Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds



Departamento de Química Orgánica and Instituto de Síntesis Orgánica (ISO), Facultad de Ciencias, Universidad de Alicante, Apdo. 99, E-03080 Alicante, Spain
*
Authors to whom correspondence should be addressed.
Catalysts 2018, 8(5), 217; https://doi.org/10.3390/catal8050217
Submission received: 27 April 2018
/
Revised: 15 May 2018
/
Accepted: 16 May 2018
/
Published: 18 May 2018
(This article belongs to the Special Issue Catalyzed Synthesis of Natural Products)
Abstract
:The enantioselective α-amination of 1,3-dicarbonyl compounds has been performed using deep eutectic solvents (DES) as a reaction media and chiral 2-amino benzimidazole-derived compounds as a catalytic system. With this procedure, the use of toxic volatile organic compounds (VOCs) as reaction media is avoided. Furthermore, highly functionalized chiral molecules, which are important intermediates for the natural product synthesis, are synthetized by an efficient and stereoselective protocol. Moreover, the reaction can be done on a preparative scale, with the recycling of the catalytic system being possible for at least five consecutive reaction runs. This procedure represents a cheap, simple, clean, and scalable method that meets most of the principles to be considered a green and sustainable process.
1. Introduction
Asymmetric organocatalysis is an extremely attractive methodology for the preparation of functionalized chiral molecules and natural products, since small organic compounds are used as catalysts under very mild and simple reaction conditions [1,2,3]. Due to the lack of a metal element in the catalyst, organocatalytic methods are often used to prepare compounds that do not tolerate metal contamination such as pharmaceutical products. Asymmetric organocatalysis has become such an effective method of maintaining sustainability in organic synthesis as it provides many advantages, such as accessibility, low molecular weight, inexpensive catalysts and reduced toxicity.
Among the limited number of available green solvents, [4,5] deep eutectic solvents (DES) [6,7,8,9,10,11,12] maintain consistency within different criteria, such as availability, non-toxicity, inexpensiveness, high recyclability and low volatility. A deep eutectic solvent is a mixture between two or more components, one acting as hydrogen bond acceptor and the other as donor, having a melting point lower than the melting point of each one of the components. This behavior is due to hydrogen bond interactions between the acceptor and donor species. The use of DES as a reaction media is considered a new and expanding topic, which further assists and advances the importance of green chemistry. Recently, the association of these reaction media with asymmetric organocatalyzed processes [13,14] has been envisaged as a new and bright approach to advance sustainable processes.
Additionally, significant developments have been reached in the asymmetric electrophilic α-amination of carbonyl compounds through metal- or organo-catalyzed processes during recent years [15,16,17,18,19]. In fact, chiral carbonyl derivatives bearing stereogenic α-amine substitution are widely distributed among pharmaceutically active compounds. In particular, the organocatalyzed asymmetric α-amination of prochiral 1,3-dicarbonyl compounds have received great interest, since the resulting functionalized chiral molecules can be further elaborated allowing the synthesis of chiral biologically active natural products. However, this process remains unexplored with DES as reaction media.
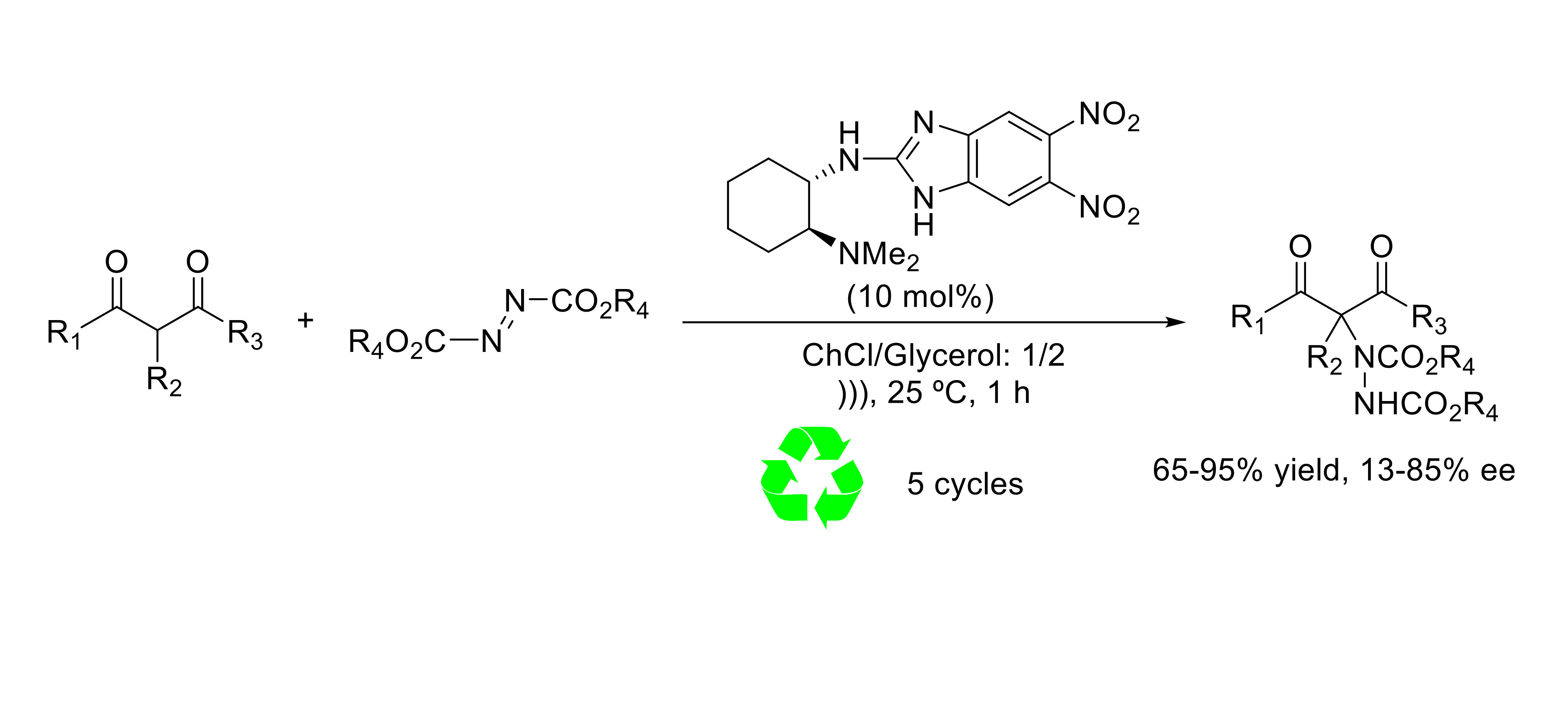
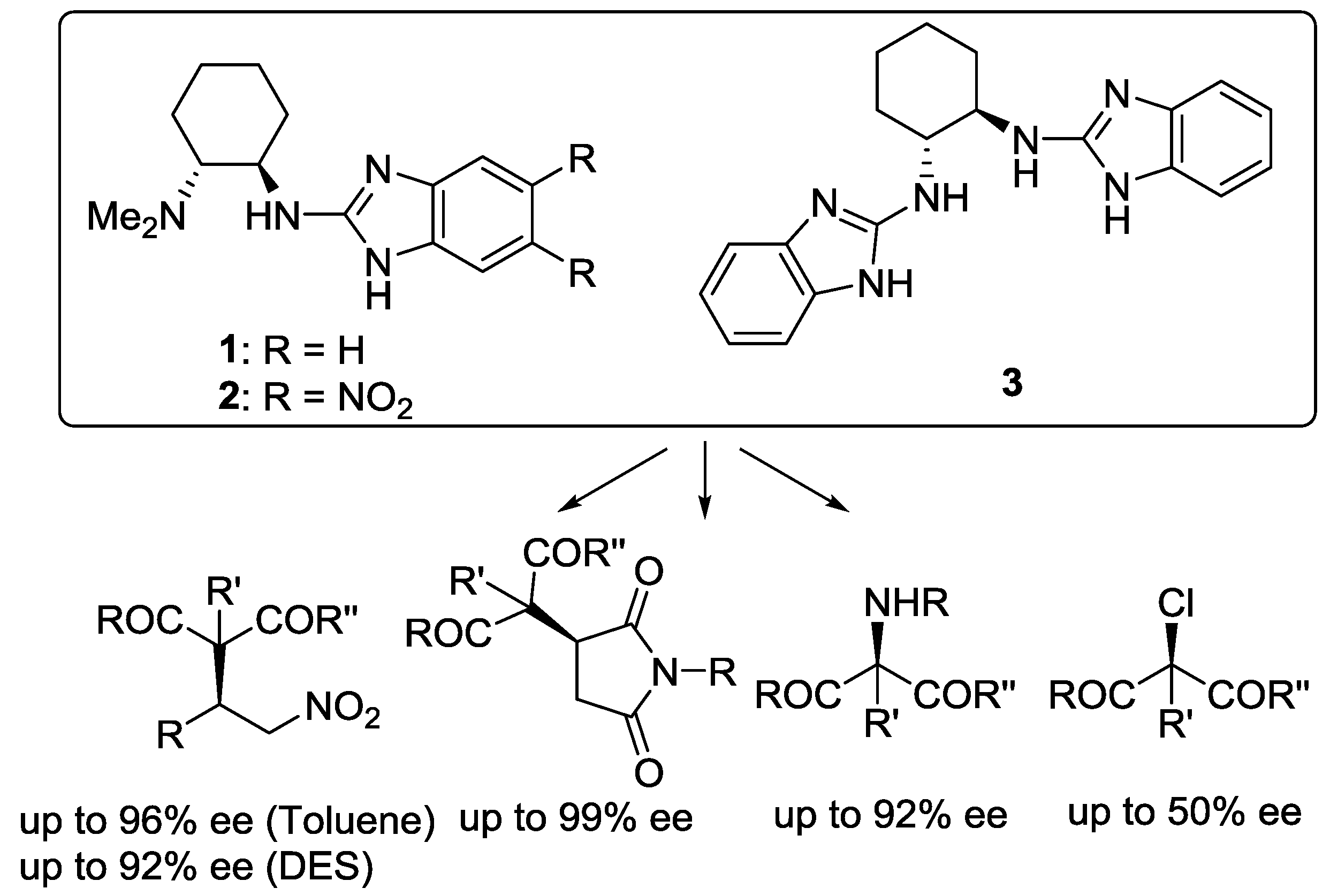
Our research group has established the practicality of bifunctional chiral 2-aminobenzimidazole derivatives [20,21] 1 and 3 (Scheme 1) as efficient organocatalysts in the asymmetric conjugate addition of 1,3-dicarbonyl compounds to nitroolefins [22] and maleimides [23,24] as well as in the α-functionalization [25,26,27,28] of these interesting nucleophiles using volatile organic solvents (VOCs) as a reaction medium. More fascinating, we have also demonstrated that the catalytic system based on the deep eutectic solvent choline chloride/glycerol and chiral 2-aminobenzimidazole organocatalysts 2 efficiently promotes the enantioselective addition of 1,3-dicarbonyl compounds to β-nitrostyrenes, avoiding the use of toxic VOC as reaction media [29].
For asymmetric organocatalyzed processes, the use of DES as a reaction medium has been barely studied, with the aldol reaction [30,31,32,33,34] and conjugated addition [13,29,35] being the main focus. For these processes a rational design of the organocatalyst and the right choice of the DES has shown to be critical to obtain good results and allow organocatalyst recycling.
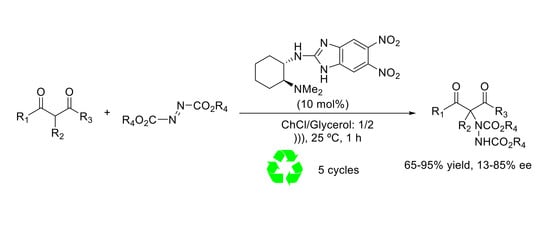
Herein, the use of chiral benzimidazole derivatives as organocatalysts for the electrophilic α-amination of 1,3-dicarbonyl compounds using DES as reaction media is presented.
2. Results and Discussion
Initially, the electrophilic α-amination of ethyl 2-oxocyclopentane-1-carboxylate with di-tert-butyl azodicarboxylate (DBAB) in the presence of catalyst 1 (10 mol %) in different choline chloride-based DES was investigated at 25 and 0 °C (Table 1). In general, good conversions and higher enantioselectivities were obtained at 0 °C, especially when using ChCl/urea (94%, 78% ee) and ChCl/glycerol (94%, 80% ee) as reaction media (Table 1, entries 2 and 6). The reaction time of the reaction was initially 4–5 h, however it was reduced to 1 h using ultrasounds (360 W) at 25 °C. The reduction in reaction time by the use of ultrasounds was previously observed in other related systems (i.e., ionic liquids), being attributed to physical-chemical effects [36,37]. As shown in Table 1, entries 12 and 13, under these conditions compound 4 was obtained with similar enantioselectivities with only a small erosion of the reaction conversion.
The optimization of the reaction medium resulted in the understanding that choline chloride/glycerol and choline chloride/urea were the best solvents to go forward with the conditions study using ultrasound irradiation at 25 °C. Next, the influence of the catalyst structure in the reaction results was studied: for this purpose, a series of several chiral benzimidazole-derived organocatalysts (as well other type of organocatalysts such as thiourea or sulphonamide derivatives, see Figure S1 in Supplementary Material) were tested in the α-amination model reaction under the optimized conditions using choline chloride/urea and choline chloride/glycerol as reaction media.
In both solvents, all chiral catalysts tested showed high performance achieving high reaction conversions (70–95%). However, different results concerning the enantioselectivities were encountered depending on the steric and/or electronic nature of the chiral organocatalyst. Chiral derivative 2, containing two strong electron-withdrawing nitro groups lead to the best results, giving product 4 in 84% ee in ChCl/urea and 82% ee in ChCl/glycerol (Table 2, entries 3 and 4). The presence of two nitro groups on the benzimidazole ring increases the hydrogen-bonding ability of 2 and as a consequence the interaction with the DES structure, leading to an improvement of the selectivity of the electrophilic amination. Conversely, a strong decrease in the enantioselectivity of the process was observed when using the sterically congested C2-symmetric chiral benzimidazoles 3 and 6, which afforded compound 4 with enantiomeric excess ranging from 33 to 44% (Table 2, entries 7–10). It can be concluded that in order to obtain good selectivity in the amination addition, it is crucial to have good correlation between the steric and electronic properties within the organocatalyst [38].
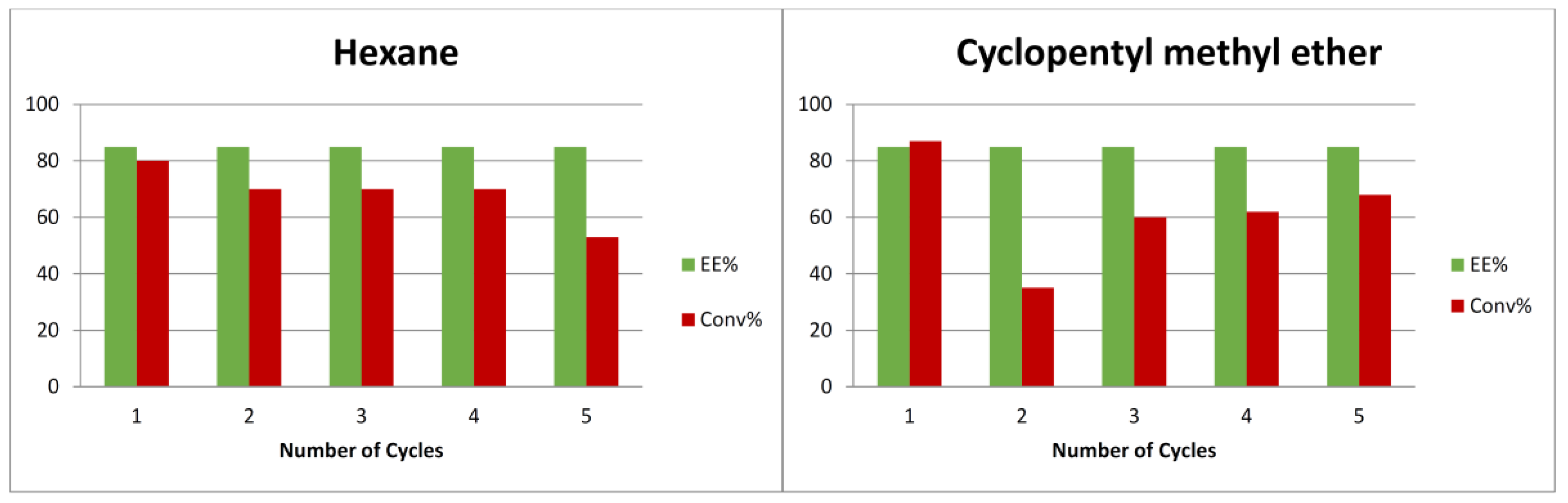
The recyclability of organocatalyst 2 and the eutectic liquid was performed in the model reaction under the optimized reaction conditions (Scheme 2). Therefore, to separate the DES/chiral organocatalyst mixture from the unreactive reagents and reaction products, hexane and cyclopentyl methyl ether were tested as extractive media. As shown in Scheme 2, the product was extracted and most of the catalyst remained in the DES, with the mixture being recovered and reused in five consecutive reaction runs, maintaining high enantioselectivity but with a decreased activity. Furthermore, vigorous stirring is mandatory when performing the extraction of the products to obtain a good recyclability results. For instance, in the second cycle of the cyclopentyl methyl ether recovering sequence (Scheme 2), a standard stirring was used and therefore a decrease in the conversion was observed. However, for the third run, again a vigorous stirring was applied and the conversion of the process was almost recovered.
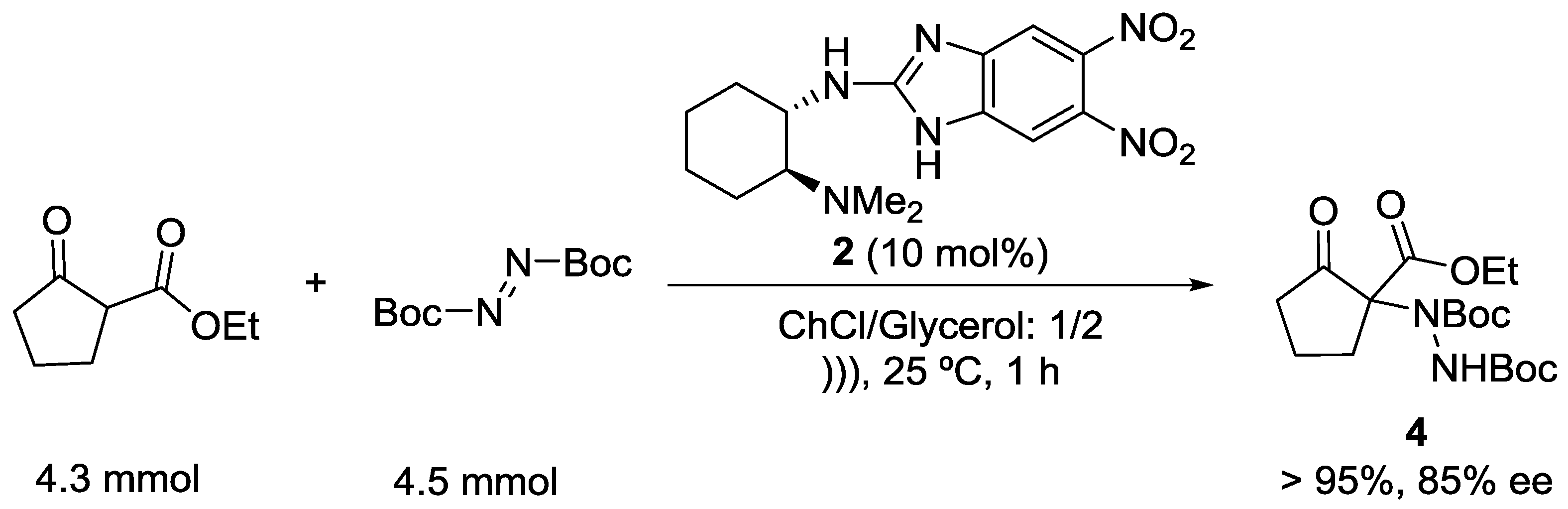
The efficiency and synthetic utility of 2 in ChCl/glycerol was further evaluated by performing a gram-scale experiment (4.3 mmol of ethyl 2-oxocyclopentane-1-carboxylate) for the synthesis of compound 4 which was obtained in a 95% yield and 85% ee (Scheme 3).
Lastly, the influence of different electrophiles and nucleophiles were assessed during the scope of the reaction. For this purpose, the different reactions were carried out under the optimized conditions using ChCl/glycerol as solvent (Table 3). Regarding the electrophile, an important steric effect was observed, being compound 4 obtained with the best enantioselectivity when using di-tert-butyl azodicarboxylate (DBAB) as electrophile (Table 3, entry 3). This electrophile was used for further studies.
The α-amination of other β-ketoesters such as, ethyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate, methyl 1-oxo-2,3-dihydro-1H-indene-2-carboxylate, methyl 1-oxo-1,2,3,4-tetrahydronaphthalene-2-carboxylate, and 3-acetyldihydrofuran-2(3H)-one was also assessed (Table 3, entries 5–7). In general, good isolated yields were obtained with low enantioselectivites (13 to 36% ee). Better enantioselection was observed in the α-amination with DBAB of 1,3-diketones, especially in the case of 2-acetylcyclopentan-1-one (Table 3, entry 9), which afforded compound 14 in a 75% isolated yield and 53% enantiomeric excess.
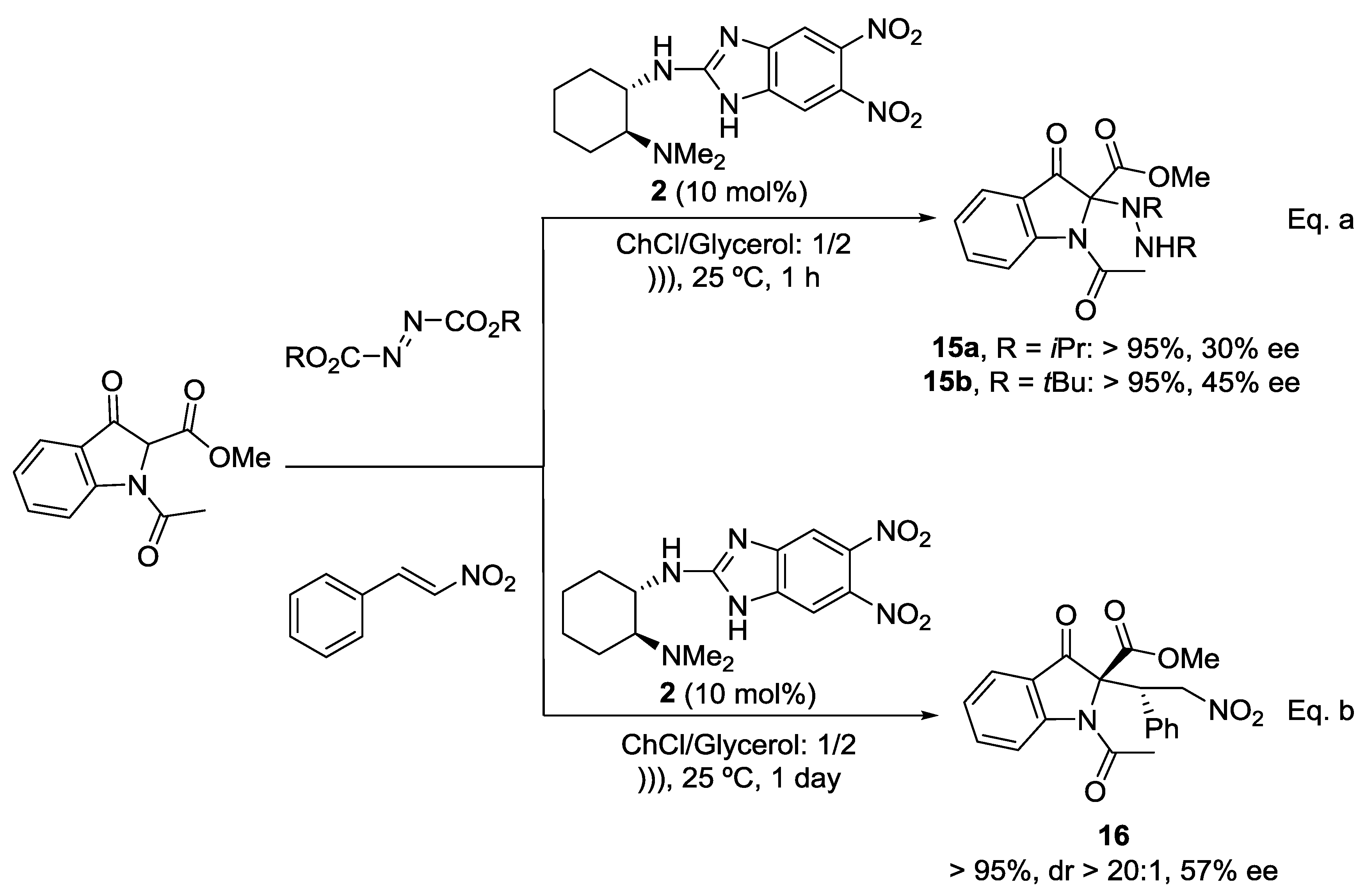
Due to accessibility, as well as green considerations, enantioselective organocatalysis has proved to be one of the most efficient approach towards the synthesis of drugs and natural products [39,40,41,42,43]. In particular, the organocatalytic functionalization of indolin-3-one has been recently studied since this type of heterocycles are commonly found in an ample range of biologically active natural alkaloids [44,45,46,47,48]. As depicted in Scheme 4 (Equation (a)), the 2-catalyzed electrophilic α-amination of methyl 1-acetyl-3-oxoindoline-2-carboxylate [49] in ChCl/glycerol (1/2) as solvent under the optimized reaction conditions gave compound 15 in excellent yields and moderate enantioselectivities (15a: R = iPr, 30% ee; 15b: R = tBu, 45% ee).
On the other hand, 2,2-disubstituted oxindole 16, which is a precursor of biologically active molecules containing indolin-3-ones with a quaternary stereocenter at the 2-position, such as Brevianamide A, Austamide, among others, has been prepared with excellent yield and diastereoselectivity and a 57% ee by the 2-catalyzed conjugate addition of methyl 1-acetyl-3-oxoindoline-2-carboxylate to β-nitrostyrene [50] (Scheme 4, Equation (b)). The use of Takemoto´s thiourea type catalyst for this transformation led to compound 16 with similar results (>95%, dr > 20:1, 63% ee).
3. Materials and Methods
3.1. General
Unless otherwise noted, all commercial reagents and solvents were used without further purification. Reactions under argon atmosphere were carried out in oven-dried glassware sealed with a rubber septum using anhydrous solvents. Melting points were determined with a hot plate apparatus and are uncorrected. 1H-NMR (300 or 400 MHz) and 13C-NMR (75 or 101 MHz) spectra were obtained on a Bruker AC-300 or AC-400(Bruke Corporation, Villerica, MA., USA), using CDCl3 as solvent and tetramethyl silane (TMS) (0.003%) as reference, unless otherwise stated. Chemical shifts (δ) are reported in ppm values relative to TMS and coupling constants (J) in Hz. Low-resolution mass spectra (MS) were recorded in the electron impact mode (EI, 70 eV, He as carrier phase) using an Agilent 5973 Network Mass Selective Detector spectrometer (Agilent Technologies, Santa Clara, CA, USA), being the samples introduced through a GC chromatograph Agilent 6890N (Agilent Technologies, Santa Clara, CA, USA) equipped with a HP-5MS column [(5%-phenyl)-methylpolysiloxane; length 30 m; ID 0.25 mm; film 0.25 mm]. IR spectra were obtained using a JASCO FT/IR 4100 spectrophotometer (Jasco Analytical Spain, Madrid, Spain) equipped with an ATR component; wavenumbers are given in cm−1. Analytical TLC was performed on Merck aluminium sheets with silica gel 60 F254. Analytical TLC was visualized with UV light at 254 nm Silica gel 60 (0.04–0.06 mm) was employed for flash chromatography whereas P/UV254 silica gel with CaSO4 (28–32%) supported on glass plates was employed for preparative TLC. Chiral High-performance liquid chromatography (HPLC) analyses were performed on an Agilent 1100 Series (Agilent Technologies, Santa Clara, CA, USA), (Quat Pump G1311A, DAD G1315B detector and automatic injector) equipped with chiral columns using mixtures of hexane/isopropanol as mobile phase, at 25 °C. The asymmetric reactions were sonicated in an ultrasounds P-Selecta instrument at 360 W.
3.2. Synthesis of Catalyst 2
Catalyst 1 [51] (50 mg, 0.2 mmol, 1 equiv.) was dissolved in concentrated H2SO4 (0.2 mL, 98%) and stirred vigorously for 5 minutes; concentrated HNO3 (0.4 mL, 65%) was then carefully added to the mixture at −20 °C. Then, the reaction was stirred at room temperature for 16 h. After this period, the mixture was treated with cold water and basified until pH 8 with a 25% aqueous solution of NH3. Finally, the aqueous phase was extracted with AcOEt (3 × 20 mL). The collected organic phases were dried over anhydrous MgSO4. After filtration, the organic solvent was removed under reduced pressure to give catalyst 2 without further purification as a red solid (74% yield, 52 mg, 0.15 mmol); mp 110–115 °C (CH2Cl2, decomposes); δH (300 MHz, CDCl3) 1.19–1.49 (m, 4H, 2 × CH2), 1.63–1.98 (m, 4H, 2 × CH2), 2.37 (s, 6H, 2 × Me), 2.51 (m, 1H, CHNMe2), 3.66 (bs, 1H, CHNH),7.49 (s, 2H, ArH); δC (75 MHz, CDCl3) 21.7, 24.4, 24.6, 33.2, 39.8, 53.8, 67.8, 108.3, 136.8, 142.0, 161.8; m/z 348 [M+, <1%] 128 (10), 126 (11), 125 (100), 124 (25), 84 (64), 71 (24), 58 (20), 44 (10).
3.3. Typical Procedure for the α-Amination Reaction
Catalyst 2 (5.22 mg, 0.015 mmol, 10 mol %) and ethyl 2-oxocyclopentane-1-carboxylate (23.4 mg, 0.15 mmol) were dissolved in a mixture of ChCl/Gly (1/2 molar ratio, 0.2 mL) and kept under stirring for 10 minutes at rt., followed by the addition of di-tert-butylazodicarboxylate (36.8 mg, 0.16 mmol). The reaction was vigorously stirred in ultrasounds for 1 h. After this period, water (3 mL) was added to the mixture and the reaction product was extracted with EtOAc (3 × 5 mL). The collected organic phases were dried over anhydrous MgSO4 and, after filtration, the solvent was evaporated under reduced pressure to give crude 4. Purification by flash column chromatography on silica gel (hexane/EtOAc: 7/3) afforded pure 4 (45.1 mg, 78% yield). δH (300 MHz, CDCl3) 1.28 (t, J = 7.1 Hz, 3H), 1.59–1.36 (m, 18H), 2.98–1.75 (m, 6H), 4.24 (m, 2H), 6.53 (br s, 1H) ppm). The enantiomeric excess of 4 was determined by chiral HPLC analysis (Chiralpack IA, hexane/EtOH: 96/04, 0.7 mL/min).
3.4. Typical Procedure for the Recovery of the Catalyst in the α-Amination Reaction
A mixture of catalyst 2 (5.22 mg, 0.015 mmol, 10 mol %) and ethyl 2-oxocyclopentane-1-carboxylate (23.4 mg, 0.15 mmol) in ChCl/Gly (1/2 molar ratio, 0.2 mL) was stirred for 10 minutes at rt. Then, di-tert-butylazodicarboxylate (36.8 mg, 0.16 mmol) was added. The reaction was vigorously stirred in ultrasounds for 1 h. After this period, the corresponding organic solvent was added (3 mL) and the mixture was stirred for 10 minutes at rt. The stirring was then stopped to allow phase separation and the upper organic layer was removed. This extractive procedure was repeated two more times and the combined organic extracts were washed with water (3 × 5 mL), dried (MgSO4), filtered, and evaporated under reduced pressure. Then, the next reaction cycle was performed with the obtained DES/2 mixture, adding fresh ethyl 2-oxocyclopentane-1-carboxylate and di-tert-butylazodicarboxylate. This reaction mixture was subjected again to the above-described procedure and further reaction cycles were repeated using the recycled deep eutectic solvent phase.
4. Conclusions
The enantioselective electrophilic α-amination of 1,3-dicarbonyl compounds with diazodicarboxylates catalyzed by the bifunctional chiral 2-aminobenzimidazole-derivative 2 has been carried out in choline chloride/glycerol or choline chloride/urea deep eutectic solvents. The protocols presented are simple, cheap, clean and scalable. Moreover, the recovery and reuse of the catalyst and reaction medium can be performed at least five times, achieving high and similar enantioselectivities. The synthesis of two natural product precursors is possible by the application of this procedure, as well as the conjugate addition to β-nitrostyrene. With these results, it has been shown that the combination of organocatalyzed enantioselective organic processes in deep eutectic solvents as a reaction media are a clear example of a green, bio-renewable and sustainable process.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/8/5/217/s1.
Author Contributions
D.R.Ñ. and P.K. performed the synthetic works. G.G. and D.A.A. designed the experiments of the project and supervised the whole studies reported in the manuscript. D.R.Ñ., P.K., G.G. and D.A.A. wrote the manuscript.
Acknowledgments
Financial support from the University of Alicante (UAUSTI16-03, UAUSTI16-10, VIGROB-173), the Spanish Ministerio de Economía, Industria y Competitividad (CTQ2015-66624-P) is acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest.
References and Note
- Mahrwald, R. Enantioselective Organocatalyzed Reactions; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis—From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Dalko, P. Enantioselective Organocatalysis; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Clarke, C.J.; Tu, W.C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Kerton, F.M. Solvent Systems for Sustainable Chemistry. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Liu, Y.; Friesen, J.B.; McAlpine, J.B.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Natural Deep Eutectic Solvents: Properties, Applications, and Perspectives. J. Nat. Prod. 2018, 81, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; De Oliveira Vigier, K.; Royera, S.; Jerome, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, J. Deep eutectic solvents and their applications as new green and biorenewable reaction media. In Handbook of Solvents; Wypych, G., Ed.; ChemTec Publishing: Toronto, ON, Canada, 2014; Volume 2, pp. 813–844. [Google Scholar]
- Guajardo, N.; Müller, C.R.; Schrebler, R.; Carlesi, C.; Domínguez de María, P. Deep eutectic solvents for organocatalysis, biotransformations, and multistep organocatalyst/enzyme combinations. ChemCatChem 2016, 8, 1020–1027. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.P.; Ramón, D.J. Deep Eutectic Solvents: The Organic Reaction Medium of the Century. Eur. J. Org. Chem. 2016, 2016, 612–632. [Google Scholar] [CrossRef]
- García-Álvarez, J. Deep Eutectic Mixtures: Promising Sustainable Solvents for Metal-Catalysed and Metal-Mediated Organic Reactions. Eur. J. Inorg. Chem. 2015, 2015, 5147–5157. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Hevia, E.; Capriati, V. Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities. Eur. J. Org. Chem. 2015, 2015, 6779–6799. [Google Scholar] [CrossRef] [Green Version]
- Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F.M. Stereoselective organocatalysed reactions in deep eutectic solvents: Highly tunable and biorenewable reaction media for sustainable organic synthesis. Green Chem. 2016, 18, 792–797. [Google Scholar] [CrossRef]
- Branco, L.C.; Faisca Phillips, A.M.; Marques, M.M.; Gago, S.; Branco, P.S. Recent advances in sustainable organocatalysis. In Recent Advances in Organocatalysis; Karame, I., Srour, H., Eds.; InTech: Rijeka, Croatia, 2016; pp. 141–182. ISBN 978-953-51-2673-7. [Google Scholar]
- Genet, J.-P.; Creck, C.; Lavergne, D. Modern Amination Methods; Ricci, A., Ed.; Wiley-VCH: Weinheim, Germany, 2000; Chapter 3. [Google Scholar]
- Amino Group Chemistry: From Synthesis to the Life Sciences; Ricci, A. (Ed.) Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Janey, J.M. Recent Advances in Catalytic, Enantioselective α-Aminations and α-Oxygenations of Carbonyl Compounds. Angew. Chem. Int. Ed. 2005, 44, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.R.; Hii, K.K. Transition Metal Catalyzed Enantioselective α-Heterofunctionalization of Carbonyl Compounds. Chem. Rev. 2011, 111, 1637–1656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liao, F.-M.; Yu, J.-S.; Zhou, J. Catalytic Asymmetric Electrophilic Amination Reactions to Form Nitrogen-Bearing Tetrasubstituted Carbon Stereocenters. Synthesis 2014, 46, 2983–3003. [Google Scholar] [CrossRef]
- Khose, V.N.; John, M.E.; Pandey, A.D.; Karnik, A.V. Chiral benzimidazoles and their applications in stereodiscrimination processes. Tetrahedron Asymmetry 2017, 28, 1233–1289. [Google Scholar] [CrossRef]
- Nájera, C.; Yus, M. Chiral benzimidazoles as hydrogen bonding organocatalysts. Tetrahedron Lett. 2015, 56, 2623–2633. [Google Scholar] [CrossRef] [Green Version]
- Almaşi, D.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Chiral 2-Aminobenzimidazoles as Recoverable Organocatalysts for the Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. J. Org. Chem. 2009, 74, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides Using a Chiral C2-Symmetric Bis(2-aminobenzimidazole) as Recyclable Organocatalyst. Org. Lett. 2011, 13, 6106–6109. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Synthesis of Succinimides by Michael Addition of 1,3-Dicarbonyl Compounds to Maleimides Catalyzed by a Chiral Bis(2-aminobenzimidazole) Organocatalyst. Eur. J. Org. Chem. 2013, 2013, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Martínez, M.; Alonso, D.A.; Pastor, I.P.; Guillena, G.; Baeza, A. Organocatalyzed Assembly of Chlorinated Quaternary Stereogenic Centers. Asian J. Org. Chem. 2016, 5, 1428–1437. [Google Scholar] [CrossRef]
- Sánchez, D.; Baeza, A.; Alonso, D. Organocatalytic Asymmetric α-Chlorination of 1,3-Dicarbonyl Compounds Catalyzed by 2-Aminobenzimidazole Derivatives. Symmetry 2016, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Trillo, P.; Gómez-Martínez, M.; Alonso, D.A.; Baeza, A. 2-Aminobenzimidazole Organocatalyzed Asymmetric Amination of Cyclic 1,3-Dicarbonyl Compounds. Synlett 2015, 26, 95–100. [Google Scholar]
- Benavent, L.; Puccetti, F.; Baeza, A.; Gómez-Martínez, M. Readily Available Chiral Benzimidazoles-Derived Guanidines as Organocatalysts in the Asymmetric α-Amination of 1,3-Dicarbonyl Compounds. Molecules 2017, 22, 1333. [Google Scholar] [CrossRef] [PubMed]
- Ñíguez, D.R.; Guillena, G.; Alonso, D.A. Chiral 2-Aminobenzimidazoles in Deep Eutectic Mixtures: Recyclable Organocatalysts for the Enantioselective Michael Addition of 1,3-Dicarbonyl Compounds to β-Nitroalkenes. ACS Sustain. Chem. Eng. 2017, 5, 10649–10656. [Google Scholar] [CrossRef]
- Muller, C.R.; Meiners, I.; Domínguez de María, P. Highly enantioselective tandem enzyme–organocatalyst crossed aldol reactions with acetaldehyde in deep-eutectic-solvents. RSC Adv. 2014, 4, 46097–46101. [Google Scholar] [CrossRef]
- Muller, C.R.; Rosen, A.; Domínguez de María, P. Multi-step enzyme-organocatalyst C–C bond forming reactions in deep-eutectic-solvents: Towards improved performances by organocatalyst design. Sustain. Chem. Process 2015, 3, 12–20. [Google Scholar] [CrossRef]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef] [Green Version]
- Fanjul-Mosteirín, N.; Concellon, C.; del Amo, V. l-Isoleucine in a choline chloride/ethylene glycol deep eutectic solvent: A reusable reaction kit for the asymmetric cross-aldol carboligation. Org. Lett. 2016, 18, 4266–4269. [Google Scholar] [CrossRef] [PubMed]
- Brenna, D.; Massolo, E.; Puglisi, A.; Rossi, S.; Celentano, G.; Benaglia, M.; Capriati, V. Towards the development of continuous, organocatalytic, and stereoselective reactions in deep eutectic solvents. Beilstein J. Org. Chem. 2016, 12, 2620–2626. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ferrándiz, J.; Chinchilla, R. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides in deep eutectic solvents. Tetrahedron Asymmetry 2017, 28, 302–306. [Google Scholar] [CrossRef]
- Mason, T.J. Ultrasound in synthetic organic chemistry. Chem. Soc. Rev. 1997, 26, 443–451. [Google Scholar] [CrossRef]
- Chatel, G.; MacFarlane, D.R. Ionic liquids and ultrasound in combination: Synergies and challenges. Chem. Soc. Rev. 2014, 43, 8132–8149. [Google Scholar] [CrossRef] [PubMed]
- For the full catalyst optimization study, see the SI.
- Merad, J.; Lalli, C.; Bernadat, G.; Maury, J.; Masson, G. Enantioselective Brønsted Acid Catalysis as a Tool for the Synthesis of Natural Products and Pharmaceuticals. Chem. Eur. J. 2018, 24, 3925–3943. [Google Scholar] [CrossRef] [PubMed]
- Dibello, E.; Gamenara, D.; Seoane, G. Organocatalysis in the Synthesis of Natural Products: Recent Developments in Aldol and Mannich Reactions, and 1,4-Conjugated Additions. Curr. Organocatal. 2015, 2, 124–149. [Google Scholar] [CrossRef]
- Sun, B.-F. Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions. Tetrahedron Lett. 2015, 2133–2140. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Chen, D.F.; Gong, L.-Z. Organocatalytic asymmetric synthesis of chiral nitrogenous heterocycles and natural products. Pure Appl. Chem. 2014, 86, 1217–1226. [Google Scholar] [CrossRef]
- Abbasov, M.E.; Romo, D. The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis. Nat. Prod. Rep. 2014, 31, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Xing, H.; Yu, D.-Q.; Liu, H.-M. Catalytic asymmetric synthesis of biologically important 3-hydroxyoxindoles: An update. Beilstein J. Org. Chem. 2016, 12, 1000–1039. [Google Scholar] [CrossRef] [PubMed]
- Macaev, F.Z.; Sucman, N.S.; Boldescu, V.V. Selective transformations of isatins to substituted 2-oxindoles. Russ. Chem. Bull. 2014, 63, 15–25. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Recent advances in organocatalytic methods for the synthesis of disubstituted 2- and 3-indolinones. Chem. Soc. Rev. 2012, 41, 7247–7290. [Google Scholar] [CrossRef] [PubMed]
- Badillo, J.J.; Hanhan, N.V.; Franz, A.K. Enantioselective synthesis of substituted oxindoles and spirooxindoles with applications in drug discovery. Curr. Opin. Drug. Discov. Dev. 2010, 13, 758–776. [Google Scholar]
- Chauhan, P.; Chimni, S.S. Organocatalytic asymmetric synthesis of 3-amino-2-oxindole derivatives bearing a tetra-substituted stereocenter. Tetrahedron Asymmetry 2013, 24, 343–356. [Google Scholar] [CrossRef]
- Yarlagadda, S.; Ramesh, B.; Reddy, C.R.; Srinivas, L.; Sridhar, B.; Subba Reddy, B.V. Organocatalytic Enantioselective Amination of 2-Substituted Indolin-3-ones: A Strategy for the Synthesis of Chiral α-Hydrazino Esters. Org. Lett. 2017, 19, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.-Y.; Wang, Y.; Liu, Y.-Z.; Shen, C.; Xu, P.-F. Organocatalytic Asymmetric Michael Addition of Oxindoles to Nitroolefins for the Synthesis of 2,2-Disubstituted Oxindoles Bearing Adjacent Quaternary and Tertiary Stereocenters. J. Org. Chem. 2012, 77, 11307–11312. [Google Scholar] [CrossRef] [PubMed]
- For the synthesis of catalyst 1 see SI.
Scheme 1.
Chiral benzimidazoles in asymmetric organocatalysis.

Scheme 2.
Recycling studies.


Scheme 3.
Gram-scale α-amination of ethyl 2-oxocyclopentane-1-carboxylate catalyzed by 2.

Scheme 4.
Asymmetric organocatalyzed functionalization of methyl 1-acetyl-3-indol-2-carboxylate in DES.
Scheme 4.
Asymmetric organocatalyzed functionalization of methyl 1-acetyl-3-indol-2-carboxylate in DES.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Asymmetric α-amination of ethyl 2-oxocyclopentane-1-carboxylate with di-tert-butyl azodicarboxylate (DBAB). Deep eutectic solvents (DES) study.
Table 1.
Asymmetric α-amination of ethyl 2-oxocyclopentane-1-carboxylate with di-tert-butyl azodicarboxylate (DBAB). Deep eutectic solvents (DES) study.

| Entry | DES | T (°C) | t (h) | Conversion (%) 1 | Ee (%) 2 |
|---|---|---|---|---|---|
| 1 | ChCl/Urea: 1/2 | 25 | 5 | 61 | 77 |
| 2 | ChCl/Urea: 1/2 | 0 | 5 | 94 | 78 |
| 3 | AcChCl/Urea: 1/2 | 25 | 5 | 64 | 72 |
| 4 | AcChCl/Urea: 1/2 | 0 | 5 | 55 | 75 |
| 5 | ChCl/Glycerol: 1/2 | 25 | 5 | 94 | 73 |
| 6 | ChCl/Glycerol: 1/2 | 0 | 5 | 94 | 80 |
| 7 | ChCl/Ethyleneglycol: 1/2 | 25 | 5 | 78 | 72 |
| 8 | ChCl/Ethyleneglycol: 1/2 | 0 | 5 | 84 | 70 |
| 9 | ChCl/Malic acid: 1/1 | 25 | 5 | <5 | nd |
| 10 | ChCl/Tartaric acid: 1/1 | 25 | 5 | 64 | 76 |
| 11 | ChCl/Tartaric acid: 1/1 | 0 | 5 | 58 | 77 |
| 12 | ChCl/Urea: 1/2 | 25 3 | 1 | 92 | 76 |
| 13 | ChCl/Glycerol: 1/2 | 25 3 | 1 | 80 | 80 |
1 Reaction conversion towards 4 determined by GC analysis. 2 Enantiomeric excess determined by chiral HPLC analysis. 3 Reaction performed under ultrasounds irradiation (360 W).
Table 2.
Asymmetric α-amination of ethyl 2-oxocyclopentane-1-carboxylate with DBAB. Catalyst study.

| Entry | Catalyst | DES | Conversion (%) 1 | Ee (%) 2 |
|---|---|---|---|---|
| 1 | 1 | ChCl/Urea: 1/2 | 92 | 76 |
| 2 | 1 | ChCl/Glycerol: 1/2 | 80 | 80 |
| 3 | 2 | ChCl/Urea: 1/2 | 85 | 84 |
| 4 | 2 | ChCl/Glycerol: 1/2 | 90 | 82 |
| 5 | 5 | ChCl/Urea: 1/2 | 95 | 74 |
| 6 | 5 | ChCl/Glycerol: 1/2 | 70 | 78 |
| 7 | 3 | ChCl/Urea: 1/2 | 90 | 40 |
| 8 | 3 | ChCl/Glycerol: 1/2 | 95 | 44 |
| 9 | 6 | ChCl/Urea: 1/2 | 92 | 40 |
| 10 | 6 | ChCl/Glycerol: 1/2 | 91 | 33 |
1 Reaction conversion towards 4 determined by GC analysis. 2 Enantiomeric excess determined by chiral HPLC analysis.
Table 3.
Asymmetric α-amination catalyzed by 3. Reaction scope.

| Entry | Dicarbonyl | Azodicarboxylate | Product | Yield (%) 1 | Ee (%) 2 |
|---|---|---|---|---|---|
| 1 |  | BocN=NBoc | 4 | 78 | 85 |
| 2 | iPrO2CN=NCO2iPr | 7 | 52 | 60 | |
| 3 | EtO2CN=NCO2Et | 8 | 76 | 65 | |
| 4 | BnO2CN=NCO2Bn | 9 | 0 | nd | |
| 5 |  | BocN=NBoc | 10 | 66 | 36 |
| 6 |  | BocN=NBoc | 11 | 65 | 35 |
| 7 |  | BocN=NBoc | 12 | 65 | 13 |
| 8 |  | BocN=NBoc | 13 | 68 | 25 |
| 9 |  | BocN=NBoc | 14 | 75 | 53 |
1 Isolated yield after flash chromatography. 2 Enantiomeric excess determined by chiral HPLC analysis.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ros Ñíguez, D.; Khazaeli, P.; Alonso, D.A.; Guillena, G. Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds. Catalysts 2018, 8, 217. https://doi.org/10.3390/catal8050217
AMA Style
Ros Ñíguez D, Khazaeli P, Alonso DA, Guillena G. Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds. Catalysts. 2018; 8(5):217. https://doi.org/10.3390/catal8050217
Chicago/Turabian StyleRos Ñíguez, Diego, Pegah Khazaeli, Diego A. Alonso, and Gabriela Guillena. 2018. "Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds" Catalysts 8, no. 5: 217. https://doi.org/10.3390/catal8050217
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.