Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow


Department of Chemistry, Wrocław University of Environmental and Life Sciences, Norwida 25, 50-375 Wrocław, Poland
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(12), 624; https://doi.org/10.3390/catal8120624
Submission received: 31 October 2018
/
Revised: 23 November 2018
/
Accepted: 25 November 2018
/
Published: 5 December 2018
Abstract
:The production of chiral sulphoxides is an important part of the chemical industry since they have been used not only as pharmaceuticals and pesticides, but also as catalysts or functional materials. The main purpose of this review is to present biotechnological methods for the oxidation of sulfides. The work consists of two parts. In the first part, examples of biosyntransformation of prochiral sulfides using whole cells of bacteria and fungi are discussed. They have more historical significance due to the low predictability of positive results in relation to the workload. In the second part, the main enzymes responsible for sulfoxidation have been characterized such as chloroperoxidase, dioxygenases, cytochrome flavin-dependent monooxygenases, and P450 monooxygenases. Particular emphasis has been placed on the huge variety of cytochrome P450 monooxygenases, and flavin-dependent monooxygenases, which allows for pure sulfoxides enantiomers effectively to be obtained. In the summary, further directions of research on the optimization of enzymatic sulfoxidation are indicated.
1. Introduction
Sulfur is an element that is about one percent of the dry mass of the human body because many sulfur-organic compounds are involved in metabolism. They are not only amino acids (cysteine, methionine) and their derivatives, but also glutathione and sulfolipids. Important enzyme cofactors also contain sulfur such as biotin, thioredoxin, lipoic acid, and coenzyme A [1]. Strong poisons can be found among the sulfur-containing compounds. For example, the consumption of Amanita mushrooms can cause fatal human poisoning and the toxin responsible for this effect is amanitin, which is an R-sulfoxide [2].
The organo-sulfur compounds are also an important group of secondary plant metabolites. They were isolated, among others, from onions, radishes, and cress. The characteristic smell and healing properties of plants of the genus Allium are attributed to sulfur-containing compounds, including chiral sulfoxides such as a compound characteristic of garlic: alliin (3-(2-propenylsulfinyl)-L-alanine) [3].
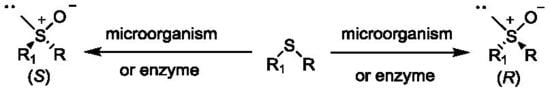
Since many compounds containing sulfur in their structure perform important biological functions, an attempt was made to develop methods for the synthesis of sulfur-based compounds, among which, the largest group are sulfoxides. It is noteworthy that the sulfur atom is a stereogenic center if it is linked to two different alkyl or aryl substituents, as well as to an oxygen atom and a lone electron pair. However, the bond between sulfur and oxygen is indirect between the coordination bond and the polarized double bond [4]. It does not form a typical π bond like between carbon and oxygen. It is possible to obtain stable enatiomers of sulfoxides because the lone electron pair is not inverted. The value of energy needed to change the configuration on sulfur is in the range of 35–43 kcal mol−1 for a typical sulfoxide [1,5]. The first examples of optically active sulfoxides were described in 1926, and the first general method for the synthesis of enantiomeric sulfoxides was reported in 1962 [6].
Since then, the chemistry of chiral sulfoxides has been very popular among researchers because these compounds have found many applications [7,8,9,10]. They are used as chiral ligands in enantioselective catalysis. They have also found wide applications as chiral auxiliaries and intermediates in asymmetric synthesis. High asymmetric induction was observed when using chiral sulfinyl. In addition, several drugs containing chirotopic sulfur in sulfoxide are used in the pharmaceutical industry, such as esomeprazole or modafinil [11].
Due to their importance, a significant effort has been made to develop efficient methods for their preparation. There are three main ways to obtain optically active sulfoxides:
- nucleophilic substitution of a chiral precursor;
- asymmetric sulphoxidation of prochiral sulfides;
- kinetic separation of racemic sulfoxides.
In this work, we will focus on discussing the biotechnological methods of oxidation of sulfides because biocatalysis is undoubtedly a method of gentle and environmentally friendly synthesis of sulfoxides [12].
2. Biotransformations in Cultures of Microorganisms
The use of whole cells of bacteria, fungi, and yeast in the asymmetric oxidation of sulfides is an excellent alternative to reactions catalyzed using pure enzymes. Such biotransformations are much cheaper and more convenient than enzymatic reactions. In addition, the costly co-factors, such as nicotinamide adenine dinucleotde/nicotinamide adenine dinucleotide phosphate (NADH/NADPH), are avoided. The costs associated with the purification of enzymes also disappear. It is also extremely important that some microorganisms can simulate the metabolism of mammalian cytochrome P450 monooxygenases (CYPs) [13,14,15,16,17].
2.1. Fungus and Yeast
The reactions of asymmetric sulfoxidation with fungi and yeasts are carried out successfully in the second half of the last century [18]. In 1982, Holland and Carter published a paper in which they presented the results of the biotransformation of aryl-substituted phenyl and benzyl-methyl sulfides using the Mortierella isabellina strain. These compounds have been oxidized to sulfoxides during enzymatic oxidation with oxygen from the atmosphere. In the studies, the authors focused mainly on determining the rate and mechanism of enzymatic oxidation [19]. In subsequent studies, the authors confirmed that benzylic hydroxylation and sulfoxidation reactions can be catalyzed using the same enzyme in M. isabellina. This enzyme has the characteristics of cytochrome P450 monooxygenase [20].
Further research by the team led by Holland showed that the phenyl-alkylated sulfoxides of the (S) configuration are the main biotransformation products of the respective sulfides catalyzed using the strain Helminthosporium sp. NRRL 4671 [21]. Subsequent studies conducted under the guidance of Holland showed that similar sulfides were also oxidized using the strain M. isabellina ATCC 4261 3 (Scheme 1). However, the formation of R-isomers was preferred here [22,23]. Further research has suggested that both strains of Helminthosporium and M. isabellina use other enzymes for the oxidation of sulfides [24].
Also, the white-rot Basidiomycetes catalyzed oxidation of aromatic pro-chiral sulfides into sulfoxides with good enantioselectivity and yield. The reactions were carried out using whole cells of Trametes rigida, Trametes versicolor, Trametes villosa, Pycnoporus sanguineus, Trichaptum byssogenum, and Irpex lacteus. These microorganisms also produced a small amount of sulfones. In the oxidation reactions of all the alkyl aryl sulfoxides, formation of (S)-enantiomers was preferred (Scheme 2). All strains of Basidiomycetes gave (S)-(phenylpropyl)sulfoxide with high enantiomeric excess (ee ≥ 99%) from the (phenylpropyl)sulfide [25].
In 2007, Pinedo-Rivilla and coworkers [26] presented the results of biotransformation of four alkyl aryl and alkyl sulfides catalyzed by Eutypa lata, Botrytis cinerea, and Trichoderma viride. Reactions were carried out both in resting and shaken cultures. It was observed that the substrates containing the aromatic ring were mainly oxidized. The alkyl sulfides were transformed with low yields, mainly to sulfones or treated as a carbon source. The best result for the oxidation of thioanisole was obtained during the transformation in a resting culture of T. viride. (R)-methylphenyl sulfoxide was obtained with 70% ee but with low yield (10%). With greater efficiency, this sulfoxide was obtained in a culture of B. cinerea (50% ee, 32% yield). In the reactions, the formation of the corresponding sulfone, which formed during the further oxidation of sulfoxides, was observed. R-sulfoxide is obtained in both reactions catalyzed using E. lata and T. viride. Oxidation in a T. viride shaking culture gives the best enantiomeric excess in good yield (ee > 95%, 60%). The optical purity of this sulfoxide is extremely high, suggesting that this substrate is well suited to the enzymatic system in which the oxidation takes place. This result indicates the potential use of T. viride in the production of this chiral sulphoxide in preparative scale.
Aspergillus strains are also capable of enantioselective oxidation of sulfides (Scheme 3). Two sulfides were selected for the tests: cyclohexyl methyl and thioanisole. All tested strains showed the ability to oxidize both alkyl sulfides and thioanisole.
The formation of R-enantiomers was preferred and full chemoselectivity was confirmed because no sulfones were detected. During the research, it was noticed that the addition of isopropyl alcohol affects both chemo- and enantioselectivity of the process. Among the tested strains of Aspergillus japonicus ICFC 744/11 biotransformed the alkyl sulfide with the best yield and stereoselectivity (100% reaction, ee > 99% (R)) [27].
Research on the oxidation of sulfides allowed to think that the fungal enzyme system can be used to simultaneously transform two functional groups. Therefore, research has been carried out on the use of Helminthosporium sp. NRRL 4671 and M. isabellina ATCC 42613 for bioconversion of substrates that contain both carbonyl and sulfide functional groups (Scheme 4).
These reactions allowed to obtain pure isomers containing two new chiral centers [28]. As in previous studies on sulfide oxidation, Helminthosporium and M. isabellina strains catalyzed the formation of sulfoxides of opposite configuration. The Helminthosporium sp. catalyzed the formation of hydroxy-sulfoxide with high optical purity if the carbonyl group had a methyl substituent (Table 1). Sulfoxide alcohols from phenylcarbonyl-containing substrates were prepared via a reaction with the M. isabellina strain.
Another research topic carried out by the Holland team was the oxidation of sulfur amino acids. The protected amino acids were biotransformed using Beauveria species strains. Only methionine derivatives were biotransformed with good yield and high enantiomeric excess. A strain of Beauveria caledonica ATCC 64970 oxidized N-phthaloyl-L-methionine to sulfoxide (SSSC) with de 90% (Figure 1). N-phthaloyl-D-methionine was converted to (SSRC) sulfoxides with a lower yield but equally high de = 92% [29]. No such good results were observed for cysteine derivatives.
However, in the oxidation reaction of N-Boc derivative of l-4-S-morpholine-2-carboxylic acid, very high conversion (above 90%) was observed with a high diasteroisomeric excess (above 95%) at the same time. It is worth noticing that strains of B. caledonica and B. bassiana [30] oxidized the substrate to compound, which is the intermediate compound in the synthesis of chondrine (Scheme 5).
In order to obtain the active form of rabeprazole, the authors [31] carried out screening tests on 650 microorganisms. The most effective biocatalyst for oxidation of 2-[4-(3-methoxypropoxy)-3-methylpyridin-2-yl]-methylthiobenzimidazole (PTBI) is the Cunninghamella echinulata MK40 strain (Scheme 6). After 48 h of transformation, the authors observed a 92% conversion without forming the sulfone as an undesired product. The main product was the S-sulfur enantiomer. During the study, it was found that the addition of glucose affected the biotransformation efficiency. On this basis, it was found that conversion of PTBI to rabeprazole may involve the regeneration of NADH or NADPH in C. echinulata MK40 supported by glucose.
Also, other Cunninghamella strains tested were just as capable of converting PTBI to rabeprazole. The efficiency of the reactions catalyzed by C. echinulata IAM 6210 and C. blakesleeana strains IAM 6219 were comparable to those of C. echinulata MK40 (4 mM and 3.9 mM, respectively, after 48 h). The lower yield of biotransformation by strain of C. echinulata IAM 6209 after 48 h was observed, because 0.8 mM rabeprazole was formed.
As well, sulfuric precursors omeprazole and lansoprazole (Figure 2) were selected for biotransformation. These compounds, like rabeprazole, are proton pump inhibitors. In reactions catalyzed using C. echinulata MK40, omeprazole and lansoprazole were also obtained, but in low yield. It is surprising that other substrates, such as DL-methionine, DL-ethionine, DL-lanthionine, 4-methylthiophenol, thioanisole, and 4-chlorophenylmethyl sulfide were not oxidized to the corresponding sulfoxides.
2.2. Bacteria
The catalytic bio-oxidation reactions can also be catalyzed using bacterial strains. Previous research [32,33] has shown that this process occurs in bacteria, as in the case of fungi, and can be catalyzed using P450 or dibenzothiophene monooxygenases [34].
Research carried out by the Ohta team [35,36] in the 1980s proved the usefulness of bacteria of the genus Rhodococcus for the oxidation of alkylaryl sulfides. For biotransformation, the authors used the Rhodococcus equi strain. As a result of the reaction, a high enantiomeric excess (ee 97–100%) of alkyl aryl sulfoxides from sulfide was obtained. The authors also observed the formation of sulfones.
One of the most commonly selected bacterial strains for the biotransformation of sulfur-containing compounds is the Rhodococcus erythropolis, which has been extensively tested for its ability to oxidize sulfur-containing compounds. This strain is also used in the biocatalytic desulfurization of fossil fuels [37]. The dibenzothiophene desulfurization (dsz) operon from R. erythropolis IGTS8 encodes three proteins, DscC, DscA, and DsaB. Two of the enzymes are cytoplasmic monooxygenases (DszA and DszC), while the third (DszB) is a desulfinase [33]. The specific role in sulfur oxidation has monooxygenase DscC, which participates in conversion of sulfide to sulfoxide and thence to sulfone. In 2003, Holland published a study [38] on the oxidation of many sulfur derivatives using the strain R. erythropolis, in which the enzymes DscA and DscB were lacking, while there were different DscC enzymes. The first group were methyl phenyl sulfides, which were biotransformed mainly to the isomer with the R configuration of a sulfur asymmetric center (Table 2). Sulfoxides having a substitution in the para position were obtained with a higher enantiomeric excess. The enantioselectivity of the process was also influenced by the group that substituted the aromatic ring. Higher optical purity has products with higher electronic withdrawing groups (e.g. p-CN, and p-NO2).
The second group contained substituted benzyl sulfides (Table 3). The authors investigated the effect of a substituent in the aromatic ring during oxidation. The best optical purity of sulfoxides was obtained with para-substituted polynuclear groups of substrates (p-F, p-Cl, p-Br, p-NO2, p-CN). Large alkyl or aryl substituents, such as C6H5CH2S(alkyl)C6H13 or p-t-C4H9C6H4CH2SCH3, C6H5C2H4SCH3, and C6H5C3H6SCH3, caused a significant decrease in enantiomeric excess [38].
The tests were also subjected to sulfur derivatives of amino acids (Table 4). In the oxidation reaction of sulfides to sulfoxides, the R configuration was preferred regardless of the amino acid stereochemistry. Similar stereochemistry was observed during the oxidation of methionine and cysteine derivatives by chloroperoxidase [39,40].
Ai-Tao and coworkers [41] presented an uncommon perception of sulfoxidation reaction. Formerly, sulfone formation was an undesirable reaction, whereas in this case the reaction was used to obtain an optically pure sulfoxide with the S configuration as a result of deracemization (Scheme 7). Whole cells of Rhodococcus sp. ECU0066 were used for asymmetric oxidation of racemic sulfoxides, which were excellent substrates for biotransformation due to their lower biotoxicity compared to sulfides.
(S)-Phenylmethylsulfoxide was obtained at higher concentrations and good enantiomeric excess (37.8 mM and 93.7% ee (S)) if the substrate was in the form of racemic sulfoxide. In comparison, asymmetric oxidation of phenylmethyl sulfide gave the product at a lower yield and enantiomeric excess (10 mM and 80% ee (S)). Rhodococcus was also characterized by a fairly good activity (yield, 22.7–43.2%, in 1–8 h) and enantioselectivity (ee (S) > 99.0%) to para substituted (methyl and chloro) phenylmethylsulfoxides and phenylethyl sulfonate.
He and colleagues realized very good results in the oxidation of phenylmethyl sulfide [42], where biotransformation reactions catalyzed using Rhodococcus sp. CCZU10-1 were carried out in a two-phase n-octane/water system: 55.3 mM (S)-phenyl-methylsulfoxide with an enantiomeric excess >99.9% was conveniently obtained.
Pseudomonas bacteria also convert sulfides into chiral sulfoxides. An example is the strain of P. monteilii CCTCC M2013683, which catalyzed the oxidation reaction of a series of aryl alkyl sulfides to sulfoxides with a yield of 54–99% and a good enantiomeric excess (63–99%). The cultivation was carried out in M9 medium with toluene as the carbon source. Oxidation reactions were carried out in resting cultures. The obtained results showed that the aromatic ring of substrates with the substituted group may influence the activity and enantioselectivity of the oxidation reaction with P. monteilii CCTCC M2013683 [43].
In turn, the use of biotransformation cultures of the genus Streptomyces and Bacillus cereus allowed for the formation of both enantiomers of cyclohexylmethyl sulfoxide, where S. badius, S. hiroshimensis, S. phaerocromogenes S. flaviviseus, and B. cereus strains produced significant amounts (>25%) of sulfoxides. The first two strains gave a racemic mixture. The other three performed the sulfoxidation reaction with different enantioselectivity. S. hiroshimensis ATCC 27429 and S. flavogriseus ATCC 33331 oxidized to the R- and S-sulfoxide species without generating sulfone. The reaction catalyzed using S. phaeochromogenes NCIMB 11741 was very interesting as there was the synchronized reaction of two enzymes that simultaneously oxidized the sulfide to the corresponding enantiomers of sulfoxide. During the first six hours of biotransformation, mainly S-sulfoxide with high enantiomeric excess (ee > 99%) was formed. After this time, the oxidation of cyclohexylmethyl sulfide to the R-isomer was also observed. Ultimately after 48 h, 55% R-cyclohexylmethyl sulfoxide with 55% ee was obtained [30].
One of the methods of optimizing biotransformation conditions carried out by other cells may be their immobilization. The use of Gordonia terrae IEGM 136 cells, which was immobilized in a cryogel of polyvinyl alcohol, significantly reduced the oxidation time of methylphenyl sulfide to (R)-sulfoxide from 144 to 24 h. The immobilized Gordonia cells were resistant to high (up to 1.5 g/L) sulfide concentrations and catalyzed a complete bioconversion to (R)-sulfoxide (95% ee) within 72 h. The immobilized biocatalyst also showed high activity against other sulfides giving the right products from 77–95% ee [44].
3. Enzymatic Sulfoxidation
3.1. Chloroperoxidase
Fungal chloroperoxidases is one of the classes of enzymes that produce natural organochlorides in the soil. The first enzyme in this class was chloroperoxidase (CPO, EC 1.11.1.10), which was isolated from the marine fungus Caldariomyces fumago and it was first described in 1959. This enzyme is a highly glycosylated heme-dependent haloperoxidase with a molecular mass of 42 kDa. In vivo, it catalyzes the oxidative chlorination of 1,3-cyclopentadione by generating in situ hypochlorite via chloride ion oxidation. In the presence of free chloride and hydrogen peroxide at an acidic pH in the range of 2.75–4.50, the CPO easily chlorinates compounds with a variety of structures, such as antipirin, barbituric acid, catechin, phenols, and many others [40]. While CPO usually catalyzes the chlorination reaction in the presence of free chlorine and low pH, it can also act as an oxidizing enzyme with cytochrome P450-like activity at higher pH values (pH > 5.0), especially in the absence of chlorides [45]. In the case of an oxidation reaction, the oxygen is transferred directly to the sulfur from the heme group of CPO [46]. Chloroperoxidase catalyzes many oxidation reactions such as olefin epoxidation, sulfoxidation, hydroxylations, oxidation of indoles, and oxidation of primary alcohols to aldehydes [47].
Activated forms of oxygen, such as hydrogen peroxide or hydroxyl radicals, can cause the suicide inactivation of CPO via oxidation of the heme group. One strategy to circumvent this problem is to ensure that the steady-state hydrogen peroxide concentration is kept as low as possible by controlling its addition to the reaction environment. This step is crucial for enzyme activity, because concentration of H2O2 even as low as 30 μM results in complete deactivation of CPO [42]. Additionally, hydrogen peroxide alone can cause oxidation of more sensitive substrates. To solve this problem, an electroenzymatic method with the participation of CPO was developed. A carbon cathode was used, which reduced dissolved oxygen to hydrogen peroxide, which in the presence of CPO from C. fumago, can enantiomerycally oxidize sulfides to sulfoxides. This method was used to react with thioanisole, which was oxidized to form a sulfoxide with very high optical purity (ee = 99%). Another way was to use the cascade reaction with Pd (0) for oxidation of the same substrate, which catalyzed the formation of the hydrogen peroxide necessary for the operation of CPO. The next method was the use of supercritical carbon dioxide (scCO2) as a medium for H2O2 production in situ with H2 and O2 using a Pd catalyst. The system gave thioanisole sulfoxide with a 34% yield and 94% ee [48].
Chloroperoxidase is also sensitive to a number of factors such as temperature, pH, organic solvents, and high salt concentrations. This problem can be solved via immobilization. Therefore, in the oxidation reaction of dibenzothiophene (DBT), classified as polycyclic aromatic hydrocarbons (PAHs) [49], to the corresponding sulfoxide was carried out using CPO from C. fumago immobilized on (1D)-γ-Al2O3 nanorods (Scheme 8). The enzymatic reaction was initiated via the addition of 1 mM hydrogen peroxide to a mixture containing 1.0 × 10−7 mol of DBT and 1.3 × 10−12 mol of CPO in 1 mL. After 120 min, the reaction yield was 42.2% [46].
Modafinil is a very effective pharmaceutical for the treatment of excessive drowsiness caused by sleep disorders, narcolepsy, and obstructive sleep apnea. Compared to racemic modafinil, the R-enantiomer of this drug has a long half-life due to its excretion and slow metabolism, which results in a longer time of exposure to the drug and consequently a longer duration of its action. For this reason, it is important to develop a method for obtaining pure modafinil R-enantiomer. This was possible via enantioselective sulfoxidation of 2-(diphenylmethylthio)acetamide using chloroperoxidase (Scheme 9). Only the use of ionic liquids (ILs) in the form of quaternary ammonium salts or polyhydric compounds introduced into the reaction environment allowed for the increase in its efficiency to 40.8%. The enantiomeric excess of 97.3% (R) was obtained at pH 5.5 in the presence of [EMIM] [Br] (v ILs/v 10% buffer) and enzyme concentration 0.013 mM. The use of an ionic liquid caused the unveiling of the heme, making the CPO active center more accessible to the substrate. In addition, ILs improved the affinity and specificity of CPO [50].
The optimal use of chloroperoxidase from C. fumago in sulfoxidation depends on the method of adding an oxidizer such as hydrogen peroxide. Due to the high sensitivity of the enzyme to H2O2 concentrations in the thioanisole oxidation reaction, glucose oxidase (GOX; EC 1.1.3.4) from Aspergillus niger was used to generate hydrogen peroxide in situ. Maximum yields and enantioselectivity of biotransformation were observed at high chloroperoxidase reaction rates, but not at low hydrogen peroxide formation rates, as would be expected based on the CPO instability at high hydrogen peroxide concentrations. It has been observed that glucose oxidase catalyzing the oxygen sulfoxylation gave racemic sulfoxide, which is an unexpected and innovative reaction. It has been attributed to the flavonoid hydroperoxide oxidation resulting from the reaction of the free flavin cofactor associated with glucose oxidase and dioxygen. The speed of this side reaction depends on the amount of co-solvent in the system, and the enantiomeric excess of the oxidation product can be improved by lowering the concentration of the co-solvent [47].
3.2. Horseradish Peroxidase
Horseradish peroxidase (HRP, EC 1.11.1.7) catalyzes the oxidation of various substrates including sulfides by hydrogen peroxide. The most abundant is isoenzyme C, which is a single polypeptide of 308 amino acid residues. The sequence of this enzyme was determined in 1976 [51].
Initial studies using horseradish peroxidase in asymmetric sulfide oxidation were disappointing. By contrast, when in sequence of HRP phenylalanine was replaced with leucine, this change significantly improved enzyme yield to obtain (S) alkyl aryl sulfoxides with high optical purity (ee > 90%). In turn, substitution of the same amino acid with threonine in HRP resulted in a decrease of enantioselectivity [48].
3.3. Dioxygenase
Dioxygenases are a heterogeneous group of enzymes that are capable of introducing two oxygen atoms into the double bond. These enzymes are involved in the aerobic degradation of aromatic compounds and they are mainly catalyzed for the cleavage of aromatic ring and the cis-dihydroxylation of arenes [52].
3.3.1. Toluene Dioxygenase
Toluene dioxygenase (TDO, EC 1.14.12.11) from Pseudomonas putida F1 was able to oxidise thioanisole mainly to the (R)-sulfoxide (ee > 98%) [53,54]. It is worth emphasizing that this biotransformation was carried out in higher scale (bioreactor 8 L, 7.8 g of substrate, glucose for the regeneration of cofactors), obtaining a 90% substrate conversion.
This enzyme was also expressed in P. putida UV4 and a biotransformation of benzo[b]thiophene (B[b]T) (Figure 3), and its 2-methyl and 3-methyl derivatives were performed. These compounds can be found in the environment, e.g., from petroleum, coal, and products of its combustion. In the case of sulfoxidation by TDO in addition to the formation of the desired sulfoxides, the formation of a variety of other products has been also observed, e.g., the formation of di- and monohydroxyls or dimers [52,55].
Although the resulting benzo[b]thiophen sulfoxide was enantiomerically enriched (predominantly in the R-enantiomer), it quickly racemized within a few hours and was thermally unstable, either dimerized or disproportionate over several days. The methyl derivatives were more stable and therefore R-sulfoxide with ee > 99% was obtained from 2-methylbenzo[b]thiophene [52,55].
3.3.2. Naphthalene Dioxygenase
For the biotransformation of benzo[b]thiophene and its 2-methyl or 3-methyl derivatives in addition to TDO, naphthalene dioxygenase (NDO, EC 1.14.12.12.) was used, which was expressed in Pseudomonas putida NCIMB 8859 (NDO2), P. putida mutant 9816/11 expressing NDO1, and E. coli F352V expressing NDO3.
As with TDO, many different products were also made from benzo[b]thiophen and its derivatives, which were more stable. NDO1,3 produced sulfoxides in 7–40% of the yield, while NDO2 provided a relative yield of 90%. In the case of NDO2, 2-methylbenzo[b]thiophene sulfoxide was obtained with the isolated yield of 26% and ee = 56%, but with the S-configuration of sulfoxide [52,55].
3.4. Monooxygenases
3.4.1. Cytochrome P450 Monooxygenases
Cytochrome P450 monooxygenases (P450, CYP) are hemoproteins found in viruses, archaea, bacteria, protists, fungi, plants, and animals. They play an important role in the oxyfunctionalization of many structurally and physiologically important chemical compounds, including fatty acids, steroids, or vitamins. What is important is that they also participate in the detoxification process of xenobiotics, and in this case, some enzymes can catalyze sulfoxidation [56].
The sulfoxidation reaction can be catalyzed using CYP116B monooxygenases classified as class VII [57]. These are self-contained enzymes because a single polypeptide chain has three distinct functional parts: the flavine mononucleotide (FMN) binding domain, the NADPH binding domain, and the [2Fe-2S] ferredoxin domain. Therefore, the redox partners transfer electrons from the reduced NADPH, through the specific domains of FMN and [2Fe-2S], to their acceptor, the heme domain [58]. They show preference for more structurally differentiated substrates from the most well-known CYP102A1 monooxygenase (P450-BM3 from class VIII). To combine this catalytic self-sufficiency with thermal stability, new P450 enzymes derived from thermophilic microorganisms have recently been characterized. The P450-TT monooxygenase from Thermus thermophilus has been shown to have exceptional thermostability within all multiple-domain enzymes and has a half-life greater than 9 h at 50 °C [56].
An example member of the CYP116B subfamily is P450RhF monooxygenase (CYP116B2), which has been isolated from Rhodococcus sp. NCIMB 9784. This enzyme is able to catalyze various reactions, including O-dealkylation, aromatic hydroxylation, epoxidation of olefins, and asymmetric sulfoxylation of a number of substituted aromatic compounds. The natural substrates for CYP116B2 are unknown; however, the incomparably high activity exhibited by the enzyme against sulfides suggests that its natural function may involve the detoxification of related compounds via sulfoxidation [59].
The pharmacokinetics of omeprazole is associated with phenotypic P450 2C19 polymorphism in humans. An easily commercially available racemic mixture of omeprazole was used as a proton pump inhibitor in the treatment of gastric acid related diseases. However, it is noteworthy that the omeprazole S-enantiomer esomeprazole was developed as an enantiomerically pure drug [60].
High catalytic activity was observed for recombinant human CYP2C19 in 5-hydroxylation of R-omeprazole, CYP2C19 of cynomolgus in hydroxylation of both R- and S-omeprazole isomers, and CYP2C19 of marmosets in 5-hydroxylation of S-omeprazole. On the other hand, human, cynomolgus, and marmoset P4503A preferentially mediated S-omeprazole sulfoxidation. Correlations and kinetic analysis have shown a high affinity of polymorphic CYP2C19 of macaques with marmoset microsomal enzymes in relation to 5-hydroxylation of R- and S-omeprazole, and high yield of cynomolgus P4503A4 and microsomal CYP2C19 marmosets for omeprazole 5-hydroxylation and sulfoxidation [60].
Recombinant E. coli cells (P450pyrI83H-GDH) co-expressing three-component P450pyrI83H monooxygenase and glucose dehydrogenase (GDH) were used for sulfoxidation of a number of compounds such as thioanisole, 4-fluorothioanisole, ethyl phenyl sulfide, methyl p-tolyl sulfide, and 4-methoxythioanisole. High R-enantioselectivity was observed. Due to the high toxicity of both substrates and biotransformation products, the phosphate buffer and ionic liquid two-phase system was used to solve this problem. [P6,6,6,14] [NTf2] showed excellent biocompatibility during the growth of E. coli cells (P450pyrI83H-GDH) and the ability to protect substrate toxicity against cells, being the ideal co-solvent in a two-phase KP/IL buffer system for efficient processes of biooxidation. The applied system contributed to the growth of both productivity and enantioselectivity of transformation. In such a two-phase system, >99% of the substrate and 35–60% of the products remained in the IL, which significantly reduced the toxicity of the substrate and partially reduced the toxicity and inhibition of the product [61].
Not only the involvement of CYP in the metabolism of drugs must be carefully studied; it is equally important to discover the contribution of liver enzymes in the detoxification of pesticides. Benfuracarb (Figure 4) is one of the carbamate pesticides. This compound is a systemic and contact insecticide used to control insect pests in various crops. Benfuracarb and its main metabolite, carbofuran, have been shown to cause neurotoxic effects. In addition, they can cross the placental barrier and have a significant impact on the maternal-placental-fetal unit. It is therefore a threat to humans, and therefore its metabolism in liver microsomes was examined.
Benfuracarb was activated via the carbamfuran metabolic pathway, and in this case, the CYP3A subfamily turned out to be the most active enzymes (mainly CYP3A4). Only CYP2C9 and CYP2C19 participated in the sulfoxidation of benfuracarb. It is important to notice that the formation of toxic products of the carbofuran metabolic pathway is faster than sulfoxidation [62].
Chlorpromazine (Figure 5) belongs to phenothiazine neuroleptics and it is a potent dopaminergic D2 receptor antagonist that is responsible for antipsychotic activity of this drug. Chlorpromazine is a blocker of α1 adrenergic receptors and muscarinic M1 receptors that may be associated with certain side effects, such as anticholinergic symptoms, hypotension, and sedation. During phase I of this drug metabolism in the liver, it undergoes a number of transformations catalyzed via cytochrome P450 (CYP) monooxygenase-associated isotactic compounds such as 5-sulfoxidation, mono-N-demethylation, and di-N-demethylation. In the studies described by Wójcikowski [63], selective CYP inhibitors, human CYP isoforms (Supersomes 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4), and liver microsomes from different donors were used in the experiments in vitro. The authors suggest that CYP1A2 is the only CYP isoform responsible for mono-N-demethylation and di-N-demethylation of chlorpromazine (100%), and it is also the main isoform that catalyzes 5-sulfoxidation (64%) at the therapeutic concentration of this drug (10 mM). In addition, 5-sulfoxidation of chlorpromazine is also carried out by CYP3A4 (34%) [63].
The same researchers also became interested in metabolism of another drug from this group, namely levomepromazine (methotrimeprazine) (Figure 5), which is used to treat schizophrenia and schizoaffective disorder. This drug has also found widespread use in symptom control in palliative care as an antipsychotic, anxiolytic, antimimetic, and sedative agent. It is also used as a pre-operative sedative, in the final control of pain, and post-operative analgesia. Its long half-life allows for administration once a day, and its cost is also beneficial. This compound is a moderate dopamine D2 receptor antagonist responsible for its antipsychotic activity, as well as α1-adrenoceptor and M1-muscarinic receptors, which are associated with some side effects of this drug (hypotension, sedation, and anticholinergic symptoms) such as the chlorpromazine mentioned above [64].
For the biotransformation of this drug in the liver, CYP3A4 is mainly responsible because it performs levomepromazine 5-sulfoxylation (72%) and N-demethylation (78%) at the therapeutic drug concentration (10 mM). CYP1A2 contributes less to levomepromazine 5-sulfoxide (20%). The role of CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP2E1 in the catalysis of the above-mentioned reactions is negligible (0.1–8%). The results obtained indicate that in case of both chlorpromazine and levomepromazine provide possible pharmacokinetic interactions between these drugs and CYP3A4 substrates (e.g., testosterone, tricyclic antidepressants, and macrolide antibiotics calcium channel blockers), and inhibitors (e.g., erythromycin, ketoconazole, and selective serotonin reuptake inhibitor (SSRI)), or inducers (e.g., carbamazepine, rifampicin) [64].
CYP3A4 is also responsible for the sulfoxidation of Prothipendil (Dominal®), which is a neuroleptic drug and tricyclic azapenothiazine derivative. Prothipendil (Figure 5) is used in patients with psychomotor and anxiety disorders [65].
Thioridazine (Figure 6) is widely used as an antipsychotic drug. The main reactions that take place in the human liver are mono-2-sulfoxidation to mesoridazine and di-2-sulfoxidation to sulforidazine. First of all, these reactions can be attributed to CYP2D6. Both products of sulfoxidation also display antipsychotic activity. The interest of thioridazine usage has increased again because it can potentially serve as an antimicrobial for the control of multidrug-resistant strains, including the treatment of drug-resistant tuberculosis [66].
In the genome of Streptomyces platensis DSM 40041, 21 cyp genes were identified. One of these P450 enzymes was classified as CYP107L and its ability to biotransform drugs compared to human metabolism was established. The conversion of thioridazine with CYP107L reached 63% after 20 h, compared to 35% with CYP2D6. CYP2D6 provided 33% of the mono-sulfoxidation product and only 1% of the di-sulfoxidation product after 20 h. CYP107L, on the other hand, provided 26% of monosulfoxidation and an additional 6% of disulfoxidation [66].
3.4.2. Flavin-Dependent Monooxygenases
Metabolism of xenobiotics in humans and other mammals often begins with oxidation. Most reactions in phase I metabolism are catalyzed via CYP450s, which are discussed in the previous section. However, beyond CYP450s, recent studies indicated that flavin-containing monooxygenases (FMOs) also play a key role in the biotransformation of xenobiotics, including drugs and natural compounds. The human proteome contains five FMO isoforms (FMO1-FMO5). All these enzymes have expression patterns and the role in human metabolism is dependent on the tissue. The major organs among which they are present, consist of the liver, kidneys, small intestine, lungs, and brain. Human FMO (hFMO) and their mammalian orthologs are usually associated with cell membranes and often thermolability, which seems to be the main reason for their problematic isolation from tissue and ineffective production in the form of a recombinant protein. FMO has been shown to be involved in the oxidation of heteroatom-containing compounds such as sulfides. Unlike CYP450, which contain heme, FMO uses flavin for oxidation, which also translates into a different mechanism of catalyzed reactions. For this reason, to distinguish whether a compound is metabolized via FMO or CYP450, specific inhibitors are often used. FMO enzymes require NADPH to reduce flavin adenine dinucleotide (FAD). The reduced flavin then reacts with molecular oxygen to produce a reactive 4α-hydroperoxiflavin. This reactive intermediate from flavin is capable of carrying out various oxidation reactions, e.g., sulfoxylation.
The wider use of mammals’ FMO in biocatalysis is difficult and little investigated due to their typical binding to membranes and thermal instability; although, some researchers have presented the involvement of these mammalian enzymes in specific chiral reactions including sulfoxidation. For example, purified FMO2 from a rabbit’s lung managed to catalyze stereoselective oxidation of methyl p-tolyl sulfide with an enantiomeric excess of 96% for (R)-sulfoxide [67,68]. Another example is Sulindac, which contains a chiral sulfoxide moiety. This nonsteroidal anti-inflammatory drug is clinically administered as a racemic mixture. Sulindac sulfoxide is reduced in vivo to sulindac sulfide, a pharmacologically active metabolite [69]. In the serum and urine of volunteers, a clear stereoselective enrichment of (R)-sulindac sulfoxide was detected. Hamman and colleagues showed that purified FMO1 from pig liver, FMO2 from rabbit lung, and human FMO3 were able to stereoselectively oxidize sulindac sulfide to the R-sulfoxide isomer. Using human FMO3, the (R)-sulindac sulfoxide was obtained with an ee of 87%. The remaining FMO were even more stereoselective in the formation of this sulfoxide because in case of FMO1 and FMO2 observed ee of 98 and 99%, respectively [68,69].
In turn, albendazole (Figure 7) and fenbendazole sulfoxides, benzimidazole derivatives with anthelmintic activity, were obtained using hepatic microsomes of sheep and cattle liver [68,70]. The authors suggest that the sulfoxidation of albendazole led to enantiomerically pure sulfoxide. Another research group [71] demonstrated that the resulting FMO1 mutants could also oxidize insecticide fenthion with excellent selectivity to (+)-sulfoxide [68].
The ability to catalyze sulfoxidation in the case of FMO5 was also investigated. This FMO, which naturally shows good expression in the small intestine, kidneys, and lungs, accounts for over 50% of all FMO transcripts in the human liver. A moderate conversion (20%) of thioanisole forming the corresponding sulfoxide was observed. The enzyme was also able to oxidize S-methyl-esonarimod and fenthion [72].
It was noted that Baeyer-Villiger Monooxygenases (BVMOs) from many microbes are sequentially associated with human FMO and exhibit similar effects; hence, they can be used as a tool to prepare metabolites in studies on the metabolism of xenobiotics [73].
3.4.3. Baeyer-Villiger Monooxygenases (BVMOs)
Baeyer-Villiger monooxygenases (BVMOs) are flavoprotein, which are dependent on nicotinamide cofactors and capable of mainly carrying out the oxidation of carbonyl compounds (Baeyer-Villiger oxidation) and also oxidation of various heteroatoms, including sulfur [52]. Many different BVMOs are capable of catalyzing sulfoxidation, which will be discussed later in this chapter. These are, for example:
- cyclohexanone monooxygenase (CHMO, EC 1.14.13.22)
- phenylacetone monooxygenase (PAMO, EC 1.14.13.92)
- 4-hydroxyacetophenone monooxygenase (HAPMO, EC 1.14.13.84)
- styrene monooxygenase (SMO).
BVMOAFL706 from Aspergillus flavus NRRL3357 readily oxidizes thioanisole with both sulfoxide and excessively oxidized sulfone being formed. The sulfone product is about 60% of the total reacted substrate after 12 h. Researchers found that it is difficult to determine the similarity of this enzyme with other known BVMOs, finding only ≈38% sequence similarity with CHMOAc [74].
Other BVMOs used for the oxidation of thioanisole were BVMOBo from Bradyrhizobium oligotrophicum and BVMOAm from Aeromicrobium marinum, which were expressed in E. coli BL21 (DE3). Evolutionary dependence and sequence analysis revealed that both BVMOs belong to the BVMO type I family and the EtaA monooxygenase mono-oxide subtype. The sequence identity is 48% between BVMOBo and BVMOAm. Both BVMO showed sequence similarity with EtaA from M. tuberculosis and with EthA from P. putida KT2440. The identity of the BVMOBo and BVMOAm sequences with EthA was 47% and 54%, respectively.
Both BVMOs are active, not only for ketones, but also for various thioether substrates. Among the studied substrates, BVMOBo and BVMOAm showed the highest activity in relation to thioanisole (0.117 and 0.025 U/mg protein). Importantly, no sulfone formation was observed. In addition, the BVMOs used exhibited pronounced activity and excellent stereoselectivity in relation to prochiral prazole thioethers, such as omeprazoles, ilaprazoles, rabeprazoles, pantoprazoles, and lansoprazoles, which is a unique feature in this BVMO family [75].
The recombinant BVMO4 from Dietzia sp. D5 was expressed in E. coli BL21-CodonPlus (DE3)-RP. Protein expression was induced in E. coli with 1 mM isopropyl b-D-1-thiogalactopyranoside (IPTG). The soluble part of the BVMO4 was obtained via centrifugation of the lysed cells [76].
BVMO4 from Dietzia sp. D5 has been tested for the oxidation of several sulfur-containing compounds. This enzyme exhibits a preference for unsubstituted aromatic sulfides, but also tolerates small substituents in the para (methyl, fluoro) or meta (chloro) positions. It is noteworthy that the presence of one additional methyl group in the substrate, i.e., ethyl phenyl sulfide, significantly increased the stereospecificity of the oxidation (ee > 99% R) compared to thioanisole (ee = 50% R), while the reaction rate was comparable [76].
3.4.4. Cyclohexanone Monooxygenase
Cyclohexanone monooxygenase (CHMO, EC 1.14.13.22) from Acinetobacter calcoaceticus NCIB 9871 is currently one of the best-studied Baeyer-Villiger monooxygenases [74,77]. As a member of BVMO type I, CHMO is dependent on FAD and NADPH as cofactors [78]. Under physiological conditions, CHMO catalyzes the key step in the biodegradation of cyclohexanol, i.e., the conversion of a cyclic ketone into a lactone, that can then be hydrolyzed to aliphatic acid [68,79]. The first CHMO was isolated from Acinetobacter spp. NCIMB 9871 in 1975 [80]. Although typical substrates are four- to eight-membered cyclic ketones, this enzyme is characterized by a broad spectrum of acceptable substrates and high enantio- and regioselectivity. These features enable its use in industrial production routes [77].
Cyclohexanone monooxygenase not only catalyzes the oxidation of Baeyer-Villiger but also sulfoxidation. Colonna et al. [81] (1996) showed that CHMO from Acinetobacter calcoaceticus NCIMB was able to oxidize alkyl aryl sulfide, disulphate, dialkyl sulfide, cyclic and acyclic 1,3-di-thioacetals, and 1,3-oxathioacetals to the corresponding mono-sulfoxides, with high ee. The researchers were interested in the synthesis of various sulfoxides, not only because of participation of those compounds in many biological activities, but also because of their pharmaceutical importance.
The method of directed evolution was used to generate mutants of CHMO from Acinobacter sp. NCIMB 9871. In this way M.T. Reetz and coworkers [82] wanted to obtain variants of CHMO that catalyzed sulfoxidation of methyl-p-methylbenzyl thioether S or R selectively, and at the same time, limit the further oxidation of sulfide to sulfone. Finally, the mutant with six mutations, namely Q92R, Y132C, P169L, F246N, V361A, and T415A, allowed the researchers to obtain S-sulfoxide with an ee value of 99.8% and the amount of undesired sulfone below 5%.
Also, in the sulfoxidation reaction, CHMOAc accepts a wide range of substrates including 3-(methylthio)aniline (3.0 mM) or albendazole (Figure 7 and Figure 8). Using the reduced form of NADPH, phosphate dehydrogenase was used to regenerate coenzymes. After 135 min of incubation, (+)-(R)-3-(methylthio)aniline sulfoxide with ee = 71% was obtained. In turn, the R-sulfoxide of albendazole was obtained with an ee of 32%. It is worth noting that both CHMOAc and BVMOMt1 formed the same product as human FMO [73].
To solve the problems of stability of this enzyme, a number of CHMO mutants were generated. Particularly noteworthy is the cyclohexanone monooxygenase mutant (CHMO-QM), which contained four distinct mutations C376L, M400I, T415C, and A463C. This resulted in increased long-term stability and higher activity at high temperatures compared to wild CHMO [77].
BVMOs from Rhodoccocus jostii (BVMORj4 and BVMORj24) were subsequently used to oxidize 3-(methylthio)aniline. A substrate at 3.0 mM was incubated with cell extracts supplemented with phosphate, NADPH, and phosphate dehydrogenase to regenerate the reduced coenzyme. A conversion of 72% was achieved with ee > 99.5% of S-(−) after 135 min. Unfortunately, the formation of sulfone in an amount not exceeding 2% was also observed. This enzyme was also able to oxidize albendazole with a conversion of 55% and ee = 95% (S) [73].
In the oxidation of 3-(methylthio)aniline using BVMO from the fungus Myceliophthora thermophila (BVMOMt1), a conversion of substrate of 97% and ee = 88% (R)-(+) was observed, thus the opposite enantiomer of 3-(methylthio)aniline sulfoxide was obtained when BVMORj4 was used for this reaction. Albendazole was even more oxidized (25%) with the predominance of R-enantiomer. It is noteworthy that this enzyme formed the same product as mammalian FMOs [73].
3.4.5. Phenylacetone Monooxygenase
Phenylacetone monooxygenase (PAMO, EC 1.14.13.92) from thermophilic bacteria Thermobifida fusca is a monomeric BVMO type I capable of catalyzing inter alia enantioselective sulfoxylation. It deserves special attention due to its good thermostability and tolerance to solvents. For this reason, it is attractive for practical use [12,83]. However, PAMO has a somewhat narrow range of substrates favoring relatively simple aromatic compounds, such as benzyl alkyl sulfides. PAMO was not suitable for the synthesis of alkyl heteroaryl sulfoxides because of its generally low activity and/or selectivity. In addition to aromatic substrates, PAMO can also moderately convert non-aromatic substrates [12].
Rodrigues et al. 2012 [84] attempted to use a regeneration system in the reaction oxidation of thioanisole catalyzed using PAMO at pH 9.0. The highest conversion was observed when glucose/GDH as a recycling system NADPH (c = 95%) was used; however, the obtained (S)-methylphenyl sulfoxide has a slightly lower optical purity (ee = 35%) than for the oxidation of ketones. When condensed CRE2-PAMO was used, 60% of (S)-methylphenyl sulfoxide was obtained. For the three enzymes, sulfoxide was obtained with an optical purity of about 40%. Variability of selectivity depending on the cofactor regeneration system is explained by the allosteric effect on enzyme behavior.
The use of the addition of organic solvents to the reaction mixture allows the range of substrates to be extended by working with hydrophobic substrates, and they are used to develop solvent engineering techniques to modify the biocatalytic properties of this BVMO. The use of PAMO with the addition of small amounts of short-chain alcohols, such as methanol or ethanol, or a water-soluble polymer, such as PEG 4000, resulted in a significant improvement in enantioselectivity in the oxidation of prochiral sulfides.
Using the I-TASSER methodology with matrix 1 W4X, it was found that PAMO from T. fusca has 86% homology with BVMO4 from Dietzia sp. D5. The difference is mainly due to the longer sequence in BVMO4 at the N-terminus compared to PAMO. The variable regions that correspond to amino acid residues 228–241, 319–334, 448–475, and 548–571 are part of the loop, and thus, can be flexible, which is consistent with the data found in the X-ray structures. The remaining part of the protein shows high homology, especially the binding sites of the cofactor, as well as the active site of the enzyme, i.e., the key sites for its catalytic activity [85].
The methionine 446 in PAMO has recently been replaced with glycine, after comparing the PAMO structure with the homologous cyclopentanone monoxygenase model (CPMO, EC 1.14.13.16). The resulting M446G PAMO protein retains the thermostability of PAMO but has an altered substrate-binding pocket, which explains the essential changes in substrate specificity and enantioselectivity in the sulfoxidation of sulfides. It has been found that this biocatalyst is active in relation to many aromatic sulfides for which the wild-type PAMO has no activity [83].
The presence of various hydrophilic organic solvents has led to an increase in the enzymatic activity of the M446G mutant of PAMO when it is used in enantioselective sulfoxidations. For example, cyclohexyl propyl sulfide was not converted by M446G PAMO in an aqueous medium, but reacted in the presence of hydrophilic solvents. In the case of wild-type PAMOs, short-chain alcohols led to reduced conversion and increase selectivity, while the presence of low (5–10% by volume) hydrophilic solvents and PEG in M446G catalyzed reactions led to increased sulfides conversion; meanwhile, the selectivity of the biocatalyst remained unchanged or slightly improved. This effect can be explained by the better solubility of the substrate and improved stability of the enzyme in sulfoxidation carried out in the presence of solvents [83].
As result of directed evolution of PAMO, several enzyme variants with reversed enantioselectivity have been obtained, which were used in asymmetric sulfoxidation of several structurally different thioethers. Whereas using wild PAMO leads to obtaining S-sulfoxide with an ee value of 90%, the evolved mutant of PAMO with a four-point mutation, namely 167Q, P440F, A442N, L443I, was R-selective (ee = 95%) [86].
Pyridine methyl sulfides containing a nitrogen atom in the 2-, 3-, or 4-position were oxidized by the use of PAMO and its mutant M446G (M-PAMO). A second enzyme, glucose-6-phosphate dehydrogenase, was used to regenerate NADPH. Reactions were carried out in a Tris-HCl 50 mM buffer (pH = 9.0) at 30 °C for 24 h [87].
When wild-type PAMO and its mutant M446G were used for the oxidation of these substrates, only (R)-3-(methylsulfinyl)pyridine was acquired while the other compounds were not reactive. In sulfoxidation which was carried out in the presence of M-PAMO led to the formation of (R)-3-(methylsulfinyl)pyridine with 54% conversion and 88% of ee was observed, whereas PAMO showed lower activity (c = 26%) and selectivity (ee = 39%). M-PAMO was also able to transform the corresponding sulfide to (R)-2-(ethylsulfinyl)pyridine with a conversion of 13% and ee of 76% after 24 h. It is important to mention that both 3-(methylsulfinyl)pyridine enantiomers can be obtained with high enantiomeric excess using different BVMO because PAMO has the preference to form (R)-sulfoxide, while HAPMO prefers the formation of (S)-sulfoxide [87].
The 2-furfuryl methyl sulfide was also transformed. The (S)-sulfoxide from this compound can be obtained with a high conversion and an ee of 81% using the wild-type PAMO. The application of M-PAMO led to lower ee values. Regarding the oxidation of 2-(methylthio)thiophene, PAMO showed lower activity and selectivity than HAPMO in the preparation of the opposite enantiomer, (R)-sulfoxide (c = 22% and ee = 34%). The M446G mutant catalyzed sulfoxidation with even lower activity.
Sulfoxidation of cyclohexyl alkyl sulfides was also carried out using PAMO, which gave (S)-sulfoxide after 24 h with a 96% conversion and moderate optical purity (ee = 35%); meanwhile, this biocatalyst was not able to oxidize the ethyl derivative. Reactions that were carried out in the presence of M-PAMO led to (S)-cyclohexylmethyl sulfoxide with a moderate conversion and enantiomeric excess, whereas (S)-cyclohexylethyl sulfoxide was obtained after 24 h with a conversion of 19% and an ee of 31%. As expected from previous results, the M446G mutant was able to catalyze cyclohexylethyl sulfide oxidation for which the wild type of PAMO was not active due to the change in the size of the active site of the enzyme [87].
For both PAMO enzymes, wild and M-PAMO, the optimal pH for sulfoxidation activity ranges from 9.0 to 10.0. Regarding the enzymatic selectivity, (S)-cyclohexyl methyl sulfoxide was gathered at the highest enantiomeric excess at pH 10.5 (ee = 43%), while pH 9.0 was more suitable for M-PAMO (ee = 49%). Further increase in pH led to a gradual loss of enantioselectivity. This observation differs from that of previous work, which presented that this biocatalyst is able to catalyze Baeyer-Villiger oxidation with high conversion to 1-isochromanone synthesis at pH 10.5.
PAMO and its mutant M446G were able to carry out moderate oxidation of alkyl butyl sulfides (c = 34–45%, ee = 36–65%) [87].
3.4.6. 4-Hydroxyacetophenone Monooxygenase
4-Hydroxyacetophenone monooxygenase (HAPMO, EC 1.14.13.84) is a BVMO isolated from Pseudomonas fluorescens ACB. The primary reaction catalyzed by this enzyme is NADPH-dependent oxidation of 4-hydroxyacetophenone to 4-hydroxyphenylacetate. This enzyme is a 145 kDa homodimer and each subunit contains a non-covalently bound FAD cofactor [88]. In addition to Baeyer-Villiger oxidation, the enzyme is also used in the oxidation of a range of aromatic sulfides including thioanisols. A second enzyme reaction catalyzed via glucose-6-phosphate dehydrogenase (G6PDH) was used in the regeneration of NADPH. The reactions were carried out in a Tris-HCl buffer (50 mM, pH 9.0) at 25 °C [88].
In general, the phenyl sulfides appear to be the best substrates for this enzyme, giving (S)-sulfoxides with high optical purity. Low enantiomeric excess was obtained using benzyl sulfides and the enantioselectivity inversion from S to R was observed for alkyl chains longer than ethyl. The inversion of the preferred enantioselectivity was also found when the sulfur atom was further away from the aromatic ring. In the case of para-substituted phenylmethyl sulfides, the enzyme showed high selectivity for electron donor groups, while strongly accepting electron groups showed a negative effect on the selectivity and efficiency of the reaction. Interestingly, in this study, the position of the substituent did not affect selectivity [88].
Pyridine methyl sulfides containing the nitrogen atom in the 2-, 3-, or 4-position were also oxidized using HAPMO. A second enzyme, glucose-6-phosphate dehydrogenase (G6PDH), was used to regenerate NADPH. Reactions were carried out in a Tris-HCl 50mM buffer (pH = 9.0) at 20 °C for 24 h [87].
HAPMO was capable of catalyzing the enzymatic sulfoxidation of the three derivatives with moderate to high conversion and excellent enantiomeric excess. In a study by Rioz-Martinez [87], the best substrate for HAPMO was 2-(methylthio)pyridine because (S)-sulfoxide with complete conversion was achieved. The use of substrates containing a nitrogen atom further away from the methylthio group contributed to lower conversions. Thus, (S)-3-(methylsulfinyl)pyridine was obtained with a conversion of 95%, and the least-oxidized 4-(methylthio)pyridine (c = 63%); however, it yielded the opposite enantiomer because (R)-sulfoxide was obtained. It is important to underline that both 3-(methylsulfinyl) pyridine enantiomers can be obtained with high enantiomeric excess using different biocatalysts, because HAPMO has the preference to form (S)-sulfoxide while PAMO both wild type and mutant M446G prefer formation (R)-sulfoxide.
HAPMO was also adept at catalyzing the sulfoxidation of ethyl, n-propyl, and allyl sulfide derivatives of 2-pyridine, and high enantioselectivity was observed. Enantiomerically pure (S)-2-(ethylsulfinyl)pyridine was synthesized with complete conversion after 24 h. When the alkyl chain length was extended to the propyl group, (S)-sulfoxide was achieved in low yield (c = 11%). This tendency was previously described for this biocatalyst in the oxidation of thioanisole derivatives [87].
The heteroaryl substrate, 2-furfuryl methyl sulfide, was also transformed to give the (R)-sulfoxide enantiomer with a conversion of 69% and ee ≥ 99%. Upon oxidation of 2-(methylthio)thiophene, HAPMO was able to transform into enantiomerically pure (S)-sulfoxide with good conversion (c = 71%) after 24 h [87].
Sulfoxidation of cyclohexyl alkyl sulfides was also carried out. In this case, HAPMO proved to be a great catalyst because the enantiomerically pure (S)-sulfoxides were obtained in the reaction of the oxidation of cyclohexyl methyl sulfide and cyclohexyl ethyl sulfide. HAPMO also efficiently oxidized tetrahydrothiophene and tetrahydro-2H-thiopyran (close to 80%). Interestingly, both compounds were not transformed by other BVMO PAMOs. HAPMO also oxidizes aliphatic sulfides. For example, n-butyl methyl and n-butyl ethyl sulfoxides were obtained in the enantiomerically pure form with complete conversion after 24 h of reaction [87].
3.4.7. Styrene Monooxygenase
Styrene monooxygenase (SMO) is a flavin-dependent enzyme from group E of BVMOs. This enzyme was first described in Pseudomonas sp. It allows for the degradation of styrene, which is a source of coal and energy for this microorganism. SMO catalyzing the conversion of styrene to (S)-styrene oxide is responsible for adapting this microorganism to an environment contaminated with this hydrocarbon [89].
SMOs are known for their ability to convert a huge variety of substrates, often with high enantioselectivity. It is worth noting that while epoxidation is limited to the production of the (S)-enantiomer, in the case of sulfoxidation, the production of S- or R-sulfoxides depends on the used SMOs and structure of the substrate. However, there is still the possibility of optimization because the enantiomeric excess of the (R)-sulfoxides obtained by these monooxygenases is often not high.
SMO usually creates a two-component system. Monooxygenase (StyA, StyA1, EC 1.14.14.11) requires a second component for its activity, oxidoreductase (StyB; EC 1.5.1.36), which produces the reduced FAD necessary for the activity of the first component [90,91]. Some of these reductases exist as self-contained natural fusion proteins (or two domain proteins, StyA2B, E2 type of SMO) composed of the domain of oxygenase (A2) and reductase (B). This prototype was first isolated from Rhodococcus opacus 1CP (RoStyA2B). Most of the described monooxygenases (StyA, StyA1) belong to the pathways of degradation and detoxification of aromatic compounds. The studies found that the organization of the gene cluster is heterogeneous, in particular with regard to regulatory and transport mechanisms. In sulfoxidation reactions, the E1-type is characterized by high activity and low enantioselectivity, while the E2-type has low activity and high enantioselectivity [90].
Monooxygenase VpStyA1 and VpStyA2B isolated from Variovorax paradoxus EPS (accession numbers: ADU39063 and ADU39062) are two-component SMOs of type 2. They are the first example of StyA2B isolated from β-proteobacteria. These enzymes are proteins with two domains as an NADH: FAD-oxidoreductase (VpStyA2B). Searching databases for styA2B-like genes/proteins similar to V. paradoxus EPS indicated that these enzymes occur mainly among Actinobacteria (e.g., Amycolatopsis, Arthrobacter, Gordonia, Mycobacterium, Nocardia, Paenarthrobacter, Paeniglutamicibacter, Pseudarthrobacter, Sciscionella, Sinomonas, and Streptomyces). Variovorax belongs to β-proteobacteria and not to Actinobacteria. The closest homologs of two Variovorax domain proteins are in Delftia, although there is no natural variant of the fusion. The similarity of the VpStyA1 and VpStyA2B sequences (74% identity of more than 404 amino acids) between the monooxygenase domains (A1 and A2) was significantly higher than among other StyAl/StyA2B systems. Due to the high sequence similarity between monooxygenase subunits, Variovorax-like enzymes form a separate branch within the phylogenetic tree and results to date suggest a convergent evolution of the StyA1/StyA2B systems [90].
The ability of VpStyA1 and VpStyA2B to transform five aryl alkyl analogues that are analogues of styrene was studied. The reaction time was 2h and the substrate concentration was 2 mM. The degree of substrate conversion for VpStyA1 ranged from 32% to 95%, and the enantiomeric excess ee = 97–99% (S). The most active VpStyA1 was 95% against benzyl methyl sulfide (BMS). It is worth noting that the enzyme oxidized sulfides more efficiently as it transformed styrene; it was 3.3 to 22 times more active in relation to sulfides than styrene. VpStyA2B was definitely less active in relation to these substrates because the conversion was 6–13% [90].
The maximum of specific activity on BMS was determined to be 3.113 U mg−1 for VpStyAl and 1.616 U mg−1 for VpStyA2B. This is around 25 to 26 times faster than the specific activity for styrene. Thus, the two-component monooxygenase from the V. paradoxus EPS strain has a preference for this sulfide. It is worth noting that high sulfide activity is comparable to other SMOs known from the literature, but the high selectivity giving almost pure (S)-enantiomers is outstanding [90].
In addition, the potential linker region was identified and mutated with a protein similar to the StyA2B region. This linker region has been shown to influence both the reductase domain (B) and monooxygenase (A2). However further structural and kinetic studies are necessary to determine the effect of the linker on individual VpStyA2B domains and related monooxygenase. So far, the results indicate that this monooxygenase from Variovorax paradoxus EPS strain is a suitable candidate for structural studies as well as for the development of an enantioselective catalyst for the sulfoxidation reaction [90].
Another interesting styrene monooxygenases are PaSMO and MlSMO. PaSMO is an SMO isolated from Paraglaciecola agarilytica NO2 (previously Glaciecola agarilytica), an agar-digesting marine bacterium isolated from marine sediment, and MlSMO is an SMO isolated from Marinobacterium litorale DSM 23545, which is example of a mesophile bacteria isolated from surface seawater [92].
The MlSMO and PaSMO amino acid sequences have been aligned with other previously known SMOs. Studies have shown the presence of conservative sequence motifs typical of the mechanistic involvement of flavins and NAD(P)H. It is worth emphasizing that both MlSMO and PaSMO have a functionally conserved Thr47 residue, which is important for catalytic activity in StyA. The FAD-binding fingerprints GXGXXG, GG, and DX6G were found in both SMOs. The GXGXXG sequence (residues 9–14) is the fingerprint sequence that is characteristic for FAD- and NAD(P)H-dependent oxidoreductases and plays an important role in the FAD binding proteins. The second FAD-binding motif contains the sequence GDX6P (residues 294–302 in MlSMO, residues 292–300 in PaSMO), where the highly conserved Asp moiety connectswith the O3’ of the ribose moiety of FAD. The DG sequence is highly conserved among all flavoproteins with hydroxylase activity but only Gly is conservative in StyA (MlSMO residue 171, PaSMO residue 169) [92].
Both SMOs were expressed in E. coli BL21 (DE3). The biotransformation reactions were carried out in a two-phase system containing 10% (v/v) addition of cyclohexane to a phosphate buffer pH = 8 and they were catalyzed using whole E. coli BL21 cells (DE3) in which MlSMO or PaSMO were expressed. In general, MlSMO consistently showed higher activity than PaSMO under the same reaction conditions. The sulfoxidation reaction of thioanisole proceeded with poor enantioselectivity (ee = 19–42% (S)) [92].
An interesting bacterium is the Gram-positive actinomycete Rhodococcus opacus 1CP, which contains three different SMOs. In the work of Riedel [93], it was found that in addition to the styA1 and styA2B gene, the third SMO gene is located within the styABCD cluster, completely separated from styA1 and styA2B. The newly discovered FAD StyB reductase from the R. opacus 1CP strain is highly active with NADH as an electron donor. However, compared to StyB homologues, the binding affinity to NADH and FAD is rather low. StyB forms a homodimer as described for other StyB enzymes, but appears to be rather unstable, which limits its further characteristics [93].
In the studies, phenyl vinyl sulfide was selected as a substrate to provide sulfoxidation as well as an epoxidation site. It was found that all three SMO isoforms convert this substrate and only sulfoxidation was determined. The activities are comparable to styrene epoxidation. It is worth noting that StyA2B showed a 2.4-fold higher sulfoxidase as epoxidase activity. It was found that all three SMO isoforms catalyze sulfoxidation in an enantioselective manner. StyA1 is distinguished by the production of the vinyl-p-sulfoxide S-enantiomer with more than 99% ee [93].
In E. coli Bl21 (DE3) cells, SMO from P. putida CA-3 was expressed. Nine different sulfur-containing compounds were biotransformed. Thioanisole was consumed at a rate of 83.3 μmol/min g dry weight of cells, which resulted mainly in the formation of R-sulfoxide of thioanisole with the ee value of 45%. The consumption rate of 2-bromo-, 2-chloro-, and 2-methylothioanisole was 2-fold lower than in the case of thioanisole. Astonishingly, the oxidation product of 2-methylthioanisole had the opposite configuration (S) to the configuration of the other 2-substituted thioanisole derivatives and had a higher ee value of 84%. The rate of the 4-substituted thioanisols sulfoxidation was higher than the corresponding 2-substituted substrates, but the ee values of the products were consistently lower (10–23%). The rate of formation of benzo[b]thiophene sulphoxide and 2-methylbenzo[b]thiophene was about 10 times lower than in the case of thioanisole. The ee values of benzo[b]thiophene sulfoxide could not be determined due to the rapidly racemizing product under the reaction conditions. E. coli cells expressing the modified SMO (SMOeng R3-11) oxidized the 2-substituted thioanisole 1.8- and 2.8-fold faster compared to cells expressing the wild-type enzyme. SMOeng R3-11 oxidized benzo[b]thiophene and 2-methylbenzo[b]thiophene 10.1 and 5.6 times faster than the wild-type of enzyme. The stereospecificity of the modified SMO has not changed compared to the wild-type enzyme. Using X-ray crystallography for P. putida S12 SMO analysis, it was determined that the entry of substrates into the active site of SMO is limited by the bottleneck of the binding cavity formed by the side chains of the carboxylamide groups Val-211 and Asn-46 [55].
4. Conclusions
It seems that despite the intensive development of research on sulfoxidation, it will not be possible to find the optimal biocatalyst for a new sulfide molecule for a long time. In this regard, one should not forget about the tedious basic research involving classic biotransformations using whole cultures of microorganisms, especially those coming from extreme environments. Despite the development of genetic methods, there are no effective and rapid screening tools so far. This problem may arise from the fact that the sulfoxidation reaction may catalyze many different enzymes, as discussed in this work.
Two streams of research can be distinguished in current research. On the one hand, research using molecular biology tools on the structure of enzymes, especially the construction of their active center. Unfortunately, so far, there are no quick and effective methods to find the optimal mutant for a given sulfoxidation reaction.
The second stream of research includes the optimization of reaction conditions via immobilization or the use of various regeneration methods or the purpose of improving substrate access for the enzyme via the use of added solvents, including ionic liquids.
Author Contributions
W.M. and K.W. writing—original draft preparation, M.G. writing—review.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bentley, R. Role of sulfur chirality in the chemical processes of biology. Chem. Soc. Rev. 2005, 34, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.S.; Hart, J.; Brems, J.J.; Goldstein, L.; Lewin, K.; Busuttil, R.W. Amanita poisoning: Treatment and the role of liver transplantation. Am. J. Med. 1989, 86, 187–193. [Google Scholar] [CrossRef]
- Thomson, M.; Ali, M. Garlic [Allium sativum]: A review of its potential use as an anti-cancer agent. Curr. Cancer Drug Targets 2003, 3, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.P.; Cooper, D.L.; Gerratt, J.; Karadakov, P.B.; Raimondi, M. Chemical bonding in oxofluorides of hypercoordinate sulfur. J. Chem. Soc. Faraday Trans. 1997, 93, 2247–2254. [Google Scholar] [CrossRef]
- Morris, D.G. Stereochemia; Wydawnictwo Naukowe PWN SA: Warszawa, Poland, 2008. [Google Scholar]
- Andersen, K.K. Synthesis of (+)-ethyl p-tolyl sulfoxide from (−)-menthyl (−)-p-toluensulfinate. Tetrahedron Lett. 1962, 3, 93–95. [Google Scholar] [CrossRef]
- De Sio, V.; Acocella, M.R.; Villano, R.; Scettri, A. New chiral imino- and amino-sulfoxides as activators of allyl trichlorosilane in the asymmetric allylation of aldehydes. Tetrahedron Asymm. 2010, 21, 1432–1435. [Google Scholar] [CrossRef]
- Qi, W.-Y.; Zhu, T.-S.; Xu, M.-H. Design of chiral sulfoxide—Olefins as a new class of sulfur-based olefin ligands for asymmetric catalysis. Org. Lett. 2011, 13, 3410–3413. [Google Scholar] [CrossRef] [PubMed]
- Leśniak, S.; Rachwalski, M.; Kiełbasiński, P. Highly enantioselective conjugate addition of diethylzinc to enones using aziridine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymm. 2010, 21, 1890–1892. [Google Scholar] [CrossRef]
- Cotton, H.; Elebring, T.; Larsson, M.; Li, L.; Sörensen, H.; Unge, S. Asymmetric synthesis of esomeprazole. Tetrahedron Asymm. 2000, 18, 3819–3825. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Janicki, I.; Kiełbasiński, P. Enzyme-promoted kinetic resolution of acetoxymethyl aryl sulfoxides. J. Mol. Catal. B Enzym. 2015, 118, 23–28. [Google Scholar] [CrossRef]
- Liang, Y.; Wei, J.; Qiu, X.; Jiao, N. Homogeneous Oxygenase Catalysis. Chem. Rev. 2018, 118, 4912–4945. [Google Scholar] [CrossRef]
- Barth, T.; Hilário, V.C.; Rocha, B.A.; Furtado, N.A.J.C.; Pupo, M.T.; de Oliveira, A.R.M. Asymmetric sulfoxidation of albendazole to ricobendazole by fungi: effect of pH. Quim. Nova 2015, 38, 944–947. [Google Scholar] [CrossRef]
- Borges, W.S.; Borges, K.B.; Bonato, P.S.; Said, S.; Pupo, M.T. Endophytic fungi: Natural products, enzymes and biotransformation reactions. Curr. Org. Chem. 2009, 13, 1137–1163. [Google Scholar] [CrossRef]
- Liu, J.H.; Yu, B.Y. Biotransformation of bioactive natural products for pharmaceutical lead compounds. Curr. Org. Chem. 2010, 14, 1400–1406. [Google Scholar] [CrossRef]
- Parshikov, I.A.; Netrusov, A.I.; Sutherland, J.B. Microbial transformation of antimalarial terpenoids. Biotechnol. Adv. 2012, 30, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Holland, H.L.; Brown, F.M.; Larsen, B.G.; Zabic, M. Biotransformation of organic sulfides. Part 7. Formation of chiral isothiocyanato sulfoxides and related compounds by microbial biotransformation. Tetrahedron Asymm. 1995, 6, 1569–1574. [Google Scholar] [CrossRef]
- Holland, H.L. Chiral Sulfoxidation by Biotransformation of Organic Sulfides. Chem. Rev. 1988, 88, 473–485. [Google Scholar] [CrossRef]
- Holland, H.L.; Carter, I.M. The mechanism of sulfide oxidation by Mortierella isabellina NRRL 1757. Can. J. Chem. 1982, 60, 2420–2426. [Google Scholar] [CrossRef]
- Holland, H.L.; Bergen, E.J.; Chenchaiah, PC.; Khan, S.H.; Munoz, B.; Ninniss, R.W.; Richards, D. Side chain hydroxylation of aromatic compounds by fungi.: 1. Products and stereochemistry. Can. J. Chem. 1987, 65, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Holland, H.L.; Brown, F.M.; Lakshmaiah, G.; Larsen, B.G.; Patel, M. Biotransformation of organic sulfides—VIII. A predictive model for sulfoxidation by Helminthosporiun species NRRL 4671. Tetrahedron Asymm. 1997, 8, 683–697. [Google Scholar] [CrossRef]
- Holland, H.L.; Allen, L.J.; Chernishenko, M.J.; Diez, M.; Kohl, A.; Ozog, J.; Gu, J.-X. Side chain oxidation of aromatic compounds by fungi. A rationale for sulfoxidation, benzylic hydroxylation and olefin oxidation by Mortierella isabellina. J. Mol. Catal. B Enzym. 1997, 3, 311–324. [Google Scholar] [CrossRef]
- Holland, H.L.; Gu, J.-X.; Kerridge, A.; Willetts, A. Biotransformation of organic sulfides. Part 11. Preparation of functionalised phenyl propyl sulfoxides using Helminthosporium species and Mortierella isabellina. Biocatal. Biotransform. 1999, 17, 305–317. [Google Scholar] [CrossRef]
- Holland, H.L. Biotransformation of organic sulfides. Nat. Prod. Rep. 2001, 18, 171–181. [Google Scholar] [CrossRef]
- Ricci, L.C.; Comasseto, J.V.; Andrade, L.H.; Capelari, M.; Cass, Q.B.; Porto, A.L.M. Biotransformations of aryl alkyl sulfides by whole cells of white-rot Basidiomycetes. Enzym. Microb. Technol. 2005, 36, 937–946. [Google Scholar] [CrossRef]
- Pinedo-Rivilla, C.; Aleu, J.; Collado, I.G. Enantiomeric oxidation of organic sulfides by the filamentous fungi Botrytis cinerea, Eutypa lata and Trichoderma viride. J. Mol. Catal. B Enzym. 2007, 49, 18–23. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Ordena, A.A.; Bisogno, F.R.; de Gonzalo, G.; Kurina-Sanz, M. Aspergillus genus as a source of new catalysts for sulfide oxidation. J. Mol. Catal. B Enzym. 2012, 82, 32–36. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Barrett, F.; French, J.; Johnson, V. Biotransformation of beta-ketosulfides to produce chiral beta-hydroxysulfoxides. J. Ind. Microbiol. Biotechnol. 2003, 30, 292–301. [Google Scholar] [CrossRef]
- Holland, H.L.; Andreana, P.R.; Brown, F.M. Biocatalytic and chemical routes to all the stereoisomers of methionine and ethionine sulfoxides. Tetrahedron Asymm. 1999, 10, 2833–2843. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Palazzolo, M.A.; Lewkowicz, E.; Kurina-Sanz, M. Expanding the toolbox for enantioselective sulfide oxidations: Streptomyces strains as biocatalysts. Biocatal. Agric. Biotechnol. 2013, 2, 399–402. [Google Scholar] [CrossRef]
- Yoshida, T.; Kito, M.; Tsujii, M.; Nagasawa, T. Microbial synthesis of a proton pump inhibitor by enantioselective oxidation of a sulfide into its corresponding sulfoxide by Cunninghamella echinulata MK40. Biotechnol. Lett. 2001, 23, 1217–1222. [Google Scholar] [CrossRef]
- French, J.B.; Holland, G.; Holland, H.L.; Gordon, H.L. A comparative molecular field analysis of the biotransformation of sulfides by Rhodococcus erythropolis. J. Mol. Catal. B Enzym. 2004, 31, 87–96. [Google Scholar] [CrossRef]
- Zhang, J.D.; Li, A.T.; Yang, Y.; Xu, J.H. Sequence analysis and heterologous expression of a new cytochrome P450 monooxygenase from Rhodococcus sp. for asymmetric sulfoxidation. Appl. Microbiol. Biotechnol. 2010, 85, 615–624. [Google Scholar] [CrossRef]
- Elkin, A.A.; Kylosovab, T.I.; Grishkoc, V.V.; Ivshinaa, I.B.; Elkin, A.A.; Kylosova, T.I.; Grishko, V.V.; Ivshina, I.B. Enantioselective oxidation of sulfides to sulfoxides by Gordonia terrae IEGM 136 and Rhodococcus rhodochrous IEGM 66. J. Mol. Catal. B Enzym. 2013, 89, 82–85. [Google Scholar] [CrossRef]
- Ohta, H.; Okamoto, Y.; Tsuchihashi, G. Asymmetric synthesis of chiral sulfoxides via microbial oxidation of sulfides. Chem. Lett. 1984, 2, 205–208. [Google Scholar] [CrossRef]
- Ohta, H.; Okamoto, Y.; Tsuchihashi, G. Microbial Oxidation of Alkyl Aryl Sulfides to Corresponding Optically Active Sulfoxides. Agric. Biol. Chem. 1985, 49, 671–676. [Google Scholar] [CrossRef]
- Gray, K.A.; Pogrebinsky, O.; Mrachko, G.T.; Xi, L.; Monticello, D.J.; Squires, C.H. Molecular mechanisms of biocatalytic desulfurization of fossil fuels. Nat. Biotechnol. 1996, 14, 1705–1709. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Kerridge, A.; Pienkos, P.; Arensdor, J. Biotransformation of sulfides by Rhodocoeccus erythropolis. J. Mol. Catal. B Enzym. 2003, 22, 219–223. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Lozada, D.; Mayne, B.; Szerminski, W.R.; van Vliet, A.J. Chloroperoxidase-catalyzed oxidation of methionine derivatives. Can. J. Chem. 2002, 80, 633–639. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Johnson, D.V.; Kerridge, A.; Mayne, B.; Turner, C.D.; van Vliet, A.J. Biocatalytic oxidation of S-alkylcysteine derivatives by chloroperoxidase and Beauveria species. J. Mol. Catal. B Enzym. 2002, 17, 249–256. [Google Scholar] [CrossRef]
- Li, A.T.; Yu, H.L.; Pan, J.; Zhang, J.D.; Xu, J.H.; Lin, G.Q. Resolution of racemic sulfoxides with high productivity and enantioselectivity by a Rhodococcus sp. strain as an alternative to biooxidation of prochiral sulfides for efficient production of enantiopure sulfoxides. Bioresour. Technol. 2011, 102, 1537–1542. [Google Scholar] [CrossRef]
- He, Y.C.; Ma, C.L.; Yang, Z.X.; Zhou, M.; Xing, Z.; Ma, J.T.; Yu, H.L. Highly enantioselective oxidation of phenyl methyl sulfide and its derivatives into optically pure (S)-sulfoxides with Rhodococcus sp. CCZU10-1 in an n-octane-water biphasic system. Appl. Microbiol. Biotechnol. 2013, 97, 10329–10337. [Google Scholar] [CrossRef]
- Chen, Y.; Zhuoa, J.; Zhenga, D.; Tiana, S.; Lib, Z. Stereoselective oxidation of sulfides to optically active sulfoxides withresting cells of Pseudomonas monteilii CCTCC M2013683. J. Mol. Catal. B Enzym. 2014, 106, 100–104. [Google Scholar] [CrossRef]
- Kylosovaa, T.I.; Elkina, A.A.; Grishkoc, V.V.; Ivshina, I.B. Biotransformation of prochiral sulfides into (R)-sulfoxides using immobilized Gordonia terrae IEGM 136 cells. J. Mol. Catal. B Enzym. 2016, 123, 8–13. [Google Scholar] [CrossRef]
- Lim, M.L.; DeWayne Brooks, M.; Boothe, M.A.; Krzmarzick, M.J. Novel bacterial diversity is enriched with chloroperoxidase-reacted organic matter under anaerobic conditions. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Juarez-Moreno, K.; de León, J.N.D.; Zepeda, T.A.; Vazquez-Duhalt, R.; Fuentes, S. Oxidative transformation of dibenzothiophene by chloroperoxidase enzyme immobilized on (1D)-γ-Al2O3 nanorods. J. Mol. Catal. B Enzym. 2015, 115, 90–95. [Google Scholar] [CrossRef]
- Pereira, P.C.; Arends, I.W.C.E.; Sheldon, R.A. Optimizing the chloroperoxidase-glucose oxidase system: The effect of glucose oxidase on activity and enantioselectivity. Process Biochem. 2015, 50, 746–751. [Google Scholar] [CrossRef]
- O’Mahony, G.E.; Kelly, P.; Lawrence, S.E.; Maguire, A.R. Synthesis of Enantioenriched Sulfoxides. ARKIVOC 2011, 2011, 1–110. [Google Scholar]
- Kadri, T.; Rouissi, T.; Brar, s.K.; Cledon, M.; Sarma, S.; Verma, M. Biodegradation of polycyclic aromatic hydrocarbons (PAHs) by fungal enzymes: A review. J. Environ. Sci. 2017, 51, 52–74. [Google Scholar] [CrossRef]
- Gao, F.; Wang, L.; Liu, Y.; Wang, S.; Jiang, Y.; Hu, M.; Li, S.; Zhai, Q. Enzymatic synthesis of (R)-modafinil by chloroperoxidase-catalyzed enantioselective sulfoxidation of 2-(diphenylmethylthio)acetamide. Biochem. Eng. J. 2015, 93, 243–249. [Google Scholar] [CrossRef]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Matsui, T.; Dekishima, Y.; Ueda, M. Biotechnological production of chiral organic sulfoxides: Current state and perspectives. Appl. Microbiol. Biotechnol. 2014, 98, 7699–7706. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; Haughey, S.A.; Kennedy, M.A.; McMurray, B.T.; Sheldrake, G.N.; Allen, C.C.R.; Dalton, H.; Sproule, K. Toluene and naphthalene dioxygenase-catalysed sulfoxidation of alkyl aryl sulfides. J. Chem. Soc. Perkin Trans. 1998, 1, 1929–1934. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; McMurray, B.; Haughey, S.A.; Allen, C.C.R.; Hamilton, J.T.G.; McRoberts, W.C.; More O’Ferrall, R.A.; Nikodinovic-Runic, J.; Coulombele, L.A.; et al. Bacterial dioxygenase- and monooxygenase-catalysed sulfoxidation of benzo[b]thiophenes. Org. Biomol. Chem. 2012, 10, 782–790. [Google Scholar] [CrossRef]
- Porter, J.L.; Sabatini, S.; Manning, J.; Tavanti, M.; Galman, J.L.; Turner, N.J.; Flitsch, S.L. Cloning, expression and characterisation of P450-Hal1 (CYP116B62) from Halomonas sp. NCIMB 172: A self-sufficient P450 with high expression and diverse substrate scope. Enzym. Microb. Technol. 2018, 113, 1–8. [Google Scholar] [CrossRef]
- Ciaramella, A.; Minerdi, D.; Gilardi, G. Catalytically self-sufficient cytochromes P450 for green production of fine chemicals. Rend. Fis. Acc. Lincei 2017, 28 (Suppl. 1), S169–S181. [Google Scholar] [CrossRef]
- Li, R.-J.; Xu, J.-H.; Chen, Q.; Zhao, J.; Li, A.-T.; Yu, H.-L. Enhancing the catalytic performance of a CYP116B monooxygenase by transdomain combination mutagenesis. ChemCatChem 2018, 10, 2962–2968. [Google Scholar] [CrossRef]
- O’Reilly, E.; Corbett, M.; Hussain, S.; Kelly, P.P.; Richardson, D.; Flitsch, S.L.; Turner, N.J. Substrate promiscuity of cytochrome P450 RhF. Catal. Sci. Technol. 2013, 3, 1490–1492. [Google Scholar] [CrossRef]
- Uehara, S.; Kawano, M.; Murayama, N.; Uno, Y.; Utoh, M.; Inoue, T.; Sasaki, E.; Yamazaki, H. Oxidation of R- and S-omeprazole stereoselectively mediated by liver microsomal cytochrome P450 2C19 enzymes from cynomolgus monkeys and common marmosets. Biochem. Pharmacol. 2016, 120, 56–62. [Google Scholar] [CrossRef]
- Gao, P.; Li, A.; Lee, H.H.; Wang, D.I.C.; Li, Z. Enhancing enantioselectivity and productivity of P450-catalyzed asymmetric sulfoxidation with an aqueous/ionic liquid biphasic system. ACS Catal. 2014, 4, 3763–3771. [Google Scholar] [CrossRef]
- Abass, K.; Reponen, P.; Mattila, S.; Rautio, A.; Pelkonen, O. Human variation and CYP enzyme contribution in benfuracarb metabolism in human in vitro hepatic models. Toxicol. Lett. 2014, 224, 300–309. [Google Scholar] [CrossRef]
- Wójcikowski, J.; Daniel, W.A. Influence of antidepressant drugs on chlorpromazine metabolism in human liver—An in vitro study. Pharmacol. Rep. 2010, 62, 1062–1069. [Google Scholar] [CrossRef]
- Wójcikowski, J.; Basińska, A.; Daniel, W.A. The cytochrome P450-catalyzed metabolism of levomepromazine: A phenothiazine neuroleptic with a wide spectrum of clinical application. Biochem. Pharmacol. 2014, 90, 188–195. [Google Scholar] [CrossRef]
- Krämer, M.; Broecker, S.; Madea, B.; Hess, C. Confirmation of metabolites of the neuroleptic drug prothipendyl using human liver microsomes, specific CYP enzymes and authentic forensic samples—Benefit for routine drug testing. J. Pharm. Biomed. Anal. 2017, 145, 517–524. [Google Scholar] [CrossRef]
- Worsch, A.; Eggimann, F.K.; Girhard, M.; von Bühler, C.J.; Tieves, F.; Czaja, R.; Vogel, A.; Grumaz, C.; Sohn, K.; Lütz, S.; et al. A novel cytochrome P450 monooxygenase from Streptomyces platensis resembles activities of human drug metabolizing P450s. Biotechnol. Bioeng. 2018, 115, 2156–2166. [Google Scholar] [CrossRef]
- Rettie, A.E.; Lawton, M.P.; Sadeque, A.J.M.; Meier, G.P.; Philpot, R.M. Prochiral sulfoxidation as a probe for multiple forms of the microsomal flavin-containing monooxygenase: Studies with rabbit FMO1, FMO2, FMO3, and FMO5 expressed in Escherichia coli. Arch. Biochem. Biophys. 1994, 311, 369–377. [Google Scholar] [CrossRef]
- Catucci, G.; Gao, C.; Sadeghi, S.J.; Gilardi, G. Chemical applications of Class B flavoprotein monooxygenases. Rend. Fis. Acc. Lincei 2017, 28 (Suppl. 1), S195–S206. [Google Scholar] [CrossRef]
- Hamman, M.A.; Haehner-Daniels, B.D.; Wrighton, S.A.; Rettie, A.E.; Hall, S.D. Stereoselective sulfoxidation of sulindac sulfide by flavin-containing monooxygenases. Biochem. Pharmacol. 2000, 60, 7–17. [Google Scholar] [CrossRef]
- Virkel, G.; Lifschitz, J.; Sallovitz, J.; Pis, A.; Lanusse, C. Comparative hepatic and extrahepatic enantioselective sulfoxidation of albendazole and fenbendazole in sheep and cattle. Drug Metab. Dispos. 2004, 32, 536–544. [Google Scholar] [CrossRef]
- Furnes, B.; Schlenk, D. Evaluation of xenobiotic N- and S-oxidation by variant flavin-containing monooxygenase 1 (FMO1) enzymes. Toxicol. Sci. 2004, 78, 196–203. [Google Scholar] [CrossRef]
- Fiorentini, F.; Geier, M.; Binda, C.; Winkler, M.; Faber, K.; Hall, M.; Mattevi, A. Biocatalytic characterization of human FMO5: Unearthing Baeyer-Villiger reactions in humans ACS Chem. Biol. 2016, 11, 1039–1048. [Google Scholar] [CrossRef]
- Gul, T.; Krzek, M.; Permentier, H.P.; Fraaije, M.W.; Bischoff, R. Microbial flavoprotein monooxygenases as mimics of mammalian flavin-containing monooxygenases for the enantioselective preparation of drug metabolites. Drug Metab. Dispos. 2016, 44, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Mthethwa, K.S.; Kassier, K.; Engel, J.; Kara, S.; Smit, M.S.; Opperman, D.J. Fungal BVMOs as alternatives to cyclohexanone monooxygenase. Enzym. Microb. Technol. 2017, 106, 11–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, F.; Xu, N.; Wu, Y.-Q.; Zheng, Y.-C.; Zhao, Q.; Lin, G.; Yu, H.-L.; Xu, J.-H. The discovery of two native Baeyer-Villiger monooxygenases for asymmetric synthesis of bulky chiral sulfoxides. Appl. Environ. Microbiol. 2018, 84, e00638-18. [Google Scholar] [CrossRef]
- Bisagni, S.; Summers, B.; Kara, S.; Hatti-Kaul, R.; Grogan, G.; Mamo, G.; Hollmann, F. Exploring the substrate specificity and enantioselectivity of a Baeyer–Villiger monooxygenase from Dietzia sp. D5: Oxidation of sulfides and aldehydes. Top. Catal. 2014, 57, 366–375. [Google Scholar] [CrossRef]
- Kohl, A.; Srinivasamurthy, V.; Böttcher, D.; Kabisch, J.; Bornscheuer, U.T. Co-expression of an alcohol dehydrogenase and a cyclohexanone monooxygenase for cascade reactions facilitates the regeneration of the NADPH cofactor. Enzym. Microb. Technol. 2018, 108, 53–58. [Google Scholar] [CrossRef]
- Torres Pazmiño, D.E.; Dudek, H.M.; Fraaije, M.W. Baeyer-Villiger monooxygenases: Recent advances and future challenges. Curr. Opin. Chem. Biol. 2010, 14, 138–144. [Google Scholar] [CrossRef]
- Sheng, D.W.; Ballou, D.P.; Massey, V. Mechanistic studies of cyclohexanone monooxygenase: Chemical properties of intermediates involved in catalysis. Biochemistry 2001, 40, 11156–11167. [Google Scholar] [CrossRef]
- Donoghue, N.A.; Trudgill, P.W. The metabolism of cyclohexanol by Acinetobacter NCIB 9871. Eur. J. Biochem. 1975, 60, 1–7. [Google Scholar] [CrossRef]
- Colonna, S.; Gaggero, N.; Pasta, P.; Ottolina, G. Enantioselective oxidation of sulfides to sulfoxides catalysed by bacterial cyclohexanone monooxygenases. Chem. Commun. 1996, 20, 2303–2307. [Google Scholar] [CrossRef]
- Reetz, M.T.; Daligault, F.; Brunner, B.; Hinrichs, H.; Deege, A. Directed Evolution of Cyclohexanone Monooxygenases: Enantioselective Biocatalysts for the Oxidation of Prochiral Thioethers. Angew. Chem. 2004, 116, 4170–4173. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Rodriguez, C.; Rioz-Martinez, A.; Gotor, V. Improvement of the biocatalytic properties of ane phenylacetone monooxygenase mutant in hydrophilic organic solvents. Enzym. Microb. Technol. 2012, 50, 43–49. [Google Scholar] [CrossRef]
- Rodriguez, C.; de Gonzalo, G.; Gotor, V. Optimization of oxidative bioconversions catalyzed by phenylacetone monooxygenase from Thermobifida fusca. J. Mol. Catal. B Enzym. 2012, 74, 138–143. [Google Scholar] [CrossRef]
- Bisagni, S.; Abolhalaj, M.; de Brevern, A.G.; Rebehmed, J.; Hatti-Kaul, R.; Mamo, G. Enhancing the activity of a Dietzia sp. D5 Baeyer-Villiger monooxygenase towards cyclohexanone by saturation mutagenesis. ChemistrySelect 2017, 2, 7169–7177. [Google Scholar] [CrossRef]
- Zhang, Z.-G.; Lonsdale, R.; Sanchis, J.; Reetz, M.T. Extreme Synergistic Mutational Effects in the Directed Evolution of a Baeyer-Villiger Monooxygenase as Catalyst for Asymmetric Sulfoxidation. J. Am. Chem. Soc. 2014, 136, 17262–17272. [Google Scholar] [CrossRef]
- Rioz-Martínez, A.; de Gonzalo, G.; Torres Pazmiño, D.E.; Fraaije, M.W.; Gotor, V. Enzymatic synthesis of novel chiral sulfoxides employing Baeyer-Villiger monooxygenases. Eur. J. Org. Chem. 2010, 6409–6416. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Torres Pazmino, D.E.; Ottolina, G.; Fraaije, M.W.; Carrea, G. 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB as an oxidative biocatalyst in the synthesis of optically active sulfoxides. Tetrahedron Asymm. 2006, 17, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Tischler, D. Microbial Styrene Degradation; Springer International Publishing: Basel, Switzerland, 2015. [Google Scholar]
- Tischler, D.; Schwabe, R.; Siegel, L.; Joffroy, K.; Kaschabek, S.R.; Scholtissek, A.; Heine, T. VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An aryl alkyl sulfoxidase rather than a styrene epoxidizing monooxygenase. Molecules 2018, 23, 809. [Google Scholar] [CrossRef]
- Heine, T.; van Berkel, W.J.H.; Gassner, G.; van Pée, K.-H.; Tischler, D. Two-component FAD-dependent monooxygenases: Current knowledge and biotechnological opportunities. Biology 2018, 7, 42. [Google Scholar] [CrossRef]
- Pu, W.; Cui, C.; Guo, C.; Wu, Z.-L. Characterization of two styrene monooxygenases from marine microbes. Enzym. Microb. Technol. 2018, 112, 29–34. [Google Scholar] [CrossRef]
- Riedel, A.; Heine, T.; Westphal, A.H.; Conrad, C.; Rathsack, P.; van Berkel, W.J.H.; Tischler, D. Catalytic and hydrodynamic properties of styrene monooxygenases from Rhodococcus opacus 1CP are modulated by cofactor binding. AMB Express 2015, 5, 30. [Google Scholar] [CrossRef]
Scheme 1.
Sulfoxidation of sulfides catalyzed by Helminthosporium sp. and M. isabellina.

Scheme 2.
Sulfoxidation of alkyl aryl sulfides catalyzed by the white-rot Basidiomycetes.

Scheme 3.
Sulfoxidation of alkyl aryl and alkyl alkyl sulfides catalyzed by Aspergillus japonicus ICFC 744/11.
Scheme 3.
Sulfoxidation of alkyl aryl and alkyl alkyl sulfides catalyzed by Aspergillus japonicus ICFC 744/11.

Scheme 4.
Biotransformation of ketosulfides catalyzed by Helminthosporium sp. NRRL 4671 and M. isabellina ATCC 42613.
Scheme 4.
Biotransformation of ketosulfides catalyzed by Helminthosporium sp. NRRL 4671 and M. isabellina ATCC 42613.

Figure 1.
The structures of products obtained in the oxidation of sulfur amino acids.

Scheme 5.
Synthesis of chondrine.

Scheme 6.
Sulfoxidation of PTBI catalyzed by Cunninghamella sp.

Figure 2.
The structures of omeprazole and lansoprazole.

Scheme 7.
Sulfoxidation of aryl alkil sulfides catalyzed using Rhodococcus sp.

Scheme 8.
Sulfoxidation of DBT catalyzed using CPO from Caldariomyces fumago.

Scheme 9.
Sulfoxidation of 2-(diphenylmethylthio) acetamide catalyzed by CPO.

Figure 3.
The structure of benzo[b]thiophenes (B[b]T).

Figure 4.
The structure of Benfuracarb.

Figure 5.
The structures of Chlorpromazine, Levomepromazine, and Prothipendyl.

Figure 6.
The structure of thioridazine.

Figure 7.
The structures of Albendazole, Fenbandazole, and Fenthion.

Figure 8.
The structure of 3-(methylthio)aniline.


{kind=link}
{kind=link}
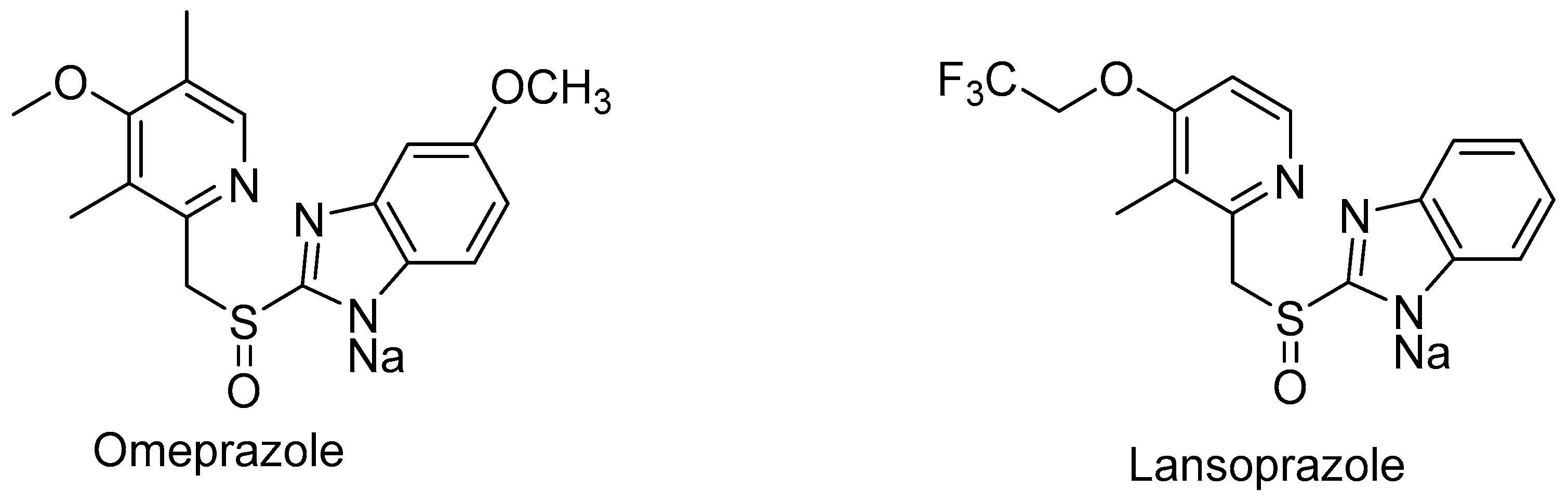{kind=link}
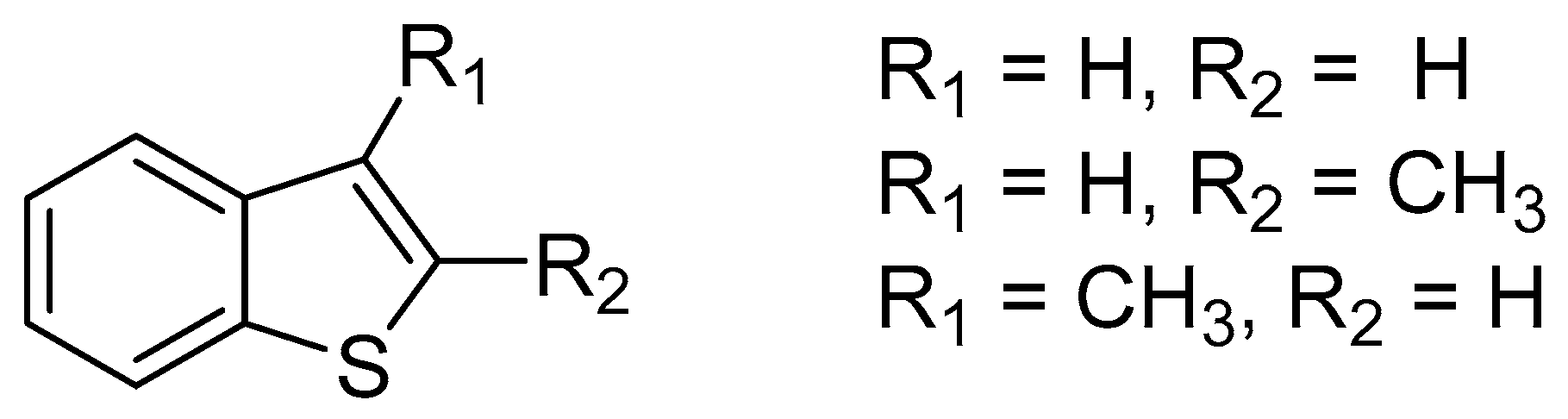{kind=link}
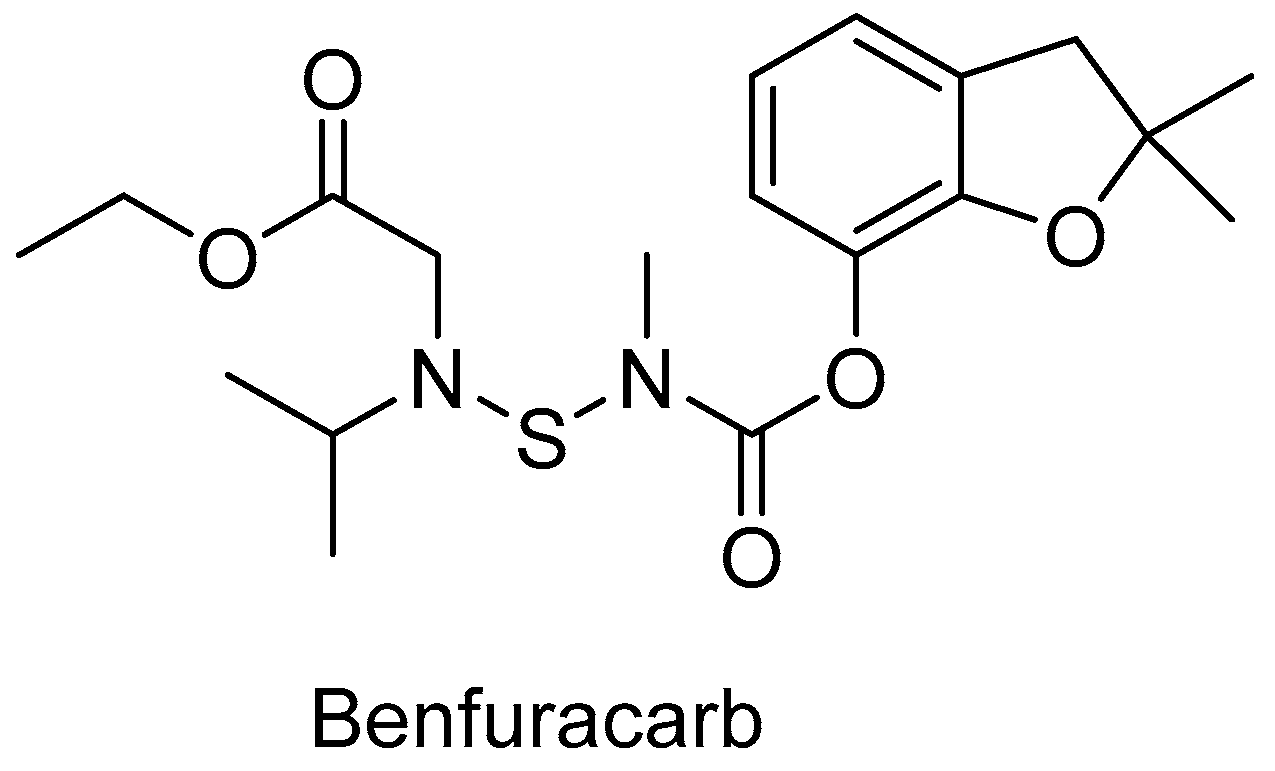{kind=link}
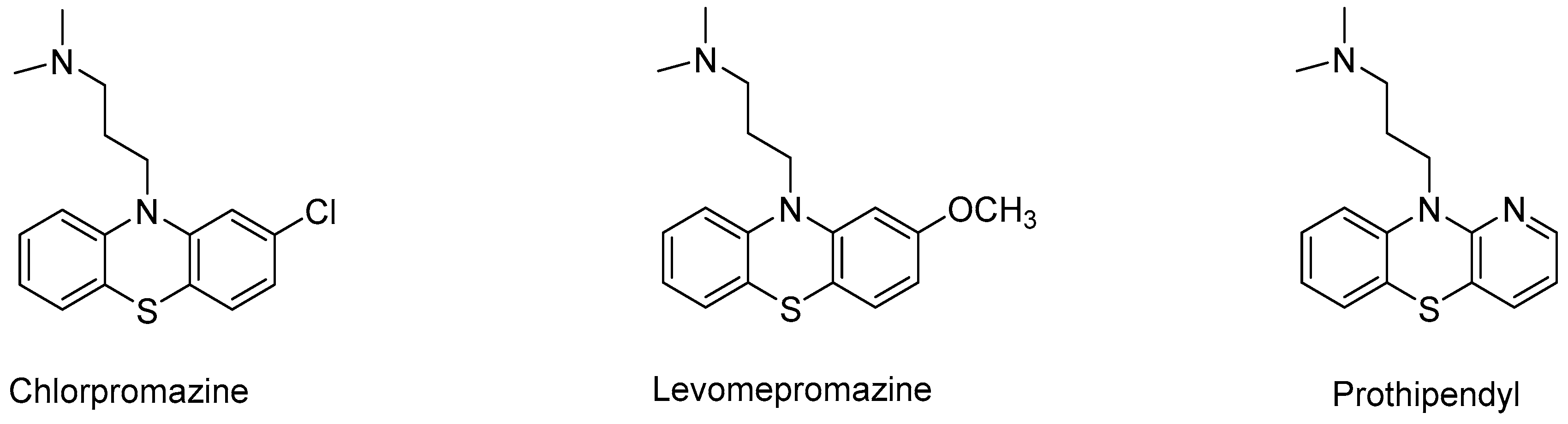{kind=link}
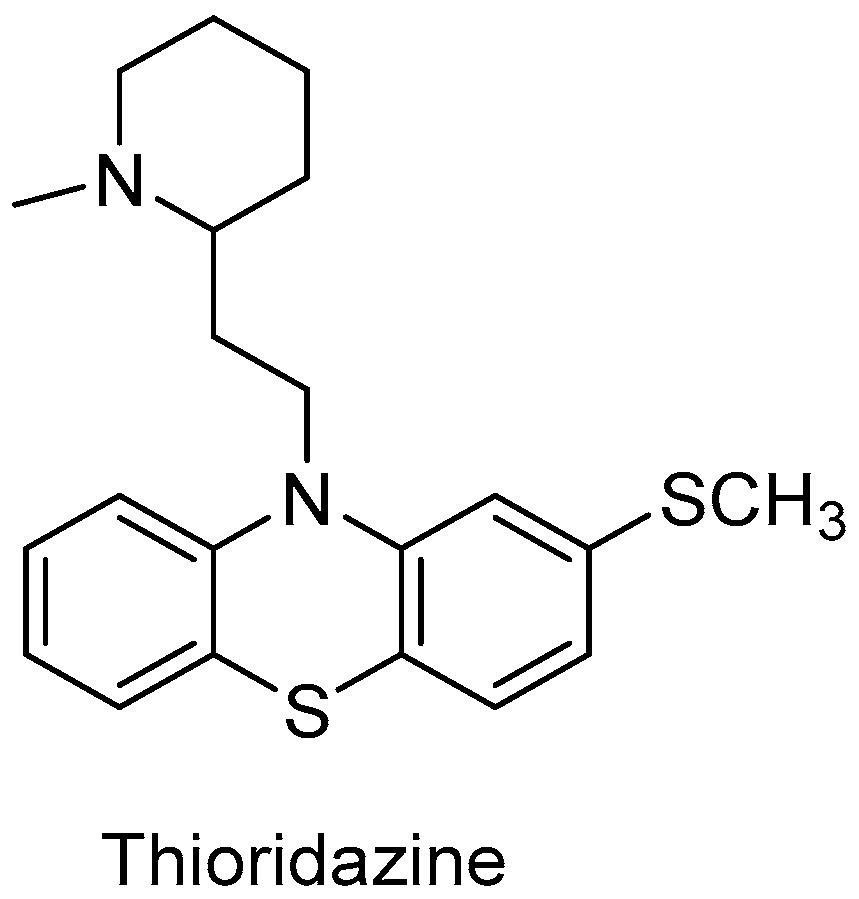{kind=link}
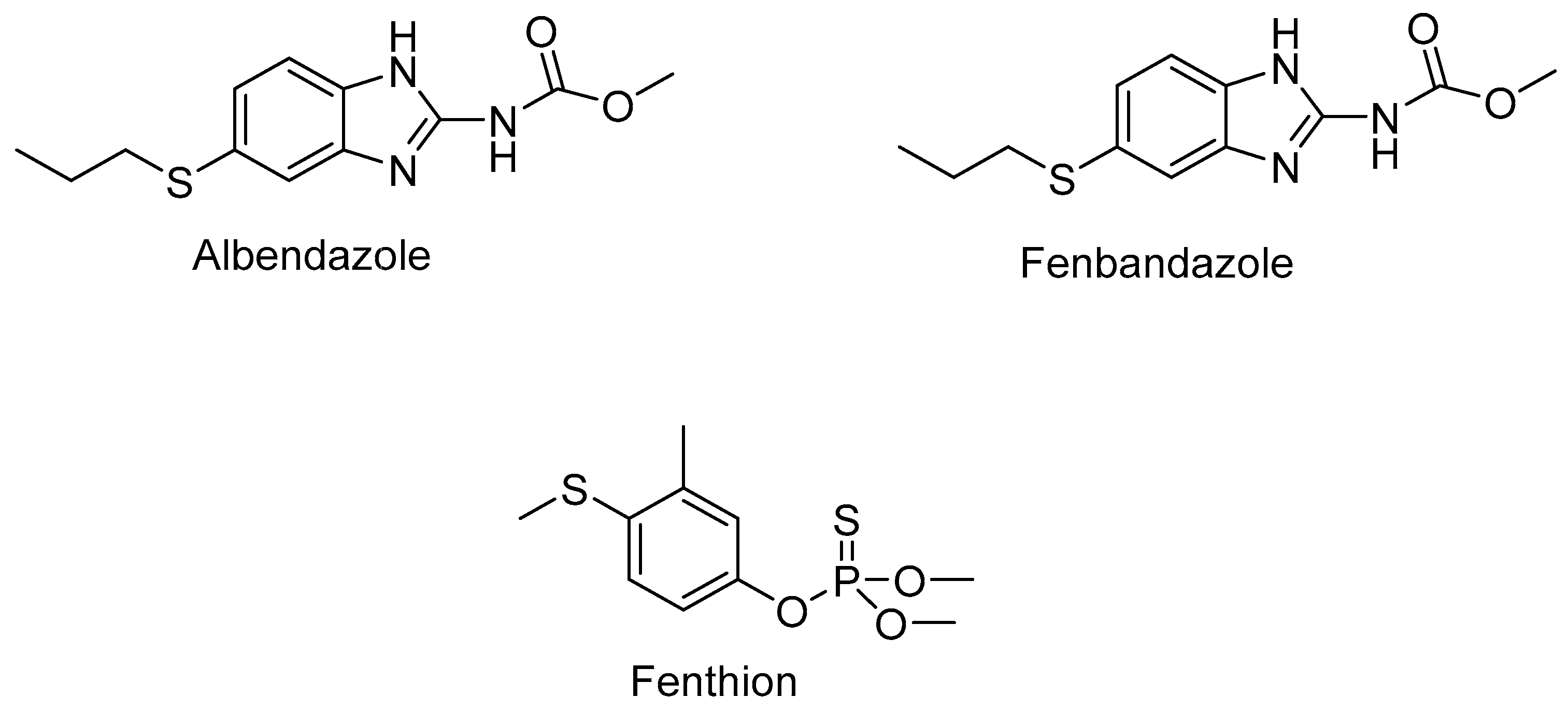{kind=link}
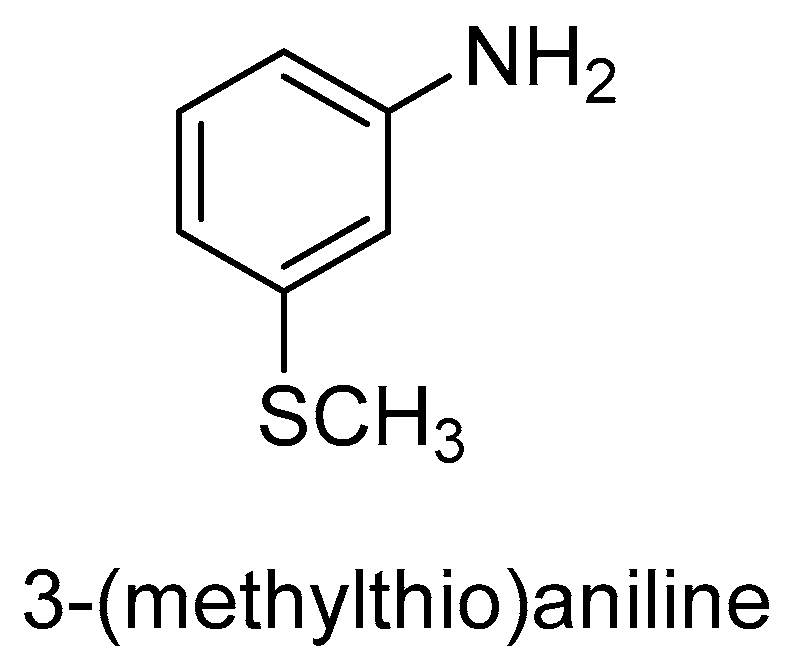{kind=link}
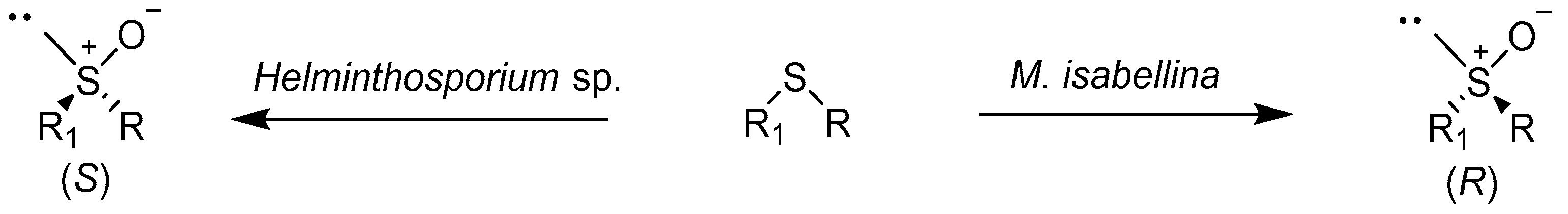{kind=link}
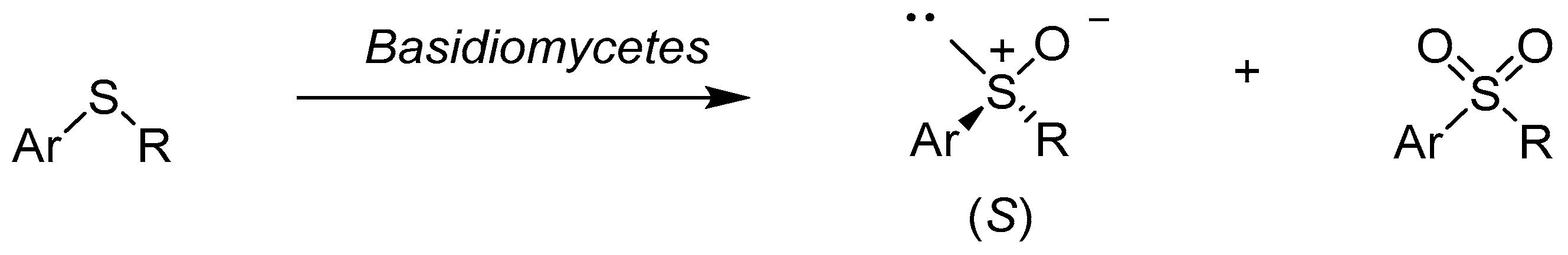{kind=link}
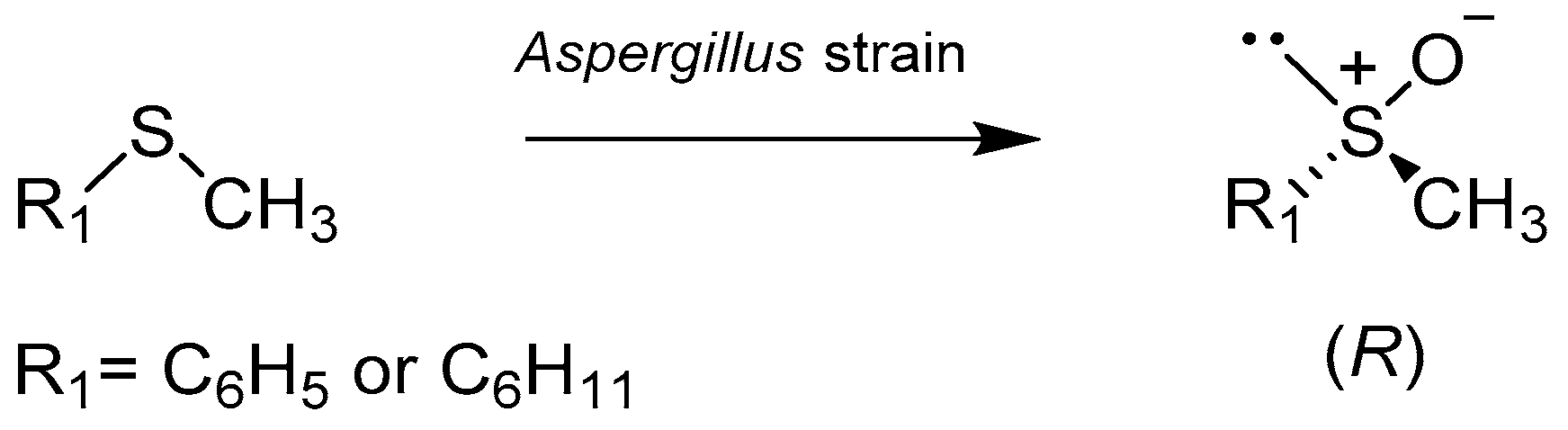{kind=link}
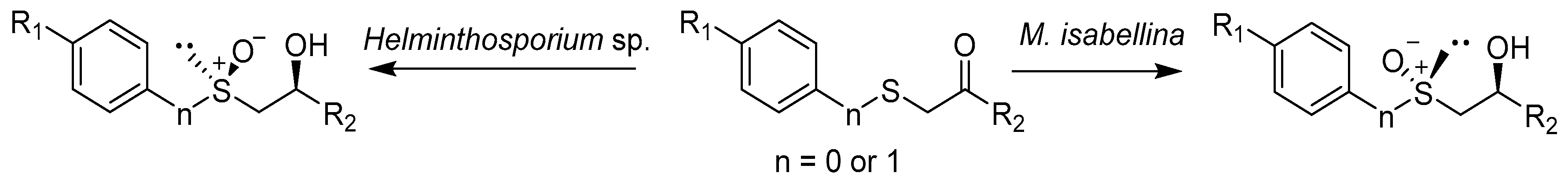{kind=link}
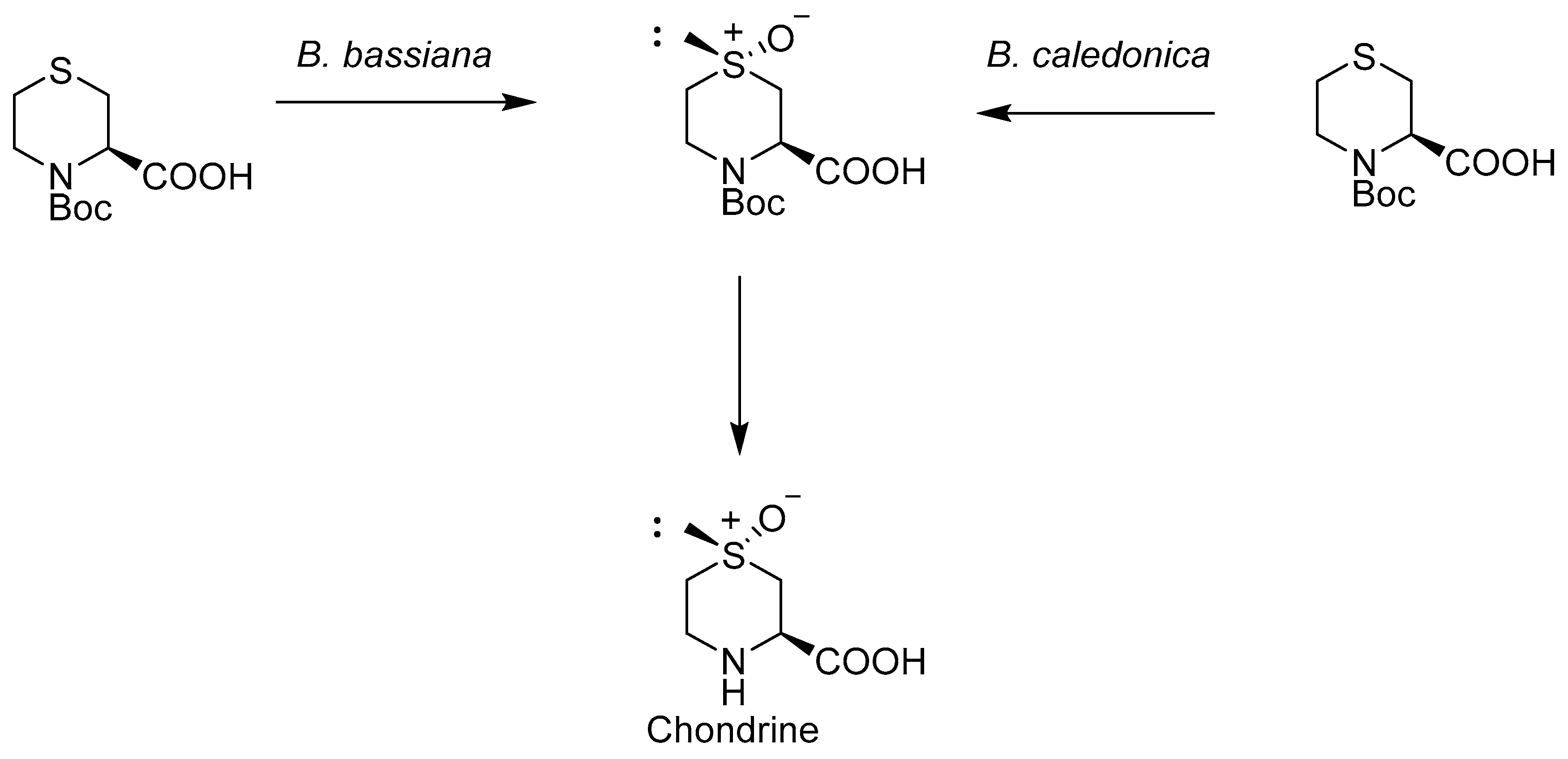{kind=link}
{kind=link}
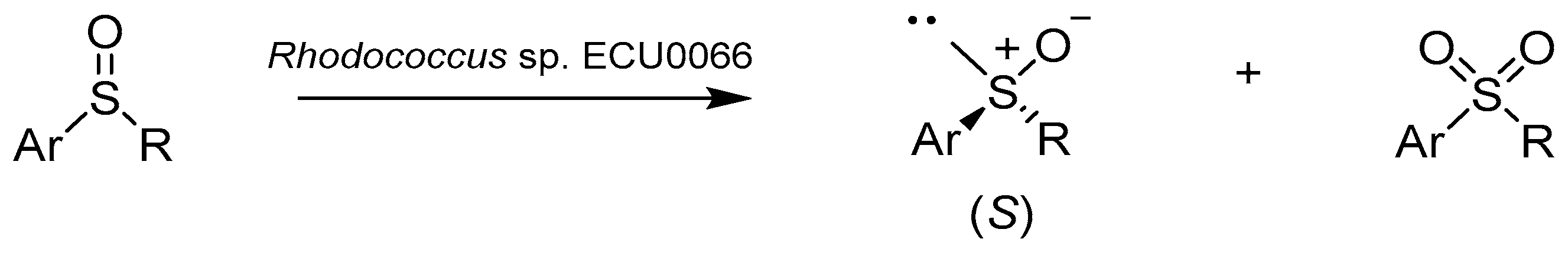{kind=link}
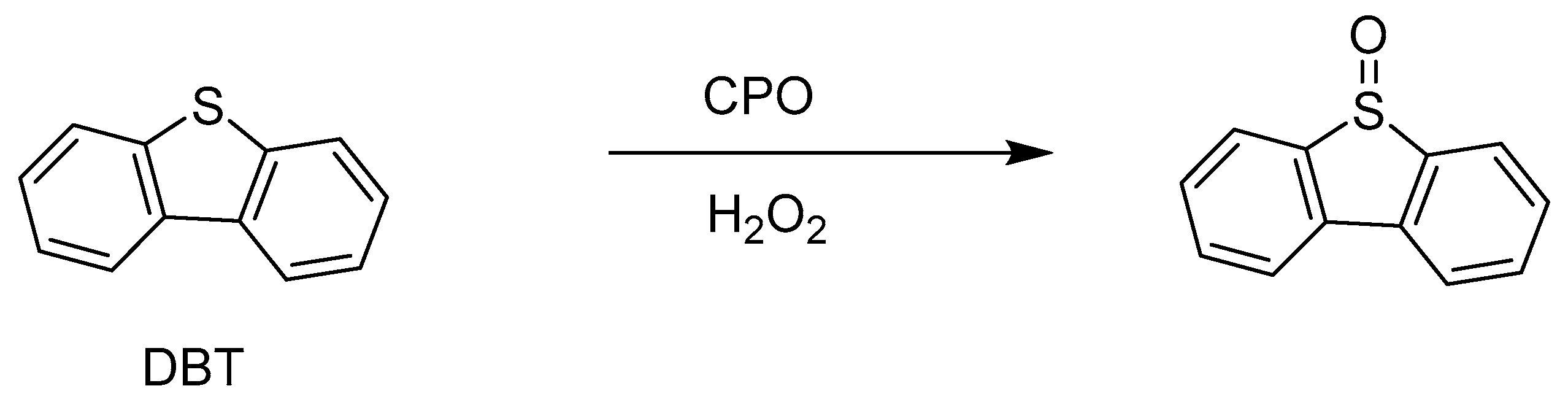{kind=link}
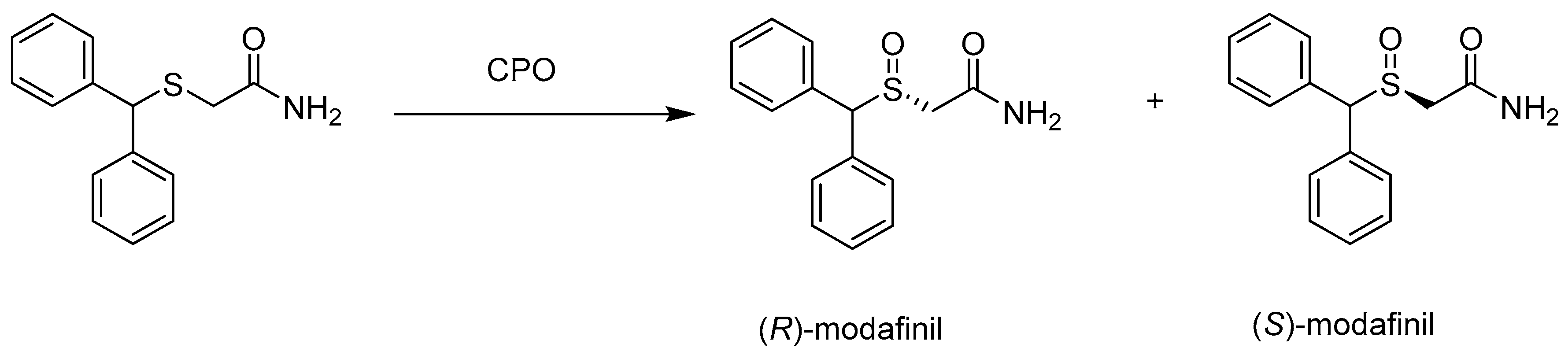{kind=link}
Table 1.
Biotransformation of the substituted sulfides catalyzed by Helminthosporium sp. and M. isabellina.
Table 1.
Biotransformation of the substituted sulfides catalyzed by Helminthosporium sp. and M. isabellina.
| Substrate | Biocatalyst | |||
|---|---|---|---|---|
| Helminthosporium sp. | M. isabellina | |||
| Yield | Stereoselectivity | Yield | Stereoselectivity | |
| R1 = H, R2 = CH3 | 90% | de 89% | 23% | de 46% |
| R1 = OCH3, R2 = CH3 | 70% | de 85% | 69% | de 52% |
| R1 = Br, R2 = CH3 | 30% | de 86% | 64% | de 50% |
| R1 = OCH3, R2 = Ph | 6% | de <5% | 48% | de 40% |
| R1 = H, R2 = CH3 | 66% | de 92% | 48% | de 40% |
| R1 = OCH3, R2 = CH3 | 72% | de 95% | 42% | de 85% |
| R1 = Br, R2 = CH3 | 72% | de 90% | 37% | de 40% |
| R1 = OCH3, R2 = Ph | 16% | de 26% | 69% | de 54% |
| R1 = Br, R2 =Ph | No products | 25% | de 28% | |
de—diastereoisomeric excess.
Table 2.
Sulfoxidation of the substituted methyl aryl sulfides catalyzed by R. erythropolis.
| Substrate | Yield (%) | Sulfoxide Configuration | ee (%) |
|---|---|---|---|
| C6H5SCH3 | 67 | rac | 2 |
| p-CH3C6H4SCH3 | 72 | R | 62 |
| m-CH3C6H4SCH3 | 60 | R | 10 |
| o-CH3C6H4SCH3 | 95 | rac | 4 |
| p-FC6H4SCH3 | 52 | R | 63 |
| p-ClC6H4SCH3 | 70 | R | 72 |
| p-BrC6H4SCH3 | 65 | R | 76 |
| p-NO2C6H4SCH3 | 83 | R | >98 |
| p-CNC6H4SCH3 | 43 | R | 85 |
Table 3.
Sulfoxidation of the substituted benzyl sulfides catalyzed using R. erythropolis.
| Substrate | Yield (%) | Sulfoxide Configuration | ee (%) |
|---|---|---|---|
| C6H5CH2SCH3 | 73 | R | 26 |
| C6H5CH2SC3H7 | 58 | R | 66 |
| C6H5C2H4SCH3 | 73 | R | 14 |
| C6H5C3H6SCH3 | 81 | rac | 3 |
| C6H5CH2SC6H5 | 65 | R | >98 |
| C6H5CH2SC3H6 C6H5 | 54 | R | 58 |
| C6H5CH2SC2H4 C6H5 | 50 | R | 78 |
| p-CH3OC6H4CH2SCH3 | 51 | R | 27 |
| m-CH3OC6H4CH2SCH3 | 54 | rac | 4 |
| o-CH3OC6H4CH2SCH3 | 82 | R | 44 |
| p-NO2C6H4CH2SCH3 | 58 | R | 76 |
| m-NO2C6H4CH2SCH3 | 15 | R | 52 |
| o-NO2C6H4CH2SCH3 | 41 | R | 70 |
| p-FC6H4CH2SCH3 | 61 | R | 62 |
| p-ClC6H4CH2SCH3 | 61 | R | 65 |
| p-BrC6H4CH2SCH3 | 60 | R | 76 |
| p-CNC6H4CH2SCH3 | 47 | R | 72 |
| p-CH3 C6H4CH2S C6H5 | 62 | R | 82 |
| p-CH3CONHC6H4CH2SCH3 | 63 | R | 39 |
| C6H5SCH2CN | 50 | S | 13 |
| p-CH3OC6H4SCH2CN | 91 | R | 88 |
| p-BrC6H4SCH2CN | 92 | R | 94 |
Table 4.
Sulfoxidation of amino acids catalyzed using R. erythropolis.
| Substrate | Yield (%) | Sulfoxide Configuration | de (%) |
|---|---|---|---|
| Methyl ester of N-MOC-S-methyl-L-Cys | 12 | R | 34 |
| Propyl ester N-MOC-S-methyl-L-Cys | 22 | R | 52 |
| Methyl ester of N-MOC-L-Met | 54 | R | 83 |
| Propyl ester of N-MOC-L-Met | 56 | R | 90 |
| Pentyl ester of N-MOC-L-Met | 59 | R | 93 |
| Heptyl ester of N-MOC-L-Met | 35 | R | >95 |
| Methyl ester of N-MOC-D-Met | 53 | R | >95 |
| Ethyl ester of N-t-Boc-D-Met | 69 | R | >95 |
| Pentyl ester of N-t-Boc-D-Met | 17 | R | >95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mączka, W.; Wińska, K.; Grabarczyk, M. Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow. Catalysts 2018, 8, 624. https://doi.org/10.3390/catal8120624
AMA Style
Mączka W, Wińska K, Grabarczyk M. Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow. Catalysts. 2018; 8(12):624. https://doi.org/10.3390/catal8120624
Chicago/Turabian StyleMączka, Wanda, Katarzyna Wińska, and Małgorzata Grabarczyk. 2018. "Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow" Catalysts 8, no. 12: 624. https://doi.org/10.3390/catal8120624
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.