Effect of Ce Doping of a Co/Al2O3 Catalyst on Hydrogen Production via Propane Steam Reforming


Abstract
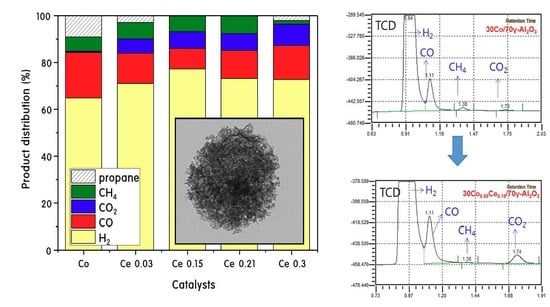
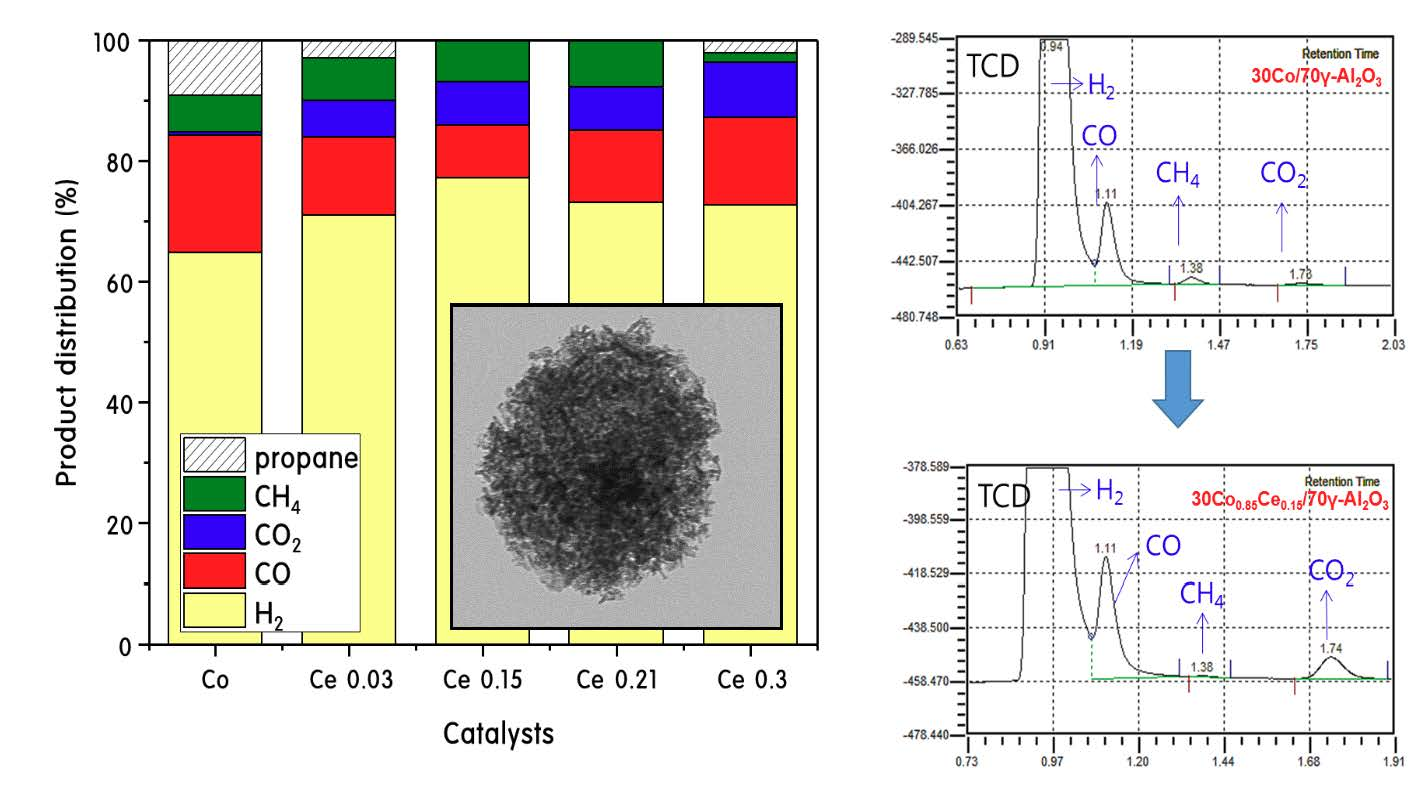
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
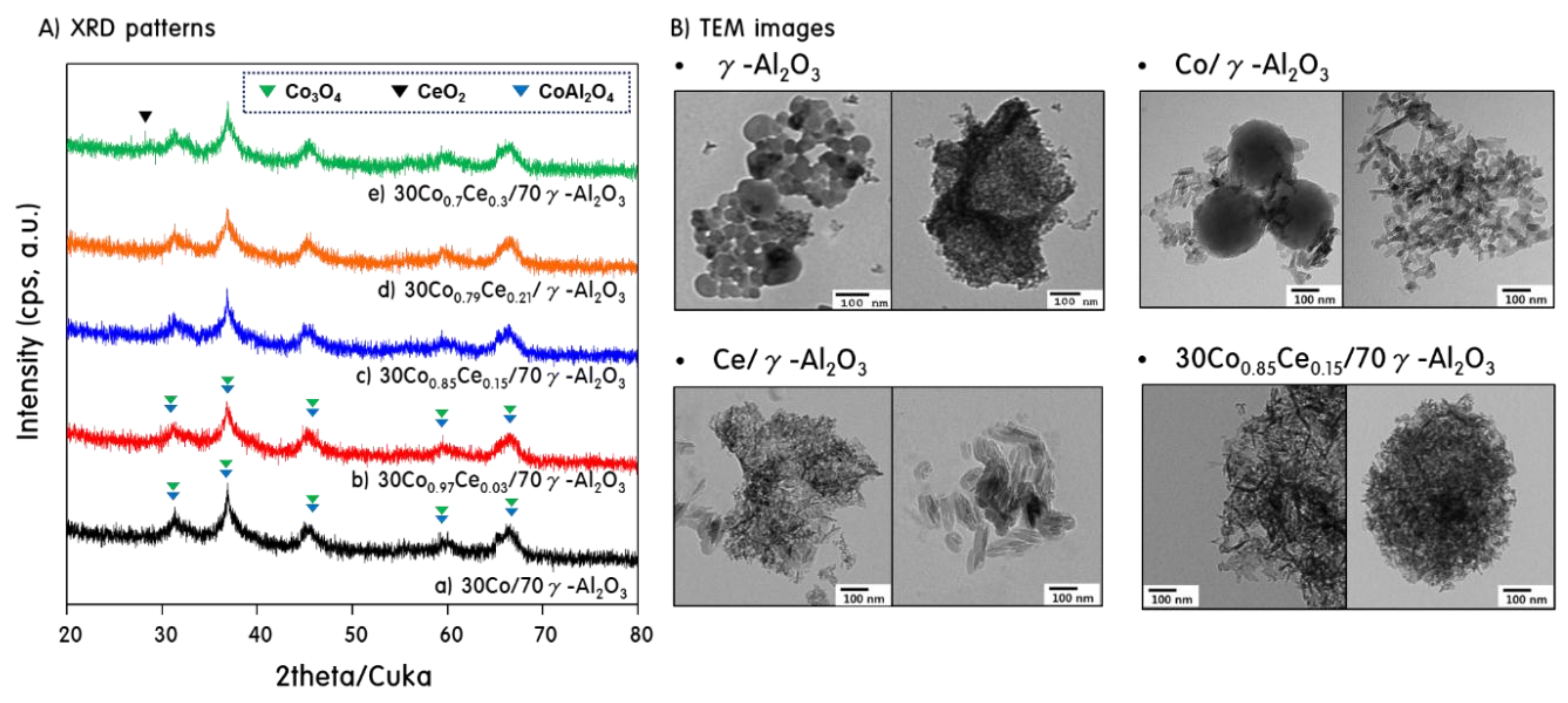
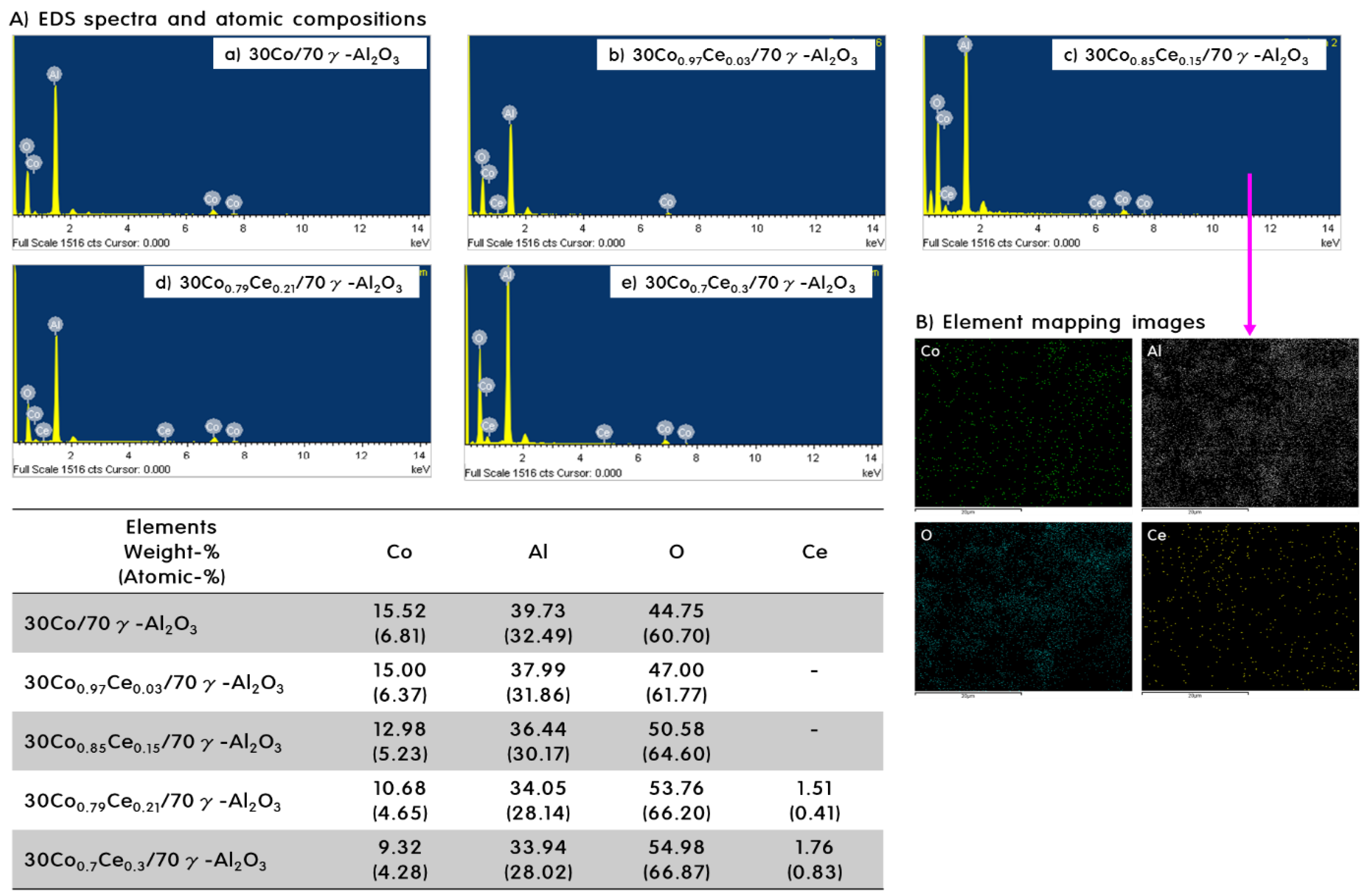
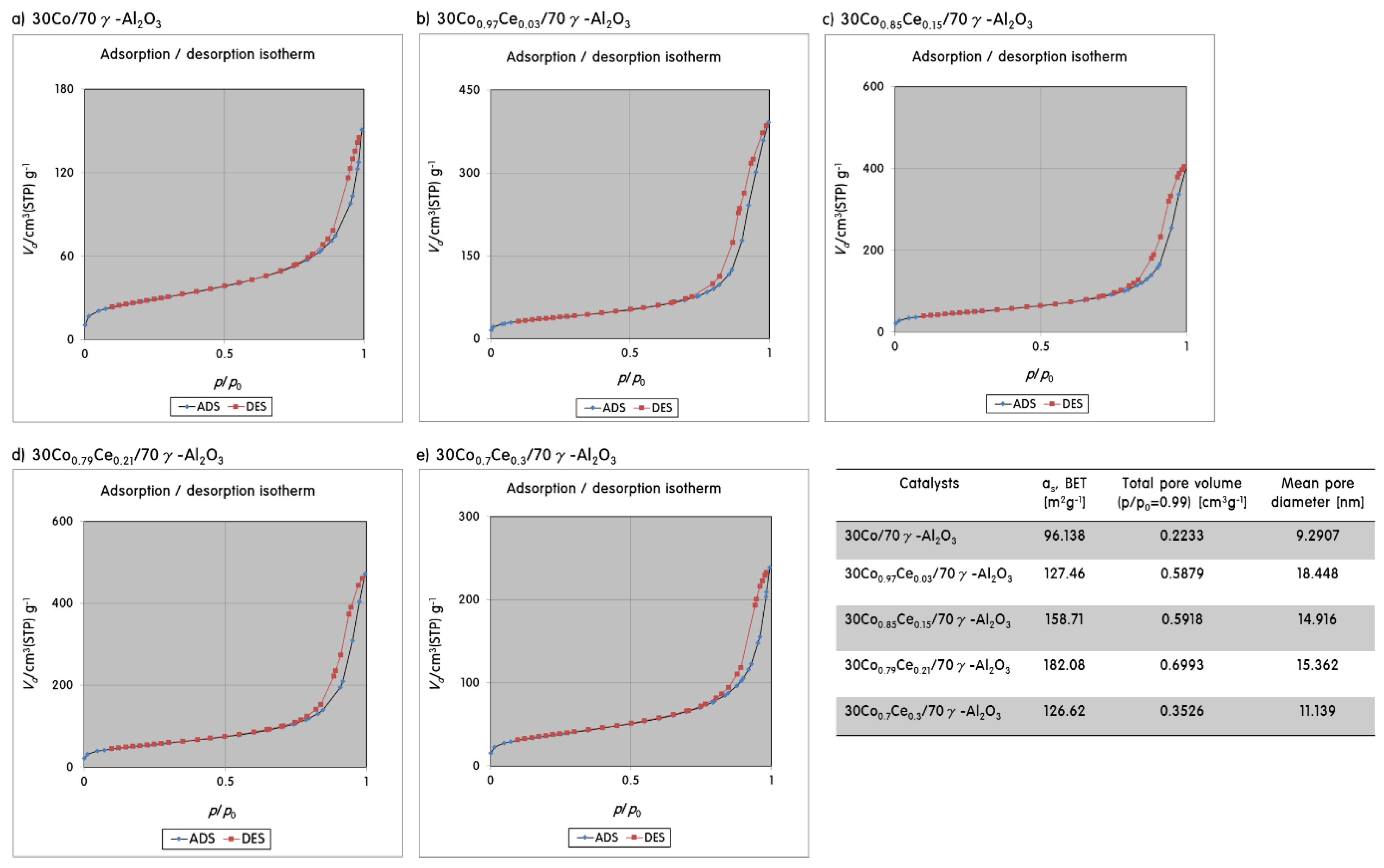
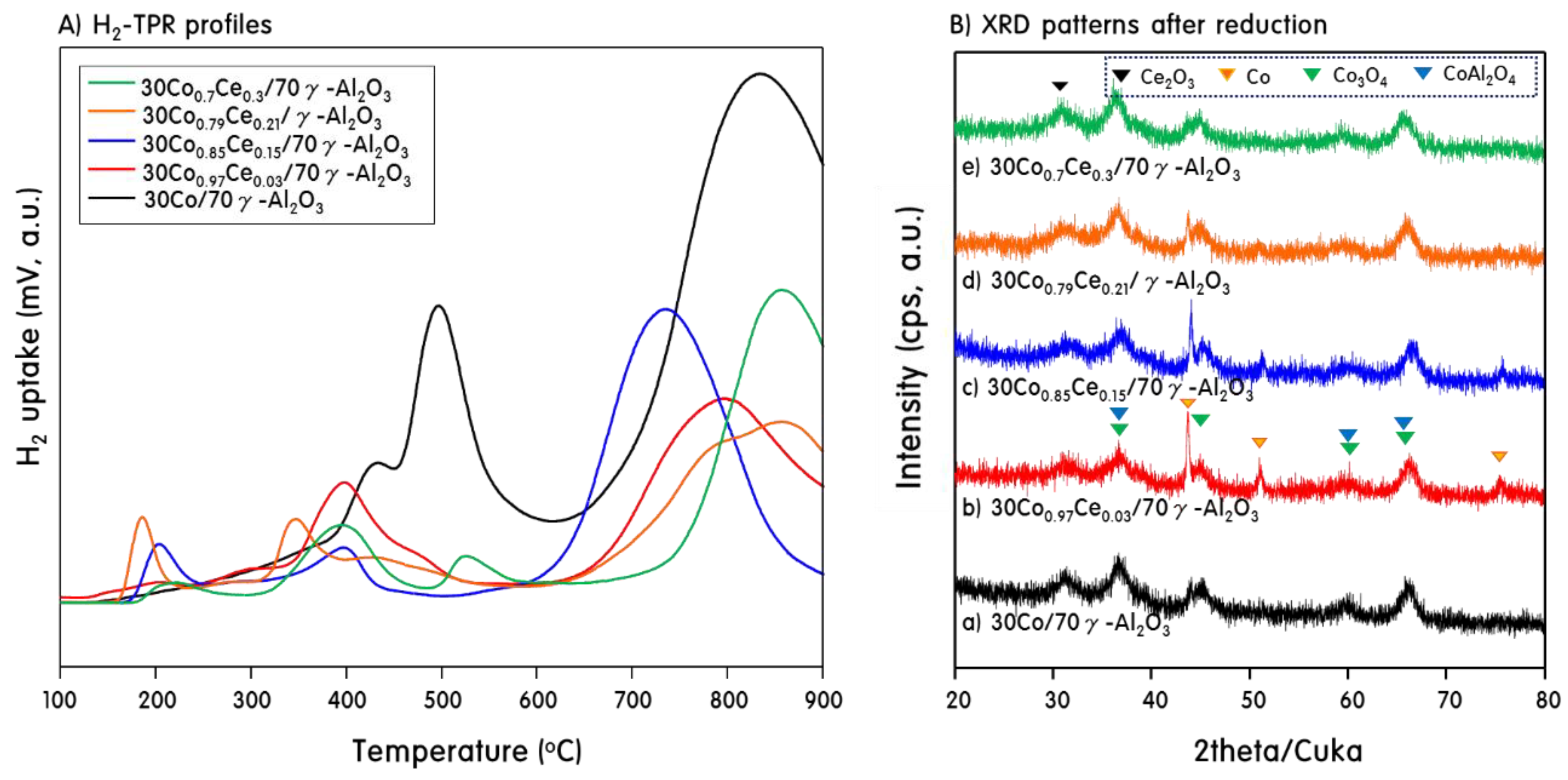
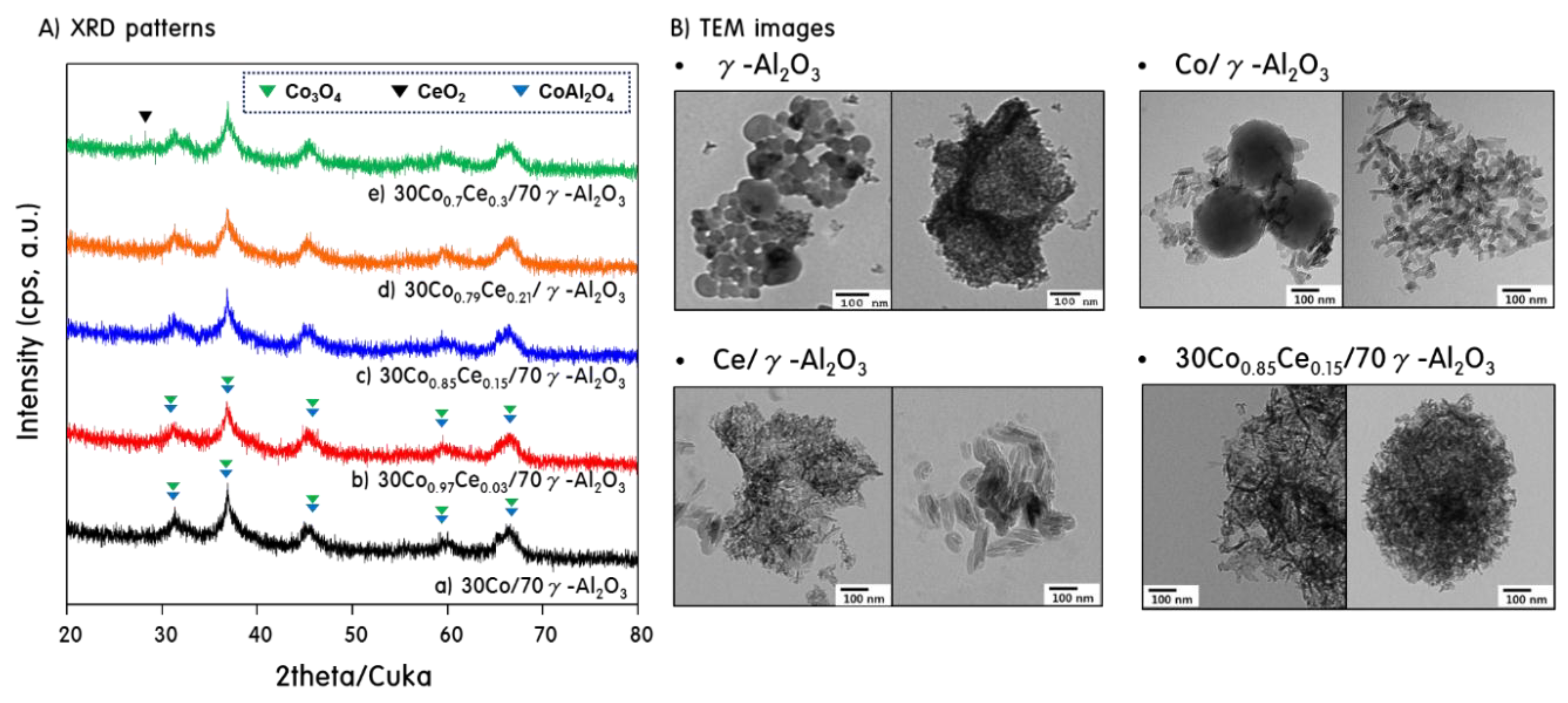
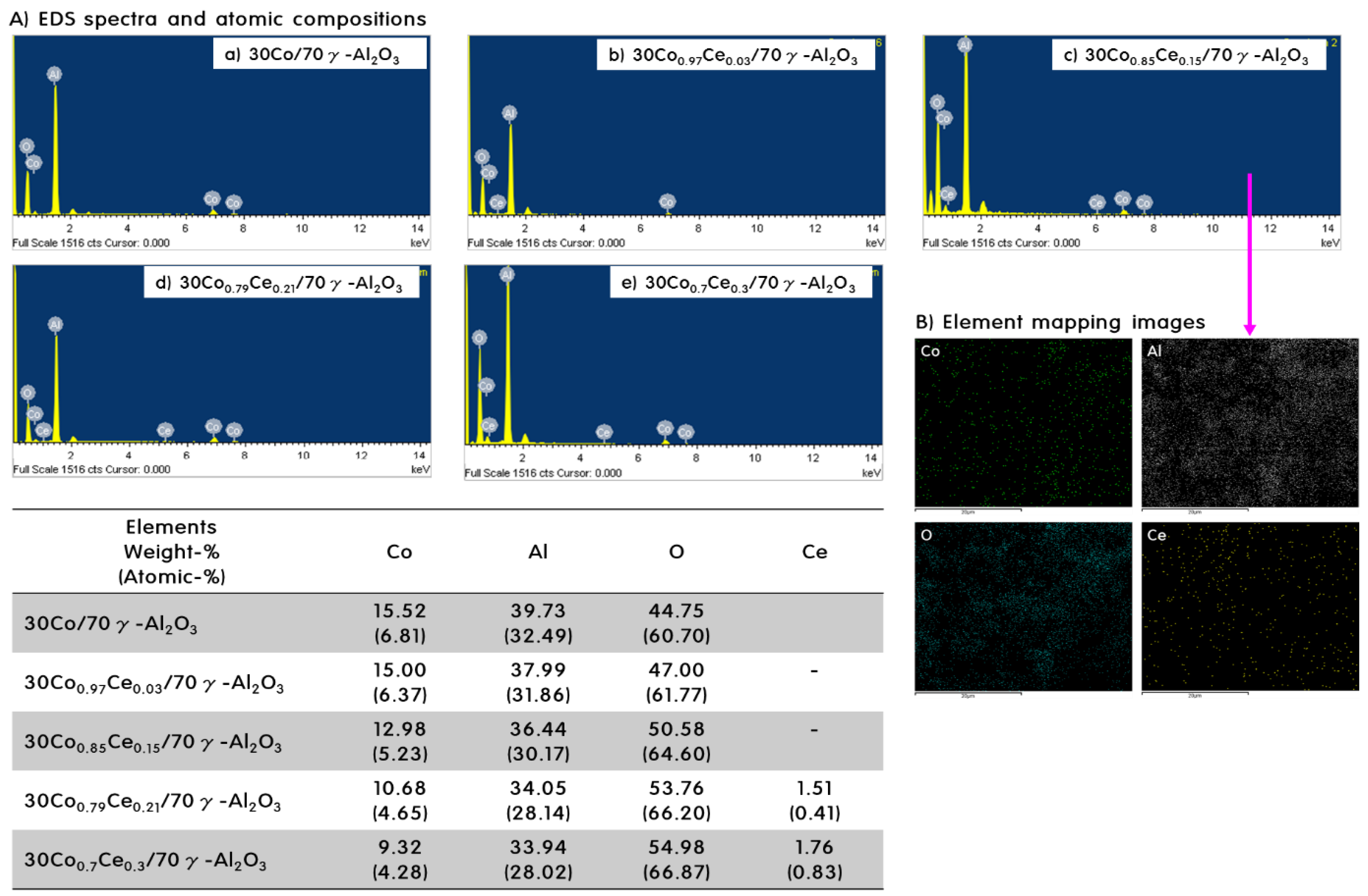
2.1. Characteristics of 30CoxCey/70γ-Al2O3 Catalysts
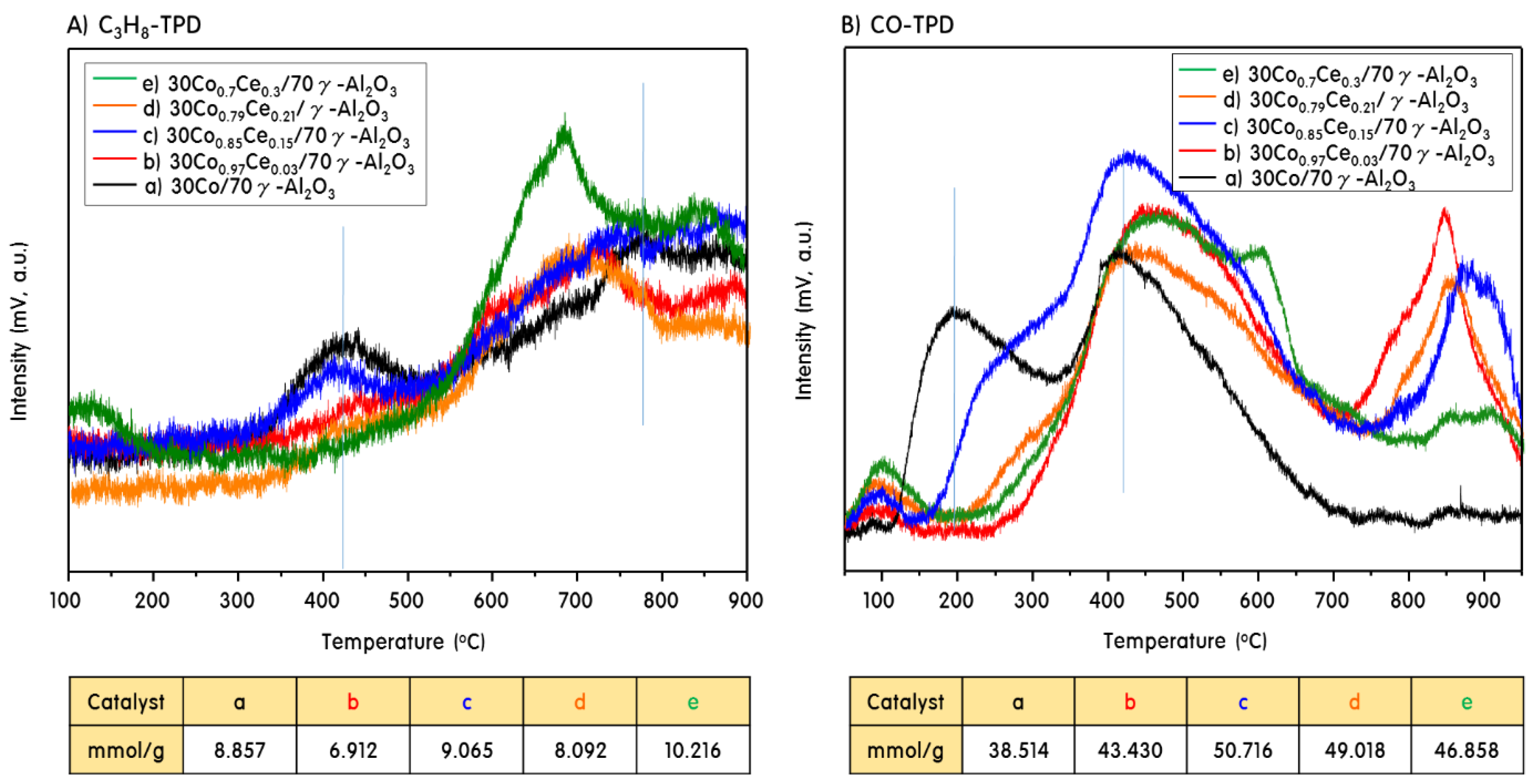
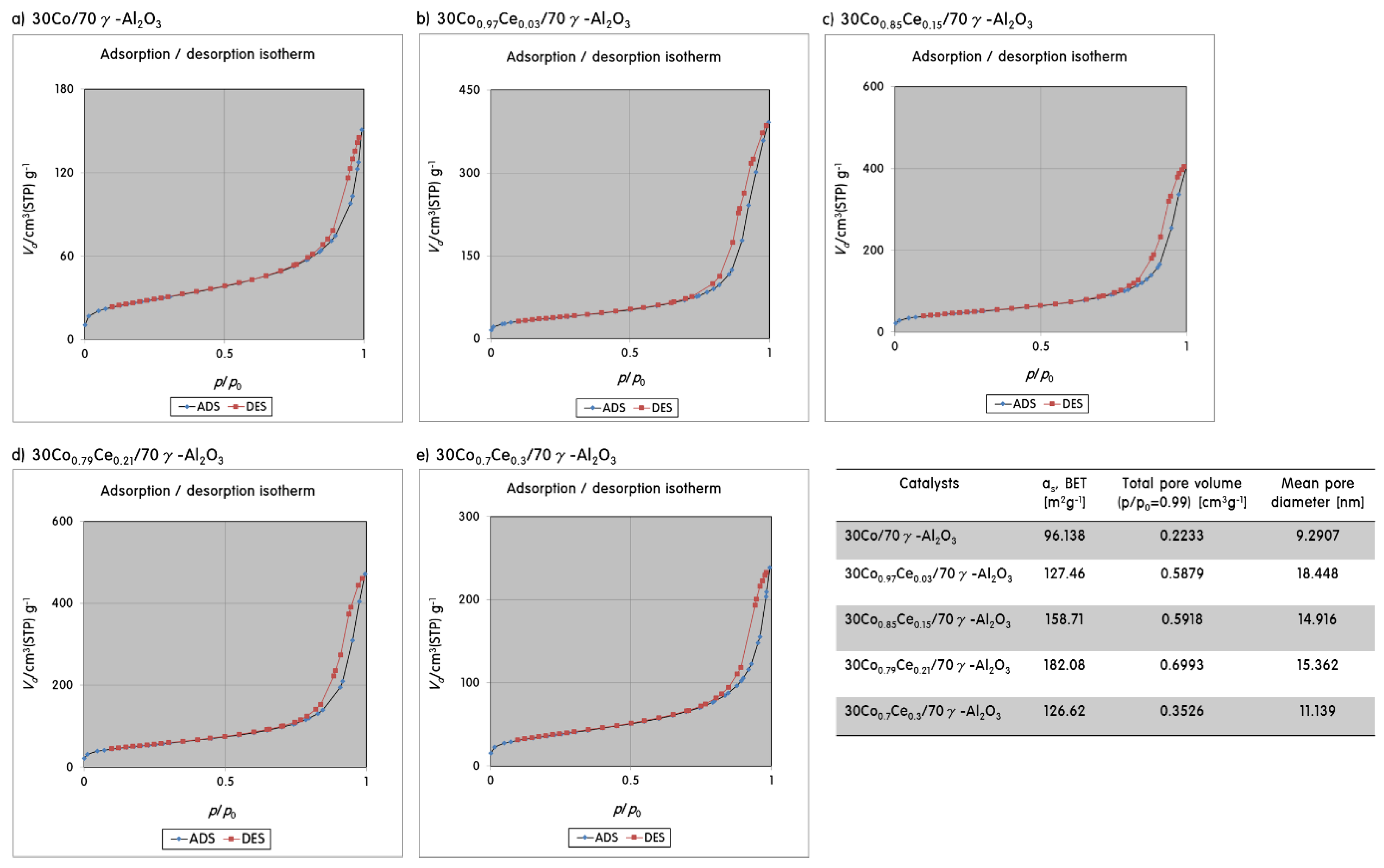
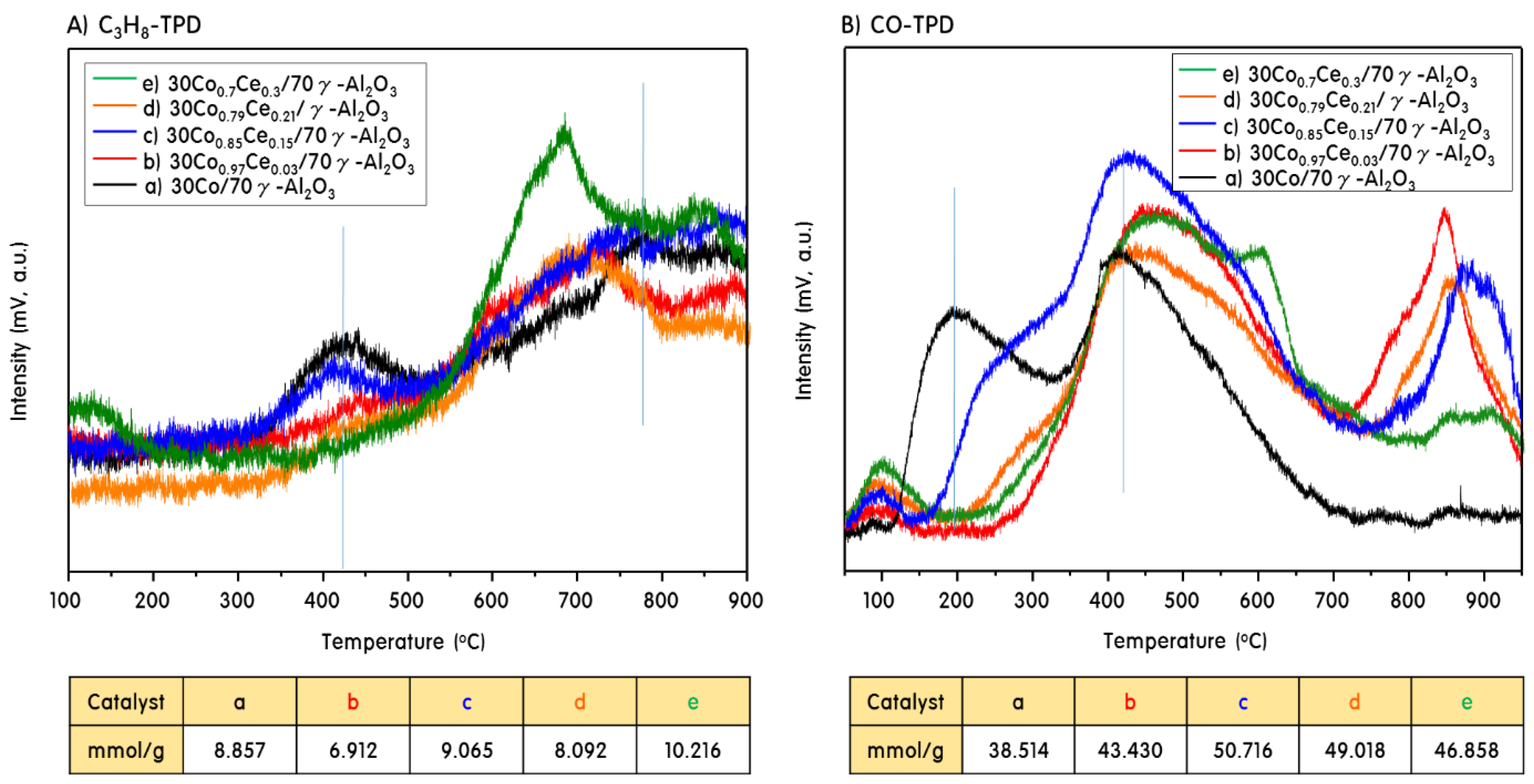
2.2. Gas Adsorption Ability of 30CoxCey/70γ-Al2O3 Catalysts
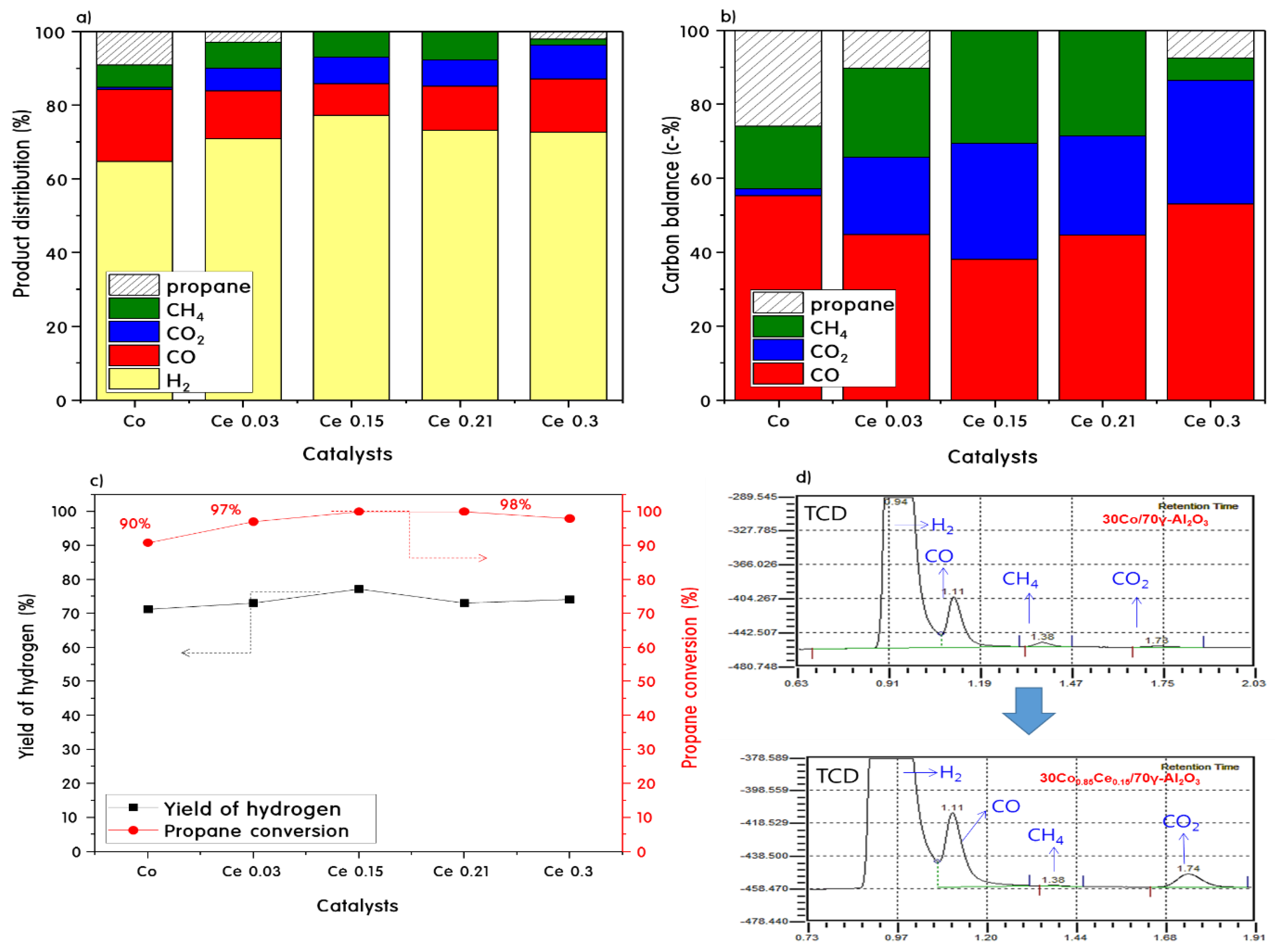
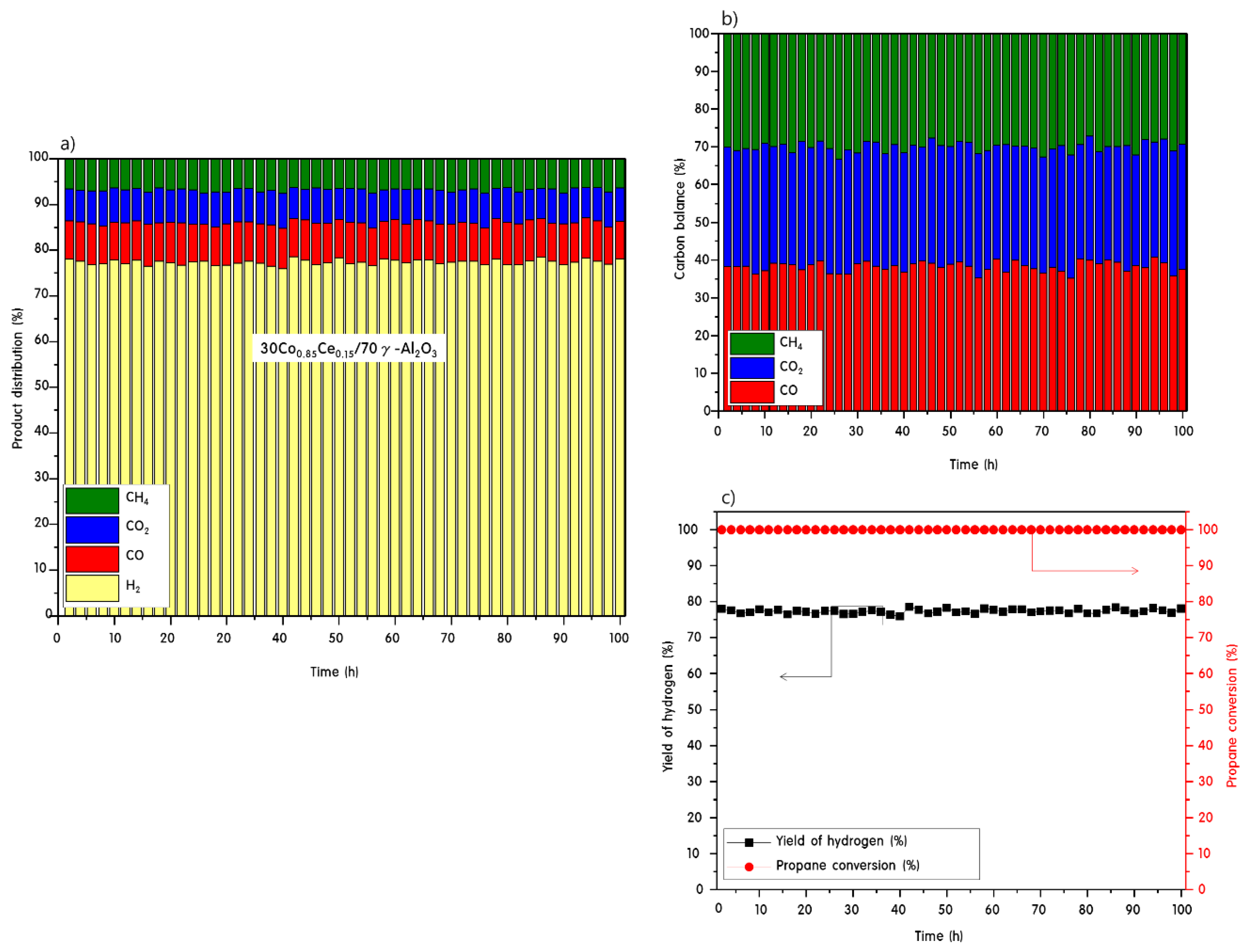
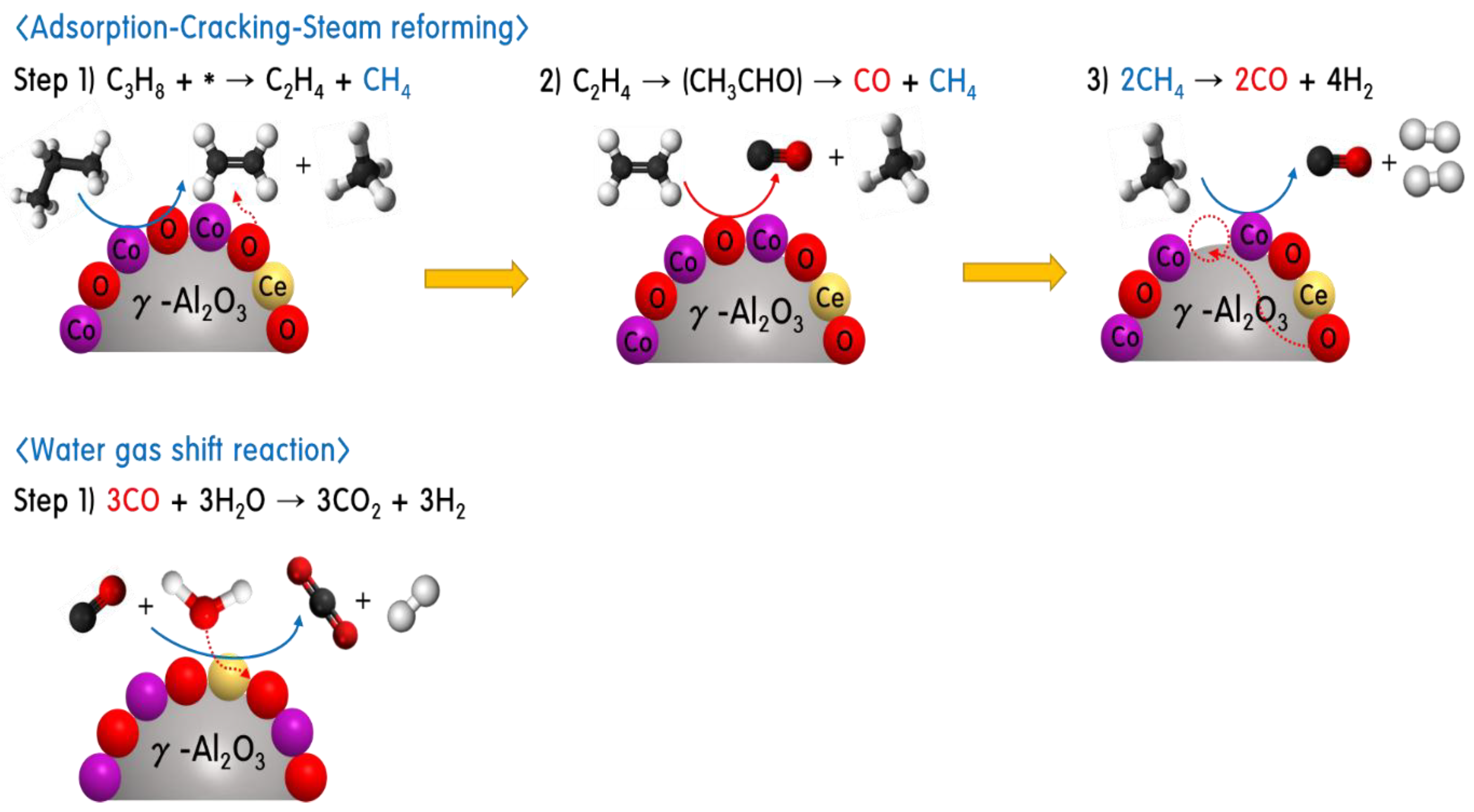
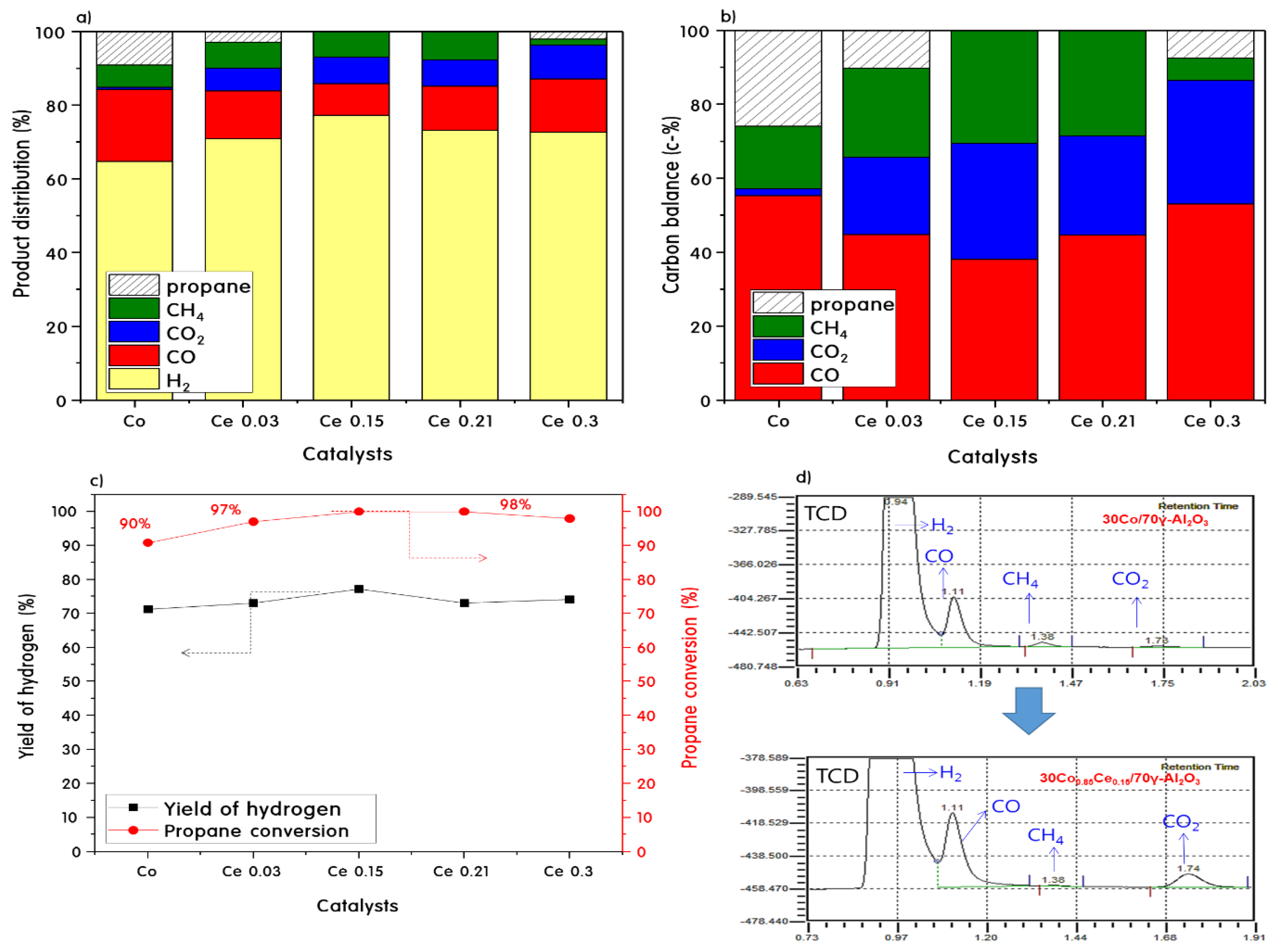
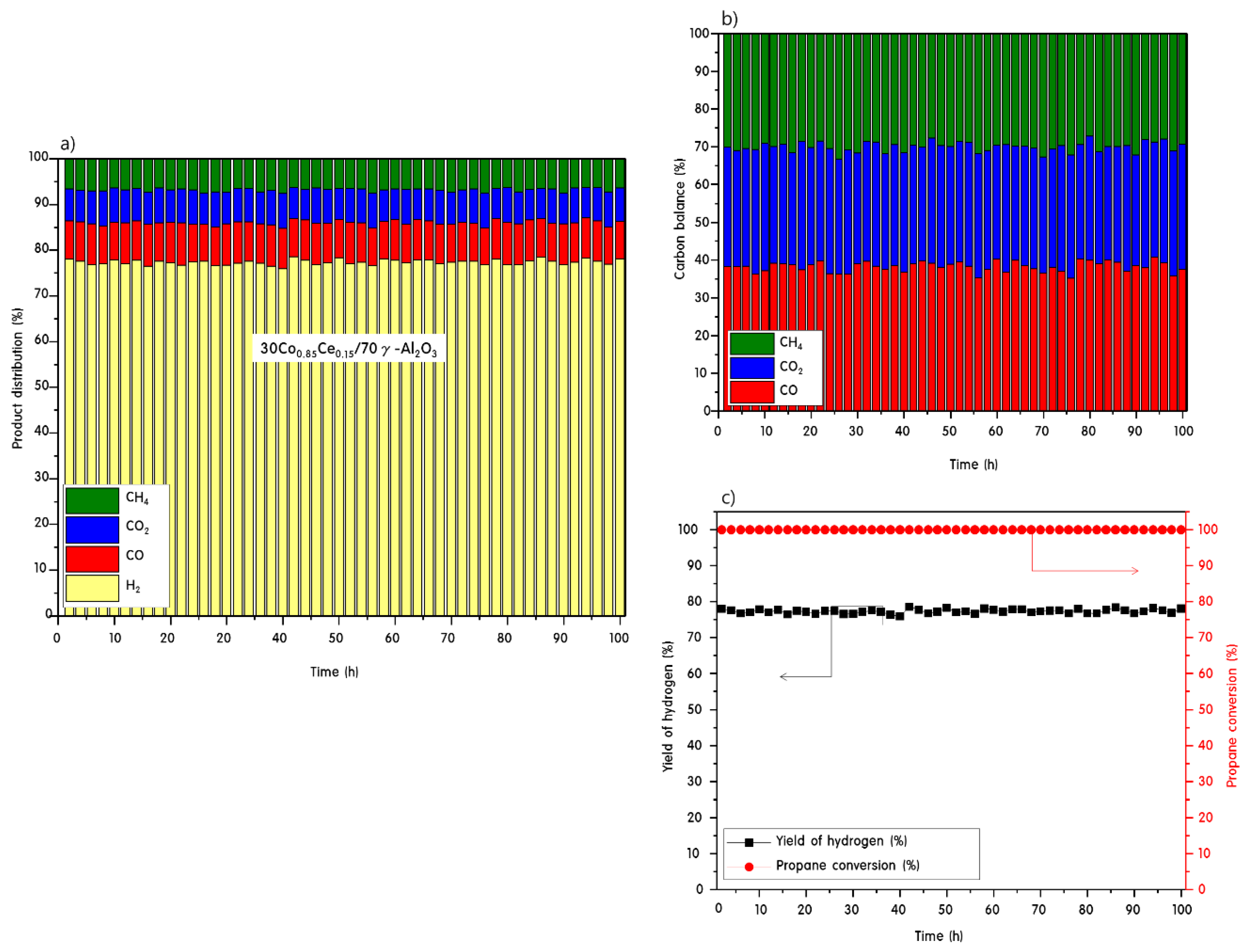
2.3. Propane Steam Reforming Reaction over 30CoxCey/70γ-Al2O3 Catalysts
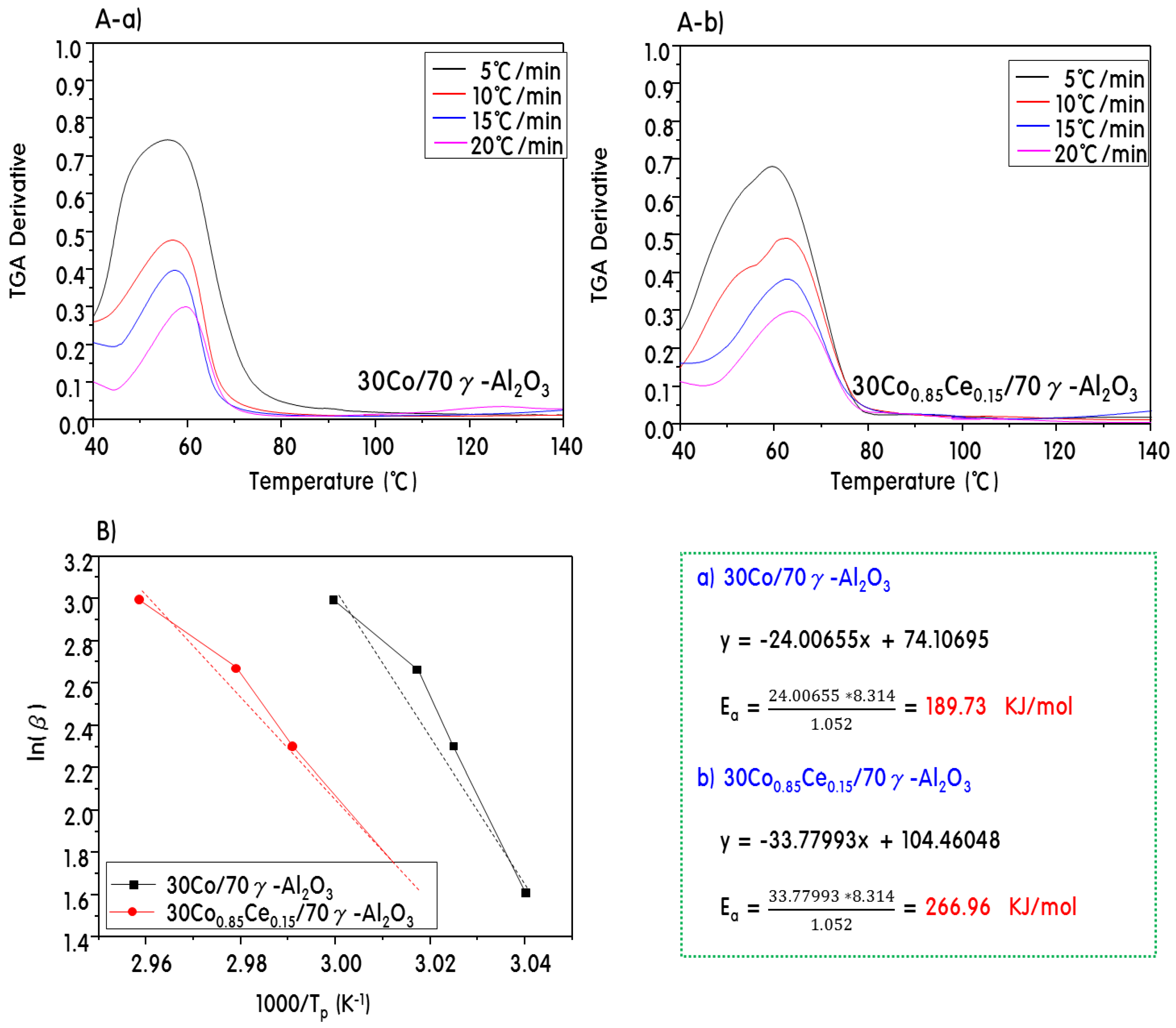
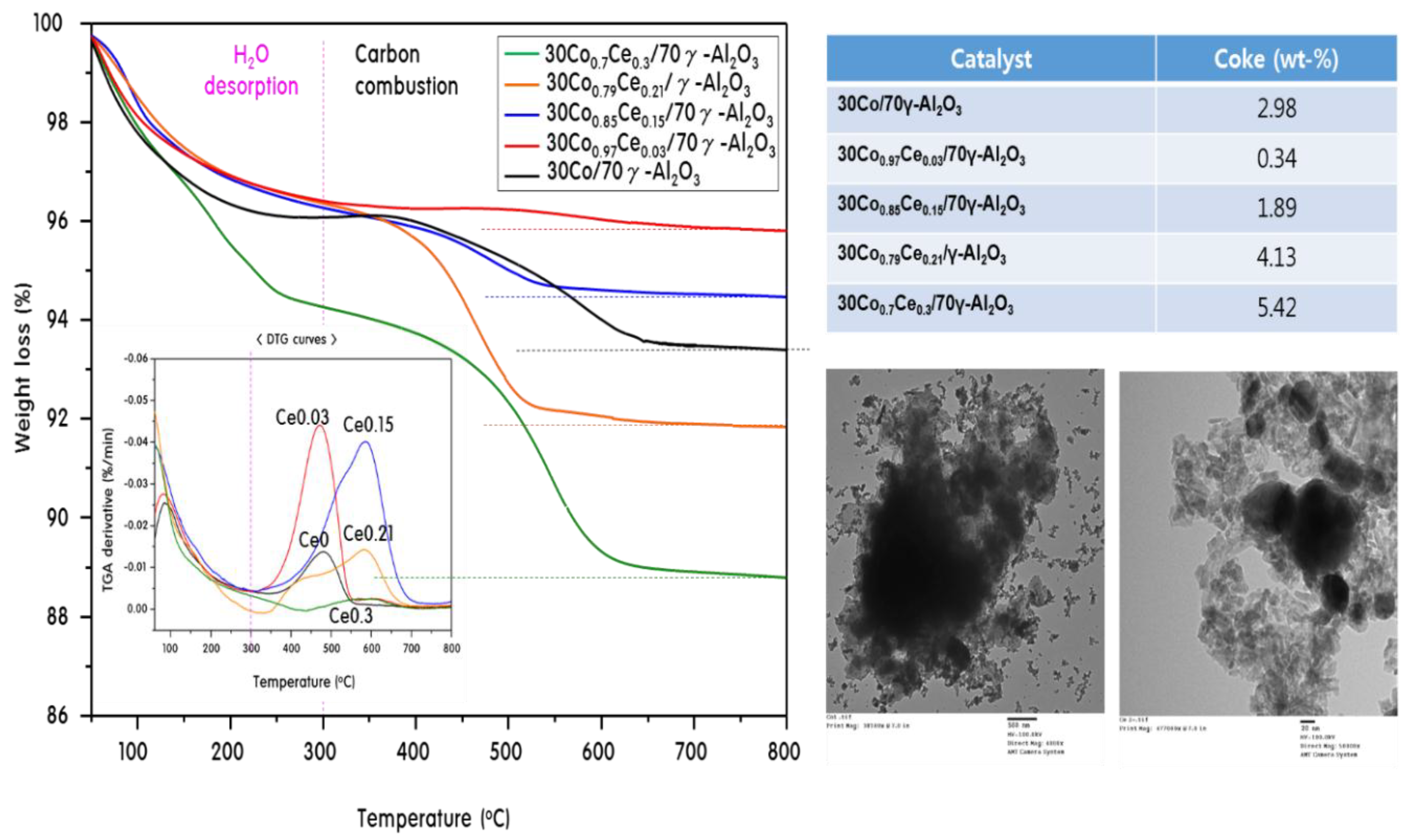
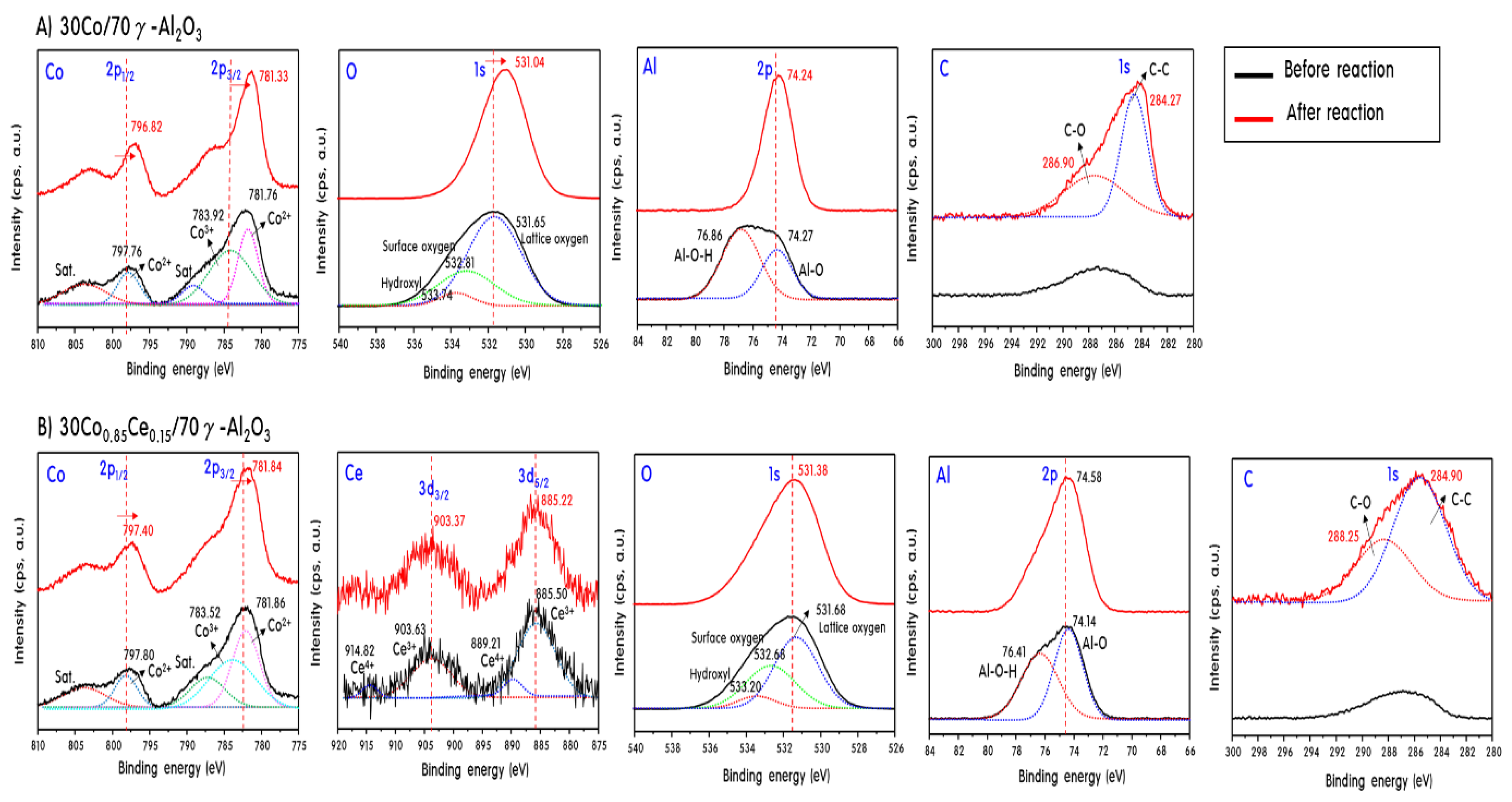
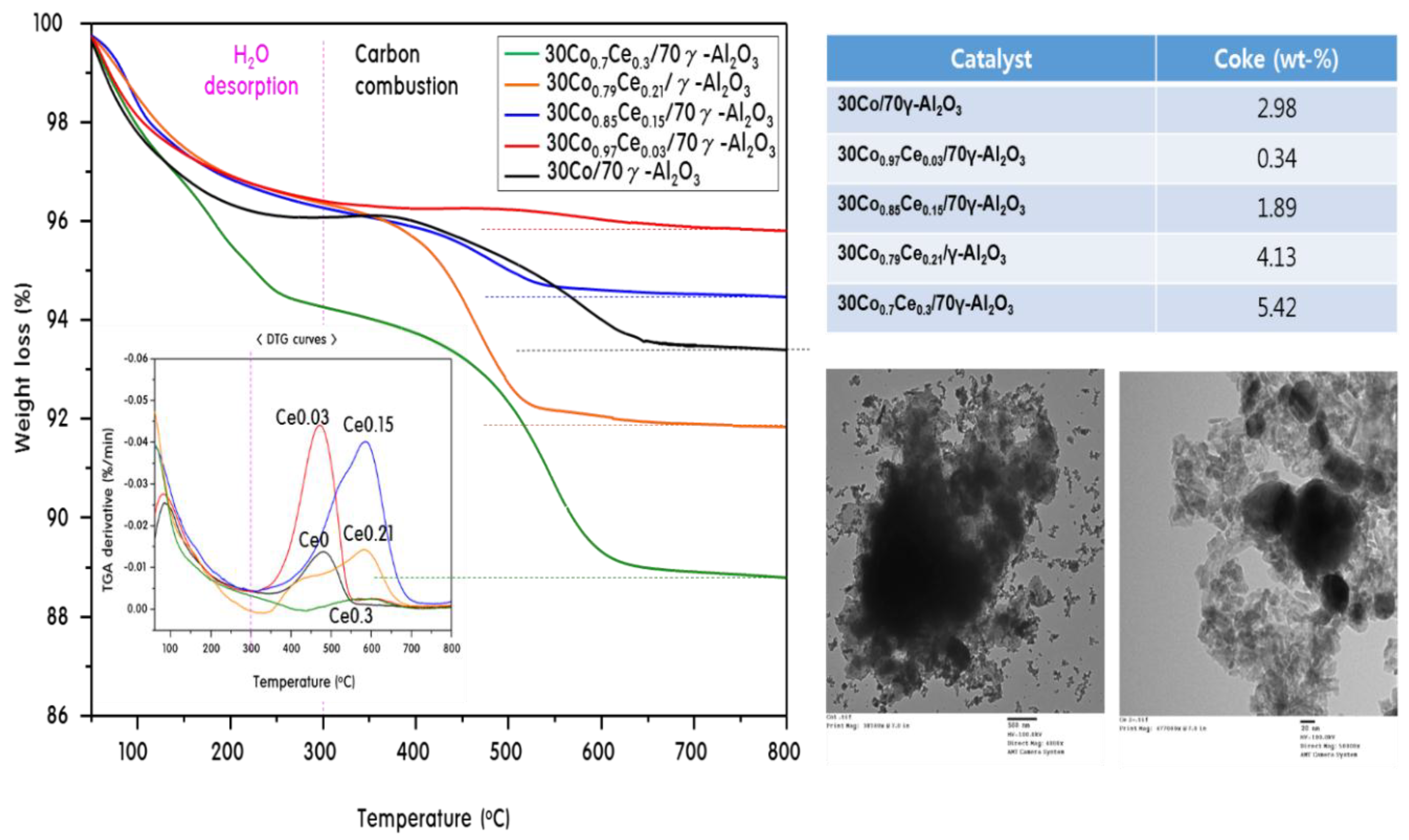
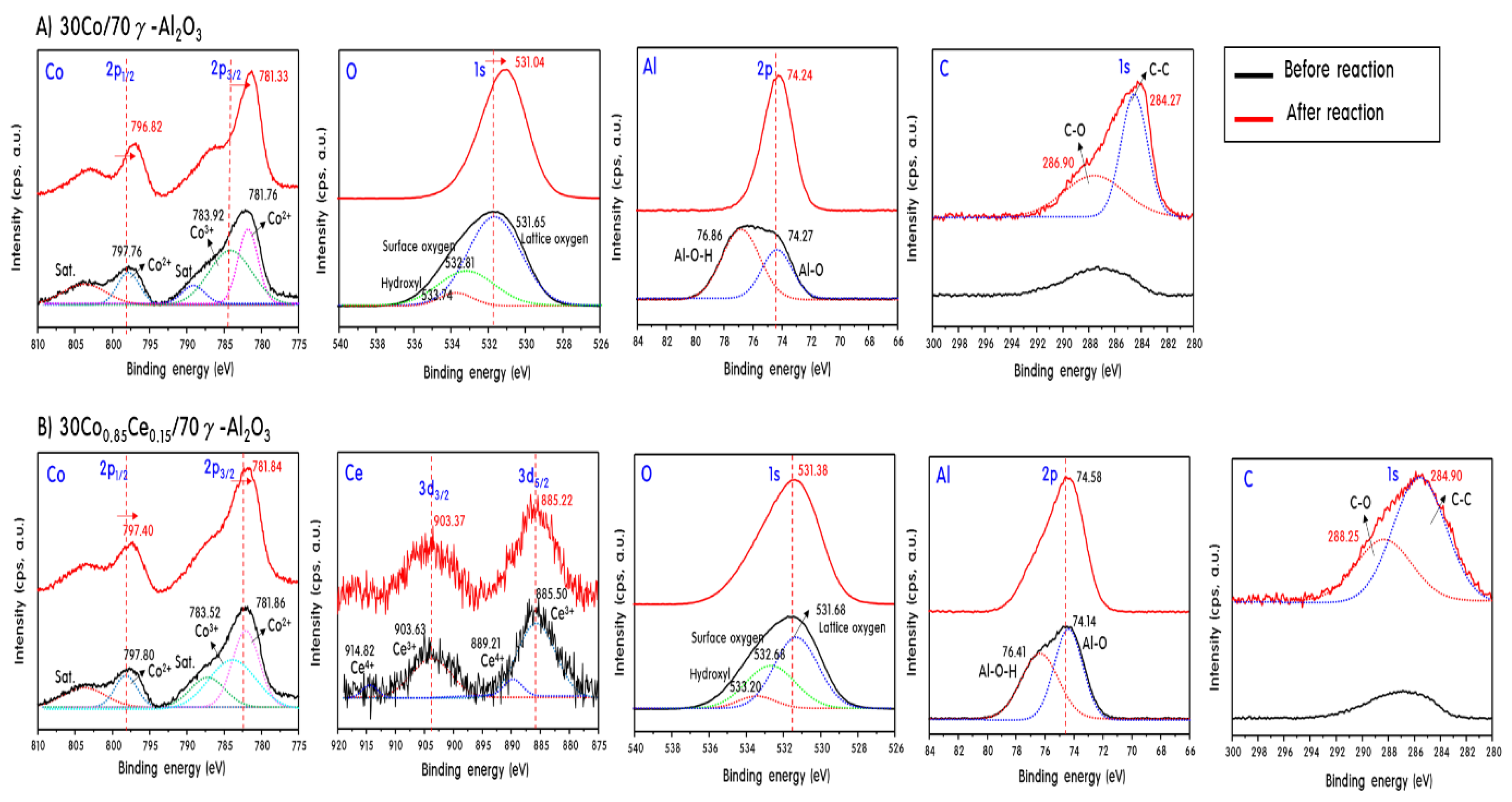
2.4. Characteristics of 30CoxCey/70γ-Al2O3 Catalysts after Propane Steam Reforming
3. Experimantal
3.1. Preparation of 30CoxCey/70 γ-Al2O3 Catalysts
3.2. Characterization
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dodds, P.E.; Staffell, L.; Hawkes, A.D.; Grunewald, P.; McDowall, W.; Ekins, P. Hydrogen and fuel cell technologies for heating: A review. Int. J. Hydrogen. Energy 2011, 40, 2065–2083. [Google Scholar] [CrossRef]
- Amin, A.M.; Croiset, E.; Epling, W. Review of methane catalytic cracking for hydrogen production. Int. J. Hydrogen. Energy 2011, 36, 2904–2935. [Google Scholar] [CrossRef]
- Enger, B.C.; Lodeng, R.; Holmen, A. A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts. Appl. Catal. A 2008, 346, 1–27. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q.; Millar, G.J. Carbon Dioxide Reforming of Methane to Produce Synthesis Gas over Metal-Supported Catalysts: State of the Art. Energy Fuels. 1996, 10, 896–904. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J. Steam reforming of hydrocarbons. A historical perspective. Stud. Surf. Sci. Catal. 2004, 147, 121–126. [Google Scholar]
- Granger, P.; Parvulescu, V.; Kaliaguine, S.; Prellier, W. Perovskites and Related Mixed Oxides: Concepts and Applications, Band 1; John Wiley & Sons: New York, NY, USA, 2015; 1056p. [Google Scholar]
- Harshini, D.; Yoon, C.Y.; Han, J.H.; Yoon, S.P.; Nam, S.W.; Lim, T.H. Catalytic steam reforming of propane over Ni/LaAlO3 catalysts: influence of preparation methods and OSC on activity and stability. Catal. Lett. 2012, 142, 205–212. [Google Scholar] [CrossRef]
- Sohn, H.; Ozkan, U.S. Cobalt-Based Catalysts for Ethanol Steam Reforming: An Overview. Energy Fuels 2016, 30, 5309–5322. [Google Scholar] [CrossRef]
- Wu, H.; Parola, V.L.; Pantaleo, G.; Puleo, F.; Venezia, A.M.; Liotta, L.F. Ni-Based Catalysts for Low Temperature Methane Steam Reforming: Recent Results on Ni-Au and Comparison with Other Bi-Metallic Systems. Catalysts 2013, 3, 563–583. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, J.G.; Jakbensen, M.; Chorkendorff, I.; Sehested, J. Methane Steam Reforming Kinetics for a Rhodium-Based Catalyst. Catal. Lett. 2010, 140, 90–97. [Google Scholar] [CrossRef]
- Zhang, X.; Gong, Y.; Yu, G.; Xie, Y. Oxygen species on NiO/Al2O3 and their reactivities. J. Mol. Catal. A-Chem. 2002, 180, 293–298. [Google Scholar] [CrossRef]
- Ashok, J.; Kathiraser, Y.; Ang, M.L.; Kawi, S. Bi-functional hydrotalcite-derived NiO–CaO–Al2O3 catalysts for steam reforming of biomass and/or tar model compound at low steam-to-carbon conditions. Appl. Catal. B 2015, 172, 116–128. [Google Scholar] [CrossRef]
- Boroujerdnia, M.; Obeydavi, A. Synthesis and characterization of NiO/MgAl2O4 nanocrystals with high surface area by modified sol-gel method. Microporous Mesoporous Mater. 2016, 228, 289–296. [Google Scholar] [CrossRef]
- Avcl, A.K.; Trimm, D.L.; Aksoylu, A.E.; Onsan, A. Hydrogen production by steam reforming of n-butane over supported Ni and Pt-Ni catalysts. Appl. Catal. A. 2004, 258, 235–240. [Google Scholar]
- Ahn, K.; Choi, S.; Lee, J.; Kim, B.; Kim, J.; Kim, H. Enhanced carbon tolerance on Ni-based reforming catalyst with Ir alloying: A DFT study. Appl. Surf. Sci. 2017, 419, 678–682. [Google Scholar] [CrossRef]
- Tian, H.; Li, X.; Chen, S.; Zeng, L.; Gong, J. Role of Sn in Ni-Sn/CeO2 Catalysts for Ethanol Steam Reforming. Chin. J. Chem. 2017, 35, 651–658. [Google Scholar]
- Wang, H.; Blaylock, W.; Dam, A.H.; Lilang, S.E.; Rout, K.R.; Zhu, Y.-A.; Green, W.H.; Homen, A.; Chen, D. Steam methane reforming on a Ni-based bimetallic catalyst: density functional theory and experimental studies of the catalytic consequence of surface alloying of Ni with Ag. Catal. Sci. Technol. 2017, 7, 1713–1715. [Google Scholar] [CrossRef]
- Park, K.S.; Son, M.; Park, M.-J.; Kim, D.H.; Kim, J.H.; Park, S.H.; Choi, J.-H.; Bae, J.W. Adjusted interactions of nickel nanoparticles with cobalt-modified MgAl2O4-SiC for an enhanced catalytic stability during steam reforming of propane. Appl. Catal. A 2018, 549, 117–133. [Google Scholar] [CrossRef]
- Resini, C.; Delgado, M.C.H.; Arrighi, L.; Alemany, L.J.; Marazza, R.; Busca, G. Propene versus propane steam reforming for hydrogen production over Pd-based and Ni-based catalysts. Catal. Commun. 2005, 6, 441–445. [Google Scholar] [CrossRef]
- Anjaneyulu, C.; Costa, L.O.; Ribeiro, M.C.; Neto, R.C.; Mattos, L.V.; Venugopal, A.; Noronha, F.B. Effect of Zn addition on the performance of Ni/Al2O3 catalyst for steam reforming of ethanol. Appl. Catal. A 2016, 519, 85–98. [Google Scholar] [CrossRef]
- Lee, G.; Kim, D.; Kwak, B.S.; Kang, M. Hydrogen rich production by ethanol steam reforming reaction over Mn/Co10Si90MCM-48 catalysts. Catal. Today 2014, 232, 139–150. [Google Scholar] [CrossRef]
- Chen, A.; Yan, Y.; Elnashaie, S. Catalyst deactivation and engineering control for steam reforming of higher hydrocarbons in a novel membrane reformer. Chem. Eng. Sci. 2004, 59, 1965–1978. [Google Scholar] [CrossRef]
- Ginsburg, J.M.; Piña, J.; Solh, T.E.; Lasa, H.I. Coke Formation over a Nickel Catalyst under Methane Dry Reforming Conditions: Thermodynamic and Kinetic Models. Ind. Eng. Chem. Res. 2005, 44, 4846–4854. [Google Scholar] [CrossRef]
- Gao, F.; Tang, X.; Yi, H.; Zhao, X.; Li, C.; Li, J.; Shi, Y.; Meng, X. A Review on Selective Catalytic Reduction of NOx by NH3 over Mn–Based Catalysts at Low Temperatures: Catalysts, Mechanisms, Kinetics and DFT Calculations. Catalysts 2017, 7, 1–32. [Google Scholar] [CrossRef]
- Obata, S.; Kato, M.; Yokoyama, H.; Iwata, Y.; Kikumoto, Y.; Sakurada, O. Synthesis of nano CoAl2O4 pigment for ink-jet printing to decorate porcelain. J. Ceram. Soc. Jpn. 2011, 119, 208–213. [Google Scholar] [CrossRef]
- Liu, K.; Schmedake, T.A.; Tsu, R. A comparative study of colloidal silica spheres: Photonic crystals versus Bragg‘s law. Phys. Lett. A 2008, 372, 4517–4520. [Google Scholar] [CrossRef]
- Cepuritis, R.; Garboczi, E.J.; Ferraris, C.F.; Jacobsen, S.; Sorensen, B.E. Measurement of particle size distribution and specific surface area for crushed concrete aggregate fines. Adv. Powder Technol. 2017, 28, 706–720. [Google Scholar] [CrossRef]
- Bakar, W.A.W.A.; Ali, R.; Mohammad, N.S. The effect of noble metals on catalytic methanation reaction over supported Mn/Ni oxide based catalysts. Arab. J. Chem. 2015, 8, 632–643. [Google Scholar] [CrossRef]
- Takenaka, S.; Orita, Y.; Umebayashi, H.; Matsune, H.; Kishida, M. High resistance to carbon deposition of silica-coated Ni catalysts in propane stream reforming. Appl. Catal. A 2008, 351, 189–194. [Google Scholar] [CrossRef]
- Wang, M.; Jin, L.; Li, Y.; Lv, J.; Wei, B.; Hu, H. In-situ catalytic upgrading of coal pyrolysis tar coupled with CO2 reforming of methane over Ni-based catalysts. Fuel Process. Technol. 2018, 177, 119–128. [Google Scholar] [CrossRef]
- Cordoba, M.; Miranda, C.; Lederhos, C.; Pascual, F.; Ardila, A.; Fuentes, G.A.; Pouilloux, Y.; Ramirez, A. Catalytic performance of Co3O4 on different activated carbon supports in the benzyl alcohol oxidation. Catalysts 2017, 7, 384–396. [Google Scholar] [CrossRef]
- Petitto, S.C.; Langell, M.A. Surface composition and structure of Co3O4 (110) and the effect of impurity segregation. J. Vac. Sci. Technol. A 2004, 22, 1690–1697. [Google Scholar] [CrossRef]
- Jin, J.; Park, J.; Shon, J.K.; Kim, J.H.; Li, Z.; Park, Y.; Kim, J.M. Low temperature CO oxidation over Pd catalysts supported on highly ordered mesoporous metal oxides. Catal. Today 2012, 185, 183–190. [Google Scholar] [CrossRef]
- Ma, D.; Lu, Z.; Tang, Y.; Li, Y.; Tang, Z.; Yang, Z. Effect of lattice strain on the oxygen vacancy formation and hydrogen adsorption at CeO2 (111) surface. Phys. Lett. A 2014, 378, 2570–2575. [Google Scholar] [CrossRef]
- Kim, K.M.; Kwak, B.S.; Park, N.-K.; Lee, T.J.; Lee, S.T.; Kang, M. Effective hydrogen production from propane steam reforming over bimetallic co-doped NiFe/Al2O3 catalyst. J. Ind. Eng. Chem. 2017, 46, 324–336. [Google Scholar] [CrossRef]
- Kim, J.; Samano, E.; Koel, B.E. CO Adsorption and Reaction on Clean and Oxygen-Covered Au (211) Surfaces. J. Phys. Chem. B 2006, 110, 17512–17517. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.R.; Mishra, B.G. Structural, redox and catalytic chemistry of ceria based materials. Bull. Catal. Soc. India 2003, 2, 122–134. [Google Scholar]
- Bobrova, L.; Andreev, D.; Ivanov, E.; Mezentseva, N.; Simonov, M.; Makarshin, L.; Gribovskii, L.; Sadykov, V. Water–Gas Shift Reaction over Ni/CeO2 Catalysts. Catalysts 2017, 7, 310–334. [Google Scholar] [CrossRef]
- Noisong, P.; Danvirutai, C. Kinetics and Mechanism of Thermal Dehydration of KMnPO4·H2O in a Nitrogen Atmosphere. Ind. Eng. Chem. Res. 2010, 49, 3146–3151. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Xie, C.; Song, C. Influence of ceria and nickel addition to alumina-supported Rh catalyst for propane steam reforming at low temperatures. Appl. Catal. A 2009, 357, 213–222. [Google Scholar] [CrossRef]
- Weerakkody, C.; Biswas, S.; Song, W.; He, J.; Wasalathanthri, N.; Dissanayake, S.; Kriz, D.A.; Dutta, B.; Sui, S.L. Controllable synthesis of mesoporous cobalt oxide for peroxide free catalytic epoxidation of alkenes under aerobic conditions. Appl. Catal. B 2018, 221, 681–690. [Google Scholar] [CrossRef]
- Pieck, C.L.; Jablonski, E.L.; Parera, J.M. Recovering of the catalytic functions of naphtha reforming catalysts by partial coke burning. Appl. Catal. 1991, 70, 19–28. [Google Scholar] [CrossRef]
- Mattos, L.V.; Rodino, E.; Resasco, D.E.; Passos, F.B.; Noronha, F.B. Partial oxidation and CO2 reforming of methane on Pt/Al2O3, Pt/ZrO2, and Pt/Ce–ZrO2 catalysts. Fuel Process. Technol. 2003, 83, 147–161. [Google Scholar] [CrossRef]
- Lu, W.; Iwasa, Y.; Ou, Y.; Jinno, D.; Kamiyama, S.; Petersen, P.M.; Ou, H. Effective optimization of surface passivation on porous silicon carbide using atomic layer deposited Al2O3. RSC Adv. 2017, 7, 8090–8097. [Google Scholar] [CrossRef]
- Deng, S.; Li, S.; Li, H.; Zhang, Yi. Oxidative dehydrogenation of ethane to ethylene with CO2 over Fe-Cr/ZrO2 catalysts. Ind. Eng. Chem. Res. 2009, 48, 7561–7566. [Google Scholar] [CrossRef]
- Xu, W.; He, H.; Yu, Y. Deactivation of a Ce/TiO2 catalyst by SO2 in the selective catalytic reduction of NO by NH3. J. Phys. Chem. C 2009, 113, 4426–4432. [Google Scholar] [CrossRef]
- Kulkarni, S.B.; Patil, U.M.; Shackery, I.; Sohn, J.S.; Lee, S.; Park, B.; Jun, S.C. High-performance supercapacitor electrode based on a polyaniline nanofibers/3D graphene framework as an efficient charge transporter. J. Mater. Chem. A 2014, 2, 498–4998. [Google Scholar] [CrossRef]
- Do, J.Y.; Lee, J.H.; Park, N.-K.; Lee, T.J.; Lee, S.T.; Kang, M. Synthesis and characterization of Ni2−xPdxMnO4/γ-Al2O3 catalysts for hydrogen production via propane steam reforming. Chem. Eng. J. 2018, 334, 1668–1678. [Google Scholar] [CrossRef]
- Jo, S.W.; Im, Y.; Do, J.Y.; Park, N.-K.; Lee, T.J.; Lee, S.T.; Cha, M.S.; Jeon, M.; Kang, M. Synergies between Ni, Co, and Mn ions in trimetallic Ni1−xCoxMnO4 catalysts for effective hydrogen production from propane steam reforming. Renew. Energy 2017, 113, 248–256. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, J.Y.; Chava, R.K.; Son, N.; Kim, J.; Park, N.-K.; Lee, D.; Seo, M.W.; Ryu, H.-J.; Chi, J.H.; Kang, M. Effect of Ce Doping of a Co/Al2O3 Catalyst on Hydrogen Production via Propane Steam Reforming. Catalysts 2018, 8, 413. https://doi.org/10.3390/catal8100413
Do JY, Chava RK, Son N, Kim J, Park N-K, Lee D, Seo MW, Ryu H-J, Chi JH, Kang M. Effect of Ce Doping of a Co/Al2O3 Catalyst on Hydrogen Production via Propane Steam Reforming. Catalysts. 2018; 8(10):413. https://doi.org/10.3390/catal8100413
Chicago/Turabian StyleDo, Jeong Yeon, Rama Krishna Chava, Namgyu Son, Junyeong Kim, No-Kuk Park, Doyeon Lee, Myung Won Seo, Ho-Jung Ryu, Jun Hwa Chi, and Misook Kang. 2018. "Effect of Ce Doping of a Co/Al2O3 Catalyst on Hydrogen Production via Propane Steam Reforming" Catalysts 8, no. 10: 413. https://doi.org/10.3390/catal8100413