1. Introduction
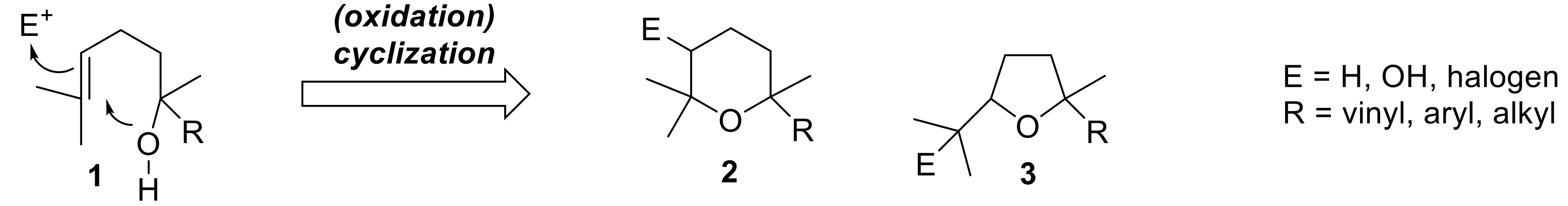
Tetrahydropyrane and tetrahydrofurane derivatives having the general framework of type
2 and
3 (
Figure 1) are quite common naturally occurring compounds. The biosynthesis of the main part of these ethers is based on the cyclization of a terpenic alcohols of type
1, and follows two main pathways, both involving the intramolecular nucleophilic addition of a hydroxyl functional group to a terminal prenyl group [
1,
2].
A first path requires the preliminary activation of the double bond through its transformation in epoxide or halonium derivatives, which easily cyclizes to give the corresponding ether derivatives bearing an additional hydroxyl or halogen functional group. Due to the general interest in this kind of chemical transformation in organic synthesis, its regio- and stereochemical outcomes were studied in depth [
3,
4,
5]. In addition, the monoterpenes possessing a hydroxyl group and a vinyl group (E = OH, R = vinyl) are well-known flavor components collectively called linalool oxides, regardless of the fact that they possess a six- or five-membered ring [
6].
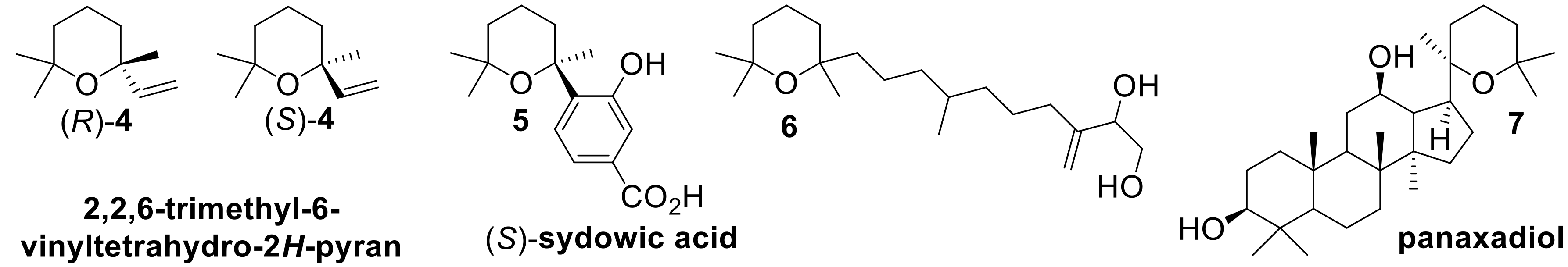
On the contrary, the cyclization path which does not involve a preliminary oxidation reaction affords tetrahydropyrane and tetrahydrofurane derivatives devoid of a hydroxyl group (E = H). Although a number of pyranoid derivatives possessing these structural frameworks were identified in nature, only few stereoselective approaches to their synthesis are described so far. For example linaloyl oxide (Compound
4) enantiomers (
Figure 2) are relevant flavor and fragrance components [
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20], sydowic acid (Compound
5) is a bioactive sesquiterpene produced by the mold
Aspergillus sidowii [
21], the diterpenic diol (Compound
6) was isolated from the African plant
Anisopappus pinnatifidus [
22], and triterpene panaxadiol (Compound
7) is a bioactive component of the ginseng extract [
23]. These terpenes share in common the same tetrahydropyranyl moiety which contains a quaternary stereocenter, whose stereoselective construction is especially demanding.
As we are involved in a number of studies describing the stereoselective synthesis of flavors and fragrances [
24,
25,
26,
27,
28], and as we recently reported a new enantioselective synthesis of pyranoid linalool oxide isomers [
29], we became interested in expanding our studies to the above-described class of compounds. More specifically, we envisaged that the enantiomeric forms of the primary alcohol (2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol (Compound
8;
Figure 3) could be regarded as potential building blocks for the stereoselective synthesis of this kind of natural product.
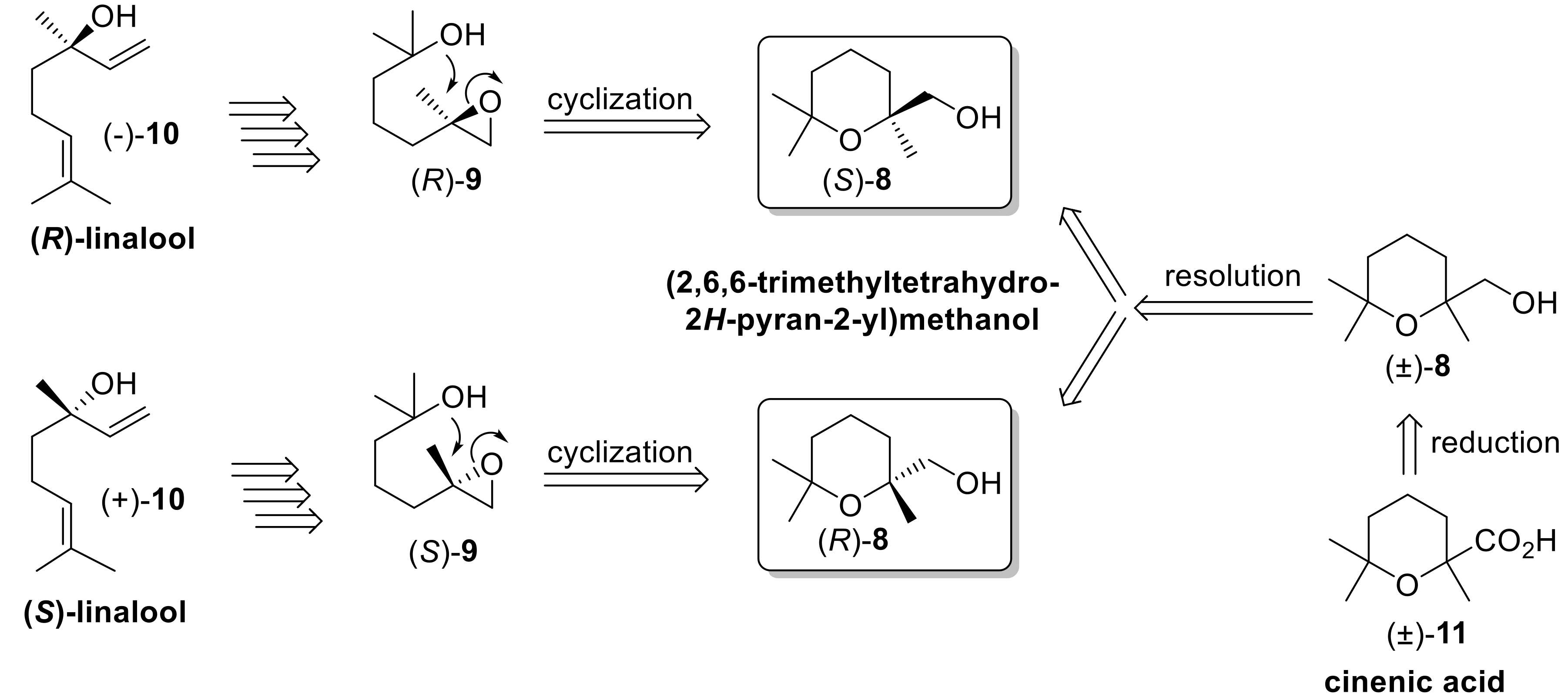
According to our retrosynthetic analysis, we devised two different synthetic methods for the stereoselective preparation of alcohol
8. Both approaches are based on an enantioselective-catalyzed reaction as key step. The first procedure takes advantage of the stereospecific (+)-10-camphorsulfonic acid (10-CSA)-catalyzed cyclization of (
R)- and (
S)-enantiomers of 2-methyl-5-(2-methyloxiran-2-yl)pentan-2-ol (Compound
9), which were in turn synthesized from (
R)- and (
S)-enantioforms of linalool (Compound
10), respectively. The latter cyclization reaction proceeds with very high stereocontrol as previously described by Vidari [
30,
31], who studied this chemical transformation using very similar epoxides as substrates. In addition, both linalool enantiomers are accessible from the chiral pool with different optical purity [
32].
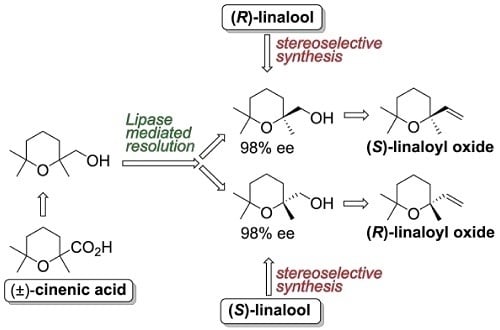
The second approach is based on the lipase-mediated resolution of the racemic alcohol
8, in turn obtained by reduction of the easily available cinenic acid (Compound
11) [
33].
Although we already reported the resolution of racemic
11 by means of the fractional crystallization of its (
R)-1-phenylethylamine salt [
34], the application of this procedure for the preparation of both enantiomers of the aforementioned acid turned out to be lengthy because of the necessary sequential preparation of both (
R)- and (
S)-phenylethylamine salts. In order to avoid the tedious chemical manipulations related to the salt formation, as well as to the number of crystallizations required, we investigated the lipase-mediated acetylation reaction of alcohol
8 using vinyl acetate as an acetyl donor. According to this approach, the enzymes are able to catalyze the esterification reaction with high enantioselectivity, allowing the separation of (2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol isomers. One enantiomer gives the corresponding acetate, and the other does not react. A simple chromatographic separation is required to complete the resolution procedure.
In the present work, we describe the accomplishment of the two above-described synthetic approaches. More specifically, we report in detail the synthetic procedure that allows transforming enantioenriched linalool into enantiopure (2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol enantiomers. Moreover, we describe a large-scale resolution procedure that exploits the opposite enantioselectivity of two different lipases in the acetylation reaction of alcohol 8. Thanks to the sequential use of both enzymes, the two enantiomeric forms of the alcohol were obtained in very high enantiomeric purity and were employed for the first stereoselective synthesis of both enantiomers of the natural flavor, linaloyl oxide (2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran).
2. Results and Discussion
As mentioned in the introduction, we selected epoxide
9 as a synthetic precursor of alcohol
8. We planned the enantioselective synthesis of both epoxide enantiomers using linalool enantiomers as starting materials. From a synthetic standpoint, the different reactivity of the two double bonds present in the linalool framework allows an outline of the regioselective preparation of
9. Indeed, the tertiary hydroxy group can be introduced via regioselective epoxidation/reduction of the trisubstituted linalool double bond (
Figure 4). Furthermore, the epoxide functional group can be introduced by oxidative cleavage of the monosubstituted linalool double bond followed by reduction to the corresponding 1,2-diol. The latter intermediate can be transformed into epoxide
9 by activation of the primary hydroxy group followed by a ring-closure reaction.
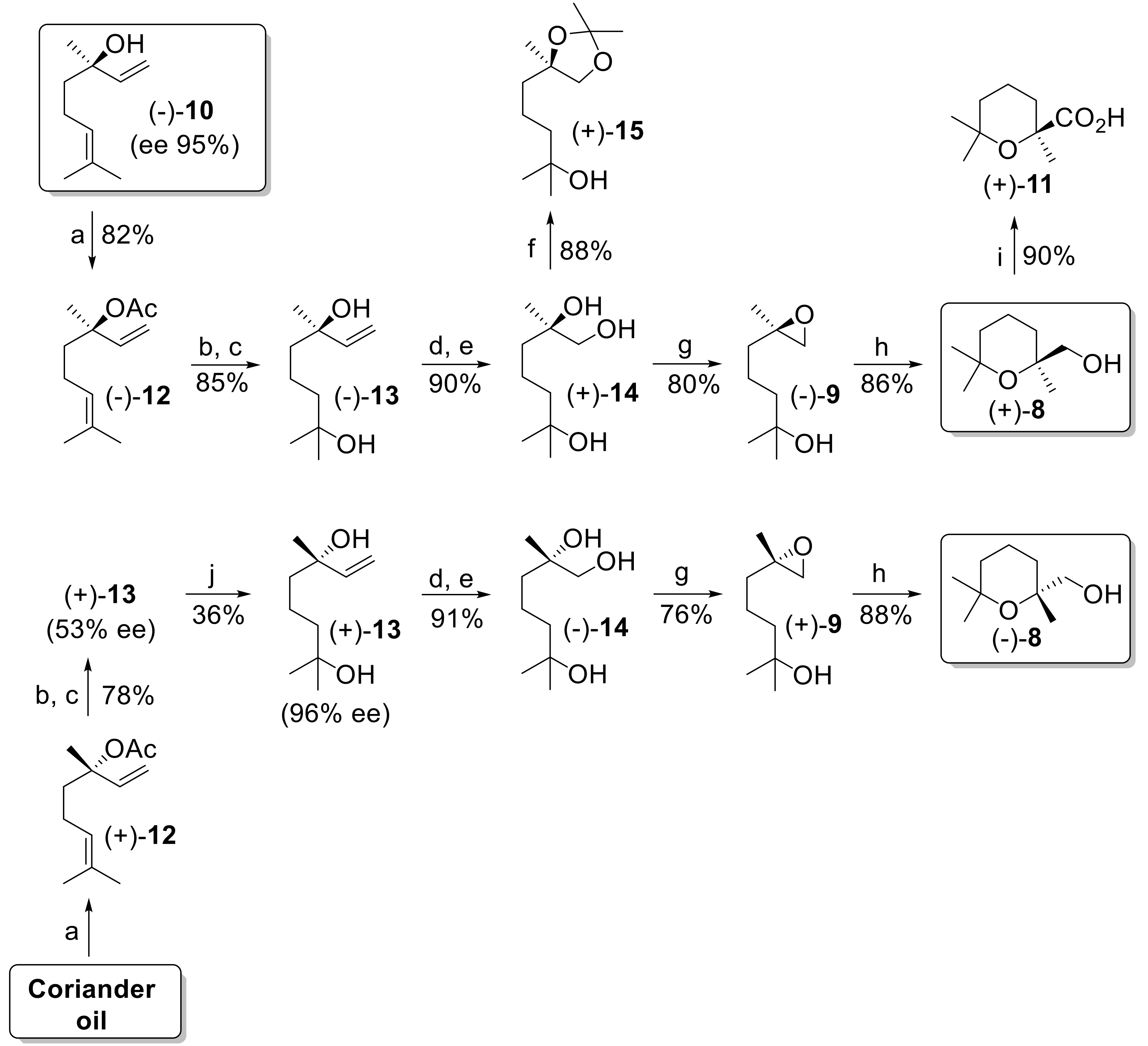
At first, we accomplished this synthetic approach starting from a sample of (−)-(
R)-linalool that is commercially available in very high enantiomeric purity by extraction from
Cinnamomun canphora (95% enantiomeric excess (ee)). Accordingly, the tertiary alcohol (−)-
10 was refluxed with acetic anhydride and pyridine (Py) in presence of catalytic amount of 4-dimethylaminopyridine (DMAP), to give the corresponding acetate (−)-
12. The latter ester was treated with
m-chloroperbenzoic acid (
mCPBA) in CH
2Cl
2 and the resulting crude epoxide was regioselectively reduced using an excess of LiAlH
4 in refluxing tetrahydrofuran (THF). The obtained diol (−)-
13 was first purified by crystallization from hexane and then treated with ozone in CH
2Cl
2/MeOH, until complete cleavage of the double bond. The following reduction with NaBH
4 afforded the triol (+)-
14 in very good yield. Compound
14 could not be characterized by NMR analysis as the strong inter- and intramolecular hydrogen bonding broadened both
1H- and
13C-NMR signals to such an extent that we could not properly describe the spectra. Since the latter compound is previously undescribed, we prepared the dioxolane (+)-
15 by reaction of (+)-
14 with 2,2-dimethoxypropane (2,2-DMP) in acetone in presence of a pyridinium
p-toluenesulfonate (PPTS) catalyst. Thus, the derivative (+)-
15 was fully characterized, and the analytical data confirmed both the regioisomeric purity and the chemical structure of triol
14. The transformation of the 1,2-diol functional group into an epoxide group was performed as described by Vidari [
30], using lithium diisopropylamide (LDA) and tosyl chloride in THF solution at −10 °C. The obtained epoxy-alcohol (−)-
9 was treated with a catalytic amount of (+)-10-CSA to give the desired (2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol (+)-
8 in good yield. The absolute configuration of (+)-
8 was confirmed to be (
S) by chemical correlation with cinenic acid. Accordingly, oxidation of (+)-
8 with Jones reagent afforded (+)-cinenic acid
11 of known (
S) configuration [
35].
In order to also obtain the compound (−)-
8, we accomplished the identical reaction sequence described above starting from (+)-linalool. The latter compound is easily available from coriander oil whose weight is made up of more than 90% (+)-
10. The direct treatment of the essential oil with acetic anhydride and pyridine in presence of a catalytic amount of DMAP, followed by purification by distillation, gave the corresponding acetate (+)-
12 in good yield. Unfortunately, the enantiomeric purity of (+)-linalool from this botanic source ranges from 45 to 85% ee [
32], which is not suitable for the preparation of the chiral building block
8. Since our synthesis makes use of the intermediate 2,6-dimethyloct-7-ene-2,6-diol
13, which can be purified by crystallization, we observed that, starting from (−)-(
R)-linalool showing 95% ee, the optical rotation value of the resulting diol was −9.9° which increased to −10.4° after crystallization. Similarly, diol (+)-
13 derived from a commercial sample of coriander oil (optical purity of the linalool of about 55% ee) showed an optical rotation value of +5.2°, which increased to +10.1° after three crystallizations from hexane. These results demonstrate that the enantiopurity of diol
13 was improved through fractional crystallization passing from 53% ee to 96% ee. Therefore, the described purification procedure allows the proper enantioselective synthesis of both enantioforms of compound
8.
Accordingly, the achieved enantiopure diol (+)-13, was transformed into alcohol (−)-8, following the experimental conditions used for the synthesis of alcohol (+)-8. Overall, both enantioforms of the building block (2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol were achieved in optical purity superior to 95% ee.
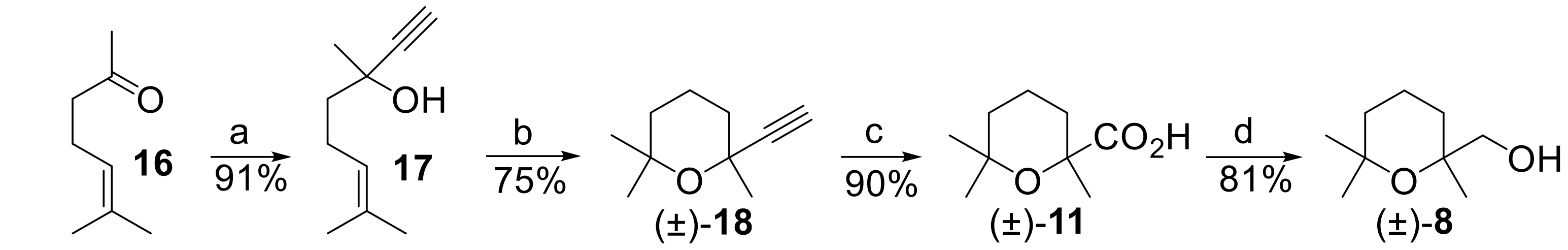
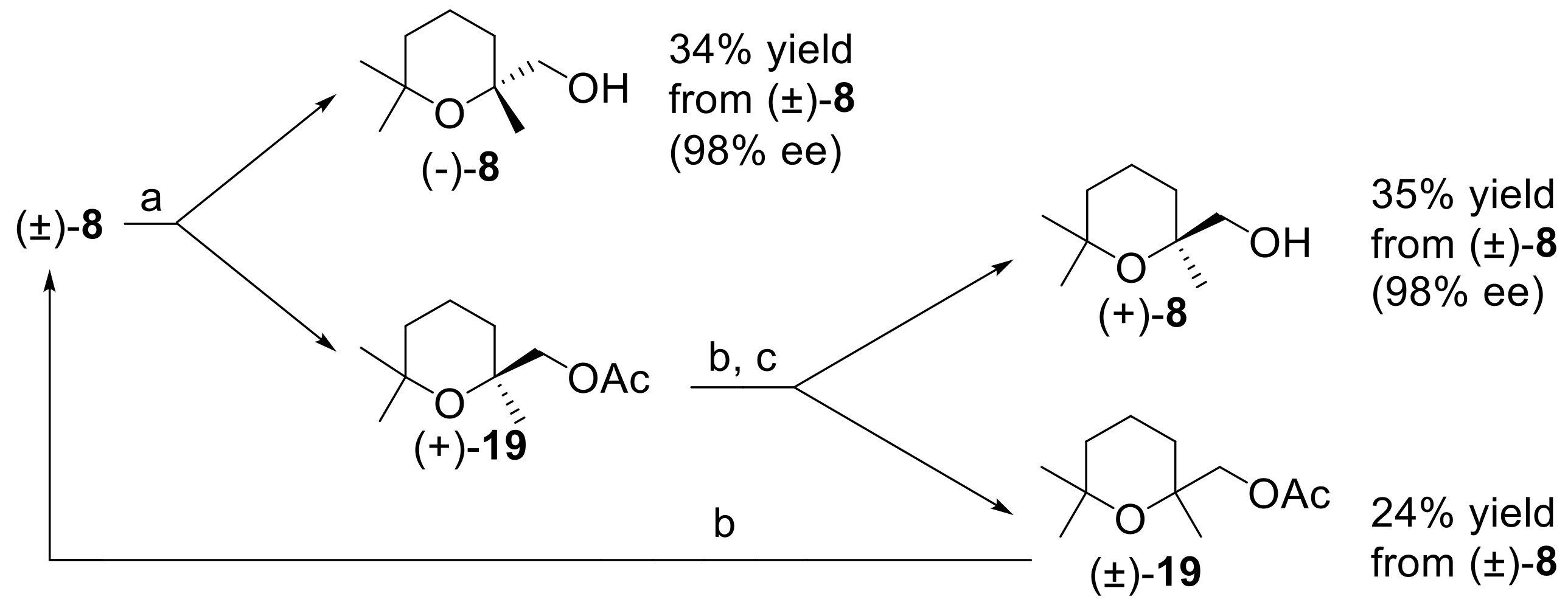
The second part of this work, namely the development of an enzyme-mediated resolution procedure of alcohol (±)-
8, first requires a valuable amount of the aforementioned racemic alcohol. Hence, we devised the preparative procedure described in
Figure 5, which improved the cinenic acid synthesis previously reported by Rupe and Lang [
33].
Accordingly, the addition of ethynylmagnesium bromide to 6-methylhept-5-en-2-one (Compound 16) afforded dehydrolinalool (Compound 17), which was then heated at reflux in formic acid/water to give 2-ethynyl-2,6,6-trimethyltetrahydro-2H-pyran (Compound 18).
The oxidation of the alkyne
18 to cinenic acid
11, performed using Rupe and Lang procedure, makes use of aqueous KMnO
4, and the yield of the isolated acid does not exceed 50%, even with complete conversion of the alkyne. Therefore, we employed a different oxidation method based on the use of NaIO
4, in the presence of a catalytic amount of RuCl
3·H
2O [
36]. As a result, cinenic acid was obtained in very good yield (90%). Finally, the transformation of the carboxylic acid functional group of
11 into the corresponding carboxy-ethyl derivative followed by reduction with NaBH
4 [
37] smoothly afforded racemic
8, in good overall yield.
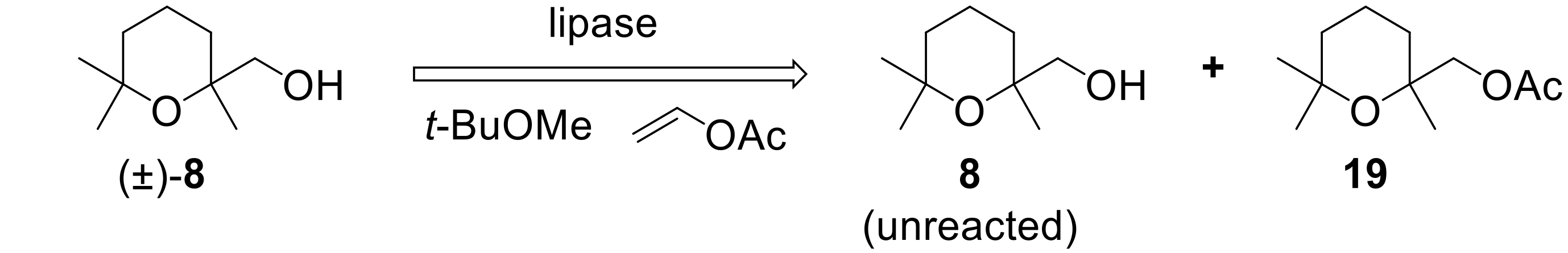
To the best of our knowledge, no enzyme-mediated resolution procedures of (2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol are reported until now. It should be noted that, usually, lipases catalyze the esterification of primary alcohols with very low enantioselectivities. Since we already described some remarkable exceptions to this behavior [
38,
39,
40,
41], we decided to investigate the reactivity of racemic
8 in the latter reaction, using a number of selected lipases. For each experiment,
8 was treated with vinyl acetate in
t-butyl-methyl ether, in the presence of the given enzyme (
Figure 6). The reactions were interrupted when the wanted conversion was achieved. The unreacted alcohol and the acetate
19 were separated by chromatography, and their enantiomeric compositions were determined by comparing their optical rotation values with those measured for the same enantiopure chemical compounds. The results are summarized in
Table 1.
The perusal of the obtained data allows drawing some relevant considerations. Firstly, we can observe that porcine pancreatic lipase (PPL) and pig liver esterase (PLE on Eupergit) were completely inactive. The reaction with PPL afforded only a trace of the acetate
19 after a long reaction time. This behavior is in sharp contrast with the results obtained in our previous studies where the same enzymes showed a remarkable catalytic activity in the acetylation of different primary alcohols. On the contrary, all the other enzymes evaluated in this work showed high activity, although with different stereoselectivity. More specifically, lipase PS and lipase AK (both from Amano Pharmaceuticals) catalyze the acetylation of (
R)-
8 in modest (E = 6.5) and good (E = 13.7) enantioselectivities, respectively. Moreover, lipase from
Candida rugosa (CRL) and Novozym 435 lipase catalyze the acetylation of (
S)-
8 in very low (E = 1.1) and good (E = 16.3) enantioselectivities, respectively. It is worth noting that CRL is a very effective catalyst, but does not show any enantioselectivity, thus making the enzyme unsuitable for any resolution process. Overall, we identified two enzymes, namely Novozym 435 and lipase AK, that are able to catalyze the acetylation of racemic alcohol
8 with good enantioselectivity. Both enzymes are suitable for setting up a resolution process, but they display opposite enantiopreference. Taking advantage of this observation, we devised a large-scale resolution procedure (
Figure 7) based on the combined and sequential use of Novozym 435 lipase and lipase AK.
Hence, racemic
8 was treated with vinyl acetate in
t-BuOMe using Novozym 435 as catalyst. The acetylation reaction was prolonged until about 65% conversion was reached. In accord with the specific lipase enantioselectivity and with the general rules of the enzyme-based kinetic resolution of racemic mixtures [
42], the unreacted alcohol (−)-(
R)-
8 was isolated in about 34% yield and in very high enantiopurity (98% ee). On the contrary, the acetate (+)-
19 possessed low optical purity and was hydrolyzed using NaOH in methanol. The resulting alcohol (+)-
8 was submitted to a second acetylation step, using lipase AK as catalyst. Indeed, the latter enzyme catalyzes the esterification of the (−)-(
R) enantiomer of alcohol
8, which was transformed in the corresponding acetate. As (−)-
8 is the minor component of the enantiomers mixture, the enzymatic reaction increased the enantiomeric purity of the unreacted alcohol. After the acetylation reaction reached a conversion of about 60%, the alcohol (+)-
8 was isolated in about 35% overall yield and with 98% ee. Nearly racemic acetate
19 was also obtained and it could be hydrolyzed to recover further alcohol, to be used in a new resolution procedure.
Overall, our resolution procedure proved to be compact, effective, and user-friendly as it can afford both enantiomeric forms of (2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol in very high optical purity by means of two commercial enzymes and without the employment of demanding experimental conditions or reagents.
As a first synthetic application of the obtained chiral building blocks, we describe here the preparation of (
R)- and (
S)-linaloyl oxide
4 starting from (
R)- and (
S)-(2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol
8, respectively. As mentioned in the introduction, this monoterpene is a relevant fragrance/flavor ingredient [
43]. More specifically, it is one of the fragrance components of geranium and lime essential oil [
7,
8] and is a trace component of the flavor of many fruits or other vegetal species [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20]. Since an effective and cheap synthesis of racemic
4 was established [
44], linaloyl oxide is currently produced and commercialized in this form, under the trade name Limetol
® (Givaudan).
We are not aware of any olfactory evaluation of the single enantiomeric forms of this monoterpene that seems to occur in essential oils in nearly racemic form [
8]. Since the organoleptic evaluation of this compound is subject to the availability of both (
R) and (
S) isomers in high enantiomeric purity, their stereoselective synthesis is highly desired. To date, only a lengthy and low-yielding synthesis of (+)-
4 was reported in the course of a study finalized to the characterization of linalool oxide [
45]. As the starting compound was (−)-linalool, a similar approach for the preparation of enantiopure (−)-
4 is not applicable, as (+)-linalool is available in low enantiomeric purity. Therefore, we synthesized (
R)- and (
S)-linaloyl oxide starting from enantiopure (
R)- and (
S)-(2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol
8, respectively, as described in
Figure 8.
Accordingly, the alcohols (−) and (+)-
8 were oxidized in good yields to the corresponding aldehydes (−) and (+)-
20, respectively, using Py·SO
3 in dimethyl sulfoxide (DMSO)/Et
3N [
46]. Then, the C
9 aldehydes
20 were homologated to the C
10 ethers
4 through a methylenation reaction. The most used method to perform this transformation, namely the Wittig reaction using triphenylphosphoniummethylene ylide, was not successful, as only the degradation of the starting aldehyde was observed. On the contrary, we found that the reagent obtained by reaction of Zn, CH
2I
2, and Me
3Al [
47] effectively converted (−) and (+)-
20 into ether (+) and (−)-
4, respectively. Overall, the described reaction sequence is high yielding and did not involve loss of optical purity. Since the starting chiral building blocks are enantiopure, the obtained linaloyl oxide enantiomers are suitable for olfactory evaluation, the study of which will be reported in due course.
3. Materials and Methods
3.1. Materials and General Methods
All moisture- and air-sensitive reactions were carried out using dry solvents under a static atmosphere of nitrogen.
All solvents and reagents were of commercial quality and were purchased from Sigma-Aldrich (St. Louis, MO, USA) with the exception of (S)-linalool acetate (+)-12 and dehydrolinalool 17 (3,7-dimethyloct-6-en-1-yn-3-ol), which were prepared by acetylation of coriander oil and by addition of ethynylmagnesium bromide to 6-methylhept-5-en-2-one 16, respectively.
(−)-(R)-Linalool, extracted from Cinnamomun canphora (L.) and possessing 99% purity by GC and = −20.9 (neat), was purchased from Sigma-Aldrich (lot MKBR2739V).
Coriander oil, = +8.3 (neat), was purchased from Sigma-Aldrich (lot MKCC6867) and was used as source of (+)-(S)-linalool.
Lipase from Porcine pancreas (PPL) type II, Sigma-Aldrich, 147 units/mg; lipase from Pseudomonas cepacia (PS), Amano Pharmaceuticals Co., Tokyo, Japan, 30 units/mg; lipase from Candida rugosa (CRL) type VII, Sigma-Aldrich, ≥700 units/g; Novozym® 435, Novozymes, ≥5000 units/g; lipase AK Amano from Pseudomonas fluorescens, Sigma-Aldrich, 20 units/mg; and PLE on Eupergit, Sigma-Aldrich, 200 units/g were employed in this work.
3.2. Analytical Methods and Characterization of the Chemical Compounds
1H and 13C-NMR spectra and DEPT (Distortionless enhancement by polarization transfer) experiments: CDCl3 solutions at room temperature (rt) using a Bruker-AC-400 spectrometer (Billerica, MA, USA) at 400, 100, and 100 MHz, respectively; 13C spectra are proton-decoupled; chemical shifts in ppm relative to internal SiMe4 (0 ppm).
Thin-layer chromatography (TLC) involved the use of Merck silica gel 60 F254 plates (Merck Millipore, Milan, Italy), while column chromatography involved the use of silica gel.
Melting points were measured on a Reichert apparatus, equipped with a Reichert microscope, and are uncorrected.
Optical rotations were measured on a Jasco-DIP-181 digital polarimeter (Jasco, Tokyo, Japan).
Mass spectra were recorded on a Bruker ESQUIRE 3000 PLUS spectrometer (ESI detector, Billerica, MA, USA) or by GC–MS analyses.
GC–MS analyses involved the use of an HP-6890 gas chromatograph equipped with a 5973 mass detector, using an HP-5MS column (30 m × 0.25 mm, 0.25-µm film thickness; Hewlett Packard, Palo Alto, CA, USA) with the following temperature program: 60° (1 min), then 6°/min to 150° (held at 1 min), then 12°/min to 280° (held 5 min); carrier gas: He; constant flow 1 mL/min; split ratio: 1/30; tR given in minutes.
The values of tR for each compound are as follows: tR(4) 5.97, tR(8) 8.78, tR(9) 10.53, tR(10) 8.86, tR(11) 12.32, tR(12) 12.42, tR(13) 11.93, tR(15) 14.58, tR(17) 8.67, tR(18) 5.81, tR(19) 12.63, and tR(20) 7.07.
3.3. Stereoselective Preparation of (S)- and (R)-(2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol Starting from (R)- and (S)-Linalool, Respectively
3.3.1. (R)-2,6-Dimethyloct-7-ene-2,6-diol (−)-13
(−)-(R)-Linalool 10 (60 g, 389 mmol), acetic anhydride (50 mL, 529 mmol), pyridine (45 mL, 559 mmol), and DMAP (1 g, 8.2 mmol) were heated at reflux under a static atmosphere of nitrogen until complete acetylation of the starting alcohol (2 h by TLC analysis). The reaction mixture was then cooled and quenched by addition to a mixture of water and crushed ice followed by extraction with diethyl ether (2 × 250 mL). The combined organic phases were washed in turn with water, and saturated with NaHCO3 solution (3 × 200 mL) and brine. The organic phase was dried (Na2SO4) and concentrated in vacuo. The residue was purified by distillation to afford colorless (R)-linalool acetate 12 (62.9 g, 82% yield, 94% purity by GC analysis).
A sample of acetate 12 (24 g, 122 mmol) was dissolved in CH2Cl2 (100 mL) and treated with m-chloroperbenzoic acid (31 g, 77% w/w, 138 mmol) stirring at 0 °C until completion of the reaction (TLC monitoring). The m-chlorobenzoic acid formed was removed by filtration and the liquid phase was washed in turn with aqueous Na2SO3 (10% w/w), aqueous NaHCO3 (saturated solution) and brine. The organic phase was dried (Na2SO4), concentrated under reduced pressure and the residue was diluted with dry THF (60 mL). The aforementioned solution was added dropwise to a stirred suspension of LiAlH4 (6 g, 158 mmol) in dry THF (200 mL). The reaction was stirred at reflux until the starting epoxide was completely transformed into the diol 13 (5 h, TLC analysis); then, the mixture was cooled (0 °C), diluted with diethyl ether (300 mL), and quenched by dropwise addition of 40% aqueous solution of NaOH (50 mL), stirring vigorously for 1 h. The resulting heterogeneous mixture was filtered on a celite pad and the organic phase was washed with brine, before being dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography using n-hexane/AcOEt (8:2–1:1) as an eluent to afford pure (R)-2,6-dimethyloct-7-ene-2,6-diol 13 (17.8 g, 85% yield) as a colorless oil which solidified on standing; = −9.9 (c 2, CHCl3). A sample of the diol was recrystallized from hexane. The collected crystals showed 98% purity by GC analysis; melting point (mp): 55–56 °C; = −10.4 (c 2.1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 5.92 (dd, J = 17.4, 10.8 Hz, 1H), 5.21 (dd, J = 17.4, 1.2 Hz, 1H), 5.05 (dd, J = 10.8, 1.2 Hz, 1H), 1.69 (br s, 2H), 1.59–1.49 (m, 2H), 1.49–1.34 (m, 4H), 1.29 (s, 3H), 1.21 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 145.1 (CH), 111.6 (CH2), 73.2 (C), 71.0 (C), 44.1 (CH2), 42.6 (CH2), 29.2 (Me), 27.7 (Me), 18.6 (CH2).
GC–MS m/z (relative intensity): 154 (M+-H2O, <1), 139 (11), 121 (19), 109 (8), 93 (10), 81 (37), 71 (100), 59 (33), 43 (46).
3.3.2. (R)-2,6-Dimethylheptane-1,2,6-triol (+)-14
An oxygen stream containing ozone (0.1 mole/hour) was bubbled into a cooled (−70 °C) solution of the diol (−)-13 (14 g, 81.3 mmol) in CH2Cl2/MeOH (3:1 v/v, 280 mL). As soon as the solution took a persistent light-blue color, the ozone addition was stopped and the oxygen stream was switched to a nitrogen stream. After a few minutes the solution became pale yellow and NaBH4 (5 g, 132.2 mmol) was added portionwise. The reaction was slowly allowed to reach rt and set aside overnight. The excess of hydride was then quenched by addition of acetic acid (50 mL). After excluding the presence of residual peroxides (negative KI/starch test) the solvents and the excess of acetic acid were removed under reduced pressure and the residue was diluted with water (80 mL) and extracted with n-butanol (4 × 100 mL). The combined organic phases were washed with brine, before being dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography eluting first with n-hexane/AcOEt (1:1) and then increasing the polarity using AcOEt/MeOH (2:1) to afford pure (+)-(R)-2,6-dimethylheptane-1,2,6-triol 14 as a thick, colorless oil (12.9 g, 90% yield).
= +2.9 (c 4.7, CHCl3); MS (ESI): 199.1 (M+ + Na)
Due to the strong intermolecular hydrogen bonding, 1H and 13C-NMR analysis of the latter compound were not clear and did not allow an unambiguous characterization of compound 14. Therefore, we prepared the derivative 15 that was fully characterized. The obtained analytical data substantiated the chemical structure of 15, and thus, the structure of triol 14. Accordingly, a sample of triol (+)-14 (200 mg, 1.13 mmol) was dissolved in acetone (10 mL) and treated with 2,2-dimethoxypropane (5 mL) and pyridinium p-toluenesulfonate (0.1 g, 0.40 mmol), and then stirred at rt for 8 h. Next, Et3N (2 mL) was added and the solvents were removed under reduced pressure. The residue was partitioned between EtOAc (50 mL) and water (50 mL) and the organic phase was washed with water and with brine, before being dried (Na2SO4) and concentrated in vacuo. The obtained pale-yellow oil was purified by chromatography using n-hexane/AcOEt (8:2–1:1) as an eluent to afford pure (+)-(R)-2-methyl-5-(2,2,4-trimethyl-1,3-dioxolan-4-yl)pentan-2-ol 15 (215 mg, 88%).
= +2.3 (c 3.2, CHCl3)
1H NMR (400 MHz, CDCl3): δ 3.79 (d, J = 8.3 Hz, 1H), 3.71 (d, J = 8.3 Hz, 1H), 1.72–1.33 (m, 7H), 1.40 (s, 3H), 1.38 (s, 3H), 1.28 (s, 3H), 1.22 (s, 6H).
13C NMR (100 MHz, CDCl3): δ 109.0 (C), 81.2 (C), 74.0 (CH2), 70.9 (C), 44.3 (CH2), 40.5 (CH2), 29.3 (Me), 29.2 (Me), 27.2 (Me), 27.1 (Me), 24.8 (Me), 19.3 (CH2).
GC–MS m/z (relative intensity): 201 (M+-Me, 31), 183 (16), 141 (9), 123 (100), 115 (92), 107 (10), 97 (19), 81 (39), 72 (52), 59 (50), 43 (75).
MS (ESI): 239.1 (M+ + Na).
3.3.3. (R)-2-Methyl-5-(2-methyloxiran-2-yl)pentan-2-ol (−)-9
A stirred solution of triol (+)-14 (8.2 g, 46.5 mmol) in dry THF (60 mL) was treated at −15 °C with freshly prepared LDA (44 mL of a 2.4 M solution in THF). After ten minutes, a solution of tosyl chloride (9.7 g, 50.9 mmol) in dry THF (30 mL) was added dropwise. The mixture was stirred at 0 °C until complete transformation of the starting triol (3 h). Then, the reaction was quenched by pouring into a mixture of saturated NH4Cl solution and crushed ice followed by extraction with diethyl ether (2 × 200 mL). The combined organic phases were washed in turn with saturated NaHCO3 solution and brine. The organic solution was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography using n-hexane/AcOEt (9:1–1:1) as an eluent to afford pure (−)-(R)-2-methyl-5-(2-methyloxiran-2-yl)pentan-2-ol 9 as a colorless oil (5.9 g, 80% yield).
= −6.2 (c 5.2, CHCl3)
1H NMR (400 MHz, CDCl3): δ 2.62 (d, J = 4.8 Hz, 1H), 2.58 (d, J = 4.8 Hz, 1H), 1.67–1.41 (m, 7H), 1.32 (s, 3H), 1.22 (s, 6H).
13C NMR (100 MHz, CDCl3): δ 70.7 (C), 56.9 (C), 53.8 (CH2), 43.6 (CH2), 36.9 (CH2), 29.3 (Me), 29.1 (Me), 20.8 (Me), 19.9 (CH2).
MS (ESI): 181.1 (M+ + Na).
3.3.4. (S)-(2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol (+)-8
A stirred solution of epoxide (−)-9 (5.6 g, 35.4 mmol) in CH2Cl2 (50 mL) was treated at 0 °C with (+)-10-camphorsulfonic acid (100 mg, 0.43 mmol). As soon as the starting epoxide was no longer detectable by TLC analysis (2 h), the reaction was quenched by addition of a saturated NaHCO3 solution (40 mL) and was extracted with CH2Cl2 (2 × 60 mL). The combined organic phases were dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography using n-hexane/Et2Ot (9:1–2:1) as an eluent to afford pure (+)-(S)-(2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol 8 as a colorless oil (4.8 g, 86% yield).
= +9.8 (c 3.4, CHCl3)
1H NMR (400 MHz, CDCl3): δ 3.32 (d, J = 10.6 Hz, 1H), 3.24 (d, J = 10.6 Hz, 1H), 2.28 (br s, 1H), 1.84–1.58 (m, 3H), 1.55–1.47 (m, 1H), 1.41–1.25 (m, 2H), 1.25 (s, 3H), 1.19 (s, 3H), 1.17 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 73.3 (C), 71.8 (C), 70.6 (CH2), 36.4 (CH2), 32.2 (Me), 30.3 (CH2), 28.1 (Me), 24.3 (Me), 16.1 (CH2).
GC–MS m/z (relative intensity): 143 (M+-Me, 2), 127 (59), 109 (100), 97 (3), 81 (4), 75 (4), 69 (49), 59 (12), 43 (45).
3.3.5. Chemical Correlation of (+)-(2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol 8 with (+)-(S)-Cinenic Acid 11
Jones reagent (5 mmol) was added dropwise to a stirred solution of the alcohol (+)-8 (0.2 g, 1.26 mmol, 95% ee) in acetone (15 mL) at 0 °C. The reaction was allowed to reach rt and stirring was prolonged since TLC analysis indicated complete transformation of the intermediate aldehyde into the corresponding acid (one hour). The reaction was then quenched by dilution with water (60 mL) and extraction with diethyl ether (2 × 70 mL). The organic phase was washed with water and with brine, before being dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by chromatography using n-hexane/ethyl acetate (9:1–7:3) as an eluent to give pure (+)-(S)-2,6,6-trimethyltetrahydro-2H-pyran-2-carboxylic acid 11 as a colorless oil which crystallized on standing (195 mg, 90% yield, 94% purity by GC–MS analysis).
= +2.6 (c 4, CHCl3)
1H NMR (400 MHz, CDCl3): δ 9.95 (br s, 1H), 2.11–2.02 (m, 1H), 1.80–1.62 (m, 2H), 1.56–1.47 (m, 3H), 1.45 (s, 3H), 1.28 (s, 3H), 1.24 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 178.5 (C), 75.2 (C), 74.4 (C), 35.9 (CH2), 32.0 (CH2), 30.1 (Me), 27.7 (Me), 27.4 (Me), 16.4 (CH2).
GC–MS m/z (relative intensity): 157 (M+-Me, 2), 139 (4), 127 (58), 109 (100), 95 (3), 69 (62), 59 (14), 43 (57).
3.3.6. (S)-2,6-Dimethyloct-7-ene-2,6-diol (+)-13
Coriander oil (60 g), acetic anhydride (50 mL, 529 mmol), pyridine (45 mL, 556 mmol), and DMAP (1 g, 8,2 mmol) were heated at reflux under a static atmosphere of nitrogen until complete acetylation of the (+)-(S)-linalool (2 h by TLC analysis). The reaction mixture was then cooled and quenched by addition to a mixture of water and crushed ice followed by extraction with diethyl ether (2 × 200 mL). The combined organic phases were washed in turn with water, before being saturated NaHCO3 solution (3 × 200 mL) and brine. The organic phase was dried (Na2SO4) and concentrated in vacuo. The residue was purified by distillation to afford colorless (+)-(S)-linalool acetate 12 (51.2 g, 91% purity by GC analysis).
A sample of acetate (+)-12 (28 g, 142.6 mmol) was dissolved in CH2Cl2 (120 mL) and treated with m-chloroperbenzoic acid (34 g, 77% w/w, 151.7 mmol) stirring at 0 °C until completion of the reaction (TLC monitoring). The m-chlorobenzoic acid formed was removed by filtration and the liquid phase was washed in turn with aqueous Na2SO3 (10% w/w), aqueous NaHCO3 (saturated solution), and brine. The organic phase was dried (Na2SO4), concentrated under reduced pressure and the residue was diluted with dry THF (60 mL). The aforementioned solution was added dropwise to a stirred suspension of LiAlH4 (6.5 g, 171.3 mmol) in dry THF (200 mL). The reaction was stirred at reflux until the starting epoxide was completely transformed in the diol (+)-13 (5 h, TLC analysis); then, the mixture was cooled (0 °C), diluted with diethyl ether (300 mL), and quenched by dropwise addition of 40% aqueous solution of NaOH (50 mL), stirring vigorously for 1 h. The resulting heterogeneous mixture was filtered on a celite pad and the organic phase was washed with brine, before being dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography using n-hexane/AcOEt (8:2–1:1) as an eluent to afford pure (+)-(S)-2,6-dimethyloct-7-ene-2,6-diol 13 (19.1 g, 78% yield) as a colorless thick oil, showing = +5.3 (c 3.5, CHCl3). The diol was then recrystallized three times from hexane. The third crystal crop (6.8 g, recrystallization yield 36%) showed 98% purity by GC analysis; mp: 52–53 °C; = +10.1 (c 3.5, CHCl3), corresponding to 96% ee. 1H-NMR, 13C-NMR, and GC–MS were superimposable to those reported for the (−)-(R) isomer.
3.3.7. (S)-2,6-Dimethylheptane-1,2,6-triol (−)-14
According to the procedure outlined for the synthesis of triol (+)-14, diol (+)-13 (96% ee, 98% chemical purity) gave, in 91% yield, (−)-14 as a colorless thick oil with = −2.8 (c 3.5, CHCl3).
3.3.8. (S)-2-Methyl-5-(2-methyloxiran-2-yl)pentan-2-ol (+)-9
According to the procedure outlined for the synthesis of epoxide (−)-9, triol (−)-14 (96% ee) gave, in 76% yield, (+)-9 as a colorless oil with = +6.0 (c 4.1, CHCl3) and 95% chemical purity by GC; 1H-NMR, 13C-NMR, and MS (ESI) were superimposable to those reported for the (−)-(R) isomer.
3.3.9. (R)-(2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol (−)-8
According to the procedure outlined for the synthesis of alcohol (+)-8, epoxide (+)-9 (96% ee, 95% chemical purity) gave, in 88% yield, (−)-8 as a colorless oil with = −10.0 (c 3.1, CHCl3) and 95% chemical purity by GC.
3.4. Synthesis of Racemic (2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol
3.4.1. 2-Ethynyl-2,6,6-trimethyltetrahydro-2H-pyran
A solution of ethynylmagnesium bromide (390 mL, 0.9 M in THF) was added dropwise at −70 °C to a stirred solution of 6-methylhept-5-en-2-one 16 (40 g, 317 mmol) in dry THF (100 mL) under a static atmosphere of nitrogen. The reaction was allowed to reach rt, and after 2 h, was poured into a mixture of crushed ice (300 g) and saturated NH4Cl solution (300 mL) followed by extraction with diethyl ether (2 × 300 mL). The combined organic phases were washed with water and with brine, before being dried (Na2SO4) and concentrated in vacuo. The residue was purified by distillation (boiling point (bp) 98 °C at 20 mmHg) to afford pure 3,7-dimethyloct-6-en-1-yn-3-ol 17 (43.8 g, 91% yield, 95% purity by GC–MS analysis).
A solution of the alkynol 17 (20 g, 131.4 mmol) in HCOOH/H2O (85:15, 25 mL) under a static atmosphere of nitrogen was heated at reflux until complete transformation of the starting propargylic alcohol (half an hour, TLC analysis). After cooling, the reaction was diluted with cool water (250 mL) and extracted with diethyl ether (2 × 150 mL). The combined organic phases were washed in turn with water, with saturated NaHCO3 solution and with brine, before being dried (Na2SO4) and concentrated in vacuo. The residue was purified by distillation (bp 65 °C at 20 mmHg) to afford pure 2-ethynyl-2,6,6-trimethyltetrahydro-2H-pyran 18 (15.1 g, 75% yield, 94% purity by GC–MS analysis) as a colorless oil.
1H NMR (400 MHz, CDCl3): δ 2.34 (s, 1H), 2.01 (qt, J = 13.5, 3.4 Hz, 1H), 1.84 (dm, J = 13.0 Hz, 1H), 1.61 (dt, J = 13.5, 3.7 Hz, 1H), 1.55 (dm, J = 13.0 Hz, 1H), 1.47 (s, 3H), 1.46 (s, 3H), 1.43–1.28 (m, 2H), 1.18 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 89.0 (C), 73.5 (C), 71.3 (CH), 67.1 (C), 38.3 (CH2), 36.4 (CH2), 33.0 (Me), 32.6 (Me), 25.2 (Me), 17.5 (CH2).
GC–MS m/z (relative intensity): 137 (M+-Me, 100), 119 (11), 109 (76), 95 (32), 79 (69), 66 (81), 56 (62), 43 (83).
3.4.2. 2,6,6-Trimethyltetrahydro-2H-pyran-2-carboxylic Acid or Cinenic Acid 11
A heterogeneous mixture of the alkyne 18 (10 g, 65.7 mmol), sodium periodate (60 g, 280.5 mmol) CCl4 (50 mL), CH3CN (50 mL), water (75 mL), and a catalytic amount of RuCl3 hydrate (40% w/w Ru, 80 mg, 0.32 mmol) was vigorously stirred at rt. When the starting alkyne was no longer detectable by TLC analysis (24 h), the reaction was diluted with water (200 mL), acidified using diluted aqueous HCl and extracted with CH2Cl2 (3 × 120 mL). The combined organic phases were dried (Na2SO4) and were concentrated in vacuo. The residue was purified by chromatography using n-hexane/AcOEt (9:1–1:1) as an eluent to afford pure 2,6,6-trimethyltetrahydro-2H-pyran-2-carboxylic acid 11 as a colorless oil which crystallized on standing (10.2 g, 90% yield, 94% purity by GC–MS analysis). A sample of the acid was recrystallized from hexane. The collected crystals showed 98% purity by GC–MS analysis; mp: 83–84 °C; 1H-NMR, 13C-NMR, and GC–MS were superimposable to those reported above for the (+)-(S)-isomer.
3.4.3. Racemic (2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol 8
Ethyl chloroformate (5.5 mL, 57.5 mmol) was added dropwise at −10 °C to a stirred solution of acid 11 (9 g, 52.3 mmol) and Et3N (8 mL, 57.4 mmol) in dry THF (60 mL). After one hour, the precipitate triethylammonium chloride was filtered and the solid was washed with cold THF (10 mL). The combined liquid phases were added dropwise to a stirred solution of NaBH4 (5 g, 132.2 mmol) in water (50 mL) keeping the temperature below 10 °C by external cooling. After complete addition, the reaction was allowed to reach rt, before being stirred at this temperature for 4 h and quenched by acidification with diluted HCl (3% in water). The obtained mixture was extracted with diethyl ether (3 × 100 mL) and the combined organic phases were washed with saturated NaHCO3 solution and with brine, before being dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography using n-hexane/AcOEt (9:1–7:3) as an eluent to afford pure (2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol 8 (6.7 g, 81% yield) as a colorless oil; 1H-NMR, 13C-NMR, and GC–MS were superimposable to those reported above for the (+)-(S) isomer.
3.5. Enzyme-Mediated Resolution of (2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol
3.5.1. Determination of the Enantioselectivity in the Lipase-Catalyzed Acetylation of Racemic Alcohol 8
A solution of the racemic alcohol
8 (0.5 g, 3.16 mmol), lipase/esterase, vinyl acetate (5 mL), and
t-BuOMe (20 mL) was stirred at rt, and the formation of the acetylated compound was monitored by TLC analysis. The reaction was stopped at the reported conversion (see
Table 1) by filtration of the enzyme and evaporation of the solvent at reduced pressure. The residue was then purified by chromatography using hexane-acetate (9:1–7:3) as an eluent. The obtained acetate and the unreacted alcohol were bulb-to-bulb distilled in order to obtain solvent free samples suitable for the accurate measurement of their optical rotation values. The enantiomeric purity of the samples was determined comparing the measured optical rotation values with those of (
S)-(2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol
8 and (
S)-(2,6,6-trimethyltetrahydro-2
H-pyran-2-yl)methanol acetate (+)-
19 possessing 95% ee,
= +9.8 (
c 3.4, CHCl
3) and
= +7.9 (
c 2.9, CHCl
3), respectively.
Data analysis for acetate (+)-19: 1H NMR (400 MHz, CDCl3): δ 4.02 (d, J = 10.9 Hz, 1H), 3.86 (d, J = 10.9 Hz, 1H), 2.08 (s, 3H), 1.76–1.58 (m, 2H), 1.56–1.32 (m, 4H), 1.23 (s, 3H), 1.21 (s, 3H), 1.18 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 170.9 (C), 71.8 (C), 71.5 (C), 70.9 (CH2), 36.3 (CH2), 31.7 (CH2), 30.8 (Me), 29.7 (Me), 25.4 (Me), 20.9 (Me), 16.0 (CH2).
GC–MS m/z (relative intensity): 185 (M+-Me, 2), 127 (71), 109 (100), 97 (3), 81 (3), 69 (36), 56 (6), 43 (54).
3.5.2. Large-Scale Resolution of (2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol 8
A solution of the racemic alcohol 8 (10 g, 63.2 mmol), Novozym® 435 lipase (3 g), vinyl acetate (10 mL), and t-BuOMe (50 mL) was stirred at rt, and the formation of the acetylated compound was monitored by TLC analysis. The reaction was stopped at 65% conversion by filtration of the enzyme and evaporation of the solvent at reduced pressure. The residue was then purified by chromatography using hexane-acetate (9:1–7:3) as an eluent. The unreacted alcohol (−)-8 (3.4 g, 34% yield) showed the following analytical data: 96% chemical purity by GC–MS, = −10.1 (c 3.6, CHCl3), corresponding to 98% ee. The obtained acetate (+)-19 was treated with NaOH (6 g, 0.15 mol) in MeOH (80 mL) at reflux for 1 h. After the work-up procedure, the obtained alcohol was submitted again to the resolution procedure using lipase AK (4 g) as a catalyst, vinyl acetate (10 mL), and t-BuOMe (50 mL) allowing the acetylation reaction to reach a conversion of about 60%. The unreacted alcohol (+)-8 (3.5 g, 35% yield) showed the following analytical data: 97% chemical purity by GC–MS, = +10.1 (c 3.0, CHCl3), corresponding to 98% ee. The remaining acetate (3.0 g, 24% yield) could be hydrolyzed to recover further alcohol with low ee that could be used in a new resolution procedure.
3.6. Synthesis of the Enantiomeric Forms of 2,2,6-Trimethyl-6-vinyltetrahydro-2H-pyran
3.6.1. (R)-2,6,6-Trimethyltetrahydro-2H-pyran-2-carbaldehyde (−)-20
A solution of Py·SO3 complex (2.7 g, 17 mmol) in dry DMSO (10 mL) was added in one portion to a stirred solution of alcohol (−)-8 (1 g, 6.32 mmol, 98% ee) and Et3N (10 mL, 72 mmol) in dry DMSO (15 mL). After complete transformation of the starting alcohol (by TLC analysis, 2 h), the reaction was quenched by addition of water (100 mL) followed by extraction with diethyl ether (2 × 80 mL). The combined organic phases were washed in turn with water, diluted HCl solution, and brine. The organic solution was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography using n-hexane/Et2O (95:5–8:2) as an eluent to afford pure (−)-(R)-2,6,6-trimethyltetrahydro-2H-pyran-2-carbaldehyde 20 (810 mg, 82% yield, 92% purity by GC–MS analysis) as a colorless oil.
= −44.7 (c 3.9, CHCl3)
1H NMR (400 MHz, CDCl3): δ 9.59 (d, J = 1.7 Hz, 1H), 2.15 (dm, J = 13.3 Hz, 1H), 1.61–1.35 (m, 4H), 1.30–1.17 (m, 1 H), 1.27 (s, 3H), 1.12 (s, 3H), 1.11 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 205.5 (C), 78.4 (C), 73.0 (C), 35.7 (CH2), 31.9 (Me), 29.2 (CH2), 25.8 (Me), 24.5 (Me), 16.6 (CH2).
GC–MS m/z (relative intensity): 141 (M+-Me, 2), 127 (60), 109 (100), 95 (3), 81 (4), 69 (75), 59 (8), 43 (54).
3.6.2. (R)-2,2,6-Trimethyl-6-vinyltetrahydro-2H-pyran (+)-4
A solution of trimethylaluminium in hexane (3.2 mL of a 1 M solution) was added dropwise, under a static atmosphere of nitrogen, to a stirred suspension of activated zinc dust (3.2 g, 48.9 mmol), CH2I2 (4.2 g, 15.7 mmol), and dry THF (20 mL). The temperature of the mixture was kept below 30 °C by external cooling until the exothermic reaction settled down. The stirring was prolonged at rt for a further 10 min; then, aldehyde (−)-20 (0.8 g, 5.12 mmol, 98% ee) in dry THF (3 mL) was added dropwise at 0 °C and the mixture was stirred for one further hour. The reaction was diluted with diethyl ether (100 mL) and was acidified with diluted HCl (3% in water). The ether was separated and the aqueous phase was extracted with further ether (60 mL). The combined organic phases were washed with water and with brine, before being dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography using n-pentane/Et2O (99:1–9:1) as an eluent to afford pure (+)-2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran 4 (0.61 g, 77% yield, 93% purity by GC–MS analysis) as a colorless oil. The bulb-to-bulb distillation (60–65 °C, 20 mmHg) of the latter compound afforded very pure (+)-4 (99% purity by GC–MS analysis).
= +8.9 (c 3.3, CHCl3)
1H NMR (400 MHz, CDCl3): δ 5.96 (ddd, J = 17.8, 11.0, 0.8 Hz, 1H), 4.99 (dd, J = 17.8, 0.8 Hz, 1H), 4.94 (dd, J = 11.0, 1.0 Hz, 1H), 1.89 (dm, J = 13.4 Hz, 1H), 1.78–1.65 (m, 1H), 1.62–1.51 (m, 1H), 1.50–1.32 (m, 3H), 1.20 (s, 3H), 1.19 (s, 3H), 1.18 (s, 3H).
13C NMR (100 MHz, CDCl3): δ 147.2 (CH), 110.2 (CH2), 73.4 (C), 72.2 (C), 36.6 (CH2), 33.0 (CH2), 32.3 (Me), 31.5 (Me), 27.5 (Me), 16.8 (CH2).
GC–MS m/z (relative intensity): 154 (M+, <1), 139 (100), 121 (42), 109 (24), 93 (11), 81 (59), 71 (64), 56 (29), 43 (47).
3.6.3. (S)-2,6,6-Trimethyltetrahydro-2H-pyran-2-carbaldehyde (+)-20
According to the procedure outlined for the synthesis of aldehyde (−)-20, alcohol (+)-8 (98% ee) gave, in 85% yield, aldehyde (+)-20 as a colorless oil with = +45.4 (c 3.1, CHCl3) and 95% chemical purity by GC; 1H-NMR, 13C-NMR, and GC–MS were superimposable to those reported for the (−)-(R)-isomer.
3.6.4 (S)-2,2,6-Trimethyl-6-vinyltetrahydro-2H-pyran (−)-4
According to the procedure outlined for the synthesis of ether (+)-4, aldehyde (+)-20 (98% ee) gave, in 70% yield, ether (−)-4 as a colorless oil with = −9.0 (c 2.1, CHCl3) and 95% chemical purity by GC; 13C-NMR, and GC–MS were superimposable to those reported for the (+)-(R)-isomer.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
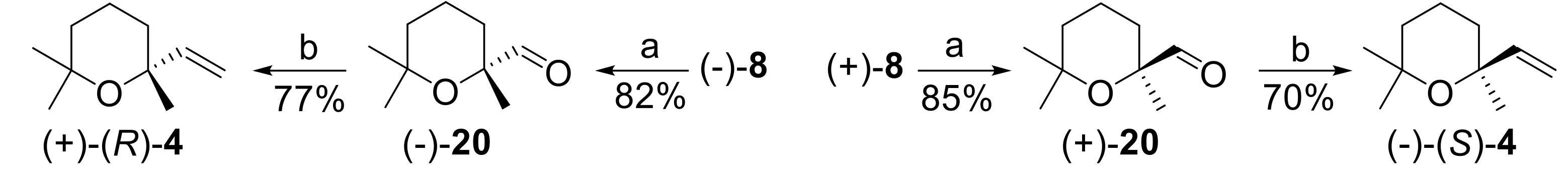{kind=link}