
Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids

Department of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102A, 10000 Zagreb, Croatia
*
Authors to whom correspondence should be addressed.
Crystals 2017, 7(12), 376; https://doi.org/10.3390/cryst7120376
Submission received: 8 November 2017
/
Revised: 6 December 2017
/
Accepted: 12 December 2017
/
Published: 14 December 2017
(This article belongs to the Special Issue Photocrystallography and Solid-State Structural Dynamics)
Abstract
:This review is focused on the dimerization and dissociation of aromatic C-nitroso compounds and their dimers, the reactions that could be used as a convenient model for studying the thermal organic solid-state reaction mechanisms. This molecular model is simple because it includes formation or breaking of only one covalent bond between two nitrogen atoms. The crystalline molecular solids of nitroso dimers (azodioxides) dissociate by photolysis under the cryogenic conditions, and re-dimerize by slow warming. The thermal re-dimerization reaction is examined under the different topotactic conditions in crystals: disordering, surface defects, and phase transformations. Depending on the conditions, and on the molecular structure, aromatic C-nitroso compounds can associate to form one-dimensional polymeric structures and are able to self-assemble on gold surfaces.
1. Introduction
Over the last decades, solid-state chemistry has emerged as a rapidly growing area of science. The importance of solid-state chemistry lies in its numerous applications in development of solvent-free synthetic methods and the design of new functional materials in areas such as the electronics and semiconductor industries, energy conversion and storage, catalysis, etc. [1,2]. In this context, it is of crucial importance to gain deeper insights into the mechanistic basis of reactions occurring in the solid state. The present review is focused on the recent research of the organic solid-state reactions in the crystalline molecular solids under thermal conditions. While the studies of photochemically-induced processes and their basic mechanistic studies initiated by Cohen, Schmidt, Buergi, and Dunitz et al. are available from the vast literature [3,4,5,6,7,8,9,10,11], the mechanisms of the thermal organic solid-state reactions are still an open issue. Although Paul and Curtin established fundamental theoretical basis of solid-state reactions already forty years ago [12], the detailed and quantitative descriptions of these processes, especially in organic chemistry, are still incomplete.
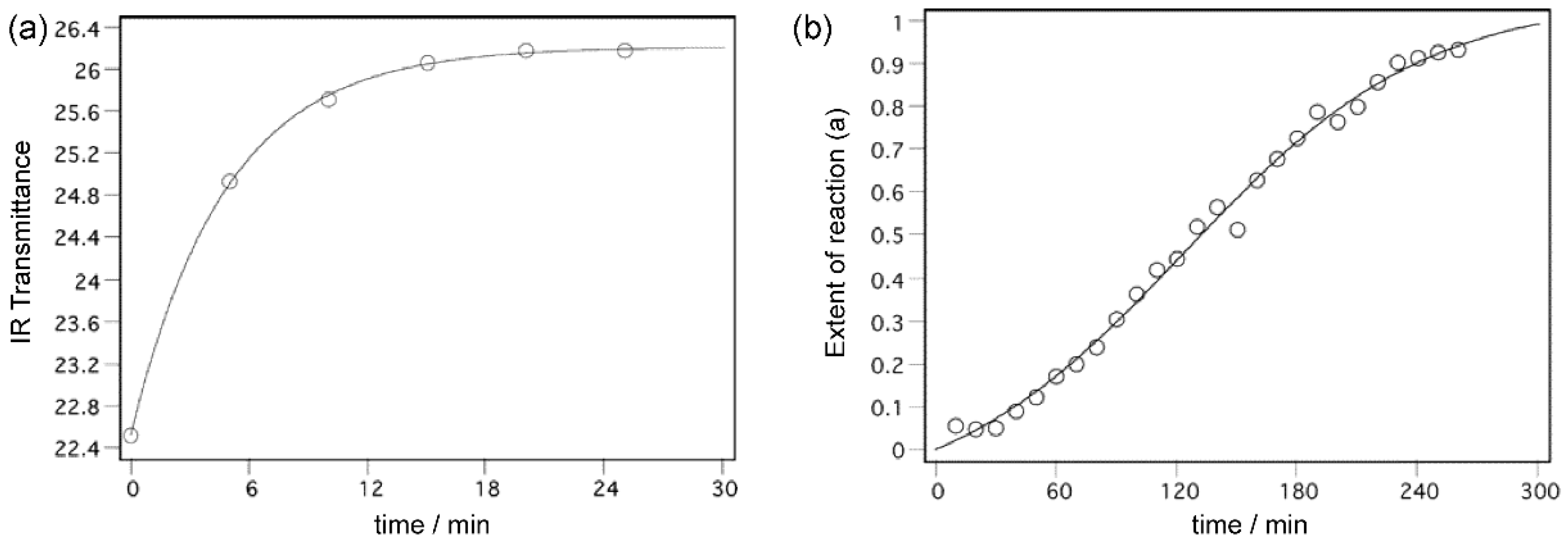
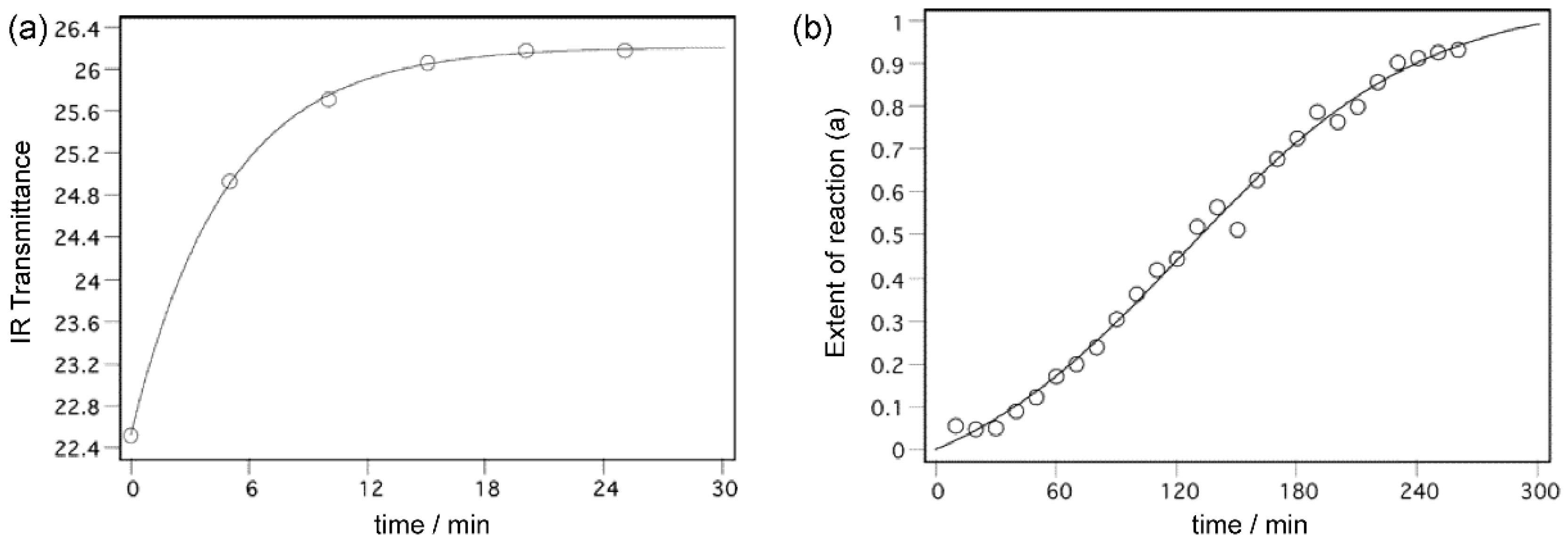
Chemical reactions, i.e., the breaking and/or formation of chemical bonds are, in crystalline molecular solids, more or less accompanied by and coupled with phase changes of the crystal lattice [7]. In the experiments with aromatic C-nitroso compounds, these two processes have been measured by two independent methods [13]. While the chemical reaction was followed by time-resolved IR spectroscopy, the corresponding phase transformation was monitored by the time-resolved X-ray powder diffraction (XRPD) [13]. In cases of some substituted nitrosobenzenes, the studied processes afford the diverse kinetic behaviours: typically, the chemical reaction is characterized by the exponential kinetic curve, and the phase transformation mostly follows the sigmoid kinetic curve (Figure 1). In principle, from the shapes of the kinetic curves it is then possible to estimate which of the processes (chemical reaction or the phase change) is dominant.
First models for the interpretation of solid-state processes known from the literature were focused mostly on the phase transformations or the reactions of inorganic ionic compounds and complexes [14]. In these molecular systems, the main process is the phase transformation. The most popular models for the solid-state kinetics of phase transformations have mathematical representations described by the Avrami-Erofeev (AE) equations [15,16,17,18]. These were recently reinterpreted by Finke and Watzky [19,20,21,22], as well as by Brown [23] with the discussion about the meaning of the Arrhenius parameters and the meaning of the Avrami parameter for the dimensionality of the reaction (parameter m).
The main difficulty in the experimental solid-state kinetics of the thermally-induced reactions is in satisfying the reproducibility of measurements [23,24]. Crystalline or polycrystalline samples differ from one experiment to another because of “individuality” of every crystal emerged from its own history of growth. Crystals are very far from the perfect molecular arrangements because of the defects distributed in the crystal lattice, and especially on the surface. In order to explain the reaction mechanisms in the crystalline phase it is of great importance to design a model, which includes a simple solid-state chemical reaction with the rate constants that can be reproducibly measured in real time under various conditions, and in which the reactants, intermediates, and products have defined molecular and crystal structures.
2. Experimental and Conceptual Model
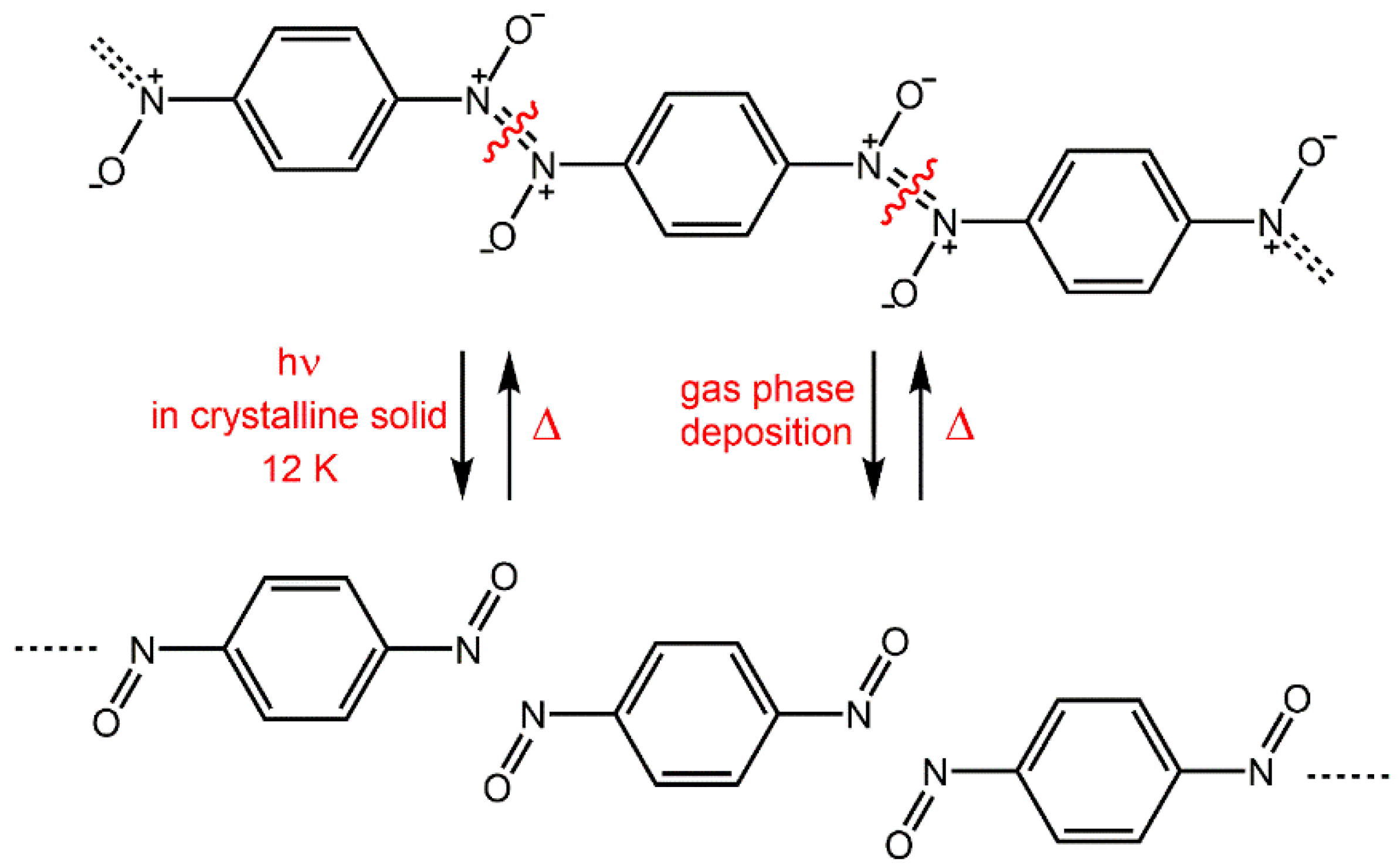
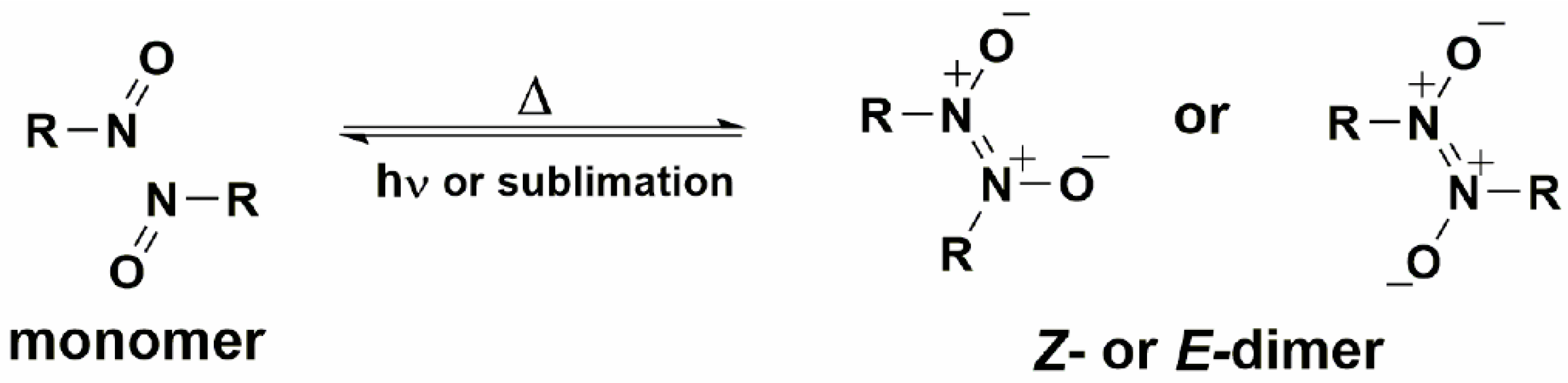
The model, which we have developed is based on the reactions of the nitrosobenzene derivatives in the crystal phase [13,25,26,27,28,29,30,31]. Such a system is simple because it includes the formation and/or breaking of only one chemical bond between two nitrogen atoms during the dimerization of C-nitroso compounds to the Z- or E-azodioxy dimers, or their subsequent dissociation to the starting monomers (Scheme 1) [32,33,34,35,36,37,38,39].
Although most of the aromatic C-nitroso compounds appear as Z- or E-azodioxides in solid state, certain nitrosobenzene derivatives, such as those with strong electron-donating substituents in the para-position (e.g., p-nitrosoanisoles and p-nitrosoanilines), prefer a monomeric form. Aromatic C-nitroso compounds which form dimers in the solid state can also be isolated as metastable monomers. Monomeric molecules can be prepared within the crystalline solids by photodissociation of azodioxy dimers. Photolysis of different azodioxides to the corresponding monomers has been observed in crystals when they are cooled to cryogenic temperatures (10–12 K), and irradiated by UV light (254 nm) [26,29,30,31,40]. The process can be followed even visually because the monomers are coloured blue or green, while most of the azodioxides are colourless or yellow. Warming of the nitroso monomer prepared as a crystalline solid above some critical temperature (usually above 100 K) causes its thermal re-dimerization to azodioxide.
Since the average bond energy of the nitrogen-nitrogen double bond in azodioxides is 120 kJ·mol−1 [41] along with the fact that its rupture results in the formation of two N=O groups, the aromatic azodioxides promptly dissociate to nitroso monomers in the gas phase. This is further supported by the thermodynamic data for the gas-phase dimerization of nitrosobenzenes. For instance, the dimerization reaction enthalpy (ΔrH°) of the parent nitrosobenzene is −22.15 kJ·mol−1 for the Z-isomer (ΔrG° = 33.39 kJ·mol−1) and −26.21 kJ·mol−1 for the E-isomer (ΔrG° = 30.08 kJ·mol−1) [42]. After deposition of the gaseous monomer molecules on the cold surface, the monomeric form is preserved in most cases. The nature of the mutual interactions of the deposited monomers depends strongly on the temperature of the cold surface. For example, the temperature of the cold finger of the laboratory sublimator of about 10 °C is low enough to stabilize the crystals of nitroso monomers that are formed after sublimation. However, removal of these crystals from the cold surface and successive warming to the room temperature causes re-formation of dimers within the crystal phase. Fortunately, the kinetics of this process are measurable in real-time, which makes the dimerization of aromatic C-nitroso compounds a good model for studying the thermally-induced chemical reactions in the crystals. On the other hand, if the surface for the deposition is cooled to the cryogenic temperature (below 20 K), the deposited molecules of nitroso monomers are randomly distributed rather than arranged in crystals and their mutual interactions are expected to be different.
In summary, depending on the experimental conditions the molecules of the starting nitroso monomers can form three different arrangements: (i) the stiff, perhaps metastable, crystal packing in which the reactants appear as an “intimate monomer pair” (obtained by cryogenic photolysis); (ii) the stable crystal phase where the reactant molecules are at the distance larger than van der Waals (produced by classical sublimation); or (iii) the more or less disordered phase (obtained by deposition on the cryogenic surface).
Here, we propose the technical term topochemical effect, which can be defined as an influence of the packing “stiffness” of the neighbouring reacting molecules on their chemical reactivity (the term topochemical effect should be distinguished from the generally accepted concept of the topochemical principle introduced earlier by Cohen, Schmidt et al. [3]). This effect can be observed only by measuring the reaction rates under the ceteris paribus conditions, i.e., by varying exclusively the topotactical intermolecular arrangements for the same chemical reaction, in our case the dimerization of nitrosobenzene molecules.
3. Dimerization Starting from the “Intimate Monomer Pair”
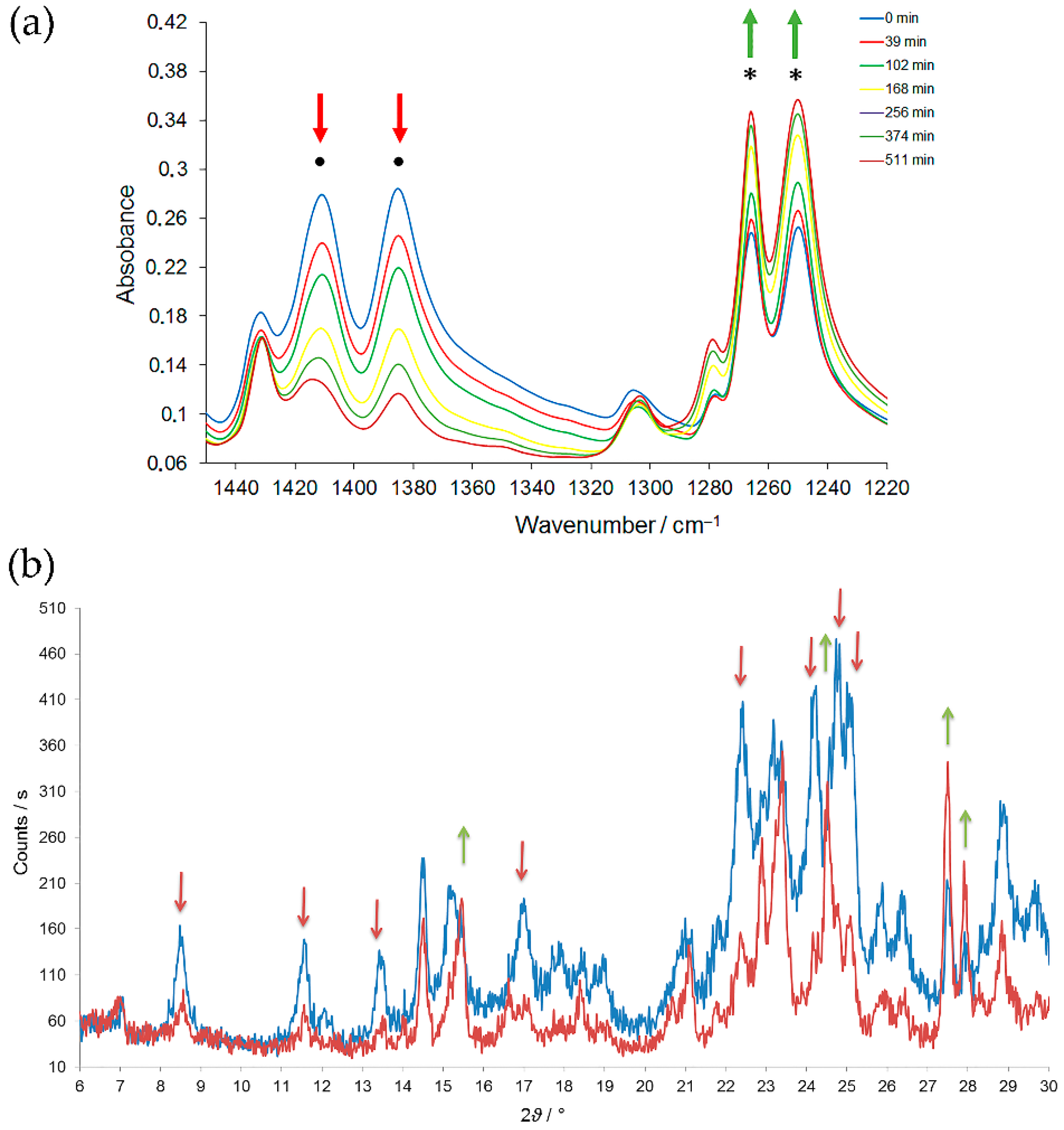
The UV irradiation of azodioxides in solution at room temperature causes their decomposition, either by extrusion of NO [43,44,45], or by molecular rearrangements [46]. There are also some indications based on the UV-VIS spectroscopy, that UV irradiated azodioxides in solution at −60 °C could dissociate to corresponding nitroso monomers [47]. Such photodissociations were finally confirmed by the experiments performed in the crystal phase under cryogenic conditions [40]. Nitroso monomers produced by cryogenic photolysis of azodioxy crystals were characterized by the appearance of a new signal in the IR spectra at about 1500 cm−1, assigned to monomer N=O stretching vibration (Figure 2). The monomers are stable only under cryogenic conditions. At temperatures higher than 100 K they readily dimerize to the starting azodioxides (Scheme 1, Figure 2).
The thermal recovery of azodioxides and re-photolysis to nitroso monomers is the efficient repetitive process (Figure 3) and, thus, the solid-state photochromic and thermochromic behaviour of aromatic C-nitroso compounds could be investigated as a potential simple OFF-ON molecular switch.
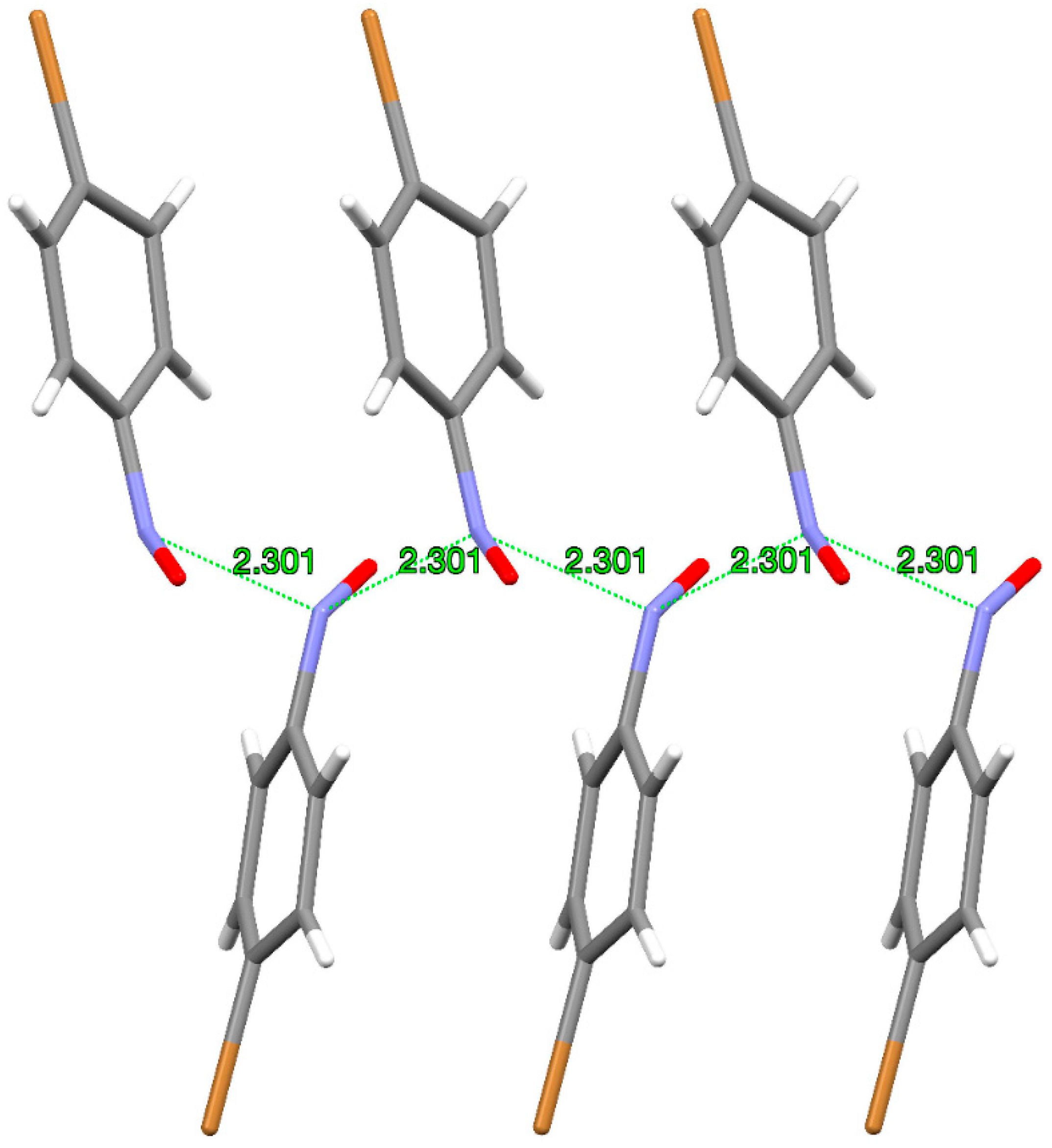
The rate coefficient of dimerization of p-bromonitrosobenzene monomers produced by photolysis (measured as an increase in the intensity of the peak at 1260 cm−1, assigned to the asymmetric stretching vibration of the E-ON=NO group) was found to be k = 2.24 × 10−4 s−1 at 170 K [30]. The dimerization reaction is very fast because the interacting nitrogen atoms of neighbouring monomer molecules obtained by cryogenic photolysis remain very close to each other. Formation of such an “intimate monomer pair” has been confirmed by the X-ray analysis of the product of photodissociation of p-bromonitrosobenzene dimer in which the azodioxide underwent the single-crystal-to-single-crystal transformation to the corresponding pair of nitroso molecules [26]. The thus-obtained crystal of monomer is a sort of metastable phase with an interesting structure in which the nitrogen–nitrogen distance between neighbouring molecules of 2.3 Å is closer by 23% than the sum of the two nitrogen van der Waals radii. Monomers in such a metastable crystal phase have repetitive molecular arrangement where the close 2.3 Å N···N distance forms a quasi-polymeric motif (Figure 4).
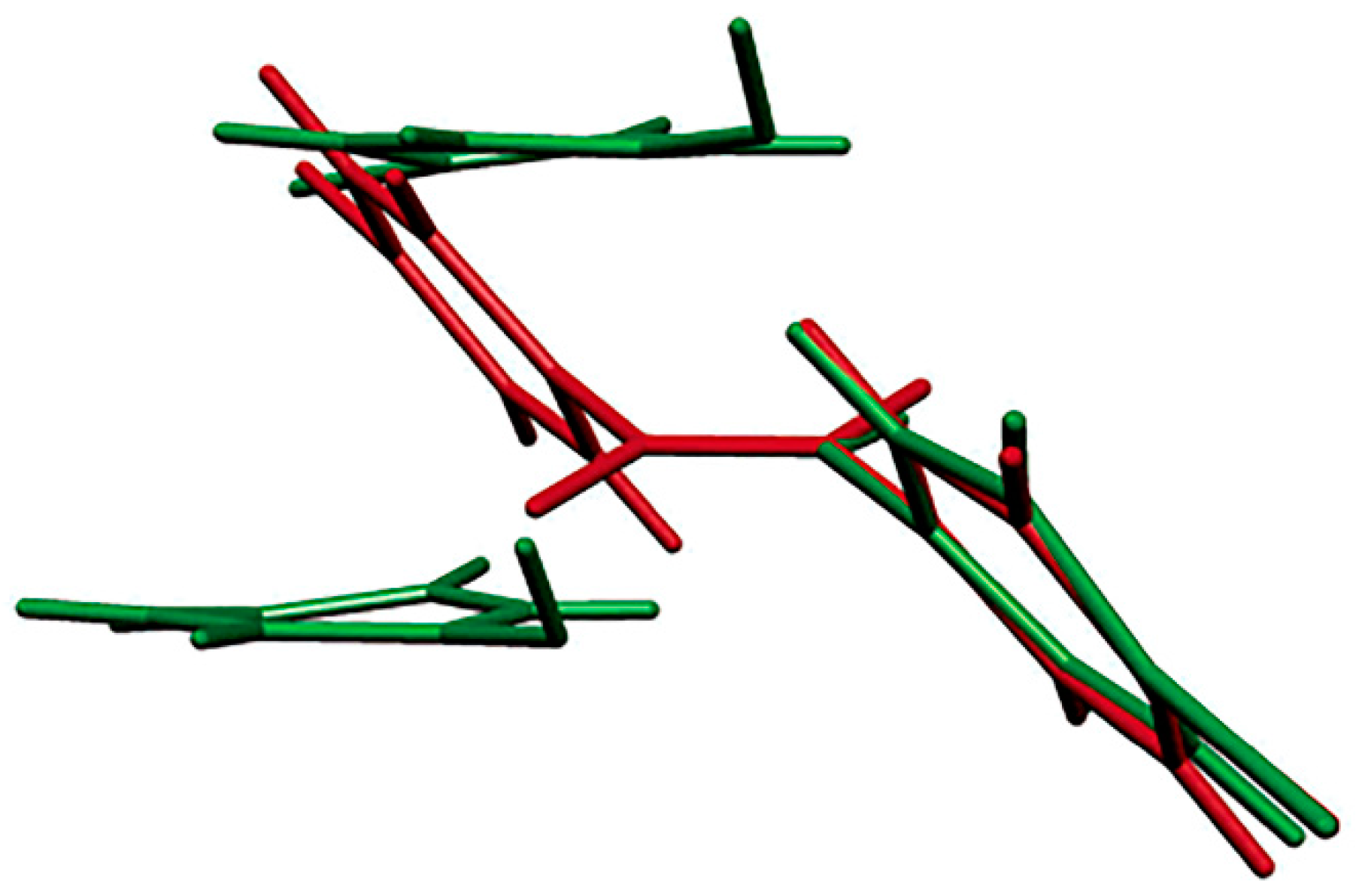
The molecular orbital analysis of the dimerization of C-nitroso compounds published earlier by Hoffmann, Gleiter, and Mallory has demonstrated that the reaction is symmetrically forbidden in the gas phase if the nitroso groups approach one another in the same plane [48]. A comparison of crystal structures of the p-bromonitrosobenzene dimer and monomer in the single-crystal-to-single-crystal transformation showed that molecular orientations changed markedly during the photodissociation reaction, suggesting that dimerization may occur because the reacting nitroso groups do not lie in the same plane (Figure 5). The process of the thermal re-dimerization is not a single-crystal-to-single-crystal conversion. Unfortunately, the photochemical single-crystal-to-single-crystal transformation failed in the experiments performed so far with the other investigated aromatic C-nitroso compounds, but work in this direction is in progress.
The very fast dimerization reaction of p-bromonitrosobenzene after cryogenic photolysis could be attributed to the strong topochemical effect where the reaction starts from the “intimate monomer pair” of the molecules at the nitrogen-nitrogen distance of 2.3 Å (the term strong topochemical effect is used as a terminus technicus describing the influence of the vicinity of the reactive centers on the reactivity in formation of covalent bond). Similar kinetic behaviour was also observed in solid-state photothermal reactions of polymeric p-dinitrosobenzene [49,50]. The kinetics of this polymerization reaction will be discussed later in more detail.
4. Dimerization Starting from the Van Der Waals Distant Monomers
The weak topochemical effect appears in the crystals of nitroso monomers prepared by the mild sublimation, when the temperature of the cold finger is about 10 °C. The reaction starts by warming the crystals of monomers to room temperature. As in the previous case, the rate of the dimerization is calculated from the change in intensity of the E-ON=NO asymmetric stretching signal at 1260 cm−1 [30]. The rate coefficients measured at different temperatures and the activation energy for the dimerization of p-bromonitrosobenzene in crystals prepared by sublimation are represented in Table 1.
The nitrogen-nitrogen distance of the reacting nitroso molecules in these sublimed monomer crystals is longer than the sum of the van der Waals radii [13]. This interatomic separation is markedly larger than the one measured in the crystals of monomers prepared by the cryogenic photolysis (2.3 Å) [26]. Such a looser packing of molecules of nitroso monomers after sublimation, i.e., the weak topochemical effect causes the decrease in the reaction rate for 108 (estimated by extrapolation of rate constants from Table 1 to 170 K) in comparison with the rate of re-dimerization starting from the “intimate monomer pair” obtained by photolysis.
In the sample of the randomly distributed nitroso monomers prepared by the vacuum deposition on the surface cooled to cryogenic temperatures, the reaction of dimerization is very slow and mostly incomplete [30]. At higher temperatures, the molecules of monomer, due to the absence of the proper topotactical orientation, do not interact to form azodioxides, but rather sublimate in the surrounding vacuum chamber.
5. Solid-State Z-E Interconversion
While the sublimations of p-halogenonitrosobenzenes under the mild conditions yield nitroso monomers, the sublimations of the meta-substituted derivatives (e.g., m-chloro- and m-bromonitrosobenzene) result in the formation of Z-azodioxides on the cold finger of sublimator. Evidently, the gaseous monomers of m-halogenonitrosobenzenes dimerize immediately after the contact with the cold surface. Surprisingly, at room temperature, the Z-isomer spontaneously rearranges to the E-isomer in crystal phase [29]. The reaction is interesting because it is one of the very rare examples of the thermally-induced Z-E interconversion in the crystal phase. Most of the examples of Z-E isomerizations in solid state known from the literature are reactions triggered by UV or VIS irradiation. The observed Z-E interconversion of m-halogenonitrosobenzene azodioxides follows the first order kinetics with the activation energy of 126.2 ± 3.0 kJ·mol−1 [29].
As it is well known from the chemistry of aromatic C-nitroso compounds, azodioxides can spontaneously dissociate to nitroso monomers in solution and in the gas phase. Consequently, the interconversion between Z- and E-isomers is the two-step process either in solution or in the gas phase [51,52,53,54,55]. The first step is dissociation to monomers, and the second is dimerization either to Z- or E-isomer. As revealed by ab initio calculations, the thermal unimolecular E-Z interconversion of azodioxides is forbidden by symmetry [54]. However, the appearance of the nitroso monomer as an intermediate during Z-E interconversion of m-halogenonitrosobenzene azodioxides in crystal phase has not been detected, either by kinetic measurements performed by following the changes in IR spectra or by time-resolved XRPD experiments (Figure 6). It could be that the solid-state Z-E isomerization reaction proceeds through the pairs of monomers which have very short lifetime. The explanation of this unusual behaviour and of underlying reaction mechanism requires additional studies. As it will be discussed later, similar thermal solid-state Z-E rearrangement was also observed in the case of polymeric p-dinitrosobenzene [49].
6. Surfaces and Formation of Crystal Defects
Thermally-induced solid-state reactions often start in the areas of crystal defects [56,57,58,59]. It is very difficult to recognize and to determine the type and the number of crystal deformations on the microscale. The most probable appearance of the defects is on the crystal surface. Dimerization of aromatic C-nitroso compounds at room temperature was found to be triggered by the off-sublimation of the starting molecules from the surface [27]. If the sublimation is suppressed either by closing the sample into the sealed ampoule or by covering it by the inert liquid (glycerol), the solid-state reaction is much slower or inhibited (Figure 7).
Appearance of such surface vacancies can induce the molecular translations within the crystal network. Such softening of the molecular arrangement is one of the necessary conditions for the thermal solid-state reaction, exactly as Paul and Curtin suggested [12]. Since the rate of off-sublimation of molecules from the surface can be controlled and measured, for instance by isothermal TGA, by using this method it has been found that the kinetics of the formation of crystal defects is correlated with the chemical reaction kinetics that occurs in the same crystal [30]. The isothermal TGA measurements showed that the critical number of defects required for the initiation of the reaction corresponds to the loss of 1.0 ± 0.5% of mass.
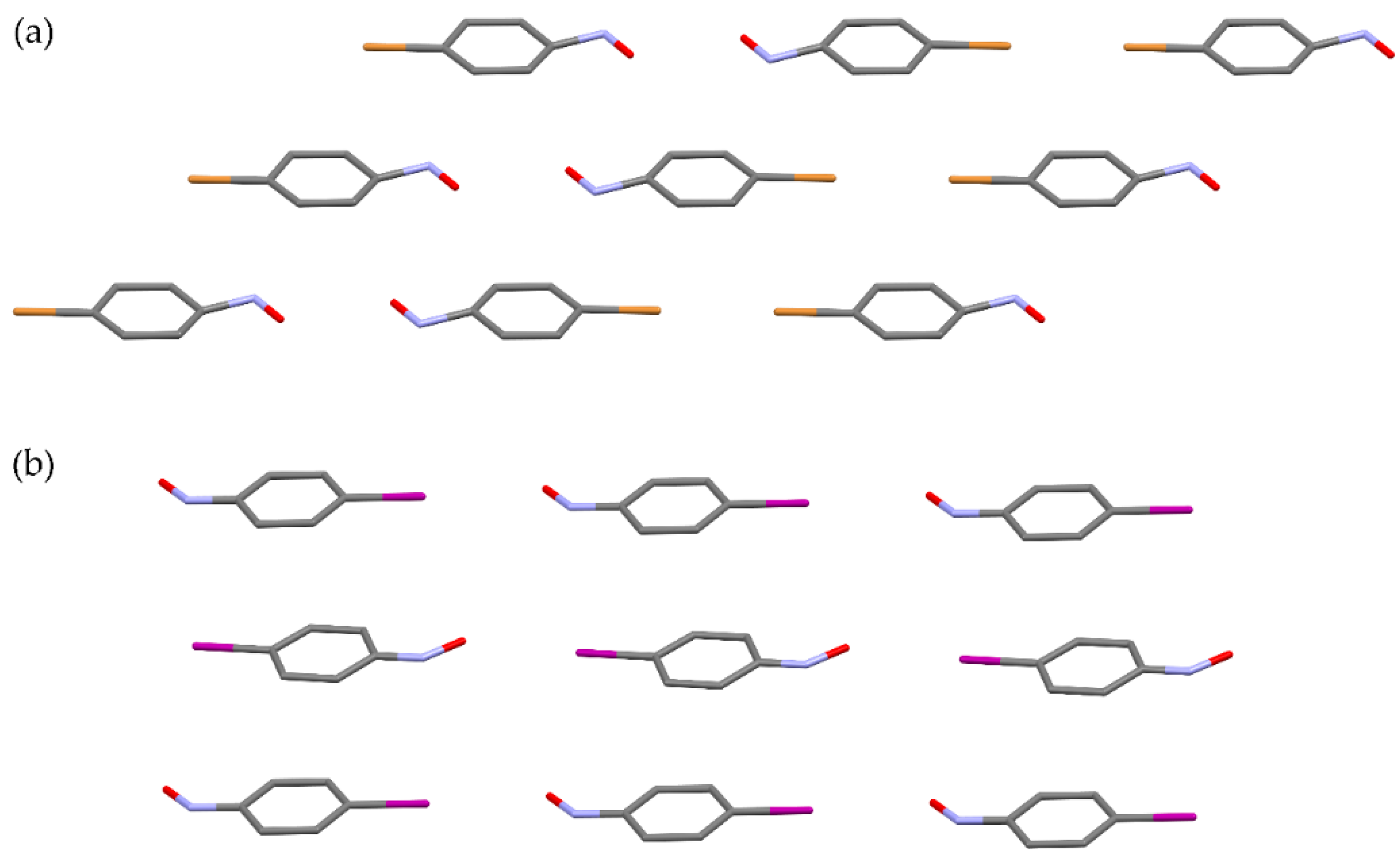
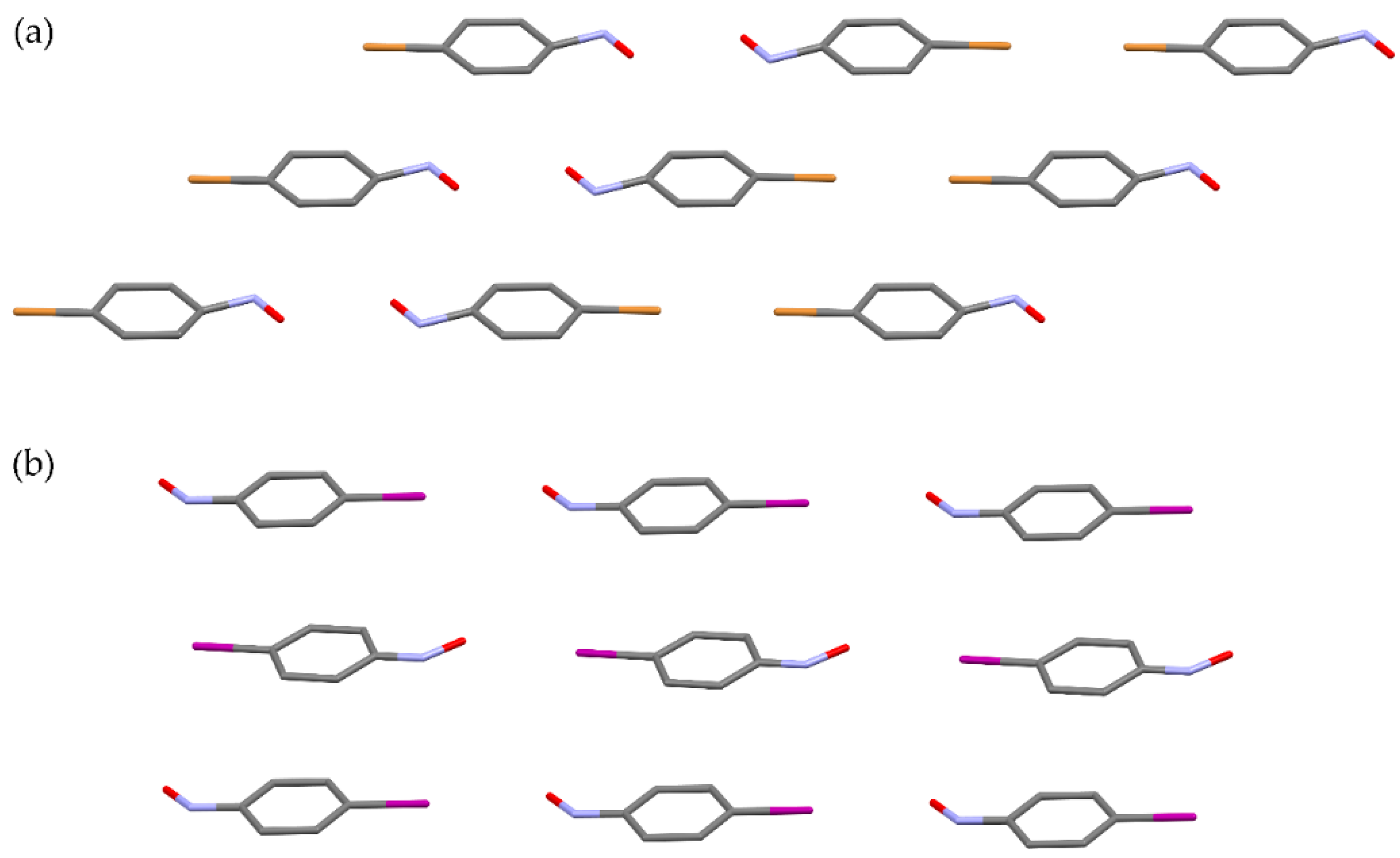
The efficiency of the solid-state dimerization of p-bromo- and p-iodonitrosobenzene is strongly influenced by the degree of the orientational disorder. While the monomers of bromo-derivative produced by sublimation dimerize readily, the dimerization reaction of the sublimed iodo-derivative is very slow and with the negligible yield. This different behaviour can be explained by comparison of crystal structures and packing of molecules of p-bromo- and p-iodonitrosobenzene after sublimation (Figure 8) [13,60]. In the solid monomeric phase of the bromo-derivative, molecules are orientationally disordered in such a way that half of monomer pairs have orientation with the nitroso nitrogen atoms in the vicinity, satisfying the topochemical condition (Figure 8a) [13]. On the other hand, in iodo-derivative the molecular arrangement is much more regular with nitroso groups lying in the neighbourhood to the iodine atoms, and, thus, no interactions between the nitroso nitrogen atoms are possible (Figure 8b) [60]. Clearly, the bromo-derivative dimerizes because of the orientational disorder of molecules, which is not the case in the iodo-derivative.
It must be pointed out that p-iodonitrosobenzene can be isolated in the form of E-dimer by crystallization from solution and that both Z- and E-dimers are present in equilibrium with monomers in solution at low temperatures [35]. However, after sublimation this compound forms exclusively the stable monomers that do not dimerize spontaneously [13,30]. As it has been described in the experimental part of the paper by Fletcher, Gowenlock et al. [35], the bright yellow crystals of E-azodioxy dimer of p-iodonitrosobenzene revert on heating to the green crystals of the monomer. This transformation could tentatively be explained by the higher stability of the monomer polymorph. Evidently, solid-state chemistry of p-iodonitrosobenzene displays an interesting and somewhat complex behaviour. For a deeper clarification of this process further detailed research has to be done.
7. Metathesis-Like Reactions
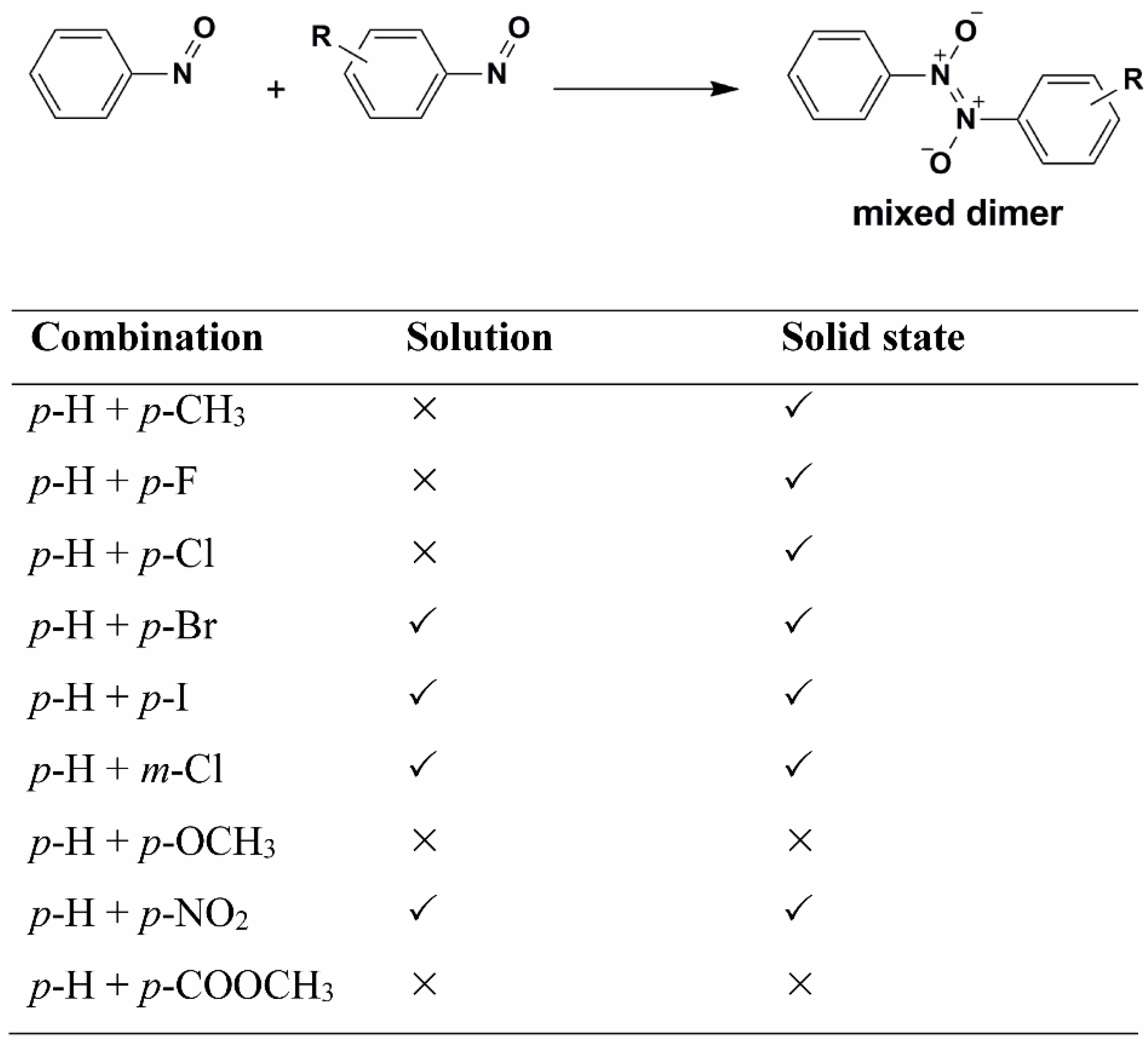
Depending on the conditions, the aromatic nitroso molecules can form mixed dimers. Namely, by combining two different nitroso compounds in addition to formation of symmetrical dimers or homodimers, asymmetrical dimers or heterodimers can also be formed in solution and in the solid state. The first heterodimer in which none of the nitroso partners is sterically crowded with large groups in o-position was obtained by reaction of p-bromo- and p-nitronitrosobenzene [61]. The selectivity in formation of heterodimers was analysed by studying cross-dimerization of several p- and m-substituted nitrosobenzene derivatives with the parent nitrosobenzene in solution and in the solid state (Scheme 2) [62]. It was found that cross-dimerization strongly depends on the selectivity properties of starting monomer molecules. In addition, in solution and in the solid state the selectivity is different. Although the selectivity is in both media influenced by the electron-donating ability of the substituent in the m- or p-position of the benzene ring, the differences can be explained by the influence of molecular arrangements in the crystal lattice. The effect of the solid phase on the intermolecular selectivity is still an open question that stimulates future investigations.
Cross-dimerization has been observed also in the solid-state reactions occurring in ball-mill reactors [28]. By labelling one of the nitrosobenzene molecule with 15N isotope, the appearance of the O14N=15NO signal in the IR spectrum indicated the formation of the corresponding heterodimer. In addition, the system has been exploited for measuring the kinetics of the cross-dimerization reactions triggered by ball milling.
8. One-Dimensional Polymerization Reactions
Aromatic dinitroso compounds, e.g., p-dinitrosobenzene, are prone to polymerization which makes them promising candidates for the building blocks of supramolecular architectures [37,38,39]. Hacker reported the first isolation of p-dinitrosobenzene by gas phase deposition on the cold surface at 10 K, and its subsequent spontaneous polymerization upon heating the film of monomer to 170 K [63].
Kinetics of polymerization of p-dinitrosobenzene to azodioxides was studied recently under two different topochemical conditions [49]. Monomers were prepared either by cryogenic photolysis of the polyazodioxide or by cryogenic vapour deposition (Figure 9). Warming the monomers, prepared by photolysis of the polymer, to 150 K produces E-azodioxides. Kinetics of re-polymerization of photolytically-obtained monomers, followed at 150 K, displays sigmoidal behaviour indicating that the dominant observable is the phase transformation. Analysis of these kinetic data by the Avrami-Erofeev model indicated a one-dimensional (1D) growth with a rate coefficient of k = 2 × 10−4 s−1 for the formation of E-form. 1D growth and fast process of polymerization of p-dinitrosobenzene could probably be explained as a consequence of the conserved linear arrangements of the monomer molecules after the cryogenic photolysis.
In contrast, the gas phase cryogenic deposition of monomers results in randomly-distributed molecules on the cold surface. Polymerization reaction of monomers prepared in such a way is less pronounced and it starts with the formation of oligomeric Z-isomers, which above 150 K rearrange to the E-form of the polymer. Kinetics of formation of Z-azodioxides below 150 K affords sigmoidal behaviour [49].
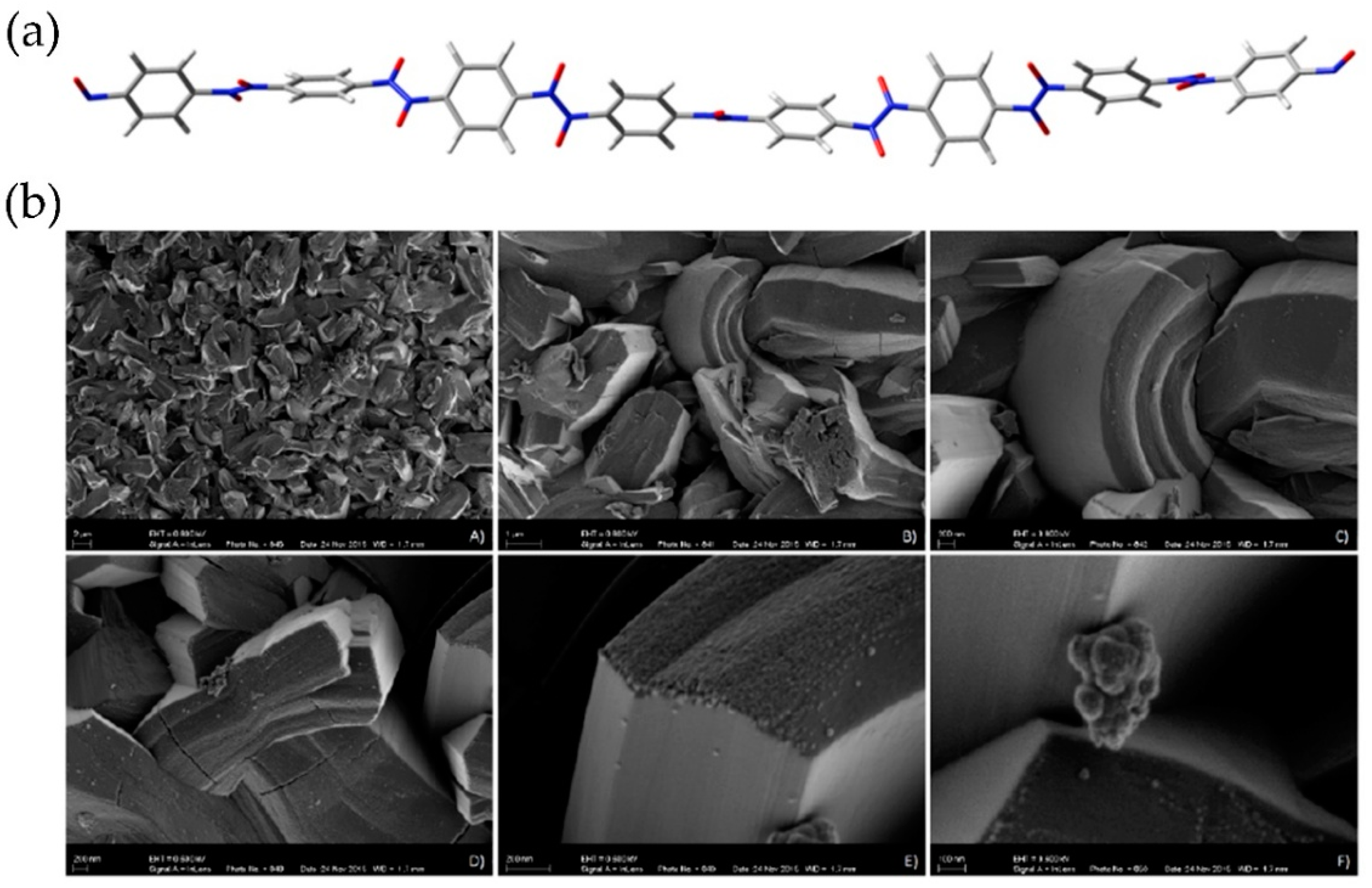
A computational study by the FMO approach of structural characteristics of oligomers constructed from p-dinitrosobenzene predicted that the most stable conformations have helical structure [49] (Figure 10a). In addition, morphology of the p-dinitrosobenzene polymer as examined by scanning electron microscopy (SEM), shows interesting features (Figure 10b) [50].
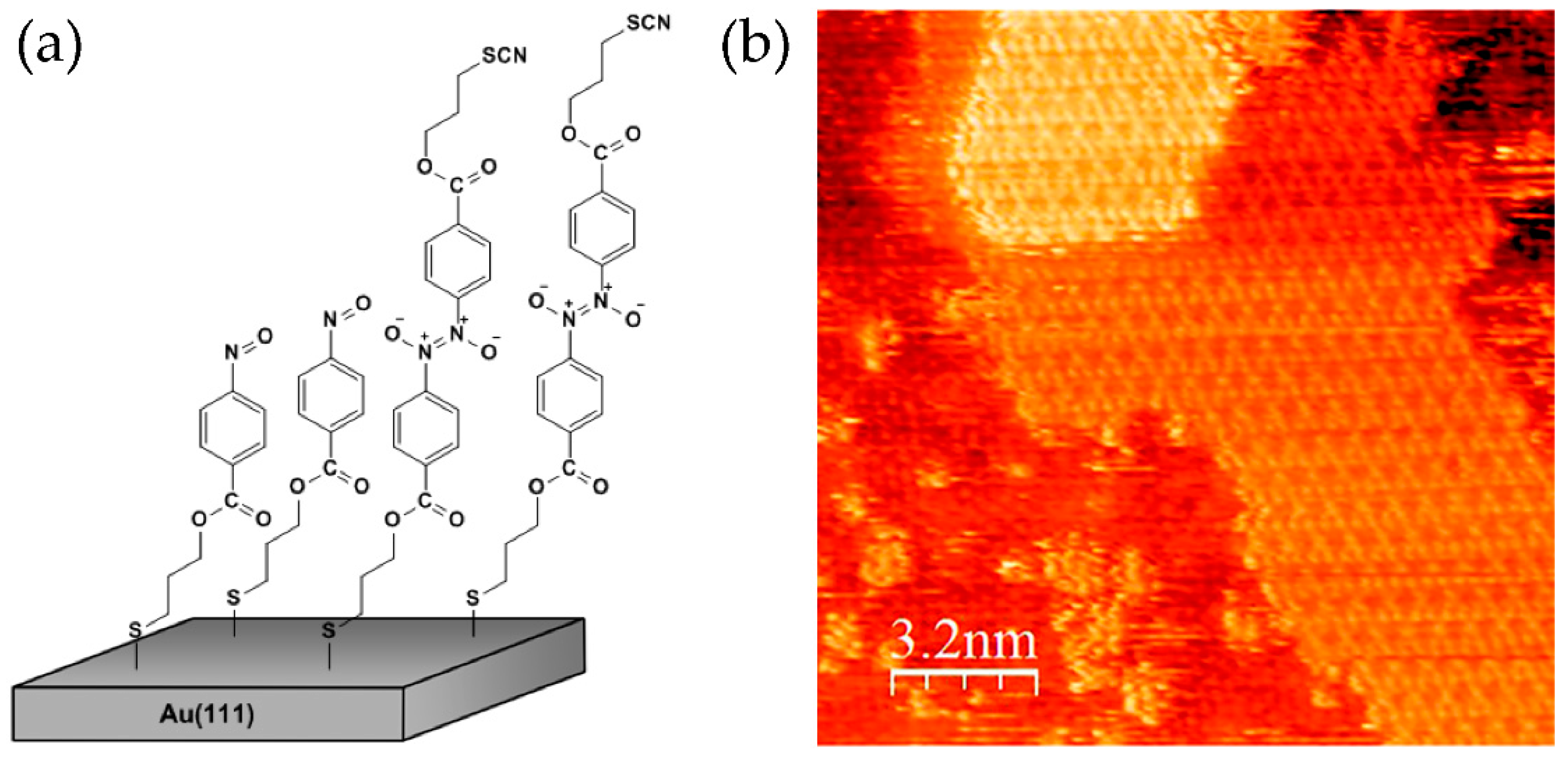
9. Self-Assembly and Dimerization on a Gold Surface
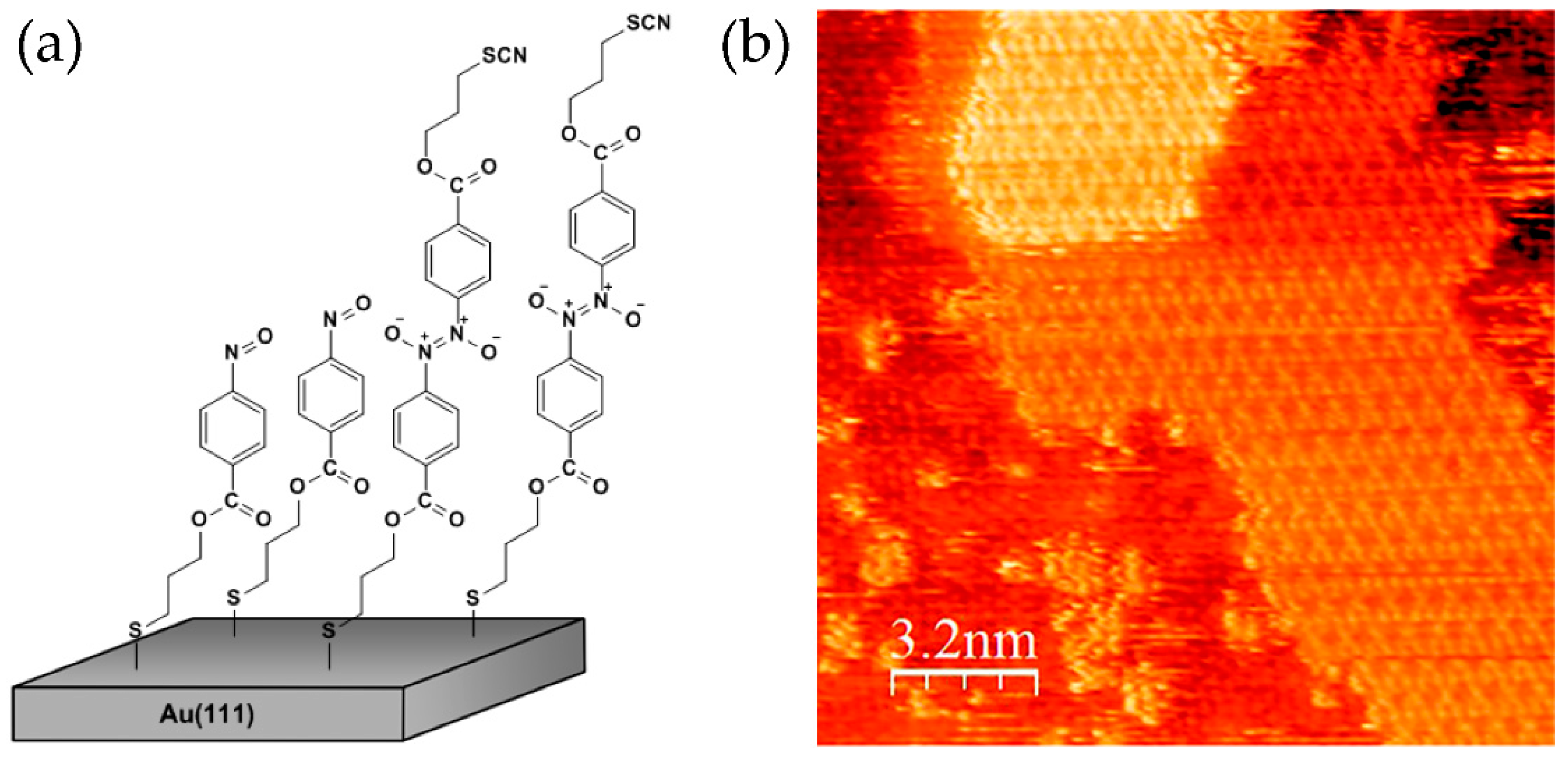
Self-assembled monolayers (SAMs) provide opportunity to study dimerization reactions of aromatic C-nitroso compounds that could occur in a confined two-dimensional (2D) space. Nitrosobenzene molecules functionalized with the alkyl chains and sulphur headgroups at the p-position can be adsorbed on an Au(111) surface by forming SAMs [64,65]. Such systems were studied by scanning tunnelling microscopy (STM) and atomic force microscopy (AFM). It was found that SAMs of nitrosobenzene-terminated molecules are well ordered with molecules arranged into a hexagonal 3√3 × 3√3 structure. However, in addition to self-assembly into monolayers, the molecules of nitrosobenzenes can also dimerize on an Au(111) surface and form ordered bilayers (Figure 11). Self-assembled bilayers (SABs) of nitrosobenzene derivatives could be formed by interactions of nitroso groups exposed at the interface of SAM and those present in solution. Within SABs, molecules are also arranged into hexagonal structures, but with somewhat lower periodicity when compared to SAM regions. Interestingly, the second layer displays better ordering with respect to the first one, which is probably due to the softer background. As revealed from high-resolution STM images, the second layer can be organized from already few molecules enabling direct insights into nucleation and 2D crystallization processes. Formation of highly-ordered SABs on an Au(111) surface shows that 2D self-organization of nitrosobenzene molecules strongly promotes dimerization reaction. Monolayer with regularly oriented nitroso groups at the interface represents a good topotactic condition for formation of 2D molecular arrangement of dimers.
Sulphur-containing derivatives of aromatic C-nitroso compounds were also adsorbed on three-dimensional (3D) surface of gold nanoparticles [65]. As revealed by IR and UV-VIS spectroscopy, and transmission electron microscopy (TEM), dimerization of aromatic C-nitroso compounds by interparticle interaction of exposed nitroso groups seem to induce the aggregation of gold nanoparticles.
10. Conclusions and Outlook
Dimerization of aromatic C-nitroso compounds and the dissociation of corresponding dimers can be used as an experimental model for studying the thermal solid-state organic reaction mechanisms. This simple model includes formation or breaking of only one covalent bond. While the photochemical reactivity in the solid state is studied within the frame of topochemical principles, the thermally-induced solid-state reactions are much more complicated because they are influenced by very different and variable topotactic conditions. By using our model, it is possible to follow the solid-state reaction starting from various intermolecular arrangements of the reactants such as the intimate molecular pairs, or van der Waals-like molecular pairs. The example of the optimal topotactic conditions for the formation of the azodioxide bond is a very close “nonbonding” contact of 2.3 Å between the reacting nitrogen atoms.
Influence of crystal disordering on the surface could play crucial role in triggering the solid-state reaction. It is demonstrated how the enhancement of the dimerization rate is induced by the appearance of the surface defects obtained by off-sublimation of the molecules from the crystal. It has been found that the nitroso monomers dimerize much faster if their crystals were exposed to the open air in comparison with the same crystals emerged in the neutral liquid which prevents the sublimation. Evidently, the thermal solid-state reaction requires gradual softening of the lattice, which is most probable on the surface. Crystals are very far from the “chemical cemetery”, how Leopold Ružička once (more than 50 years ago) remarked to Jack Dunitz.
The topotactic influence on the organic reactivity is clearly demonstrated in the case of polymerization of p-dinitrosobenzene. The re-polymerization of the monomers obtained by photolysis of the E-oriented polymer produces only the E-form. On the other hand, the polymerization of the gas-phase deposited p-dinitrosobenzene molecules with randomly-oriented monomers yield the Z-isomeric oligomers in the first step, which transform to the more stable E-polymer. The thermal solid-state Z-E transformation found during polymerization of p-dinitrosobenzene has been also observed for meta-substituted azodioxides. The details of this solid-state reaction mechanism are still unclear, and represent the topic of the further investigation.
A detailed study of polymerizations of dinitroso compounds, as well as the surface organization and formation of the self-assembled bilayers (SABs) on the gold surface opens new possibilities not only in fundamental research, but also in the design of new intelligent materials.
Acknowledgments
This work was supported by the Croatian Science Foundation, grant no. 7444, ORGMOL.
Author Contributions
Hrvoj Vančik conceived the topic of this review. Hrvoj Vančik and Ivana Biljan wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- West, A.R. Solid State Chemistry and Its Applications, 2nd ed.; Wiley-Blackwell: Chichester, UK, 2014. [Google Scholar]
- Smart, L.E.; Moore, E.A. Solid State Chemistry: An Introduction, 4th ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Cohen, M.D.; Schmidt, G.M.J. 383. Topochemistry. Part I. A survey. J. Chem. Soc. 1964, 383, 1996–2000. [Google Scholar] [CrossRef]
- Kaupp, G. Solid-state molecular syntheses: Complete reactions without auxiliaries based on the new solid-state mechanism. CrystEngComm 2003, 5, 117–133. [Google Scholar] [CrossRef]
- Kaupp, G. Organic solid-state reactions with 100% yield. Top. Curr. Chem. 2005, 254, 95–183. [Google Scholar]
- Schmidt, G.M.J. Photodimerization in the solid state. Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar] [CrossRef]
- Schmidt, G.M.J. Solid State Photochemistry; Verlag Chemie: Weinheim, NY, USA, 1976. [Google Scholar]
- Toda, F. Thermal and photochemical reactions in the solid state. Top. Curr. Chem. 2005, 254, 1–40. [Google Scholar]
- Bürgi, H.B. Structure correlation and chemistry. Acta Crystallogr. A 1998, 54, 873–885. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Dunitz, J.D. From crystal statics to chemical dynamics. Acc. Chem. Res. 1983, 16, 153–161. [Google Scholar] [CrossRef]
- Dunitz, J.D. Phase transitions in molecular crystals from a chemical viewpoint. Pure Appl. Chem. 1991, 63, 177–185. [Google Scholar] [CrossRef]
- Paul, D.; Curtin, Y. Thermally induced organic reactions in the solid state. Acc. Chem. Res. 1973, 6, 217–225. [Google Scholar] [CrossRef]
- Vančik, H.; Šimunić-Mežnarić, V.; Meštrović, E.; Halasz, I. Nitrosobenzene Dimerizations as a model system for studying solid-state reaction mechanisms. J. Org. Chem. 2004, 69, 4829–4834. [Google Scholar] [CrossRef] [PubMed]
- Galwey, A.K.; Brown, M.E. Thermal Decomposition of Ionic Solids, Volume 86: Chemical Properties and Reactivities of Ionic Crystalline Phases (Studies in Physical and Theoretical Chemistry), 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Avrami, M. Kinetics of phase change. I General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II Transformation-Time relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure kinetics of phase change. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Erofeev, B.V. Generalized equation of chemical kinetics and its application in reactions involving solids. Compt. Rend. Acad. Sci. USSR 1946, 52, 511–514. [Google Scholar]
- Finney, E.E.; Finke, R.G. Nanocluster nucleation and growth kinetic and mechanistic studies: A review emphasizing transition-metal nanoclusters. J. Colloid Interface Sci. 2008, 317, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Finney, E.E.; Finke, R.G. Is there a minimal chemical mechanism underlying classical Avrami-Erofeev treatments of phase-transformation kinetic data? Chem. Mater. 2009, 21, 4692–4705. [Google Scholar] [CrossRef]
- Finney, E.E.; Finke, R.G. Fitting and interpreting transition-metal nanocluster formation and other sigmoidal-appearing kinetic data: A more thorough testing of dispersive kinetic vs chemical-mechanism-based equations and treatments for 4-step type kinetic data. Chem. Mater. 2009, 21, 4468–4479. [Google Scholar] [CrossRef]
- Watzky, M.A.; Finke, R.G. Transition metal nanocluster formation kinetic and mechanistic studies. A new mechanism when hydrogen is the reductant: Slow, continuous nucleation and fast autocatalytic surface growth. J. Am. Chem. Soc. 1997, 119, 10382–10400. [Google Scholar] [CrossRef]
- Brown, M.E. Steps in a minefield. J. Therm. Anal. 1997, 49, 17–32. [Google Scholar] [CrossRef]
- Galwey, A.K. What can we learn about the mechanisms of thermal decompositions of solids from kinetic measurements? J. Therm. Anal. Calorim. 2008, 92, 967–983. [Google Scholar] [CrossRef]
- Vančik, H. Aromatic C-Nitroso Compounds; Springer: New York, NY, USA, 2013. [Google Scholar]
- Halasz, I.; Meštrović, E.; Čičak, H.; Mihalić, Z.; Vančik, H. Solid-State reaction mechanisms in monomer–dimer interconversions of p-Bromonitrosobenzene. single-crystal-to-single-crystal photodissociation and formation of new non-van der Waals close contacts. J. Org. Chem. 2005, 70, 8461–8467. [Google Scholar] [CrossRef] [PubMed]
- Halasz, I.; Vančik, H. Surface nucleation in solid-state dimerisation of nitrosobenzenes promoted by sublimation. CrystEngComm 2011, 13, 4307–4310. [Google Scholar] [CrossRef]
- Maganjić, A.; Šolić, I.; Milovac, S.; Halasz, I.; Biljan, I.; Vančik, H. Mechanochemically induced cross-dimerizations of nitrosobenzenes. Kinetics and solid-state isotope effects. J. Phys. Org. Chem. 2014, 27, 177–182. [Google Scholar] [CrossRef]
- Varga, K.; Vančik, H. Topochemical effect in thermal E-Z isomerization of azodioxides in solid state. J. Phys. Org. Chem. 2016, 29, 214–220. [Google Scholar] [CrossRef]
- Varga, K.; Volarić, J.; Vančik, H. Crystal disordering and organic solid-state reactions. CrystEngComm 2015, 17, 1434–1438. [Google Scholar] [CrossRef]
- Knežević, A.; Medančić, T.; Milovac, S.; Biljan, I.; Halasz, I.; Vančik, H. Photothermal reactions of nitrosobenzene and halonitrosobenzenes in solid-state. Croat. Chem. Acta 2011, 84, 21–24. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G. Structural investigations of C-nitrosobenzenes. Part 1. Solution state 1H NMR studies. J. Chem. Soc. Perkin Trans. 2 1997, 2201–2206. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G. Structural investigations of C-nitrosobenzenes. Part 2.1 NMR studies of monomer-dimer equilibria including restricted nitroso group rotation in monomers. J. Chem. Soc. Perkin Trans. 2 1998, 797–804. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G.; Šik, V. Dynamic NMR study of the factors governing nitroso group rotation in p-nitrosoanilines in the solution and solid states. Magn. Reson. Chem. 1995, 33, 561–569. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Gowenlock, B.G.; Orrell, K.G.; Šik, V.; Hibbs, D.E.; Hursthouse, M.B.; Malik, A.K.M. 4-Iodonitrosobenzene. Structural and spectroscopic studies of the monomeric solid and of previously unreported dimers. J. Chem. Soc. Perkin Trans. 2 1996, 191–197. [Google Scholar] [CrossRef]
- Gowenlock, B.G.; Maidment, M.J.; Orrell, K.G.; Šik, V.; Mele, G.; Vasapollo, G.; Hursthouse, M.B.; Abdul Malik, K.M. The solid- and solution-state structures of 2-nitrosopyridine and its 3- and 4-methyl derivatives. J. Chem. Soc. Perkin Trans. 2 2000, 2280–2286. [Google Scholar] [CrossRef]
- Gowenlock, B.G.; Richter-Addo, G.B. Dinitroso and polynitroso compounds. Chem. Soc. Rev. 2005, 34, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, D.; Maris, T.; Wuest, J.D. Constructing monocrystalline covalent organic networks by polymerization. Nat. Chem. 2013, 5, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, D.; Wuest, J.D. Dimerization of aromatic C-nitroso compounds. Chem. Rev. 2016, 116, 258–286. [Google Scholar] [CrossRef] [PubMed]
- Vančik, H.; Šimunić-Mežnarić, V.; Meštrović, E.; Milovac, S.; Majerski, K.; Veljković, J. Solid state photochromism and thermochromism in nitroso monomer-dimer equilibrium. J. Phys. Chem. B 2002, 106, 1576–1580. [Google Scholar] [CrossRef]
- Glaser, R.; Murmann, R.K.; Barnes, C.L. Why do nitroso compounds dimerize while their oxime tautomers do not? A structural study of the trans-dimer of 2-chloro-2-methyl-3-nitrosobutane and higher level ab initio study of thermodynamic stabilities and electronic structures of isomers of diazene dioxides. J. Org. Chem. 1996, 61, 1047–1058. [Google Scholar]
- Novak, I. Computational thermochemistry of C-nitroso compounds. Struct. Chem. 2016, 27, 1395–1401. [Google Scholar] [CrossRef]
- Rassat, A.; Rey, P. Nitroxides: Photochemical synthesis of trimethylisoquinuclidine N-Oxyl. J. Chem. Soc. D 1971, 1161–1162. [Google Scholar] [CrossRef]
- Ullman, E.; Call, L.; Tseng, S.S. Stable free radicals. XII. Direct and sensitized nitronyl nitroxide photochemistry in aprotic solvent. J. Am. Chem. Soc. 1973, 95, 1677–1679. [Google Scholar] [CrossRef]
- Ullman, E.F.; Singh, P. 3,3,4,4-Tetramethyl-1,2-diazetine 1,2-dioxide, a useful low-energy triplet quencher. J. Am. Chem. Soc. 1972, 94, 5077–5078. [Google Scholar] [CrossRef]
- Dunkin, I.R.; Lynch, M.A.; Boulton, A.J.; Henderson, N. 1,2-Dinitrosobenzene in argon matrices at 14 K. J. Chem. Soc. Chem. Commun. 1991, 1178–1179. [Google Scholar] [CrossRef]
- Azoulay, M.; Fischer, E. Low-temperature proton nuclear magnetic resonance and ultraviolet absorption spectra and photochemistry of the system nitrosobenzene-azodioxybenzene and its methyl derivatives. J. Chem. Soc. Perkin Trans. 2 1982, 637–642. [Google Scholar] [CrossRef]
- Hoffmann, R.; Gleiter, R.; Mallory, F.B. Non-least-motion potential surfaces. Dimerization of methylenes and nitroso compounds. J. Am. Chem. Soc. 1970, 92, 1460–1466. [Google Scholar] [CrossRef]
- Bibulić, P.; Rončević, I.; Varga, K.; Mihalić, Z.; Vančik, H. Structure and topochemistry of azodioxide oligomers in solid state. J. Mol. Struct. 2016, 1104, 85–90. [Google Scholar] [CrossRef]
- Bibulić, P.; Rončević, I.; Bermanec, V.; Vančik, H. Polymerization of 1,4-dinitrosobenzene: Kinetics and submicrocrystal structure. Croat. Chem. Acta 2017, in press. [Google Scholar]
- Gowenlock, B.G.; Trotman, J. Geometrical isomerism of dimeric nitrosomethane. J. Chem. Soc. 1955, 4190–4196. [Google Scholar] [CrossRef]
- Chaudhry, A.U.; Gowenlock, B.G. Thermal cis→trans isomerization of two dimeric nitrosoalkanes. J. Chem. Soc. B 1968, 1083–1084. [Google Scholar] [CrossRef]
- Wajer, T.A.J.; de Boer, T.J. Chemistry of small ring compounds. Part 16: Kinetics and mechanism of the isomerization of some 1,1-cyclopropanedicarboxylic acids in water and in aqueous sulfuric acid. Recl. Trav. Chim. Pays-Bas 1972, 91, 657–666. [Google Scholar]
- Minato, T.; Yamabe, S.; Oda, H. A theoretical study on the cis/trans-isomerization of azodioxymethane. Can. J. Chem. 1982, 60, 2740–2748. [Google Scholar] [CrossRef]
- Orrell, K.G.; Šik, V.; Stephenson, D. Study of the monomer-dimer equilibrium of nitrosobenzene using multinuclear one- and two-dimensional NMR techniques. Magn. Reson. Chem. 1987, 25, 1007–1011. [Google Scholar] [CrossRef]
- Mnyukh, Y. Mechanism and kinetics of phase transitions and other reactions in solids. AJCMP 2013, 3, 89–103. [Google Scholar]
- Rao, C.N.R.; Gopalakrishnan, J. New Directions in Solid State Chemistry, 2nd ed.; Cambridge University Press: Cambridge, UK, 1997. [Google Scholar]
- Shalaev, E.; Shalaeva, M.; Zografi, G. The effect of disorder on the chemical reactivity of an organic solid, tetraglycine methyl ester: Change of the reaction mechanism. J. Pharm. Sci. 2002, 91, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Stowell, J.G.; Cao, W.; Morris, K.R.; Byrn, S.R.; Carvajal, M.T. Effect of milling and compression on the solid-state maillard reaction. J. Pharm. Sci. 2005, 94, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.S. An X-ray examination of the crystal structure of p-iodonitrosobenzene. J. Chem. Soc. 1956, 2841–2845. [Google Scholar] [CrossRef]
- Halasz, I.; Biljan, I.; Novak, P.; Meštrović, E.; Plavec, J.; Mali, G.; Smrečki, V.; Vančik, H. Cross-dimerization of nitrosobenzenes in solution and in solid state. J. Mol. Struct. 2009, 918, 19–25. [Google Scholar] [CrossRef]
- Biljan, I.; Cvjetojević, G.; Smrečki, V.; Novak, P.; Mali, G.; Plavec, J.; Babić, D.; Mihalić, Z.; Vančik, H. Nitrosobenzene cross-dimerization: Structural selectivity in solution and in solid state. J. Mol. Struct. 2010, 979, 22–26. [Google Scholar] [CrossRef]
- Hacker, N.P. Investigation of the polymerization of 1,4-dinitrosobenzene by low-temperature infrared and UV absorption spectroscopy. Macromolecules 1993, 26, 5937–5942. [Google Scholar] [CrossRef]
- Biljan, I.; Kralj, M.; Mišić Radić, T.; Svetličić, V.; Vančik, H. Dimerization of nitrosobenzene derivatives on an Au(111) Surface. J. Phys. Chem. C 2011, 115, 20267–20273. [Google Scholar] [CrossRef]
- Biljan, I.; Medančić, T.; Kralj, M.; Mišić Radić, T.; Svetličić, V.; Vančik, H. Nitrosoarene dimerization on two- and three-dimensional gold surfaces. Croat. Chem. Acta 2013, 86, 83–94. [Google Scholar] [CrossRef]
Figure 1.
Kinetics of the solid-state dimerization of p-bromonitrosobenzene followed by (a) time-resolved IR spectroscopy (exponential kinetic curve) and by (b) time-resolved XRPD (sigmoidal kinetic curve). Reproduced with permission from [13]. Copyright 2004 American Chemical Society.
Figure 1.
Kinetics of the solid-state dimerization of p-bromonitrosobenzene followed by (a) time-resolved IR spectroscopy (exponential kinetic curve) and by (b) time-resolved XRPD (sigmoidal kinetic curve). Reproduced with permission from [13]. Copyright 2004 American Chemical Society.
Scheme 1.
Dimerization of C-nitroso compounds and dissociation of azodioxy dimers under various conditions.
Scheme 1.
Dimerization of C-nitroso compounds and dissociation of azodioxy dimers under various conditions.

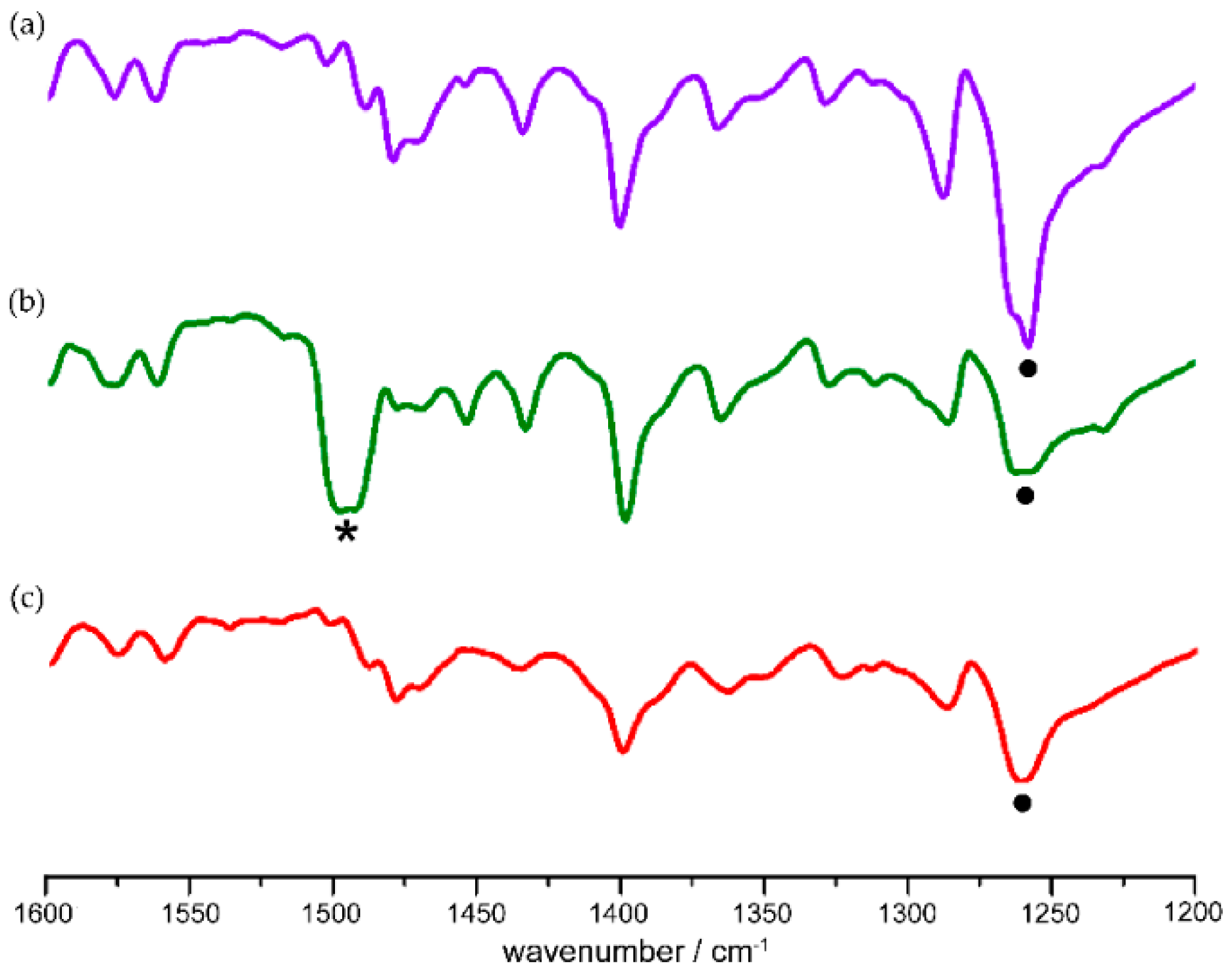
Figure 2.
FTIR spectra of p-bromonitrosobenzene in KBr pellet (a) before photolysis at 12 K, (b) after 30 min of photolysis by a low-pressure Hg lamp, and (c) after warming to 170 K. The signal of E-ON=NO asymmetric stretching vibration is labelled with •, while the signal of monomeric N=O stretching vibration is labelled with *.
Figure 2.
FTIR spectra of p-bromonitrosobenzene in KBr pellet (a) before photolysis at 12 K, (b) after 30 min of photolysis by a low-pressure Hg lamp, and (c) after warming to 170 K. The signal of E-ON=NO asymmetric stretching vibration is labelled with •, while the signal of monomeric N=O stretching vibration is labelled with *.
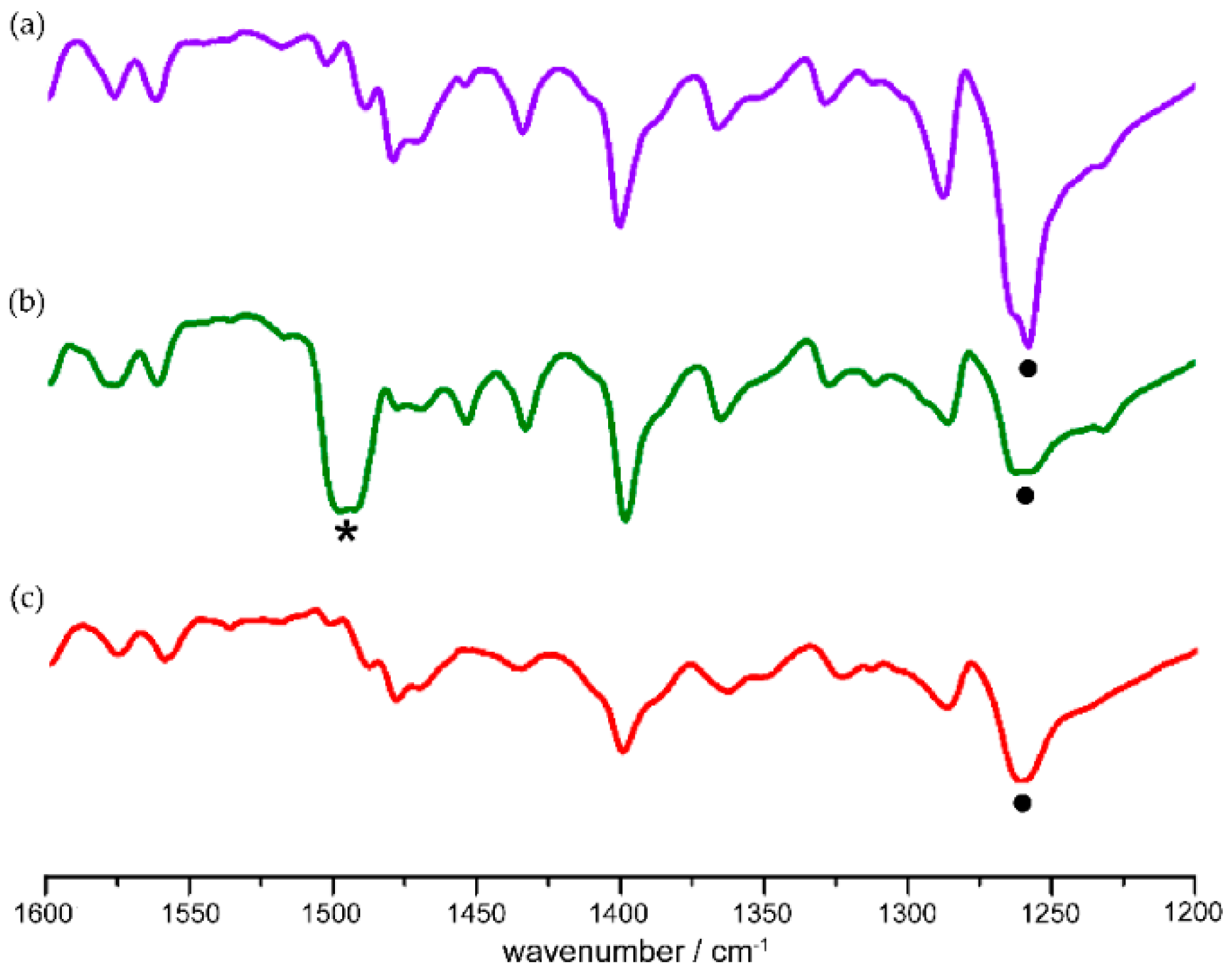
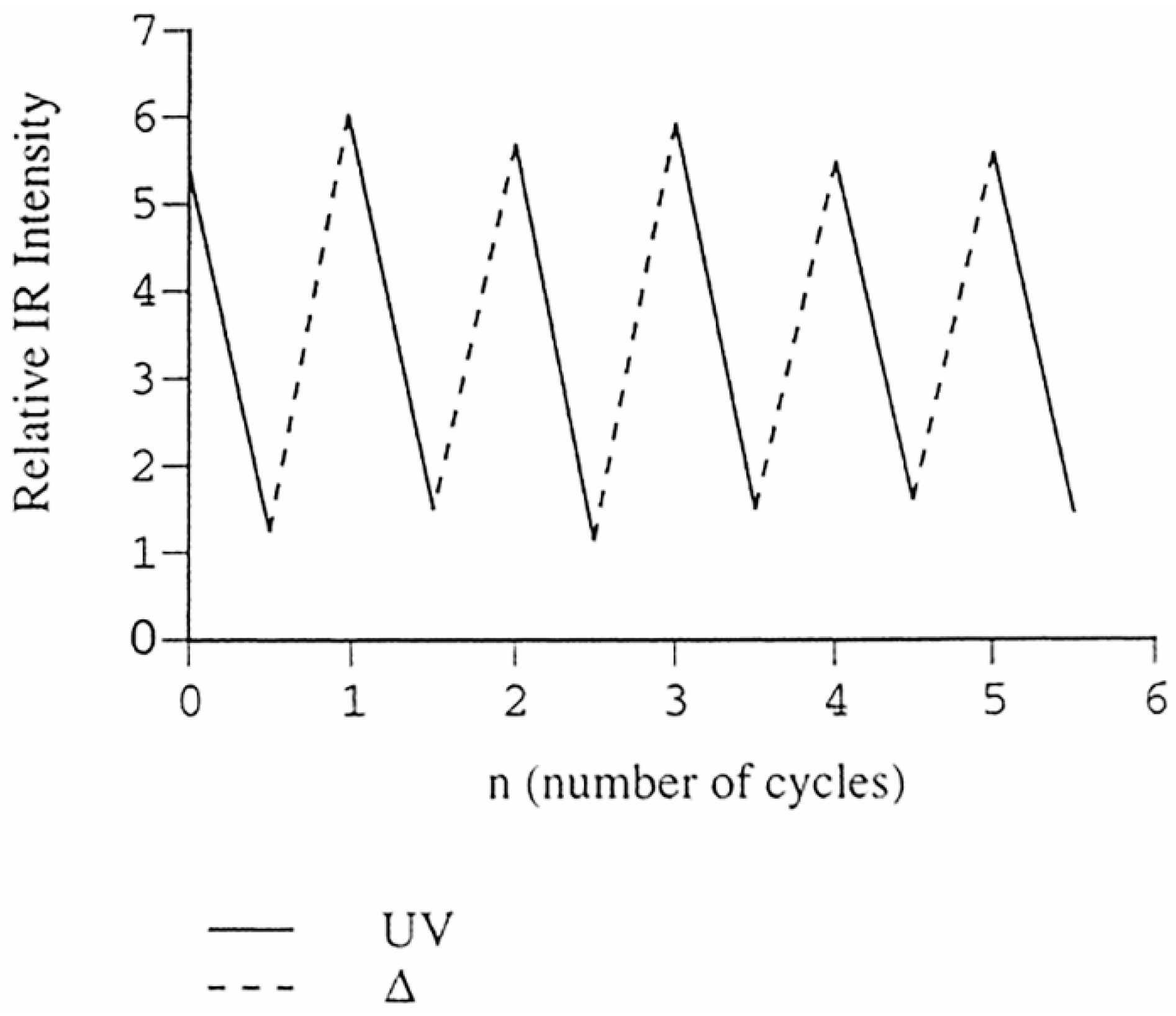
Figure 3.
Reversible changes in IR intensity of E-ON=NO stretching vibration of dimer of p-bromonitrosobenzene obtained by consecutive photolysis (solid line) and thermal reactions (dotted line). Reproduced with permission from [40]. Copyright 2002 American Chemical Society.
Figure 3.
Reversible changes in IR intensity of E-ON=NO stretching vibration of dimer of p-bromonitrosobenzene obtained by consecutive photolysis (solid line) and thermal reactions (dotted line). Reproduced with permission from [40]. Copyright 2002 American Chemical Society.
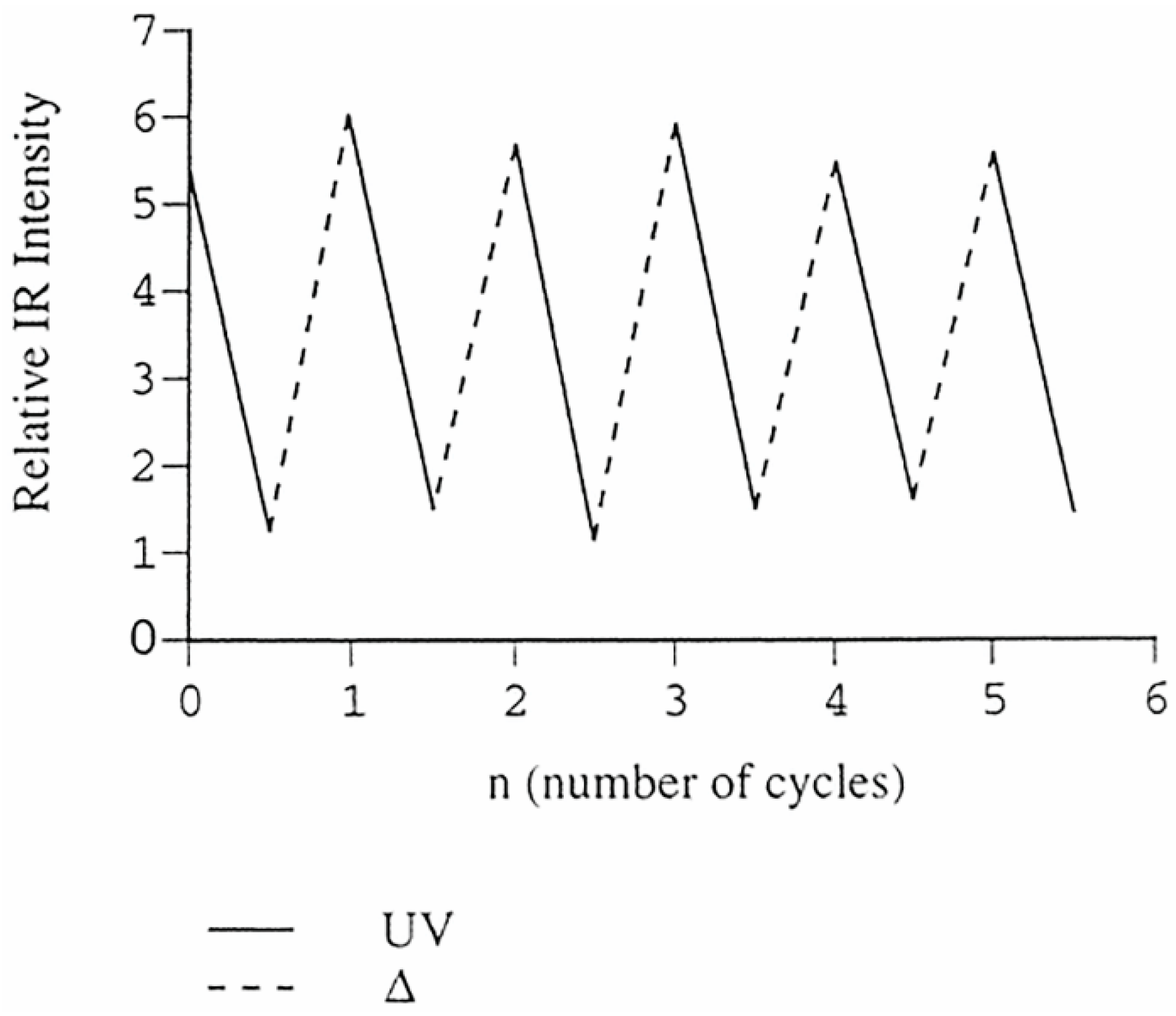
Figure 4.
Molecular arrangement of p-bromonitrosobenzene monomers in the metastable crystal after the photochemical single-crystal-to-single-crystal transformation at 100 K.
Figure 4.
Molecular arrangement of p-bromonitrosobenzene monomers in the metastable crystal after the photochemical single-crystal-to-single-crystal transformation at 100 K.
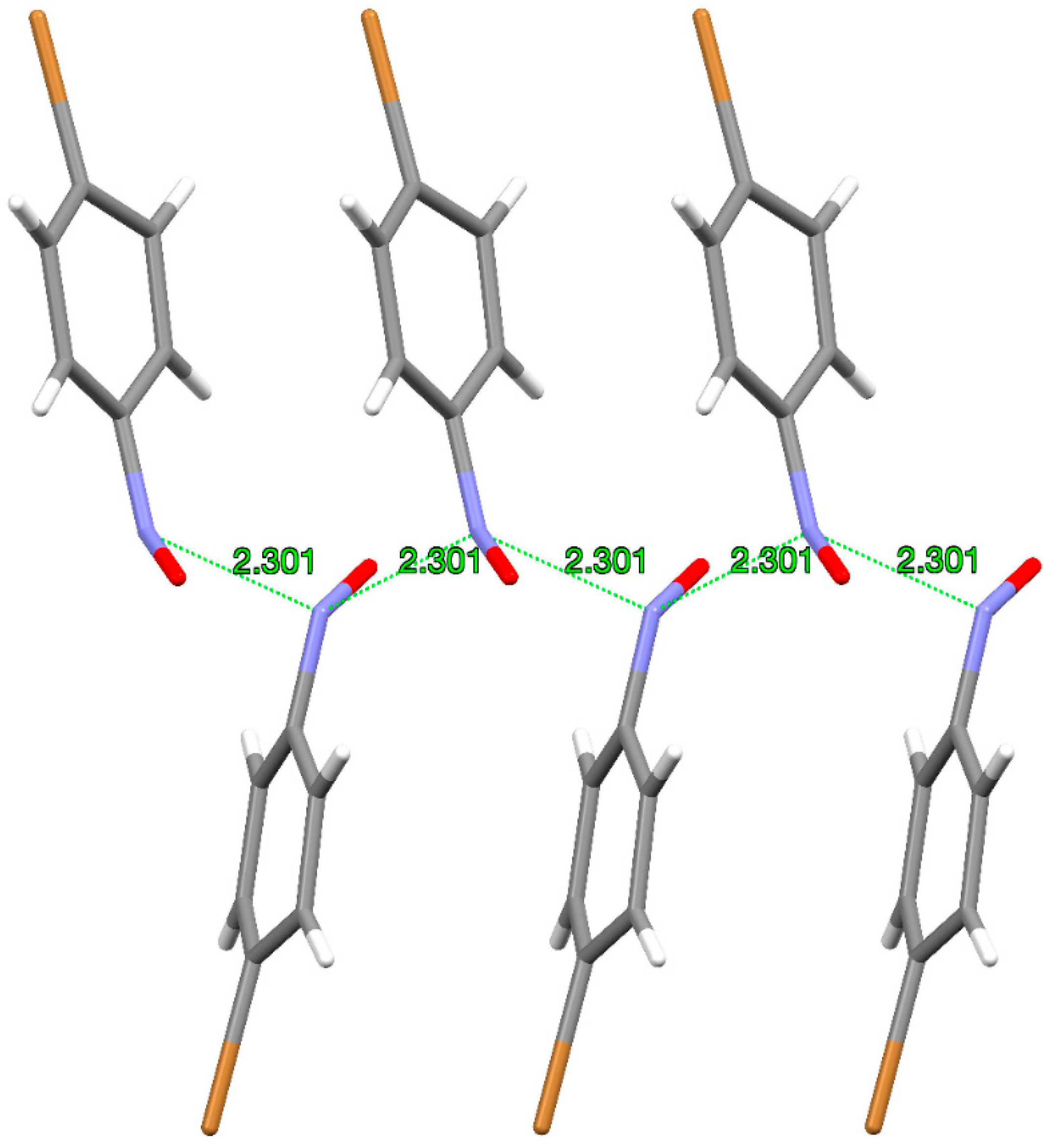
Figure 5.
Superposition of molecular orientations of the starting p-bromonitrosobenzene dimer (red) and the monomer product (green) after the photochemically-induced single-crystal-to-single-crystal conversion. Reproduced with permission from [26]. Copyright 2005 American Chemical Society.
Figure 5.
Superposition of molecular orientations of the starting p-bromonitrosobenzene dimer (red) and the monomer product (green) after the photochemically-induced single-crystal-to-single-crystal conversion. Reproduced with permission from [26]. Copyright 2005 American Chemical Society.
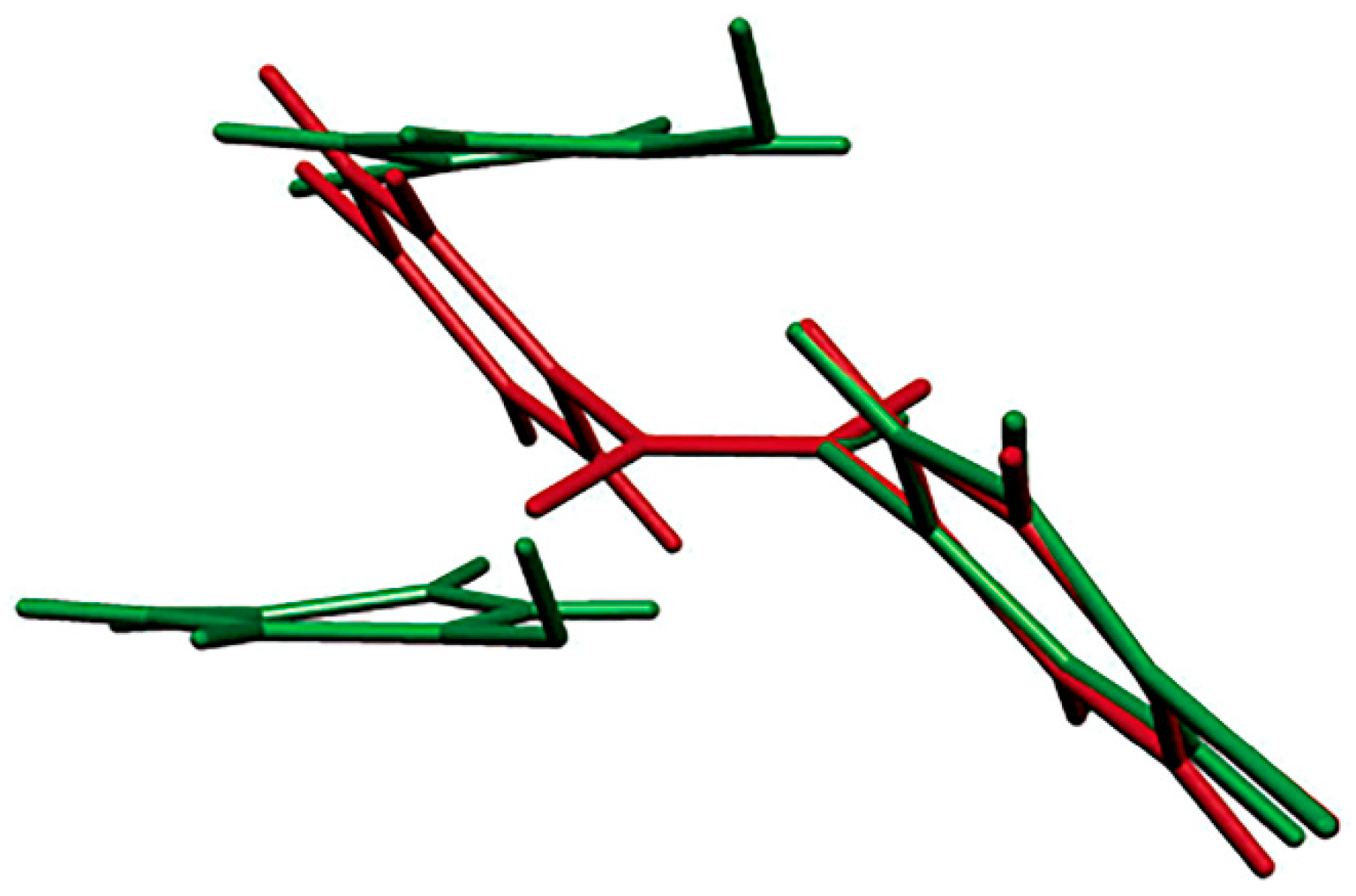
Figure 6.
Solid-state Z-E interconversion of azodioxides of m-chloronitrosobenzene monitored by (a) FTIR spectroscopy at 299 K (the characteristic signals of Z- and E-isomers are labeled with • and *, respectively) and by (b) XRPD at 298 K (X-ray diffraction patterns at the beginning of the reaction are shown in blue and after 18 h in red). Reproduced with permission from [29]. Copyright 2016 Wiley-VCH.
Figure 6.
Solid-state Z-E interconversion of azodioxides of m-chloronitrosobenzene monitored by (a) FTIR spectroscopy at 299 K (the characteristic signals of Z- and E-isomers are labeled with • and *, respectively) and by (b) XRPD at 298 K (X-ray diffraction patterns at the beginning of the reaction are shown in blue and after 18 h in red). Reproduced with permission from [29]. Copyright 2016 Wiley-VCH.
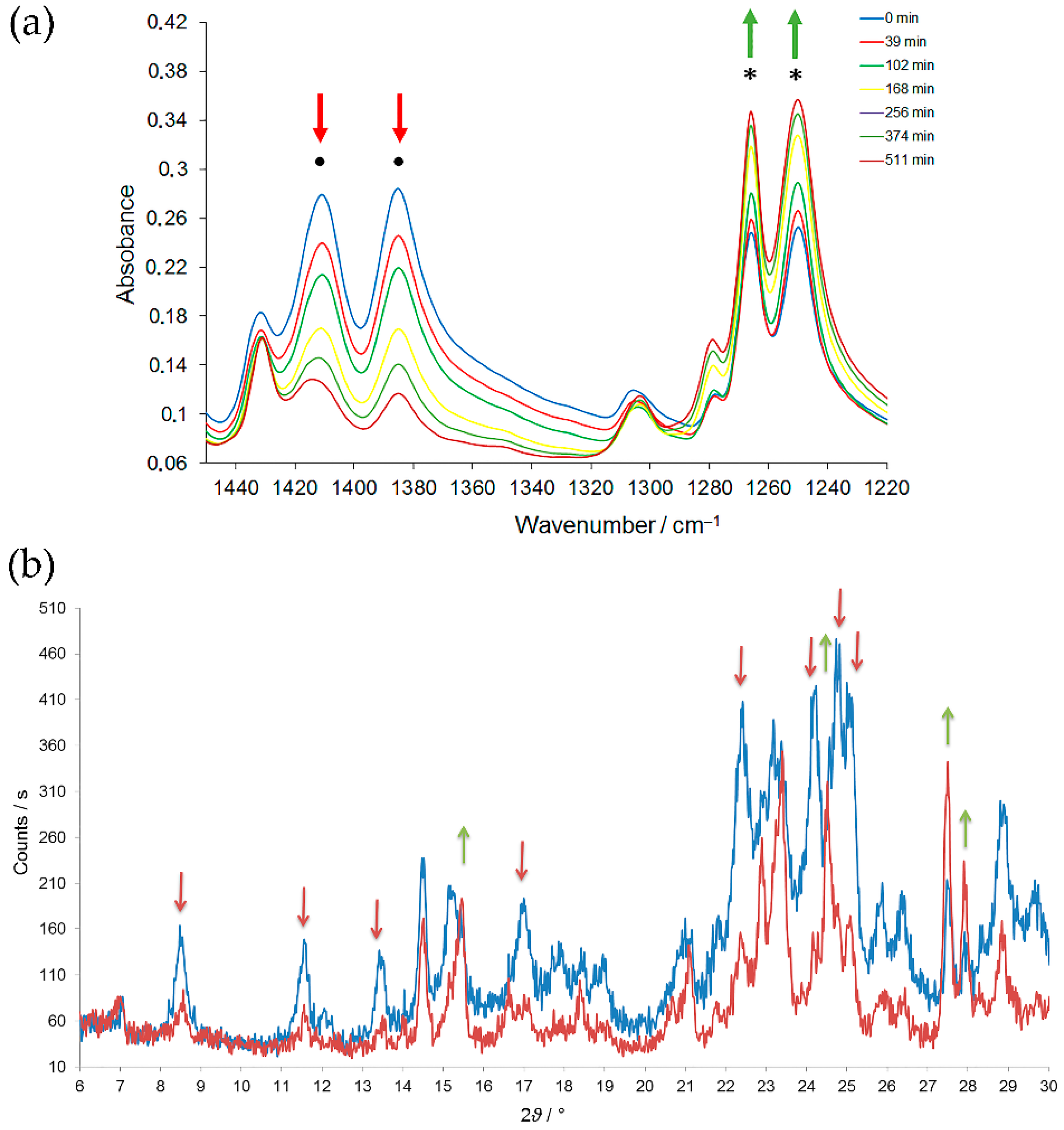
Figure 7.
Comparison of dimerization rates of p-bromonitrosobenzene in air, and immersed in glycerol for (a) 2 min, (b) 75 min and (c) 22 h. The monomer crystals are green, and the dimer phase is white. Reproduced with permission from [27]. Copyright 2011 Royal Society of Chemistry.
Figure 7.
Comparison of dimerization rates of p-bromonitrosobenzene in air, and immersed in glycerol for (a) 2 min, (b) 75 min and (c) 22 h. The monomer crystals are green, and the dimer phase is white. Reproduced with permission from [27]. Copyright 2011 Royal Society of Chemistry.

Figure 8.
Crystal packing of the sublimed monomer of (a) p-bromo- [13] and (b) p-iodonitrosobenzene [60].
Scheme 2.
Cross-dimerization of nitrosobenzene derivatives in solution and in solid state.
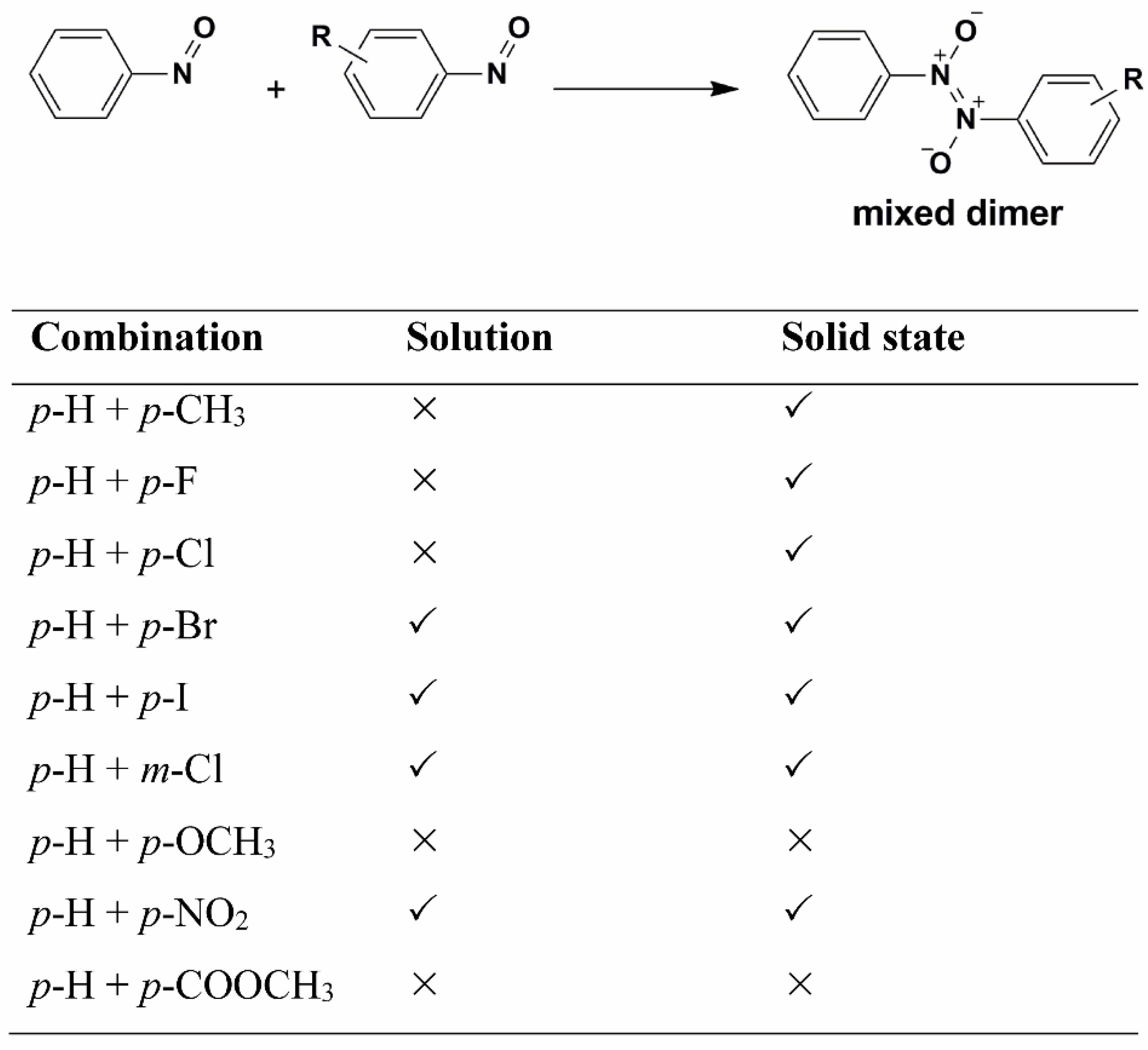
Figure 9.
Preparation of p-dinitrosobenzene by cryogenic photolysis of the corresponding polymer and by the gas phase cryogenic deposition.
Figure 9.
Preparation of p-dinitrosobenzene by cryogenic photolysis of the corresponding polymer and by the gas phase cryogenic deposition.
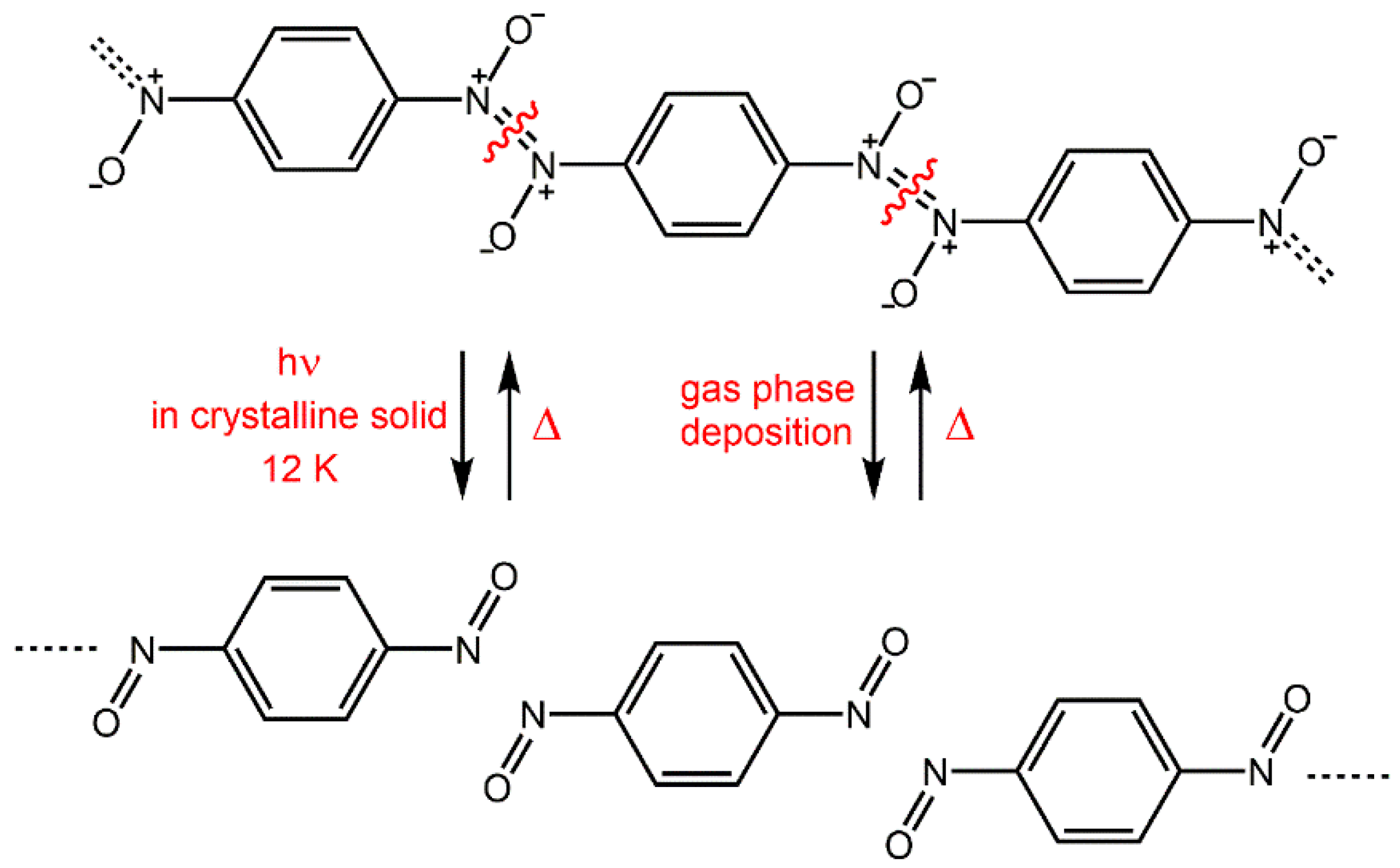
Figure 10.
(a) Calculated helical structure of the p-dinitrosobenzene octamer. Reproduced with permission from [49]. Copyright 2016 Elsevier. (b) The morphology of the p-dinitrosobenzene polymer obtained by SEM microscopy [50].
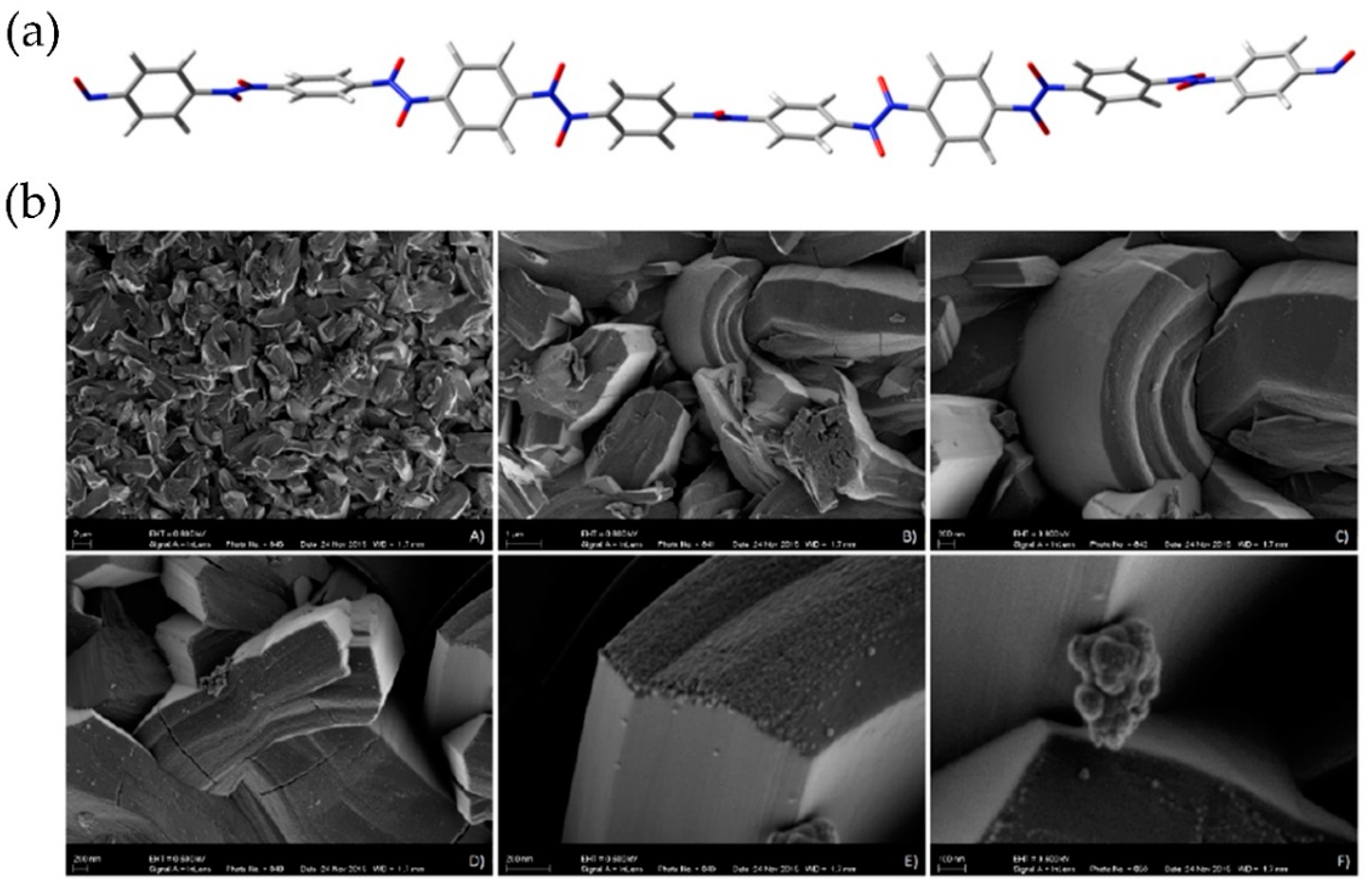
Figure 11.
(a) Model and (b) high-resolution STM image of mono- and bilayer domains of nitrosobenzene molecules on an Au(111) surface. Reproduced with permission from [64]. Copyright 2011 American Chemical Society.
Figure 11.
(a) Model and (b) high-resolution STM image of mono- and bilayer domains of nitrosobenzene molecules on an Au(111) surface. Reproduced with permission from [64]. Copyright 2011 American Chemical Society.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The rate coefficients and the activation energy for the solid-state dimerization of the p-bromonitrosobenzene after sublimation (T—temperature, Ea—activation energy, k—rate coefficient).
Table 1.
The rate coefficients and the activation energy for the solid-state dimerization of the p-bromonitrosobenzene after sublimation (T—temperature, Ea—activation energy, k—rate coefficient).
| T/K | 104 k/s−1 | Ea/kJ mol−1 |
|---|---|---|
| 293 | 1.78 ± 0.37 a | 59.27 ± 0.44 |
| 298 | 3.33 ± 0.15 | |
| 303 | 4.41 ± 0.10 | |
| 308 | 7.51 ± 0.63 | |
| 313 | 8.21 ± 0.31 |
a Standard deviation.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Biljan, I.; Vančik, H. Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids. Crystals 2017, 7, 376. https://doi.org/10.3390/cryst7120376
AMA Style
Biljan I, Vančik H. Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids. Crystals. 2017; 7(12):376. https://doi.org/10.3390/cryst7120376
Chicago/Turabian StyleBiljan, Ivana, and Hrvoj Vančik. 2017. "Aromatic C-Nitroso Compounds and Their Dimers: A Model for Probing the Reaction Mechanisms in Crystalline Molecular Solids" Crystals 7, no. 12: 376. https://doi.org/10.3390/cryst7120376
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.