macrocycle from three independent determinations. J. Am. Chem. Soc. 1972, 94, 4151–4157. [Google Scholar] [CrossRef] [PubMed]
- Lauher, J.W.; Ibers, J.A. Structure of Ocaethylporphyrin. A comparison with other free base porphyrins. J. Am. Chem. Soc. 1973, 95, 5148–5152. [Google Scholar] [CrossRef] [PubMed]
- Caughey, W.S.; Ibers, J.A. Crystal and molecular structure of the free base porphyrin, protoporphyrin IX dimethyl ester. J. Am. Chem. Soc. 1977, 99, 6639–6645. [Google Scholar] [CrossRef] [PubMed]
- Jentzen, W.; Trowska-Tyrk, I.; Scheidt, W.R.; Shetnutt, J.A. Planar solid-state and solution structures of (porphinato) nickel(II) as determined by X-ray diffraction and Raman spectroscopy. Inorg. Chem. 1996, 35, 3559–3567. [Google Scholar] [CrossRef]
- Devillers, C.H.; Dime, A.K.D.; Cattey, H.; Lucas, D. Crystallographic, spectroscopic and electrochemical characterization of pyridine adducts of magnesium(II) and zinc(II) porphine complexes. C. R. Chim. 2013, 16, 540–549. [Google Scholar] [CrossRef]
- Mavridis, A.; Tulinsky, A. Crystal and molecular structure of dimethoxy-porphinatogermanium (IV). Inorg. Chem. 1976, 15, 2723–2727. [Google Scholar] [CrossRef]
- Fleischer, E.B. The structure of porphyrins and metalloporphyrins. Acc. Chem. Res. 1970, 3, 105–112. [Google Scholar] [CrossRef]
- Arnold, D.P.; Goh, M.S.; Harper, S.R.; McMurtie, J.C. Oligoporphyrins with one- and two-atom covalent bridges: Synthesis, and reactivity, solid-state, solution and electronic structures, spectroscopy and applications. In Handbook of Porphyrin Science; Kadish, W., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Singapore, 2014; Volume 31, pp. 277–302. [Google Scholar]
- Esdaile, L.J.; Jensen, P.; McMurtie, J.C.; Arnold, D.P. Azoporphyrin: The porphrin analogue of azobenzene. Angew. Chem. Int. Ed. 2007, 46, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.; Johnson, A.W.; Winter, M. Bis-porphyrin derivatives. Part 1. Reaction of meso-hydroxymethyl-porphyinatometal derivatives with acids. J. Chem. Soc. Perkin Trans. 1977, 14, 1643–1647. [Google Scholar] [CrossRef]
- Senge, M.; Vicente, M.G.; Gerzevske, K.R.; Forsyth, T.P.; Smith, K.M. Models for the photosynthetic reaction center: Preparation, spectroscopy, and crystal and molecular structures of cofacial bisporphyrins linked by cis-1-2- and trans-1-2-ethene bridges and of 1,1-carbinol-bridged bisporphyrins. Inorg. Chem. 1994, 33, 5625–5638. [Google Scholar] [CrossRef]
- Crossley, M.J.; Burn, P.L. An approach to porphyrin-based molecular wires: Synthesis of a bis(porphyrin)tetraone and its conversion to a linearly conjugated tetrakisporphyrin system. J. Chem. Soc. Chem. Commun. 1991, 21, 1569–1571. [Google Scholar] [CrossRef]
- Perlovich, G.L. Thermodynamics of porphyrin sublimation. J. Porphyr. Phthalocyan. 2000, 4, 699–706. [Google Scholar] [CrossRef]
- Jurow, M.; Schuckman, A.; Batteas, J.D.; Drain, C.M. Porphyrins as molecular components of functional devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Aüwarter, W.; Écija, D.; Klappenberger, F.; Varth, J.V. Porphyrins at interfaces. Nat. Chem. 2015, 7, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Kubatkin, S.; Danilov, A.; Hjort, M.; Cornil, J.; Brédas, J.-L.; Stuhr-Hanson, N.; Hedegard, P.; Bjornholm, T. Single-electron transistor of a single organic molecule with access to several redox states. Nature 2003, 425, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Averin, D.V.; Likharev, K.K. Single electronics: A correlated transfer of single electrons and Cooper pairs in systems of small tunnel junctions. In Mesoscopic Phenomena in Solids; Altshuler, B.L., Lee, P.A., Webb, R.A., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 173–270. [Google Scholar]
- Wang, N.; Liu, H.; Zhan, J.; Cui, Y.; Xu, Z.; Ye, Y.; Kiguchi, M.; Murakoshi, K. Theoretical investigation on the electron transport path through the porphyrin molecules and chemisorption of CO. J. Phys. Chem. C 2009, 113, 7416–7423. [Google Scholar] [CrossRef]
- Galloni, P.; Vecchi, A.; Coletti, A.; Gatto, E.; Floris, B.; Conte, V. Porphyrins as active components for electrochemical and photoelectrochemical devices. In Handbook of Porphyrin Science; Kadish, W., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Singapore, 2014; Volume 33, pp. 225–415. [Google Scholar]
- Reimers, J.R.; Lu, T.X.; Crossley, M.J.; Hush, N.S. Molecular electronic properties of fused rigid porphyrin-oligomer molecular wires. Nanotechnology 1996, 7, 424–429. [Google Scholar] [CrossRef]
- Aviram, A. A view of the future of molecular electronics. Mol. Cryst. Liq. Cryst. 1993, 234, 13–28. [Google Scholar] [CrossRef]
- Yoon, D.H.; Lee, S.B.; Yoo, K.-H.; Kim, J.; Lim, J.K.; Aratani, N.; Tsuda, A.; Osuka, A.; Kim, D. Electrical conduction through linear porphyrin arrays. J. Am. Chem. Soc. 2003, 125, 11062–11064. [Google Scholar] [CrossRef] [PubMed]
- Sedghi, C.; Esdaile, L.J.; Anderson, H.L.; Martin, S.; Bethell, D.; Higgins, S.J.; Nichols, R.J. Comparison of the conductance of three types of porphyrin-based molecular wires: β,meso,β-fused tapes, meso-butdiyne-linked and twisted meso-meso linked oligomers. Adv. Mater. 2012, 24, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Medforth, C.J.; Shelnutt, J.A. Porphyrin nanotubes by ionic self-assembly. J. Am. Chem. Soc. 2004, 126, 15954–15955. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Yang, C.-J.; Chen, W.-H.; Lin, K.-J. Towards the development of electrical conduction and lithium-ion transport in a tetragonal porphyrin wire. Angew. Chem. Int. Ed. 2003, 42, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lee, L.L.; Lin, K.J. Electrical and optical properties of porphyrin single crystals. J. Macromol. Sci. Part B Phys. 2008, 47, 955–966. [Google Scholar] [CrossRef]
- Schramm, C.J.; Stojakovic, D.R.; Hoffman, B.M.; Maarks, T.J. New low-dimensional molecular metals: Single-crystal electrical conductivity of nickel phthalocyanine iodide. Science 1978, 200, 47–48. [Google Scholar] [CrossRef] [PubMed]
- Livshits, V.A.; Blyumenfel’d, L.A. Semiconductor properties of porphyrins. J. Struct. Chem. 1968, 8, 383–388. [Google Scholar] [CrossRef]
- Kobayashi, N.; Nevin, W.A.; Mizunuma, S.; Awaji, H.; Yamaguchi, M. Ring-expanded porphyrins as an approach towards highly conductive molecular semiconductors. Chem. Phys. Lett. 1993, 205, 51–54. [Google Scholar] [CrossRef]
- Jones, R.; Tredgold, R.H.; Hoorfar, A. Electrical conductivity in Langmuir-Blodgett films of porphyrins: In-plane and thorough-the-film studies. Thin Solid Films 1984, 113, 115–128. [Google Scholar] [CrossRef]
- Lui, C.-Y.; Pan, H.-L.; Fox, M.A.; Bard, A.J. High-density nanosecond charge trapping in thin films of the photoconductor ZnODEP. Science 1993, 261, 897–899. [Google Scholar]
- Wood, D.W.; Andersen, T.N.; Eyring, H. Electrical properties of some porphyrins under high pressure. J. Phys. Chem. 1966, 70, 360–366. [Google Scholar] [CrossRef]
- Collman, J.P.; McDevitt, J.T.; Yee, G.T.; Leidner, C.R.; McCullough, L.G.; Little, W.A.; Torrance, J.B. Conductive polymers derived from iron, ruthenium, and osmium metalloporphyrins: The shish-kebab approach. Proc. Natl. Acad. Sci. USA 1986, 83, 4581–4585. [Google Scholar] [CrossRef] [PubMed]
- Collman, J.P.; McDevitt, J.T.; Leidner, C.R.; Yee, G.T.; Torrance, J.B.; Little, W.A. Synthetic, electrochemical, optical and conductivity studies of coordination polymers of iron, ruthenium, and osmium octaethylporphyrin. J. Am. Chem. Soc. 1987, 109, 4606–4614. [Google Scholar] [CrossRef]
- Suslick, K.S.; Chen, C.-T. Polymeric metalloporphyrins for field responsive materials. Polym. Mater. Sci. Eng. 1990, 63, 272–278. [Google Scholar]
- Chen, C.-T.; Suslick, K.S. One-dimensional coordination polymers: Applications to materials science. Coord. Chem. Rev. 1993, 128, 293–322. [Google Scholar] [CrossRef]
- Aratani, N.; Osuka, A. Synthetic strategies toward multiporphyrinic architectures. In Handbook of Porphyrin Science; Kadish, W., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Singapore, 2010; Volume 1, pp. 1–132. [Google Scholar]
- Gao, W.-Y.; Chrzanowski, M.; Ma, S. Metal-metalloporphyrinframeworks: A resurging class of functional materials. Chem. Soc. Rev. 2014, 43, 5841–5866. [Google Scholar] [CrossRef] [PubMed]
- Frampton, M.J.; Akda, H.; Cowley, A.R.; Rogers, J.E.; Slagle, J.E.; Fleitz, P.A.; Drobizhev, M.; Rebane, A.; Anderson, H.L. Synthesis, crystal structure, and nonlinear optical behavior of β-unsubstituted meso-meso-E-vinylene-linked porphyrin dimers. Org. Lett. 2005, 7, 5365–5368. [Google Scholar] [CrossRef] [PubMed]
- Imahori, H.; Fukuzumi, S. Porphyrin- and fullerene-based molecular photovoltaic devices. Adv. Funct. Mater. 2004, 14, 525–536. [Google Scholar] [CrossRef]
- Hisaki, I.; Hiroto, S.; Kim, K.S.; Noh, S.B.; Kim, D.; Shinokubo, H.; Osuka, A. Synthesis of doubly β-to-β 1,3-butadiene-bridged diporphyrins: Enforced planar structures and large two-photon absorption cross sections. Angew. Chem. Int. Ed. 2007, 46, 5125–5128. [Google Scholar] [CrossRef] [PubMed]
- Stulz, E.; Scott, S.M.; Bond, A.D.; Teat, S.J.; Sanders, J.M. Selection and amplification of mixed-metal porphyrin cages from dynamic combinatorial libraries. Chem. Eur. J. 2003, 6039–6048. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-Y.; Tashiro, K.; Hirabayashi, Y.; Kinbara, K.; Saigo, K.; Aida, T.; Sakamoto, S.; Yamaguchi, K. Cyclic dimers of metalloporphyrins as tunable hosts for fullerenes: A remarkable effect of rhodium(III). Angew. Chem. Int. Ed. 2001, 40, 1858–1861. [Google Scholar] [CrossRef]
- Deng, Y.; Chang, C.J.; Nocera, D.G. Direct observation of “Pac-Man” effect from dibenzofuran-bridged cofacial bisporphyrins. J. Am. Chem. Soc. 2000, 122, 410–411. [Google Scholar] [CrossRef]
- Tokuji, S.; Yurino, T.; Aratani, N.; Shinokubo, H.; Osuka, A. Palladium-catalyzed dimerization of meso-bromoporphyrins: Highly regioselective meso-β coupling through unprecedented remote C-H bond cleavage. Chem. Eur. J. 2009, 15, 12208–12211. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jang, S.Y.; Yamaguchi, S.; Sankar, J.; Hiroto, S.; Aratani, N.; Shin, J.Y.; Easwaramoorthi, A.; Kim, K.S.; Kim, D.; et al. 2,5-thienylene-bridged triangular and linear porphyrin trimers. Angew. Chem. Int. Ed. 2008, 47, 6004–6007. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, A.; Nakamura, T.; Sakamoto, S.; Yamaguchi, K.; Osuka, A. A self-assembled porphyrin box from meso-meso-linked bis [5-p-pyridyl-15-(3,5-di-octyloxyphenyl)porphyrinato zinc(II)]. Angew. Chem. Int. Ed. 2002, 41, 2817–2821. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Katoh, T.; Shinokubo, H.; Osuka, A. Pt(II)- and Pt(IV)-bridged cofacial diporphyrins via carbon—transition metal σ-bonds. J. Am. Chem. Soc. 2008, 130, 14440–14441. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Yamazaki, T.; Nishimura, Y.; Yamazaki, I.; Osuka, A. Three-dimensionally arranged windmill and grid porphyrin arrays by AgI-promoted meso-meso block oligomers. Chem. Eur. J. 2000, 6, 3254–3271. [Google Scholar] [CrossRef]
- Hori, T.; Peng, X.; Aratani, N.; Takagi, A.; Matsumoto, T.; Kawai, T.; Yoon, Z.S.; Yoon, M.C.; Yang, J.; Kim, D.; et al. Synthesis of nanometer-scale porphyrin wheels of variable size. Chem. Eur. J. 2008, 14, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamaguchi, S.; Osuka, A.; Shinokubo, H. Platinum(II) and Platinum(IV) porphyrin pincer complexes: Synthesis, structures, and reactivity. Organometallics 2010, 29, 3997–4000. [Google Scholar] [CrossRef]
Figure 1.
The structure of porphine, from data of [7,9]. Dark gray = C, light blue = N, white = H. (Note: This is the color scheme used from here on, unless otherwise described).
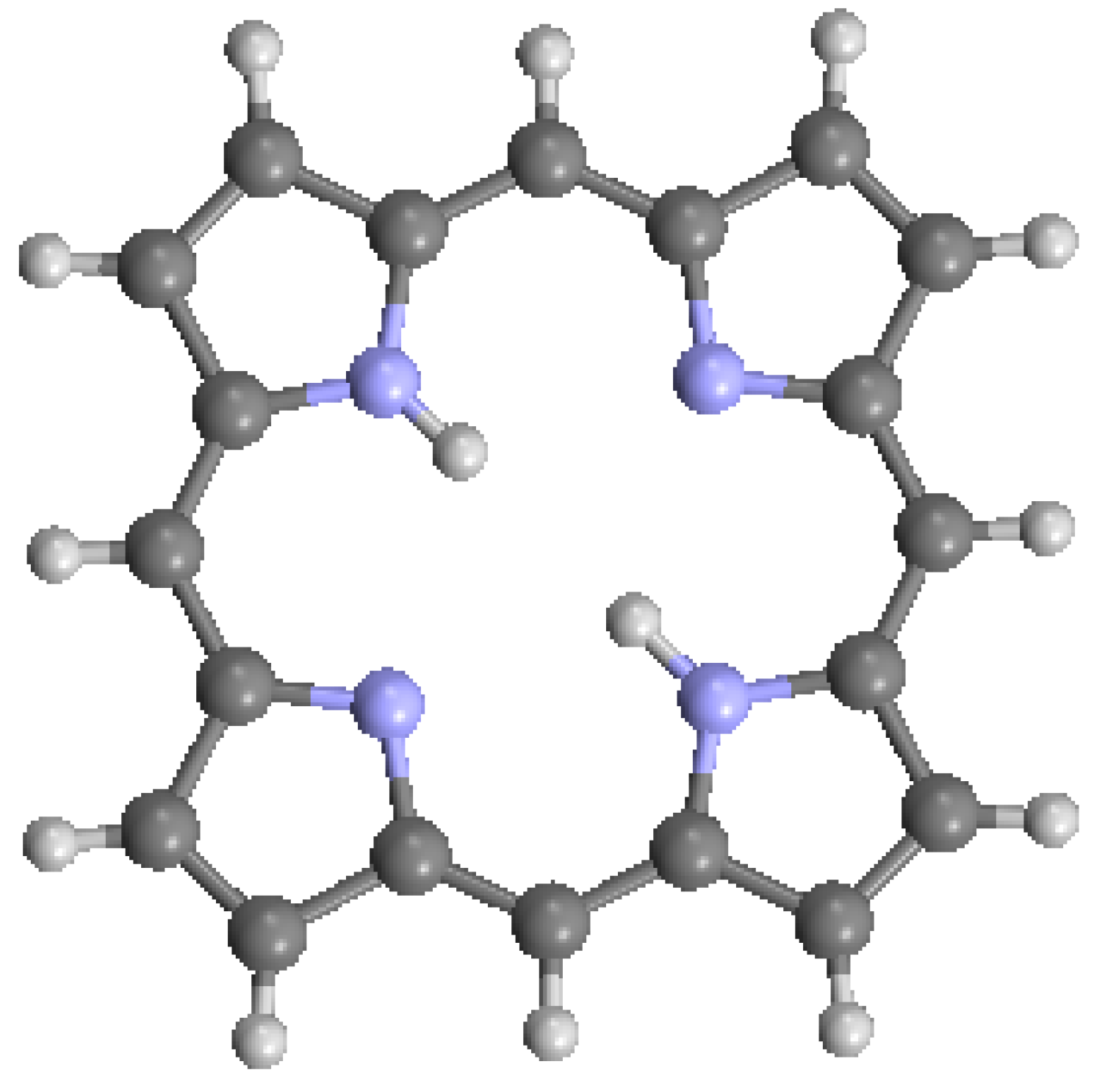
Figure 2.
Nomenclature of macrocycle atomic positions [8]: (a) numerical designation; and (b) site designation.
Figure 2.
Nomenclature of macrocycle atomic positions [8]: (a) numerical designation; and (b) site designation.
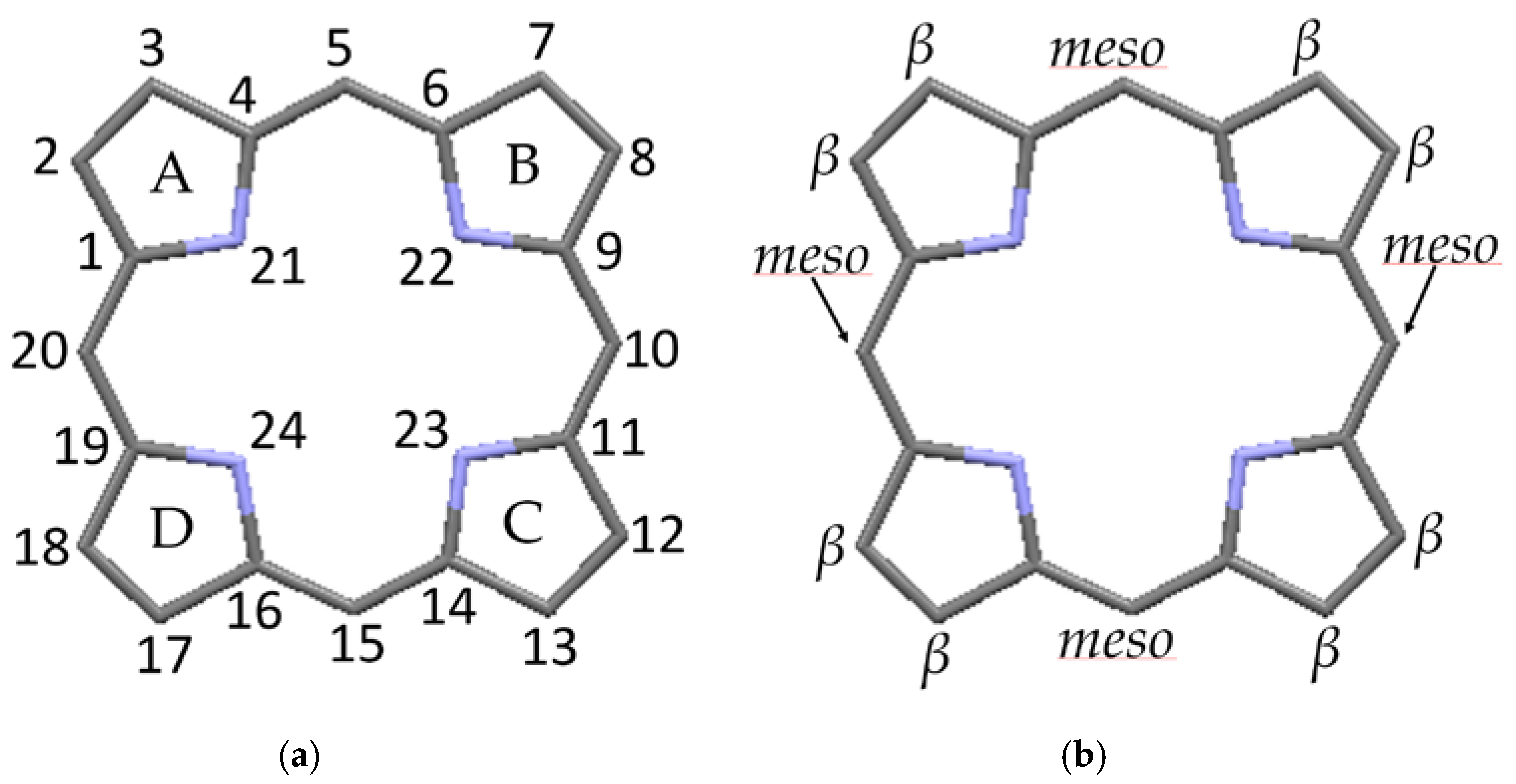
Figure 3.
Mean bond distances and angles in the porphine macrocycle, after [10], with permission from World Scientific Publishing C.o. Note that the numbering system is the mirror image of that in Figure 2a, with the exception of the N positions.
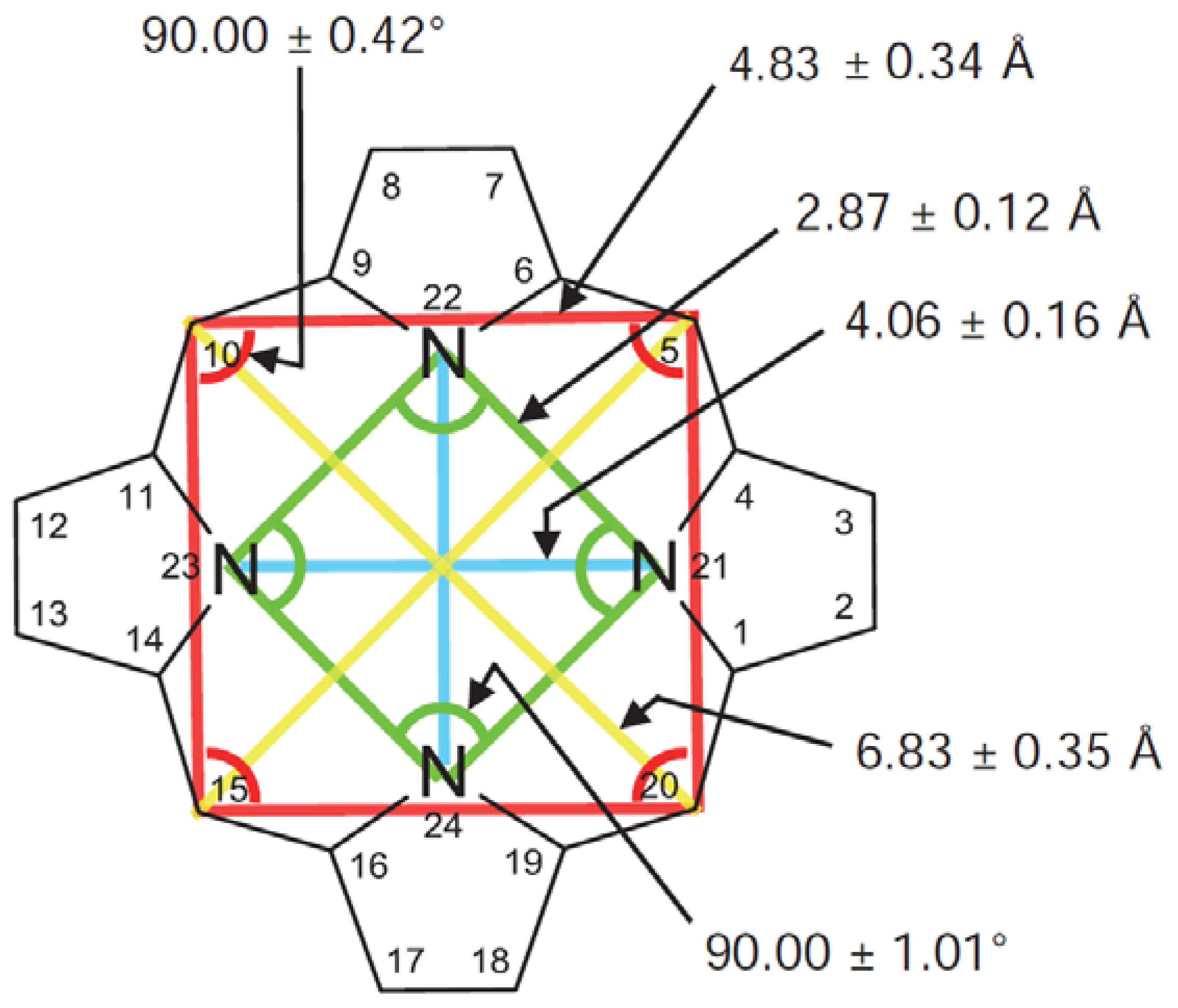
Figure 4.
Examples of substituents: (a) TPP; (b) TPrP; (c) OEP; and (d) PP9; from data of [7,12,13,14,15]. Shown without hydrogens. Red = O.
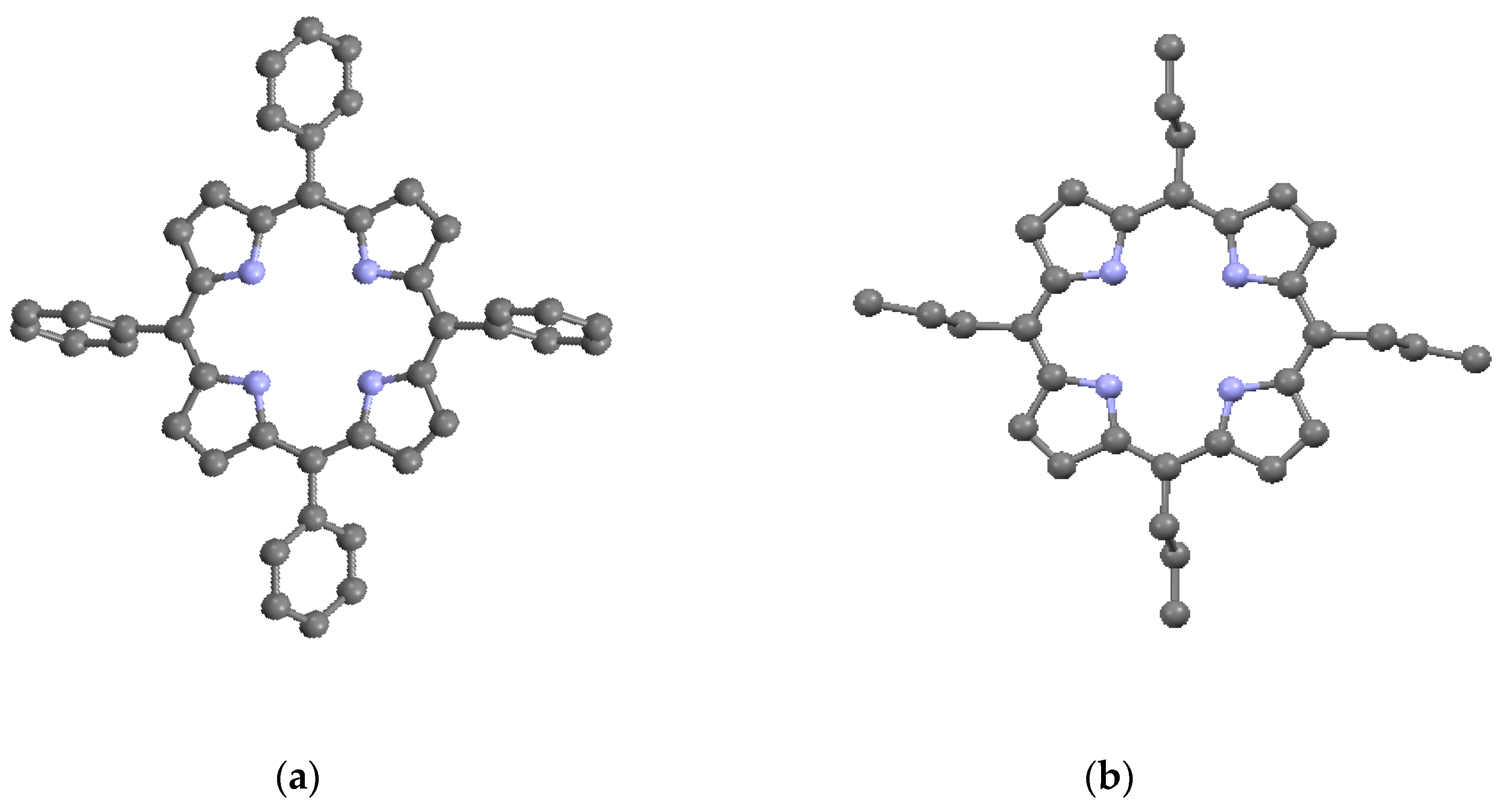
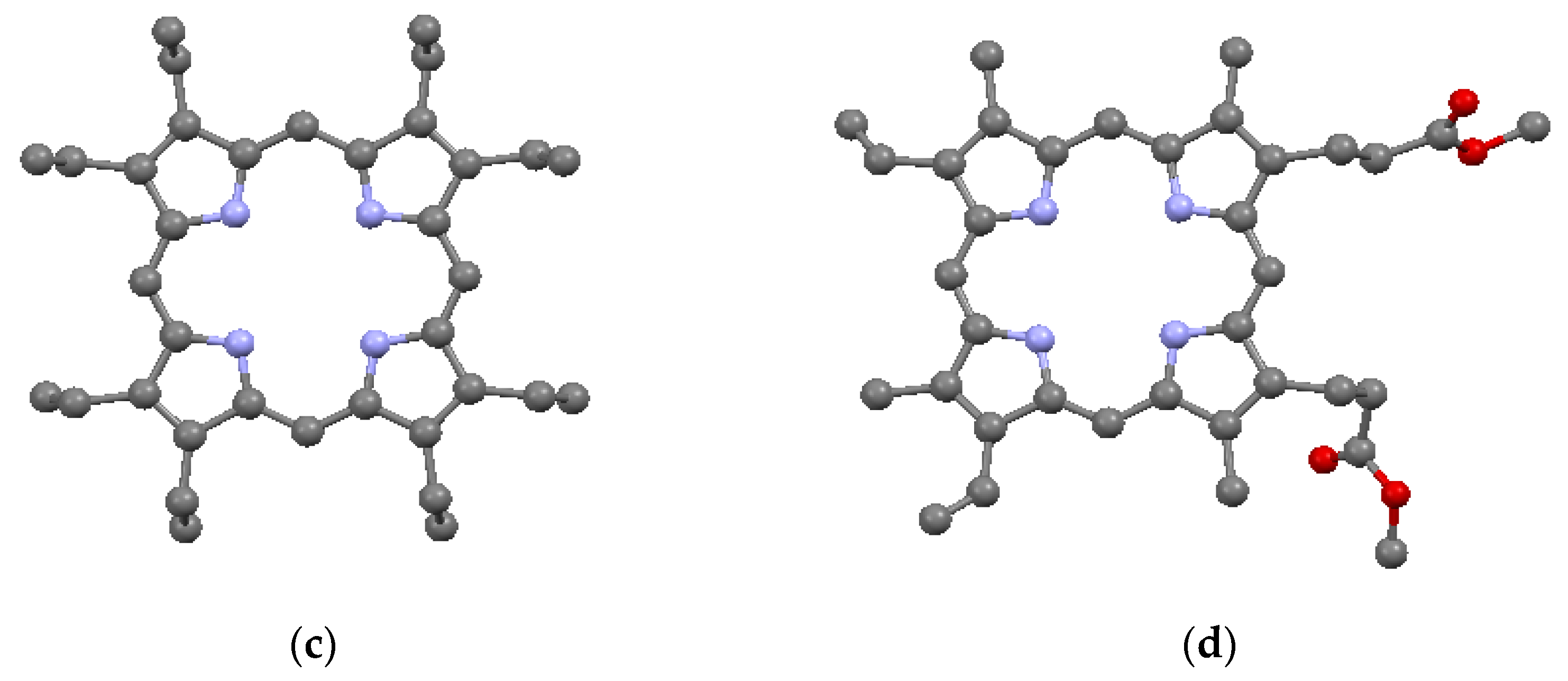
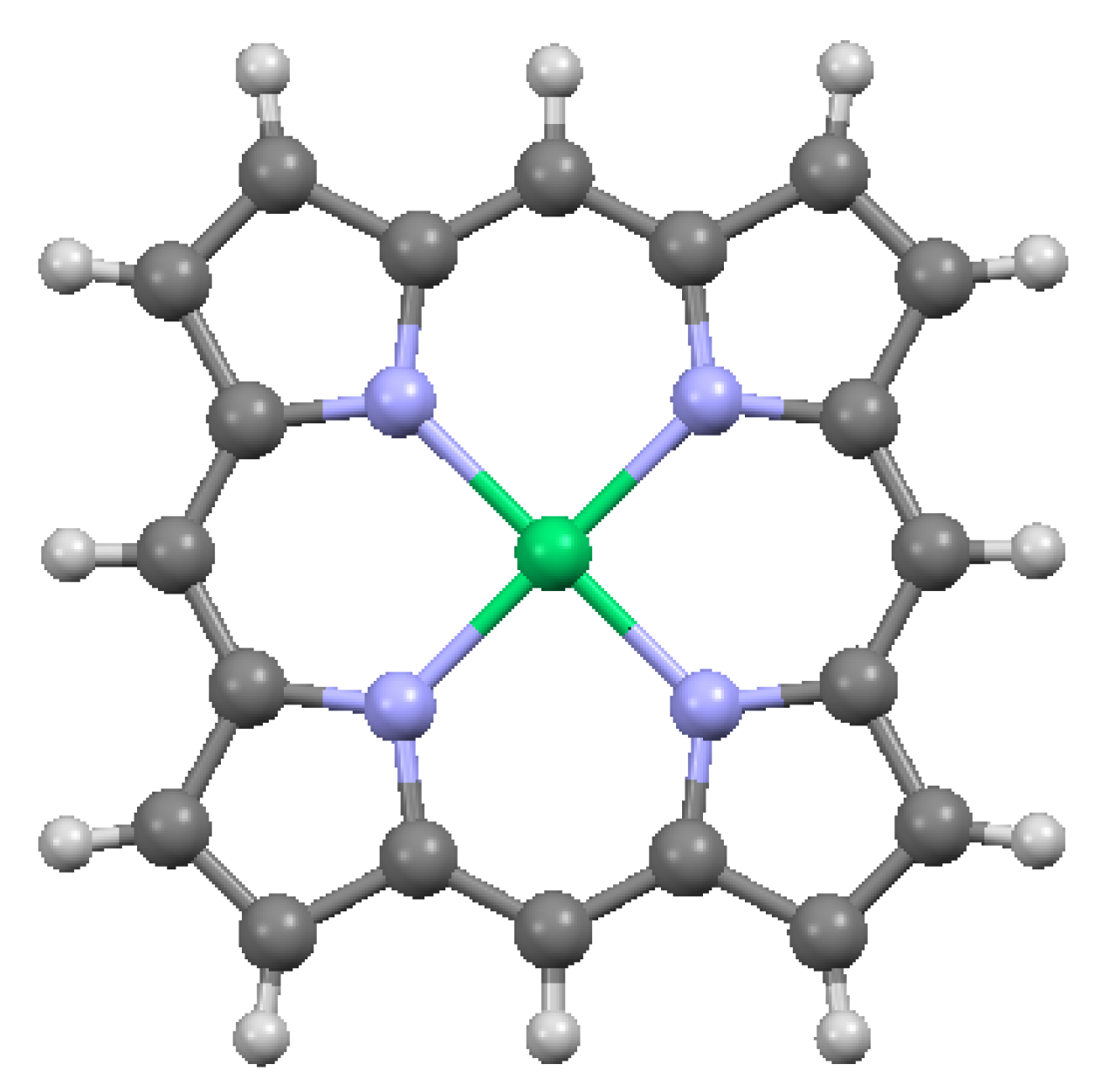
Figure 6.
Mg-porphine, from data of [7,17]: (a) single molecule; and (b) crystal packing. Light green = Mg.

Figure 7.
Zn-porphine, from data of [7,17]: (a) top view, with hydrogens; and (b) side view, without hydrogens. Dark blue = Zn.
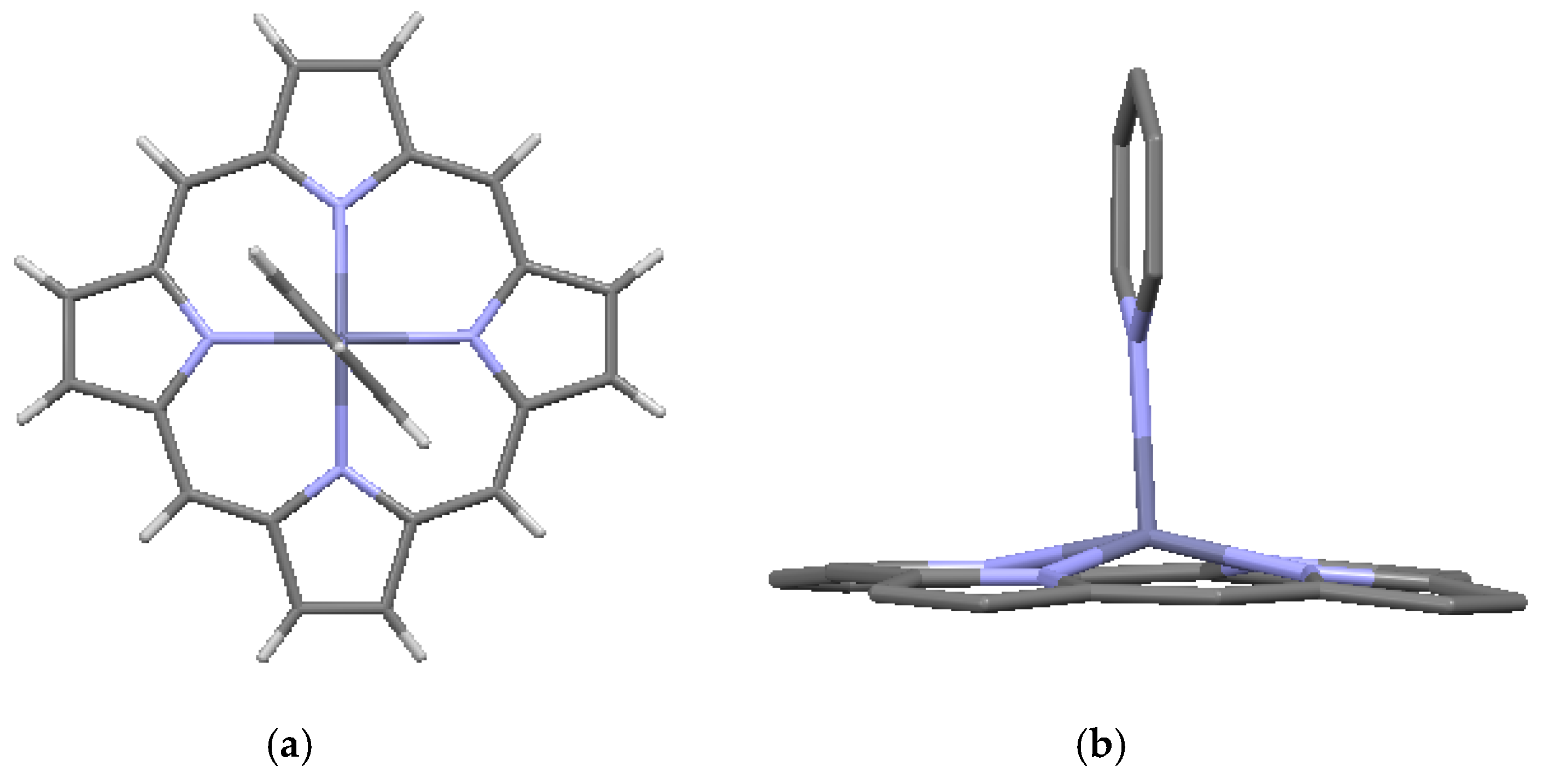
Figure 8.
Ge-porphine, after data of [7,18]: (a) top view; and (b) side view. Dark green = Ge, red = O.


Figure 10.
–C(OH)– covalent β-β linkage in OEP-based dimer. From data of [7,22,23]. Peripheral substituents are omitted for clarity. Green = Ni, red = O.
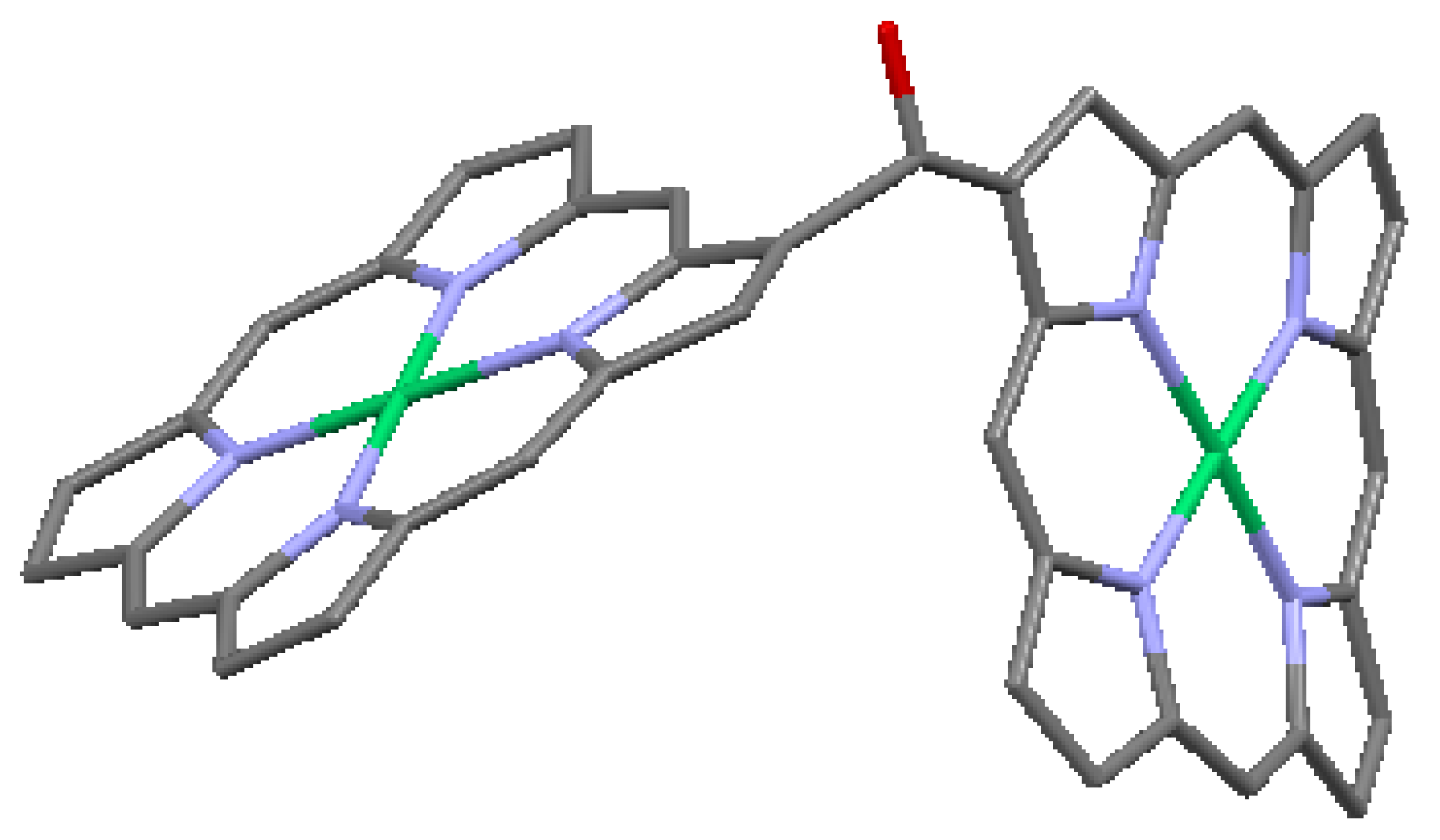
Figure 11.
Theoretically possible electron transport pathways in a porphyrin macrocycle with: (a) meso–meso connections; and (b) with β–β connections [30]; diagram after [31], with permission from World Scientific Publishing Co.
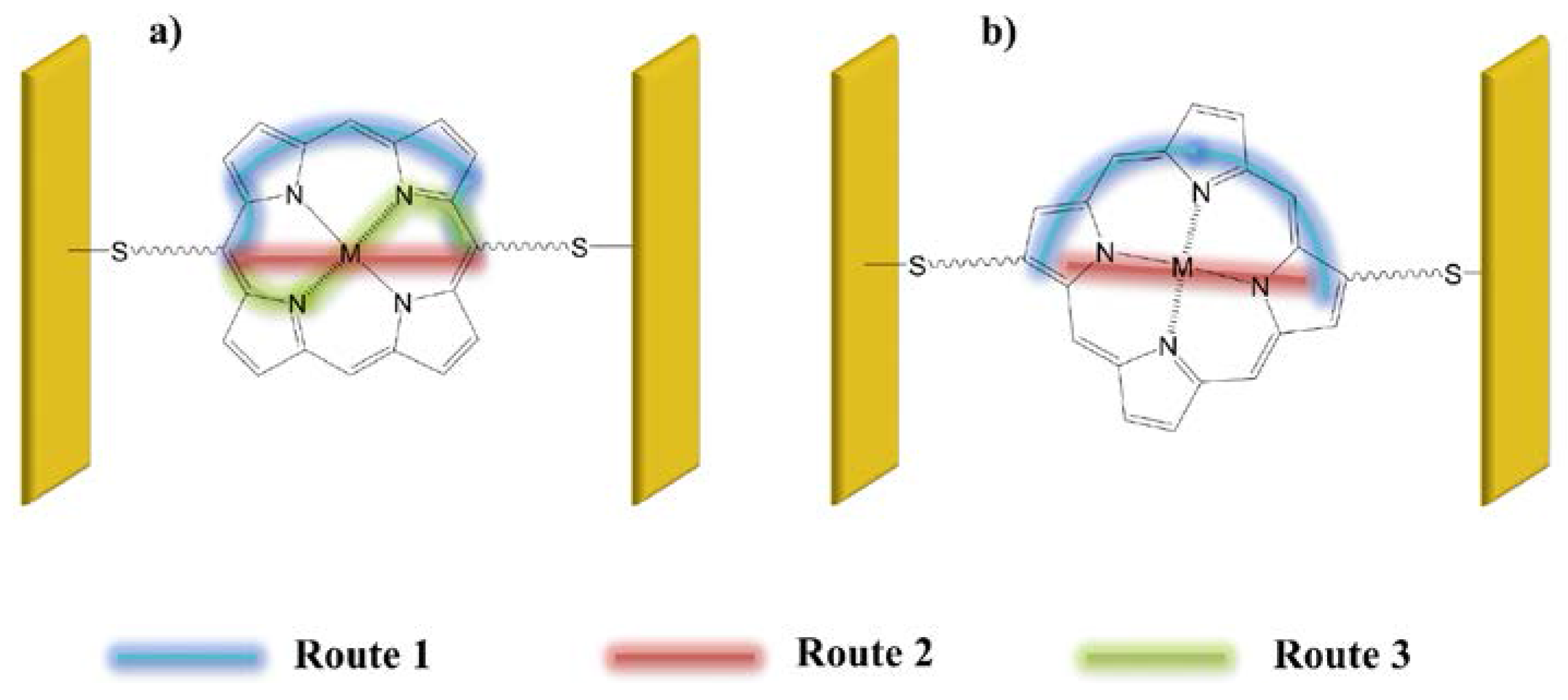
Figure 12.
An anthracene-conjugated tetrakisporphyrin with tert-butyl substituents, after [24], with permission from the Royal Society of Chemistry .
Figure 12.
An anthracene-conjugated tetrakisporphyrin with tert-butyl substituents, after [24], with permission from the Royal Society of Chemistry .

Figure 13.
An oligoporphyrin-containing molecular switch, after [32], with permission from IOP Publishing Co. “+” indicates location of electron hole on oligoporphyrin side chain.
Figure 13.
An oligoporphyrin-containing molecular switch, after [32], with permission from IOP Publishing Co. “+” indicates location of electron hole on oligoporphyrin side chain.
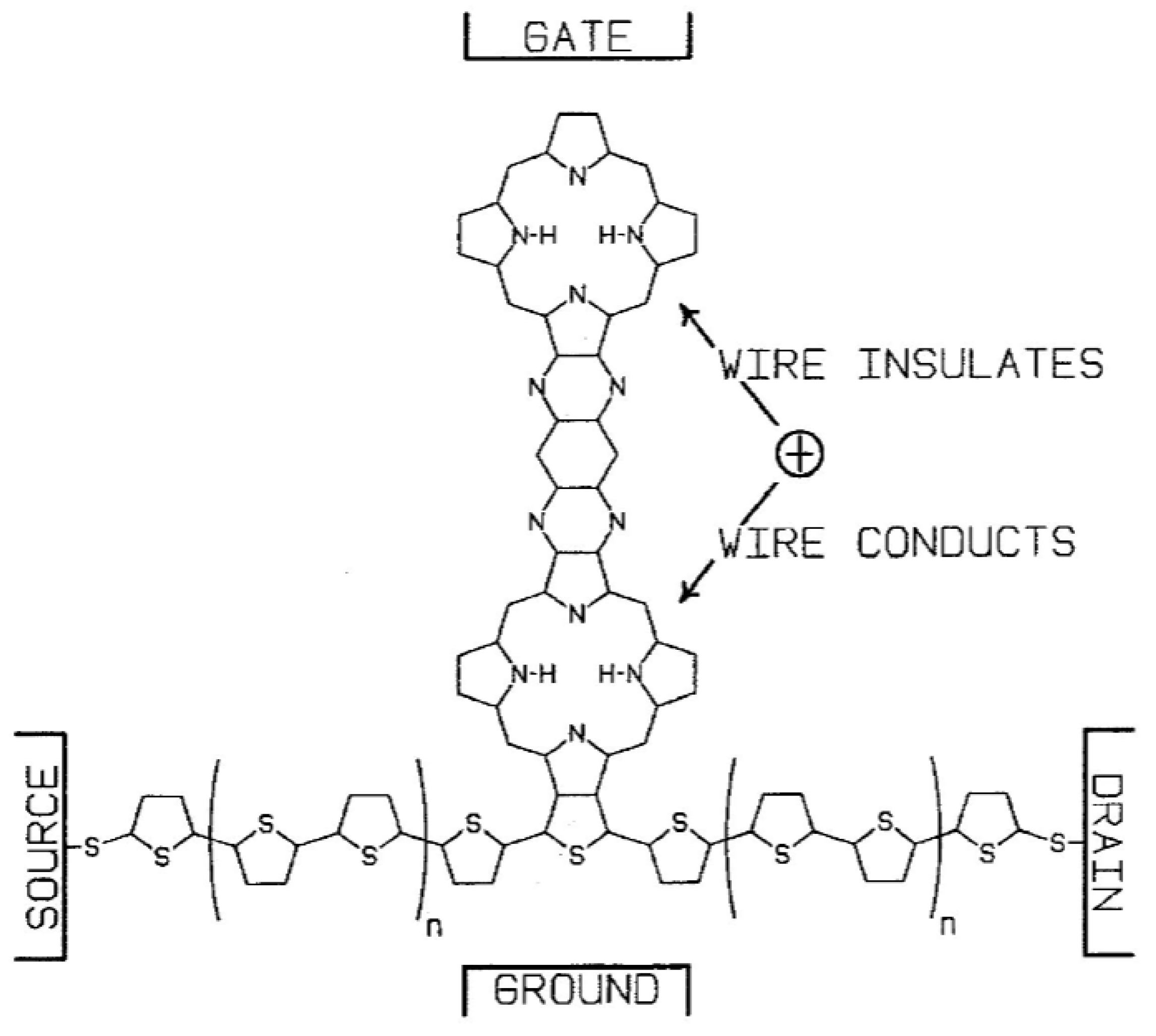
Figure 14.
Electrical conductivity in a triclinic porphyrin single crystal (after Ref. [27], with permission from Taylor and Francis): (a) conductivity perpendicular to the a-axis; and (b) parallel with the a-axis.
Figure 14.
Electrical conductivity in a triclinic porphyrin single crystal (after Ref. [27], with permission from Taylor and Francis): (a) conductivity perpendicular to the a-axis; and (b) parallel with the a-axis.
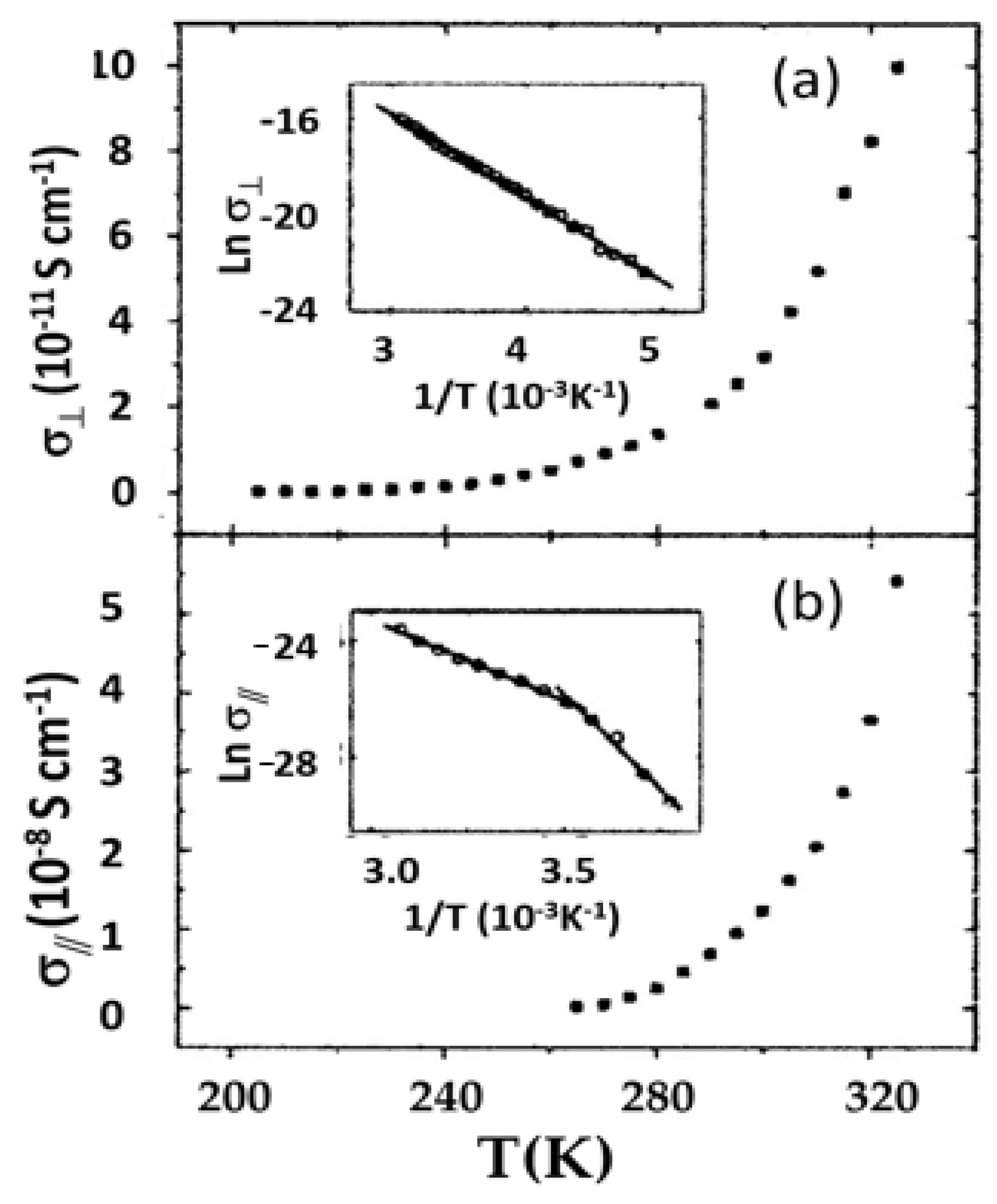
Figure 15.
Structure of the photoconductor ZnODEP, after [43], with permission from the American Association for the Advancement of Science.
Figure 15.
Structure of the photoconductor ZnODEP, after [43], with permission from the American Association for the Advancement of Science.
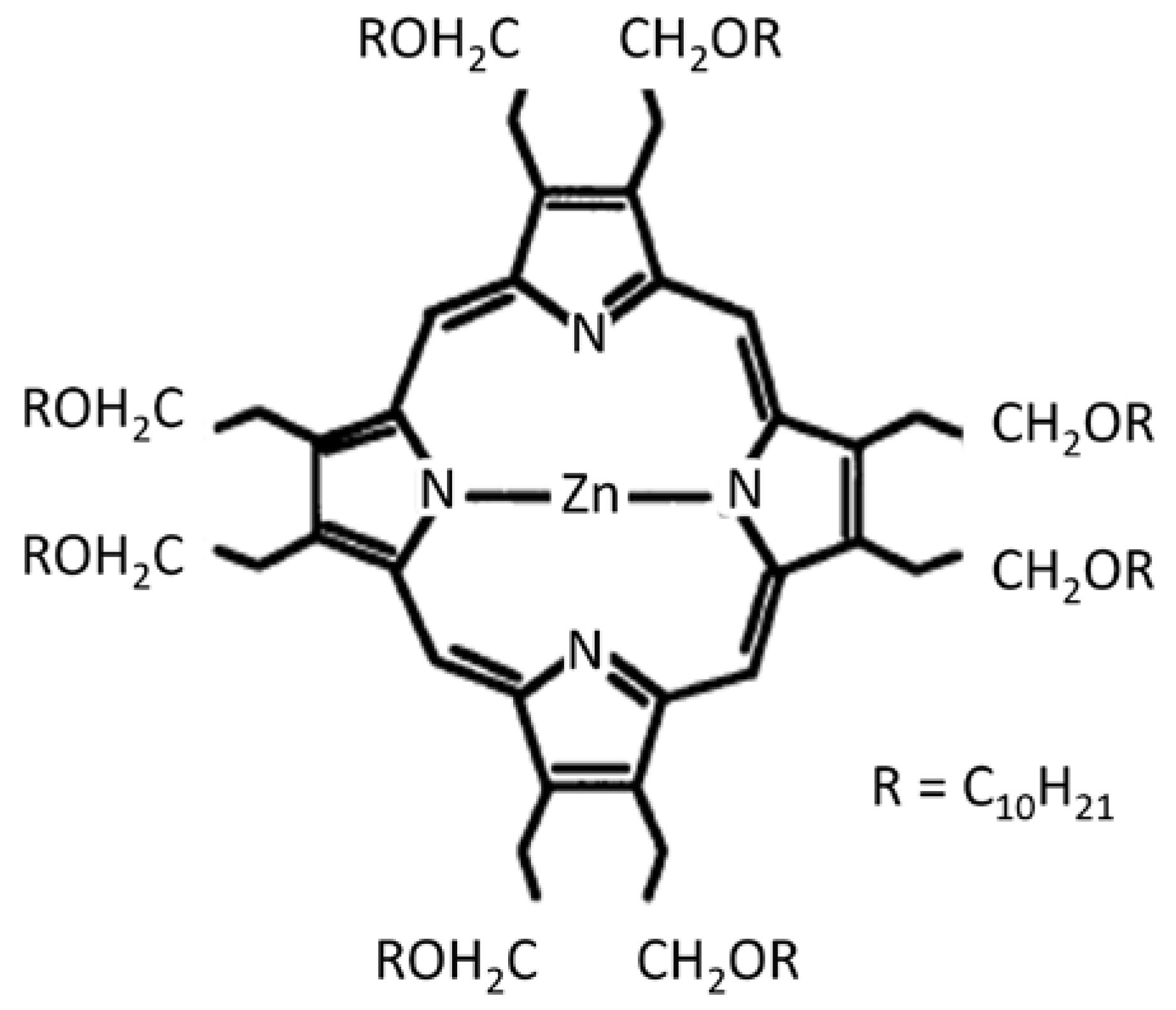
Figure 16.
Schematic representation of dipole moment switching in a ferroelectric porphyrin polymer. D = donor, X = acceptor. After Ref. [48], with permission from Elsevier.
Figure 16.
Schematic representation of dipole moment switching in a ferroelectric porphyrin polymer. D = donor, X = acceptor. After Ref. [48], with permission from Elsevier.
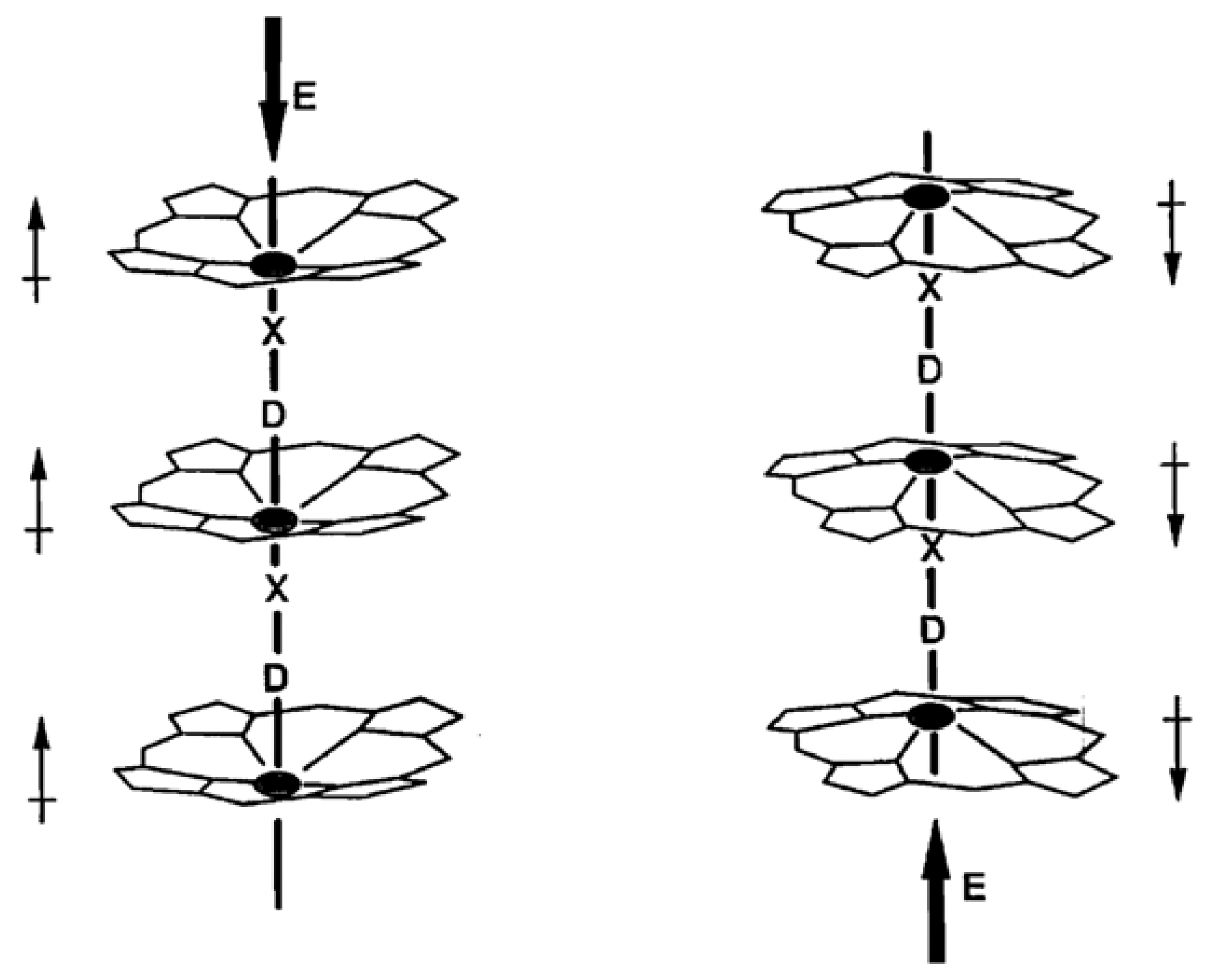
Figure 17.
A doubly β-to-β 1,3-butadiyne-bridged TPP-based dimer, from data in [7,53]. Each zinc has one axially bound isopropanol ligand. Each phenyl ring has two tert-butyl groups at 3,5 positions. Solvates and peripheral groups are omitted in this and the following diagrams as necessary for clarity. Dark blue = Zn.
Figure 17.
A doubly β-to-β 1,3-butadiyne-bridged TPP-based dimer, from data in [7,53]. Each zinc has one axially bound isopropanol ligand. Each phenyl ring has two tert-butyl groups at 3,5 positions. Solvates and peripheral groups are omitted in this and the following diagrams as necessary for clarity. Dark blue = Zn.
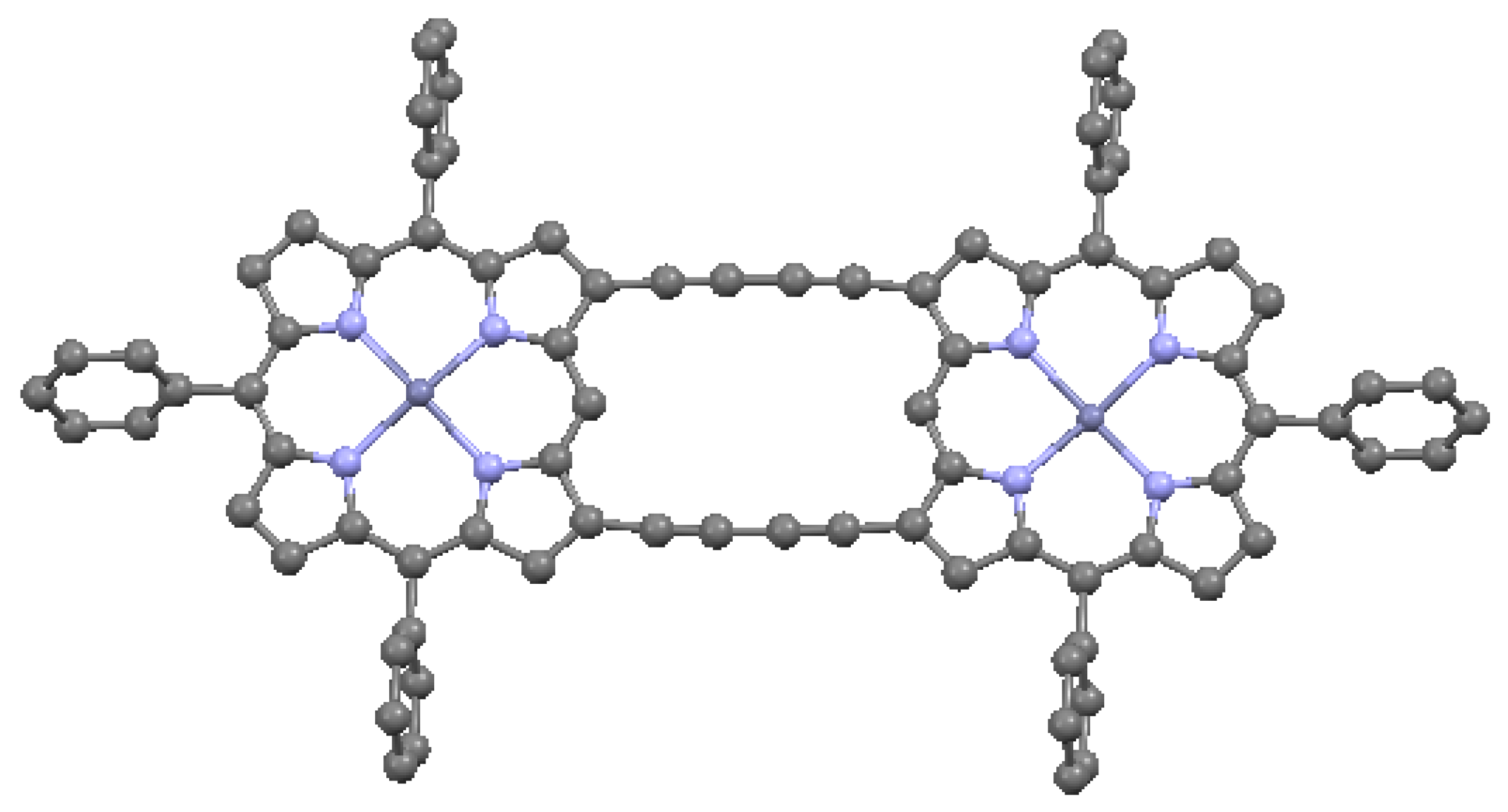
Figure 18.
Mixed metalloporphyrin cage, from data in [7,54]. Dark blue = Zn, dark green = Rh, orange = P. Cl is not shown.
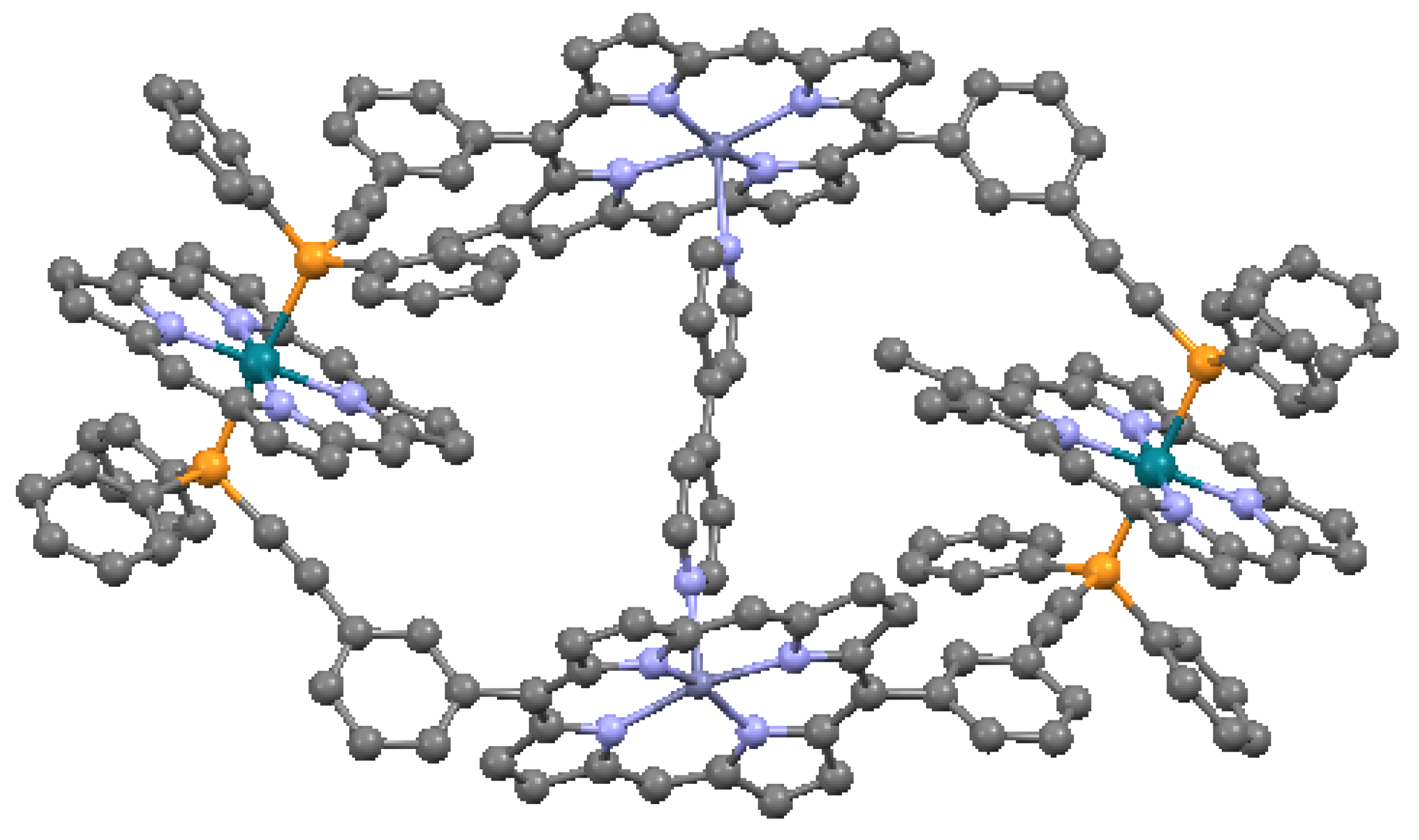
Figure 19.
Inclusion complex of C60 in cyclic Zn-porphyin dimer, from data in [7,55]. Dark blue = Zn, red = O.
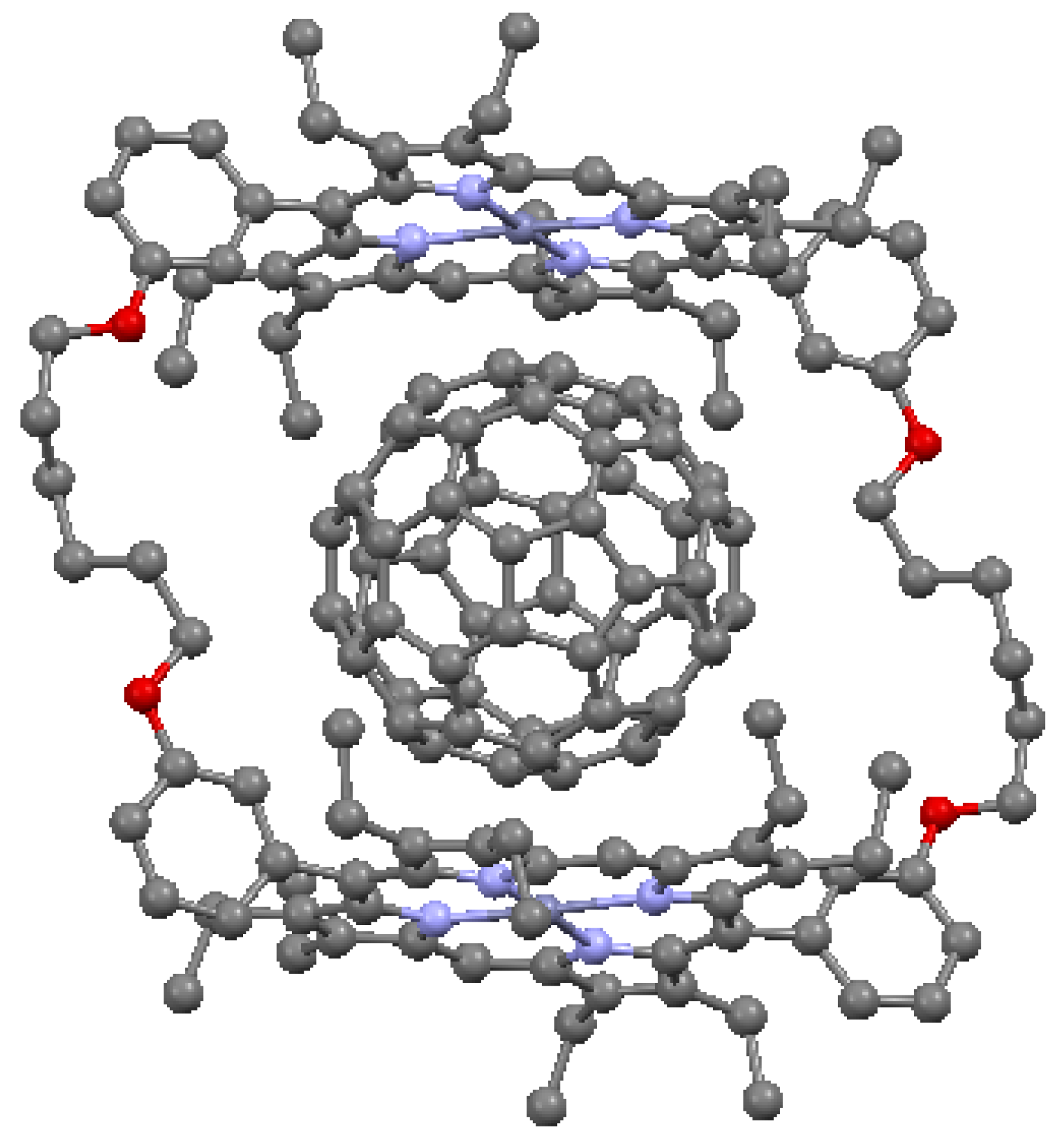
Figure 20.
Dibenzofuran-bridged cofacial bisporphyrin, from data in [7,56]. Dark blue = Zn, red = O.

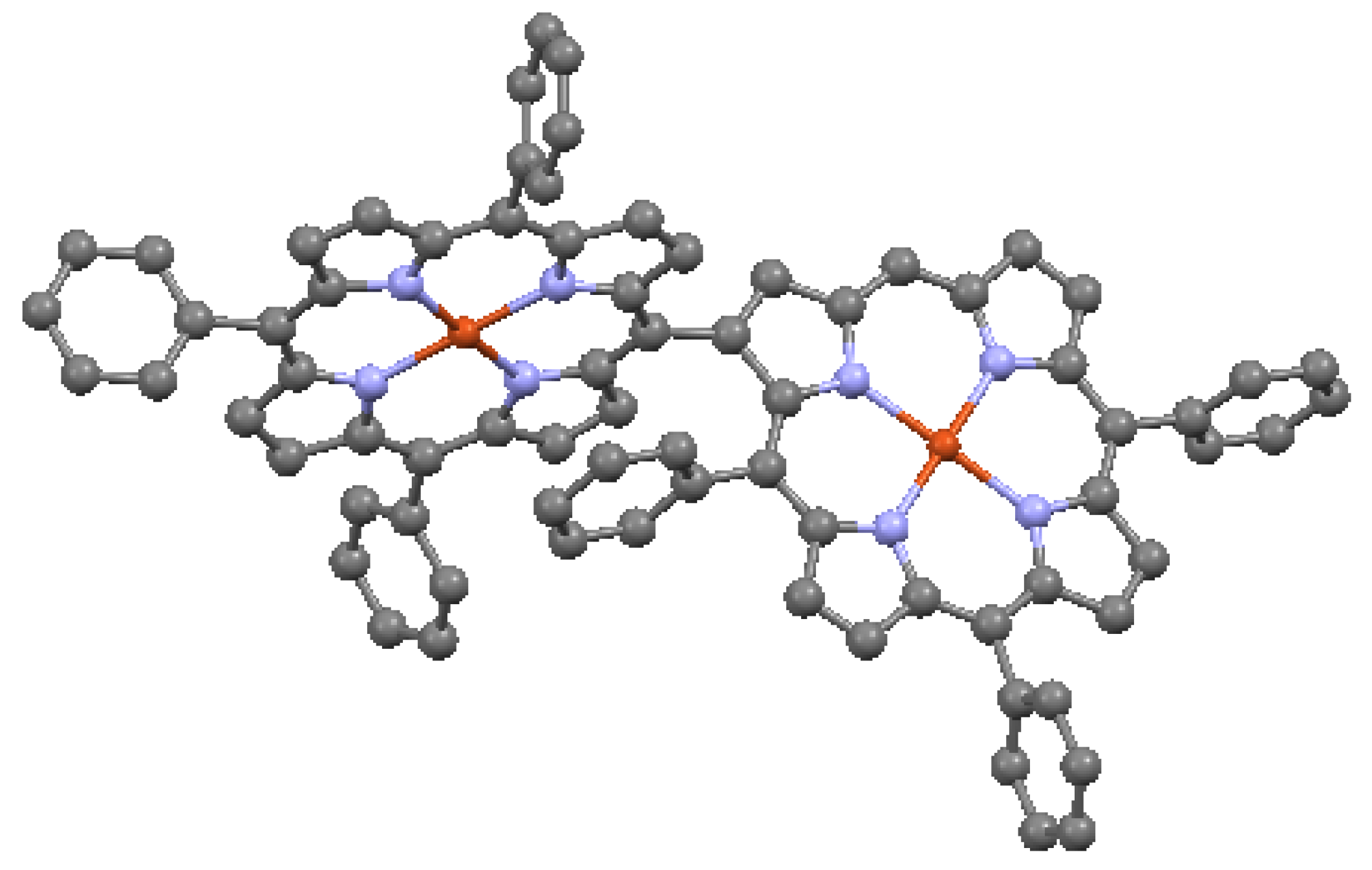
Figure 22.
Thienylene-bridged triangular Ni-porphyrin tetramer, from data in [7,58]. Green = Ni, yellow = S.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic Properties of Porphine [9].
Table 1.
Crystallographic Properties of Porphine [9].
| CSD No. | 609953 (Figure 1) |
|---|---|
| Name | porphine |
| Formula | C14H20N4 |
| System | monoclinic |
| Space gr. | P 21/c |
| A (Å) | 10.2262(3) |
| B (Å) | 11.9060(5) |
| C (Å) | 12.3853(4) |
| α (deg.) | 90.0 |
| β (deg.) | 101.711(3) |
| γ (deg.) | 90.00 |
| V (Å3) | 1476.56 |
| Z | 4 |
| ρ (g/cm3) | 1.396 |
Table 2.
Crystallographic Properties of Selected Substituted Porphines (no metalation).
| CSD No. | 1275315 (Figure 4a) | 1051569 (Figure 4b) | 1225729 (Figure 4c) | 1238138 (Figure 4d) |
|---|---|---|---|---|
| Name | Tetraphenyl porphyrin (TPP) | Tetrapropyl porphyrin (TPrP) | Octaethyl porphyrin (OEP) | protoporphyrin IX (PP9) |
| Formula | C44H30N4 | C32H38N4 | C36H46N4 | C36H38N4O4 |
| System | triclinic | monoclinic | triclinic | triclinic |
| Space gr. | P −1 | P 21/n | P −1 | P −1 |
| a (Å) | 6.44 (1) | 5.0843 (2) | 9.791 (2) | 11.303 (5) |
| b (Å) | 10.42 (1) | 11.6074 (6) | 10.771 (2) | 22.553 (10) |
| c (Å) | 12.41 (1) | 22.1695 (11) | 7.483 (2) | 6.079 (3) |
| α (deg.) | 96.06 (5) | 90.00 | 97.43 (1) | 91.38 (2) |
| β (deg.) | 99.14 (5) | 93.53 (2) | 106.85 (1) | 94.08 (2) |
| γ (deg.) | 101.12 (5) | 90.00 | 93.25 (1) | 81.96 (1) |
| V (Å3) | 798.747 | 1305.86 (11) | 745.23 | 1530.36 |
| Z | 1 | 2 | 1 | 2 |
| ρ (g/cm3) | 1.278 | 1.217 | 1.192 | 1.282 |
| Temp. | 295 | 293 (2) | 295 | 295 |
| Ref. | [7,12] | [7,13] | [7,14] | [7,15] |
Table 3.
Crystallographic Properties of Selected Metalated Porphines (no substituents).
| CSD No. | 1268870 (Figure 5) | 912742 (Figure 6a,b) | 912743 (Figure 7a,b) | 1296339 (Figure 8a,b) |
|---|---|---|---|---|
| Name | Ni(porphine) | Mg(porphine) (C5H5N)2 | Zn(porphine) (C5H5N) | Ge(porphine) (OCH3)2 |
| Formula | C20H12NiN4 | C20H12MgN4 (C5H5N)2 | C20H12ZnN4 (C5H5N) | C20H12GeN4 (OCH3)2 |
| System | monoclinic | monoclinic | monoclinic | monoclinic |
| Space gr. | P 21/c | C 2/c | P 21/c | P 21/c |
| A (Å) | 10.1066 (7) | 12.7579 (9) | 9.5746 (4) | 15.013 (5) |
| B (Å) | 11.945 (9) | 15.0501 (12) | 14.6945 (6) | 14.441 (5) |
| C (Å) | 12.229 (2) | 12.3850 (8) | 14.6410 (6) | 8.414 (4) |
| α (deg.) | 90.0 | 90.00 | 90.0 | 90.0 |
| β (deg.) | 101.56 (3) | 92.071 (4) | 105.542 (2) | 91.85 (2) |
| γ (deg.) | 90.0 | 90.00 | 90.0 | 90.0 |
| V (Å3) | 1446.38 | 2376.46 | 1984.44 | 1823.23 |
| Z | 4 | 4 | 4 | 4 |
| ρ (g/cm3) | 1.686 | 1.372 | 1.516 | 1.614 |
| Displ. (Å) | 0.0191 | 0.0242 | 0.0873 | 0.0157 |
| Ref. | [7,16] | [7,17] | [7,17] | [7,18] |
Table 4.
Crysallographic Properties of –N=N– and –C(OH)– Covalently Bonded Porphyrin Dimers.
| CSD No. | 627200 (Figure 9) | 1230159 (Figure 10) |
|---|---|---|
| Formula | C81H51N11Ni2 | C67H80N8Ni2O |
| System | monoclinic | monoclinic |
| Space gr. | P 2/c | C 2/c |
| A (Å) | 13.0693 (3) | 40.31 (2) |
| B (Å) | 9.7304 (2) | 14.997 (7) |
| C (Å) | 24.1344 (5) | 21.954 (11) |
| α (deg.) | 90.00 | 90 |
| β (deg.) | 104.2710 (10) | 108.6 (4) |
| γ (deg.) | 90.00 | 90 |
| V (Å3) | 2974.45 (11) | 12578.6 |
| Z | 2 | 8 |
| ρ (g/cm3) | 1.447 | 1.309 |
| Temp. | 150 (2) | 130 |
| Ref. | [7,21] | [7,22,23] |
Table 5.
Crystallographic Properties of Selected Multiporphyrins.
| CSD No. | 635463 (Figure 17) | 213358 (Figure 18) | 150074 (Figure 19) | 141402 (Figure 20) |
| F. wt. | 2947.42 | 5186.85 | 2449.64 | 1397.20 |
| Formula | C331H179N8O6Zn2 | C386H291CI2N18P4Rh2Zn2 | C168H124N8O4Zn2 | --- |
| System | triclinic | monoclinic | triclinic | monoclinic |
| Space gr. | P −1 | P 21/n | P −1 | C 2/c |
| A (Å) | 9.287 (5) | 25.1838 (9) | 13.6550 (11) | 23.0808 (2) |
| B (Å) | 20.381 (5) | 16.5456 (6) | 15.3346 (12) | 24.9458 (9) |
| C (Å) | 21.529 (5) | 32.5020 (11) | 16.2865 (13) | 13.4593 (5) |
| α (deg.) | 77.218 (5) | 90.00 | 109.187 (2) | 90.0 |
| β (deg.) | 80.967 (5) | 93.902 (2) | 107.904 (2) | 110.503 (2) |
| γ (deg.) | 80.500 (5) | 90.00 | 97.732 (3) | 90.0 |
| V (Å3) | 3889 (2) | 13511.6 (8) | 2957.7 (4) | 7549.5 (4) |
| Z | 1 | 2 | 1 | 4 |
| ρ (g/cm3) | 1.259 | 1.275 | 1.375 | 1.229 |
| Temp. | 90 (2) | 150 (2) | 163.2 | 183 (2) |
| Ref. | [7,53] | [7,54] | [7,55] | [7,56] |
| CSD No. | 728863 (Figure 21) | 685451 (Figure 22) | 173782 (Figure 23) | 712832 (Figure 24) |
| F. wt. | 1310.84 | 3308.31 | 1825.03 | --- |
| Formula | C82H51C1Cu2N8 | C200H216C17N12Ni3S3 | C120H112N10Zn2 | --- |
| System | orthorhombic | triclinic | monoclinic | monoclinic |
| Space gr. | Pbca | P−1 | P21/n | C 2/c |
| A (Å) | 26.455 (5) | 18.846 (5) | 13.7485 (2) | 38.482 (6) |
| B (Å) | 13.095 (5) | 27.950 (5) | 18.3200 (3) | 27.205 (5) |
| C (Å) | 35.226 (5) | 27.989 (5) | 40.8739 (7) | 17.098 (5) |
| α (deg.) | 90.000 (5) | 112.376 (5) | 90 | 90.0 |
| β (deg.) | 90.000 (5) | 103.171 (5) | 84.1947 (3) | 111.967 (5) |
| γ (deg.) | 90.000 (5) | 103.126 (5) | 90 | 90.0 |
| V (Å3) | 12203 (5) | 12445 (5) | 10242.2 (2) | 15306.3 |
| Z | 8 | 2 | 8 | --- |
| ρ (g/cm3) | 1.427 | 0.883 | 2.367 | --- |
| Temp. | 90 (2) | 90 (2) | 296.2 | --- |
| Ref. | [7,57] | [7,58] | [7,59] | [7,60] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cook, L.P.; Brewer, G.; Wong-Ng, W. Structural Aspects of Porphyrins for Functional Materials Applications. Crystals 2017, 7, 223. https://doi.org/10.3390/cryst7070223
AMA Style
Cook LP, Brewer G, Wong-Ng W. Structural Aspects of Porphyrins for Functional Materials Applications. Crystals. 2017; 7(7):223. https://doi.org/10.3390/cryst7070223
Chicago/Turabian StyleCook, Lawrence P., Greg Brewer, and Winnie Wong-Ng. 2017. "Structural Aspects of Porphyrins for Functional Materials Applications" Crystals 7, no. 7: 223. https://doi.org/10.3390/cryst7070223
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.