Insights in the Rhodium-Catalyzed Tandem Isomerization-Hydroformylation of 10-Undecenitrile: Evidence for a Fast Isomerization Regime



Abstract
:1. Introduction
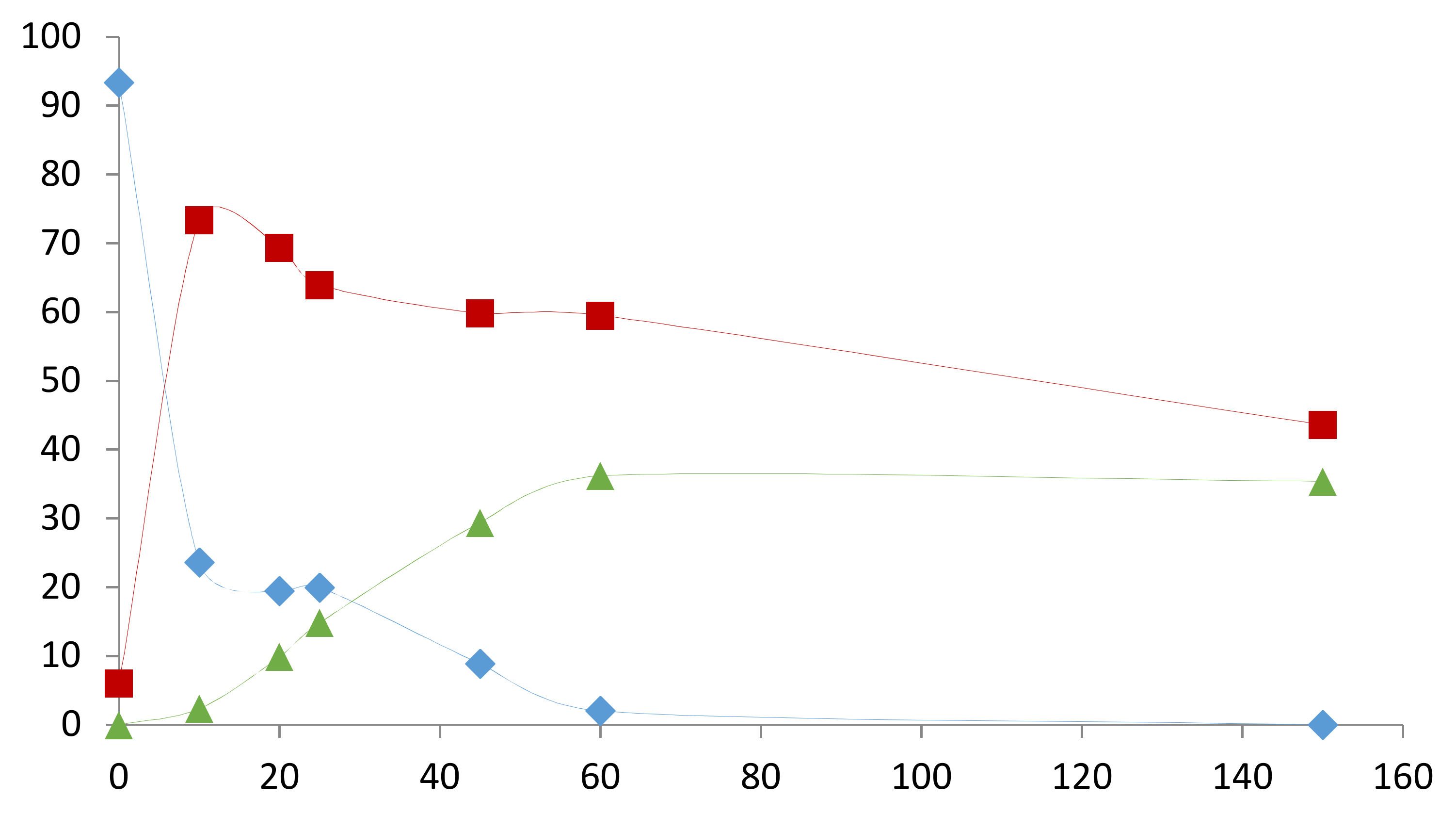
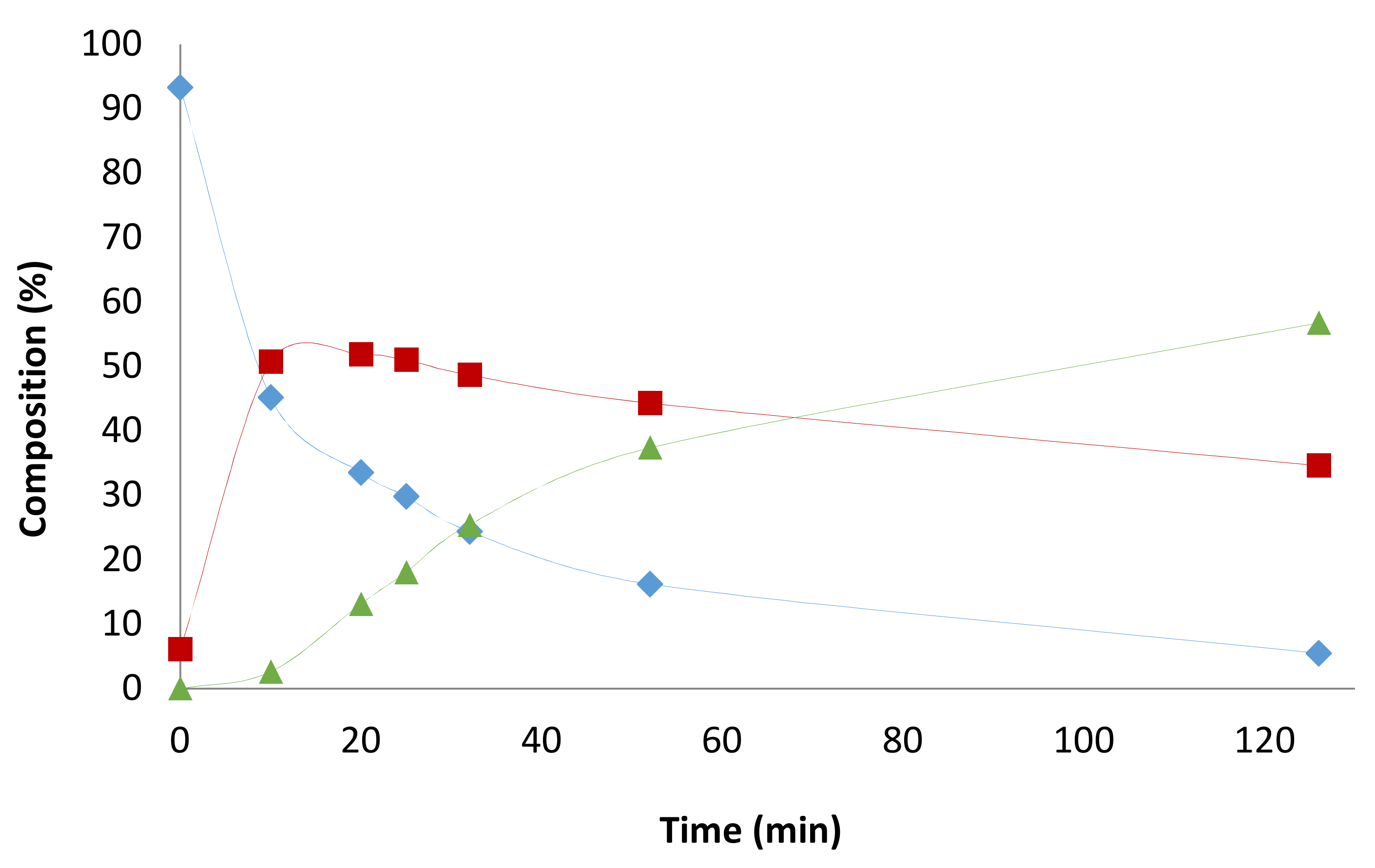
2. Results and Discussion
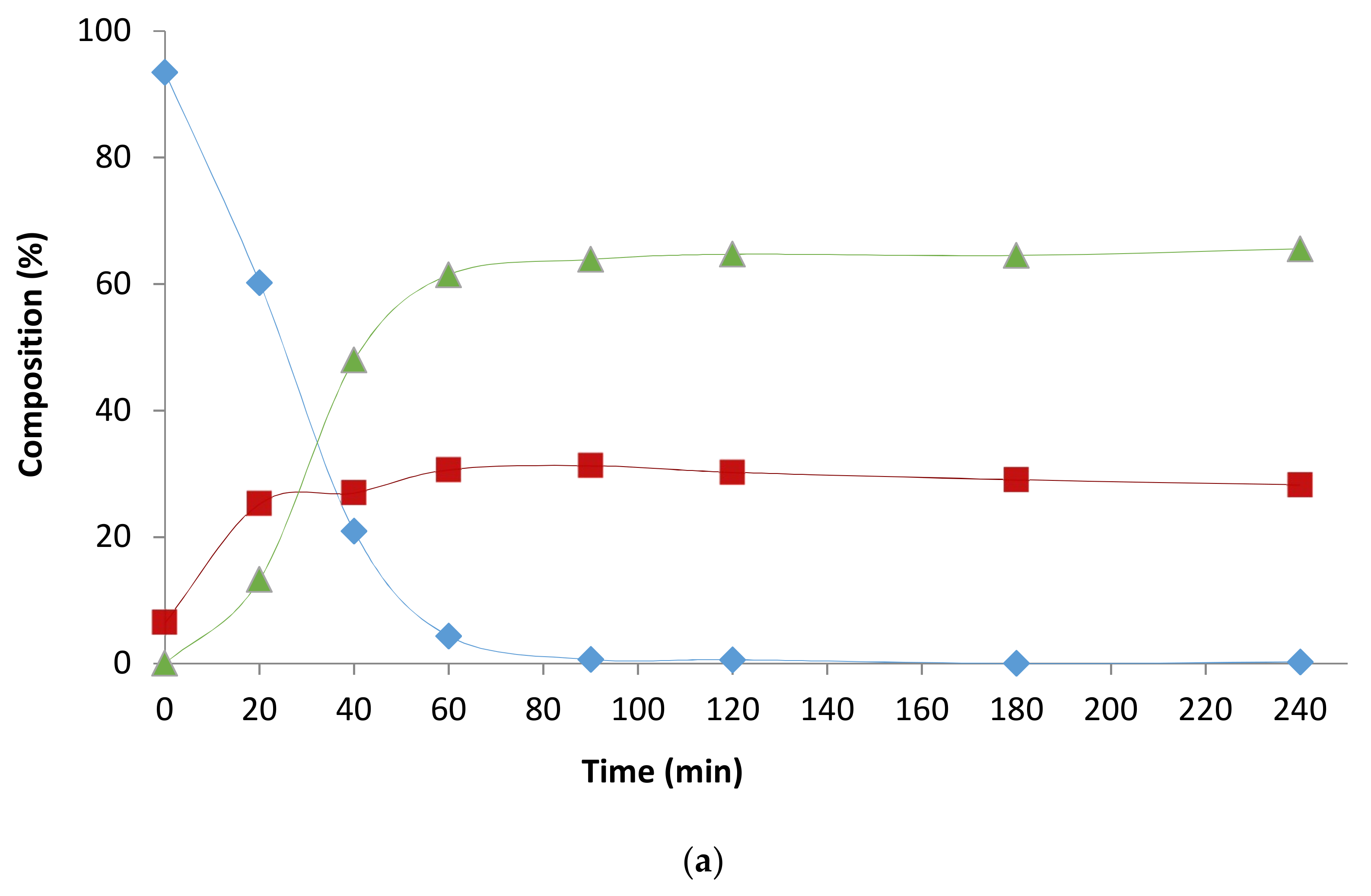
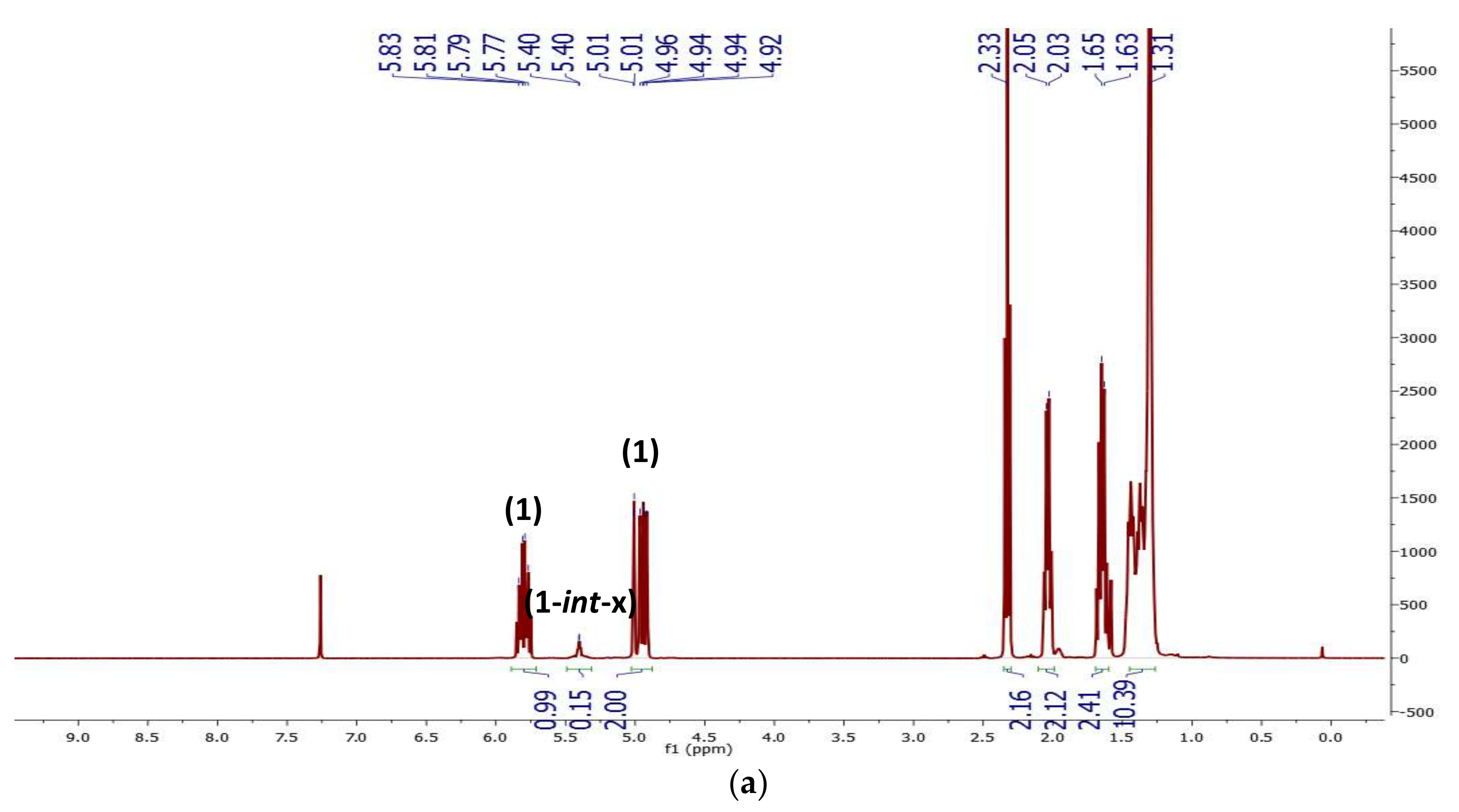
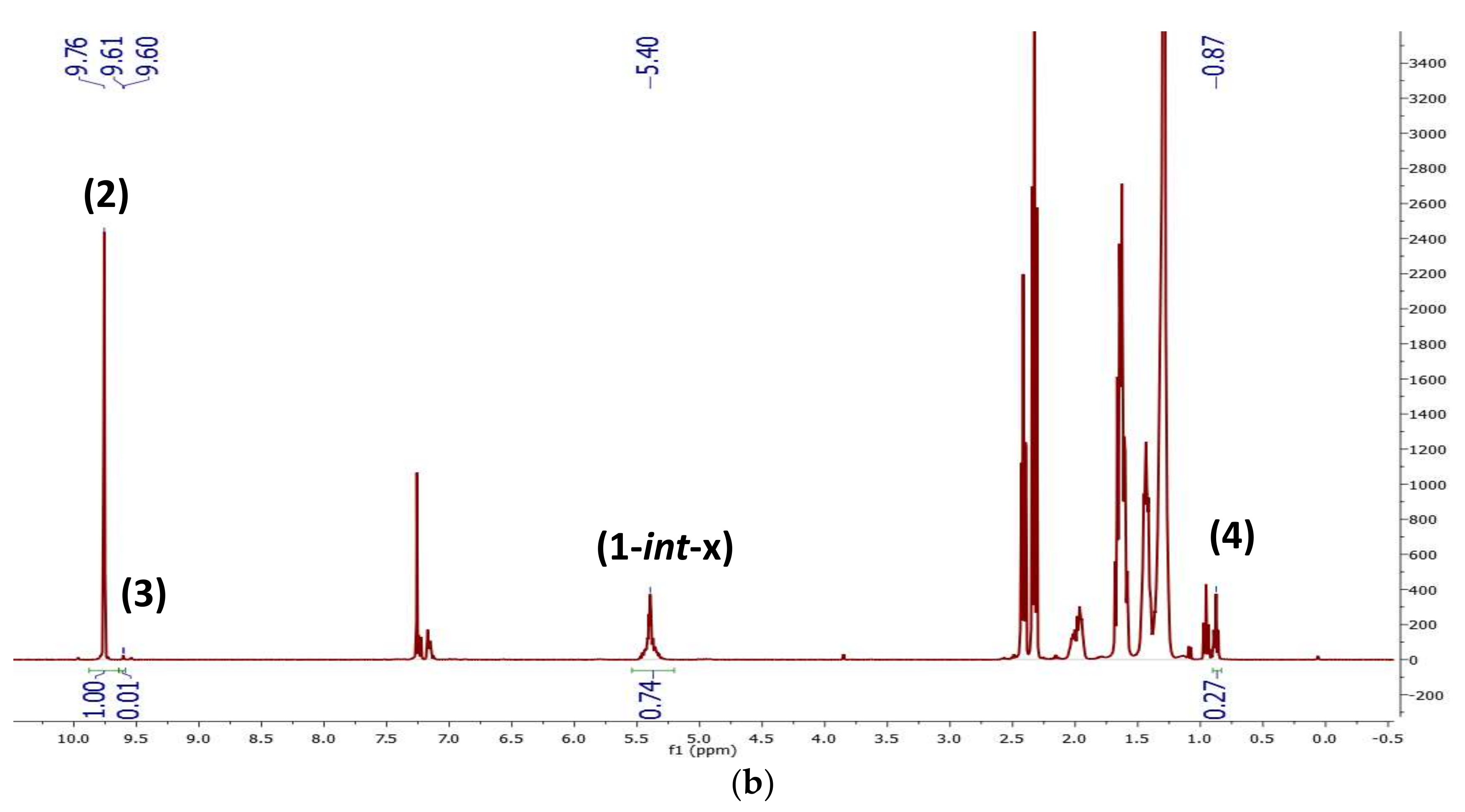
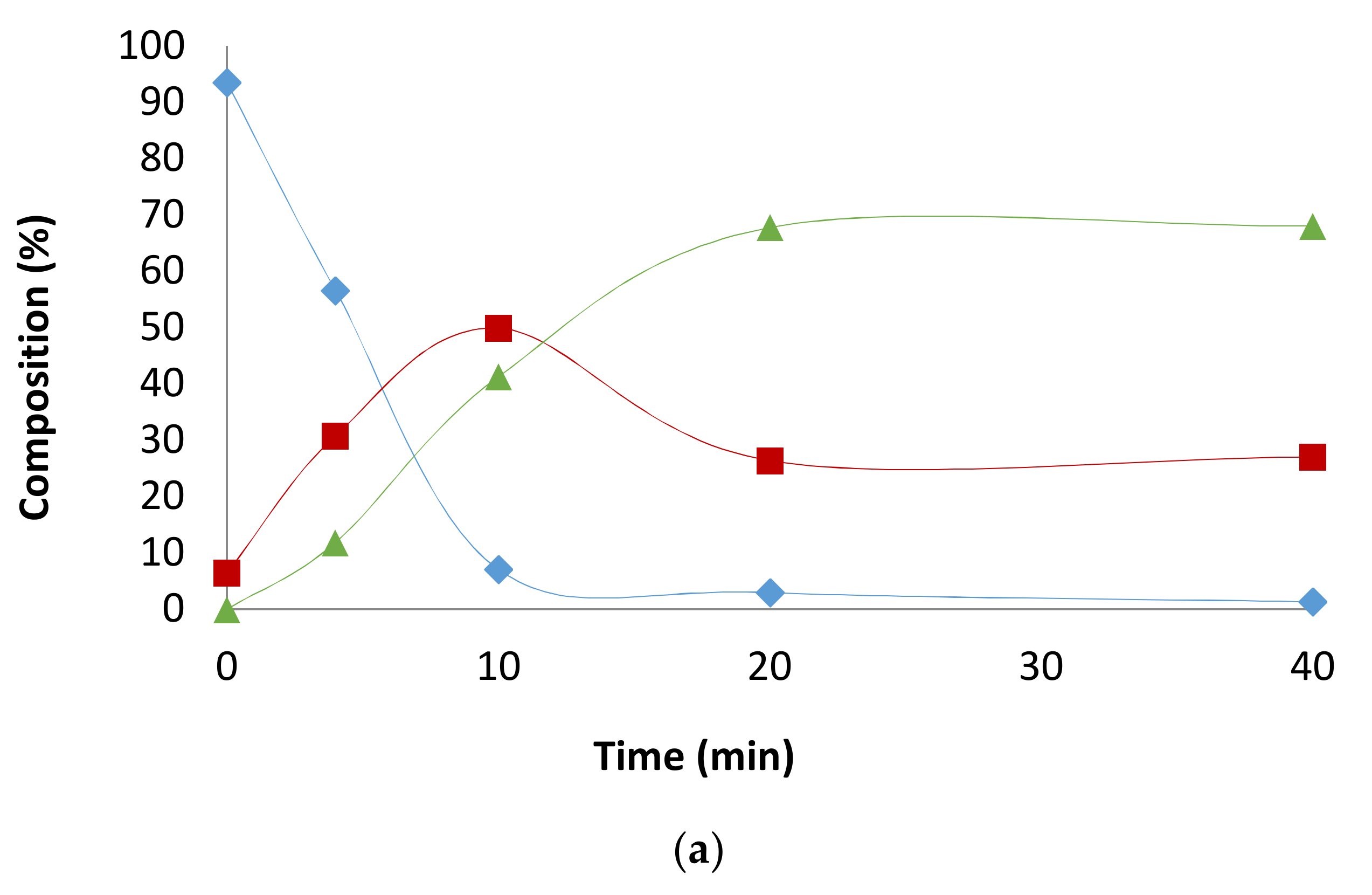
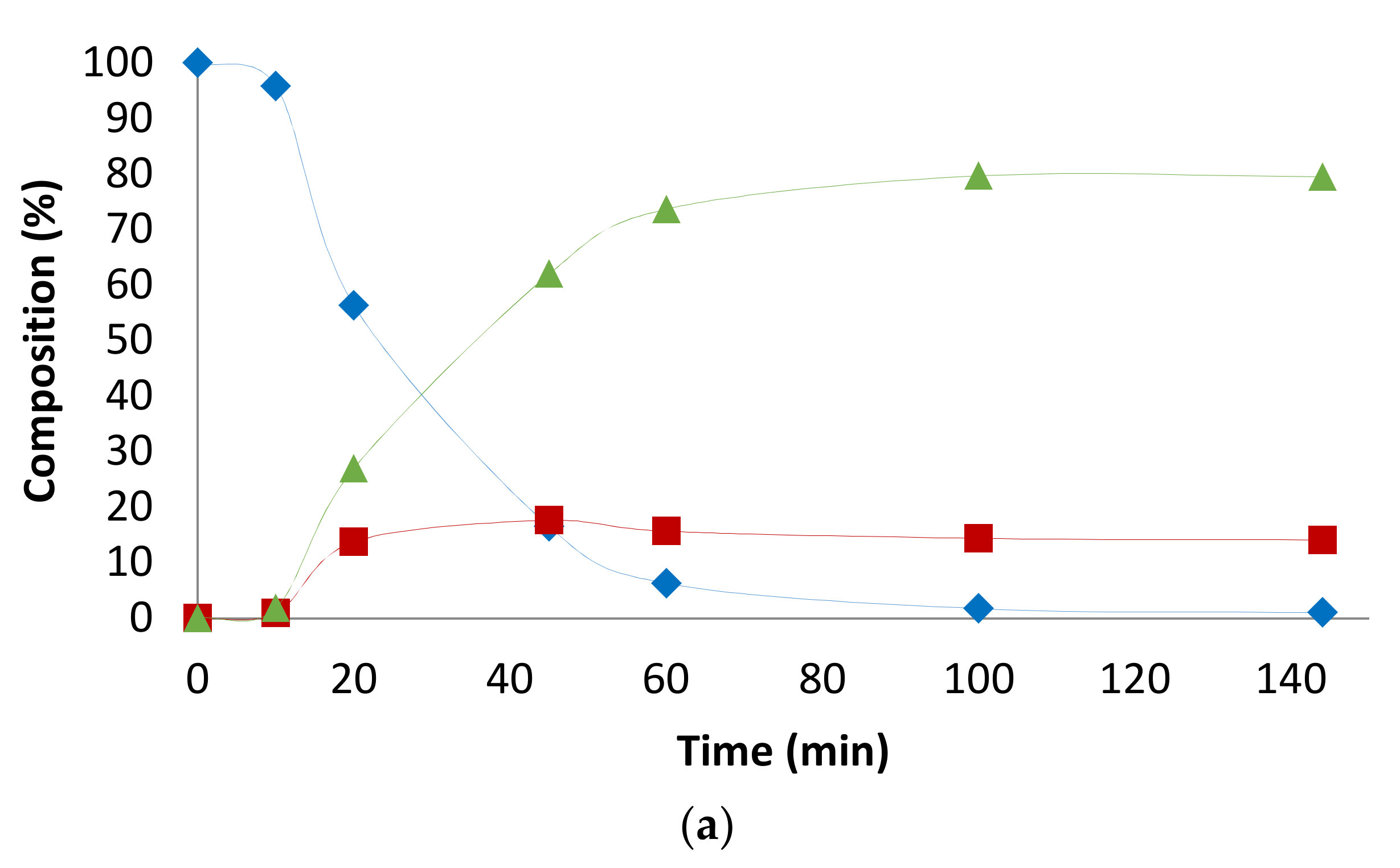
2.1. Preliminary Notes
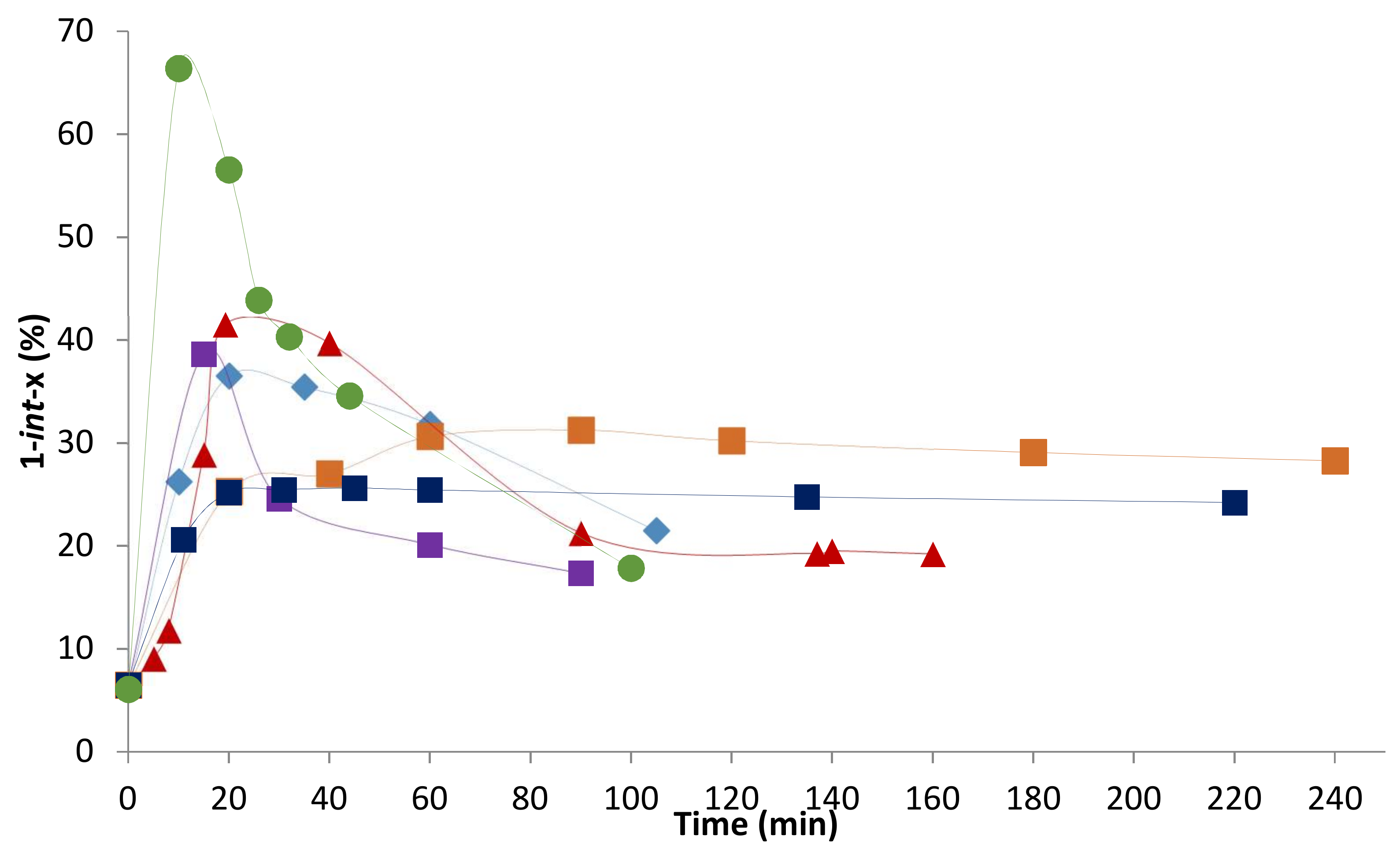
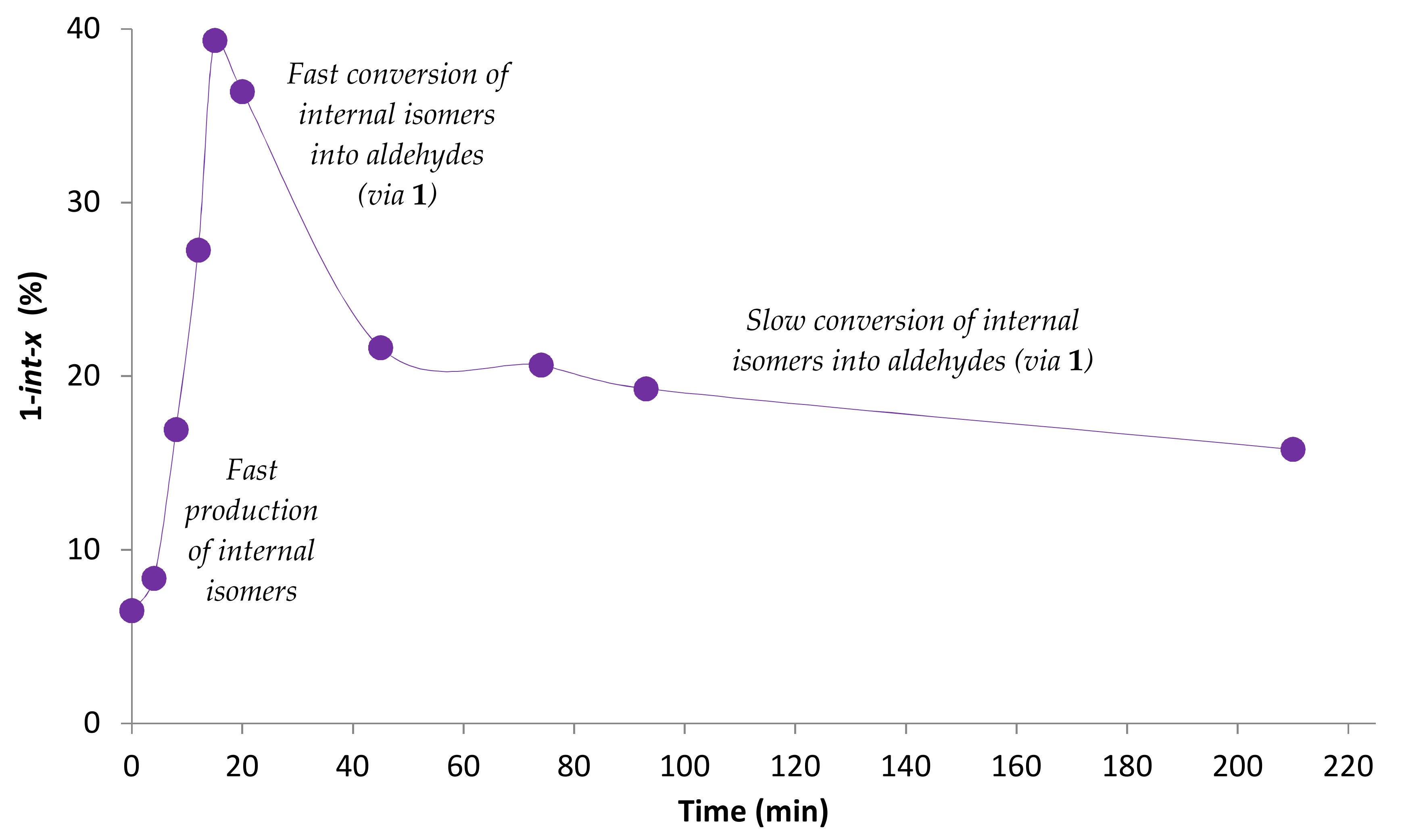
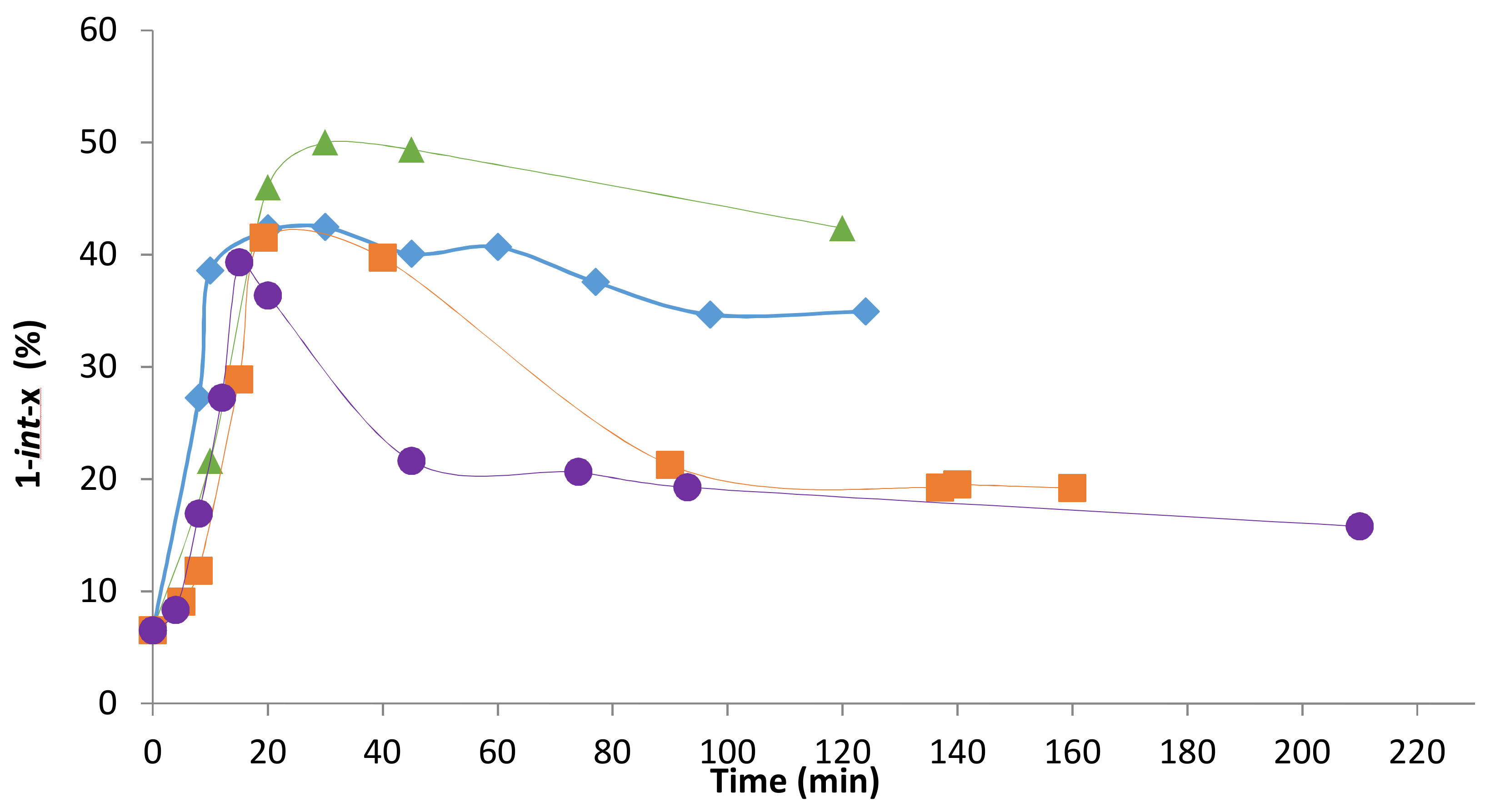
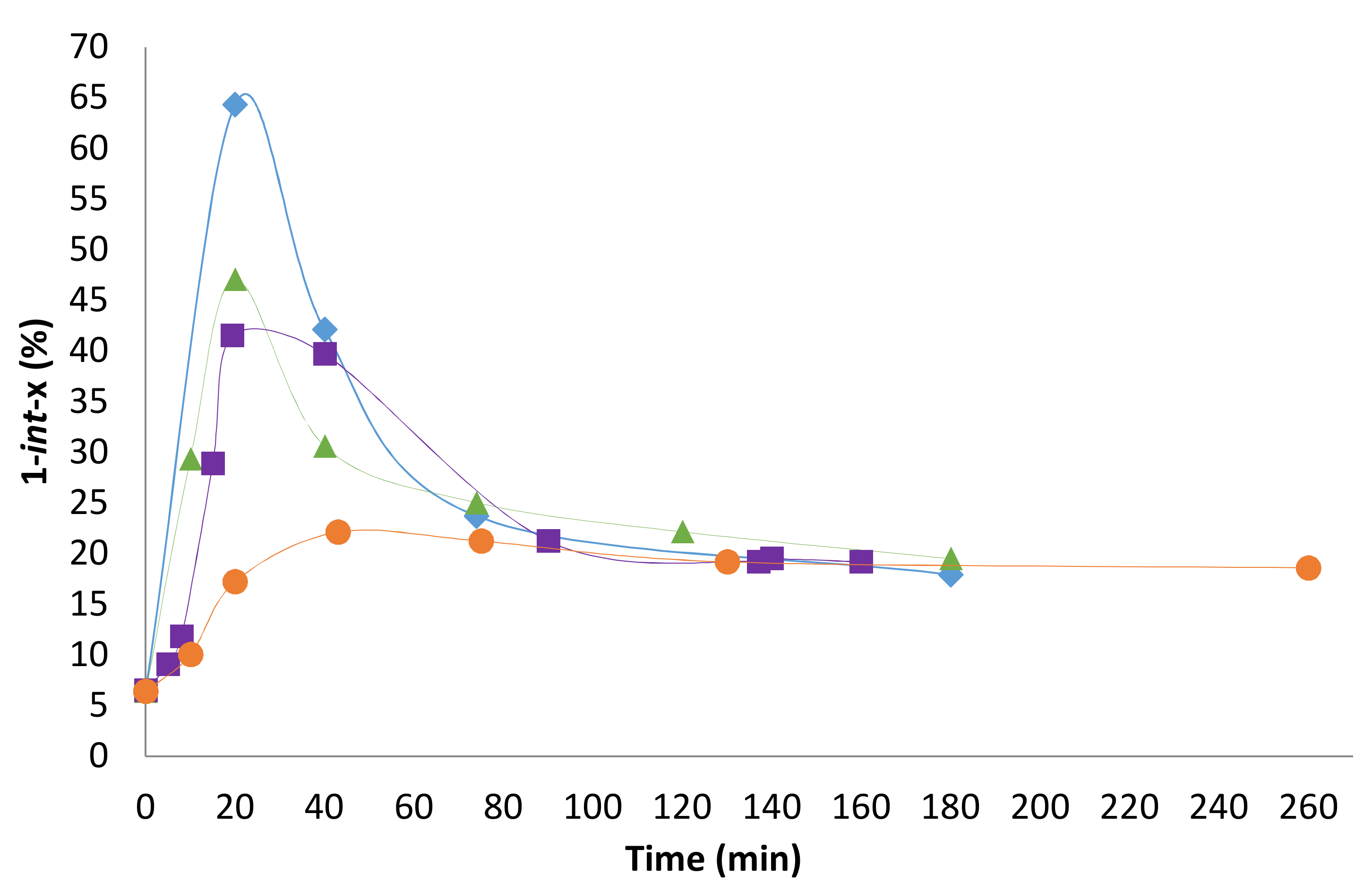
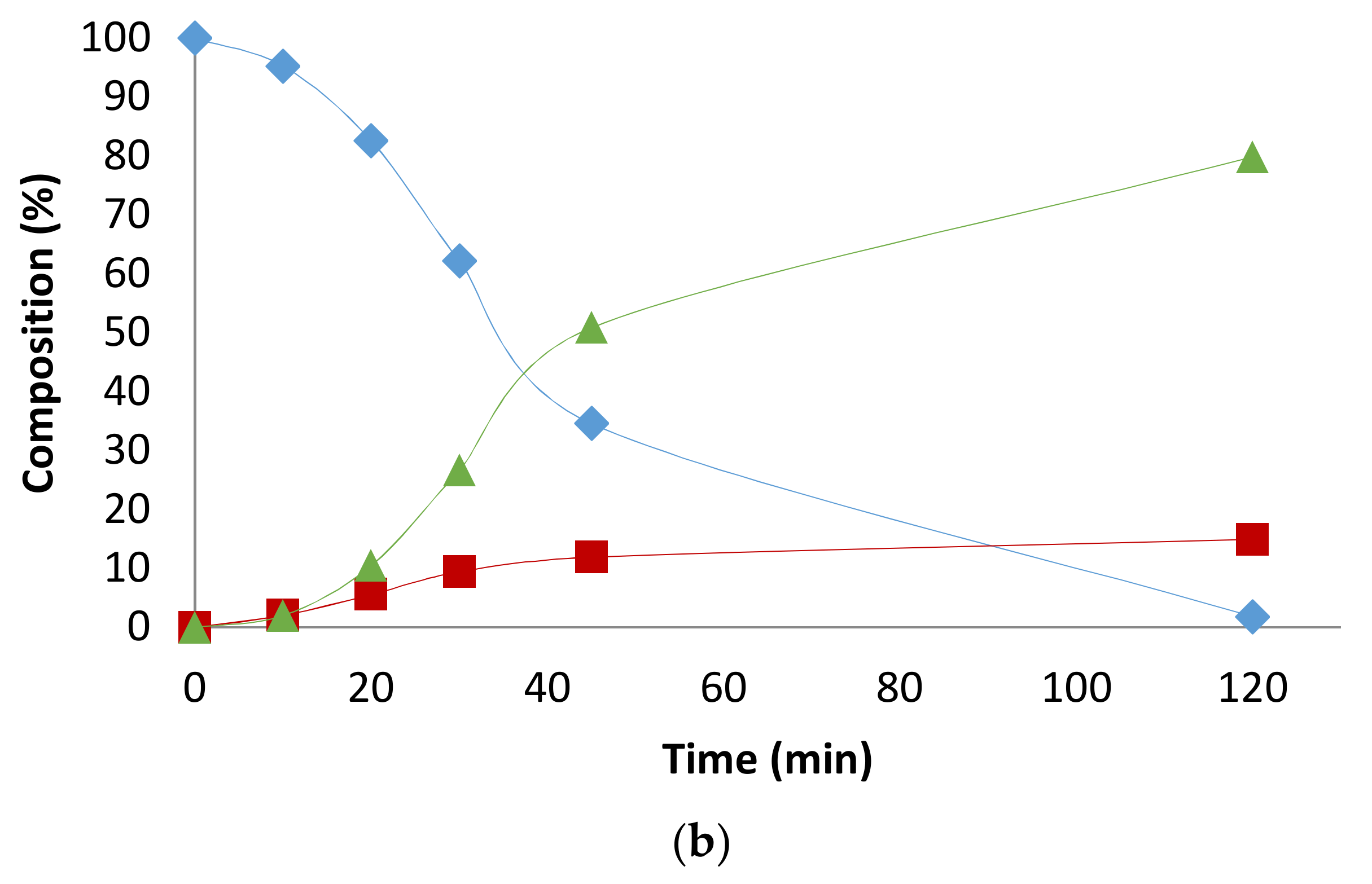
2.2. Influence of the [1]0/[Rh] Ratio
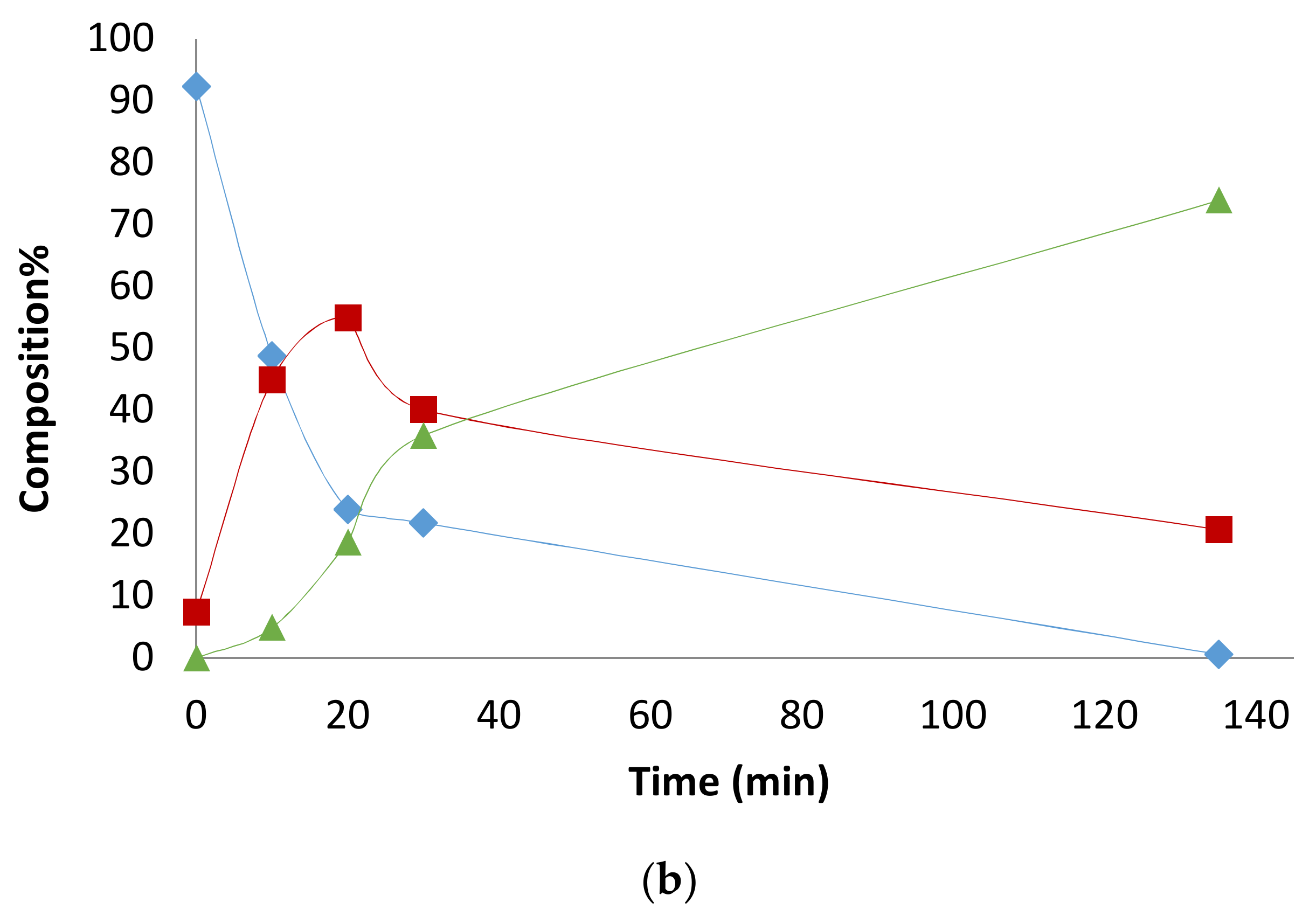
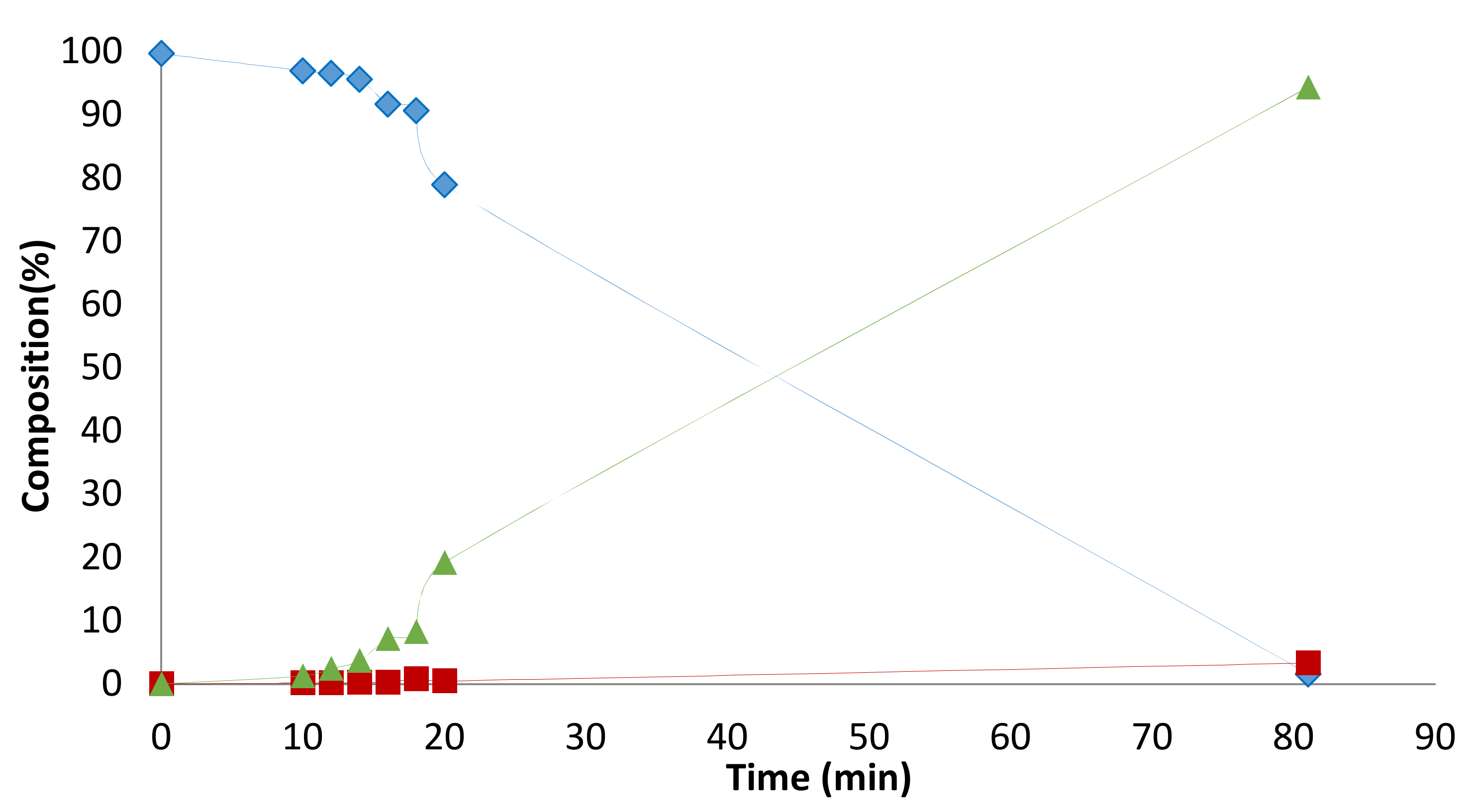
2.3. Solvent Effects
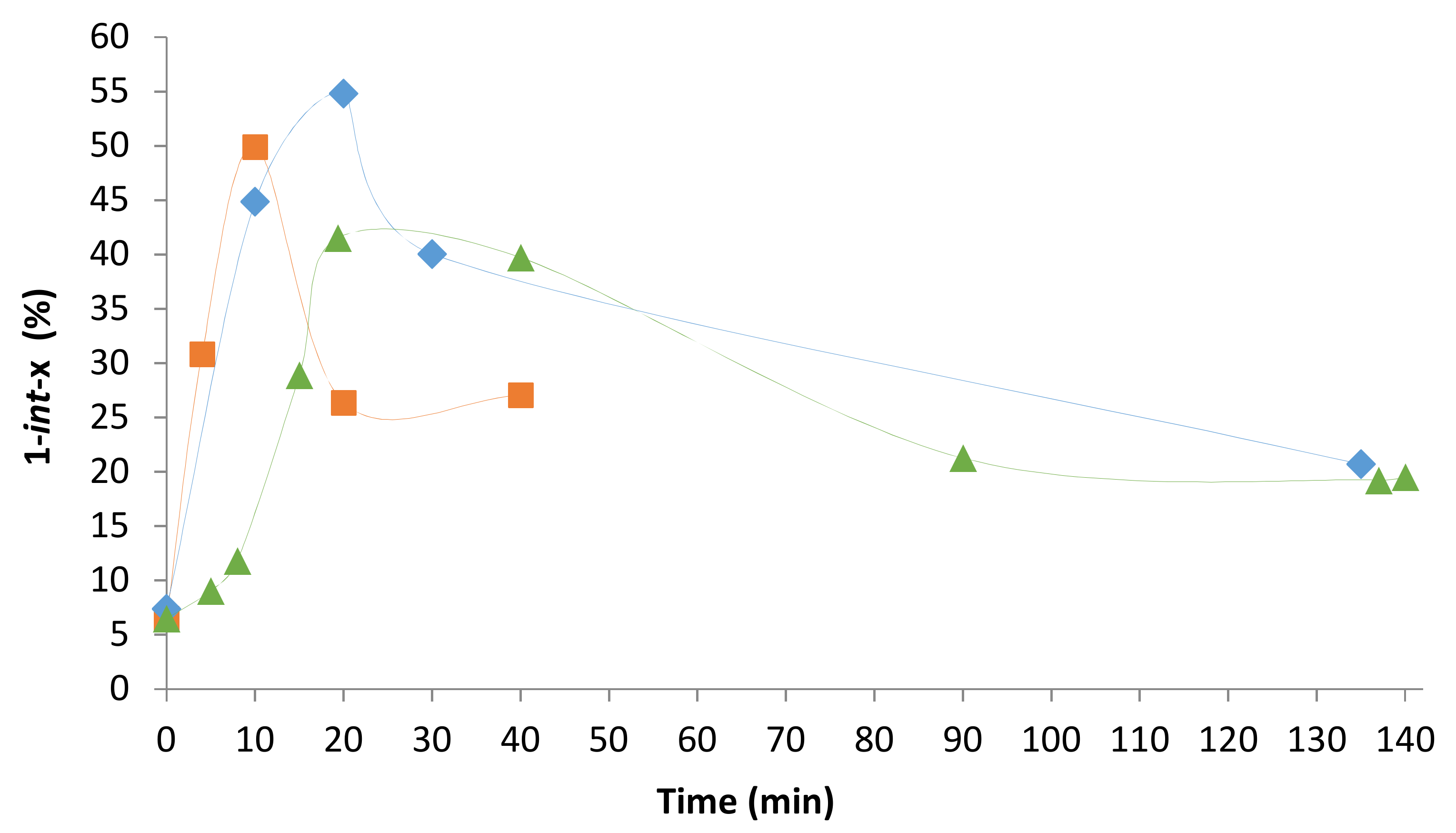
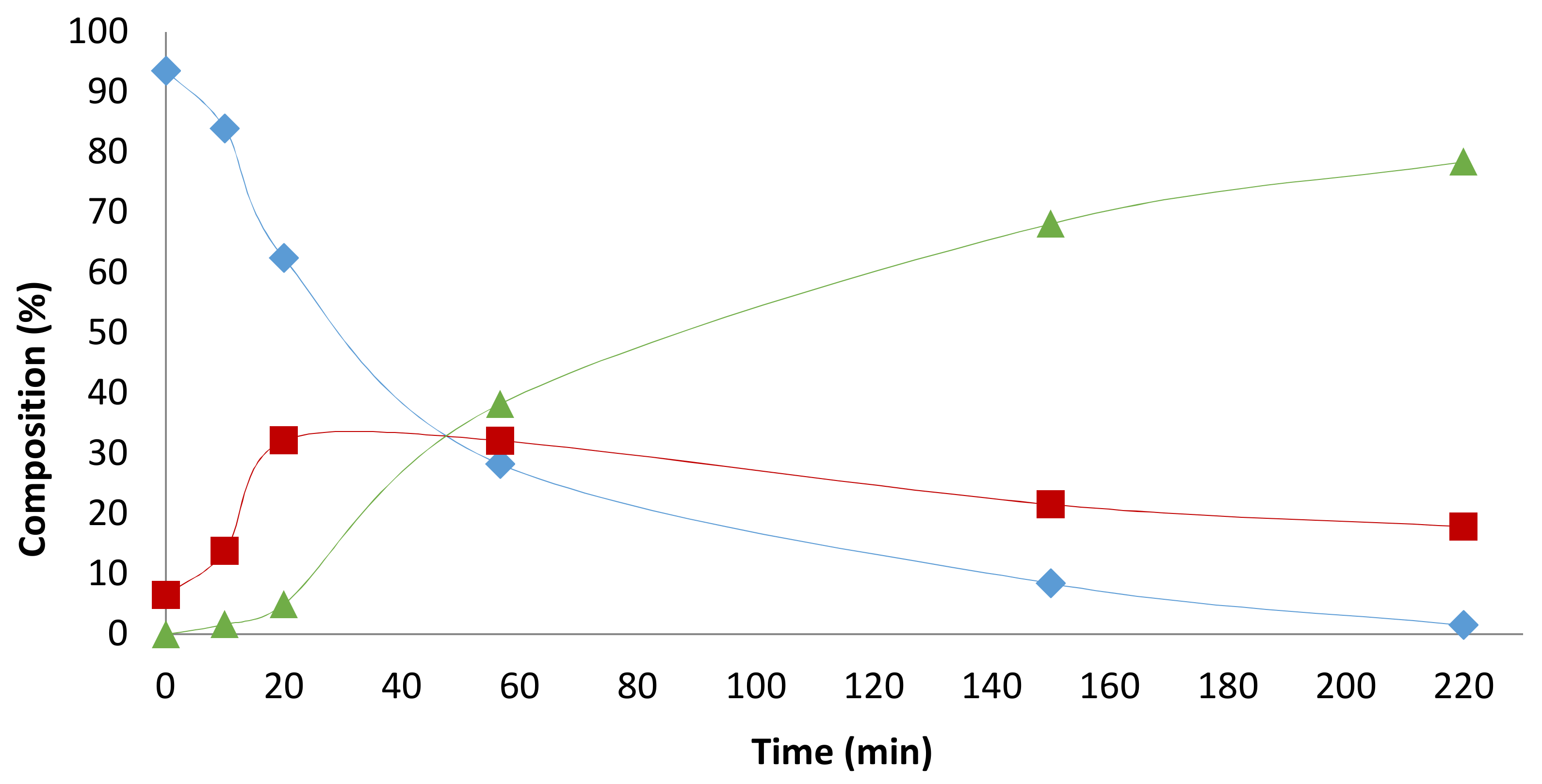
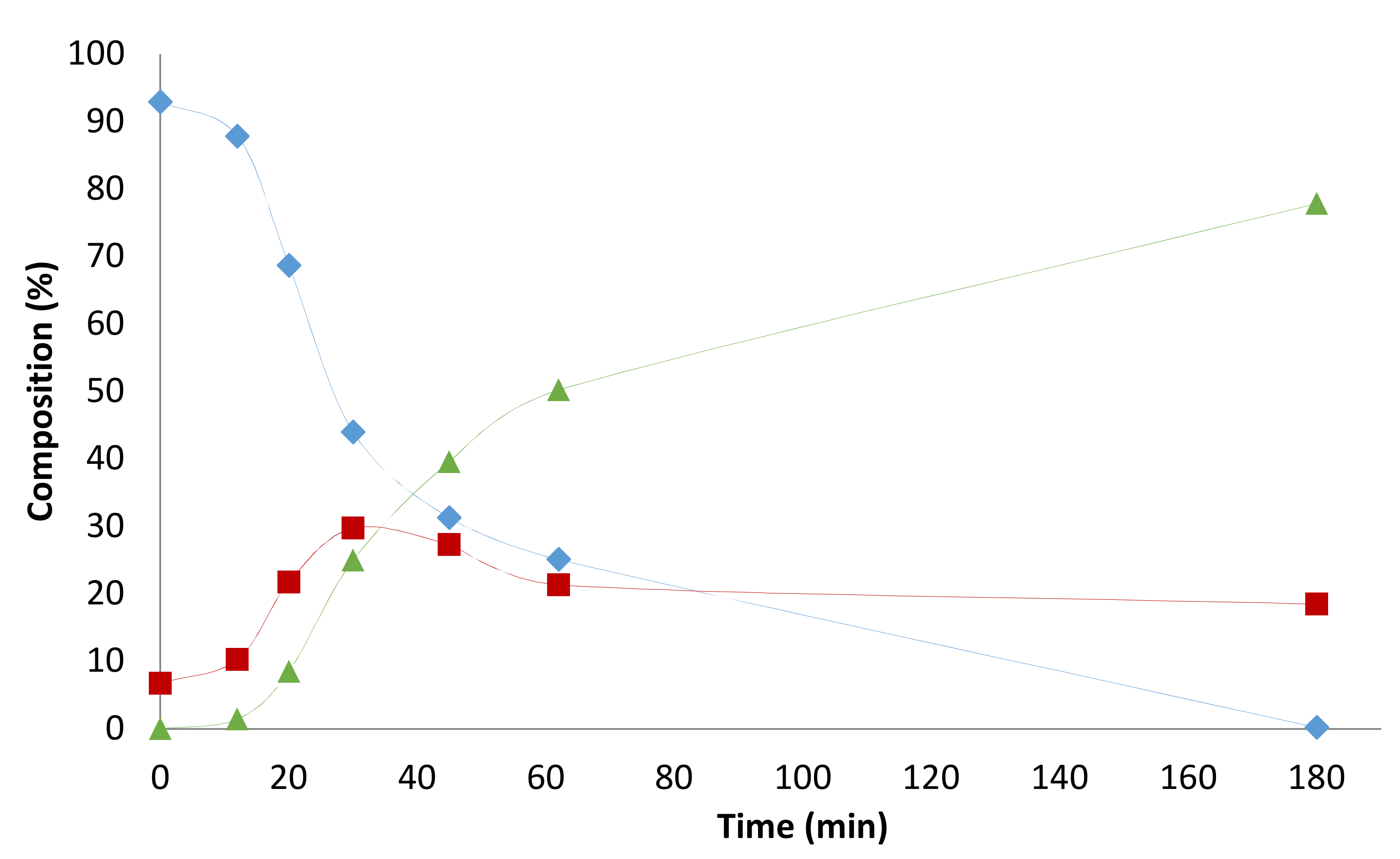
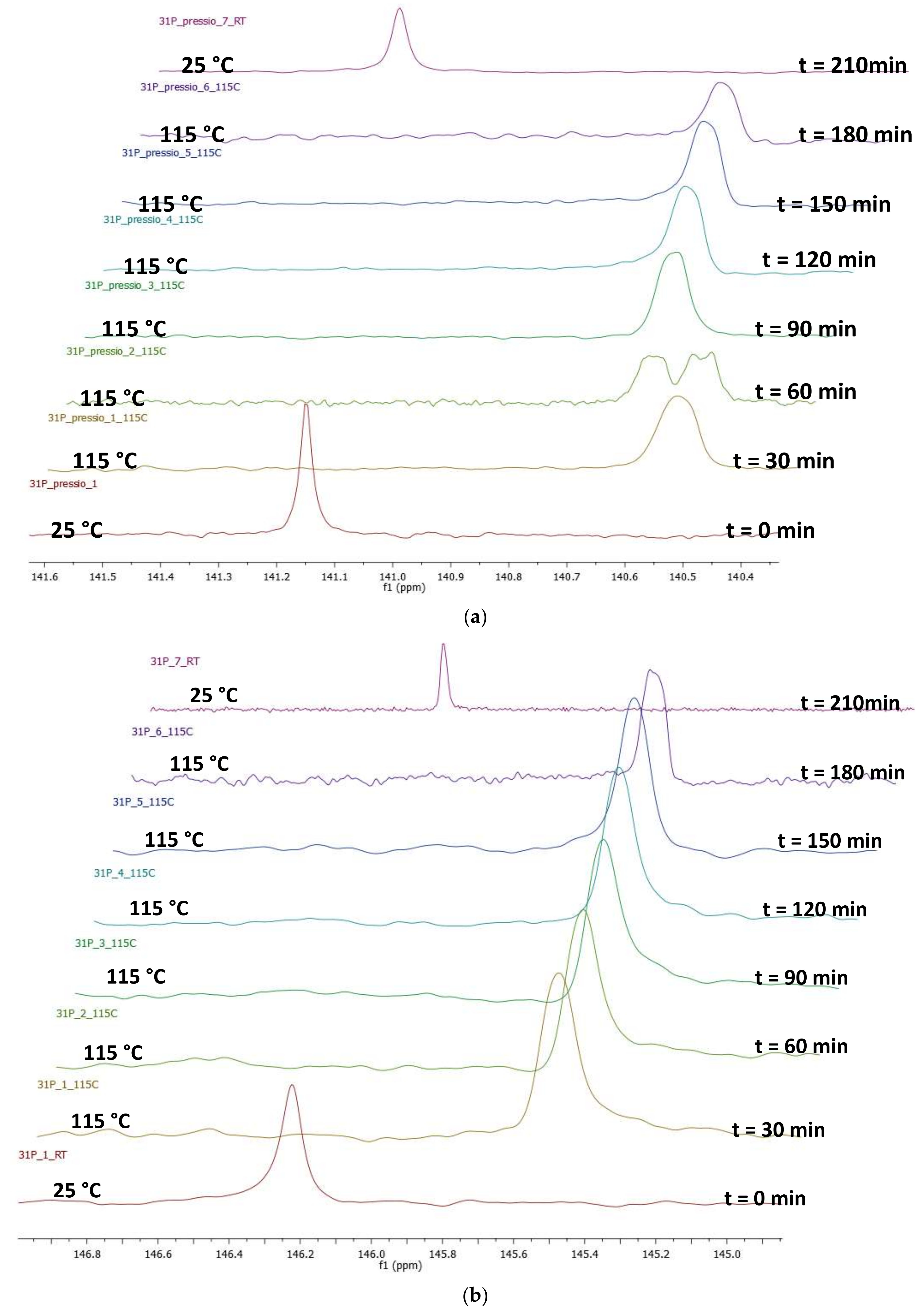
2.4. Pressure Variation
2.5. [Biphephos]/[Rh] Variation
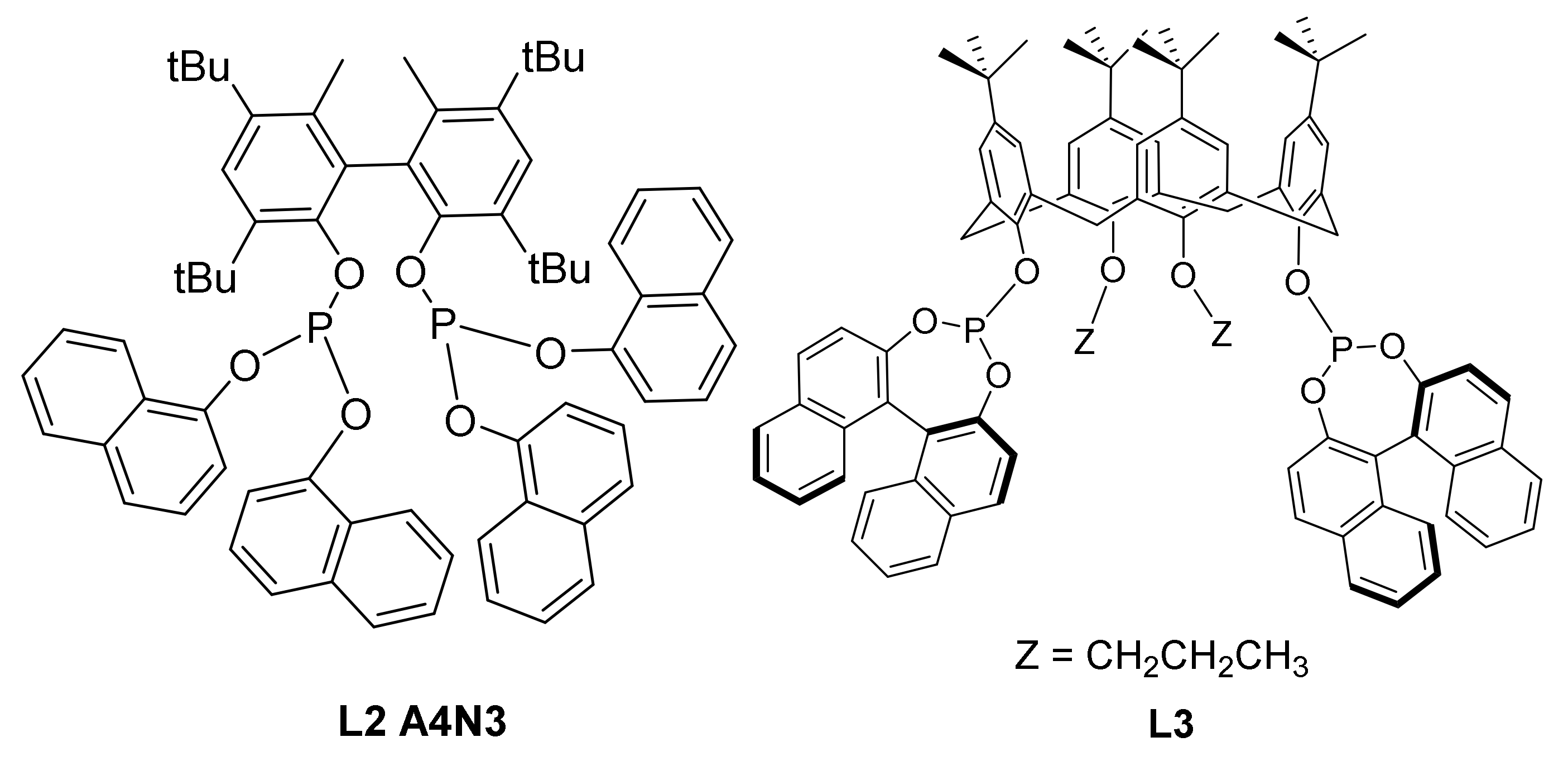
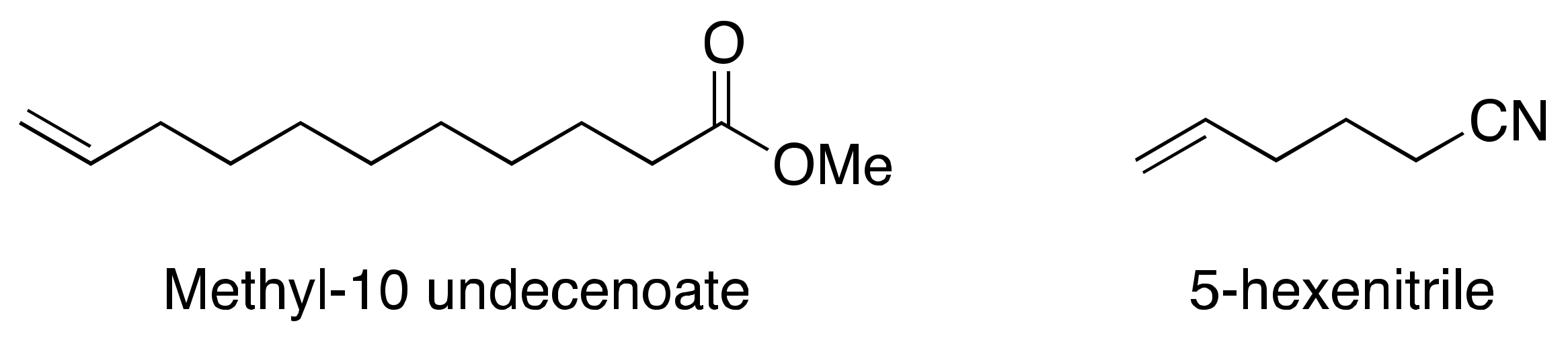
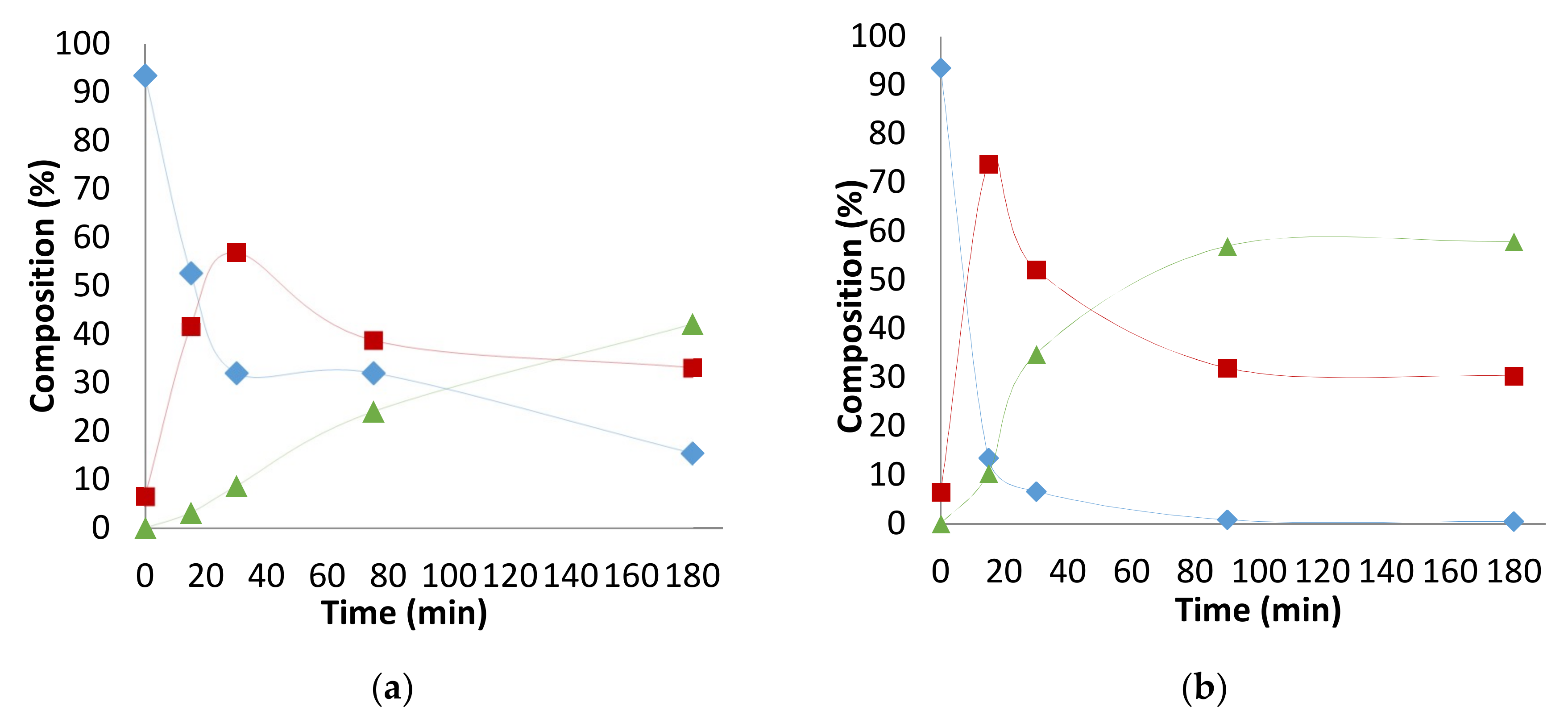
2.6. Other Effects: Ligands, Substrates
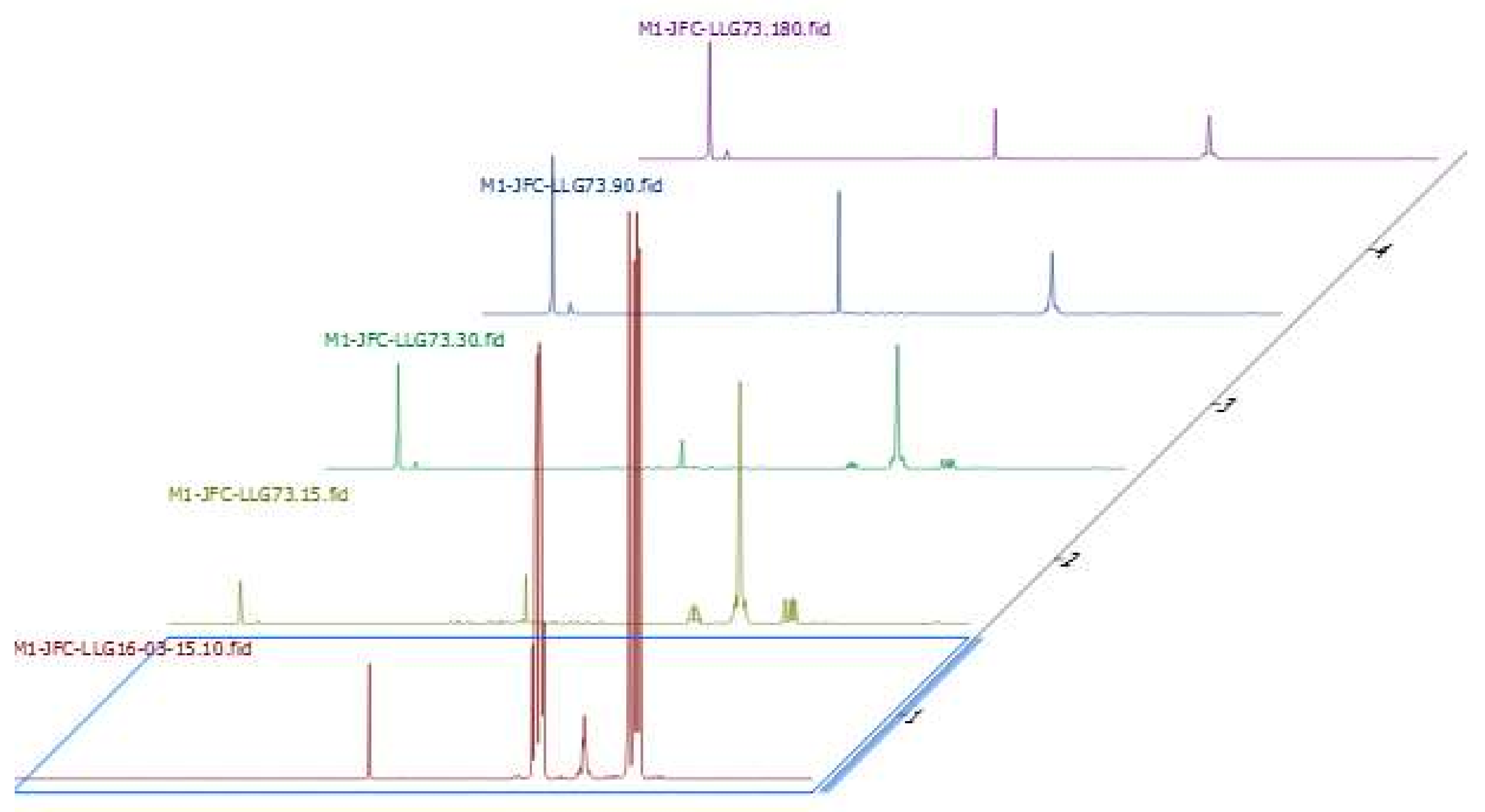
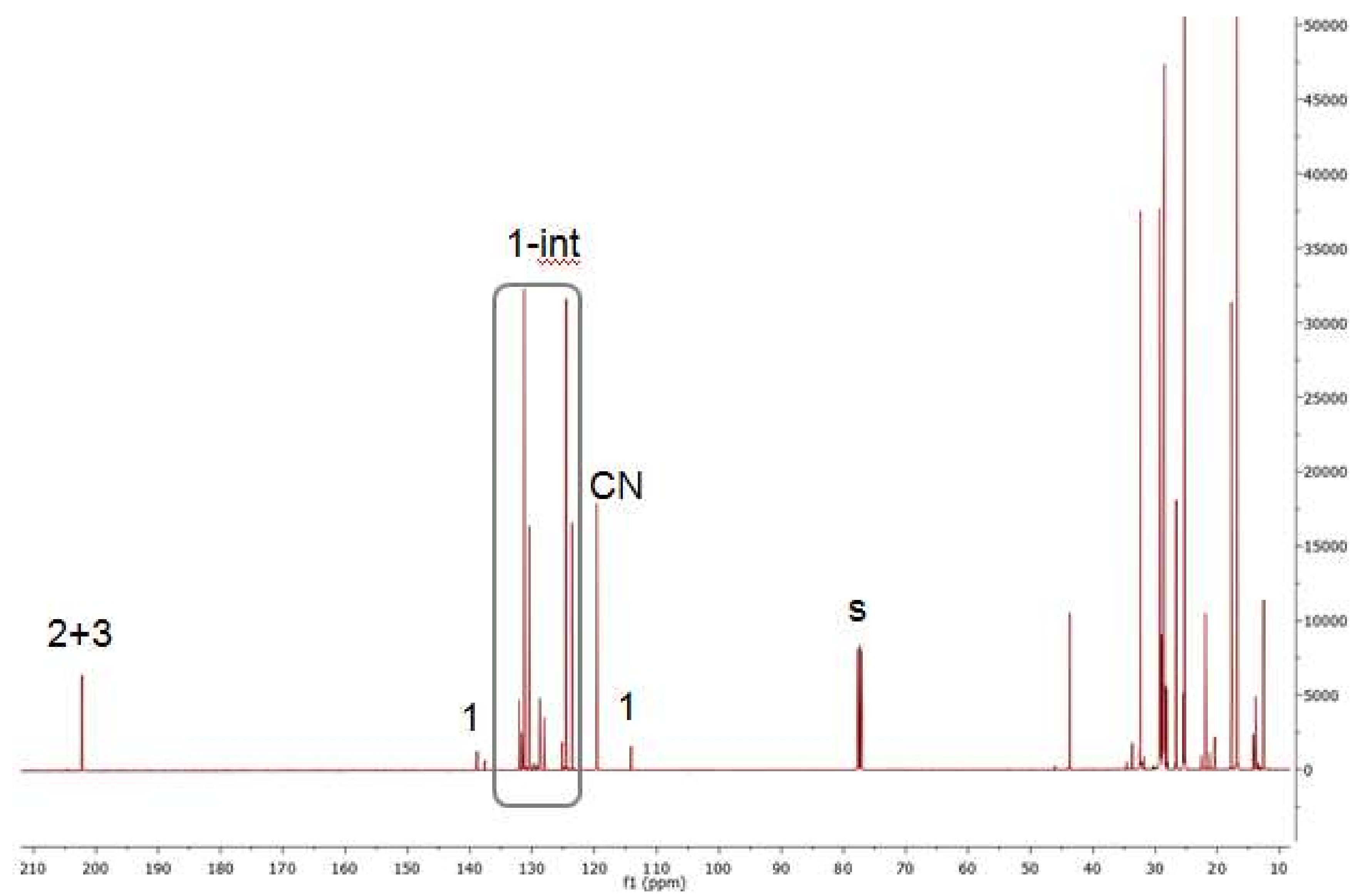
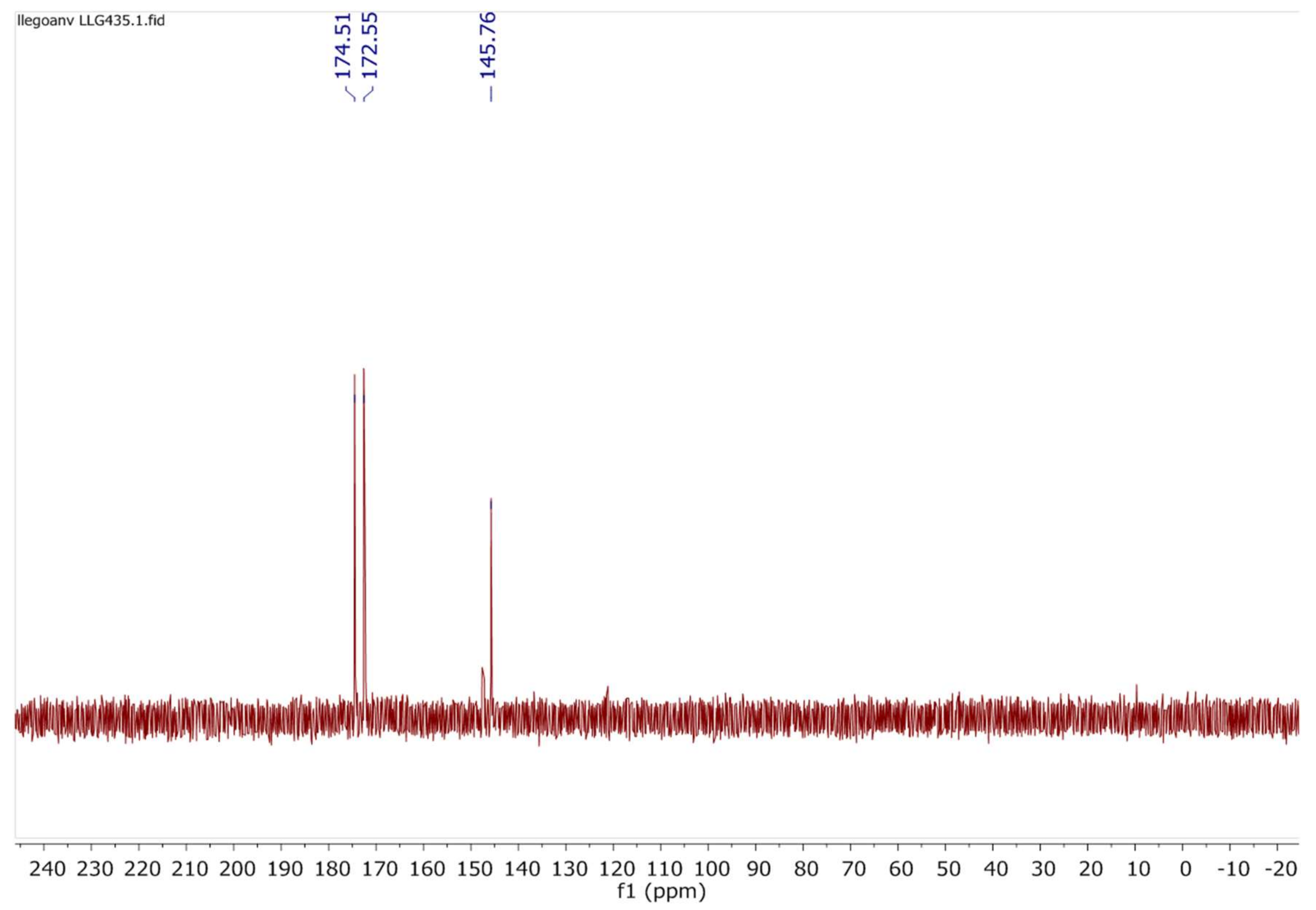
2.7. Investigations on the Catalytic Species
3. Materials and Methods
3.1. General Considerations
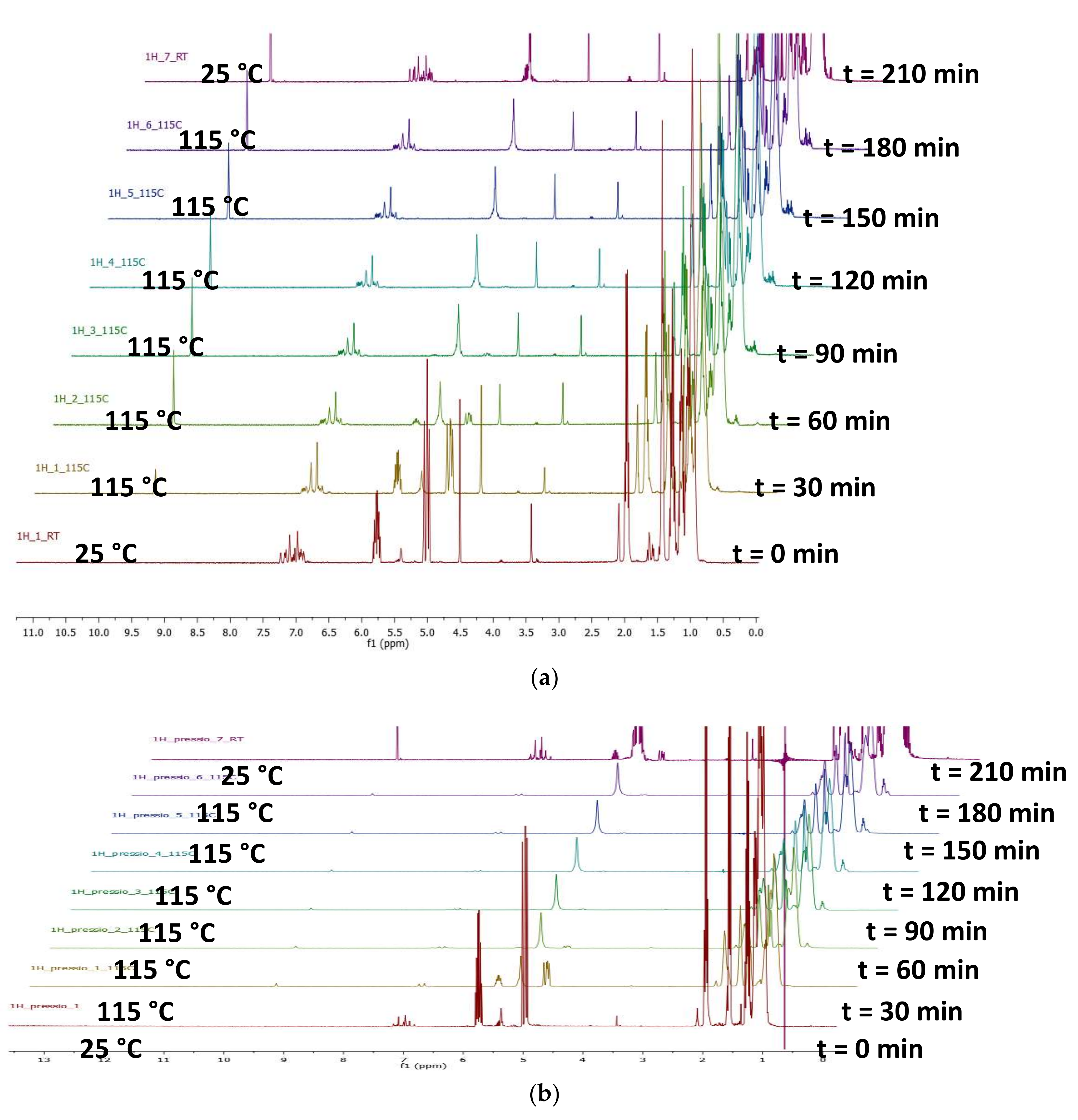
3.2. General Procedure for Monitoring of Hydroformylation Reactions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A



















References
- Kohls, E.; Stein, M. The thermochemistry of long chain olefin isomers during hydroformylation. New J. Chem. 2017, 41, 7347–7355. [Google Scholar] [CrossRef]
- Vilches-Herrera, M.; Domke, L.; Börner, A. Isomerization-Hydroformylation tandem reactions. ACS Catal. 2014, 4, 1706–1724. [Google Scholar] [CrossRef]
- Goldbach, V.; Roesle, P.; Mecking, S. Catalytic isomerizing ω-functionalization of fatty acids. ACS Catal. 2015, 5, 5951–5972. [Google Scholar] [CrossRef]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R.; Union Carbide Corporation. Transition Metal Complex Catalyzed Processes. U.S. Patent 4668651, 5 September 1985. [Google Scholar]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R.; Union Carbide Corporation. Transition Metal Complex Catalyzed Processes. European Patent 214622, 4 September 1986. [Google Scholar]
- Billig, E.; Abatjoglou, A.G.; Bryant, D.R.; Union Carbide Corporation. Transition Metal Complex Catalyzed Processes. U.S. Patent 4769498, 6 September 1988. [Google Scholar]
- Yan, Y.; Zhang, X.; Zhang, X. A tetraphosphorus ligand for highly regioselective isomerization-hydroformylation of internal olefins. J. Am. Chem. Soc. 2006, 128, 16058–16061. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chie, Y.; Guan, Z.; Zhang, X. Highly regioselective isomerization-hydroformylation of internal olefins to linear aldehyde using Rh complexes with tetraphosphorus ligands. Org. Lett. 2008, 10, 3469–3472. [Google Scholar] [CrossRef] [PubMed]
- Sémeril, D.; Matt, D.; Toupet, L. Highly regioselective hydroformylation with hemispherical chelators. Chem. Eur. J. 2008, 14, 7144–7155. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Obst, D.; Schulte, C.; Schosser, T. Highly selective tandem isomerization-hydroformylation reaction of trans-4-octene to n-nonanal with rhodium-Biphephos catalysis. J. Mol. Catal. A Chem. 2003, 206, 179–184. [Google Scholar] [CrossRef]
- Behr, A.; Obst, D.; Schulte, C. Kinetik der isomerisierenden Hydroformylierung von trans-4-Octen. Chem. Ing. Tech. 2004, 76, 904–910. [Google Scholar] [CrossRef]
- Behr, A.; Obst, D.; Westfechtel, A. Isomerizing hydroformylation of fatty acid esters: Formation of ω-aldehydes. Eur. J. Lipid Sci. Technol. 2005, 107, 213–219. [Google Scholar] [CrossRef]
- Furst, M.R.L.; Korkmaz, V.; Gaide, T.; Seidensticker, T.; Behr, A.; Vorholt, A.J. Tandem reductive hydroformylation of castor oil derived substrates and catalyst recycling by selective product crystallisation. ChemCatChem 2017, 9, 4319–4323. [Google Scholar] [CrossRef]
- Gaide, T.; Bianga, J.; Schlipköter, K.; Behr, A.; Vorholt, A.J. Linear selective isomerization/hydroformylation of unsaturated fatty acid methyl esters: A bimetallic approach. ACS Catal. 2017, 7, 4163–4171. [Google Scholar] [CrossRef]
- Pandey, S.; Chikkali, S.H. Highly regioselective isomerizing hydroformylation of long-chain internal olefins catalyzed by a rhodium bis(phosphite) complex. ChemCatChem 2015, 7, 3468–3471. [Google Scholar] [CrossRef]
- Pandey, S.; Shinde, D.R.; Chikkali, S.H. Isomerizing hydroformylation of cashew nut shell liquid. ChemCatChem 2017, 9, 3997–4004. [Google Scholar] [CrossRef]
- Yuki, Y.; Takahashi, K.; Tanaka, Y.; Nozaki, K. Tandem isomerization-hydroformylation-hydrogenation of internal alkenes to n-alcohols using Rh/Ru dual- or ternary-catalyst systems. J. Am. Chem. Soc. 2013, 135, 17393–17400. [Google Scholar] [CrossRef] [PubMed]
- Ternel, J.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Rhodium-catalyzed tandem isomerization- hydroformylation of the bio-sourced 10-Undecenenitrile: Selective and productive catalysts for production of polyamide-12 precursor. Adv. Synth. Catal. 2013, 355, 3191–3204. [Google Scholar] [CrossRef]
- Le Goanvic, L.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Ruthenium-catalyzed hydroformylation of the functional unsaturated fatty nitrile 10-undecenitrile. J. Mol. Catal. A Chem. 2016, 417, 116–121. [Google Scholar] [CrossRef]
- Dubois, J.-L.; Gillet, J.-P. Coproduction of Cyclic Carbonates and of Nitriles and/or Fatty Amines. WO Patent 2008/145941A2, 4 December 2008. [Google Scholar]
- Ternel, J.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Rhodium versus iridium catalysts in the controlled tandem hydroformylation–isomerization of functionalized unsaturated fatty substrates. ChemCatChem 2015, 7, 513–520. [Google Scholar] [CrossRef]
- Claver, C.; van Leeuwen, P.W.N.M. Rhodium Catalyzed Hydroformylation; Springer: Berlin, Germany, 2008. [Google Scholar]
- Monnereau, L.; Sémeril, D.; Matt, D. Solvent-free olefin hydroformylation using hemispherical diphosphites. Eur. J. Org. Chem. 2010, 16, 3068–3073. [Google Scholar] [CrossRef]
- Gunstone, F.D.; Pollard, M.R.; Scrimgeour, C.M.; Vedanayagam, H.S. 13C nuclear magnetic resonance studies of olefinic fatty acids and esters. Chem. Phys. Lipids 1977, 18, 115–129. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | [1]0/[Rh] | [1]0 | Time | 1 | 1-int-x | [2] + [3] | [2]/[3] (l/b) 3 | 4 | Conv. 1 | HF | TOF | TOF0iso | TOF0HF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ratio | [mol·L−1] | [h] | [%]2 | [%] 2 | [%] 2 | [%] 2 | [%] 4 | [%] 5 | [h−1] 6 | [h−1] 7 | [h−1] 7 | ||
| entry 1 | 20,000 | 1.00 | 1.5 | 1 | 25 | 68 | 99.0:1.0 | 6 | 99 | 74 | 28,250 | 7900 | 17,050 |
| entry 2 | 7000 | 0.59 | 1.75 | 9 | 21 | 66 | 99.2:0.8 | 3 | 90 | 79 | 10,100 | 8500 | 1600 |
| entry 3 | 3000 | 0.40 | 2.33 | 2 | 19 | 75 | 99.2:0.8 | 4 | 99 | 82 | 7850 | 4650 | 2600 |
| entry 4 | 1000 | 0.23 | 1.5 | 0 | 17 | 76 | 99.1:0.9 | 7 | 100 | 81 | 3450 | 1300 | 1750 |
| entry 5 | 335 | 0.12 | 1.67 | 0 | 18 | 78 | 99.1:0.9 | 4 | 100 | 84 | 1800 | 1200 | 450 |
| entry 6 | 20,000 | 0.40 | 3.67 | 2 | 24 | 69 | 99.1:0.9 | 5 | 98 | 76 | 20,100 | 8650 | 15,500 |
| Entry | Solvent | Time | 1 | 1-int-x | 2 + 3 | [2]/[3] (l/b) 3 | 4 | Conv. 1 | HF | TOF | TOF0iso | TOF0HF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [h] | [%] 2 | [%] 2 | [%] 2 | [%] 2 | [%] 4 | [%] 5 | [h−1] 6 | [h−1] 7 | [h−1] 7 | |||
| entry 7 | toluene | 2.33 | 2 | 19 | 75 | 99.2:0.8 | 4 | 99 | 82 | 7850 | 4650 | 2600 |
| entry 8 | MeCN | 0.67 | 1 | 27 | 68 | 99.4:0.6 | 4 | 99 | 74 | 17,800 | 10,950 | 7450 |
| entry 9 | 1,4-dioxane | 2.25 | 1 | 21 | 74 | 99.2:0.8 | 5 | 99 | 81 | 6650 | 6750 | 2150 |
| Entry | P | CO/H2 | Time | 1 | 1-int-x | 2 + 3 | [2]/[3] (l/b) 3 | 4 | Conv. 1 | HF | TOF | TOF0iso | TOF0HF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [bar] | Ratio | [h] | [%] 2 | [%] 2 | [%] 2 | [%] 2 | [%] 4 | [%] 5 | [h−1] 6 | [h−1] 7 | [h−1] 7 | ||
| entry 10 | 4 | 1:1 | 2 | 2 | 42 | 50 | 99.1:0.9 | 6 | 99 | 54 | 6650 | 3550 | 2450 |
| entry 11 | 8 | 1:1 | 2 | 1 | 35 | 58 | 99.0:1.0 | 6 | 99 | 63 | 1140 | 5800 | 4300 |
| entry 12 | 20 | 1:1 | 2.67 | 2 | 19 | 75 | 99.2:0.8 | 3 | 98 | 82 | 7850 | 4650 | 2600 |
| entry 13 | 40 | 1:1 | 3.5 | 0 | 16 | 79 | 98.6:1.4 | 5 | 100 | 85 | 6750 | 3950 | 2950 |
| entry 14 | 40 | 3:1 | 3.67 | 1 | 18 | 78 | 98.8:1.2 | 2 | 98 | 85 | 3000 | 2300 | 1400 |
| entry 15 | 40 | 1:3 | 2.5 | 2 | 18 | 71 | 99.1:0.9 | 9 | 97 | 75 | 6000 | 4500 | 1700 |
| Entry | [L]/[M] | Time | 1 | 1-int-x | 2 + 3 | [2]/[3] (l/b) 3 | 4 | Conv. 1 | HF | TOF | TOF0iso | TOF0HF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ratio | [h] | [%] 2 | [%] 2 | [%] 2 | [%] 2 | [%] 4 | [%] 5 | [h−1] 6 | [h−1] 6 | [h−1] 7 | ||
| entry 16 | 5 | 3 | 1 | 18 | 77 | 99.3:0.7 | 4 | 99 | 84 | 7850 | 5200 | 2300 |
| entry 17 | 10 | 3 | 1 | 20 | 76 | 99.2:0.8 | 5 | 99 | 82 | 5050 | 4150 | 1650 |
| entry 18 | 20 | 2.7 | 2 | 19 | 75 | 99.2:0.8 | 3 | 98 | 82 | 7850 | 4650 | 2600 |
| entry 19 | 50 | 4.3 | 3 | 19 | 75 | 99.1:0.9 | 3 | 97 | 83 | 2350 | 1000 | 1450 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Goanvic, L.; Couturier, J.-L.; Dubois, J.-L.; Carpentier, J.-F. Insights in the Rhodium-Catalyzed Tandem Isomerization-Hydroformylation of 10-Undecenitrile: Evidence for a Fast Isomerization Regime. Catalysts 2018, 8, 148. https://doi.org/10.3390/catal8040148
Le Goanvic L, Couturier J-L, Dubois J-L, Carpentier J-F. Insights in the Rhodium-Catalyzed Tandem Isomerization-Hydroformylation of 10-Undecenitrile: Evidence for a Fast Isomerization Regime. Catalysts. 2018; 8(4):148. https://doi.org/10.3390/catal8040148
Chicago/Turabian StyleLe Goanvic, Lucas, Jean-Luc Couturier, Jean-Luc Dubois, and Jean-François Carpentier. 2018. "Insights in the Rhodium-Catalyzed Tandem Isomerization-Hydroformylation of 10-Undecenitrile: Evidence for a Fast Isomerization Regime" Catalysts 8, no. 4: 148. https://doi.org/10.3390/catal8040148