
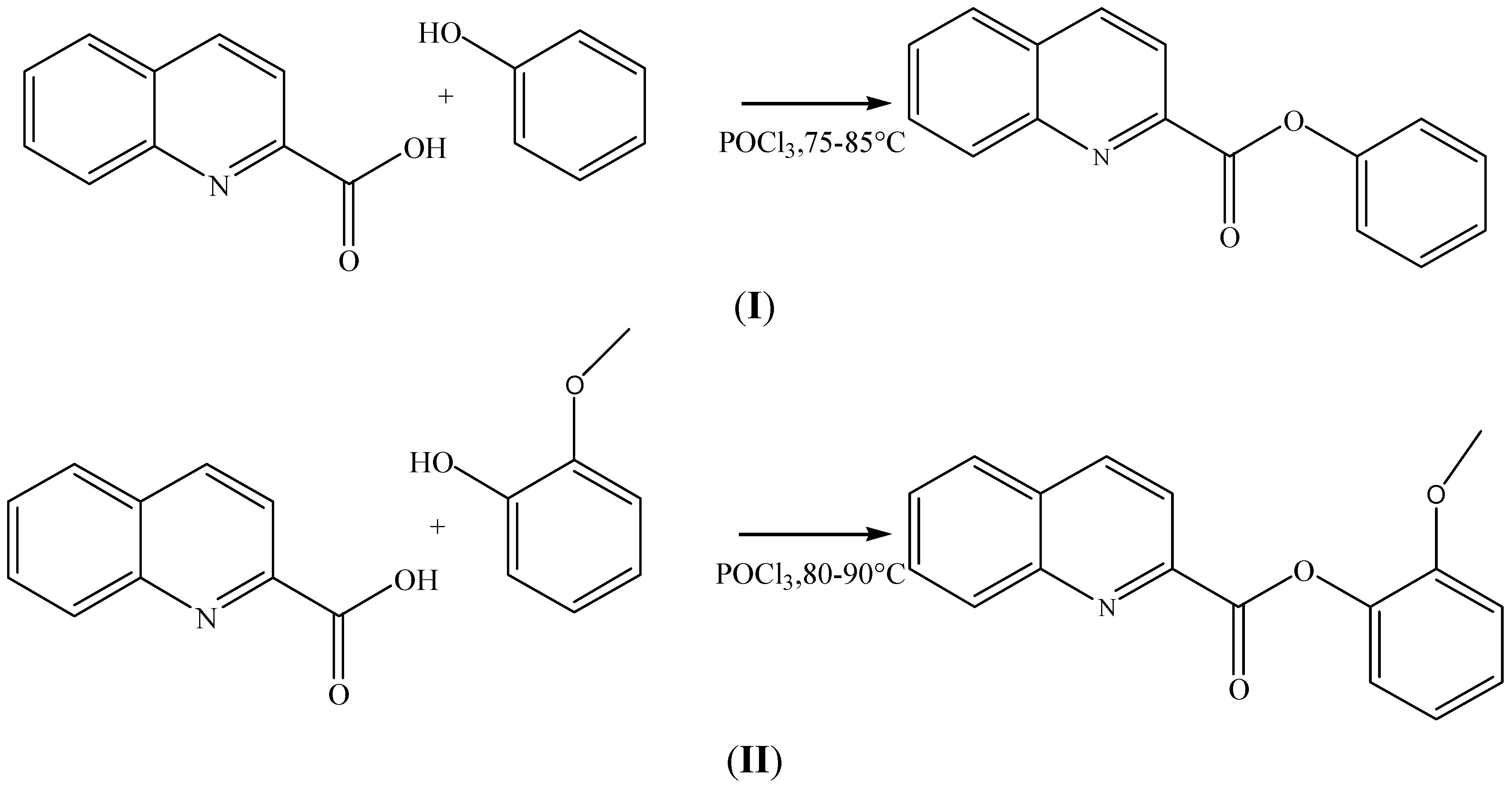
Synthesis, Crystal and Molecular Structure Studies and DFT Calculations of Phenyl Quinoline-2-Carboxylate and 2-Methoxyphenyl Quinoline-2-Carboxylate; Two New Quinoline-2 Carboxylic Derivatives

Abstract
:1. Introduction
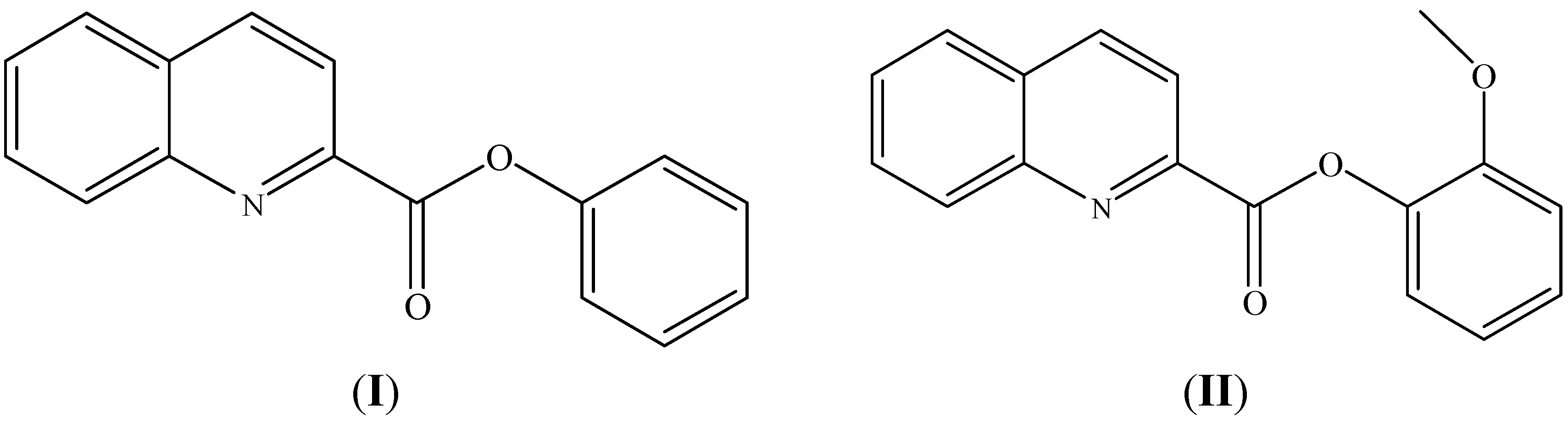
2. Results and Discussion
2.1. Structural Study of (I): Phenyl Quinoline-2-Carboxylate
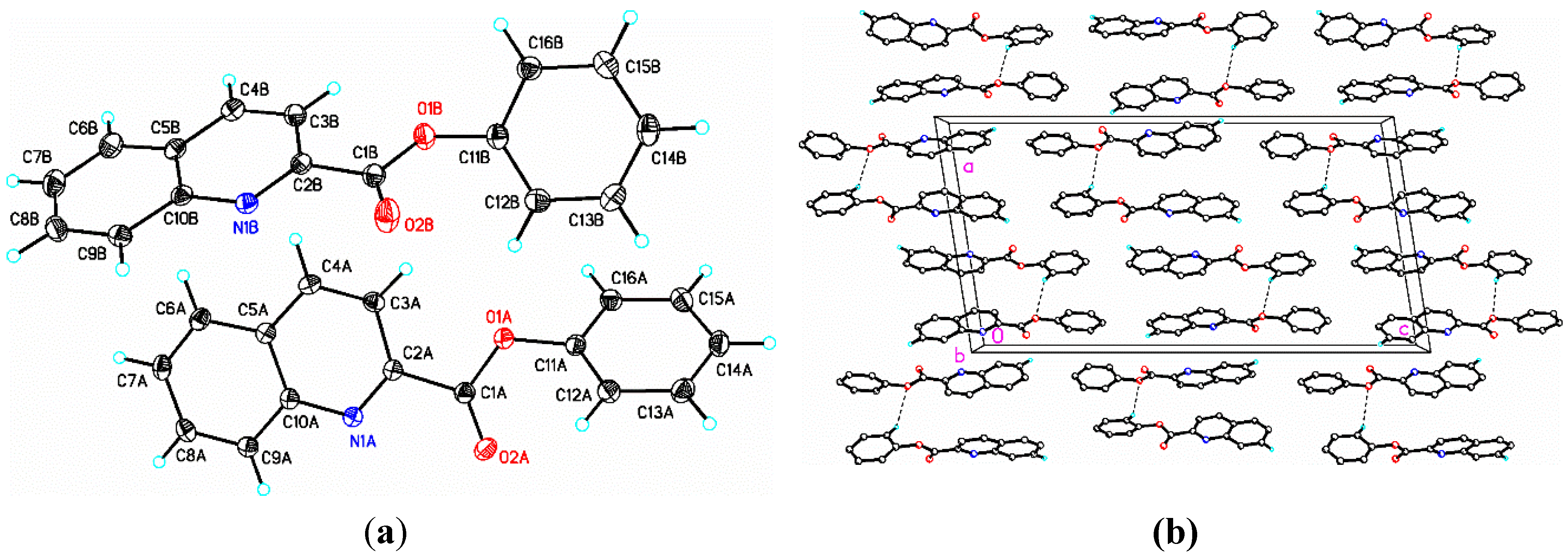

{kind=link}
{kind=link}
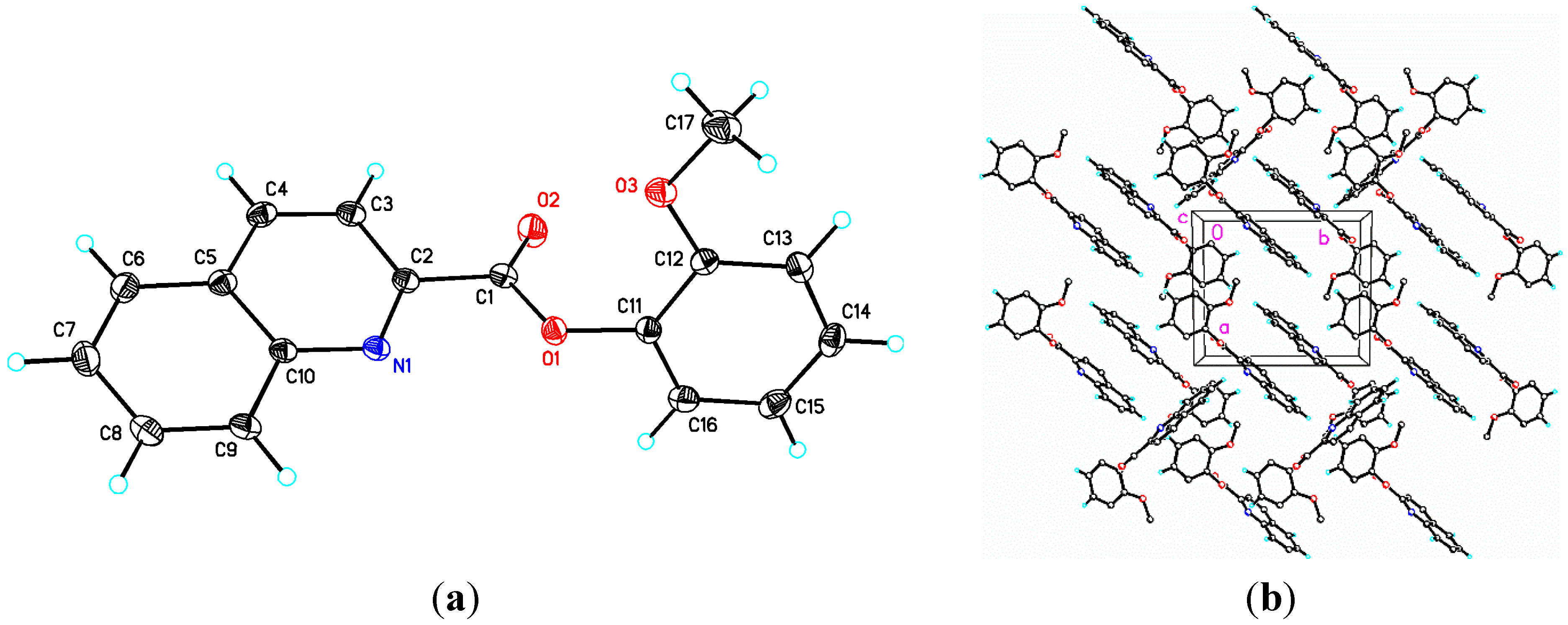{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms(I) | Distance(I) | DFT(I) | Atoms(I) | Distance(I) | DFT(I) |
|---|---|---|---|---|---|
| O1A–C1A | 1.354(3) | *1.373 | O2A–C1A | 1.192(3) | *1.204 |
| O1A–C11A | 1.408(2) | *1.397 | N1A–C2A | 1.313(3) | *1.321 |
| N1A–C10A | 1.369(3) | *1.359 | C1A–C2A | 1.510(3) | *1.509 |
| C2A–C3A | 1.416(3) | *1.422 | C3A–C4A | 1.362(3) | *1.374 |
| C6A–C7A | 1.361(3) | *1.377 | C8A–C9A | 1.373(3) | *1.375 |
| C9A–C10A | 1.414(3) | *1.422 | C12A–C13A | 1.384(3) | *1.395 |
| C14A–C15A | 1.390(3) | *1.396 | O1B–C1B | 1.354(3) | *1.397 |
| O2B–C1B | 1.197(3) | *1.204 | O1B–C11B | 1.410(2) | *1.397 |
| N1B–C2B | 1.316(3) | *1.321 | N1B–C10B | 1.369(3) | *1.359 |
| C13B–C14B | 1.387(4) | *1.396 | C15B–C16B | 1.389(3) | *1.395 |
| Atoms(I) | Angles(I) | DFT(I) | Atoms(I) | Angles(I) | DFT(I) |
| C1A–O1A–C11A | 120.80(16) | *121.01 | C2A–N1A–C10A | 117.27(18) | *118.24 |
| O1A–C1A–O2A | 124.48(19) | *124.54 | O1A–C1A–C2A | 109.42(17) | *110.02 |
| O2A–C1A–C2A | 126.08(19) | *126.44 | N1A–C2A–C1A | 114.84(18) | *114.75 |
| N1A–C2A–C3A | 124.48(18) | *123.74 | C3A–C2A–C1A | 120.65(18) | *121.51 |
| C4A–C3A–C2A | 118.52(19) | *118.54 | C3A–C4A–C5A | 119.70(19) | *119.76 |
| C4A–C5A–C6A | 123.30(19) | *123.68 | C7A–C6A–C5A | 120.2(2) | *120.30 |
| C9A–C8A–C7A | 120.9(3) | *120.57 | N1A–C10A–C5A | 122.32(18) | *122.55 |
| N1A–C10A–C9A | 118.32(19) | *118.31 | C9A–C10A–C5A | 119.36(18) | *119.14 |
| C12A–C11A–O1A | 122.01(19) | *122.70 | C12A–C11A–C16A | 122.26(19) | *121.25 |
| C16A–C11A–O1A | 115.47(18) | *115.93 | C11A)–12A–C13A | 118.5(3) | *118.65 |
| C16A–C15A–C14A | 120.3(2) | *120.16 | C1B–O1B–C11B | 119.21(16) | *121.01 |
| C2B–N1B–C10B | 117.52(18) | *118.24 | O2B–C1B–O1B | 124.19(19) | *125.44 |
| O1B–C1B–C2B | 109.75(79) | *110.02 | O2B–C1B–C2B | 126.05192) | *126.44 |
| N1B–C2B–C1B | 114.12(18) | *114.75 | N1B–C2B–C3B | 124.43(19) | *123.74 |
| C8B–C9B–C10B | 120.1(2) | *120.26 | N1B–C10B–C5B | 121.97(19) | *122.55 |
| N1B–C10B–C9B | 118.69(19) | *118.31 | C12B–C11B–O1B | 120.97(19) | *122.70 |
| Atoms(I) | Torsion(I) | DFT(I) | Atoms(I) | Torsion(I) | DFT(I) |
| O1A–C1A–C2A–N1A | 175.65(17) | *178.71 | O1A–C1A–C2A–C3A | −2.9(3) | *−1.51 |
| O2A–C1A–C2A–N1A | −2.8(3) | *1.07 | O2A–C1A–C2A–C3A | 178.7(2) | *178.71 |
| O1A–C11A–C12A–C13A | −174.10(19) | *−176.19 | O1A–C11A–C16A–C15A | 175.39(18) | *176.26 |
| N1A–C2A–C3A–C4A | 0.8(3) | *0.11 | C1A–O2A–C11A–C12A | −57.0(3) | *−47.75 |
| C1A–O2A–C11A–C16A | 128.7(2) | *136.27 | C2A–N1A–C10A–C9A | 178.75(17) | *179.87 |
| C4A–C5A–C10A–N1A | 0.6(3) | *0.03 | C10A–N1A–C2A–C1A | −178.67(16) | *−170.93 |
| O1B–C1B–C2B–N1B | 175.70(17) | *178.71 | O1B–C1B–C2B–C3B | −4.5(3) | *−1.51 |
| O2B–C1B–C2B–N1B | −4.1(3) | *−1.07 | O2B–C1B–C2B–C3B | 175.7(2) | *178.71 |
| O1B–C11B–C12B–C13B | 175.20(18) | *176.19 | O1B–C11B–C16B–C15B | −176.16(18) | *−176.19 |
| C4B–C5B–C10B–N1B | −2.4(3) | *−0.03 | C10B–N1B–C2B–C3B | 1.5(3) | *0.15 |
| D---H | d(D---H) | d(H…A) | d(D…A) | <(DHA) |
|---|---|---|---|---|
| C8A---H8A…O2A #1 | 0.95 | 2.64 | 3.418(3) | 139.6 |
| C8B---H8B…O2B #2 | 0.95 | 2.95 | 3.38132) | 141.5 |
| C12B---H12B…O1A | 0.95 | 2.69 | 3.498(3) | 143.5 |
| Cg1---Cg1 #3 | – | – | – | 3.8913(11) |
| Cg1---Cg2 #3 | – | – | – | 3.7842(12) |
| Cg5---Cg5 #4 | – | – | – | 3.8576(11) |
| Cg5---Cg6 #4 | – | – | – | 3.9654(12) |
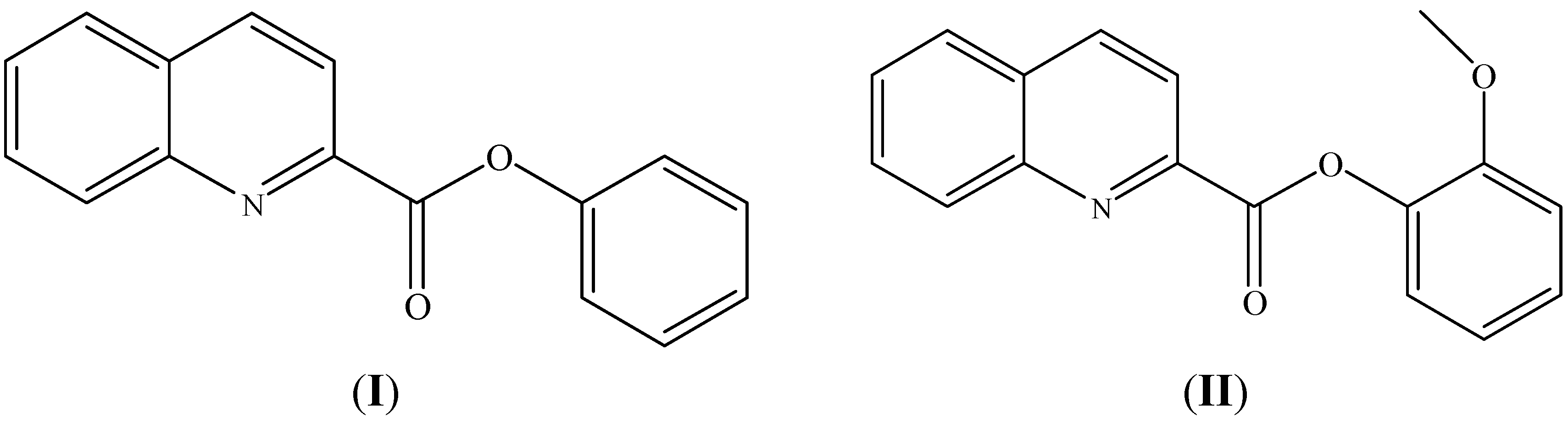
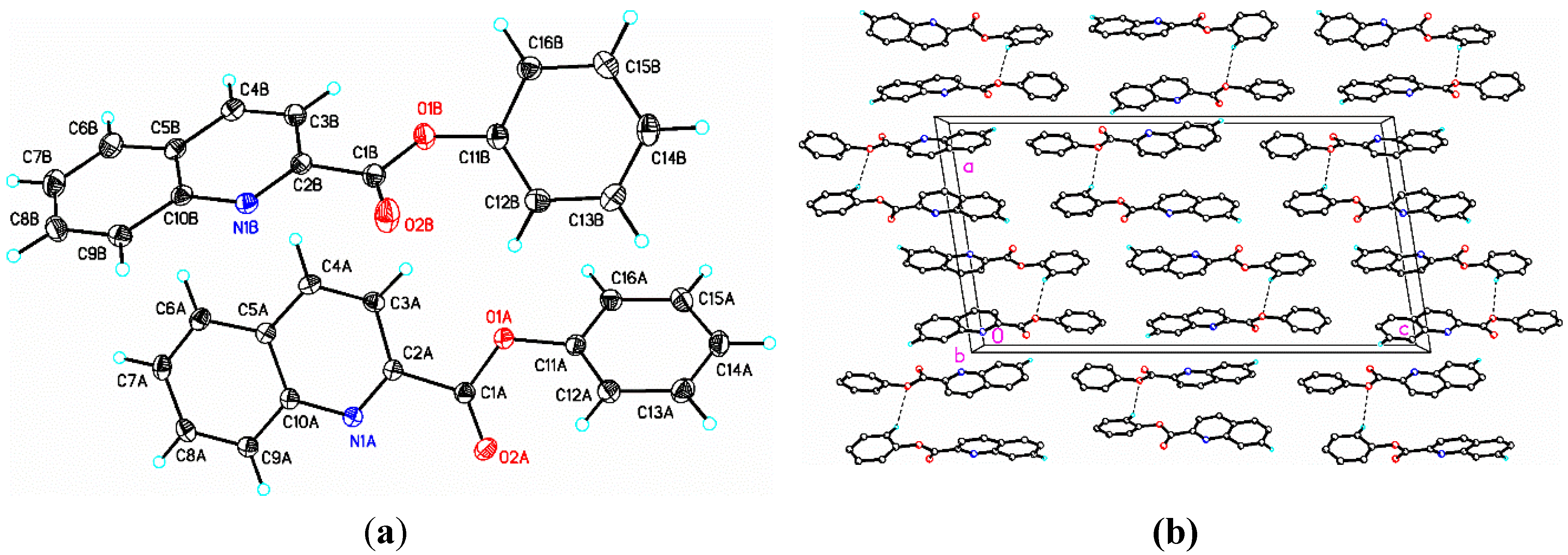
2.2. Structural Study of (II): 2-Methoxyphenyl Quinoline-2-Carboxylate
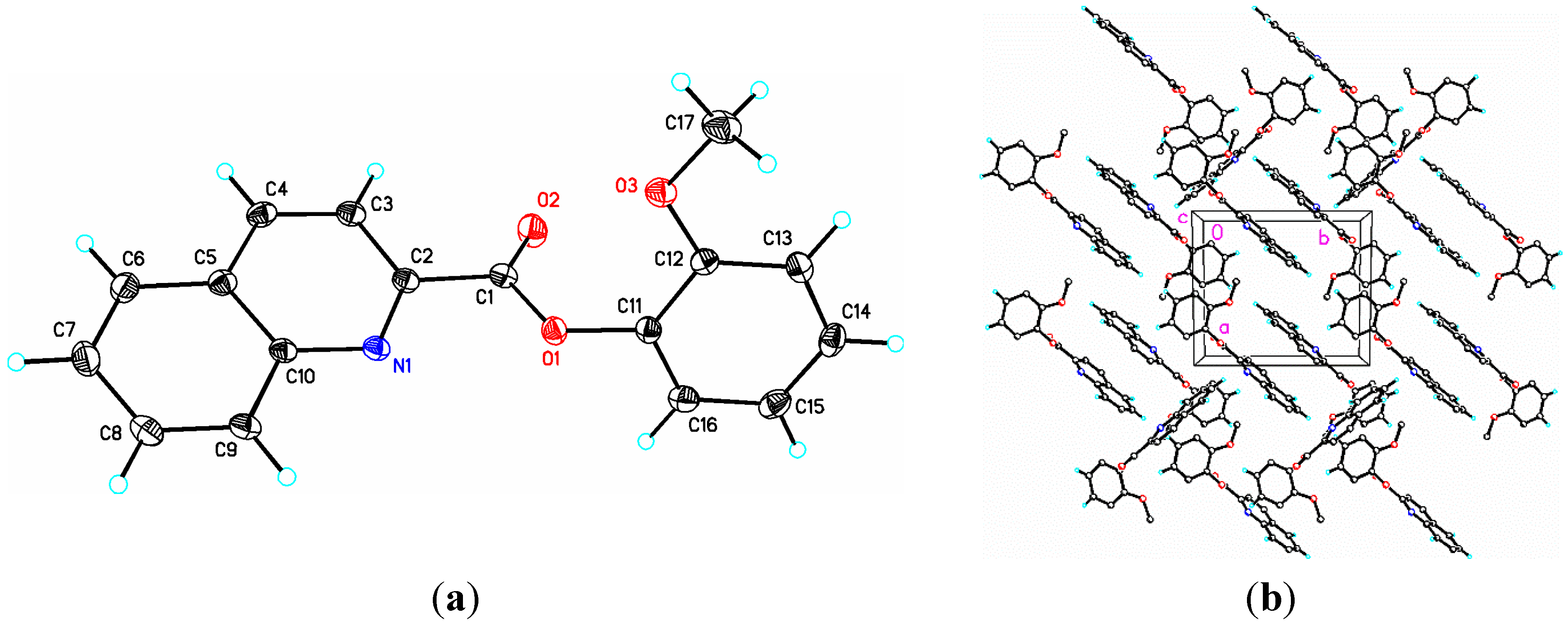
| Atoms(II) | Distance(II) | DFT(II) | Atoms(II) | Distance(II) | DFT(II) |
|---|---|---|---|---|---|
| O2–C1 | 1.1997(16) | *1.210 | O1–C1 | 1.3494(15) | *1.358 |
| O1–C11 | 1.4062(15) | *1.358 | O3–C12 | 1.3626(16) | *1.360 |
| O3–C17 | 1.4283(17) | *1.419 | N1–C2 | 1.3183(16) | *1.320 |
| N1–C10 | 1.3668(16) | *1.361 | C1–C2 | 1.5021(18) | *1.504 |
| C2–C3 | 1.4131(18) | *1.421 | C3–C4 | 1.3596(19) | *1.373 |
| C4–C5 | 1.4140(19) | *1.418 | C5–C6 | 1.4178(19) | *1.419 |
| C5–C10 | 1.4194(18) | *1.433 | C6–C7 | 1.363(2) | *1.377 |
| C7–C8 | 1.412(2) | *1.419 | C8–C9 | 1.3660(2) | *1.376 |
| C9–C10 | 1.4197(18) | *1.422 | C11–C12 | 1.3950(18) | *1.407 |
| C11–C16 | 1.3757(19) | *1.366 | C12–C13 | 1.3930(18) | *1.398 |
| C13–C14 | 1.387(2) | *1.399 | C14–C15 | 1.379(2) | *1.392 |
| Atoms(II) | Angles(II) | DFT(2) | Atoms(II) | Angles(II) | DFT(II) |
| C1–O1–C11 | 117.72(10) | *117.76 | C2–N1–C10 | 117.26(11) | *117.91 |
| O1–C1–O2 | 124.39(12) | *124.52 | O1–C1–C2 | 111.51(10) | *112.25 |
| O2–C1–C2 | 124.39(12) | *123.22 | N1–C2–C1 | 117.83(11) | *118.95 |
| N1–C2–C3 | 124.33(12) | *124.10 | C3–C2–C1 | 117.82(11) | *116.95 |
| C4–C3–C2 | 118.66(12) | *118.54 | C4–C5–C6 | 123.26(12) | *123.56 |
| C4–C5–C10 | 117.54(12) | *117.30 | C6–C5–C1 | 119.18(12) | *119.14 |
| C8–C9–C10 | 120.29(12) | *120.36 | N1–C10–C5 | 122.69(11) | *122.58 |
| N1–C10–C9 | 118.33(11) | *118.31 | C12–C11–O1 | 119.51(11) | *119.76 |
| C16–C11–O1 | 118.66(12) | *118.90 | C2–N1–C10 | 117.26(11) | *117.91 |
| Atoms(II) | Torsion(II) | DFT(II) | Atoms(II) | Torsion(II) | DFT(II) |
| O1–C1–C2–N1 | −13.46(16) | *−0.14 | O1–C1–C2–C3 | 164.87(11) | *179.72 |
| O2–C1–C2–N1 | 167.42(13) | *179.29 | O2–C1–C2–C3 | −14.25(19) | *−0.56 |
| O2–C1–O1–C11 | 1.74(19) | *4.08 | O1–C11–C16–C15 | −173.64(12) | *−176.06 |
| N1–C2–C3–C4 | 0.5(2) | *0.15 | C1–O1–C11–C12 | −104.49(14) | *−72.68 |
| C1–O1–C11–C16 | −104.49(14) | *−111.70 | C2–N1–C10–C5 | 0.68(17) | *0.07 |
| C2–N1–C10–C9 | −177.81(11) | *−180 | C4–C5–C10–N1 | 0.30(18) | *0.11 |
| C11–O1–C1–O2 | 1.74(19) | *4.08 | C11–O1–C1–C2 | 1.74(19) | *176.78 |
| D---H | d(D---H) | d(H…A) | d(D…A) | <(DHA) |
|---|---|---|---|---|
| C9---H9…O2 #1 | 0.95 | 2.61 | 3.4207(16) | 143.8 |
| Cg1---Cg1 #2 | – | – | – | 3.7719(7) |
| Cg1---Cg2 #2 | – | – | – | 3.6878(8) |
2.3. Computational Details
2.4. Theoretical Density Functional Theory (DFT) Calculations for (I) and (II)
| λmax (nm/eV) | Experimental (MO Contributions) | f* | λmax (nm/eV) | Calculated (MO Contributions) |
|---|---|---|---|---|
| 322/3.85 | HOMO→LUMO | 0.87 | 267/4.64 | HOMO→LUMO |
| 322/3.85 | HOMO−1→LUMO | 0.87 | 261/4.75 | HOMO−1→LUMO |
| 322/3.85 | HOMO→LUMO+1 | 0.87 | 224/5.54 | HOMO→LUMO+1 |
| 322/3.85 | HOMO−1→LUMO+1 | 0.87 | 219/5.66 | HOMO−1→LUMO+1 |
| 322/3.85 | HOMO→LUMO+2 | 0.87 | 193/6.42 | HOMO→LUMO+2 |
| 322/3.85 | HOMO→LUMO+3 | 0.87 | 190/6.53 | HOMO−1→LUMO+2 |
| 322/3.85 | HOMO−1→LUMO+2 | 0.87 | 189/6.56 | HOMO→LUMO+3 |
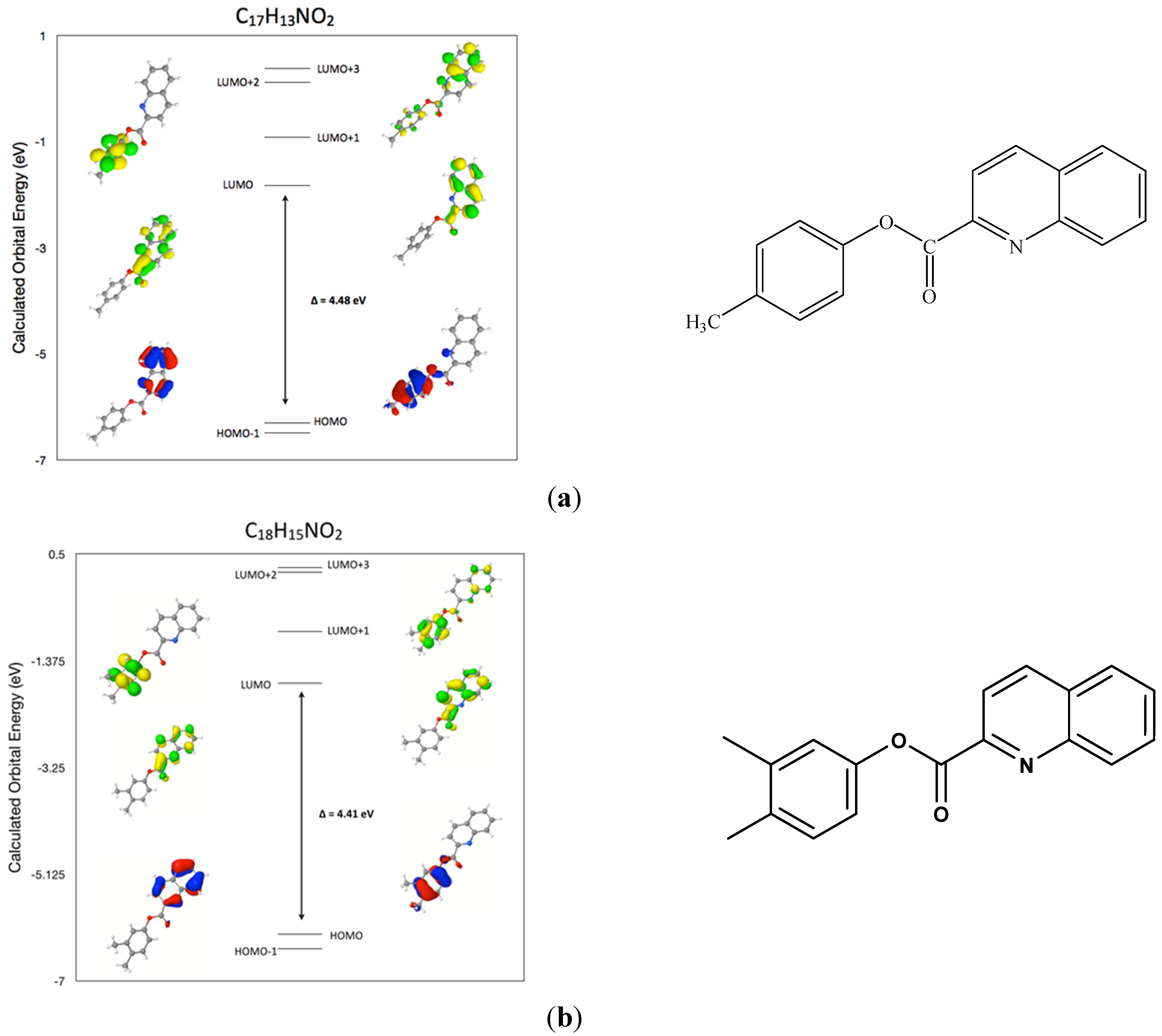
2.5. DFT Frontier Molecular Orbitals for (I)
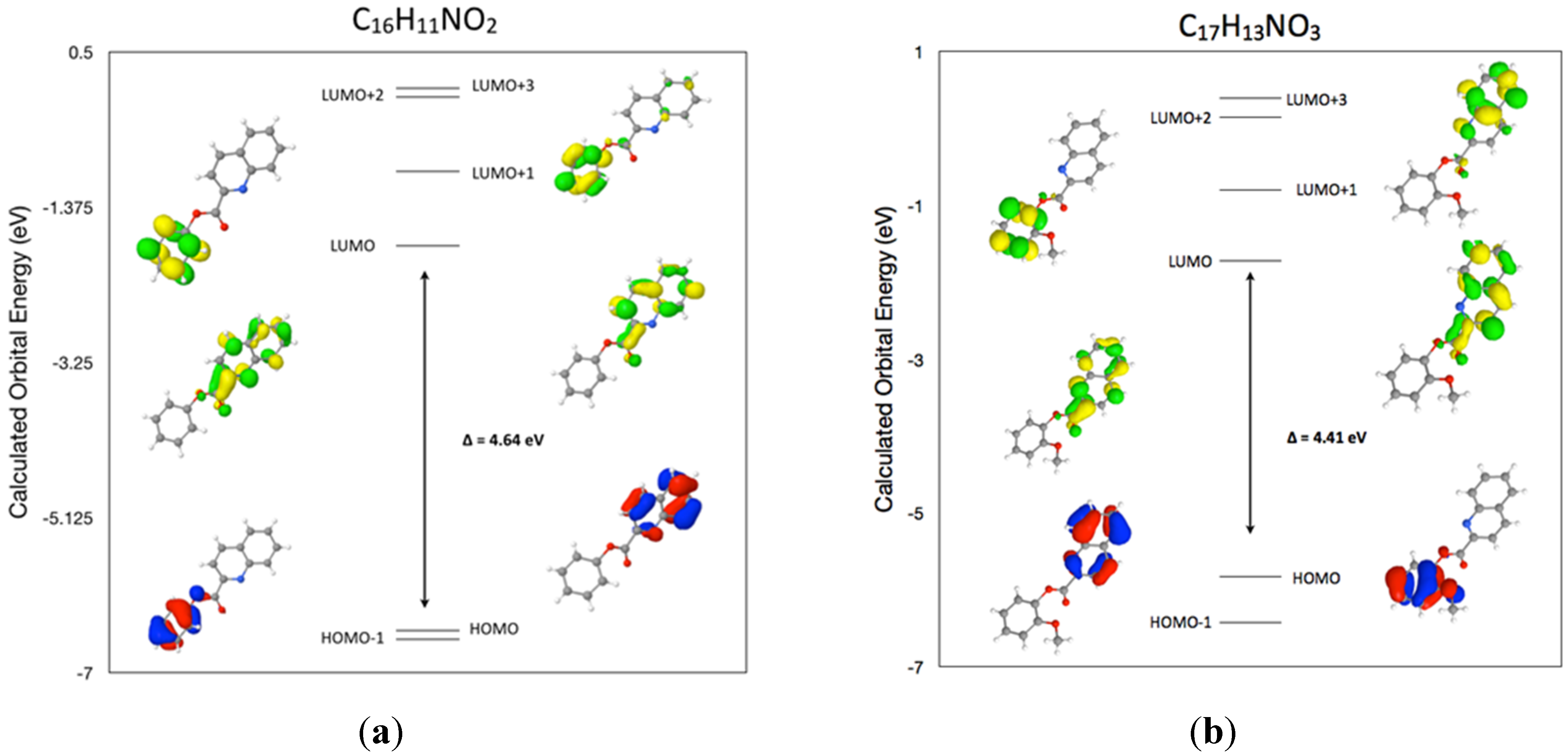
2.6. DFT Frontier Molecular Orbitals for (II)
| λmax (nm/eV) | Experimental (MO Contributions) | f* | λmax (nm/eV) | Calculated (MO Contributions) |
|---|---|---|---|---|
| 319/3.88 | HOMO→LUMO | 1.97 | 302/4.11 | HOMO→LUMO |
| 319/3.88 | HOMO−1→LUMO | 1.97 | 263/4.71 | HOMO−1→LUMO |
| 319/3.88 | HOMO→LUMO+1 | 1.97 | 246/5.04 | HOMO→LUMO+1 |
| 254/4.88 | HOMO−1→LUMO+1 | 0.72 | 220/5.64 | HOMO−1→LUMO+1 |
| 254/4.88 | HOMO→LUMO+2 | 0.72 | 207/5.99 | HOMO→LUMO+2 |
| 254/4.88 | HOMO→LUMO+3 | 0.72 | 199/6.23 | HOMO→LUMO+3 |
| 254/4.88 | HOMO−1→LUMO+2 | 0.72 | 189/6.56 | HOMO−1→LUMO+2 |
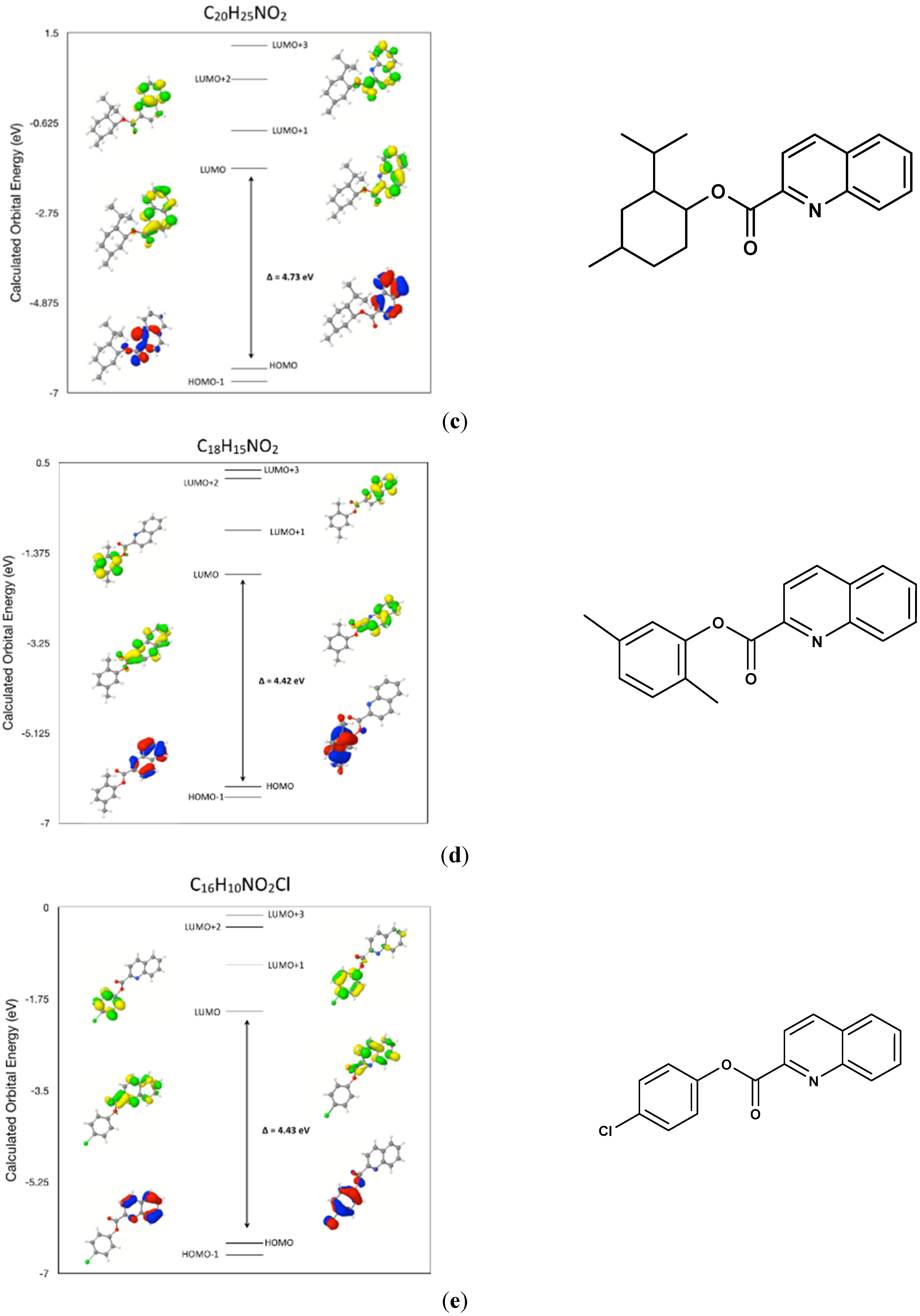
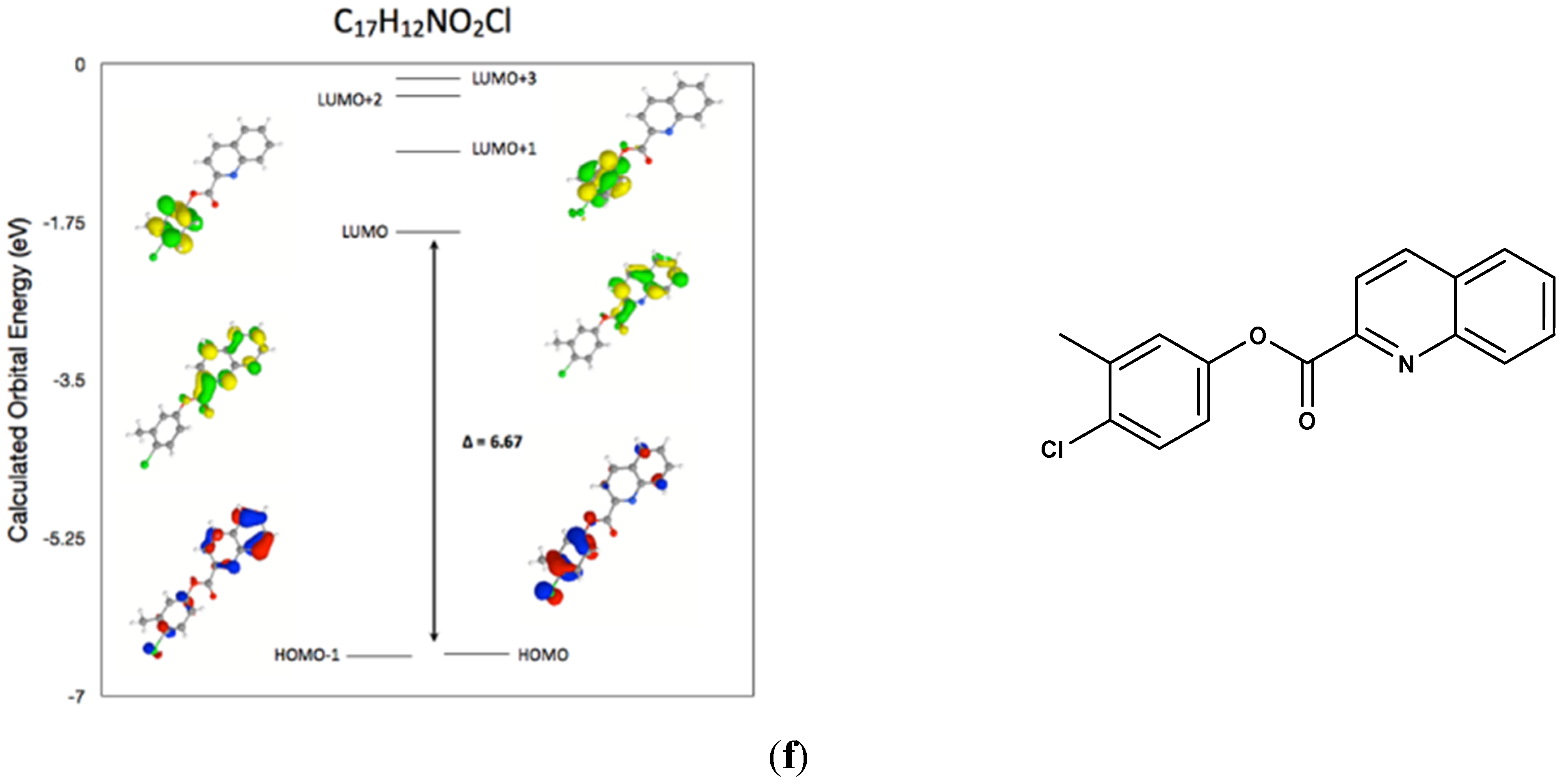
2.7. Comparison of the Frontier Molecular Orbitals from Six Related Quinoline-2-Carboxylate Derivatives to the Title Compounds
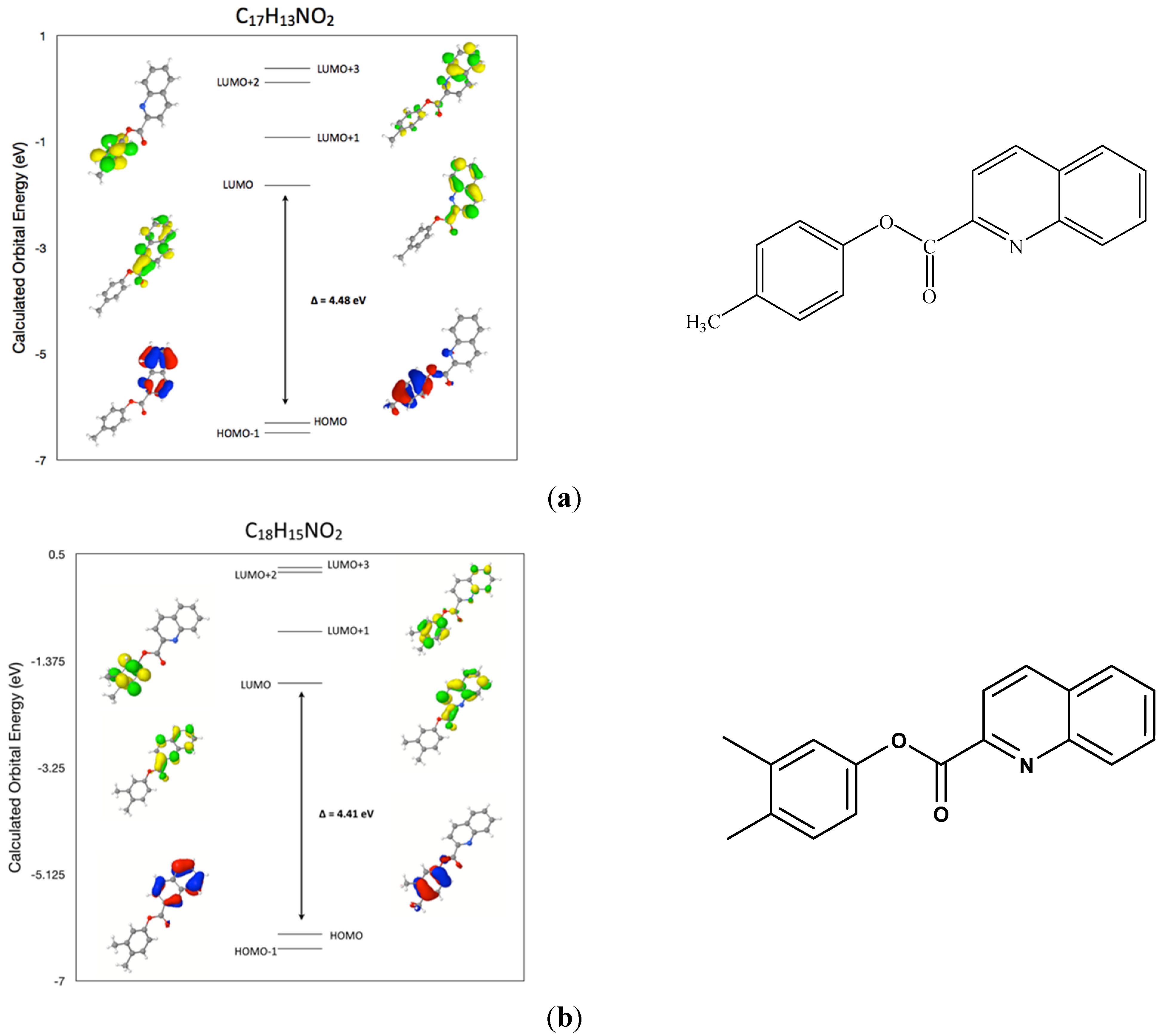
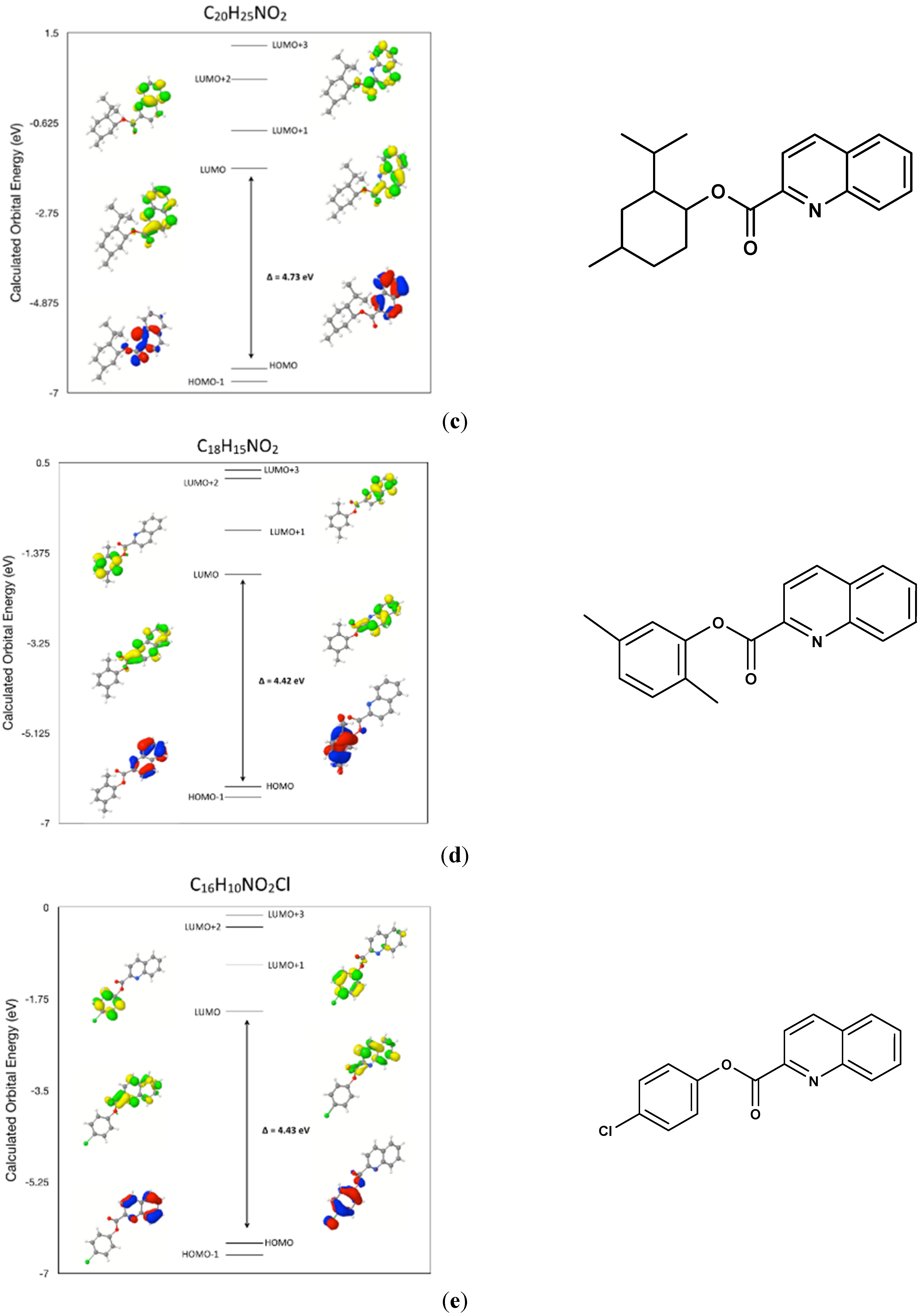
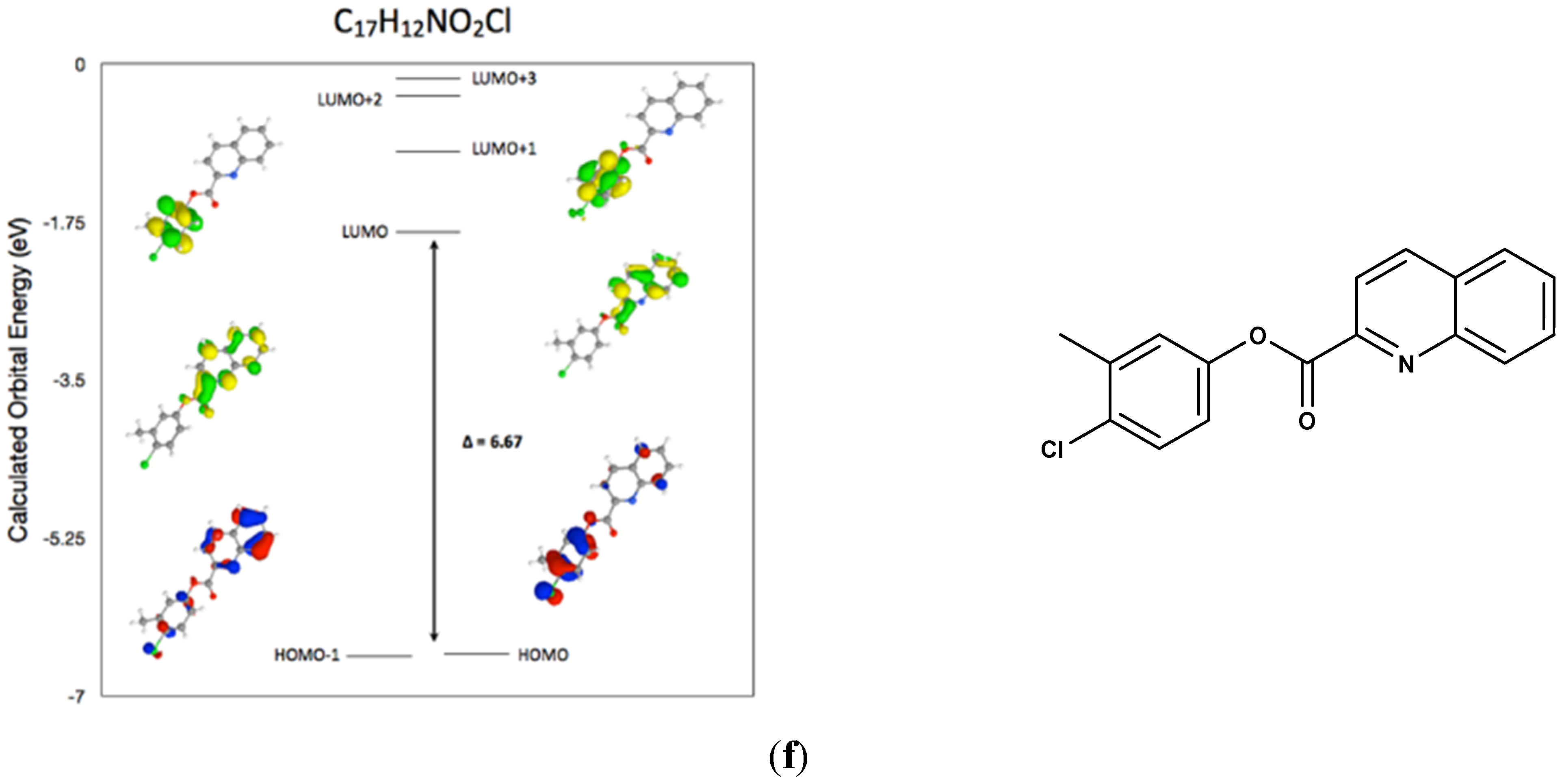
3. Experimental Section
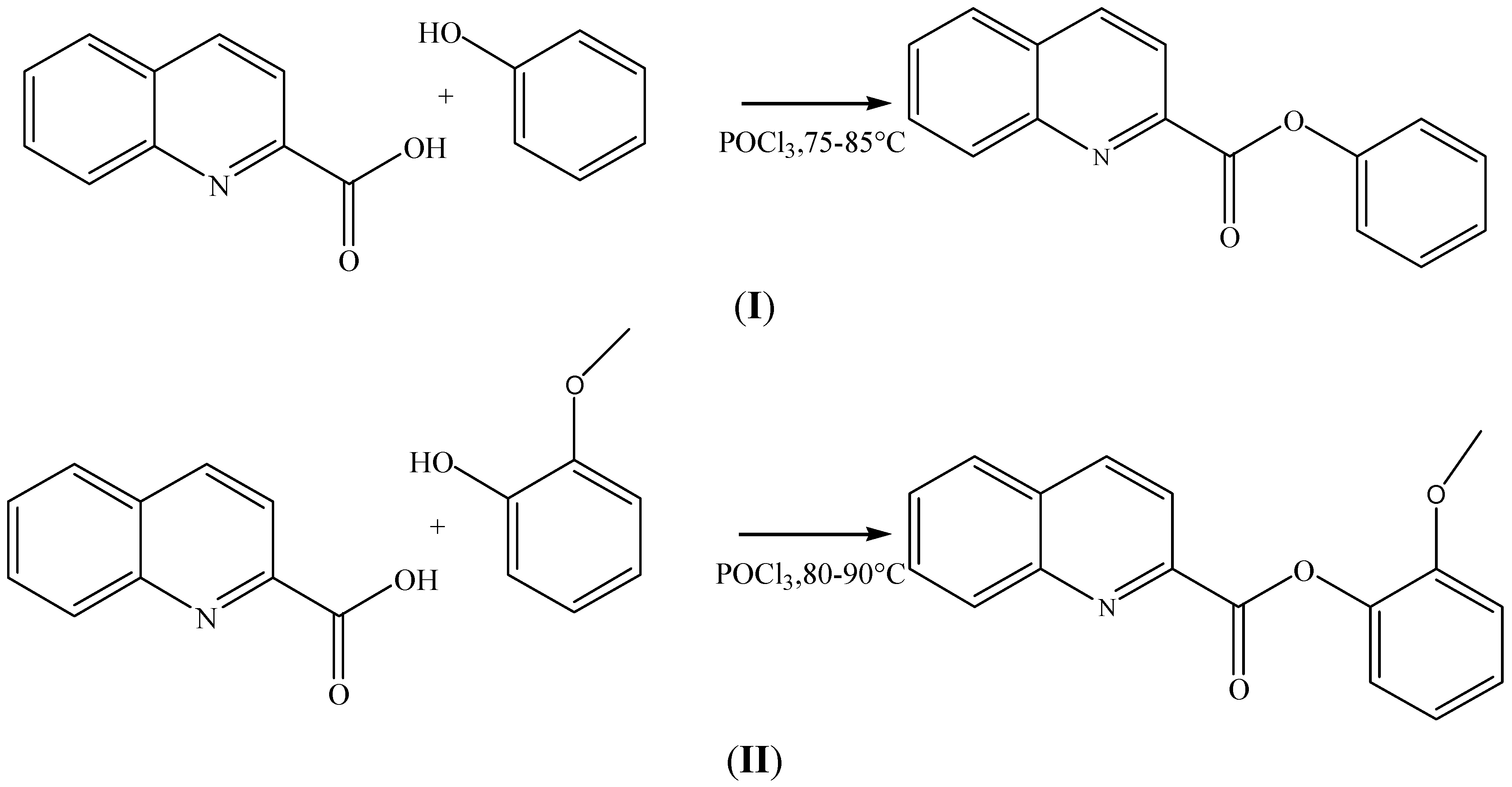
3.1. Synthesis of Phenyl Quinoline-2-Carboxylate and 2-Methoxyphenyl Quinoline-2-Carboxylate
3.2. Data Collection and Refinement
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morimoto, Y.; Matsuda, F.; Shirahama, H. Total Synthesis of (±)-Virantmycin and Determination of Its Stereochemistry. Synlett 1991, 3, 202–203. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Padwa, A.; Brodney, M.A.; Liu, B.; Satake, K.; Wu, T. Cycloaddition Approach toward the Synthesis of Substituted Indolines and Tetrahydroquinolines. J. Org. Chem. 1999, 64, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Markees, D.G.; Dewey, V.C.; Kidder, G.W. Antiprotozoal 4-aryloxy-2-aminoquinolines and related compounds. J. Med. Chem. 1970, 13, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.F.; Hardstone, J.D.; Palmer, M.J. 2,4-Diamino-6,7-Dimethoxyquinoline Derivatives as α1-Adrinoceptor Antagonists and Antihypertensive Agents. J. Med. Chem. 1988, 31, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R.; Magdziarz, T.; Kurczyk, A. Quinoline Scaffold as a Privileged Substructure in Antimicrobial Drugs. In Science against Microbial Pathogens: Communicating Current Research and Technological Advances; Formatex Research Center: Badajoz, Spain, 2011. [Google Scholar]
- Solomon, V.R.; Lee, H. Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Curr. Med. Chem. 2011, 18, 1488–1508. [Google Scholar] [CrossRef] [PubMed]
- Fazal, E.; Jasinski, J.P.; Krauss, S.T.; Sudha, B.S.; Yathirajan, H.S. 4-Methylphenyl quinoline-2-carboxylate. Acta Cryst. 2012, E68, o3231–o3232. [Google Scholar]
- Fazal, E.; Kaur, M.; Sudha, B.S.; Nagarajan, S.; Jasinski, J.P. 4-Chloro-3-methylphenyl quinoline-2-carboxylate. Acta Cryst. 2013, E69, o1842–o1843. [Google Scholar]
- Fazal, E.; Kaur, M.; Sudha, B.S.; Nagarajan, S.; Jasinski, J.P. 4-Chlorophenyl quinoline-2-carboxylate. Acta Cryst. 2013, E69. [Google Scholar] [CrossRef]
- Fazal, E.; Kaur, M.; Sudha, B.S.; Nagarajan, S.; Jasinski, J.P. 3,4-Dimethylphenyl quinoline-2-carboxylate. Acta Cryst. 2014, E69, o1853–o1854. [Google Scholar]
- Fazal, E.; Kaur, M.; Sudha, B.S.; Nagarajan, S.; Jasinski, J.P. 2,5-Dimethylphenyl quinoline-2-carboxylate. Acta Cryst. 2014, E70. [Google Scholar] [CrossRef]
- Fazal, E.; Jasinski, J.P.; Anderson, B.J.; Sudha, B.S.; Nagarajan, S. 2-Isopropyl-5-methylcyclohexyl quinoline-2-carboxylate. Acta Cryst. E 2013, 70, o35–o36. [Google Scholar] [CrossRef]
- Jasinski, J.P.; Butcher, R.J.; Mayekar, A.N.; Yathirajan, H.S.; Narayana, B.; Sarojini, B.K. Synthesis, Crystal Structures and Theoretical Studies of Four Schiff Bases Derived from 4-Hydrazinyl-8-(trifluoromethyl) quinoline. J. Mol. Struct. 2010, 980, 172–181. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths Determined by X-ray and Neutron Diffraction. Part I. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Schmidt, J.R.; Polik, W.F. WebMO Pro Homepage, version 8.0.01e. Available online: http://www.webmo.net (accessed on 6 December 2014).
- Frisch, M.J. Gaussian 03; Revision C01; Wallingford: New Haven, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Random, L.; Schleyer, P.R.; Pople, J.A. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Georgakopoulous, S.; Grondelle, R.V.; Zwan, G.V.D. Circular Dichroism of Carotenoides in Bacterial Light-harvesting Complexes: Experimental and Modeling. J. Biophys. 2005, 87, 3010–3022. [Google Scholar] [CrossRef]
- Guzin, A. Derivative Spectrophotometric Determination of Caffeine in Some Beverages. Turk J. Chem. 2002, 26, 295–302. [Google Scholar]
- IGOR Pro (1988–2009), WaveMetrics. Available online: www.wavemetrics.com (accessed on 21 January 2015).
- Oxford Diffraction. CrysAlis PRO and CrysAlis RED; Oxford Diffraction Ltd.: Abingdon, UK, 2010. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Smtbx. cif: A Comprehensive CIF Toolbox. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON-A Multipurpose Crystallographic Tool; Ultrecht University: Ultrecht, The Netherlands, 2001. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fazal, E.; Jasinski, J.P.; Anderson, B.J.; Kaur, M.; Nagarajan, S.; Sudha, B.S. Synthesis, Crystal and Molecular Structure Studies and DFT Calculations of Phenyl Quinoline-2-Carboxylate and 2-Methoxyphenyl Quinoline-2-Carboxylate; Two New Quinoline-2 Carboxylic Derivatives. Crystals 2015, 5, 100-115. https://doi.org/10.3390/cryst5010100
Fazal E, Jasinski JP, Anderson BJ, Kaur M, Nagarajan S, Sudha BS. Synthesis, Crystal and Molecular Structure Studies and DFT Calculations of Phenyl Quinoline-2-Carboxylate and 2-Methoxyphenyl Quinoline-2-Carboxylate; Two New Quinoline-2 Carboxylic Derivatives. Crystals. 2015; 5(1):100-115. https://doi.org/10.3390/cryst5010100
Chicago/Turabian StyleFazal, Edakot, Jerry P. Jasinski, Brian J. Anderson, Manpreet Kaur, Subban Nagarajan, and Belgur Satyanarayana Sudha. 2015. "Synthesis, Crystal and Molecular Structure Studies and DFT Calculations of Phenyl Quinoline-2-Carboxylate and 2-Methoxyphenyl Quinoline-2-Carboxylate; Two New Quinoline-2 Carboxylic Derivatives" Crystals 5, no. 1: 100-115. https://doi.org/10.3390/cryst5010100