Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review


1
Department of Chemical Engineering, Kansas State University, Manhattan, KS 66506, USA
2
College of Medicine, Pennsylvania State University, Hershey, PA 17033, USA
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(2), 44; https://doi.org/10.3390/catal8020044
Submission received: 16 December 2017
/
Revised: 5 January 2018
/
Accepted: 6 January 2018
/
Published: 24 January 2018
(This article belongs to the Special Issue Glycerol Conversion by Heterogeneous Catalysis)
Abstract
:Glycerol has been utilized in an extremely diversified manner throughout human civilization—ranging from food, to various consumer products, to pharmaceuticals, and even explosives. Large surplus in glycerol supply thanks to biodiesel production and biomass processing has created a demand to further boost its utility. One growing area is to expand the use of glycerol as an alternative feedstock to supplement fuels and chemicals production. Various catalytic processes have been developed. This review summarizes catalytic materials for glycerol reforming, hydrodeoxygenation, and oxidation. In particular, rationale for catalyst selection and new catalyst design will be discussed aided by the knowledge of reaction mechanisms. The role of theoretical density functional theory (DFT) in elucidating complex glycerol conversion chemistries is particularly emphasized.
1. Glycerol Uses and Production
The use of glycerol can be traced back to early civilized human society, where animal fats and vegetable oils were hydrolyzed in alkaline conditions for soap manufacturing [1]. During World War I, glycerol was used on a large scale, by momentarily overtaking soap manufacturing industry, for the production of nitroglycerin—a base for dynamite and cordite [2,3]. Around World War II, feedstocks for glycerol production have shifted to petroleum-based sources such as propene or propylene. For more than half a century, synthetic glycerol accounts for approximately 25% of global glycerol demand [4]. More recently, with the prospect of dwindling petroleum resources, and the increased cost of propylene, biological resources has regained favor as the feedstock for glycerol production.
By 2011, the annual global glycerol production reached almost 1.6 million metric tons (see Figure 1 [5]), and is expected to triple by 2024 [5,6]. A number of comprehensive reviews on glycerol production from various biological or chemical means [3,5,7,8,9,10,11], and applications [1,4,5,12,13,14,15,16,17,18,19,20,21,22,23], have been published. The boom in glycerol production in the past ten years or so is largely a consequence of booming biofuel production activities. Specifically, biodiesel production accounts for the largest driving force (at approximately 64%) [7], followed by fatty acid conversions (at 21%), according to the 2009 statistics [4]. These figures reflect a global trend pursuing chemical and fuel sources from non-fossil-based feedstocks. The upward trend in glycerol production is accompanied by the fall of crude glycerol price (Figure 1). Therefore, it is understandable to note ongoing efforts to explore and expand the usage of glycerol, and to maintain its commodity chemical status.
The current leading applications of glycerol include drugs and pharmaceutical manufacturing, personal care products, polyols and polyether, food, and so on, as shown in Figure 2 [13]. Emerging opportunities give rise to production of value-added fine chemicals, especially those that traditionally rely on petrochemical feedstocks. Relevant topics have been recently discussed by Quipse [5], Rahmat [19], and their coworkers. It is particularly encouraging to note the diversity and range of glycerol-derived products that can be obtained by exploiting the rich chemistry. The key to success depends on the availability of active and selective catalysts.
Studies in the realm of glycerol reforming, hydrogenolysis, and oxidation, and representative catalysts for each type of catalytic processes have been surveyed based on literatures published in the past twenty years. As continuous progress in developing new catalysts and processes for glycerol conversion is being achieved, studies included in this review are far from complete. Instead, it is our goal to reveal the fundamental relationship between catalyst performance and fundamental reaction pathways. Along this line of thought, recent theoretical developments, based on quantum mechanical density functional theory (DFT) calculations, will be highlighted and presented as an effective tool to unravel the catalyst functionality-reaction activity and selectivity relationship.
2. Chemistries in Catalytic Glycerol Conversions
A roadmap for glycerol catalytic conversion, depending on the catalysts, is illustrated in Figure 3. Hydrogen, syngas (a mixture of CO and H2) can be acquired from steam or aqueous phase reforming on transition metals or metal alloys, which prioritize C–H, O–H, and C–C bond cleavages. Often, water-gas shift (WGS) reaction occurs concomitantly, producing CO2 and H2. It is also possible for glycerol to undergo C–O bond cleavage on certain metals and alloys, in the form of hydrogenolysis (when H2 is present), which further opens up product distributions to propanediols, alcohols, and alkanes. It is common that a second metal in an alloy modulates the catalyst’s capability for C–O bond cleavage. Catalysts with acid functionalities promote glycerol dehydration, and can effectively shift glycerol conversion toward hydrogenolysis as well. Oxidations of glycerol to aldehyde and ketone can occur when noble metals such as Au, Pt, or Pd are used. Electrooxidation of glycerol is a particularly promising method to obtain sustainable chemicals with tunable selectivity. Deeper oxidation products such as organic acids or even complete oxidation products can also be achieved. In the following subsections, each of the above catalytic routes will be discussed with more details.
2.1. Catalytic Glycerol Reforming for Hydrogen Production
2.1.1. Aqueous and Vapor Phase Hydrogen Production
Global hydrogen supply has reached ~700 billion Nm3 in 2009 [25], obtained primarily (~96%) from fossil fuels such as natural gas (CH4), oil refining products, and coal [26,27]. For the long term, the fate of hydrogen production can be affected by dwindling reserves, increasing energy costs, and environmental consequences, and all of these issues need to be addressed [25].
The overall reactions for glycerol decomposition, water-gas shift (WGS) reaction, and glycerol steam reforming are expressed by Equations (1)–(3).
Reforming of glycerol in either aqueous or vapor phase has been extensively investigated, as presented in recent reviews [22,28].
Dumesic and coworkers [8,24,29,30,31,32,33,34] led and demonstrated feasibility of glycerol aqueous phase reforming (APR) as an alternative solution to sustainable hydrogen production in a series of studies. Main technological advantages of APR include low operating temperature (<270 °C) [24,29], and thus low production cost. However, relatively high pressure (25–30 bar) must be maintained.
2.1.2. Catalysts for Glycerol Reforming
Ideally, APR catalysts should be effective for C–H, O–H, C–C bond cleavage [24]. With the main challenge to H2 selectivity stemming from parallel pathways involving C–O bond cleavage, mechanisms enabling such events should be avoided or minimized. Representative catalysts for glycerol reforming are summarized in Table 1.
Pt exhibits good activity under typical aqueous phase reforming conditions, with satisfactory selectivity (up to 75%) [24,40,41,42], which can be attributed to the fact that Pt is effective for C–H, O–H, and C–C bond cleavages, while being sluggish for C–O bond cleavage. Modeling of glycerol decomposition elementary steps showed that the H2 evolution process begins with C–H or O–H bond activations. As glycerol is sufficiently dehydrogenated, decarbonylation, a C–C bond cleavage step, could become competitive, producing C2 species, and CO. Under APR conditions, the latter may participates in WGS, which is a beneficial secondary reaction (see Figure 3), supported on Pt.
On Pt/C catalysts, ethylene glycol (EG) and propylene glycol (PG) are the main liquid phase products. EG is almost certainly produced from decarbonylation. On Pt, the energetics and energy barriers for C–O bond are uncompetitive throughout the decomposition process, according to DFT calculations [59,60]. Hence, PG is more likely from either dehydration of glyceraldehyde, or direct dehydration of glycerol (through an acetol intermediate), completed with hydrogenation [42]. If alumina or carbon black supports—creating more acidic conditions—are used, dehydration can be further enhanced.
Pure Pt catalysts are considered too expensive for industrial scale use in APR or steam reforming. Hence, a number of Pt-based bimetallic catalysts, allowing reduced Pt usage, were studied [40,45,46,47,48,49,50,51,52,55]. More importantly, additional catalytic functionality can be introduced this way. High-throughput screening of Pt, Pd-based bimetallic catalysts have been performed by Dumesic and his coworkers [61] using EG, which can be regarded as a polyol analog compound. The EG conversion trends acquired could be transferable to understand the glycerol conversion behaviors.
For glycerol, King et al. [40] employed Pt-Re catalysts in an APR setting, and found that the addition of Re has significantly increased glycerol conversion. Even higher glycerol conversion (89%) can be achieved eventually with additional base (e.g., KOH) [40]. The route for hydrogen production is proposed to follow a sequence of: glycerol → glyceraldehyde → EG → glycolaldehyde → methanol → syn gas.
Zhang et al. [48] aimed at catalytic functionalities of Pt-Re/C catalysts for glycerol APR by varying Re loadings, and revealed that the active site is linked to oxidized Pt-Re, with local acidity on surface. A relationship between hydrogen selectivity and C–O/C–C bond cleavage was established based on respective product ratios—products associated with C–C bond cleavage include CO2 (via WGS reaction), EG, methanol, and CO, while C–O bond cleavage produces PG. In particular, the authors noted that the Re/C catalysts alone have no activity, and the hydrogenation functionality provided by Pt is necessary for PG formation.
Pt-Mo/C catalysts prepared by Dietrich and coworkers [51,52] showed that the H2 turnover frequency (TOF) becomes 4 times higher than Pt/C. The reduced bimetallic catalyst structures were probed and revealed to have a Pt-core and Mo-rich shell, with some Mo oxides present on the surface. During the reaction, the metallic Mo content increases, indicating that these surface Mo oxide clusters are catalytically functional. This hypothesis has been confirmed with DFT calculations conducted by Liu et al. [62]. During experiments, Pt-Mo catalyst particles can retain their Pt-rich core Mo-rich shell morphology [51]. The hydrogen selectivity is estimated to be around 50%, with CH4 as the major by-product. Similar to Pt-Re catalysts, high H2 selectivity depends on the control of catalyst acidity [51]. A general rule is that low acidity is preferred in order for catalysts to be selective for H2 production. As for Pt-Re, the Mo:Pt ratio can be varied to tune H2 selectivity and yields.
Catalyst supports can influence the performance of metal sites, and even participate in reactions. Wawrzetz et al. [42] indicated that Pt supported on γ-Al2O3 behaves as a bifunctional catalyst. A Tishchenko/Cannizaro disproportionation mechanism has been proposed to be responsible for the observed acid product, which can further goes through decarboxylation reactions, producing alkanes or alcohols.
A series of supported Pt catalysts on SiO2, Al2O3, ZrO2, and Ce4Zr1α have been tested for glycerol steam reforming by Pompeo et al. [39]. It is found that Pt supported on neutral supports such as SiO2 carries out the usual dehydrogenation and C–C bond cleavage reactions. However, dehydration, dehydrogenation, hydrogenolysis, and condensation reactions are promoted on more acidic supports such as γ-Al2O3, ZrO2, and Ce4Zr1a.
Ni is known to be an effective reforming catalyst, which is the main component in hydrocarbon reforming catalysts for industrial hydrogen production [27]. Nickel is earth abundant and significantly (~1500 times) cheaper than Pt. Ni-based catalysts can be used in both aqueous phase and steam reforming of glycerol for hydrogen production [44]. Hirai et al. [56] showed that activities of metals from Group 8–10 on Y2O3 supports follow an order of Ru ≈ Rh > Ni > Ir > Co > Pt > Pd > Fe at 600 °C. At even higher temperature (i.e., 900 °C), Adhikari et al. [35] showed that glycerol conversion is in the order of Ni > Ir > Pd > Rh > Pt > Ru, on Al2O3. However, the deterioration of nickel catalysts is a serious issue, due to ongoing hydrocarbon decomposition, Boudouard reaction, and methanation reactions.
Huber et al. [29,32] used Sn-promoted Raney-nickel catalysts, which is believed to overcome some of those issues, for glycerol APR, and achieved a glycerol conversion of more than 90% and a H2 selectivity of above 70%. The performance of this inexpensive catalyst is comparable to those of Pt/Al2O3. The formation of the Ni3Sn-phase alloy is attributed to the high H2 selectivity. The addition of Sn is shown to significantly decrease methane formation, while maintaining the activity of C–C bond cleavage.
Besides Sn, the performance of Ni catalysts can be tuned with various oxide supports (e.g., MgO, CeO2, and TiO2) [36,37,63], with the maximum H2 yield obtained on MgO for glycerol steam reforming [36,37]. Iriondo et al. [44] introduced Ce, La, Mg, or Zr as promoters to Ni supported on Al2O3. For glycerol APR, the addition of these species is found to improve initial glycerol conversion compared to undoped Ni catalysts. Temperature programmed reduction and X-ray photoelectron spectroscopy (XPS) analyses showed that these metal ions have been incorporated into catalyst systems and can interact closely with the active Ni phase. These promoters function differently [44]. Some modify interactions between Ni and the Al2O3 support. La and Ce species will form mixed oxide phases with support. Moreover, Iriondo et al. [45] pointed out that La2O3-Al2O3 can improve the structure stability.
Pt-Ni alloys are also good reforming catalysts. Doukkali et al. [46] showed that Pt-Ni alloys prepared using sol-gel methods under basic condition (known as SGB) can be more active than either Pt or Ni for glycerol APR. Pt-Ni alloys are also effective for WGS reaction.
He et al. [47] used multi-walled carbon nanotubes (MWCNT) as supports for Pt-Ni alloys to improve glycerol APR. They deducted that the primary H2 production pathway proceed with initial glycerol dehydrogenation forming glyceraldehyde. Glyceraldehyde is then converted to lactic acid (LA), which can undergo decarboxylation. Base additives (e.g., CaO) increase LA selectivity. Methanol, from C–C bond cleavage, was also found in the liquid phase.
Among other metals, Zhang et al. [38] found that CeO2-supported Ir and Co catalysts which were also very active and selective for H2 production in steam reforming. The highest catalytic activity was achieved on Ir/CeO2 (100% conversion), with >85% H2 selectivity. The superior performance of Ir/CeO2 can attributed to strong interactions between Ir and the support.
2.1.3. Understanding Molecular Behaviors for Glycerol Conversion on Transition Metals with DFT
To understand trends exhibited on transition metals during polyol conversions, EG is considered ideal and studied as a model compound [30,64,65,66]. Some key findings have been summarized in Figure 4 by Davda et al. [64]. Thanks to extensive experimental screening, it is found that the activity of SiO2-supported monometallic catalysts (in terms of CO2 turnover frequency, or TOF) decreases in the order of Pt > Ni > Ru > Rh >> Pd. The H2 selectivity is ranked in the decreasing order of Pd > Pt > Ni > Ru > Rh.
Nevertheless, it also became evident that the detection and grathering information on key reaction intermediates for pathway elucidation is far from straightforward. DFT calculations can be employed to provide information to fill the gap between fundamental chemistry and catalyst performance. If properly used, the modeling can be utilized to predict and tune catalytic properties for catalyst developments.
Liu and Greeley [60,62,67,68] investigated adsorptions of glycerol and its dehydrogenation intermediates on transition metals. Adsorption patterns of glycerol conversion intermediates were investigated on close-packed surfaces of Pt, Ni, Pd, Rh, and Cu.
The behavior to describe glycerol dehydrogenation thermochemistry, transition states of C–H/O–H, C–C, and C–O bond cleavage on Pt(111) can be summarized in a unified free energy diagram on Pt(111) [60,67], as shown in Figure 5. DFT calculations showed that intermediates from C–H bond cleavage are more thermodynamically stable than those from O–H bond cleavage. Energy barriers for C–H/O–H bond cleavage steps are significantly lower than those for C–C or C–O bond cleavage steps (by more than 0.5 eV). Hence, it can be concluded that, on Pt(111), early in the decomposition process, both C–C and C–O bond cleavage will be kinetically slow compared to dehydrogenation reactions. Moreover, much lower C–C bond cleavage energy barriers are possible, via decarbonylation, after several steps of dehydrogenation.
Similar analyses can be performed on other transition metals or even bimetallic alloys [62,68]. It should be mentioned that acquiring full detail of the entire glycerol decomposition network is extremely, computationally expensive. In Section 4.1, analytical approaches such as scaling relationship, thermodynamic Group Additivity, and Brønsted-Evans-Polanyi method can potentially eliminate the necessity of performing full explicit DFT calculations, and are frequently employed for mechanistic elucidation and trend prediction.
2.2. Catalysts for Glycerol Hydrogenolysis
Glycerol reforming is always accompanied by hydrogenolysis, dehydration, and/or methanation—all involving C–O bond cleavage and H2 consumption. Despite the loss of H2 as product, 1,2-propanediol (12-PDO), 1,3-propanediol (13-PDO), 1-propanol (1-PO), 2-propanol (2-PO), EG, LA, methanol, can be obtained. Product selectivity is a primary interest in catalyst selection for glycerol hdyrogenolysis. Complicated by different catalyst behaviors and environmental conditions, the exact hydrogenolysis pathways are still being debated currently.
Montassier and co-workers [69,70,71] proposed a widely accepted scheme for glycerol hydrogenolysis over silica-supported transition metals (i.e., Pt, Ru, Rh, Ir), under basic conditions. In their scheme, C–C bond cleavage occurs through base-catalyzed retro-aldol reaction, whereas C–O bond cleavage occurs through base-catalyzed dehydration. Dehydrogenation takes place on metal sites.
Maris and Davis [72] proposed an adjusted mechanism, as shown in Figure 6, which resembles to Montassier’s original scheme, from a series of tests on carbon supported Pt and Ru monometallic catalysts under neutral or basic conditions (using NaOH or CaO).
According to the adjusted scheme (Figure 6), glycerol dehydrogenation (to glyceraldehyde) is proposed to be the first step on the metal (Ru, Pt) site. Under a neutral condition, Ru/C is more active than Pt/C for hydrogenolysis, but the selectivity over PDO is higher on Pt/C [72]. NaOH or CaO were added to elucidate base-catalyzed routes (versus metal-catalyzed reactions at neutral conditions). Under basic condition, the activity of Pt/C is enhanced more than Ru/C [72]. Lactate formation became significant higher on both Pt/C and Ru/C catalysts at high pH.
EG is formed via C–C bond cleavage catalyzed by metal because the rate of EG formation remains comparable to that under neutral condition. It was further deduced that EG cannot be formed via hydrogenolysis from PDOs, but from glycerol directly instead. Therefore, the likely route for EG formation proceeds through glyceraldehyde, followed by C–C bond cleavage, similar to the retro-aldol reaction.
Under acidic condition, King et al. [40] proposed that C–C bond cleavage proceeds through glyceraldehyde decarbonylation. CO2 in the gas phase product, which is rapidly converted from CO via WGS reaction at the metal sites, can be analyzed to monitor this pathway.
Figure 7 depicts a schematic network with multiple pathways leading to 12-PDO and 13-PDO, adapted from Reference [73]. Each of the six primary intermediates—1,2-enol, acetol, 1,2-aldol, 2,3-enol, hydroxylpropanal, and 1,3-enol—can be formed from dehydration, which is then followed by hydrogenation (of C=C or C=O bond) to 12-PDO or 13-PDO. Activities and selectivities are very sensitive to the transition metal catalyst involved, and are especially linked to the relative stability of the dehydrated intermediates. Isomerizations between different pairs of intermediates are possible (as indicated by two-headed arrows in Figure 7), cross-linking different pathways. Representative metal-based catalysts for glycerol hydrogenolysis are summarized in Table 2.
Anneau et al. [74] used DFT calculations to generate detailed glycerol hydrogenolysis pathways on Rh(111). Both dehydration and dehydrogenation were investigated as the first step for 12-PDO and LA formation. As a result, the authors confirmed that glycerol prefers to dehydrogenate to form glyceraldehyde first, and then dehydrates to form an energetically stable enol intermediate (an unsaturated alcohol as shown in Figure 7). Enols can isomerize quickly into acetol or aldol. Finally, 12-PDO is formed via hydrogenation.
As the relative stability among glycerol dehydration intermediates differs from metal to metal. Coll et al. [73] suggested that this character can be used to determine desired product selectivity. For example, on Rh(111), enols (consisting of a C=C bond) will be stabilized more than aldols (consisting of a C=O bond by up to 0.4 eV). Hence, the route corresponding to dehydration at the central carbon, forming 1,3-enol, becomes competitive. On Pd(111), the aldol structure becomes even less stable, while 1,2-enol is the most stable intermediate. On Ni(111), the energetic difference between 1,3-enol and 1,2-aldol, is 0.36 eV, much higher than that on Rh (only 0.16 eV). The larger energetic difference explains why very little 13-PDO is obtained on Ni. Supported by theory, catalyst selection can be guided by theory following a principle based on the relative stability on different metals so that 12-PDO and 13-PDO selectivity can be modified. Another useful indicator is the energy difference between 13-PDO versus 12-PDO.
The role of catalysts is also present in influencing glycerol hydrogenolysis selectivity. Huang et al. [77] tested Ni/Al2O3 and Cu/ZnO/Al2O3 for hydrogenolysis of glycerol in both aqueous and vapor phase. On Ni/Al2O3, the catalyst is more selective toward CH4 and CO, rather than 12-PDO. Cu/ZnO/Al2O3, however, is much more selective for 12-PDO and acetol formation. The 12-PDO production of was even more favored at higher H2 pressure (64 bar), reaching a selectivity of 92% at 100% glycerol conversion.
The use of catalytic promoters has also been attempted. Jimenez-Morales et al. [78] showed Ce-promoted Ni/SBA-15 catalysts are much more active for 12-PDO production than unpromoted Ni/SBA catalysts. This is because Ce species increase surface acidity that promotes the formation of acetol. The 7.5 Ce-Ni-SBA catalyst is most active among the materials tested.
Bimetallic Pt-Ru and Au-Ru have been prepared by Maris and Davis [83], by depositing respective Pt or Au onto C-supported Ru nanoparticles. Both alloys behave similarly to pure Ru during aqueous-phase hydrogenolysis. However, Pt-Ru alloys are more active than either of its pure Pt or Ru component. It is possible that the presence of Pt is able to help dissociate H2, whereas Ru provided sites for glycerol adsorption and C–C cleavage. Pt-Ru appeared to be more stable than Au-Ru, in which Au is found disintegrated from Ru onto the support. In addition, the addition of Au will decrease hydrogenolysis activity. The formation of EG is favored over PDOs by both Pt-Ru/C and Au-Ru/C under neutral condition [83]. Also, in the presence of base, bimetallic catalysts will favor the production of lactate and PDO over EG.
Pt-Re alloys were tested by Daniel et al. [81], and were found to be more active than either Pt or Re. For instance, the 13-PDO selectivity can go up to 34%. Catalyst performance can be further improved after sintering at high temperatures, benefitting from better mixing. Re is more oxophilic and is capable of coordinating with OH. For this reason, the authors suggested that surface Re–OH species is mainly responsible for C–O bond activation and cleavage.
Liu et al. [84] showed that Ru-Cu bimetallic alloys are also active for glycerol hydrogenolysis. In particular, the synergy between Ru and Cu has been identified to be crucial. Ru-Cu alloys are selective for 12-PDO production (at 84%, and no 13-PDO). In addition, catalyst supports also influence the performance of catalysts. Especially, ZrO2 enables the best performance for 12-PDO production. Acidic supports such as NaY and HY promote 1-PO formation. Carbonium ions are believed to convert 12-PDO further into 1-PO under acidic conditions.
Ma et al. [85] added Re to Ru to promote glycerol conversion of glycerol and the selectivities of PDOs. In this case, Re is shown to prevent Ru particles from aggregation. In addition, the Re component (ReOx) acts as a solid acid to promote dehydration. Ru-Re favors 12-PDO over EG formation. Despite some acid functionality being present, Ru-Re catalysts are effective to inhibit conversions of 12-PDO and 13-PDO to 1-PO and 2-PO, a beneficial property to maintain high PDO selectivities.
On Ir-Re alloys, Nakagawa et al. [86] pointed out that glycerol hydrogenolysis occurs at its terminal position near the ReOx cluster to form 2,3-dihydroxypropoxide. Hydrogen at the Ir site approaches the 2-position of 2,3-dihydroxypropoxide, producing 3-hydroxypropoxide that is then hydrolyzed into 13-PDO. Due to structure sensitivity, the size of ReOx cluster is a key factor. To achieve high 13-PDO selectivity, the cluster should be sufficiently large so that steric effect for the preferential terminal alkoxide formation can be induced.
2.3. Metal–Acid Bifunctional Catalysts for Glycerol Hydrogenolysis
It has become apparent that the acid functionality plays a dominant role in catalyzing the C–O bond cleavage. A group of catalysts, which are bifunctional by possessing both metal and acid sites, are effective for both dehydration and hydrogenation. Particularly, catalytic synergies are unique and extremely beneficial.
A series of bifunctional catalysts based on Ru have been investigated by Kusunoki et al. [75] (Table 3). The addition of solid acids to Ru is able to broadly boost glycerol conversion and promote 12-PDO selectivity even at a mild temperature (e.g., 120 °C). The study confirmed that 1-hydroxyacetone, resulting from dehydration, is the intermediate for 12-PDO formation. On the other hand, 13-PDO is formed via 3-hydroxypropanal. It is also shown that the reactivity of 12-PDO is lower than both glycerol and 13-PDO in the presence of solid acids, contributing to the enhanced selectivity.
With amorphous silicate aluminate (ASA)-supported Pt catalysts, Gandarias et al. [80] noted that acetol is formed from glycerol dehydration at the acid site. Meanwhile, Pt plays the role of hydrogenating acetol to 12-PDO. This study also suggested that the glycerol-to-acetol pathway is enhanced when Pt and ASA coexist. In addition, Pt can also reduce coke formation by facilitating hydrogen spillover. Pt/ASA is also catalytically active for C–C bond cleavage and the hydrogenolysis of PDOs to 1-PO, and thus may lower the selectivity of PDO productions.
Miyazawa et al. [87,88,89] performed a series of glycerol hydrogenolysis studies using Ru-based catalysts containing ion-exchanged resins (e.g., Amberlyst). Compared to Rh/C, Pt/C, and Pd/C, such a metal-acid combination is effective for glycerol hydrogenolysis under acidic conditions and mild temperatures (120 °C). By comparing the yields of PDOs on Ru/C + Amberlyst and Ru/C catalysts, it is deduced that 12-PDO is formed via dehydration over the Brønsted acid sites of Amberlyst. The intermediate, i.e., acetol, results from proton attacking the OH bonded to terminal carbons. Acetol hydrogenation to 12-PDO is a fast step. 13-PDO is first formed through dehydration to 3-hydroxypropionaldehyde at the Ru site, and then hydrogenated at the Ru sites as well. Both 1-PO and 2-PO are formed via 13-PDO catalyzed by Ru/C. EG is formed directly from glycerol (not observed from PDO conversions). Ethanol and CH4 are from the degradation of PDOs. Ethanol can also be formed during hydrogenolysis of EG. Methanol is formed via C2 (i.e., EG) degradation.
As shown above, Ru is often the metal by choice for hydrogenolysis as it is much more active than other noble metals. Surface OH species on Ru is believed to assist Ru for dehydration. An OH-assisted glucose dehydration mechanism on Ru has been suggested by Tyrlik et al. [91]. Glycerol can be catalyzed by Ru–OH to form 3-hydroxypropionaldehyde, as OH can attack H atoms on terminal carbons.
A number of optimizations of Ru/C + Amberlyst catalysts have been attempted by Miyazawa et al. [88], who indicated that glycerol hydrogenolysis can be sensitive to Ru structures. For instance, on Ru5/C(I), hydrogenolysis selectivity was lower on smaller Ru particles [88]. They also pointed out that the metal component in Ru/C + Amberlyst is susceptible to sulfur poisoning by SO2 and H2S. Miyazawa et al. [89] also demonstrated the heat-resistant Amberlyst70 (or A70) can be employed to improve the heat tolerance (up to 190 °C) of the catalyst, which is still selective for 12-PDO production.
Zhu et al. [90] showed that heteropolyacids (HPAs) can be used to modify Pt/ZrO2 catalysts for glycerol hydrogenolysis to obtain 13-PDO. Moreover, acidity can improve the selectivity to 13-PDO. In this work, highest selectivity to 13-PDO was achieved on Pt-HSiW/ZrO2, compared to Pt-HPW/ZrO2 and Pt-HPMo/ZrO2. These studies also reflected that the yield of 13-PDO (via acrolein) was proportional to the concentration of the Brønsted acid sites, whereas the yield of 12-PDO (via acetol) is proportional to the concentration of Lewis acid sites. Products such as 1-PO and 2-PO are mainly formed via 12-PDO, and the formation of EG is from glyceraldehyde decarbonylation.
2.4. Catalysts for Glycerol Oxidation
Glycerol oxidation reactions can also produce many unique products (glyceraldehyde, dihydroxyacetone (DHA), and organic acids). Some of these chemicals are values medicinally, with applications related to metabolite in glycolysis. A general glycerol oxidation reaction network can be illustrated by Figure 8, adapted from Reference [92].
Representative catalysts for glycerol oxidation are summarized in Table 4. Supported noble metals, Pt, Pd, and Au, are commonly used [92,93,94,95,96].
Oxidation of glycerol on Pd and Pt in the liquid phase was investigated by Garcia et al. [94]. Glyceric acid, via oxidation of the terminal OH group in glycerol, was obtained as the main product on Pd/C catalysts (with selectivity up to 70% at pH = 11). On Pt/C, glyceric acid can be achieved at 55% selectivity in neutral media. Demirel-Gülen et al. [92] also investigated liquid-phase glycerol oxidation, and found that Au/carbon black catalysts are catalytically active.
In recent years, electrochemical techniques for glycerol oxidation have been developed. In order to elucidate oxidation pathways, high-performance liquid chromatography (HPLC), coupled with rapid online sample collection method, were developed and implemented by Kwon et al. [97,98] to detect product from electrooxidation of glycerol.
Besides catalytic materials, support, pH values, or even applied potential voltage can influence catalyst activity and main product distributions. Glycerol electrooxidation mechanisms on polycrystalline Au and Pt have been probed using HPLC-MS at different pH values [98]. Therein, more detailed reaction networks at neutral, acidic, and basic conditions were reported. In alkaline media, glyceric acid is the main product with glyceraldehyde as the intermediate on Pt. On Au, glyceric acid is the initial primary oxidation product, which is then rapidly oxidized into glycolic acid and formic acid (Figure 8). Higher applied potentials are usually necessary on Au. At such voltages, Au becomes more active than Pt as the Pt surface is susceptible to oxidation, forming PtOx. On the oxidized Pt surface, glycerol is more likely to be oxidized to glyceric and formic acids, via respective glyceraldehyde and glycolic acid. At lower pH, activities on both Au and Pt electrodes decrease, and Au will completely lose its activity under acidic conditions. Under neutral condition, the selectivity toward glyceraldehyde is high for both Au and Pt.
Using a combined experimental and
computational approach, Garcia et al. [99] established the understanding that the activity and selectivity of glycerol oxidation is very structure sensitive, as illustrated in Figure 9. Overall, small Au particles (~2.7 nm) are selective for glycolic acid formation. For larger Au particles (~3.7 nm), the selectivity to glyceric acid increases. Catalyst structure sensitivity has also been observed on Pt catalysts as well. For instance, the main glycerol oxidation products are primary and secondary alcohol oxidation on Pt(111), with respect to Pt(100).
Doping Pt with small amounts of Bi can improve activity, selectivity, as well as catalyst stability [93,95]. Kimura et al. [93] showed that Pt modified by Bi enables the oxidation of the secondary OH group of glycerol to obtain DHA, yielding 50% DHA selectivity.
Garcia et al. [95] further showed that Bi on Pt(111) can improve glycerol electrooxidation activity, by weakening the adsorption of CO. An increase of DHA selectivity was also observed. Kwon et al. [100] reasoned that this is because the Bi adatom species can block the active sites for primary alcohol oxidation on Pt/C. Enediol is the key intermediate during the isomerization between glyceraldehyde and DHA. It is produced from glycerol C–H bond cleavage, which prefers Pt(111), as illustrated in Figure 9 in purple (left). Bi adatom is able to stabilize this intermediate by interacting with the two hydroxyl groups of enediol, but not on Pt(100). Hence, glyceraldehyde is the primary product on this surface. DFT calculations are well suited to test the key hypothesis to elucidate reaction pathways eventually.
Recently, Xiao et al. [96] prepared a series of Pt and Pt-Bi catalysts loaded on MCM and ZSM-5 zeolitic supports, such as Pt-Bi/AC, Pt-Bi/ZSM-5, Pt/MCM-41, Pt-Bi/MCM-41, and Pt/Bi-MCM-41. Kinetic measurements showed that bimetallic Pt-Bi loaded on MCM-41 exhibits the highest DHA yield. Aided by DFT calculations, it was found that the Bi2O-Pt(111) complex is formed from O2 dissociative adsorption to stabilize glycerol adsorption. With this complex formation, the energy barrier to directly form DHA from glycerol can be lowered as the dehydrogenation at the middle C and middle O of glycerol occurs simultaneously under Bi2O and Pt cooperation. On the other hand, glyceraldehyde is formed from dehydrogenation of the terminal OH group at the Bi2O site. Because DHA, which is a secondary OH oxidation product, is more stable than glyceraldehyde, it will be the primary oxidation product, which is consistent with experimental work.
Kwon et al. [101] also demonstrated that Sb, Pb, Sn, or In can be introduced to Pt/C as surface adatom species to enhance the activity and selectivity of glycerol oxidation in acidic solution (e.g., H2SO4).
In recent years, glycerol electro-oxidation techniques, based on Pt, Pd, and Au catalysts, have been developed by Li’s group [102,103] to obtain sustainable commercial chemicals. Moreover, electricity power can be cogenerated using anion-exchange membrane fuel cell (AEMFC) devices. In order to further advance this technique for practical applications, understanding of the activity and selectivity governing reaction mechanism and robust catalytic materials will be the key.
3. Catalysts Selections Guided by First-Principles Methods
3.1. Linear Scaling Relationship to Estimate the Binding Energies of C3HxO3
Computational expenses increase rapidly as the number of atoms in a molecule rises, for instance, glycerol versus methanol. Hierarchical modeling methodology [59] that allows accurate kinetic parameter calculations for glycerol decomposition need to be established, so that the necessary computational cost reduction can be accomplished.
The concept and early work on the use of empirical formulations to describe chemisorption have been introduced by Shustorovich [104,105,106], known as the Unity Bond Index-Quadratic Exponential Potential (UBI-QEP). Similar work has been carried out by Kua et al. [107] to compute the heat of formation of certain gas phase adsorbates.
Both of the above approach relies, to some extent, on analyzing nearest neighbors of the central binding sites of adsorbed intermediate species. Methods adopting such concepts will be elaborated. For the purpose of discussion, binding energies of glycerol dehydrogenation intermediates (), representing C3HxO3, (x = 0–8) generically, are defined in Equation (4), by Liu and Greeley [67].
Such a definition is helpful to be related to the energy needed to decompose vapor phase glycerol into C3HxO3 and the stoichiometric amount of H2(g), and the adsorption of C3HxO3 on the substrate.
All the terms on the right side of Equation (4) can be obtained directly from DFT. The scaling scheme is, in turn, defined by Equations (5) and (6).
Variables and in Equation (5) represent the degree of undersaturation, defined by Equation (6), for all C and O atoms in C3HxO3. is 4 for carbon and 2 for oxygen, while is the total neighbors (H, C, and O), to which the central C/O atom is bonded. , , , and in Equation (5) are parameterized from DFT calculations. is the binding energy of gas phase glycerol on the substrate.
If the binding energy of a given intermediate is known for a reference metal (e.g., Pt), the binding energies of this species on different metal surfaces (M) can be computed according to Equation (7) using binding energies of atomic C and O on M.
When combined with Equation (4), Equation (7) can be modified into a more compact form as in Equation (8) [108].
More recently, Garcia-Muelas and Lopez [109] studied 14 mono and poly alcohols up to four carbon atoms on clean Pt(111) and Pd(111) surfaces, and proposed a multifactorial linear-scaling relationship, as in Equation (9). Terms in Equation (9), account for contributions from OH group (), CHx group (), and H-bonds ( and ), and will be parameterized from periodic DFT calculations.
A Group Additivity (GA) approach has been adapted by Salciccioli and coworkers [110,111] to predict thermodynamic properties of adsorbed alcohols and polyols. Different from Benson’s original approach [112], contributions from oxygenate groups (-CHxO-, x = 1, 2) center around carbon-oxygen-metal interactions. Moreover, a number of rules have been developed. Similar to the scaling relationship (as in Equations (5) and (6)), bond order rules (i.e., H = 1, C = 4, and O = 2) in the gas phase were followed. Zero-point energy correction to heat of adsorption and the vibrational frequencies associated with the intermediates were considered. The GA approach provides correlations at similar numerical quality.
3.2. Prediction of Catalyst Activity for Glycerol Decomposition Using Scaling Relationships
To deal with the numerous steps and complex reaction pathways, effective strategies have to be able to reduce the number of conventional DFT calculations. Predictive tools based on correlations associated with catalytic descriptors become critical. However, they need to be straightforward, numerically reliable, and chemically meaningful. Ferrin and coworkers [113] formulated an approach to analyze ethanol decomposition networks on a broad range of transition metals, and demonstrated initial success in connecting minima with maxima in the potential energy surface in conjunction with a scaling relation, based on binding energies of complex adsorbates with those of C and O. With this strategy, understanding key catalytic trends is essentially reduced to performing DFT calculations to retrieve binding energies of C and O, a much easier and reliable computational task.
Typical interpretations of free energy diagram for Pt have been introduced in Section 2.1.3. By executing the scaling relationship, every level of glycerol dehydrogenation on the close-packed surface of Pt, Pd, Rh, Ni, and Cu can be quickly generated, as illustrated in Figure 10. The main impact of this modeling approach is to enable a very computationally convenient means to examining a large number of plausible transition metals, equivalent to modeling-based high-throughput screening to some degree.
It becomes evident from Figure 10 that, on Pt(111), Rh(111), and Ni(111), dehydrogenation steps will not be limited by reaction thermodynamics, at least before the decarbonylation becomes competitive; on Pd(111), dehydrogenation becomes more endothermic, which is consistent with the trend observed in Figure 4 (i.e., much smaller CO2 TOF). On Cu(111), the thermodynamics for glycerol decomposition is the least favorable. The process, represented by Equation (1), is more than 2.5 eV endothermic.
Moreover, qualitative understanding of the preference for C–H versus O–H bond cleavage can be obtained using the relative binding strengths of C and O on different metals [68]. For example, on Pt and Pd, C binds much more strongly than atomic O, corresponding that intermediates from C–H bond cleavage are favored. For Rh and Ni, O–H bond cleavage becomes gradually competitive regarding stronger O binding. Cu shows the strongest preference for O–H bond cleavage due to the strongest O binding on Cu(111) relative to that of C.
Kinetic considerations for C–H, O–H, C–C, and C–O bond cleavage can be readily included in the analysis as well. Although DFT-based methods, such as Climbing Image-Nudged Elastic Band (CI-NEB) [114,115] and dimer [116], can be straightforwardly applied, the main obstacle is that such calculations will be extremely time consuming, even with the most advanced modern computing clusters. Liu and Greeley [60,67,68] showed that the so-called Brønsted-Evans-Polanyi (BEP) relationship, once reliably established from DFT, can be used to predict all the elementary steps in glycerol decomposition network. Now, it is, in principles, possible to perform full analysis (similar to Figure 5) on a given single crystal metal surfaces with minimal amount of calculations.
Most recently, periodic DFT calculations, coupled with fast energetic and kinetic analysis, have been employed to investigate glycerol decomposition on bimetallic Pt-Mo alloys [62]. It is concluded that on the Pt skin of Pt-Mo bimetallic alloys, CO binding becomes weaker and is able to promote reforming activity due to mitigated CO poisoning. In addition, surface Mo species have been found to promote CO oxidation via WGS reaction pathways. It is also likely that bifunctional site enabling synergistic interactions also exist to influence overall catalyst performance. In this regard, more sophisticated models are needed. More importantly, an integrated modeling and experimental approach (providing clues on catalyst structures, active sites, and intrinsic activity information) is the key to success.
4. Summary, Challenges, and Research Opportunities
Powered by fast advancements in parallel computing facilities, technique, and algorithms, significant progress has been made. Nevertheless, several directions—including: (1) development of efficient and accurate modeling tools to describe glycerol conversion in solutions; (2) establishment and predicting trends of bifunctional model catalysts; and (3) development of algorithms to generate reaction pathways of complex glycerol reaction network—will make further impact on catalytic glycerol conversions. Each of these topics represent one emerging research area.
4.1. Modeling Glycerol Conversion in Solutions
It can be seen that glycerol conversion into chemicals frequently takes place in the aqueous phase. Currently, consideration of the physical and chemical behaviors of glycerol in aqueous phase have been tackled by three main approaches: (1) DFT-based implicit solvation model [117,118]; (2) ab initio molecular dynamics (AIMD) [119]; and (3) combined DFT and classical molecular dynamics (MD) method [120].
Faheem et al. [118] proposed a simple implicit solvation model for solid surfaces (iSMS,) approach to model reactions at solid−liquid interfaces with implicit solvation models. The basic concept of iSMS is to account for the long-range metal interactions using slab models without solvent molecules; and the liquid effect of a local perturbation of free energy described by an embedded cluster model in an implicit continuum model. Other computational techniques, such as VASPsol (implemented in plane wave Vienna ab initio Simulation Package (VASP) code) [121], are also important tools. A comparison between implicit and explicit solvation models for surface adsorption free energies for DFT calculations was recently published by Steinmann et al. [122].
Callam et al. [119] performed G2- and CBS-QB3-level of ab initio molecular dynamics (AIMD) simulations to map the potential energy surfaces of glycerol in aqueous phases. The important and low energy conformers can be identified.
Bodenschatz et al. [120] introduced a multiple-step strategy to describe liquid phase configurations during the adsorptions of glycerol and its intermediates (C3H7O3) on Pt(111). In their approach, DFT is used to optimize surface-adsorbed species, partially relaxed H2O molecules are then used to initiate MD simulations using LAMMPS [123], with Pt atoms and other adsorbates were fixed in the positions determined from prior DFT calculations.
4.2. Modeling Synergistic Bifunctional Catalysts for Glycerol Conversion
Bifunctional catalytic materials are ubiquitous and play an important role in catalysis, including catalytic glycerol conversion. Many bimetallic alloys [34,40,48,49,62,81,83], metal-acid/support catalysts [75] can all establish multi-functional systems to influence reaction activity and product formation pathways.
In Section 2.4, Greeley and coworkers [96] modeled Pt-Bi alloys in glycerol oxidation and revealed the working principles of glycerol dehydrogenation in a synchronized manner at the PtOx and Bi sites. Such information is valuable to understand the intrinsic property related to product selectivity, and would be rather elusive to acquire without the aid of molecular modeling.
Moreover, Yun et al. [124] used DFT calculations to design W-incorporated MoVW mixed oxide catalysts for glycerol oxydehydration to obtain acrylic acid, acrolein, acetic acid, along with CO2 and acetaldehyde. It is revealed that the incorporated W species are able to induce neighboring Mo, V, and O, to a reduced state for Mo and V (e.g., Mo5+ and V4+), and those more nucleophilic O, which are necessary for selective acrylic acid production.
However, our treatments of bifunctional catalysts are far less mature than the modeling of catalysts with single and uniform active sites on surface. It is also important to point out that the success of this research direction will strongly depend on cooperative interactions between experiment and theory.
4.3. Development Tools to Generate Reaction Pathways
A number of algorithms are already available to automatically generate and predict reaction steps. Herein, a few examples are mentioned to show that there is a growing field focused on this direction. For instance, a simple protocol was developed by Suleimanov and Green [125] to allow fully automated discovery of elementary chemical reaction steps. Zimmerman et al. [126] applied the ‘Growing String Method’ to reliably find reaction path and transition state. Vinu and Broadbelt [127] presented a comprehensive review on the topics of reaction pathway elucidation using various algorithms for the purpose of reaction kinetics modeling of complex systems.
Lastly, as an alternative approach, the p-graph (or process graph) is a graph-theoretic approach to facilitate the synthesis of a process system. The concept was originally proposed and developed by Friedler, Fan, and their co-workers [128], to be exhaustive and computationally efficient. Although it has originally developed for process simulations and optimizations, recent developments have shown that this approach can be adapted for catalytic reactions such as WGS reaction [129], and hydrogenation reaction [130].
Acknowledgments
This work is supported as part of the Institute for Atom-Efficient Chemical Transformations (IACT), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences; B.L. also thanks the Start-up fund provided by Kansas State University, the National Science Foundation under Award No. EPS-0903806, and matching support from the State of Kansas through the Kansas Board of Regents for financial support. The author is also grateful for the supercomputing resources and services from the Center for Nanoscale Materials (CNM) supported by the Office of Science of the US Department of Energy under the contract No. DE-AC02-06CH11357; the Beocat Research Cluster at Kansas State University, which is funded in part by NSF grants CNS-1006860; and the National Energy Research Scientific Computing Center (NERSC) under the contract No. DE-AC02-05CH11231.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Miller, S. The Soapmaker’s Companion: A Comprehensive Guide with Recipes, Techniques & Know-How; Storey Books: North Adams, MA, USA, 1997; pp. 1–281. [Google Scholar]
- Prescott, S.C.; Dunn, C.G. Industrial Microbiology, 3rd ed.; McGraw-Hill: New York City, NY, USA, 1959; pp. 208–217. [Google Scholar]
- Wang, Z.; Zhuge, J.; Fang, H.; Prior, B.A. Glycerol production by microbial fermentation. Biotechnol. Adv. 2001, 19, 201–223. [Google Scholar] [CrossRef]
- Ciriminna, R.; Della Pina, C.; Rossi, M.; Pagliaro, M. Understanding the glycerol market, Eur. J. Lipid Sci. Technol. 2014, 116, 1432–1439. [Google Scholar] [CrossRef]
- Quispe, C.A.G.; Coronado, C.J.R.; Carvalho, J.A. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- LMC International. Feedstocks for Bio-Based Chemicals; LMC International: Oxford, UK, 2013. [Google Scholar]
- Thompson, J.C.; He, B.B. Characterization of crude glycerol from biodiesel production from multiple feedstocks. Appl. Eng. Agric. 2006, 22, 261–265. [Google Scholar] [CrossRef]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.J.; Corma, A.; De Frutos, P.; Iborra, S.; Noy, M.; Velty, A.; Concepcion, P. Chemicals from biomass: Synthesis of glycerol carbonate by transesterification and carbonylation with urea with hydrotalcite catalysts. The role of acid-base pairs. J. Catal. 2010, 269, 140–149. [Google Scholar] [CrossRef]
- Ale, C.E.; Farias, M.E.; de Saad, A.M.S.; Pasteris, S.E. Glycerol production by Oenococcus oeni during sequential and simultaneous cultures with wine yeast strains. J. Basic Microbiol. 2014, 54, S200–S209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, F.; Ni, J.; Li, C.; Xu, P. Production of C3 platform chemicals from CO2 by genetically engineered cyanobacteria. Green Chem. 2015, 17, 3100–3110. [Google Scholar] [CrossRef]
- Wilson, E.K. Biodiesel revs up—Fuel made from vegetable oil leads the pack of alternatives to petroleum products. Chem. Eng. News 2002, 80, 46–49. [Google Scholar] [CrossRef]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. From glycerol to value-added products. Angew. Chem. Int. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- Wen, G.D.; Xu, Y.P.; Ma, H.J.; Xu, Z.S.; Tian, Z.J. Production of hydrogen by aqueous-phase reforming of glycerol. Int. J. Hydrogen Energy 2008, 33, 6657–6666. [Google Scholar] [CrossRef]
- Zhou, C.H.C.; Beltramini, J.N.; Fan, Y.X.; Lu, G.Q.M. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Brandner, A.; Lehnert, K.; Bienholz, A.; Lucas, M.; Claus, P. Production of Biomass-Derived Chemicals and Energy: Chemocatalytic Conversions of Glycerol. Top. Catal. 2009, 52, 278–287. [Google Scholar] [CrossRef]
- Vaidya, P.D.; Rodrigues, A.E. Glycerol reforming for hydrogen production: A review. Chem. Eng. Technol. 2009, 32, 1463–1469. [Google Scholar] [CrossRef]
- Rahmat, N.; Abdullah, A.Z.; Mohamed, A.R. Recent progress on innovative and potential technologies for glycerol transformation into fuel additives: A critical review. Renew. Sustain. Energy Rev. 2010, 14, 987–1000. [Google Scholar] [CrossRef]
- Tanksale, A.; Beltramini, J.N.; Lu, G.M. A review of catalytic hydrogen production processes from biomass. Renew. Sustain. Energy Rev. 2010, 14, 166–182. [Google Scholar] [CrossRef]
- Stelmachowski, M. Utilization of glycerol, a by-product of the transestrification process of vegetable oils: A review. Ecol. Chem. Eng. S 2011, 18, 9–30. [Google Scholar]
- Lin, Y.C. Catalytic valorization of glycerol to hydrogen and syngas. Int. J. Hydrogen Energy 2013, 38, 2678–2700. [Google Scholar] [CrossRef]
- Tran, N.H.; Kannangara, G.S.K. Conversion of glycerol to hydrogen rich gas. Chem. Soc. Rev. 2013, 42, 9454–9479. [Google Scholar] [CrossRef] [PubMed]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.; Wietschel, M. The future of hydrogen—Opportunities and challenges. Int. J. Hydrogen Energy 2009, 34, 615–627. [Google Scholar] [CrossRef]
- Ewan, B.C.R.; Allen, R.W.K. A figure of merit assessment of the routes to hydrogen. Int. J. Hydrogen Energy 2005, 30, 809–819. [Google Scholar] [CrossRef]
- Alves, H.J.; Bley, C.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.S.; Coimbra-Araujo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrogen Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- Schwengber, C.A.; Alves, H.J.; Schaffner, R.A.; da Silva, F.A.; Sequinel, R.; Bach, V.R.; Ferracin, R.J. Overview of glycerol reforming for hydrogen production. Renew. Sustain. Energy Rev. 2016, 58, 259–266. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Dumesic, J.A. Raney Ni-Sn catalyst for H2 production from biomass-derived hydrocarbons. Science 2003, 300, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Shabaker, J.W.; Davda, R.R.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts. J. Catal. 2003, 215, 344–352. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Dumesic, J.A. Kinetics of aqueous-phase reforming of oxygenated hydrocarbons: Pt/Al2O3 and Sn-modified Ni catalysts. Ind. Eng. Chem. Res. 2004, 43, 3105–3112. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Dumesic, J.A. Aqueous-phase reforming of oxygenated hydrocarbons over Sn-modified Ni catalysts. J. Catal. 2004, 222, 180–191. [Google Scholar] [CrossRef]
- Huber, G.W.; Dumesic, J.A. An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery. Catal. Today 2006, 111, 119–132. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Simonetti, D.A.; Dumesic, J.A.; Pyrz, W.D.; Murillo, L.E.; Chen, J.G.G.; Buttrey, D.J. The role of rhenium in the conversion of glycerol to synthesis gas over carbon supported platinum-rhenium catalysts. J. Catal. 2008, 260, 164–177. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.; Haryanto, A. Production of hydrogen by steam reforming of glycerin over alumina-supported metal catalysts. Catal. Today 2007, 129, 355–364. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.D.; Haryanto, A. Hydrogen production from glycerin by steam reforming over nickel catalysts. Renew. Energy 2008, 33, 1097–1100. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.D.; To, S.D.F.; Bricka, R.M.; Steele, P.H.; Haryanto, A. Conversion of glycerol to hydrogen via a steam reforming process over nickel catalysts. Energy Fuels 2008, 22, 1220–1226. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, X.; Li, Y.; Xu, Y.; Shen, W. Hydrogen production from steam reforming of ethanol and glycerol over ceria-supported metal catalysts. Int. J. Hydrogen Energy 2007, 32, 2367–2373. [Google Scholar] [CrossRef]
- Pompeo, F.; Santori, G.; Nichio, N.N. Hydrogen and/or syngas from steam reforming of glycerol. Study of platinum catalysts. Int. J. Hydrogen Energy 2010, 35, 8912–8920. [Google Scholar] [CrossRef]
- King, D.L.; Zhang, L.; Xia, G.; Karim, A.M.; Heldebrant, D.J.; Wang, X.; Peterson, T.; Wang, Y. Aqueous phase reforming of glycerol for hydrogen production over Pt-Re supported on carbon. Appl. Catal. B Environ. 2010, 99, 206–213. [Google Scholar] [CrossRef]
- Luo, N.J.; Fu, X.W.; Cao, F.H.; Xiao, T.C.; Edwards, P.P. Glycerol aqueous phase reforming for hydrogen generation over Pt catalyst—Effect of catalyst composition and reaction conditions. Fuel 2008, 87, 3483–3489. [Google Scholar] [CrossRef]
- Wawrzetz, A.; Peng, B.; Hrabar, A.; Jentys, A.; Lemonidou, A.A.; Lercher, J.A. Towards understanding the bifunctional hydrodeoxygenation and aqueous phase reforming of glycerol. J. Catal. 2010, 269, 411–420. [Google Scholar] [CrossRef]
- Guo, Y.; Azmat, M.U.; Liu, X.H.; Wang, Y.Q.; Lu, G.Z. Effect of support’s basic properties on hydrogen production in aqueous-phase reforming of glycerol and correlation between WGS and APR. Appl. Energy 2012, 92, 218–223. [Google Scholar] [CrossRef]
- Iriondo, A.; Barrio, V.L.; Cambra, J.F.; Arias, P.L.; Gueemez, M.B.; Navarro, R.M.; Sanchez-Sanchez, M.C.; Fierro, J.L.G. Hydrogen production from glycerol over nickel catalysts supported on Al2O3 modified by Mg, Zr, Ce or La. Top. Catal. 2008, 49, 46–58. [Google Scholar] [CrossRef]
- Iriondo, A.; Cambra, J.F.; Barrio, V.L.; Guemez, M.B.; Arias, P.L.; Sanchez-Sanchez, M.C.; Navarro, R.M.; Fierro, J.L.G. Glycerol liquid phase conversion over monometallic and bimetallic catalysts: Effect of metal, support type and reaction temperatures. Appl. Catal. B Environ. 2011, 106, 83–93. [Google Scholar] [CrossRef]
- El Doukkali, M.; Iriondo, A.; Arias, P.L.; Requies, J.; Gandarias, I.; Jalowiecki-Duhamel, L.; Dumeignil, F. A comparison of sol-gel and impregnated Pt and/or Ni based gamma-alumina catalysts for bioglycerol aqueous phase reforming. Appl. Catal. B Environ. 2012, 125, 516–529. [Google Scholar] [CrossRef]
- He, C.; Zheng, J.; Wang, K.; Lin, H.; Wang, J.-Y.; Yang, Y. Sorption enhanced aqueous phase reforming of glycerol for hydrogen production over Pt-Ni supported on multi-walled carbon nanotubes. Appl. Catal. B Environ. 2015, 162, 401–411. [Google Scholar] [CrossRef]
- Zhang, L.; Karim, A.M.; Engelhard, M.H.; Wei, Z.; King, D.L.; Wang, Y. Correlation of Pt-Re surface properties with reaction pathways for the aqueous-phase reforming of glycerol. J. Catal. 2012, 287, 37–43. [Google Scholar] [CrossRef]
- Ciftci, A.; Ligthart, D.; Sen, A.O.; van Hoof, A.J.F.; Friedrich, H.; Hensen, E.J.M. Pt-Re synergy in aqueous-phase reforming of glycerol and the water-gas shift reaction. J. Catal. 2014, 311, 88–101. [Google Scholar] [CrossRef]
- Ciftci, A.; Eren, S.; Ligthart, D.; Hensen, E.J.M. Platinum-Rhenium Synergy on Reducible Oxide Supports in Aqueous-Phase Glycerol Reforming. ChemCatChem 2014, 6, 1260–1269. [Google Scholar] [CrossRef]
- Dietrich, P.J.; Lobo-Lapidus, R.J.; Wu, T.; Sumer, A.; Akatay, M.C.; Fingland, B.R.; Guo, N.; Dumesic, J.A.; Marshall, C.L.; Stach, E.; et al. Aqueous Phase Glycerol Reforming by PtMo Bimetallic Nano-Particle Catalyst: Product Selectivity and Structural Characterization. Top. Catal. 2012, 55, 53–69. [Google Scholar] [CrossRef]
- Dietrich, P.J.; Wu, T.; Sumer, A.; Dumesic, J.A.; Jellinek, J.; Delgass, W.N.; Ribeiro, F.H.; Miller, J.T. Aqueous Phase Glycerol Reforming with Pt and PtMo Bimetallic Nanoparticle Catalysts: The Role of the Mo Promoter. Top. Catal. 2013, 56, 1814–1828. [Google Scholar] [CrossRef]
- Tuza, P.V.; Manfro, R.L.; Ribeiro, N.F.P.; Souza, M. Production of renewable hydrogen by aqueous-phase reforming of glycerol over Ni-Cu catalysts derived from hydrotalcite precursors. Renew. Energy 2013, 50, 408–414. [Google Scholar] [CrossRef]
- Sanchez, E.A.; D’Angelo, M.A.; Comelli, R.A. Hydrogen production from glycerol on Ni/Al2O3 catalyst. Int. J. Hydrogen Energy 2010, 35, 5902–5907. [Google Scholar] [CrossRef]
- Iriondo, A.; Barrio, V.L.; Cambra, J.F.; Arias, P.L.; Guemez, M.B.; Navarro, R.M.; Sanchez-Sanchez, M.C.; Fierro, J.L.G. Influence of La2O3 modified support and Ni and Pt active phases on glycerol steam reforming to produce hydrogen. Catal. Commun. 2009, 10, 1275–1278. [Google Scholar] [CrossRef]
- Hirai, T.; Ikenaga, N.; Miyake, T.; Suzuki, T. Production of hydrogen by steam reforming of glycerin on ruthenium catalyst. Energy Fuels 2005, 19, 1761–1762. [Google Scholar] [CrossRef]
- Dou, B.L.; Wang, C.; Song, Y.C.; Chen, H.S.; Xu, Y.J. Activity of Ni-Cu-Al based catalyst for renewable hydrogen production from steam reforming of glycerol. Energy Convers. Manag. 2014, 78, 253–259. [Google Scholar] [CrossRef]
- Cheng, C.K.; Foo, S.Y.; Adesina, A.A. Glycerol Steam Reforming over Bimetallic Co-Ni/Al2O3. Ind. Eng. Chem. Res. 2010, 49, 10804–10817. [Google Scholar] [CrossRef]
- Chen, Y.; Salciccioli, M.; Vlachos, D.G. An efficient reaction pathway search method applied to the decomposition of glycerol on platinum. J. Phys. Chem. C 2011, 115, 18707–18720. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. Density Functional Theory Study of Selectivity Considerations for C–C versus C–O Bond Scission in Glycerol Decomposition on Pt(111). Top. Catal. 2012, 55, 280–289. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Evans, S.T.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported Pt and Pd bimetallic catalysts. Appl. Catal. B Environ. 2006, 62, 226–235. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, M.; Chan, M.K.Y.; Greeley, J.P. Understanding polyol decomposition on bimetallic Pt-Mo catalysts-a DFT study of glycerol. ACS Catal. 2015, 5, 4942–4950. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.; Haryanto, A. A comparative thermodynamic and experimental analysis on hydrogen production by steam reforming of glycerin. Energy Fuels 2007, 21, 2306–2310. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts. Appl. Catal. B Environ. 2003, 43, 13–26. [Google Scholar] [CrossRef]
- Davda, R.R.; Alcala, R.; Shabaker, J.; Huber, G.; Cortright, R.D.; Mavrikakis, M.; Dumesic, J.A. DFT and experimental studies of C–C and C–O bond cleavage in ethanol and ethylene glycol on Pt catalysis. Stud. Surf. Sci. Catal. 2003, 145, 79–84. [Google Scholar]
- Shabaker, J.W.; Huber, G.W.; Davda, R.R.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported platinum catalysts. Catal. Lett. 2003, 88, 1–8. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. Decomposition pathways of glycerol via C–H, O–H and C–C bond scission on Pt(111), a density functional theory study. J. Phys. Chem. C 2011, 115, 19702–19709. [Google Scholar] [CrossRef]
- Liu, B.; Greeley, J. A Density Functional Theory Analysis of Trends in Glycerol Decomposition on Close-Packed Transition Metal Surfaces. Phys. Chem. Chem. Phys. 2013, 15, 6475–6485. [Google Scholar] [CrossRef] [PubMed]
- Sohounloue, D.K.; Montassier, C.; Barbier, J. Catalytic hydrogenolysis of sorbitol. React. Kinet. Catal. Lett. 1983, 22, 391–397. [Google Scholar] [CrossRef]
- Montassier, C.; Giraud, D.; Barbier, J. Polyol Conversion by Liquid Phase Heterogeneous Catalysis over Metals; Elsevier Science Publishers: Amsterdam, The Netherlands, 1988; p. 165. [Google Scholar]
- Montassier, C.; Menezo, J.C.; Hoang, L.C.; Renaud, C.; Barbier, J. Aqueous polyol conversions on ruthenium and on sulfur-modified ruthenium. J. Mol. Catal. 1991, 70, 99–110. [Google Scholar] [CrossRef]
- Maris, E.P.; Davis, R.J. Hydrogenolysis of glycerol over carbon-supported Ru and Pt catalysts. J. Catal. 2007, 249, 328–337. [Google Scholar] [CrossRef]
- Coll, D.; Delbecq, F.; Aray, Y.; Sautet, P. Stability of intermediates in the glycerol hydrogenolysis on transition metal catalysts from first principles. Phys. Chem. Chem. Phys. 2011, 13, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Auneau, F.; Michel, C.; Delbecq, F.; Pinel, C.; Sautet, P. Unravelling the Mechanism of Glycerol Hydrogenolysis over Rhodium Catalyst through Combined Experimental-Theoretical Investigations. Chem. A Eur. J. 2011, 17, 14288–14299. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, Y.; Miyazawa, T.; Kunimori, K.; Tomishige, K. Highly active metal-acid bifunctional catalyst system for hydrogenolysis of glycerol under mild reaction conditions. Catal. Commun. 2005, 6, 645–649. [Google Scholar] [CrossRef]
- Chaminand, J.; Djakovitch, L.; Gallezot, P.; Marion, P.; Pinel, C.; Rosier, C. Glycerol hydrogenolysis on heterogeneous catalysts. Green Chem. 2004, 6, 359–361. [Google Scholar] [CrossRef]
- Huang, L.; Zhu, Y.L.; Zheng, H.Y.; Li, Y.W.; Zeng, Z.Y. Continuous production of 1,2-propanediol by the selective hydrogenolysis of solvent-free glycerol under mild conditions. J. Chem. Technol. Biotechnol. 2008, 83, 1670–1675. [Google Scholar] [CrossRef]
- Jimenez-Morales, I.; Vila, F.; Mariscal, R.; Jimenez-Lopez, A. Hydrogenolysis of glycerol to obtain 1,2-propanediol on Ce-promoted Ni/SBA-15 catalysts. Appl. Catal. B Environ. 2012, 117, 253–259. [Google Scholar] [CrossRef]
- Balaraju, M.; Rekha, V.; Prasad, P.S.S.; Prasad, R.B.N.; Lingaiah, N. Selective Hydrogenolysis of Glycerol to 1,2-Propanediol Over Cu-ZnO Catalysts. Catal. Lett. 2008, 126, 119–124. [Google Scholar] [CrossRef]
- Gandarias, I.; Arias, P.L.; Requies, J.; Guemez, M.B.; Fierro, J.L.G. Hydrogenolysis of glycerol to propanediols over a Pt/ASA catalyst: The role of acid and metal sites on product selectivity and the reaction mechanism. Appl. Catal. B Environ. 2010, 97, 248–256. [Google Scholar] [CrossRef]
- Daniel, O.M.; DeLaRiva, A.; Kunkes, E.L.; Datye, A.K.; Dumesic, J.A.; Davis, R.J. X-ray Absorption Spectroscopy of Bimetallic Pt-Re Catalysts for Hydrogenolysis of Glycerol to Propanediols. ChemCatChem 2010, 2, 1107–1114. [Google Scholar] [CrossRef]
- Ma, L.; He, D.H.; Li, Z.P. Promoting effect of rhenium on catalytic performance of Ru catalysts in hydrogenolysis of glycerol to propanediol. Catal. Commun. 2008, 9, 2489–2495. [Google Scholar] [CrossRef]
- Maris, E.P.; Ketchie, W.C.; Murayama, M.; Davis, R.J. Glycerol hydrogenolysis on carbon-supported PtRu and AuRu bimetallic catalysts. J. Catal. 2007, 251, 281–294. [Google Scholar] [CrossRef]
- Liu, H.Z.; Liang, S.G.; Jiang, T.; Han, B.X.; Zhou, Y.X. Hydrogenolysis of glycerol to 1,2-propanediol over Ru-Cu bimetals supported on different supports. CLEAN-Soil Air Water 2012, 40, 318–324. [Google Scholar] [CrossRef]
- Ma, L.; He, D.H. Hydrogenolysis of Glycerol to Propanediols over Highly Active Ru-Re Bimetallic Catalysts. Top. Catal. 2009, 52, 834–844. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Shinmi, Y.; Koso, S.; Tomishige, K. Direct hydrogenolysis of glycerol into 1,3-propanediol over rhenium-modified iridium catalyst. J. Catal. 2010, 272, 191–194. [Google Scholar] [CrossRef]
- Miyazawa, T.; Kusunoki, Y.; Kunimori, K.; Tomishige, K. Glycerol conversion in the aqueous solution under hydrogen over Ru/C plus an ion-exchange resin and its reaction mechanism. J. Catal. 2006, 240, 213–221. [Google Scholar] [CrossRef]
- Miyazawa, T.; Koso, S.; Kunimori, K.; Tomishige, K. Development of a Ru/C catalyst for glycerol hydrogenolysis in combination with an ion-exchange resin. Appl. Catal. Gen. 2007, 318, 244–251. [Google Scholar] [CrossRef]
- Miyazawa, T.; Koso, S.; Kunimori, K.; Tomishige, K. Glycerol hydrogenolysis to 1,2-propanediol catalyzed by a heat-resistant ion-exchange resin combined with Ru/C. Appl. Catal. Gen. 2007, 329, 30–35. [Google Scholar] [CrossRef]
- Zhu, S.H.; Qiu, Y.N.; Zhu, Y.L.; Hao, S.L.; Zheng, H.Y.; Li, Y.W. Hydrogenolysis of glycerol to 1,3-propanediol over bifunctional catalysts containing Pt and heteropolyacids. Catal. Today 2013, 212, 120–126. [Google Scholar] [CrossRef]
- Tyrlik, S.K.; Szerszen, D.; Olejnik, M.; Danikiewicz, W. Concentrated water solutions of salts as solvents for reaction of carbohydrates. Part 2. Influence of some magnesium salts and some ruthenium species on catalysis of dehydration of glucose. J. Mol. Catal. Chem. 1996, 106, 223–233. [Google Scholar] [CrossRef]
- Demirel-Gulen, S.; Lucas, M.; Claus, P. Liquid phase oxidation of glycerol over carbon supported gold catalysts. Catal. Today 2005, 102, 166–172. [Google Scholar] [CrossRef]
- Kimura, H.; Tsuto, K.; Wakisaka, T.; Kazumi, Y.; Inaya, Y. Selective oxidation of glycerol on a platinum bismuth catalyst. Appl. Catal. Gen. 1993, 96, 217–228. [Google Scholar] [CrossRef]
- Garcia, R.; Besson, M.; Gallezot, P. Chemoselective catalytic-oxidation of glycerol with air on platinum metals. Appl. Catal. Gen. 1995, 127, 165–176. [Google Scholar] [CrossRef]
- Garcia, A.C.; Birdja, Y.Y.; Tremiliosi, G.; Koper, M.T.M. Glycerol electro-oxidation on bismuth-modified platinum single crystals. J. Catal. 2017, 346, 117–124. [Google Scholar] [CrossRef]
- Xiao, Y.; Greeley, J.; Varma, A.; Zhao, Z.J.; Xiao, G.M. An experimental and theoretical study of glycerol oxidation to 1,3-dihydroxyacetone over bimetallic Pt-Bi catalysts. AIChE J. 2017, 63, 705–715. [Google Scholar] [CrossRef]
- Kwon, Y.; Koper, M.T.M. Combining voltammetry with HPLC: Application to electro-oxidation of glycerol. Anal. Chem. 2010, 82, 5420–5424. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Schouten, K.J.P.; Koper, M.T.M. Mechanism of the catalytic oxidation of glycerol on polycrystalline gold and platinum electrodes. ChemCatChem 2011, 3, 1176–1185. [Google Scholar] [CrossRef]
- Garcia, A.C.; Kolb, M.J.; Sanchez, C.V.Y.; Vos, J.; Birdja, Y.Y.; Kwon, Y.; Tremiliosi-Filho, G.; Koper, M.T.M. Strong impact of platinum surface structure on primary and secondary alcohol oxidation during electro-oxidation of glycerol. ACS Catal. 2016, 6, 4491–4500. [Google Scholar] [CrossRef]
- Kwon, Y.; Birdja, Y.; Spanos, I.; Rodriguez, P.; Koper, M.T.M. Highly selective electro-oxidation of glycerol to dihydroxyacetone on platinum in the presence of bismuth. ACS Catal. 2012, 2, 759–764. [Google Scholar] [CrossRef]
- Kwon, Y.; Hersbach, T.J.P.; Koper, M.T.M. Electro-oxidation of glycerol on platinum modified by adatoms: Activity and selectivity effects. Top. Catal. 2014, 57, 1272–1276. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xin, L.; Li, W.Z. Electrocatalytic oxidation of glycerol on Pt/C in anion-exchange membrane fuel cell: Cogeneration of electricity and valuable chemicals. Appl. Catal. B Environ. 2012, 119, 40–48. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xin, L.; Qi, J.; Chadderdon, D.J.; Li, W.Z. Supported Pt, Pd and Au nanoparticle anode catalysts for anion-exchange membrane fuel cells with glycerol and crude glycerol fuels. Appl. Catal. B Environ. 2013, 136, 29–39. [Google Scholar] [CrossRef]
- Shustorovich, E. Activation Barrier for Adsorbate Surface Diffusion, Heat of Chemisorption, and Adsorbate Registry: Theoretical Interrelations. J. Am. Chem. Soc. 1984, 106, 6479–6481. [Google Scholar] [CrossRef]
- Van Santen, R.A. On shustorovich bond-order conservation method as applied to chemisorption. Recl. Trav. Chim. Pays-Bas 1990, 109, 59–63. [Google Scholar] [CrossRef]
- Shustorovich, E.; Sellers, H. The UBI-QEP method: A practical theoretical approach to understanding chemistry on transition metal surfaces. Surf. Sci. Rep. 1998, 31, 1–119. [Google Scholar] [CrossRef]
- Kua, J.; Faglioni, F.; Goddard, W.A. Thermochemistry for hydrocarbon intermediates chemisorbed on metal surfaces: CHn-m(CH3)m with n = 1, 2, 3 and m ≤ n on Pt, Ir, Os, Pd, Ph, and Ru. J. Am. Chem. Soc. 2000, 122, 2309–2321. [Google Scholar] [CrossRef]
- Greeley, J. Theoretical Heterogeneous Catalysis: Scaling Relationships and Computational Catalyst Design. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Muelas, R.; Lopez, N. Collective descriptors for the adsorption of sugar alcohols on Pt and Pd(111). J. Phys. Chem. C 2014, 118, 17531–17537. [Google Scholar] [CrossRef]
- Salciccioli, M.; Chen, Y.; Vlachos, D.G. Density Functional Theory-Derived Group Additivity and Linear Scaling Methods for Prediction of Oxygenate Stability on Metal Catalysts: Adsorption of Open-Ring Alcohol and Polyol Dehydrogenation Intermediates on Pt-Based Metals. J. Phys. Chem. C 2010, 114, 20155–20166. [Google Scholar] [CrossRef]
- Salciccioli, M.; Edie, S.M.; Vlachos, D.G. Adsorption of Acid, Ester, and Ether Functional Groups on Pt: Fast Prediction of Thermochemical Properties of Adsorbed Oxygenates via DFT-Based Group Additivity Methods. J. Phys. Chem. C 2012, 116, 1873–1886. [Google Scholar] [CrossRef]
- Benson, S.W. Thermochemical Kinetics. In Methods for the Estimation of Thermochemical Data and Rate Parameters, 2nd ed.; John Wiley and Sons, Inc.: New York, NY, USA, 1976; pp. 1–320. [Google Scholar]
- Ferrin, P.; Simonetti, D.; Kandoi, S.; Kunkes, E.; Dumesic, J.A.; Nørskov, J.K.; Mavrikakis, M. Modeling ethanol decomposition on transition metals: A combined application of scaling and Brønsted-Evans-Polanyi relations. J. Am. Chem. Soc. 2009, 131, 5809–5815. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Johannesson, G.; Jonsson, H. Methods for Finding Saddle Points and Minimum Energy Paths. In Theoretical Methods in Condensed Phase Chemistry; Springer: Dordrecht, The Netherlands, 2000; Volume 5, pp. 269–302. [Google Scholar]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jonsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Jacob, T.; Goddard, W.A. Water formation on Pt and Pt-based alloys: A theoretical description of a catalytic reaction. ChemPhysChem 2006, 7, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Suthirakun, S.; Heyden, A. New implicit solvation scheme for solid surfaces. J. Phys. Chem. C 2012, 116, 22458–22462. [Google Scholar] [CrossRef]
- Callam, C.S.; Singer, S.J.; Lowary, T.L.; Hadad, C.M. Computational analysis of the potential energy surfaces of glycerol in the gas and aqueous phases: Effects of level of theory, basis set, and solvation on strongly intramolecularly hydrogen-bonded systems. J. Am. Chem. Soc. 2001, 123, 11743–11754. [Google Scholar] [CrossRef] [PubMed]
- Bodenschatz, C.J.; Sarupria, S.; Getman, R.B. Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. J. Phys. Chem. C 2015, 119, 13642–13651. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.N.; Sautet, P.; Michel, C. Solvation free energies for periodic surfaces: Comparison of implicit and explicit solvation models. Phys. Chem. Chem. Phys. 2016, 18, 31850–31861. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast parallel algorithms for short-range molecular-dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Yun, Y.S.; Lee, K.R.; Park, H.; Kim, T.Y.; Yun, D.; Han, J.W.; Yi, J. Rational Design of a Bifunctional Catalyst for the Oxydehydration of Glycerol: A Combined Theoretical and Experimental Study. ACS Catal. 2015, 5, 82–94. [Google Scholar] [CrossRef]
- Suleimanov, Y.V.; Green, W.H. Automated Discovery of Elementary Chemical Reaction Steps Using Freezing String and Berny Optimization Methods. J. Chem. Theory Comput. 2015, 11, 4248–4259. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Zimmerman, P.M. Reliable and Efficient Reaction Path and Transition State Finding for Surface Reactions with the Growing String Method. J. Comput. Chem. 2017, 38, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Vinu, R.; Broadbelt, L.J. Unraveling Reaction Pathways and Specifying Reaction Kinetics for Complex Systems. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Friedler, F.; Tarjan, K.; Huang, Y.W.; Fan, L.T. Graph-theoretic approach to process synthesis—Axioms and theorems. Chem. Eng. Sci. 1992, 47, 1973–1988. [Google Scholar] [CrossRef]
- Fan, L.T.; Lin, Y.C.; Shafie, S.; Hohn, K.L.; Bertok, B.; Friedler, F. Graph-theoretic and energetic exploration of catalytic pathways of the water-gas shift reaction. J. Chin. Inst. Chem. Eng. 2008, 39, 467–473. [Google Scholar] [CrossRef]
- Fan, L.T.; Lin, Y.C.; Shafie, S.; Bertok, B.; Friedler, F. Exhaustive identification of feasible pathways of the reaction catalyzed by a catalyst with multiactive sites via a highly effective graph-theoretic algorithm: Application to ethylene hydrogenation. Ind. Eng. Chem. Res. 2012, 51, 2548–2552. [Google Scholar] [CrossRef]
Figure 1.
Annual glycerol production (in million metric tons), and crude glycerol price (in US$/ton) between 2001 and 2011. Reproduced from Reference [5]. Copyright Year 2013, Elsevier.
Figure 1.
Annual glycerol production (in million metric tons), and crude glycerol price (in US$/ton) between 2001 and 2011. Reproduced from Reference [5]. Copyright Year 2013, Elsevier.
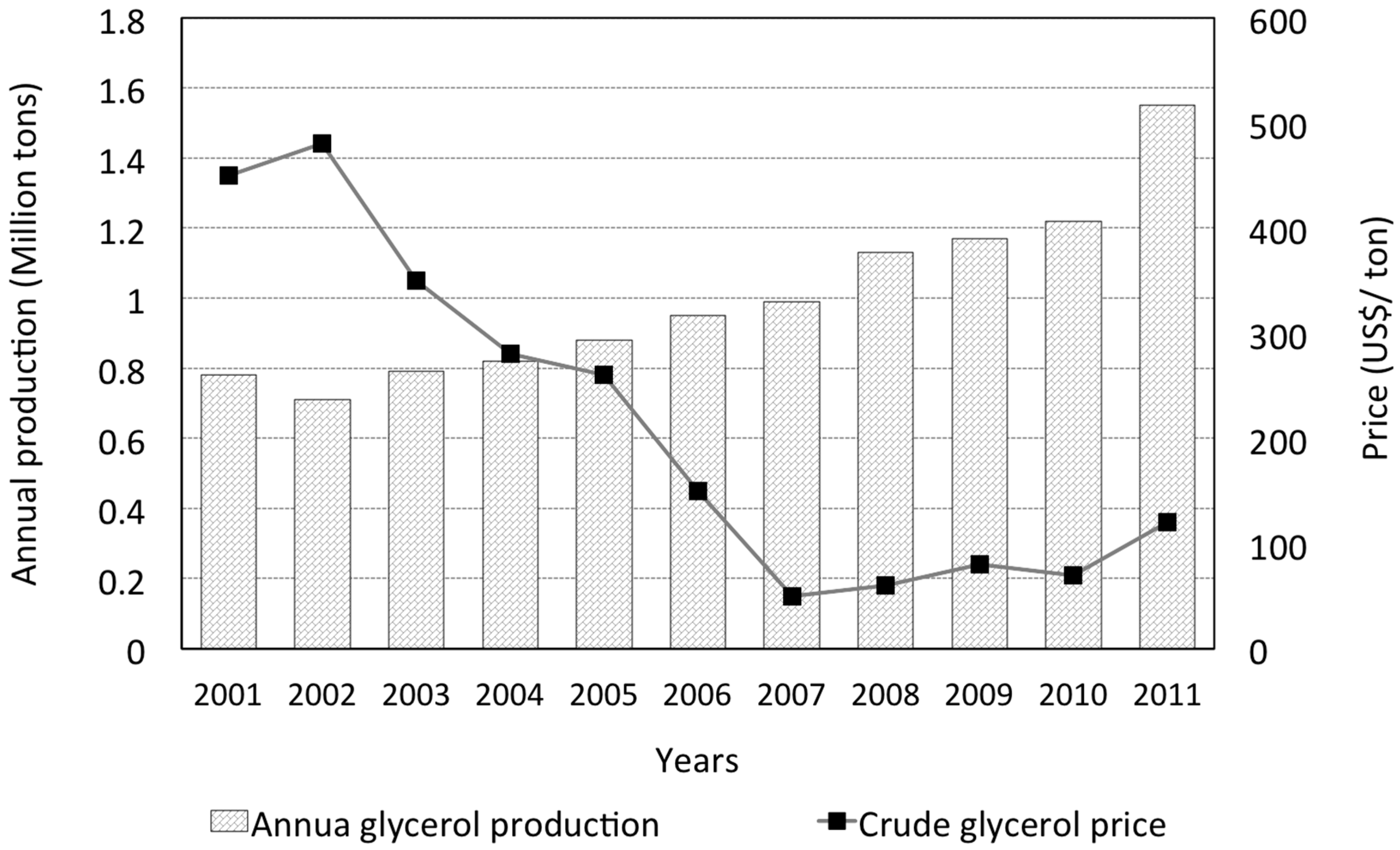
Figure 2.
Glycerol usage as a commodity chemical. Reproduced from Reference [13], Copyright Year 2007, Wiley.
Figure 2.
Glycerol usage as a commodity chemical. Reproduced from Reference [13], Copyright Year 2007, Wiley.
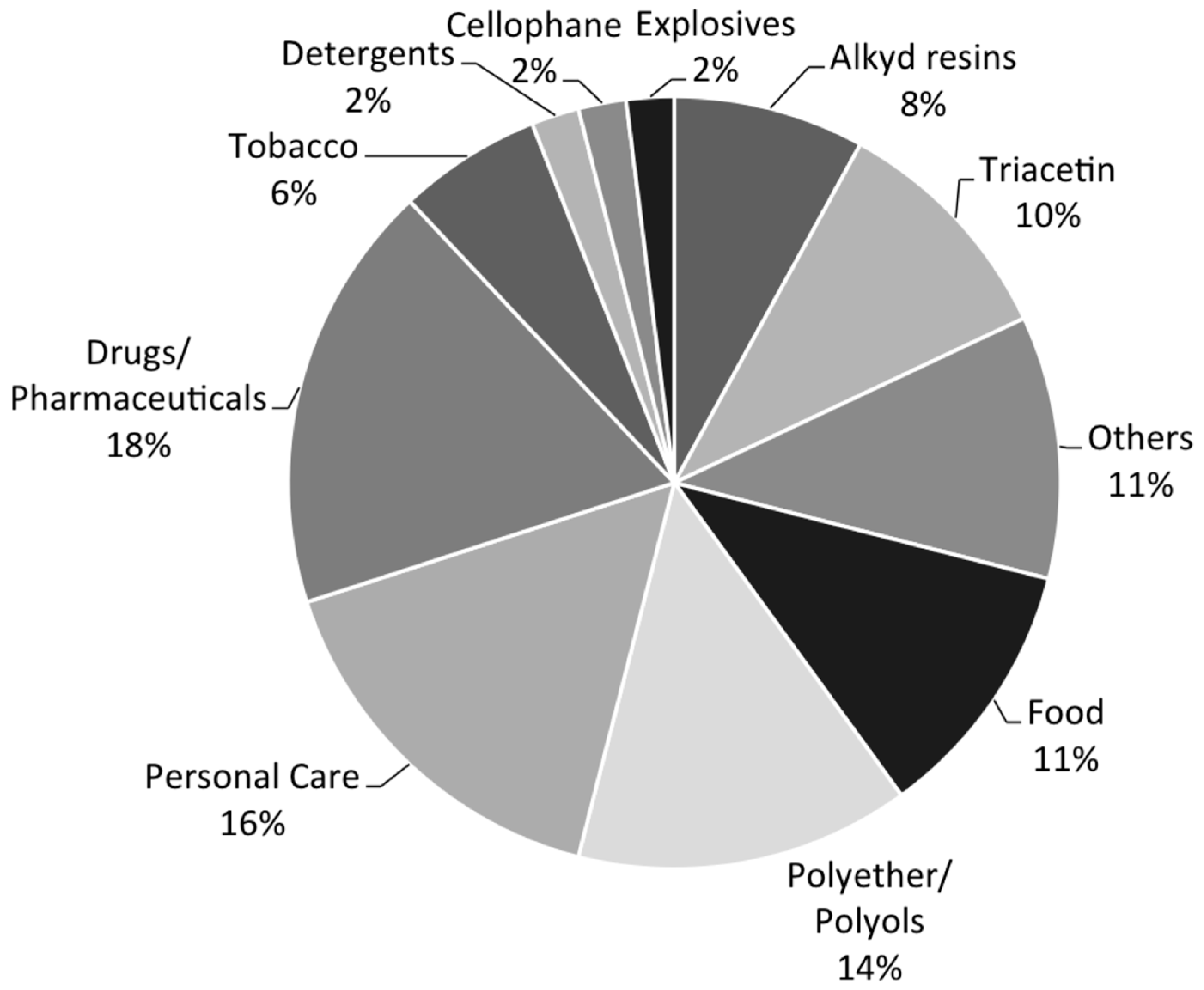
Figure 3.
A schematic roadmap for glycerol conversions when different types of catalysts (i.e., metal or alloy, acid, or metal-acid bifunctional catalytic materials in different color codes) are used. Main products from various pathways are underlined. C, O, and H atoms are depicted in black, red and white, based on Reference [24].
Figure 3.
A schematic roadmap for glycerol conversions when different types of catalysts (i.e., metal or alloy, acid, or metal-acid bifunctional catalytic materials in different color codes) are used. Main products from various pathways are underlined. C, O, and H atoms are depicted in black, red and white, based on Reference [24].
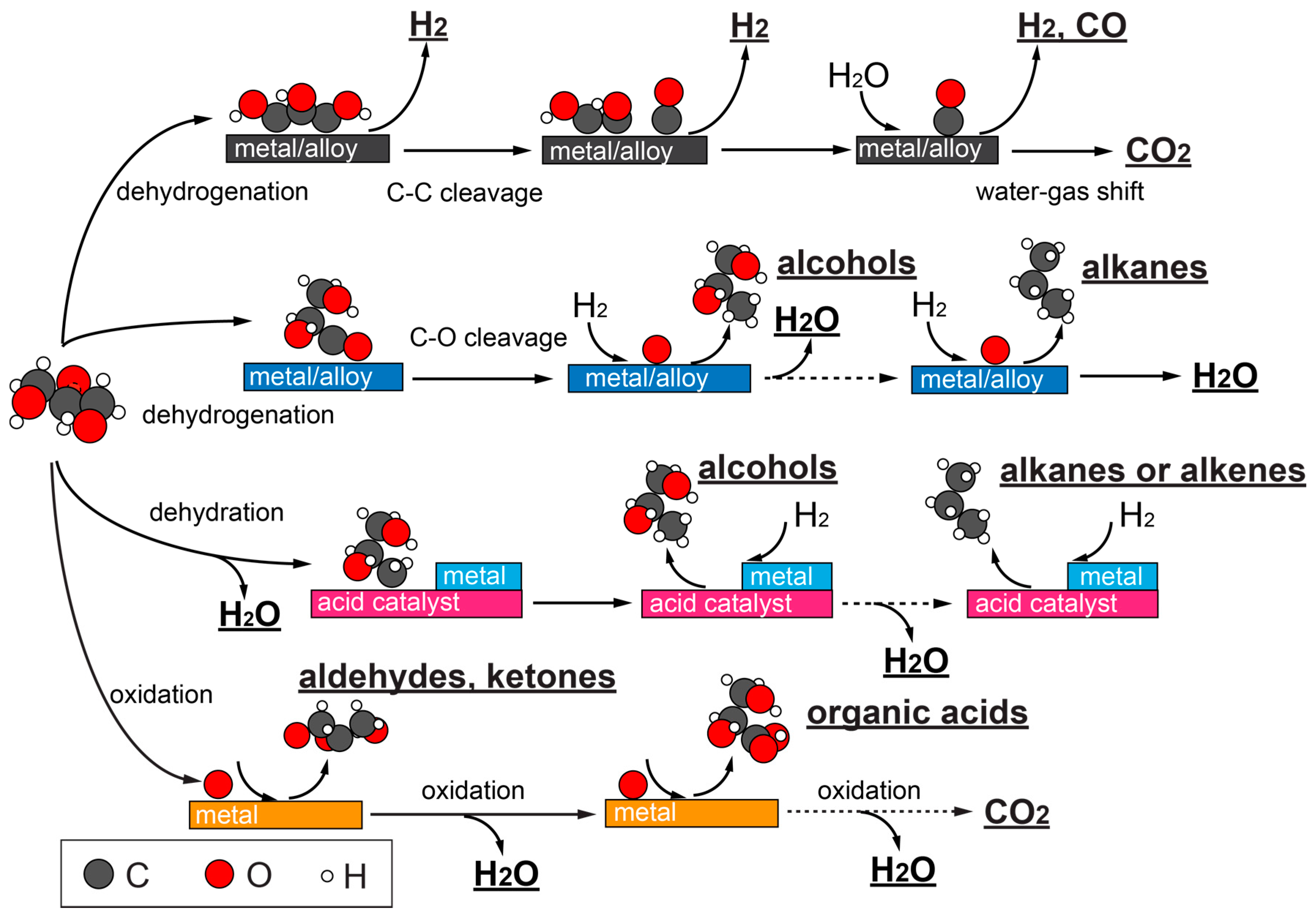
Figure 4.
Trends in ethylene glycol (EG) aqueous phase reforming (APR) on selected metals, turnover frequency (TOF) (CO2, in × 103 min−1), alkane selectivity (%), and H2 selectivity (%) are colored in green, blue, and yellow, respectively. Reproduced from Reference [64]. Copyright Year 2003, Elsevier.
Figure 4.
Trends in ethylene glycol (EG) aqueous phase reforming (APR) on selected metals, turnover frequency (TOF) (CO2, in × 103 min−1), alkane selectivity (%), and H2 selectivity (%) are colored in green, blue, and yellow, respectively. Reproduced from Reference [64]. Copyright Year 2003, Elsevier.
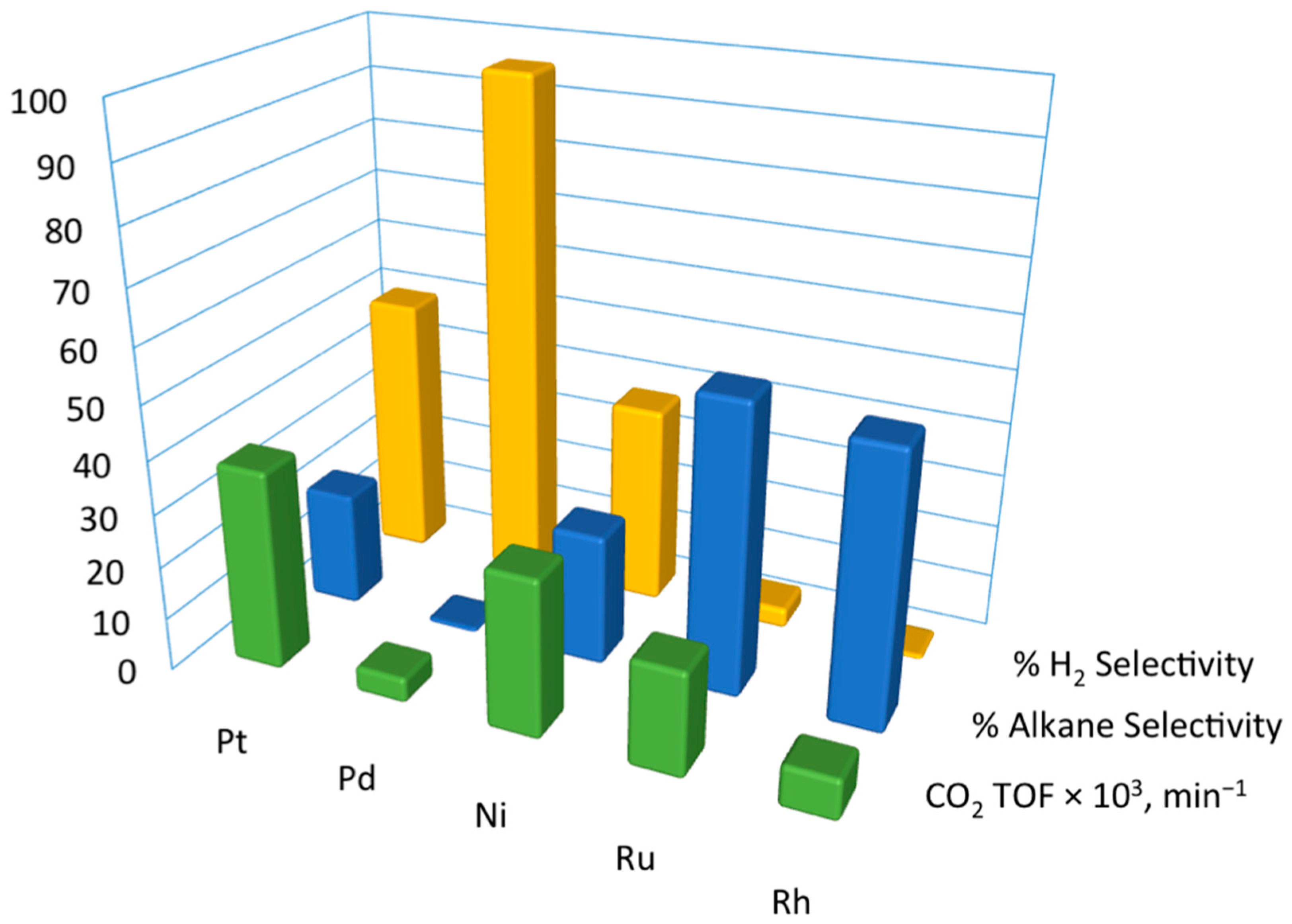
Figure 5.
Adsorption thermochemistry (estimated at 210 °C and 1 bar) of the most stable glycerol dehydrogenation intermediates (black squares), the most stable transition states for C–H/O–H, C–C, and C–O bond cleavage (represented by blue diamonds, red triangles, and purple circles, respectively) on Pt(111) are compiled in a unified free energy diagram. Reproduced from Reference [60]. Copyright Year 2011, American Chemical Society.
Figure 5.
Adsorption thermochemistry (estimated at 210 °C and 1 bar) of the most stable glycerol dehydrogenation intermediates (black squares), the most stable transition states for C–H/O–H, C–C, and C–O bond cleavage (represented by blue diamonds, red triangles, and purple circles, respectively) on Pt(111) are compiled in a unified free energy diagram. Reproduced from Reference [60]. Copyright Year 2011, American Chemical Society.

Figure 6.
Glycerol hydrogenolysis pathways on Ru and Pt-based catalysts under neutral or basic conditions. Reproduced from Reference [72]. Copyright Year 2007, Elsevier.
Figure 6.
Glycerol hydrogenolysis pathways on Ru and Pt-based catalysts under neutral or basic conditions. Reproduced from Reference [72]. Copyright Year 2007, Elsevier.
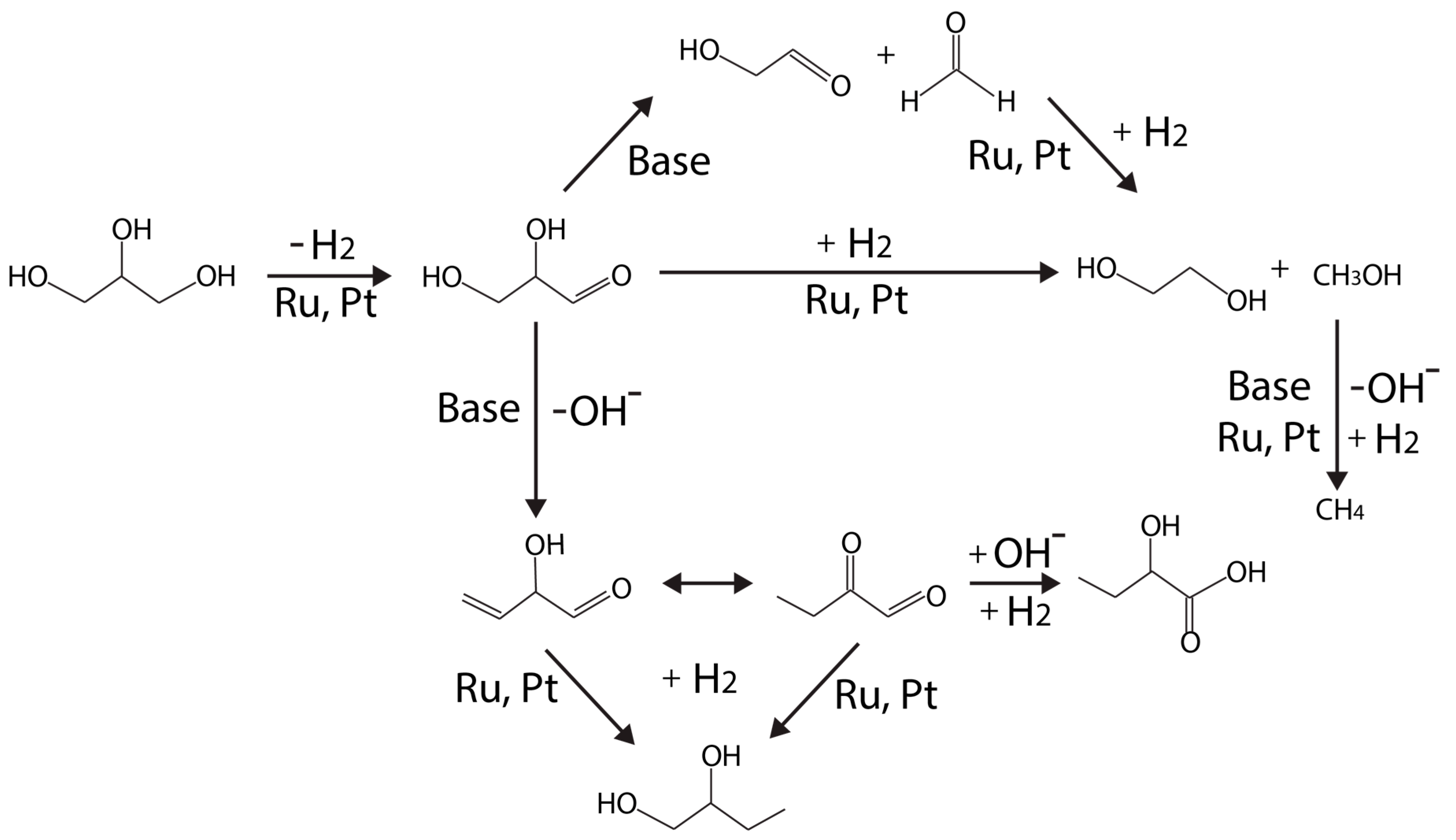
Figure 7.
Reaction network depicting glycerol conversions to 12-PDO and 13-PDO, with primary reaction intermediates. Reproduced from Reference [73]. Copyright Year 2011, Royal Society of Chemistry.
Figure 7.
Reaction network depicting glycerol conversions to 12-PDO and 13-PDO, with primary reaction intermediates. Reproduced from Reference [73]. Copyright Year 2011, Royal Society of Chemistry.
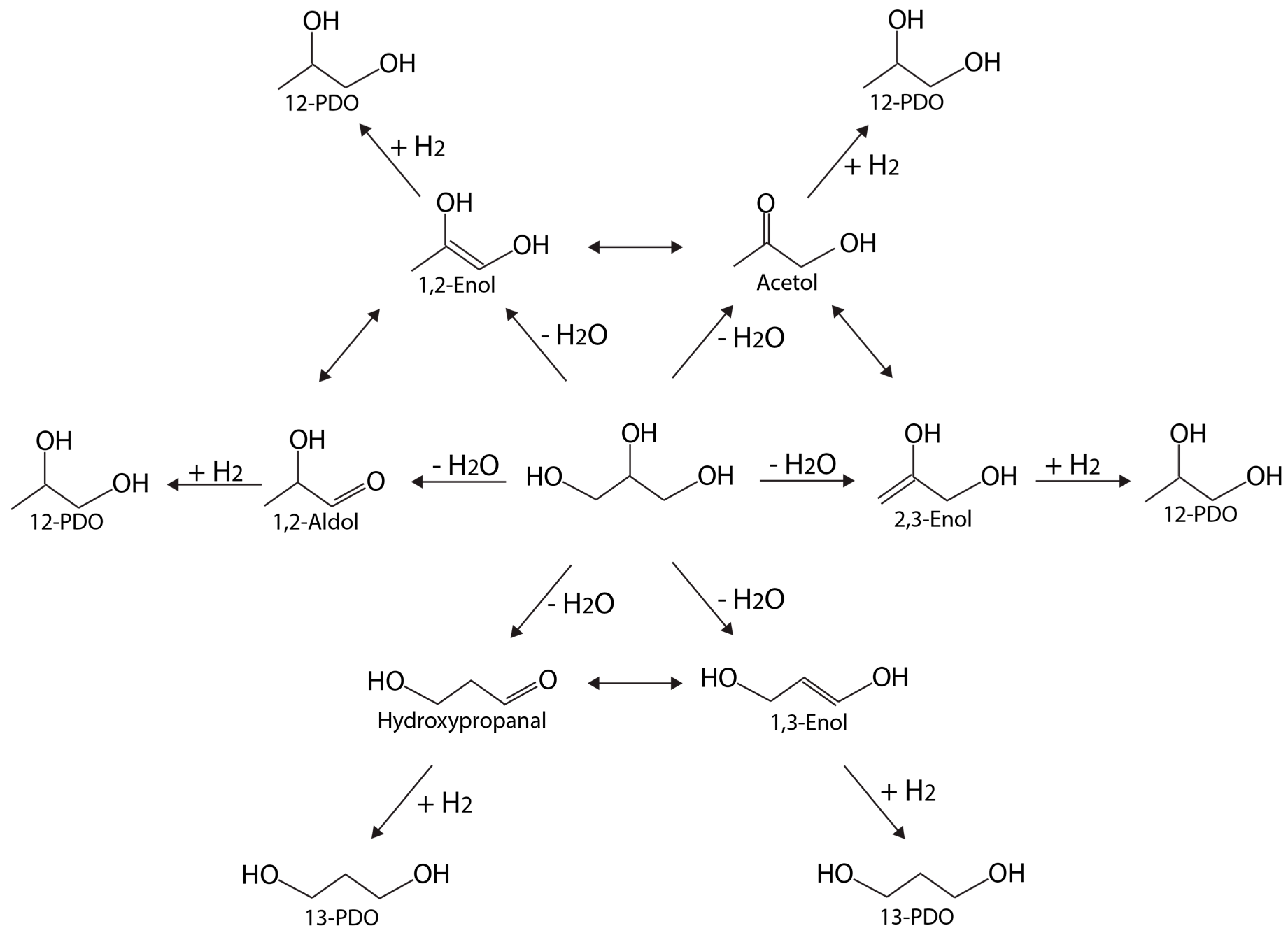
Figure 8.
Glycerol oxidation reaction network and main products. Reproduced from Reference [92]. Copyright Year 2005, Elsevier.
Figure 8.
Glycerol oxidation reaction network and main products. Reproduced from Reference [92]. Copyright Year 2005, Elsevier.
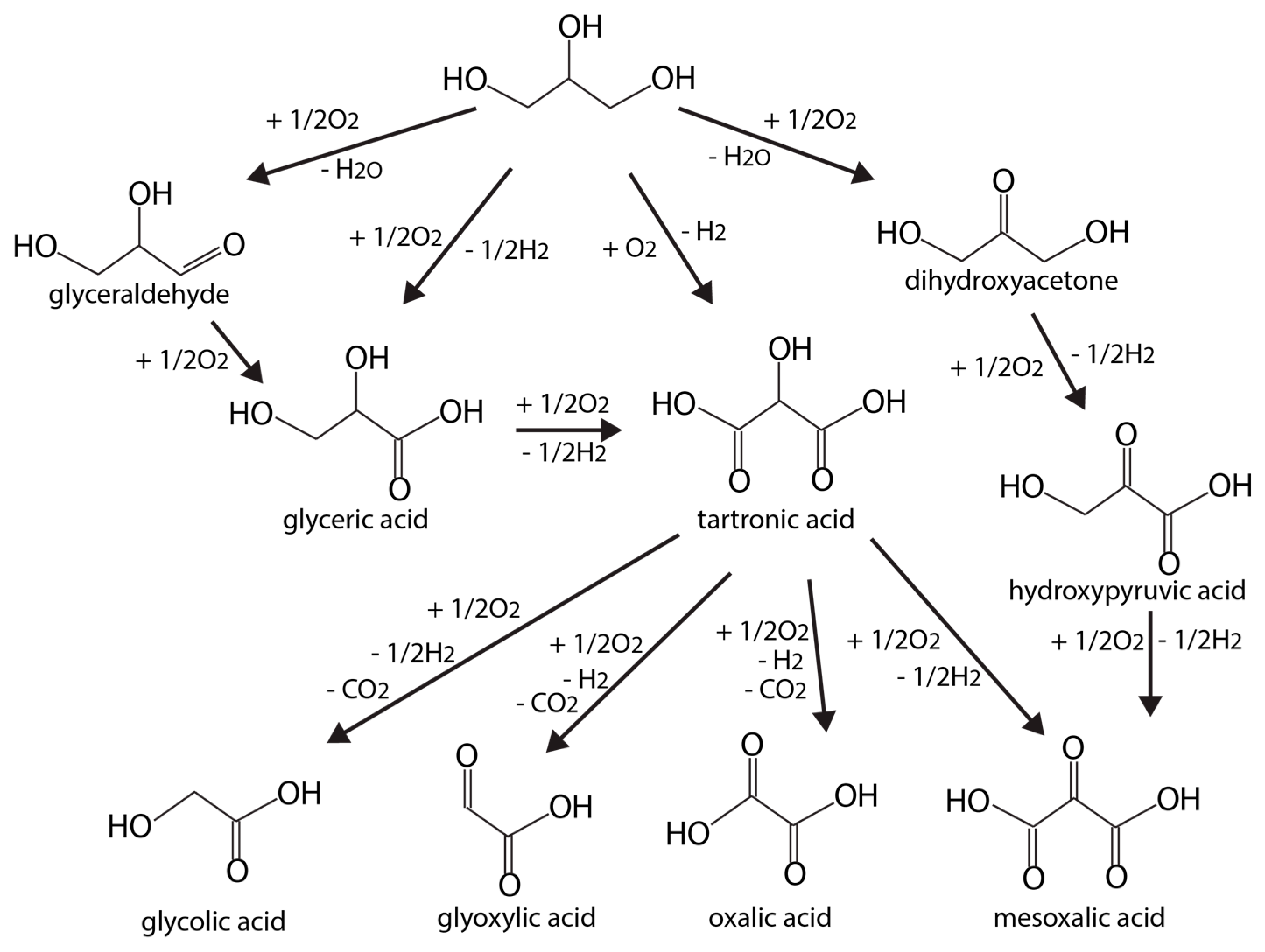
Figure 9.
Glycerol oxidation pathways on Pt(111) and Pt(100) in acidic solution. Reproduced from Reference [99], Copyright Year 2016, American Chemical Society. Purple and blue are used to indicate pathways on Pt(111) and Pt(100), respectively.
Figure 9.
Glycerol oxidation pathways on Pt(111) and Pt(100) in acidic solution. Reproduced from Reference [99], Copyright Year 2016, American Chemical Society. Purple and blue are used to indicate pathways on Pt(111) and Pt(100), respectively.
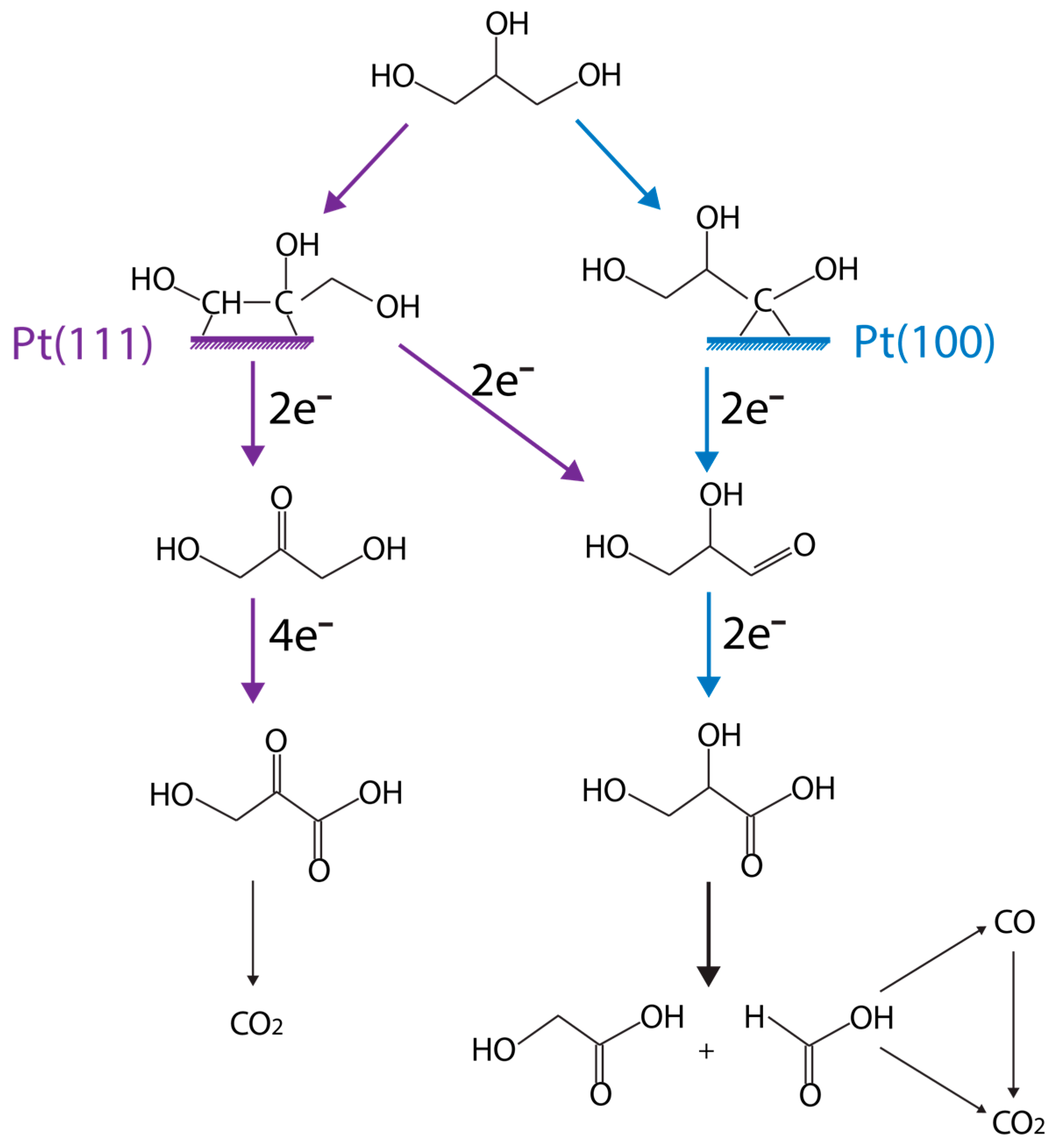
Figure 10.
Free energy diagrams showing most stable intermediates at each level of glycerol dehydrogenation on close-packed Cu, Pt, Pd, Rh, and Ni surfaces (at 483 K and 1 bar). Reproduced from Reference [68]. Copyright year 2013, Royal Society of Chemistry.
Figure 10.
Free energy diagrams showing most stable intermediates at each level of glycerol dehydrogenation on close-packed Cu, Pt, Pd, Rh, and Ni surfaces (at 483 K and 1 bar). Reproduced from Reference [68]. Copyright year 2013, Royal Society of Chemistry.
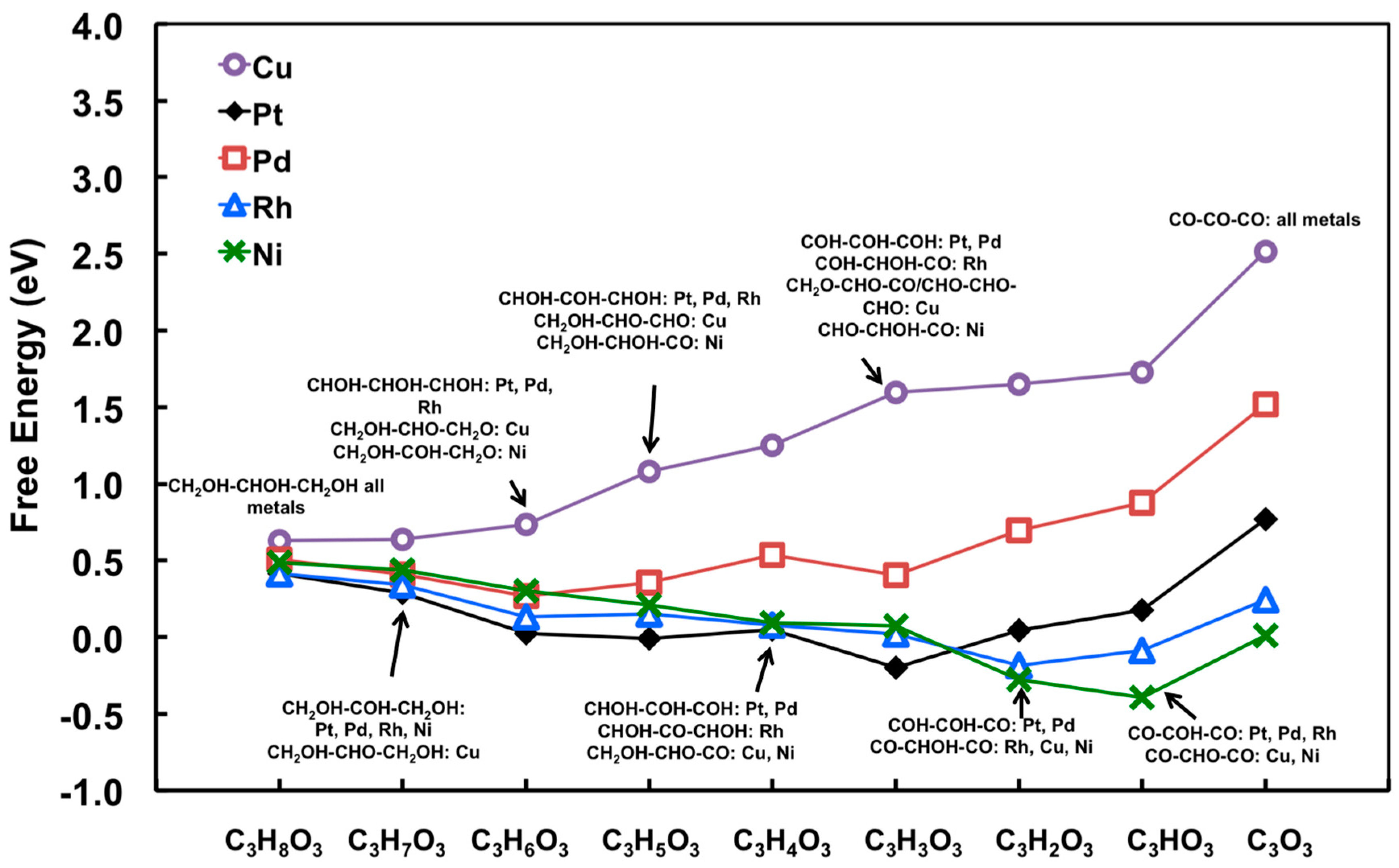

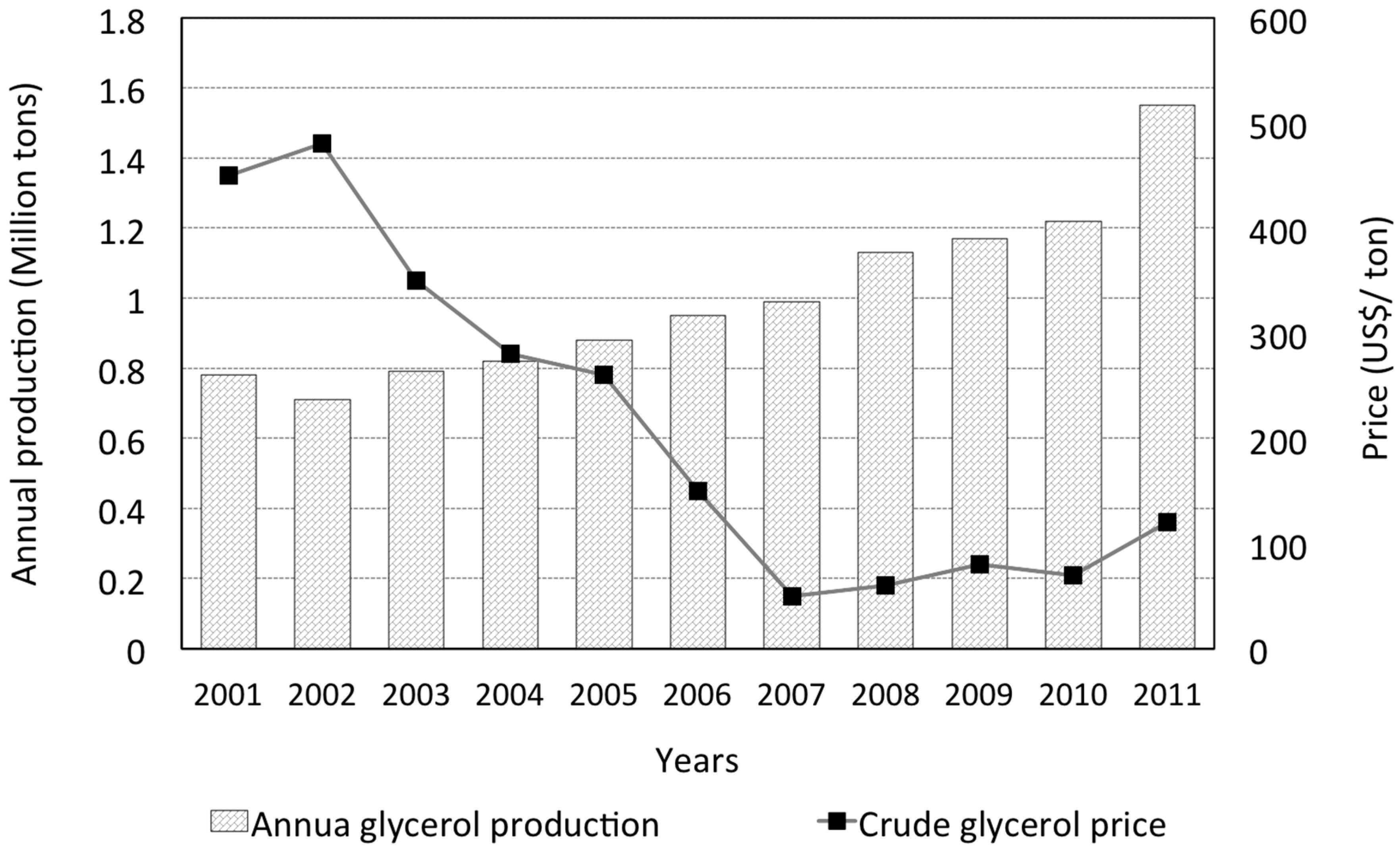{kind=link}
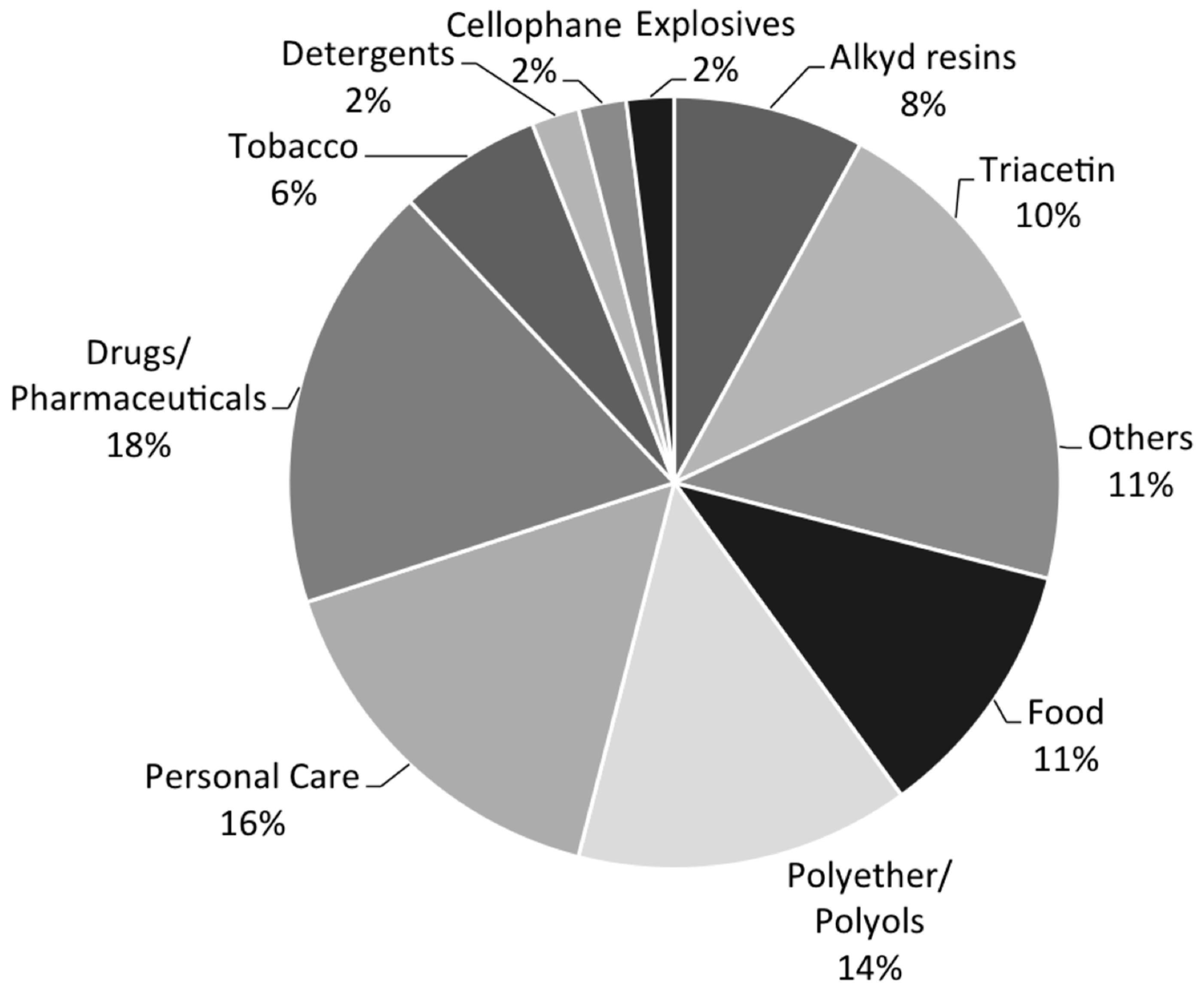{kind=link}
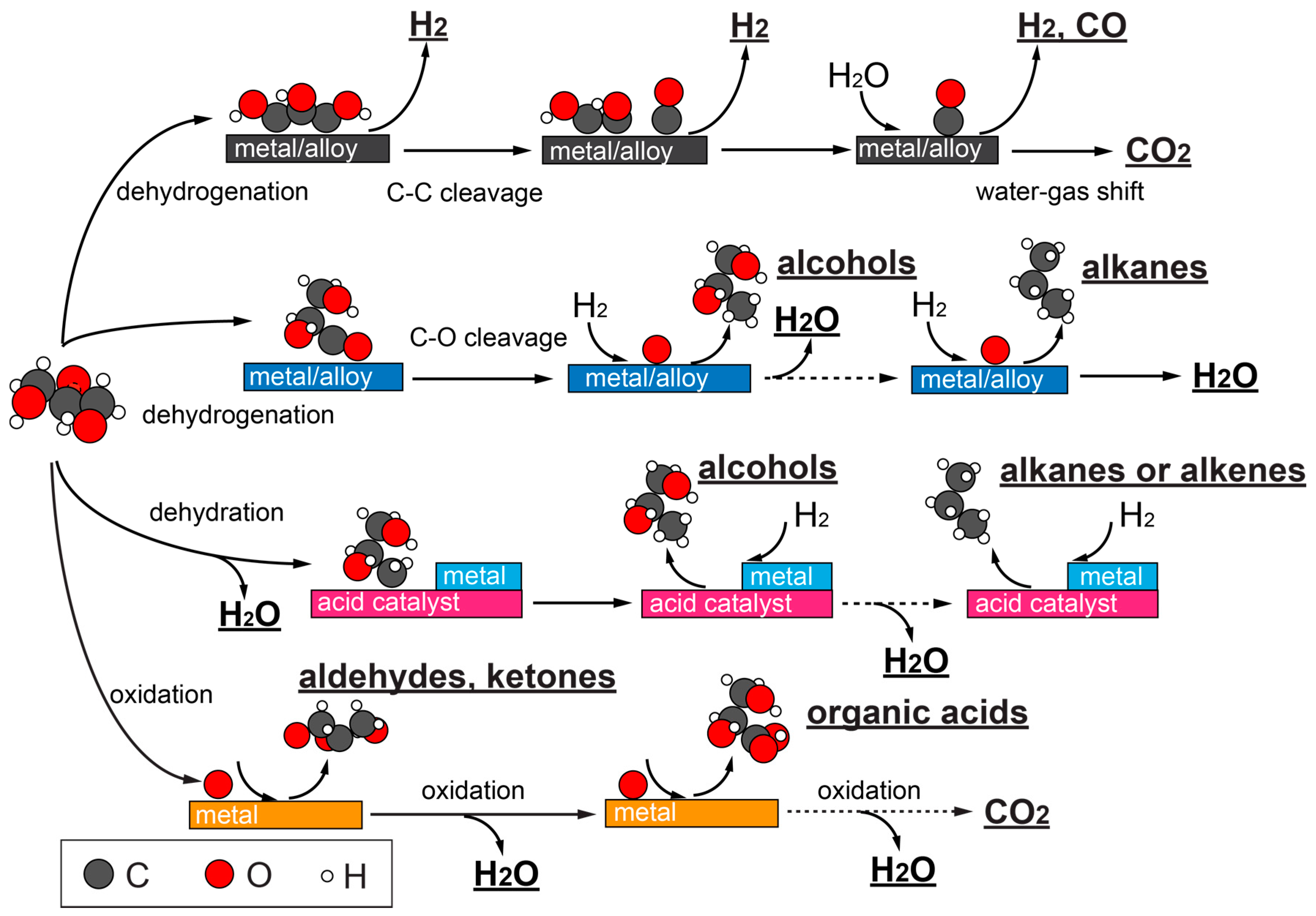{kind=link}
{kind=link}
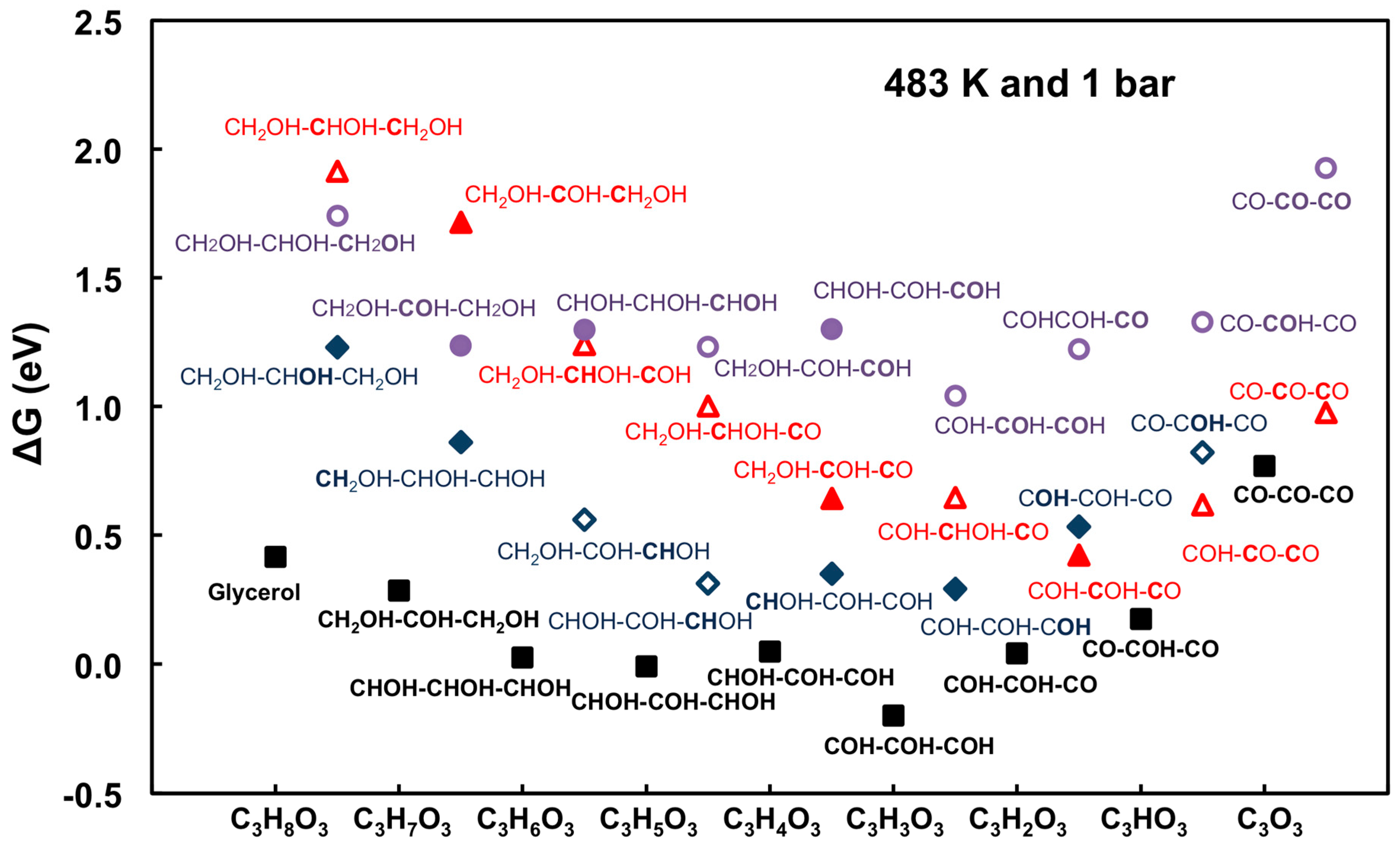{kind=link}
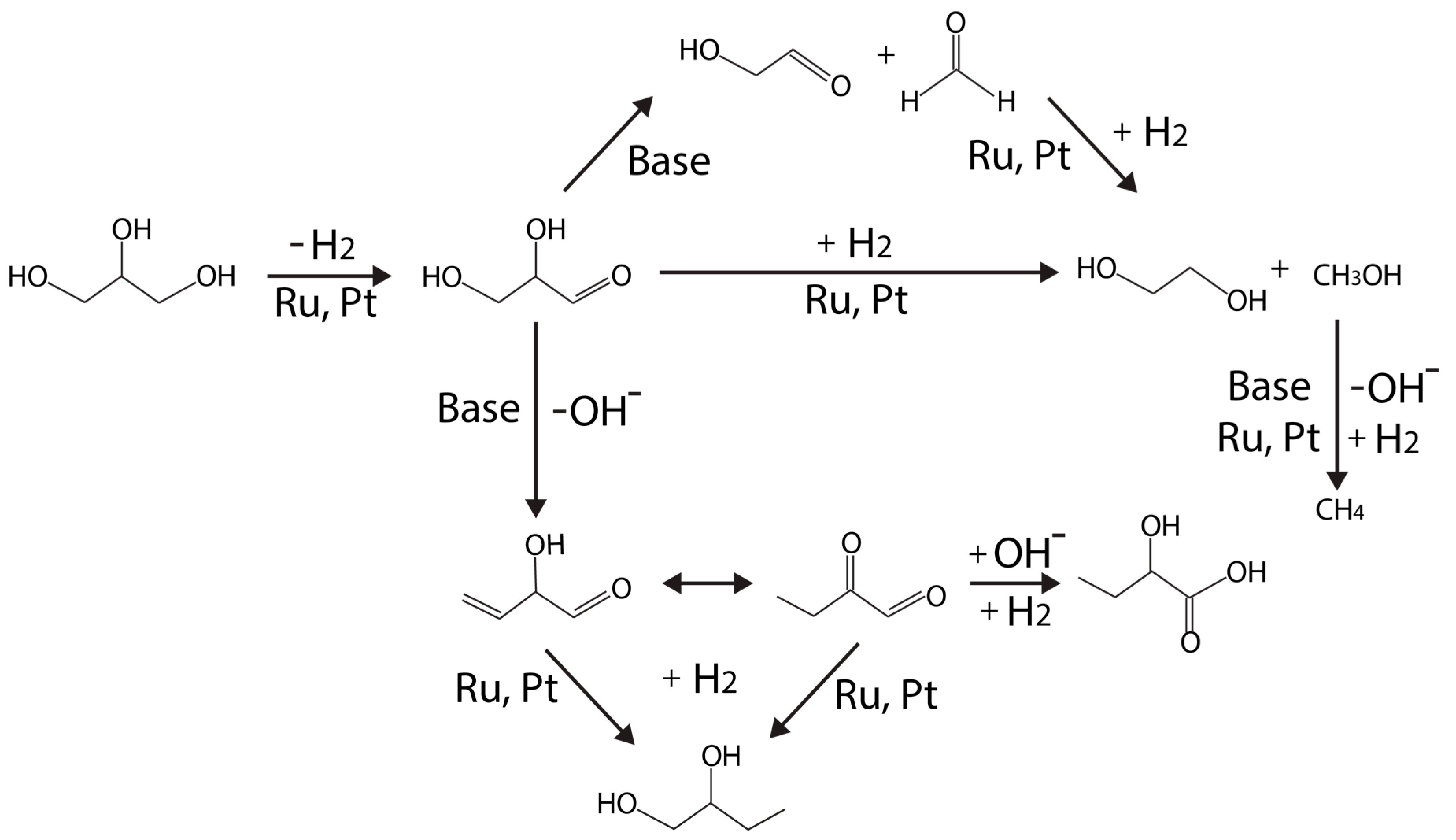{kind=link}
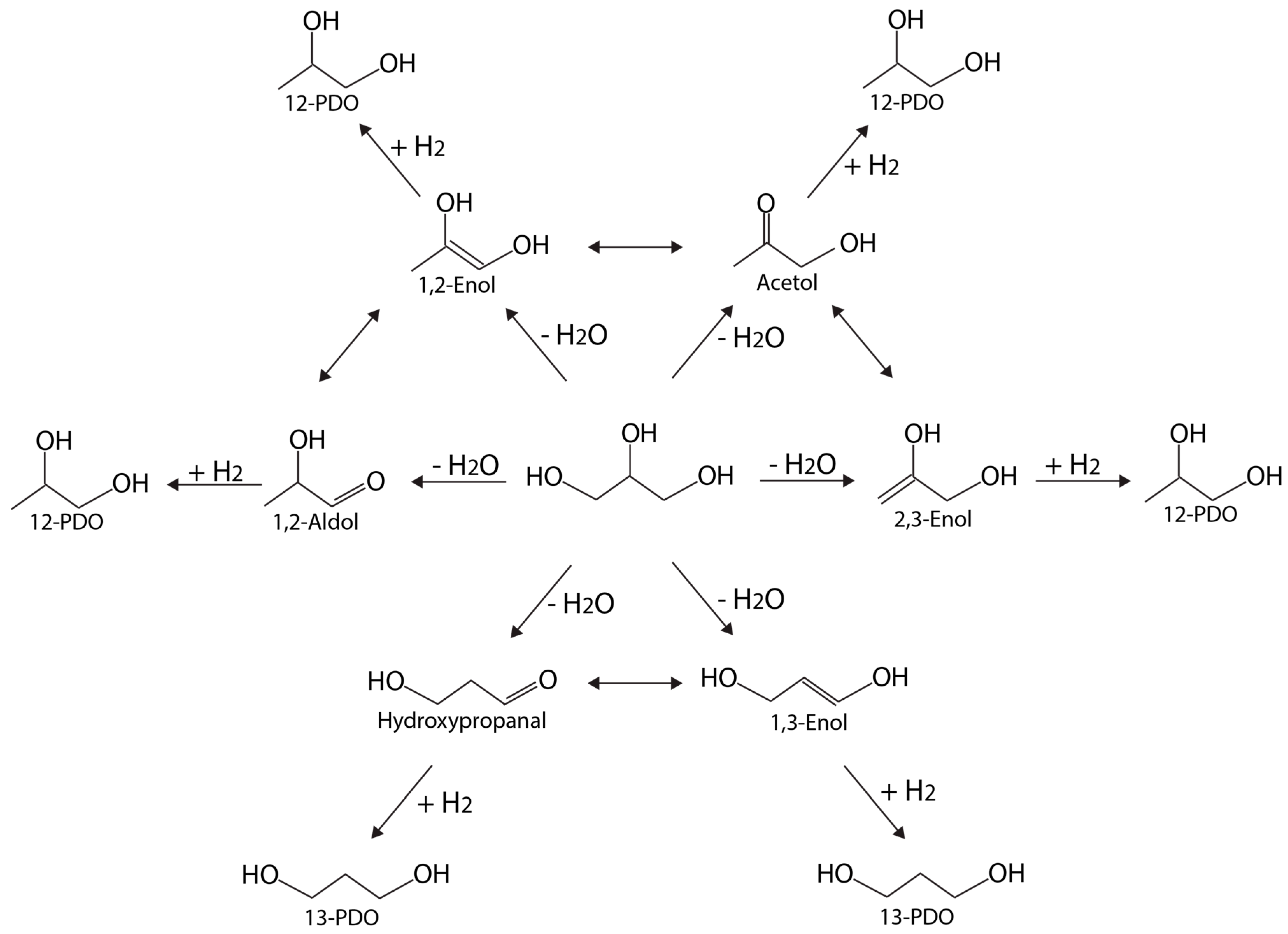{kind=link}
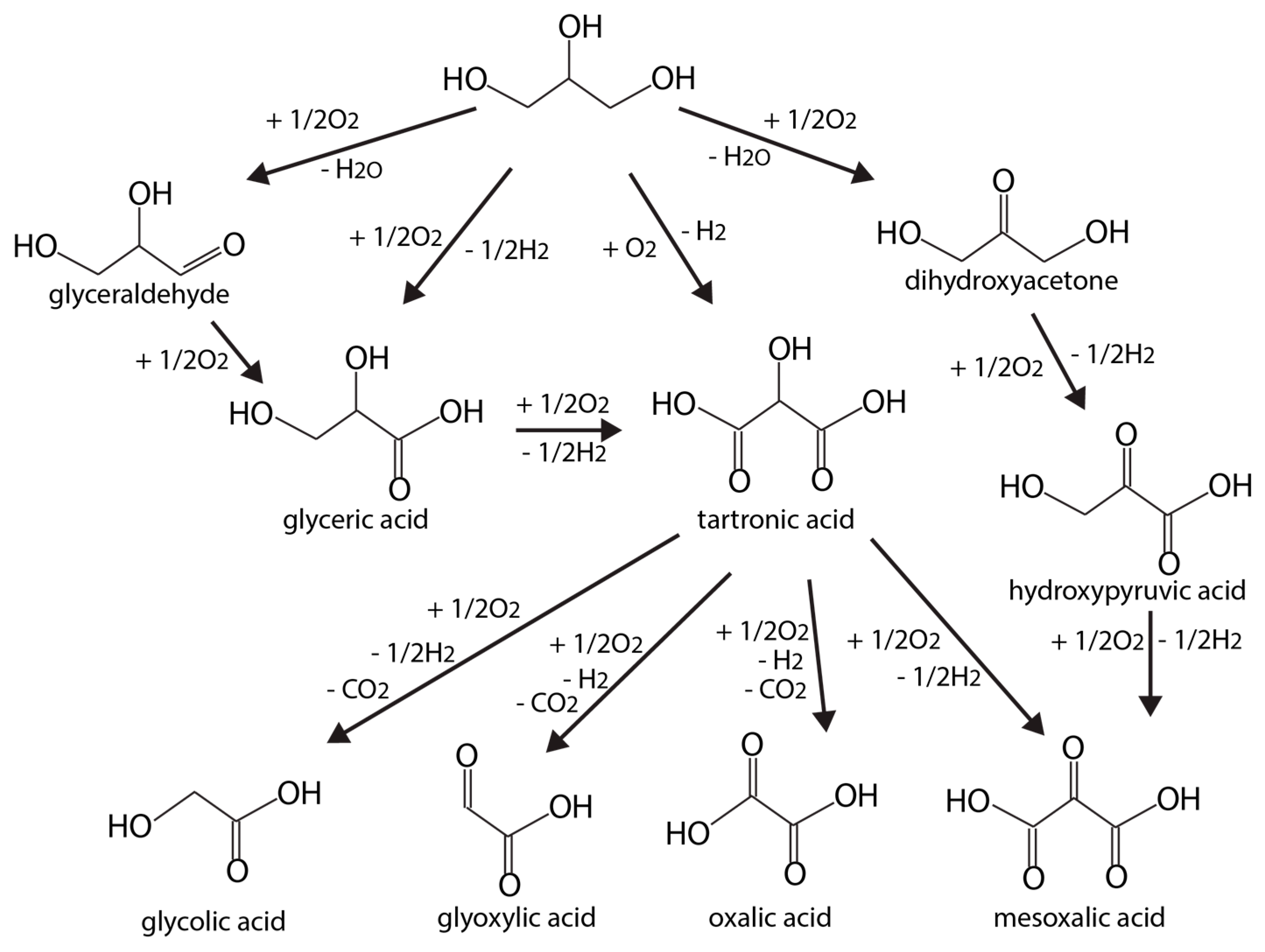{kind=link}
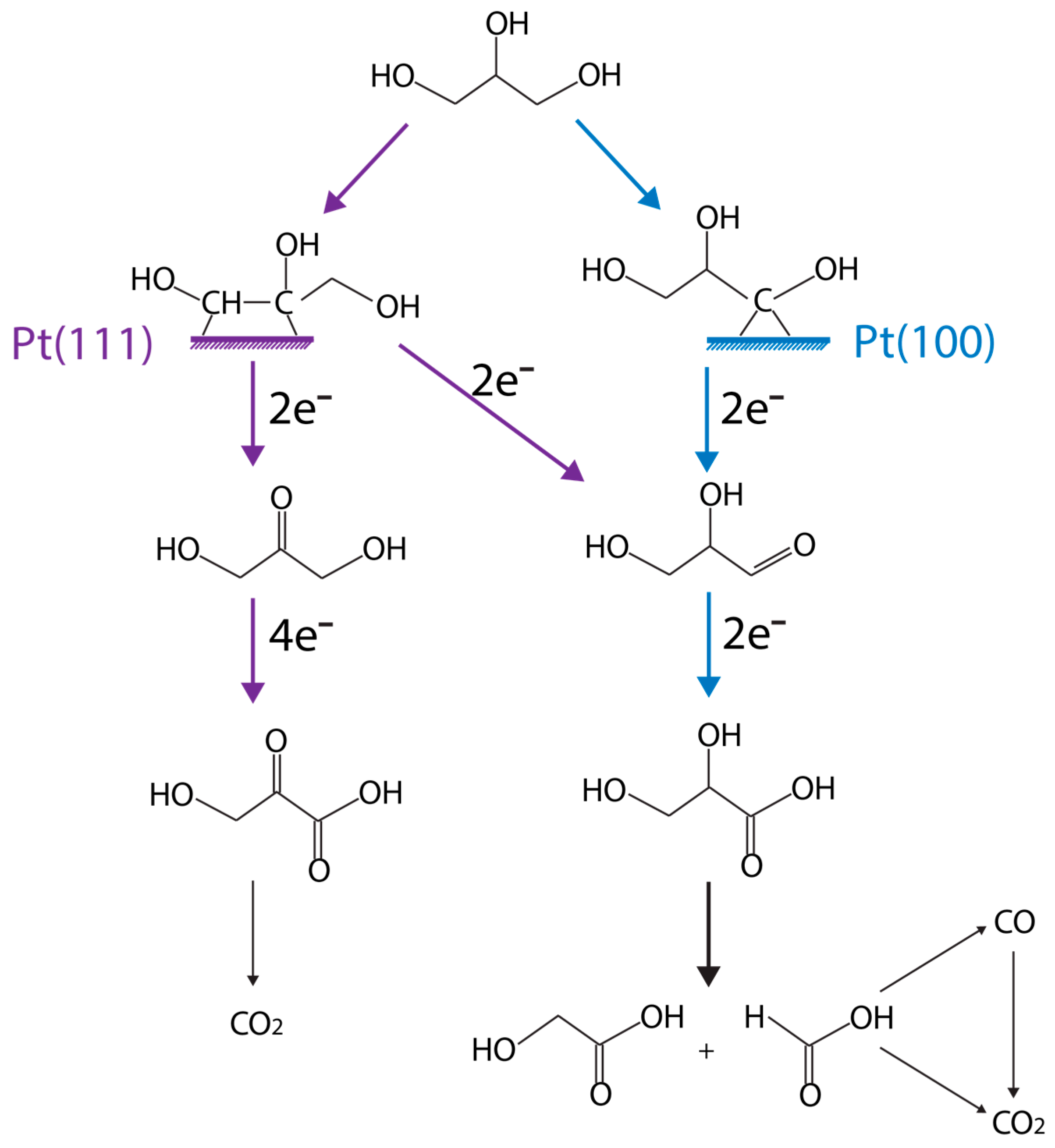{kind=link}
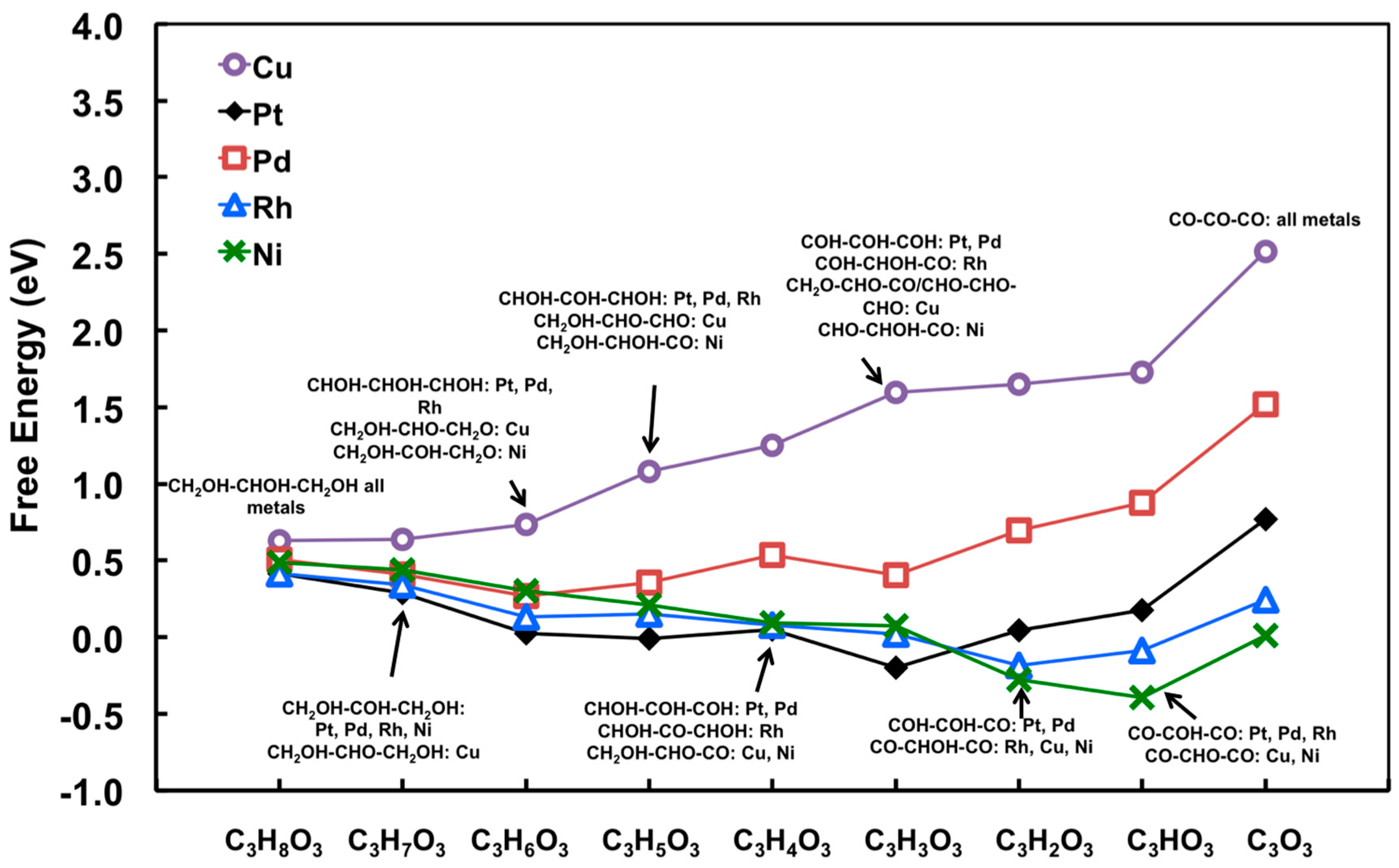{kind=link}
Table 1.
Catalysts and performance for hydrogen production from glycerol aqueous phase (APR) or steam reforming.
Table 1.
Catalysts and performance for hydrogen production from glycerol aqueous phase (APR) or steam reforming.
| Catalyst | Pressure, Temp. | Process | Gly. Conversion, % | Yield (H2) | Selectivity (H2), % | Reference |
|---|---|---|---|---|---|---|
| 3 wt.% Pt/C | 29 bar, 225 °C | APR | 5 | 68 | 57 | [40] |
| 3 wt.% Pt/γ-Al2O3 | 29–56 bar, 225–265 °C | APR | 65, 57 | 75, 51 | [24] | |
| 0.9 wt.% Pt/γ-Al2O3 | 25 bar, 220 °C | APR | 65 1 | 45 | 70 | [41] |
| 3 wt.% Pt/γ-Al2O3 | 29 bar, 225 °C | APR | ~50 | 20 2 | 20 3 | [42] |
| Pt/MgO, Al2O3, CeO2, TiO2, SiO2 | 27.6 bar, 225 °C | APR | 20–48 | 56–70 | 14–28 | [43] |
| Ni/MxOy-Al2O3 (M = Zr, Ce, La, Mg) | 30 bar, 225 °C | APR | 15–37 | 32–48 | [44] | |
| 3 wt.% Pt-10 wt.% Ni/γ-Al2O3 | 30 bar, 230 °C | APR | 75 | 54 | [45] | |
| 3.2 wt.% Pt-8 wt.% Ni/γ-Al2O3 | 50 bar, 250 °C | APR | 74 | 51 | [46] | |
| Pt-Ni/CNT | 30 bar, 230 °C | APR | 70–90 | 18–62 | [47] | |
| 3 wt.% Pt-3 wt.% Re/C | 29 bar, 225 °C, in KOH | APR | 89 | 67 | 26 | [40] |
| 3%Pt-(1–4.5%)Re/C | 29 bar, 225 °C | APR | 100 | ~70 | [48] | |
| (2.5–4 wt.%)Pt-Re (1:5 ratio)/C | 25 bar, 225 °C | APR | <45% | [49] | ||
| (2.5–3 wt.%)Pt-Re (1:2 ratio)/ZrO2, CeO2, TiO2 | 25 bar, 225 °C | APR | <35% | [50] | ||
| Pt-Mo (Pt:Mo ratio = 1:1) | 31 bar, 210–240 °C | APR | 0.5–26 | ~50 | [51,52] | |
| Ni-Sn | 25.8–51.4 bar, 225–265 °C | APR | >90 | 68–78 | [29,32] | |
| Ni-Cu | 38–52 atm, 250–270 °C | APR | 60 | >80 | [53] | |
| Pt/γ-Al2O3 | 1 bar, 350 °C | Steam | 8 | 61 | [39] | |
| Pt/SiO2, ZrO2, Ce4Zr1α | 1 bar, 350 °C | Steam | 100, 16, 78 | 69, 62, 73 | [39] | |
| 5.8 wt.% Ni/Al2O3 | 1 bar, 600–700 °C | Steam | >99 | ~100 | [54] | |
| Ni, Ir, Co./CeO2 | 1 bar, 550 °C 4 | Steam | 100 | 91, 93, 94 | [38] | |
| Ni/MxOy-Al2O3 (M = Zr, Ce, La, Mg) | 1 bar, 600 °C | Steam | 100 | 60–70 | [44] | |
| Ni/MgO | 1 bar, 550–650 °C | Steam | 100 | 43–57 | 61–66 | [36,37] |
| Ni/TiO2 | 1 bar, 550–650 °C | Steam | 96–98 | 31–47 | 44–62 | [36,37] |
| Ni/CeO2 | 1 bar, 550–650 °C | Steam | 72–98 | 31–44 | 54–67 | [36,37] |
| Pt-Ni/La2O3 | 1 bar, 600 °C | Steam | 100 | 53 | [55] | |
| 3 wt.% Ru/Y2O3 | 1 bar, 550–650 °C | Steam | 100 | ~90 | [56] | |
| Ru/Al2O3 | 900 °C | Steam | 58 | 42 | [35] | |
| Ni-Cu-Al | 1 bar, 500–600 °C | Steam | 70 | ~75 | [57] | |
| Ni-Co./Al2O3 | 1 bar, 500–550 °C | Steam | 5–6 | 65 | [58] |
1 The conversion, as in Reference [41], was reported in terms of carbon converted to gas products. 2 H2 yield is based on ca. 50% conversion, as defined in Reference [42]. 3 H2 selectivity was obtained using a 30 wt.% glycerol concentration, as defined in Reference [42]. 4 Temperature corresponding to highest H2 selectivity is listed [38].
Table 2.
Metal-based catalysts for glycerol hydrogenolysis.
| Catalyst | H2 Pressure, Temp., and Solution | Glycerol Conversion, % | Yield, % | Main Products and Selectivity, % | Reference |
|---|---|---|---|---|---|
| Ru/C | 40 bar, 200 °C, neutral | 20 | 32 (PDO) 68 (EG) | [72] | |
| Rh/C | 30 bar, 180 °C + NaOH (1 M) | 22 | 9 (12-PDO) 5 (LA) | [74] | |
| 5 wt.% Rh/C | 80 bar, 180 °C | 0.3 | 58.6 (12-PDO) 3.4 (13-PDO) | [75] | |
| Rh/C, with H2WO4 | 80 bar, 180 °C | 10 | 5.2 (12-PDO) 2.6 (13-PDO) | 52 (12-PDO) 26 (13-PDO) | [76] |
| Rh/Al2O3 | 80 bar, 180 °C | 21 | 12.4 (12-PDO) 3.1 (13-PDO) | 45 (12-PDO) 12 (13-PDO) | [76] |
| Rh/Nafion | 80 bar, 180 °C | 8 | 4.3 (12-PDO) 1.5 (13-PDO) | 54 (12-PDO) 19 (13-PDO) | [76] |
| Ni/Al2O3 | 1 bar, 190 °C | 92 | 43.6 (12-PDO) | [77] | |
| Ni/SBA-15 | 30 bar, 450 °C | 50 | 24 (12-PDO) | 30 (12-PDO) | [78] |
| Pd/C, with H2WO4 | 80 bar, 180 °C | 3 | 2 | 100 (12-PDO) | [76] |
| Pd/C | 80 bar, 180 °C | 0.7 | 93.1 (12-PDO) 1.4 (13-PDO) | [75] | |
| Cu-ZnO (Cu:Zn ratio 50:50) | 20 bar, 120–220 °C | 37 | >93 (12-PDO), 3.4 (Acetol), 3 (EG) | [79] | |
| Cu/ZnO/Al2O3 | 6.4 bar, 190 °C | 96 | 92.2 (12-PDO) | [77] | |
| CuO/ZnO, with H2WO4 | 80 bar, 180 °C | 21 | 17 | 100 (12-PDO) | [76] |
| Pt/amorphous silico alumina | 45 bar, 220 °C | 20 | 6.3 (12-PDO) | 35.3 (12-PDO), 1.2 (13-PDO) | [80] |
| Pt/C | 40 bar, 200 °C, neutral | 13 | 79 (PDO) 17 (EG) | [72] | |
| Pt/C | 40 bar, 200 °C, with 0.8 M NaOH | 20 | 30 (PDO) 62 (LA) 2 (EG) | [72] | |
| Pt/C | 80–90 bar, 160 °C | 20 | 43 (12-PDO) | [81] | |
| Pt/C | 80 bar, 180 °C | 1.1 | 87.6 (12-PDO) 1.9 (13-PDO) | [75] | |
| Ru/C | 40 bar, 200 °C, with 0.8 M NaOH | 20 | 37 (PDO) 47 (LA) 12 (EG) | [72] | |
| Ru/C | 80 bar, 160 °C | 29.7 1 | 50.9 (12-PDO) 0.8 (13-PDO) 22.9 (1-PO) 3.2 (2-PO) | [82] | |
| Ru/C | 80 bar, 180°C | 6.3 | 17.9 (12-PDO) 0.5 (13-PDO) | [75] | |
| Pt-Ru/C | 40 bar, 200 °C, with 0.8 M NaOH | 22 | 37 (PDO) 41 (LA) 15 (EG) | [83] | |
| Au-Ru/C | 40 bar, 200 °C, with 0.8 M NaOH | 21 | 25 (PDO) 60 (LA) 10 (EG) | [83] | |
| Pt-Re/C sintered | 80–90 bar, 200 °C | 20 | 33 (12-PDO) 34 (13-PDO) | [81] | |
| Cu-Ru/ZrO2 (Cu:Ru ratio = 1:10) | 80 bar, 180 °C | 100 | 78.5 (12-PDO) | 84 (12-PDO) 6.4 (1-PO) 9.3 (EG) | [84] |
| Ru-Re/ZrO2 | 80 bar, 160 °C | 57 | 47.2 (12-PDO) 5.5 (13-PDO) 27.2 (1-PO) 8.1 (2-PO) | [85] | |
| Ir-ReOx/SiO2 | 80 bar, 160 °C | 60 2 | 5 (12-PDO) 54 (13-PDO) 31 (1-PO) 4 (2-PO) | [86] |
1 Conversion is defined in Reference [85]. 2 Conversion is achieved after 20 h of experiment.
Table 3.
Metal-acid bifunctional catalysts for glycerol hydrogenolysis.
| H2 Pressure, and Temp. | Glycerol Conversion, % | Main Product Selectivity, % | Reference | |
|---|---|---|---|---|
| Ru/C + Amberlyst | 80 bar, 140 °C | 41 | 43.1 (12-PDO) 1.0 (13-PDO) 18.2 (1-PO) 2.9 (2-PO) | [75] |
| Ru/C + H2SO4 | 80 bar, 140 °C | 3.2 | 47.4 (12-PDO) 5.4 (13-PDO) 19.6 (1-PO) 1.6 (2-PO) | [75] |
| Pt/amorphous silico alumina | 45 bar, 220 °C | 20 | 35.3 (12-PDO) 1.2 (13-PDO) | [80] |
| Pt/C + Amberlyst | 80 bar, 140 °C | 0.5 | 4.4 (12-PDO) 4.4 (13-PDO) 52.7 (1-PO) 14.7 (2-PO) | [75] |
| Pd/C + Amberlyst | 80 bar, 140 °C | 0.3 | 6.1 (12-PDO) 51.5 (1-PO) 15.2 (2-PO) | [75] |
| Rh/C + Amberlyst | 80 bar, 140 °C | 6.4 | 19.5 (12-PDO) 7.2 (13-PDO) 53.2 (1-PO) 14.7 (2-PO) | [75] |
| Ru/C +Amberlyst | 80 bar, 120 °C | 13 | 55.4 (12-PDO) 4.9 (13-PDO) 14.1 (1-PO) 0.9 (2-PO) | [87] |
| Ru5/C(I) | 80 bar, 120 °C | 21 | 76.7 (12-PDO) 1.5 (13-PDO) 2.5 (1-PO) 0.5 (2-PO) | [88] |
| h-Ru/C + A70 | 80 bar, 180 °C | 49 | 70.2 (12-PDO) 1.3 (13-PDO) 7.1 (1-PO) 1.0 (2-PO) | [89] |
| Pt-HSiW/ZrO2 | 50 bar, 180 °C | 24 | 16.5 (12-PDO) 48.1 (13-PDO) 21.8 (1-PO) 4.5 (2-PO) | [90] |
| Pt-HPW/ZrO2 | 50 bar, 180 °C | 25.5 | 10.9 (12-PDO) 32.9 (13-PDO) 37.9 (1-PO) 5.2 (2-PO) | [90] |
| Pt-HPMo/ZrO2 | 50 bar, 180 °C | 27 | 39.2 (12-PDO) 7.8 (13-PDO) 30.4 (1-PO) 3.2 (2-PO) | [90] |
Table 4.
Catalysts for liquid phase glycerol oxidation reactions.
| Catalyst | Experimental Conditions | Selectivity, % | Reference |
|---|---|---|---|
| 5 wt.% Pt-1 wt.% Bi/C | With air in acidic media at 323 K and normal pressure | 20 (DHA) | [93] |
| Pd/C | pH = 11 in air | 70 (Glyceric acid) 1 8 (DHA)) | [94] |
| Pt/C | pH = 7 in air | 55 (Glyceric acid) 1 12 (DHA) | [94] |
| Pt-Bi/C | pH = 11 in air | 50 (DHA) 1 | [94] |
| Au/AC | (10 bar), 60 °C in NaOH solution | 75 (Glyceric acid) 2 15 (Glycolic acid) 2 | [92] |
| Pt(111), Pt(100) | 0.5 M HClO4, CV performed at 293 K in the range of 0.0–1.0 V | 80 3 (Glyceraldehyde) ~20 3 (DHA) | [95] |
| Pt(111)/Bi, Pt(100)/Bi | 0.5 M HClO4, CV performed at 293 K in the range of 0.0–1.0 V | 80 4 (Glyceraldehyde) ~20 4 (DHA) 90 4 (Glyceraldehyde) ~10 4 (DHA) | [95] |
| Pt/MCM-41 | (30 psi), 75 °C | 15.25 (DHA) 5 | [96] |
| Pt-Bi/AC | (30 psi), 75 °C | 77.12 (DHA) 5 | [96] |
| Pt-Bi/ZSM-5 | (30 psi), 75 °C | 41.07 (DHA) 5 | [96] |
| Pt-Bi/MCM-41 | (30 psi), 75 °C | 65.26 (DHA) 5 | [96] |
| Pt/Bi-MCM-41 | (30 psi), 75 °C | 33.73 (DHA) 5 | [96] |
1 Selectivity on Pt/C and Pd/C is reported for 90% glycerol conversion, and 70% conversion on Pt-Bi/C. 2 Selectivity is based on Au_1/BP catalyst 3.7 nm mean diameter, and 30% conversion. 3 Results are taken from Pt(111) at 0.6 V [95]. 4 Results are taken from Pt(111)/Bi [95], and Pt(100)/Bi with an applied potential of 0.6 V, respectively [95]. 5 Highest DHA selectivity reported during 5 h of experiments [96].
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, B.; Gao, F. Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review. Catalysts 2018, 8, 44. https://doi.org/10.3390/catal8020044
AMA Style
Liu B, Gao F. Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review. Catalysts. 2018; 8(2):44. https://doi.org/10.3390/catal8020044
Chicago/Turabian StyleLiu, Bin, and Feng Gao. 2018. "Navigating Glycerol Conversion Roadmap and Heterogeneous Catalyst Selection Aided by Density Functional Theory: A Review" Catalysts 8, no. 2: 44. https://doi.org/10.3390/catal8020044
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.