
Two Closely Related Organic Charge-Transfer Complexes Based on Tetrathiafulvalene and 9H-fluorenone Derivatives. Competition between Hydrogen Bonding and Stacking Interactions


Abstract
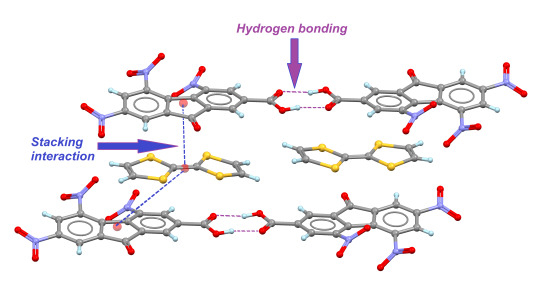
:
1. Introduction
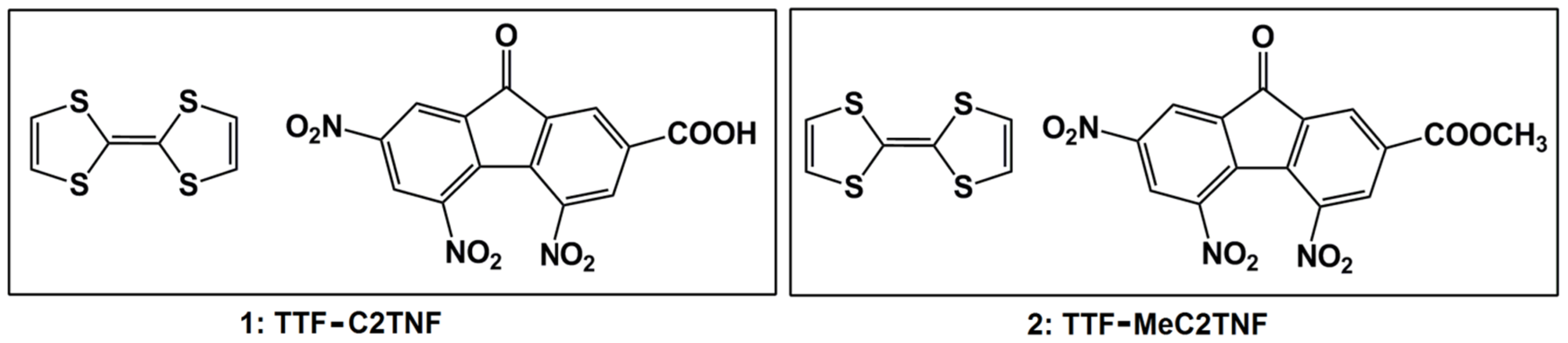
2. Results and Discussion
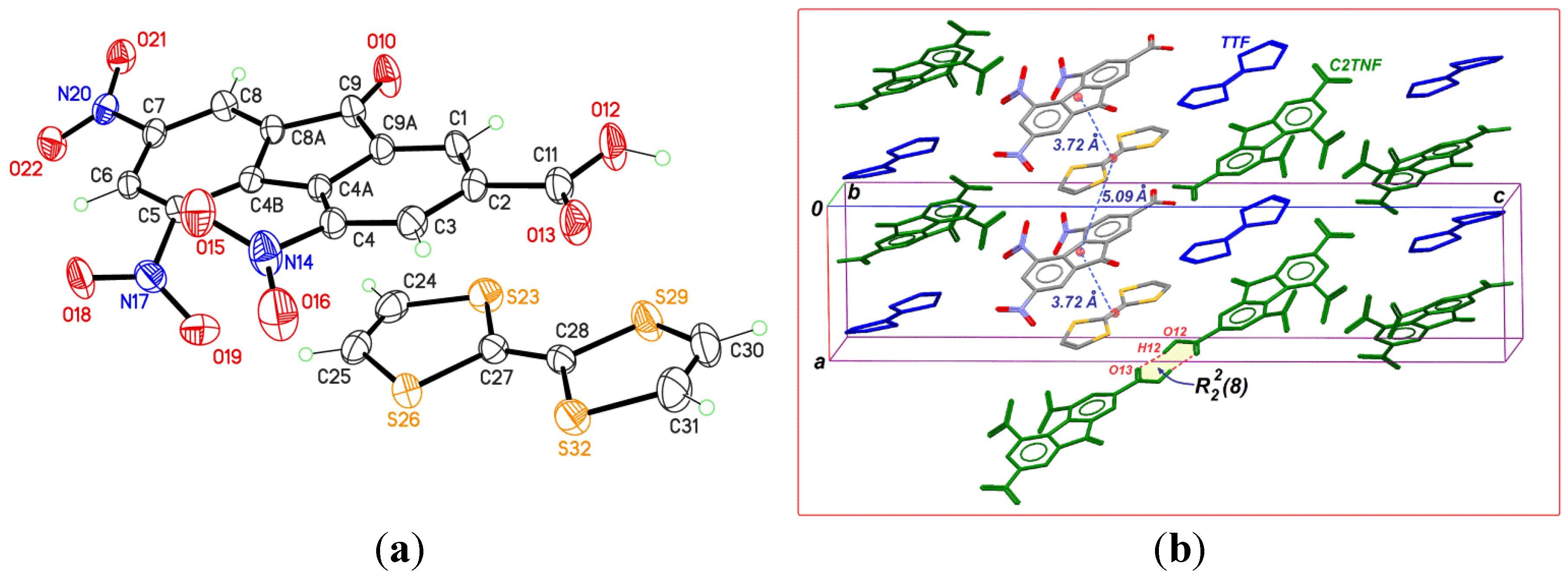
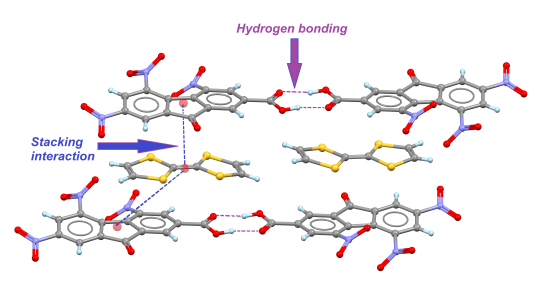
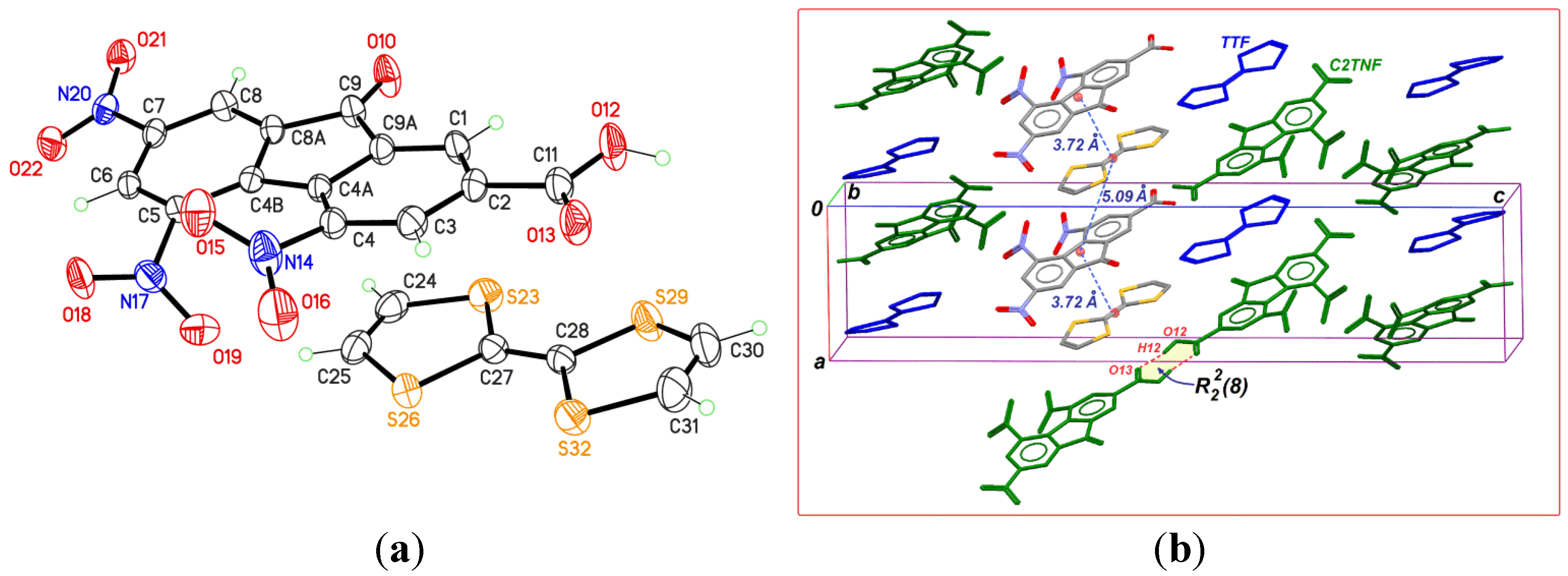
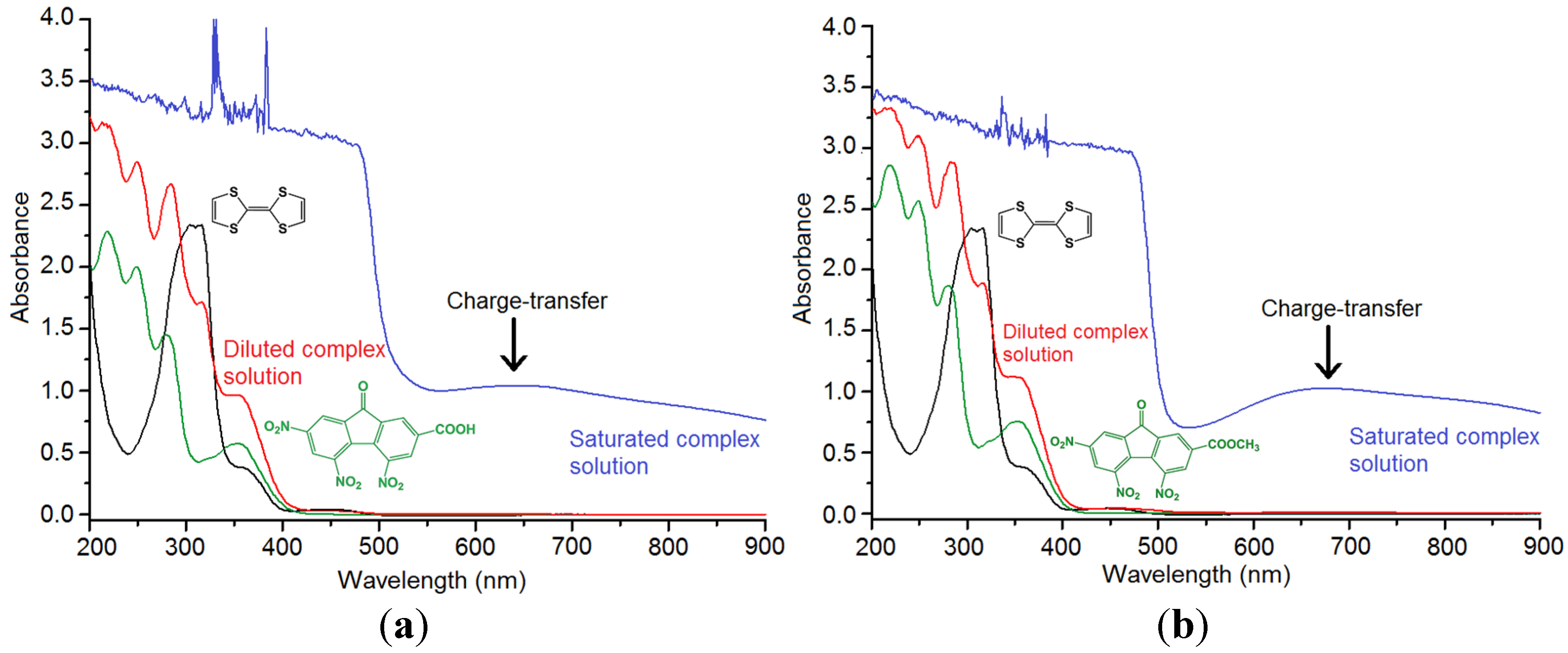
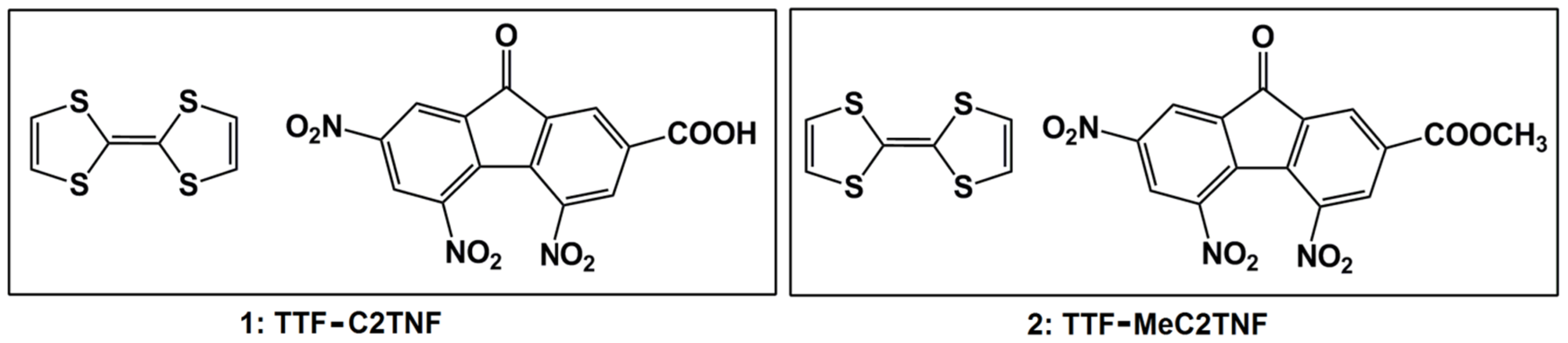
2.1. TTF-C2TNF (1)
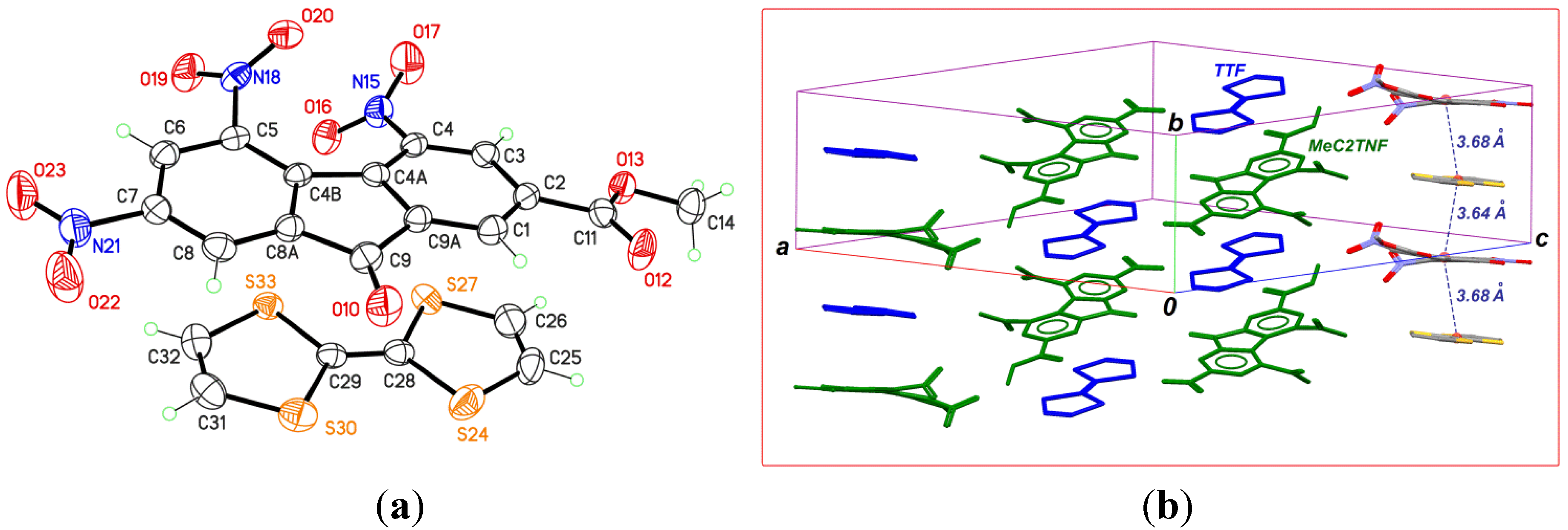
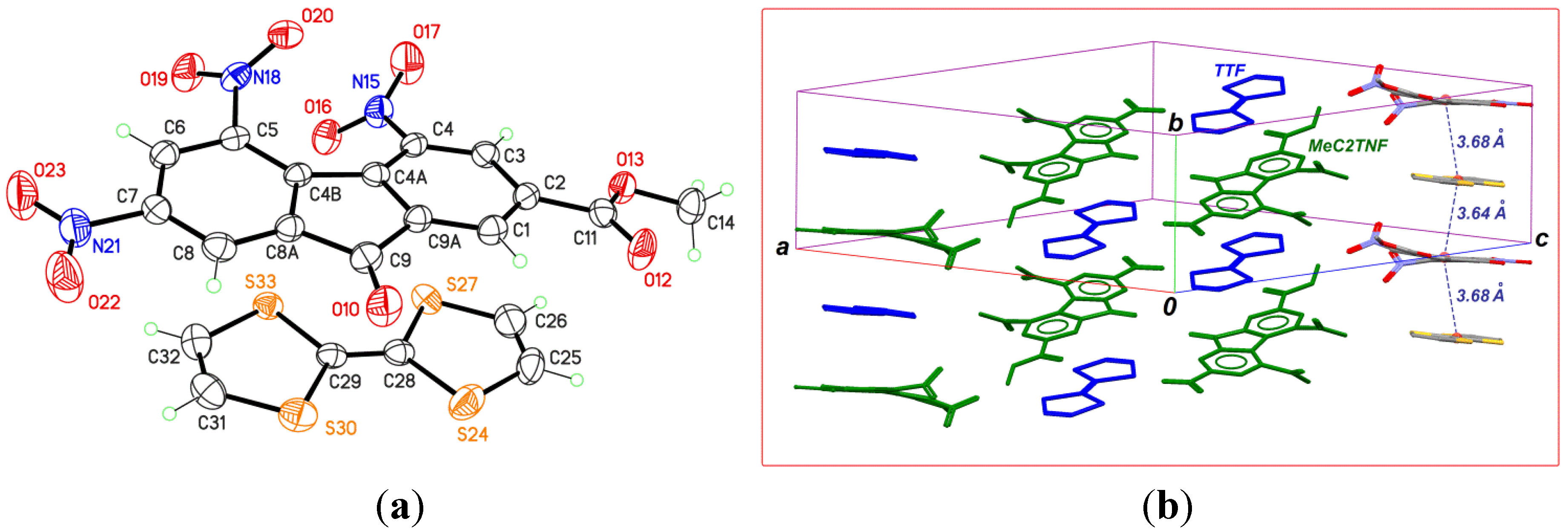
2.2. TTF-MeC2TNF (2)
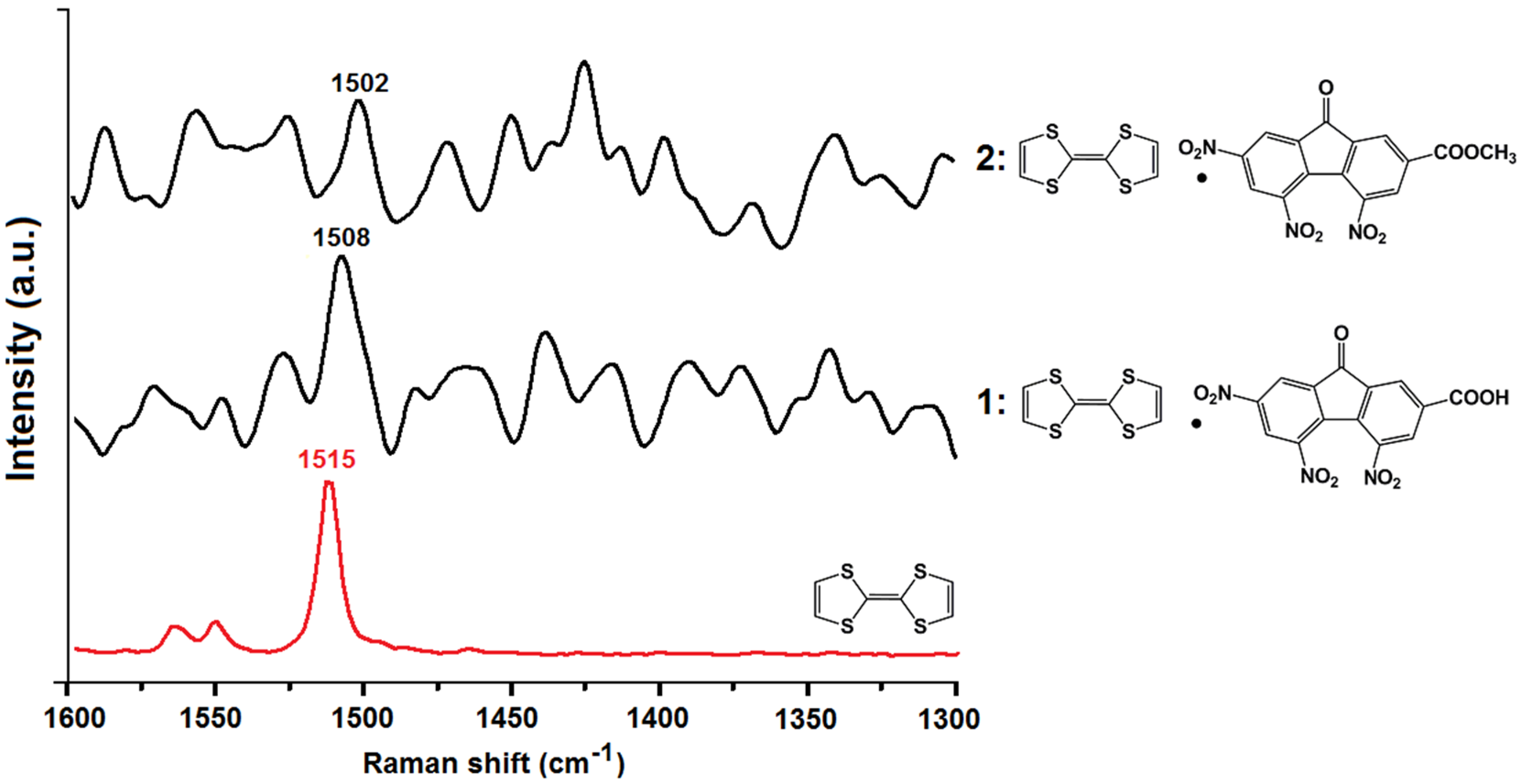
2.3. Vibrational Spectroscopy and Estimation of the Degree of Charge Transfer in 1 and 2
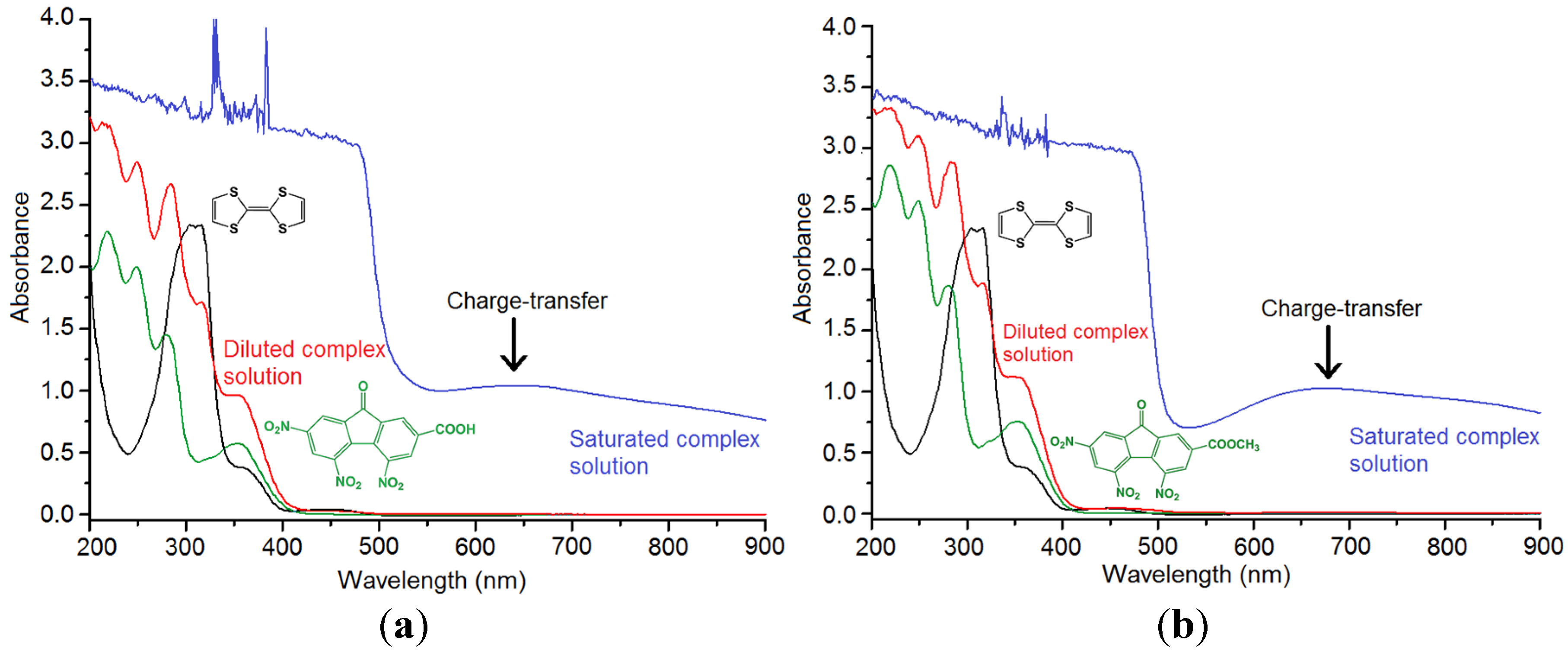
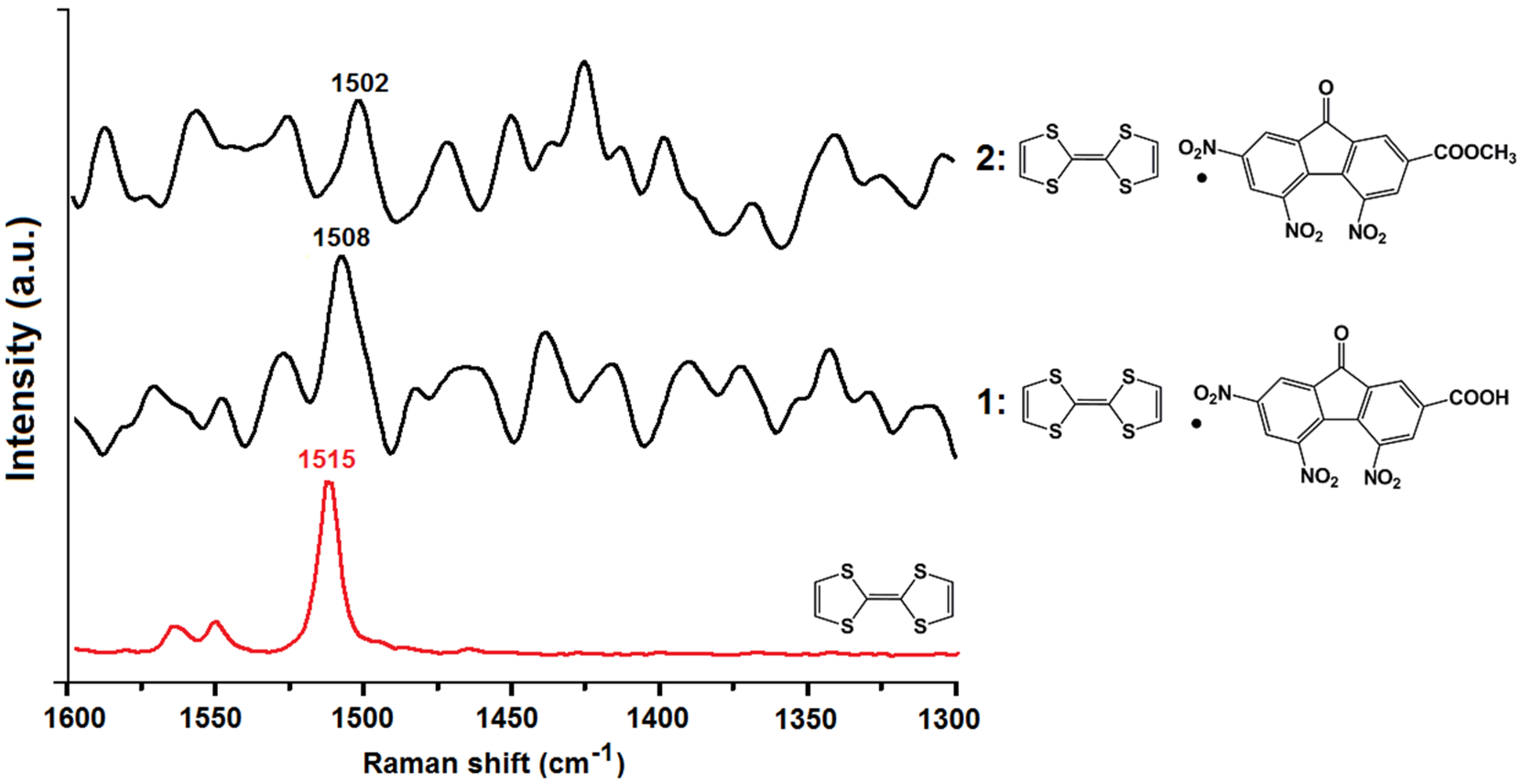
3. Experimental Section

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 |
|---|---|---|
| CCDC deposition | CCDC-1049471 | CCDC-1049472 |
| Empirical formula | C20H9N3O9S4 | C21H11N3O9S4 |
| Formula weight | 563.54 | 577.57 |
| Color, habit | Dark green, irregular | Dark brown, plate |
| Crystal size [mm] | 0.60 × 0.40 × 0.08 | 0.60 × 0.32 × 0.04 |
| Space group | P21/n | P21/n |
| a [Å] | 8.0275(10) | 19.078(5) |
| b [Å] | 7.8676(13) | 7.2068(14) |
| c [Å] | 34.961(5) | 19.443(4) |
| β [°] | 90.106(9) | 117.833(12) |
| V (Å3) | 2208.0(5) | 2364.0(9) |
| Z | 4 | 4 |
| ρcalcd. [g·cm−3] | 1.695 | 1.623 |
| μ [mm−1] | 0.492 | 0.462 |
| 2θ range [°] | 4–50 | 4–50 |
| Reflections collected | 5529 | 5681 |
| Independent reflections (Rint) | 3897 (0.026) | 4165 (0.033) |
| Transmission factors [min., max.] | 0.475–0.514 | 0.656–0.703 |
| Final R indices [I ≥ 2σ(I)] R1, wR2 | 0.049, 0.120 | 0.048, 0.097 |
| Final R indices (all data) R1, wR2 | 0.087, 0.146 | 0.106, 0.116 |
| Goodness-of-fit on F2 | 1.037 | 1.002 |
| Data/restraints/parameters | 3897/0/326 | 4165/0/336 |
| Largest difference peak/hole [e Å−3] | 0.61, −0.38 | 0.23, −0.24 |
4. Conclusions
Acknowledgments
Author Contributions
Supplementary Information
Conflicts of Interest
References
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, Oligoacenenes, and Their Buckminsterfullerene Derivatives: The Brick and Mortar of Organic Electronics. Chem. Rev. 2004, 104, 4891–4945. [Google Scholar] [CrossRef] [PubMed]
- McCoy, H.N.; Moore, W.C. Organic amalgams: Substances with metallic properties composed in part of non-metallic elements. J. Am. Chem. Soc. 1911, 33, 273–292. [Google Scholar] [CrossRef]
- Bechgaard, K.; Carneiro, K.; Rasmussen, F.B.; Olsen, M.; Rindorf, G.; Jacobsen, C.S.; Pedersen, H.J.; Scott, J.C. Superconductivity in an organic solid. Synthesis, structure, and conductivity of bis(tetramethyltetraselenafulvalenium) perchlorate, (TMTSF)2ClO4. J. Am. Chem. Soc. 1981, 103, 2440–2442. [Google Scholar] [CrossRef]
- Pouget, J.P. Structural aspects of the Bechgaard and Fabre salts: An update. Crystals 2012, 2, 466–520. [Google Scholar] [CrossRef]
- Chappell, J.S.; Bloch, A.N.; Bryden, W.A.; Maxfield, M.; Poehler, T.O.; Cowan, D.O. Degree of charge transfer in organic conductors by infrared absorption spectroscopy. J. Am. Chem. Soc. 1981, 103, 2442–2443. [Google Scholar] [CrossRef]
- Wheland, R.C.; Gillson, J.L. Synthesis of electrically conductive organic solids. J. Am. Chem. Soc. 1976, 98, 3916–3925. [Google Scholar] [CrossRef]
- Salmerón-Valverde, A.; Bernès, S.; Robles-Martínez, J.G. Structure and degree of charge transfer of two polymorphs of a 1:1 molecular complex: [2,2′-bis-1,3-dithiole][9-dicyanomethylene-4,5,7-trinitrofluorene-2-carboxylic acid methyl ester] (TTF-MeDC2TNF). Acta Crystallogr. 2003, B59, 505–511. [Google Scholar] [CrossRef]
- Salmerón-Valverde, A.; Bernès, S. Caractérisation structurale et étude spectroscopique du transfert de charge pour deux phases polymorphes du complexe (TTF)3(TNF)2, avec TTF = tétrathiofulvalène et TNF = 2,4,7-trinitro-9-fluorénone. C. R. Chim. 2005, 8, 1017–1023. (In French) [Google Scholar] [CrossRef]
- Ellern, A.; Bernstein, J.; Becker, J.Y.; Zamir, Sh.; Shahal, L.; Cohen, S. A New Polymorphic Modification of Tetrathiafulvalene. Crystal Structure, Lattice Energy and Intermolecular Interactions. Chem. Mater. 1994, 6, 1378–1385. [Google Scholar] [CrossRef]
- Batsanov, A.S. Tetrathiafulvalene revisited. Acta Crystallogr. 2006, C62, o501–o504. [Google Scholar]
- Chetkina, L.A.; Semidetko, O.V.; Belsky, V.K.; Sobolev, A.N.; Andrievsky, A.M. Structure of 2,4,5,7-tetranitro-9-fluorenone. Acta Crystallogr. 1987, C43, 931–933. [Google Scholar] [CrossRef]
- Baughman, R.G. The structure of 2,4,5,7-tetranitro-9-fluorenone. Acta Crystallogr. 1987, C43, 933–936. [Google Scholar] [CrossRef]
- Reinheimer, E.W.; Galán-Mascarós, J.R.; Dunbar, K.R. Synthesis and structure of charge transfer salts of tetrathiafulvalene (TTF) and tetramethyl-TTF with 2,4,7-trinitro and 2,4,5,7-tetranitro-9-fluorenone. Synth. Met. 2009, 159, 45–51. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Fábián, L.; Motherwell, W.D.S.; Allen, F.H.; Feeder, N. Knowledge-based model of hydrogen-bonding propensity in organic crystals. Acta Crystallogr. 2007, B63, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Jérome, D. Organic Conductors: From Charge Density Wave TTF−TCNQ to Superconducting (TMTSF)2PF6. Chem. Rev. 2004, 104, 5565–5592. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, J.E.; Wang, H.H.; Kini, A.M.; Schlueter, J.A. Assignment of the Raman spectra of some deuterated-BEDT-TTF superconductors. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2002, 58, 2237–2243. [Google Scholar] [CrossRef]
- Yakushi, K. Infrared and Raman Studies of Charge Ordering in Organic Conductors, BEDT-TTF Salts with Quarter-Filled Bands. Crystals 2012, 2, 1291–1346. [Google Scholar] [CrossRef]
- Van Duyne, R.P.; Cape, T.W.; Suchanski, M.R.; Siedle, A.R. Determination of the extent of charge transfer in partially oxidized derivatives of tetrathiafulvalene and tetracyanoquinodimethan by resonance Raman spectroscopy. J. Phys. Chem. 1986, 90, 739–743. [Google Scholar] [CrossRef]
- Clemente, D.A.; Marzotto, A. Structure of two polymorphs of the TTF-TCNE charge-transfer complex and the degree of ionicity. J. Mater. Chem. 1996, 6, 941–946. [Google Scholar] [CrossRef]
- Salmerón-Valverde, A. Síntesis y estudio del grado de transferencia de carga de complejos moleculares de tetratiafulvaleno (TTF) con derivados de fluoreno. Ph.D. Thesis, Universidad Autónoma de Puebla, 2008. [Google Scholar]
- Loufty, R.O.; Hsiao, C.K.; Ong, B.S.; Keoshkerian, B. Electrochemical evaluation of electron acceptor materials. Can. J. Chem. 1984, 62, 1877–1885. [Google Scholar]
- Wheland, R.C. Correlation of Electrical Conductivity in Charge-Transfer Complexes with Redox Potentials, Steric Factors, and Heavy Atom Effects. J. Am. Chem. Soc. 1976, 98, 3926–3930. [Google Scholar] [CrossRef]
- Sulzberg, T.; Cotter, R.J. Electron acceptors derived from fluorenecarboxylic acids and their charge-transfer complexes. J. Org. Chem. 1970, 35, 2762–2769. [Google Scholar] [CrossRef]
- Siemens. XSCANS; Siemens Analytical X-ray Instruments Inc.: Madison, WI, USA, 1996. [Google Scholar]
- North, A.C.T.; Phillips, D.C.; Mathews, F.S. A semi-empirical method of absorption correction. Acta Crystallogr. 1968, A24, 351–359. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Morita, Y.; Murata, T.; Nakasuji, K. Cooperation of Hydrogen-Bond and Charge-Transfer Interactions in Molecular Complexes in the Solid State. Bull. Chem. Soc. Jpn. 2013, 86, 183–197. [Google Scholar] [CrossRef]
- Fourmigué, M.; Batail, P. Activation of Hydrogen- and Halogen-Bonding Interactions in Tetrathiafulvalene-Based Crystalline Molecular Conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef] [PubMed]
- El-Ghayoury, A.; Mézière, C.; Simonov, S.; Zorina, L.; Cobián, M.; Canadell, E.; Rovira, C.; Náfrádi, B.; Sipos, B.; Forró, L.; et al. A Neutral Zwitterionic Molecular Solid. Chem. Eur. J. 2010, 16, 14051–14059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isono, T.; Kamo, H.; Ueda, A.; Takahashi, K.; Nakao, A.; Kumai, R.; Nakao, H.; Kobayashi, K.; Murakami, Y.; Mori, H. Hydrogen bond-promoted metallic state in a purely organic single-component conductor under pressure. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmerón-Valverde, A.; Bernès, S. Two Closely Related Organic Charge-Transfer Complexes Based on Tetrathiafulvalene and 9H-fluorenone Derivatives. Competition between Hydrogen Bonding and Stacking Interactions. Crystals 2015, 5, 283-293. https://doi.org/10.3390/cryst5030283
Salmerón-Valverde A, Bernès S. Two Closely Related Organic Charge-Transfer Complexes Based on Tetrathiafulvalene and 9H-fluorenone Derivatives. Competition between Hydrogen Bonding and Stacking Interactions. Crystals. 2015; 5(3):283-293. https://doi.org/10.3390/cryst5030283
Chicago/Turabian StyleSalmerón-Valverde, Amparo, and Sylvain Bernès. 2015. "Two Closely Related Organic Charge-Transfer Complexes Based on Tetrathiafulvalene and 9H-fluorenone Derivatives. Competition between Hydrogen Bonding and Stacking Interactions" Crystals 5, no. 3: 283-293. https://doi.org/10.3390/cryst5030283