
Synthesis, Crystal and Electronic Structures of the Pnictides AE3TrPn3 (AE = Sr, Ba; Tr = Al, Ga; Pn = P, As)


Abstract
:1. Introduction
2. Results and Discussion
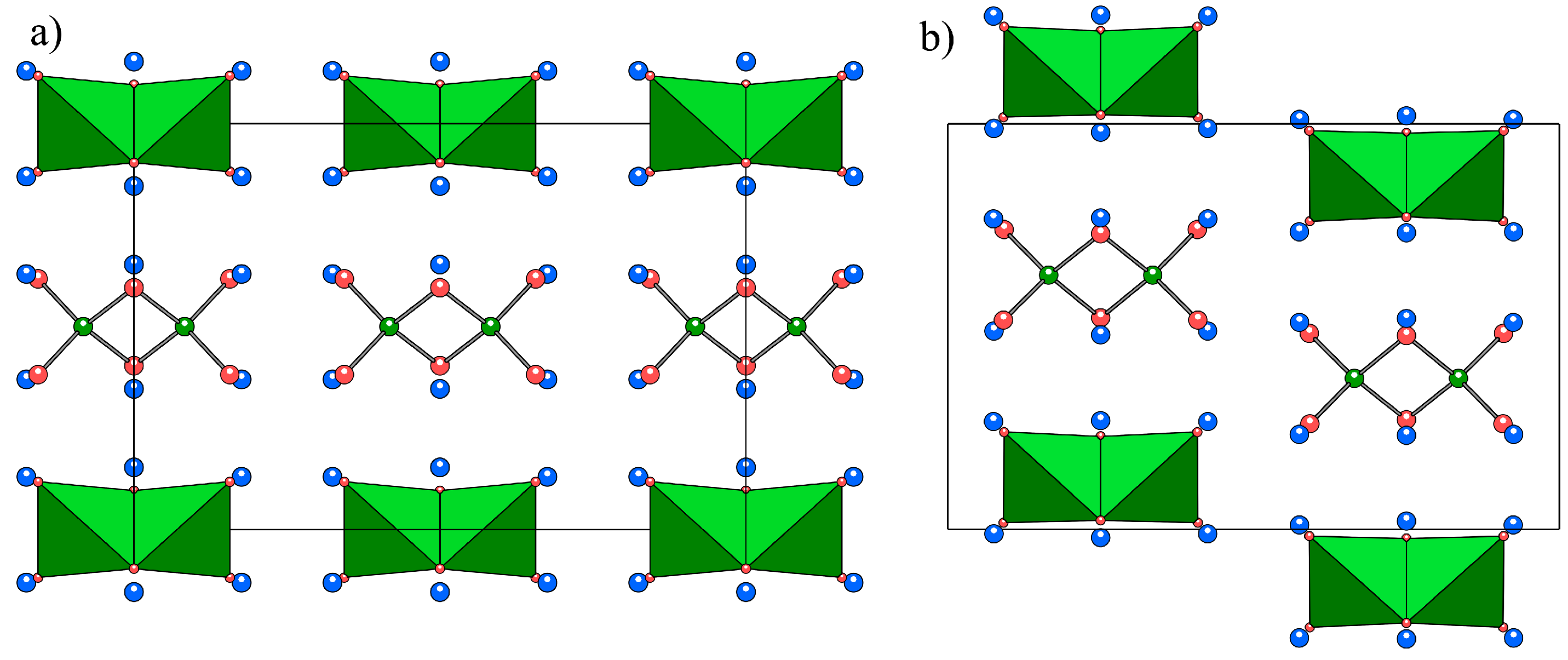
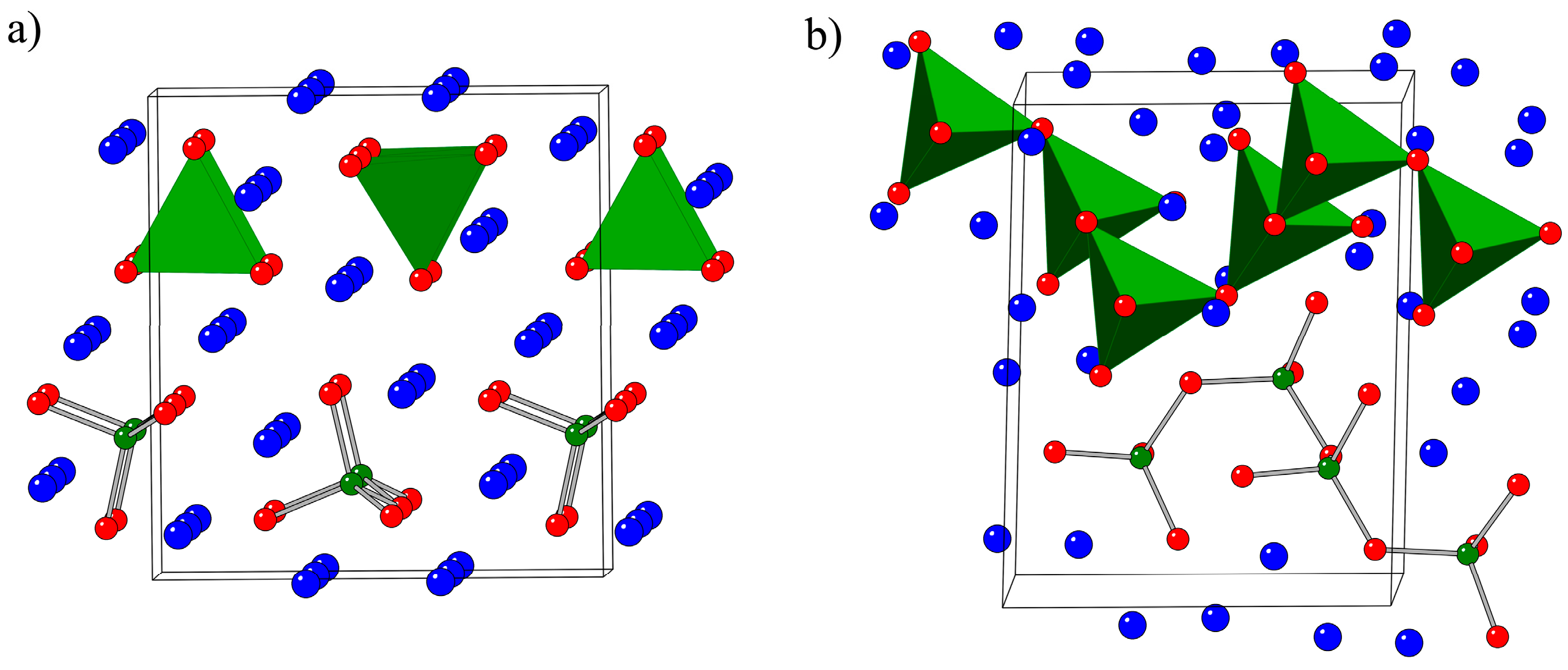
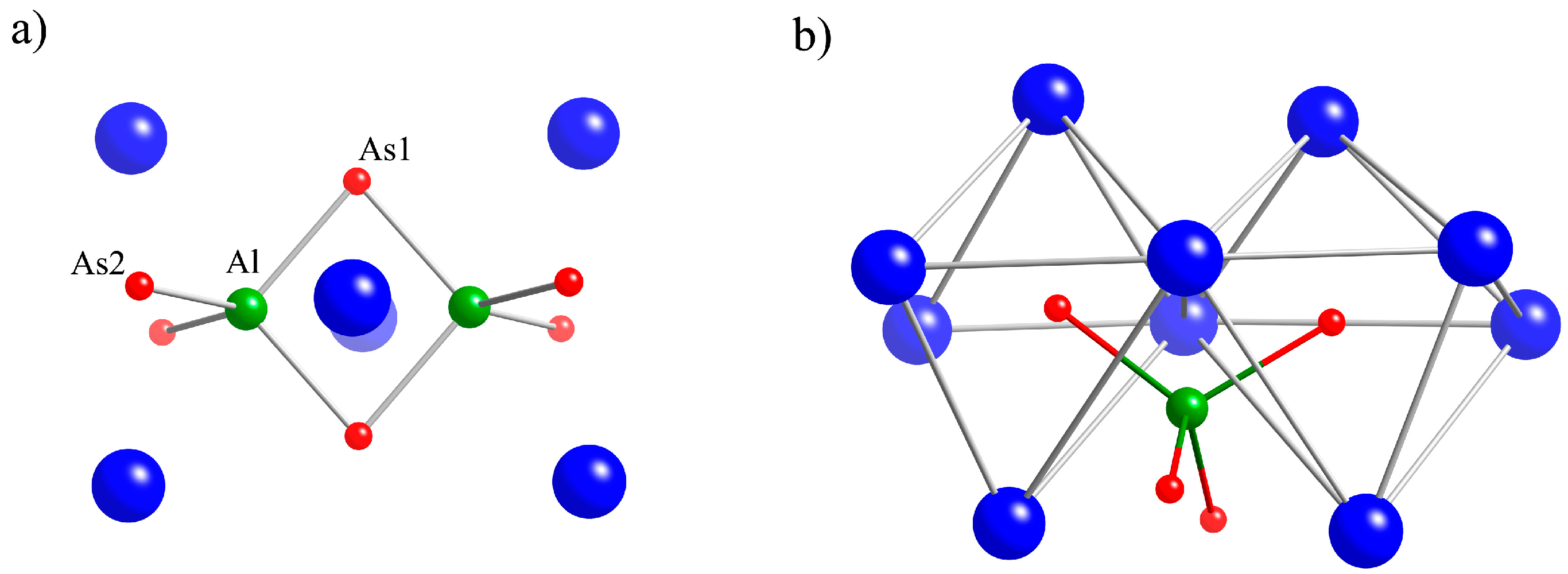
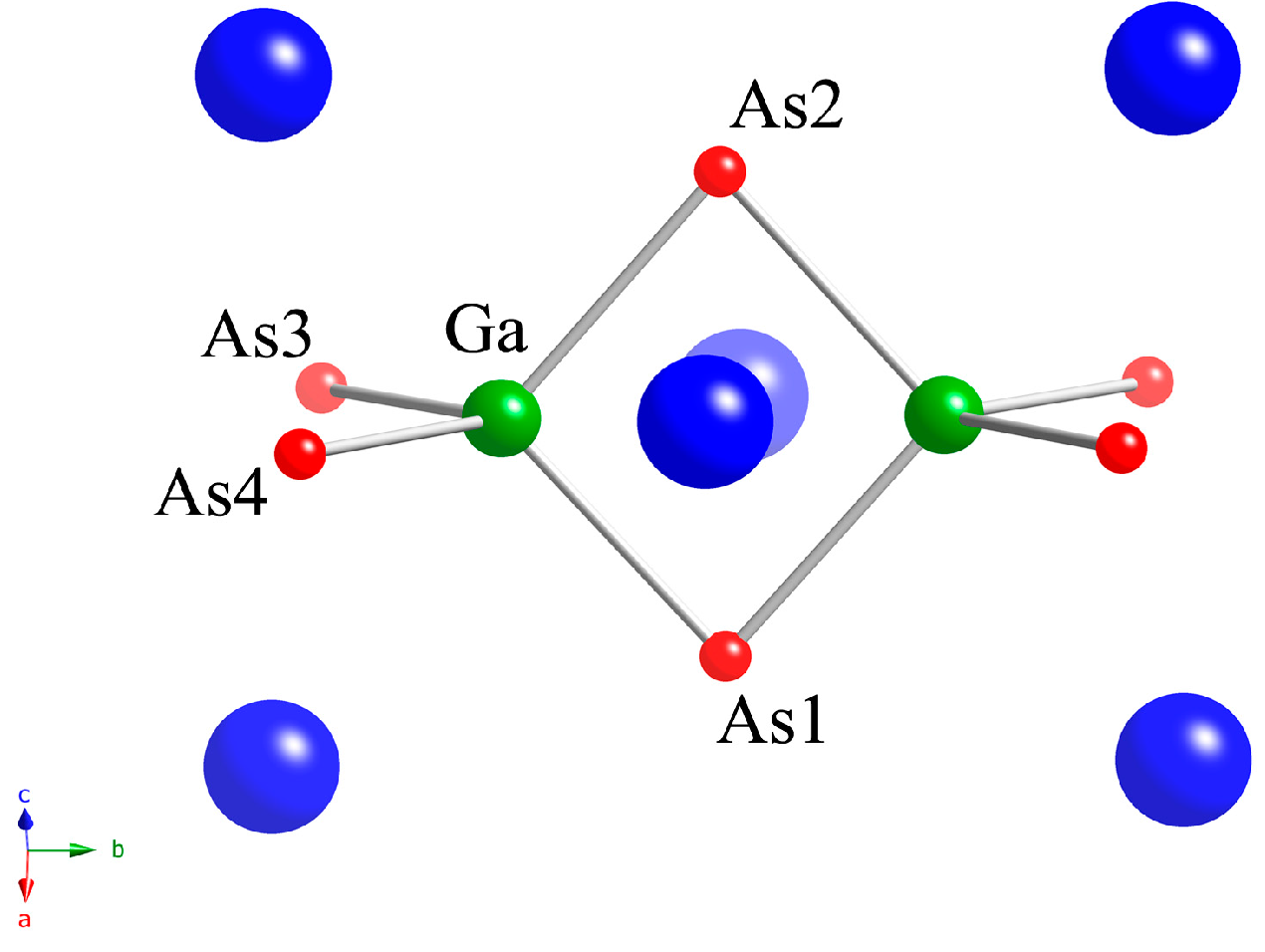
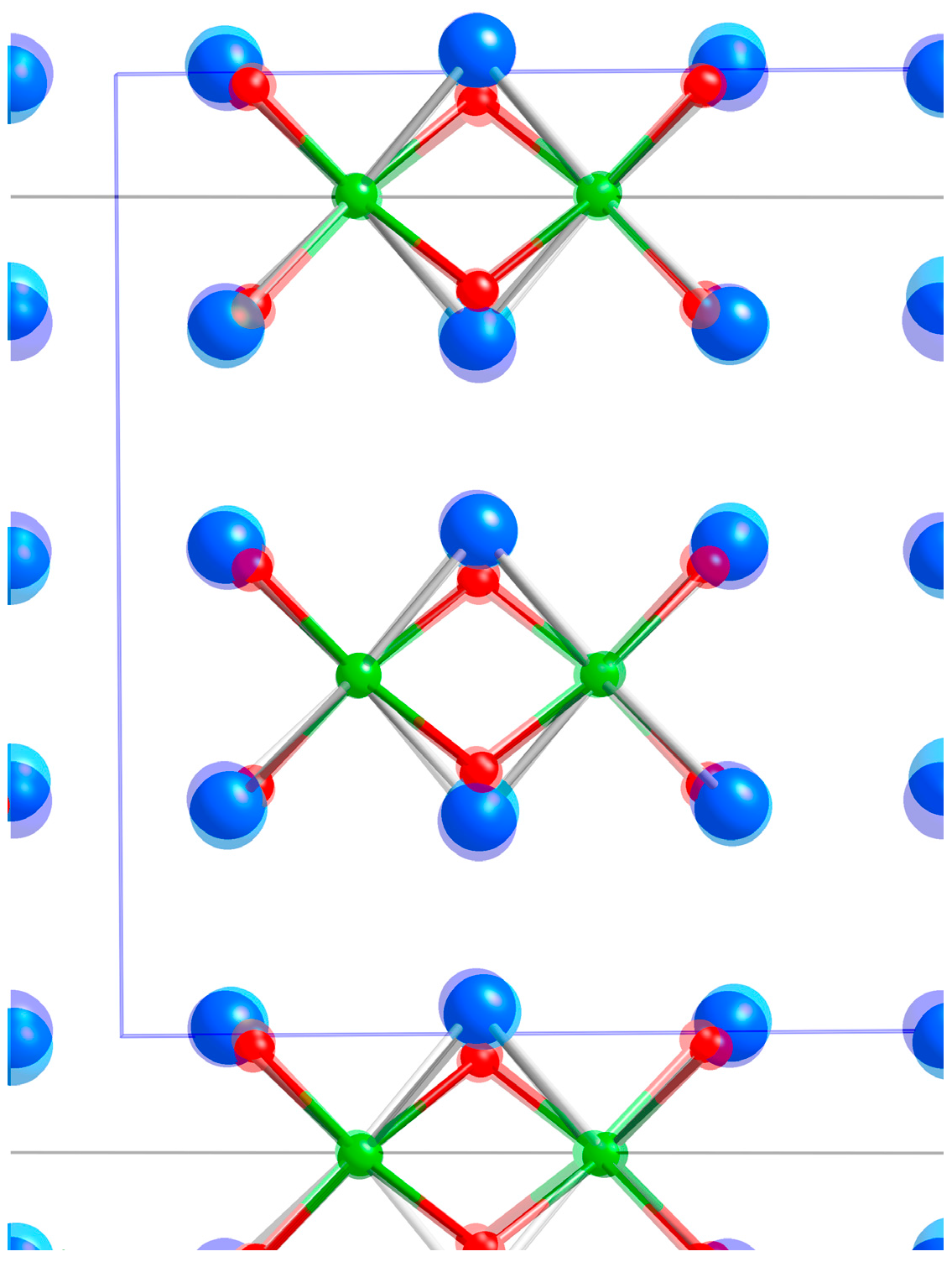
2.1. Crystal Structures




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Sr3AlAs3 | Ba3AlAs3 | Sr3GaAs3 | Ba3GaAs3 |
|---|---|---|---|---|
| Formula weight | 514.60 | 663.76 | 557.34 | 706.50 |
| Space group | Cmce (No. 64) | Cmce (No. 64) | Pnma (No. 62) | Pnma (No. 62) |
| a (Å) | 19.149(2) | 19.854(3) | 12.757(6) | 13.3589(10) |
| b (Å) | 6.5652(8) | 6.8636(9) | 19.268(9) | 19.9788(15) |
| c (Å) | 12.6871(15) | 13.2849(17) | 6.503(3) | 6.8008(5) |
| V (Å3) | 1595.0(3) | 1810.3(4) | 1598.6(13) | 1815.1(2) |
| Z | 8 | 8 | 8 | 8 |
| T (K) | 200(2) | 200(2) | 200(2) | 200(2) |
| ρcalcd (g·cm−3) | 4.29 | 4.87 | 4.63 | 5.17 |
| Crystal size (m) | 30 × 40 × 60 | 20 × 30 × 50 | 30 × 40 × 110 | 40 × 80 × 135 |
| Radiation | Graphite-monochromated Mo Kα, λ = 0.71073 Å | |||
| μ(Mo Kα) (cm−1) | 323.5 | 237.9 | 354.6 | 265.4 |
| Transmission factors | 0.260–0.424 | 0.414–0.648 | 0.112–0.397 | 0.116–0.395 |
| 2θ (Mo Kα) limits | 4.26°–56.52° | 4.10°–56.54° | 4.22°–61.78° | 4.08°–61.62° |
| Data collected | –25 ≤ h ≤ 25, –8 ≤ k ≤ 8, –16 ≤ l ≤ 16 | –26 ≤ h≤ 26, –9 ≤ k ≤ 9, –17 ≤ l ≤ 17 | –18 ≤ h≤ 17, –27 ≤ k ≤ 27, –9 ≤ l ≤ 9 | –19 ≤ h≤ 19, –28 ≤ k ≤ 28, –9 ≤ l ≤ 9 |
| No. of data collected | 7323 | 8248 | 16886 | 18478 |
| No. of unique data, including Fo2 < 0 | 1029 (Rint = 0.058) | 1151 (Rint = 0.037) | 2511 (Rint = 0.066) | 2830 (Rint = 0.028) |
| No. of unique data, with Fo2 > 2σ(Fo2) | 884 | 1031 | 1844 | 2527 |
| No. of variables | 37 | 37 | 71 | 71 |
| R(F) for Fo2 > 2σ(Fo2)a | 0.023 | 0.017 | 0.027 | 0.016 |
| Rw(Fo2) b | 0.041 | 0.036 | 0.046 | 0.031 |
| Goodness of fit | 1.074 | 1.003 | 1.054 | 1.114 |
| (Δρ)max, (Δρ)min (eÅ−3) | [0.77, –1.01] | [0.77, –1.07] | [1.28, –1.33] | [0.89, –0.82] |
| Atom | Wyckoff Position | x | y | z | Ueq (Å2) a |
|---|---|---|---|---|---|
| Sr3AlAs3 | |||||
| Sr1 | 8f | 0 | 0.17463(8) | 0.34891(4) | 0.0102(1) |
| Sr2 | 16g | 0.17691(2) | 0.31222(6) | 0.13126(3) | 0.0104(1) |
| Al | 8d | 0.08500(1) | 0 | 0 | 0.0086(4) |
| As1 | 8f | 0 | 0.20879(9) | 0.10250(5) | 0.0095(1) |
| As2 | 16g | 0.34135(2) | 0.29606(6) | 0.12022(3) | 0.0095(1) |
| Ba3AlAs3 | |||||
| Ba1 | 8f | 0 | 0.17015(4) | 0.34685(2) | 0.0116(1) |
| Ba2 | 16g | 0.17563(1) | 0.31318(3) | 0.13014(2) | 0.0116(1) |
| Al | 8d | 0.08294(7) | 0 | 0 | 0.0099(3) |
| As1 | 8f | 0 | 0.20319(7) | 0.09613(4) | 0.0109(1) |
| As2 | 16g | 0.34345(2) | 0.30905(5) | 0.11805(3) | 0.0111(1) |
| -- | Sr3AlAs3 | Ba3AlAs3 |
|---|---|---|
| AE1–As1 | 3.1199(9) | 3.2933(7) |
| AE1–As1 | 3.1342(9) | 3.3386(7) |
| AE1–As1 (× 2) | 3.1652(6) | 3.2843(5) |
| AE1–As1 | 3.3072(9) | 3.4239(7) |
| AE1–As1 | 3.5606(9) | 3.7362(7) |
| AE2–As2 | 3.1538(7) | 3.3358(6) |
| AE2–As2 | 3.1742(7) | 3.3668(6) |
| AE2–As2 | 3.1988(7) | 3.4232(6) |
| AE2–As2 | 3.2874(7) | 3.4283(6) |
| AE2–As2 | 3.4096(7) | 3.4845(6) |
| AE2–As2 | 3.4744(6) | 3.5962(5) |
| Al–As2 (× 2) | 2.472(1) | 2.5126(9) |
| Al–As1 (× 2) | 2.494(1) | 2.507(1) |
| AE1–Al (× 2) | 3.299(1) | 3.4607(7) |
| AE2–Al | 3.174(1) | 3.3161(8) |
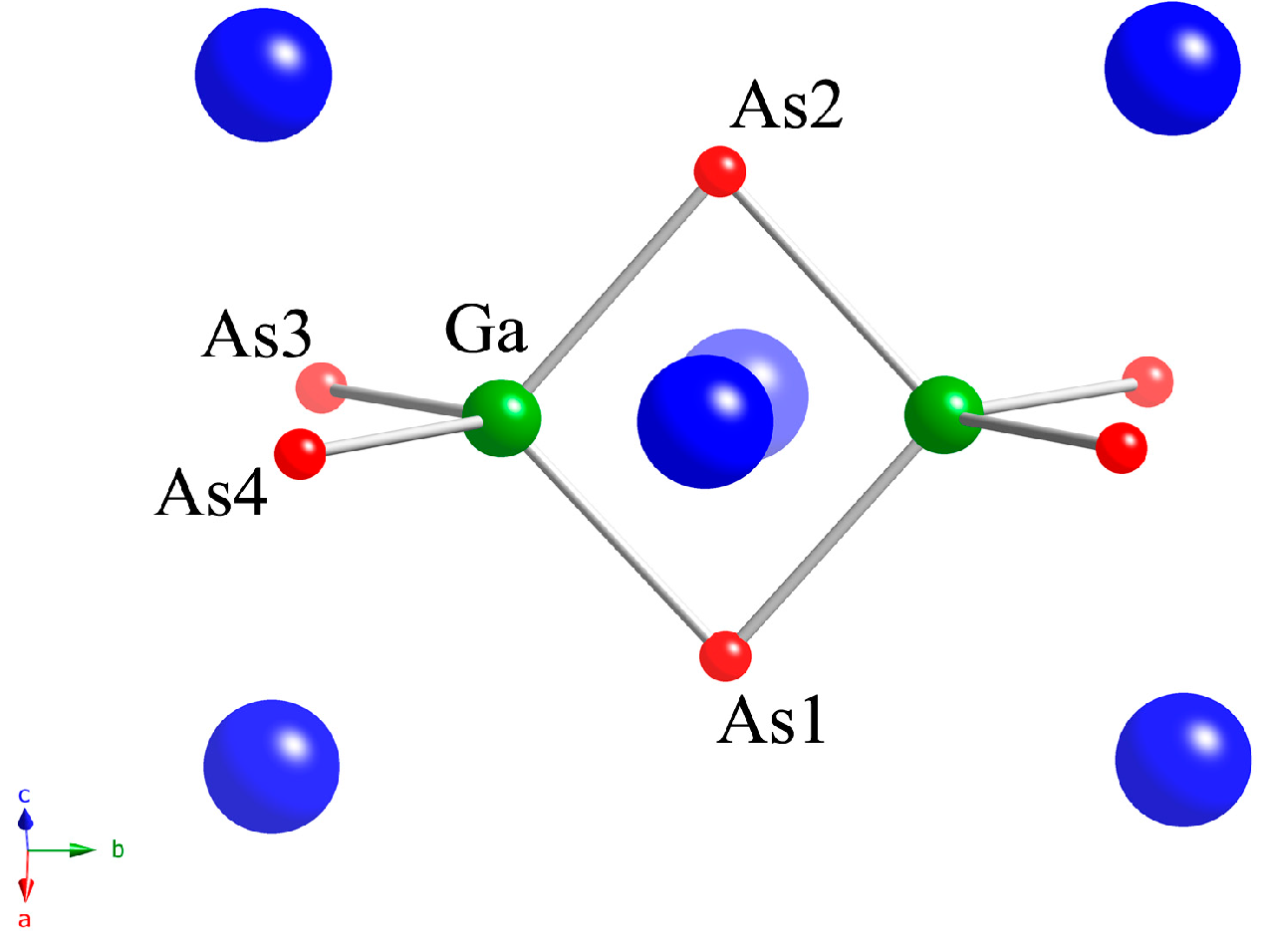
| Atom | Wyckoff Position | x | y | z | Ueq (Å2) a |
|---|---|---|---|---|---|
| Sr3GaAs3 | |||||
| Sr1 | 8d | 0.01274(4) | 0.57417(2) | 0.27769(6) | 0.0100(1) |
| Sr2 | 8d | 0.26474(3) | 0.07335(2) | 0.30854(6) | 0.0098(1) |
| Sr3 | 4c | 0.26816(5) | 1/4 | 0.68712(9) | 0.0094(1) |
| Sr4 | 4c | 0.48050(5) | 1/4 | 0.16924(9) | 0.0097(1) |
| Ga | 8d | 0.12642(4) | 0.16332(3) | 0.01367(7) | 0.0085(1) |
| As1 | 4c | 0.23468(5) | 1/4 | 0.21000(1) | 0.0090(1) |
| As2 | 4c | 0.01681(5) | 1/4 | 0.81317(9) | 0.0092(1) |
| As3 | 8d | 0.01121(4) | 0.08982(3) | 0.23326(6) | 0.0090(1) |
| As4 | 8d | 0.25889(4) | 0.58990(3) | 0.29433(7) | 0.0092(1) |
| Ba3GaAs3 | |||||
| Ba1 | 8d | 0.01098(1) | 0.575295(8) | 0.27503(2) | 0.0096(1) |
| Ba2 | 8d | 0.26589(1) | 0.074780(9) | 0.31222(2) | 0.0096(1) |
| Ba3 | 4c | 0.26836(2) | 1/4 | 0.68616(3) | 0.0090(1) |
| Ba4 | 4c | 0.47937(2) | 1/4 | 0.16338(3) | 0.0093(1) |
| Ga | 8d | 0.12737(2) | 0.16495(1) | 0.01690(4) | 0.0079(1) |
| As1 | 4c | 0.22915(3) | 1/4 | 0.20661(5) | 0.0085(1) |
| As2 | 4c | 0.02242(3) | 1/4 | 0.82548(5) | 0.0084(1) |
| As3 | 8d | 0.01687(2) | 0.09121(1) | 0.22609(4) | 0.0092(1) |
| As4 | 8d | 0.26014(2) | 0.59211(2) | 0.30814(4) | 0.0091(1) |
| -- | Sr3GaAs3 | Ba3GaAs3 | -- | Sr3GaAs3 | Ba3GaAs3 |
|---|---|---|---|---|---|
| AE1–As4 | 3.157(2) | 3.3530(4) | AE3–As2 | 3.310(2) | 3.4195(5) |
| AE1–As3 | 3.173(2) | 3.3441(4) | AE3–As1 | 3.427(2) | 3.5780(5) |
| AE1–As3 | 3.209(2) | 3.4279(4) | AE4–As1 | 3.147(2) | 3.3556(5) |
| AE1–As4 | 3.286(2) | 3.4150(4) | AE4–As2 | 3.171(2) | 3.3740(5) |
| AE1–As3 | 3.351(2) | 3.4430(4) | AE4–As3 (× 2) | 3.175(2) | 3.2985(4) |
| AE1–As2 | 3.460(2) | 3.5846(3) | AE4–As1 | 3.337(2) | 3.4519(5) |
| AE2–As4 | 3.148(2) | 3.3353(4) | AE4–As2 | 3.398(2) | 3.5234(5) |
| AE2–As3 | 3.172(2) | 3.3789(4) | Ga–As4 | 2.4855(9) | 2.5281(4) |
| AE2–As4 | 3.190(2) | 3.4082(4) | Ga–As3 | 2.4911(9) | 2.5245(4) |
| AE2–As3 | 3.287(2) | 3.3937(4) | Ga–As1 | 2.515(1) | 2.5299(4) |
| AE2–As4 | 3.373(2) | 3.4631(4) | Ga–As2 | 2.538(1) | 2.5589(4) |
| AE2–As1 | 3.485(2) | 3.6071(3) | AE1–Ga | 3.113(1) | 3.2505(4) |
| AE3–As1 | 3.132(2) | 3.3032(5) | AE2–Ga | 3.130(1) | 3.2715(3) |
| AE3–As2 | 3.172(2) | 3.3948(5) | AE3–Ga (× 2) | 3.251(1) | 3.3903(4) |
| AE3–As4 (× 2) | 3.182(2) | 3.2839(4) | AE4–Ga (× 2) | 3.241(1) | 3.3948(4) |

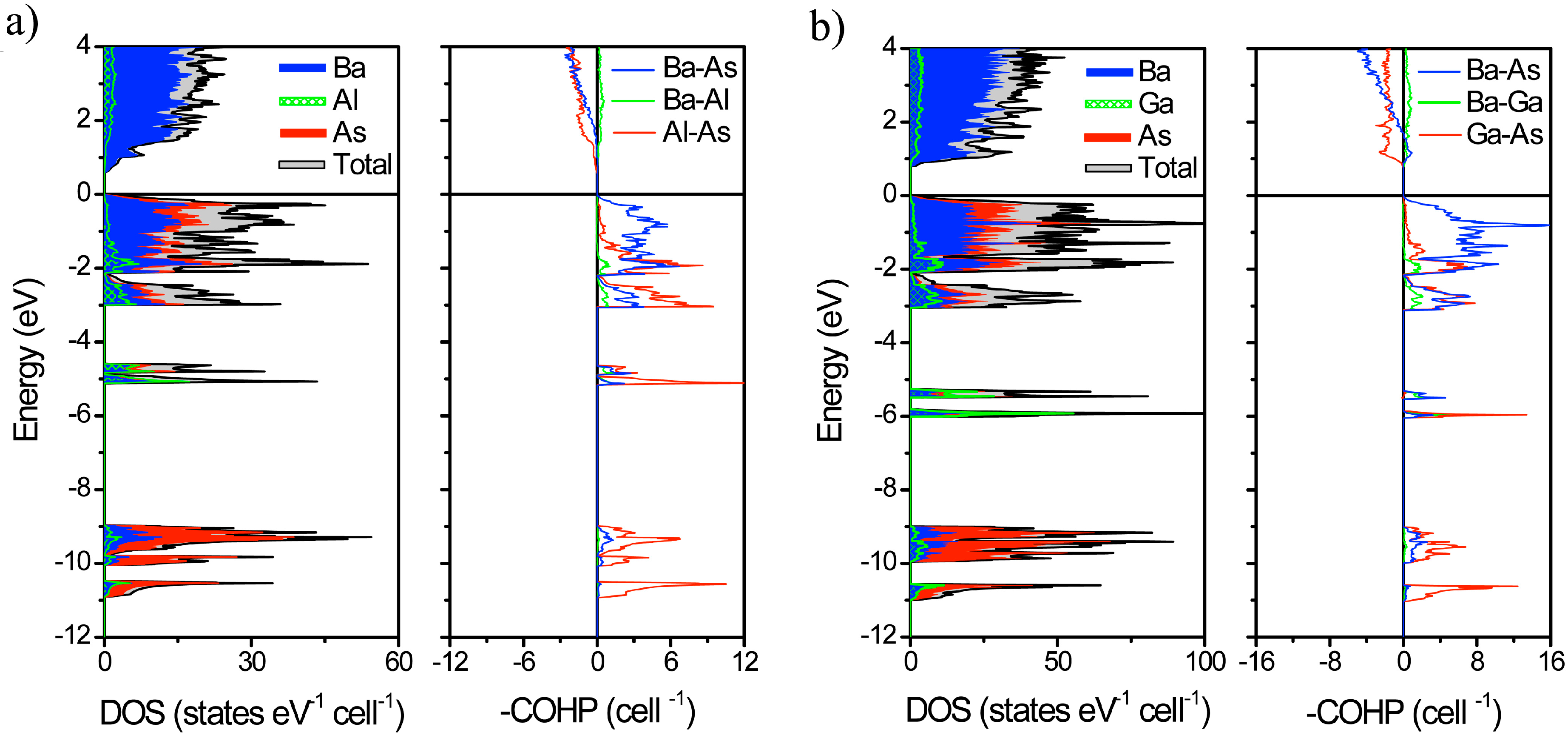
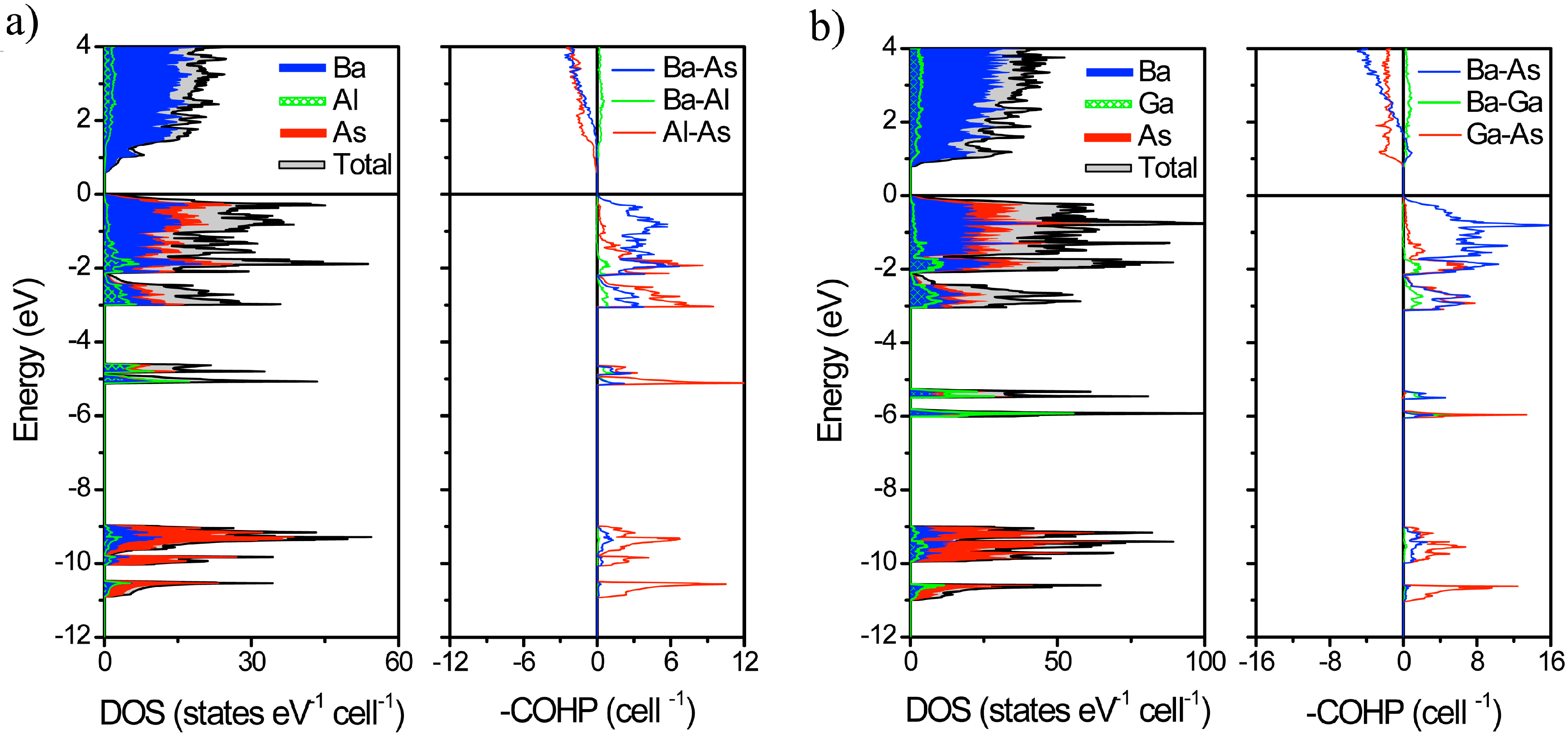
2.2. Electronic Structure
3. Experimental Section
3.1. Synthesis
| AE | Ca | Sr | Ba | AE | ||
| Tr | Pn | |||||
| Al | – | – | Ba3AlP3 [this work] | P | ||
| Ca3AlAs3 [21] | Sr3AlAs3 [this work] | Ba3AlAs3 [this work] | As | |||
| Ca3AlSb3 [31] | Sr3AlSb3 [32] | Ba3AlSb3 [21] | Sb | |||
| Ga | – | Sr3GaP3 [this work] | Ba3GaP3 [33] | P | ||
| Ca3GaAs3 [22] | Sr3GaAs3 [this work] | Ba3GaAs3 [this work] | As | |||
| – | Sr3GaSb3 [25] | Ba3GaSb3 [22] | Sb | |||
| In | Ca3InP3 [22] | Sr3InP3 [25] | – | P | ||
| – | – | Ba3InAs3 [34] | As | |||
| – | – | – | Sb | |||
3.2. X-ray Diffraction
3.3. Electronic Structure Calculations
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bobev, S.; Fritsch, V.; Thompson, J.D.; Sarrao, J.L.; Eck, B.; Dronskowski, R.; Kauzlarich, S.M. Synthesis, structure and properties of the new rare-earth Zintl phase Yb11GaSb9. J. Solid State Chem. 2005, 178, 1071–1079. [Google Scholar]
- Hullmann, J.; Xia, S.-Q.; Bobev, S. Sr11InSb9 grown from molten In. Acta Crystallogr. Sect. E 2007, 63, 178. [Google Scholar] [CrossRef]
- Xia, S.-Q.; Hullmann, J.; Bobev, S.; Ozbay, A.; Nowak, E.R.; Fritsch, V. Synthesis, crystal structures, magnetic and electric transport properties of Eu11InSb9 and Yb11InSb9. J. Solid State Chem. 2007, 180, 2088–2094. [Google Scholar] [CrossRef]
- Xia, S.-Q.; Hullmann, J.; Bobev, S. Gallium substitutions as a means to stabilize alkaline-earth and rare-earth metal pnictides with the cubic Th3P4 type: Synthesis and structure of A7Ga2Sb6 (A = Sr, Ba, Eu). J. Solid State Chem. 2008, 181, 1909–1914. [Google Scholar] [CrossRef]
- Bobev, S.; Hullmann, J.; Harmening, T.; Pöttgen, R. Novel ternary alkaline-earth and rare-earth metal antimonides from Gallium or Indium flux. Synthesis, structural characterization and 121Sb and 151Eu Mössbauer spectroscopy of the series A7Ga8Sb8 (A = Sr, Ba, Eu) and Ba7In8Sb8. Dalton Trans. 2010, 39, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Cordier, G.; Schäfer, H.; Stelter, M. Perantimonidogallate undindate: Zur kenntnis von Ca5Ga2Sb6, Ca5In2Sb6 und Sr5In2Sb6: The first perantimonidogallate and-indates. Zeitschrift für Naturforschung B 1985, 40, 5–8. [Google Scholar]
- Kim, S.-J.; Hu, S.; Uher, C.; Kanatzidis, M.G. Ba4In8Sb16: Thermoelectric properties of a new layered Zintl phase with infinite zigzag Sb chains and pentagonal tubes. Chem. Mater. 1999, 11, 3154–3159. [Google Scholar] [CrossRef]
- Kim, S.-J.; Kanatzidis, M.G. A Unique framework in BaGa2Sb2: A new Zintl phase with large tunnels. Inorg. Chem. 2001, 40, 3781–3785. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Choi, E.S.; Kang, W.; Kim, S.-J. Eu5In2Sb6, Eu5In2–xZnxSb6: Rare earth Zintl phases with narrow band gaps. J. Mater. Chem. 2002, 12, 1839–1843. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, S.-J.; Kanatzidis, M.G. Ga–Ga bonding and tunnel framework in the new Zintl phase Ba3Ga4Sb5. J. Solid State Chem. 2003, 175, 310–315. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, S.-J.; Kanatzidis, M.G. Eu7Ga6Sb8: A Zintl phase with Ga–Ga bonds and polymeric gallium antimonide chains. J. Solid State Chem. 2004, 177, 2867–2874. [Google Scholar] [CrossRef]
- Yi, T.; Cox, C.A.; Toberer, E.S.; Snyder, G.J.; Kauzlarich, S.M. High-temperature transport properties of the Zintl phases Yb11GaSb9 and Yb11InSb9. Chem. Mater. 2010, 22, 935–941. [Google Scholar] [CrossRef]
- Kanatzidis, M.G.; Pöttgen, R.; Jeitschko, W. The metal flux: A preparative tool for the exploration of intermetallic compounds. Angew. Chem. Int. Ed. 2005, 44, 6996–7023. [Google Scholar] [CrossRef] [PubMed]
- Canfield, P.C.; Fisk, Z. Growth of single crystals from metallic fluxes. Philos. Mag. B 1992, 65, 1117–1123. [Google Scholar] [CrossRef]
- Tobash, P.H.; Lins, D.; Bobev, S.; Lima, A.; Hundley, M.F.; Thompson, J.D.; Sarrao, J.L. Crystal growth, structural and property studies on a family of ternary rare-earth phases RE2InGe2 (RE = Sm, Gd, Tb, Dy, Ho, Yb). Chem. Mater. 2005, 17, 5567–5573. [Google Scholar] [CrossRef]
- He, H.; Stearrett, R.; Nowak, E.R.; Bobev, S. BaGa2Pn2 (Pn = P, As): New semiconducting phosphides and arsenides with layered structures. Inorg. Chem. 2010, 49, 7935–7940. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Stearrett, R.; Nowak, E.R.; Bobev, S. Gallium pnictides of the alkaline earth metals, synthesized by means of the flux method: Crystal structures and properties of CaGa2Pn2, SrGa2As2, Ba2Ga5As5, and Ba4Ga5Pn8 (Pn = P or As). Eur. J. Inorg. Chem. 2011, 14, 4025–4036. [Google Scholar] [CrossRef]
- He, H.; Tyson, C.; Saito, M.; Bobev, S. Synthesis and structural characterization of the ternary Zintl phases AE3Al2Pn4 and AE3Ga2Pn4 (AE = Ca, Sr, Ba, Eu; Pn = P, As). J. Solid State Chem. 2012, 188, 59–65. [Google Scholar] [CrossRef]
- He, H.; Tyson, C.; Saito, M.; Bobev, S. Synthesis, crystal and electronic structures of the new Zintl phases Ba3Al3Pn5 (Pn = P, As) and Ba3Ga3P5. Inorg. Chem. 2013, 52, 499–505. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Stoyko, S.; Bobev, S. New insights into the application of the valence rules in Zintl phases-crystal and electronic structures of Ba7Ga4P9, Ba7Ga4As9, Ba7Al4Sb9, Ba6CaAl4Sb9, and Ba6CaGa4Sb9. J. Solid State Chem. 2015. [Google Scholar] [CrossRef]
- Cordier, G.; Savelsberg, G.; Schäfer, H. Zintl phasen mit komplexen anionen: Zur kenntnis von Ca3AlAs3 und Ba3AlSb3. Zeitschrift für Naturforschung B 1982, 37, 975–980. [Google Scholar] [CrossRef]
- Cordier, G.; Schäfer, H.; Steher, M. Neue Zintl phasen: Ba3GaSb3, Ca3GaAs3 und Ca3InP3. Zeitschrift für Naturforschung B 1985, 40, 1100–1104. [Google Scholar] [CrossRef]
- Skriver, H.L. The LMTO Method; Cardona, M., Fulde, P., Queisser, H.-J., Eds.; Springer: Berlin, Germany, 1984. [Google Scholar]
- Villars, P. Pearson’s Handbook of Crystallographic Data for Intermetallic Compounds, 2nd ed.; Calvert, L.D., Ed.; American Society for Metals: Materials Park, OH, USA, 1991. [Google Scholar]
- Cordier, G.; Schäfer, H.; Stelter, M. Sr3GaSb3 und Sr3InP3, zwei neue Zintl phasen mit komplexen anionen. Zeitschrift für Naturforschung B 1982, 42, 1268–1272. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Cordier, G.; Ochmann, H. Na3AlAs2, eine Zintl phase mit SiS2-isosterer anionen teilstruktur. Zeitschrift für Naturforschung B 1988, 43, 1538–1540. [Google Scholar]
- Kauzlarich, S.M. Chemistry, Structure and Bonding in Zintl Phases and Ions, 1st ed.; VCH: New York, USA, 1996. [Google Scholar]
- Schäfer, H.; Eisenmann, B.; Müller, W. Zintl Phases: Transitions between metallic and ionic bonding. Angew. Chem. Int. Ed. Engl. 1973, 12, 694–712. [Google Scholar] [CrossRef]
- Nesper, R. Bonding patterns in intermetallic compounds. Angew. Chem. Int. Ed. Engl. 1991, 30, 789–817. [Google Scholar] [CrossRef]
- Cordier, G.; Schäfer, H.; Stelter, M. Ca3AlSb3 und Ca5Al2Bi6, zwei neue Zintl phasen mit kettenförmigen anionen. Zeitschrift für Naturforschung B 1984, 39, 727–732. [Google Scholar] [CrossRef]
- Cordier, G.; Stelter, M.; Schäfer, H. Zintl phasen mit komplexen anionen: Zur kenntnis von Sr6Al2Sb6. J. Less-Common. Met. 1984, 98, 285–290. [Google Scholar] [CrossRef]
- Peters, K.; Carrillo-Cabrera, W.; Somer, M.; von Schnering, H.-G. Crystal structure of hexabarium di-μ-phosphido-bis(diphosphidogallate(III)), Ba6[Ga2P6]. Zeitschrift fur Kristallographie 1996, 211, 53. [Google Scholar] [CrossRef]
- Somer, M.; Carrillo-Cabrera, W.; Peters, K.; von Schnering, H.-G.; Cordier, G. Crystal structure of tribarium triarsenidoindate, Ba3InAs3. Zeitschrift fur Kristallographie 1996, 211, 632. [Google Scholar] [CrossRef]
- SMART & SAINT, Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- Sheldrick, G.M. SADABS, version 2.10; University of Göttingen: Göttingen, Germany, 2003.
- Sheldrick, G.M. SHELXTL, University of Göttingen: Göttingen, Germany, 2001.
- Gelato, L.M.; Parthé, E. Structure Tidy—A computer program to standardize crystal structure data. J. Appl. Crystallogr. 1987, 20, 139–146. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoyko, S.S.; Voss, L.H.; He, H.; Bobev, S. Synthesis, Crystal and Electronic Structures of the Pnictides AE3TrPn3 (AE = Sr, Ba; Tr = Al, Ga; Pn = P, As). Crystals 2015, 5, 433-446. https://doi.org/10.3390/cryst5040433
Stoyko SS, Voss LH, He H, Bobev S. Synthesis, Crystal and Electronic Structures of the Pnictides AE3TrPn3 (AE = Sr, Ba; Tr = Al, Ga; Pn = P, As). Crystals. 2015; 5(4):433-446. https://doi.org/10.3390/cryst5040433
Chicago/Turabian StyleStoyko, Stanislav S., Leonard H. Voss, Hua He, and Svilen Bobev. 2015. "Synthesis, Crystal and Electronic Structures of the Pnictides AE3TrPn3 (AE = Sr, Ba; Tr = Al, Ga; Pn = P, As)" Crystals 5, no. 4: 433-446. https://doi.org/10.3390/cryst5040433