
Olefin Metathesis with Ru-Based Catalysts Exchanging the Typical N-Heterocyclic Carbenes by a Phosphine–Phosphonium Ylide


Abstract
:
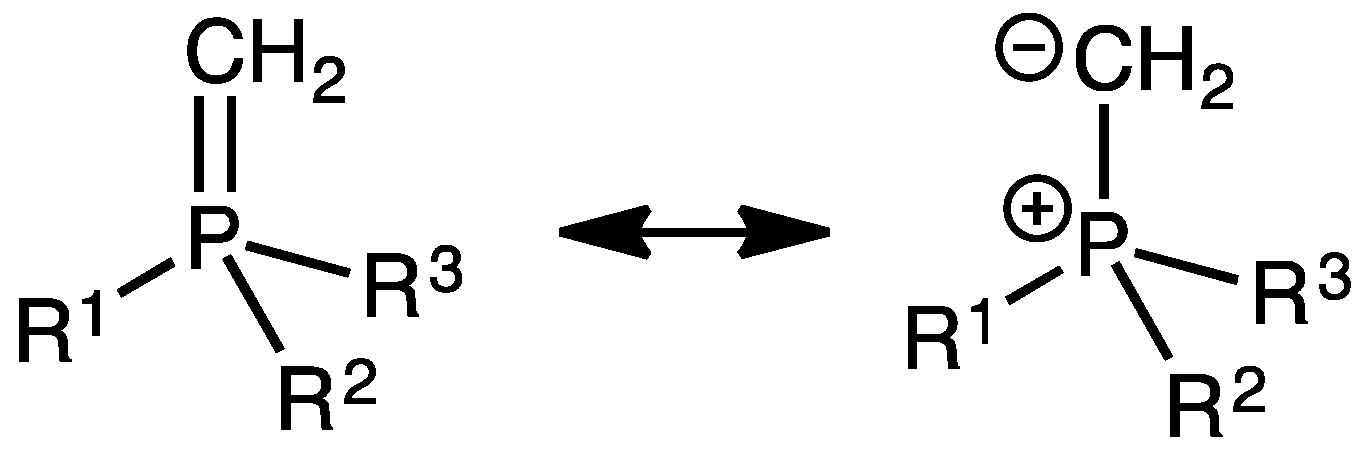
1. Introduction
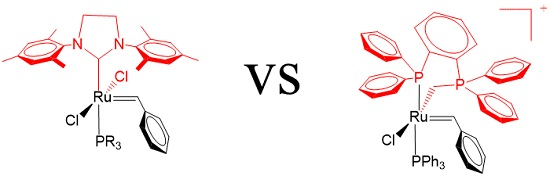
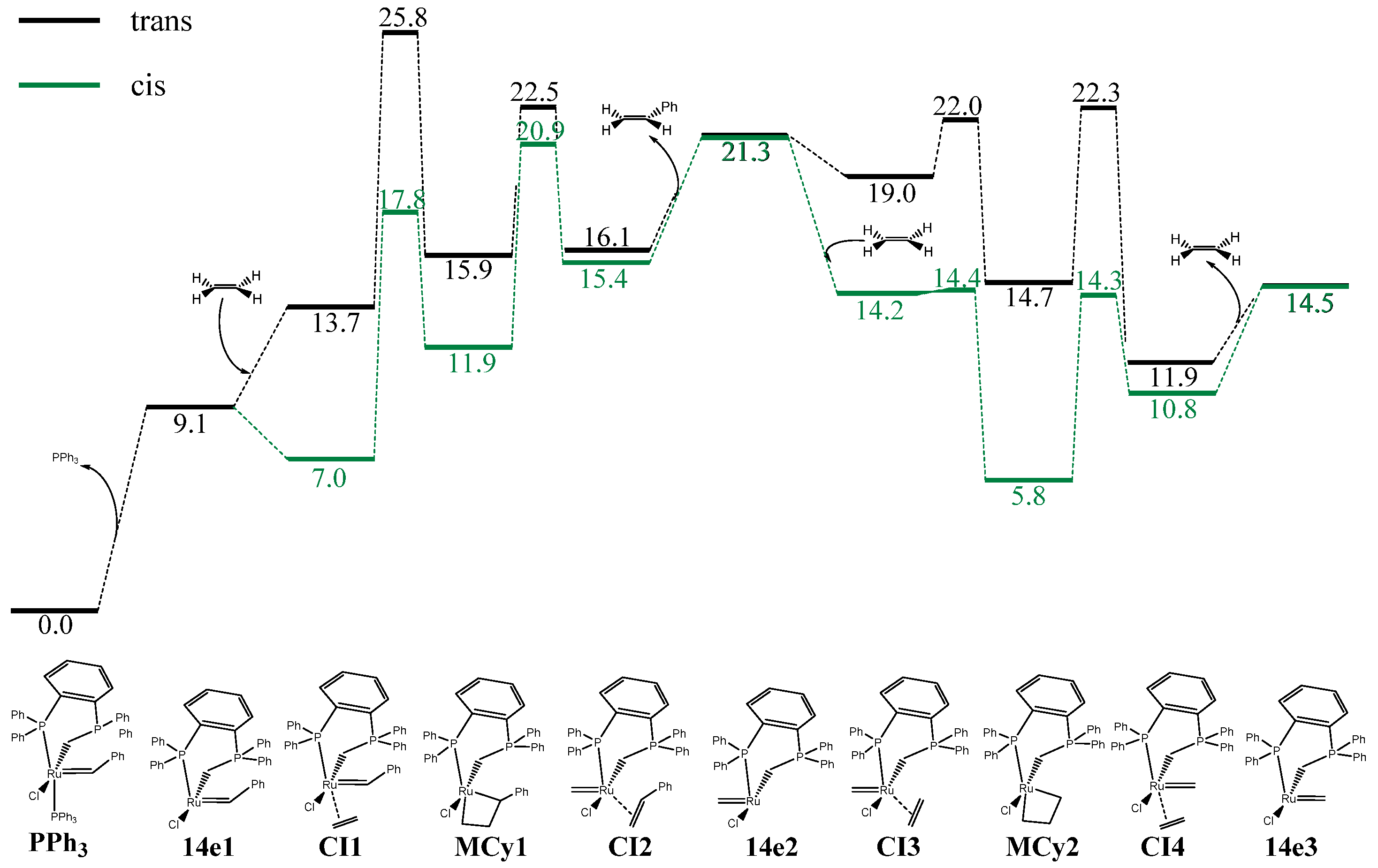
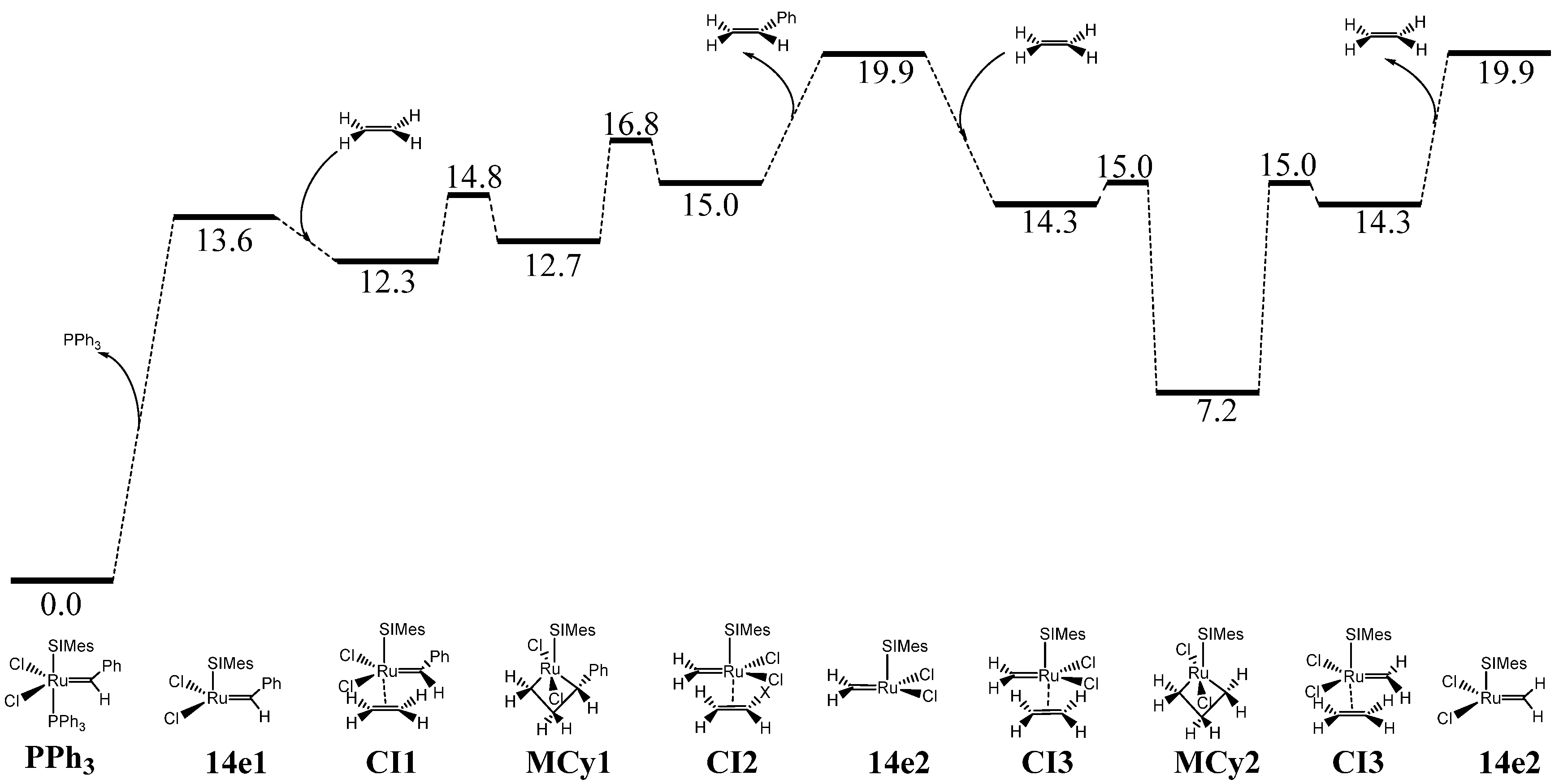
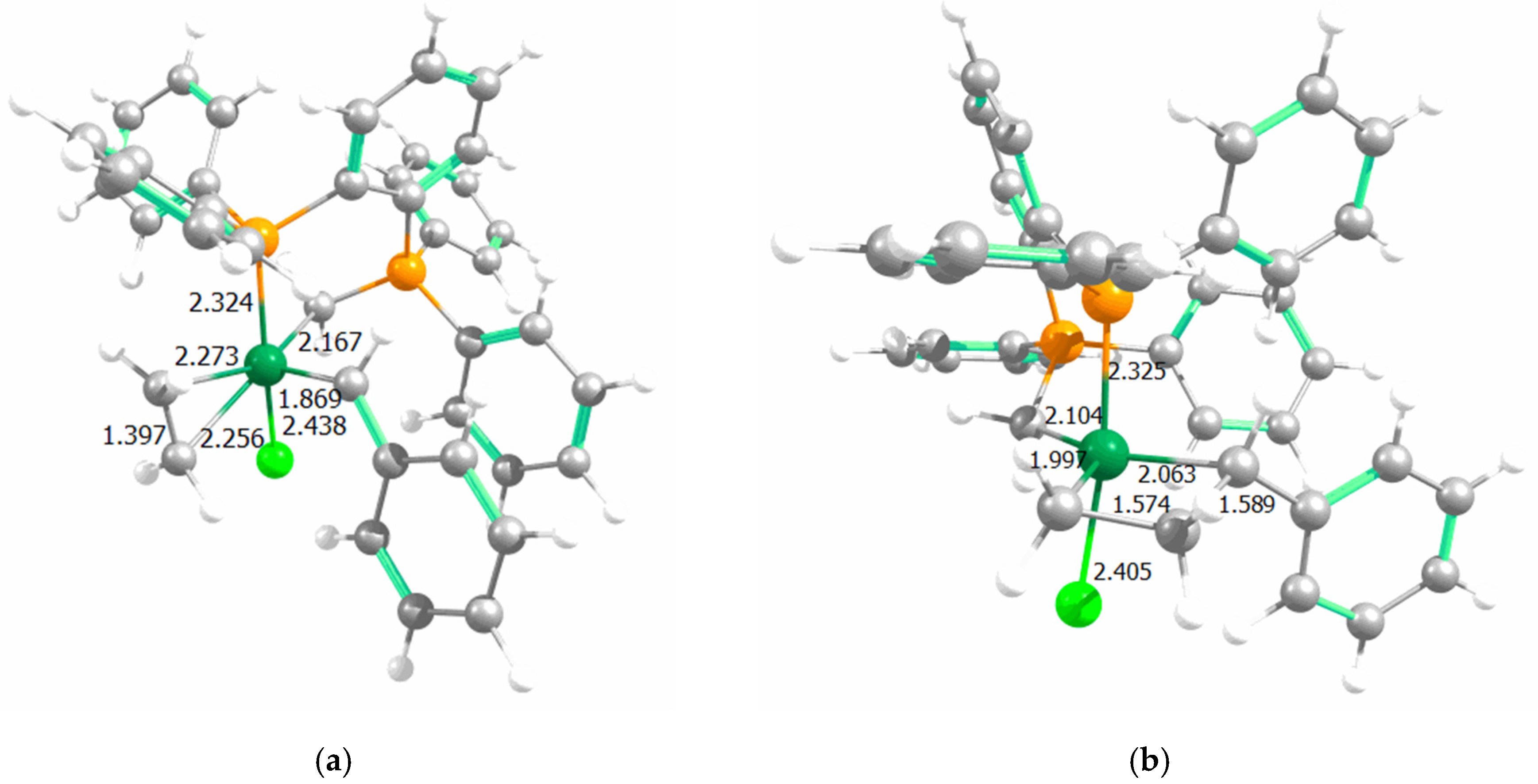
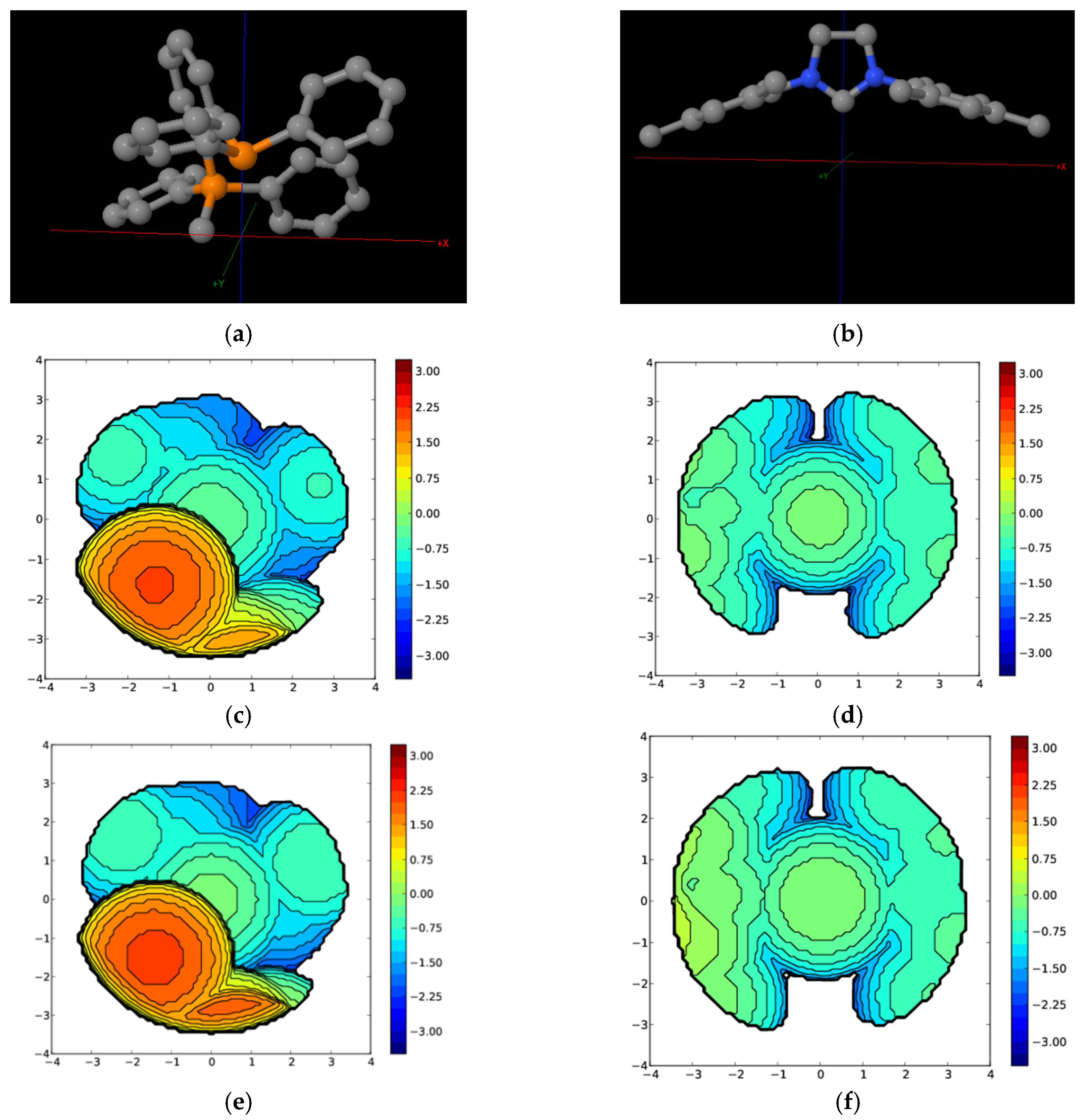
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, A.W.; Merckling, N.G. Polymeric Bicycle [2.2.1]-2-heptene. Du Pont de Nemours & Co. U.S. Patent 2,721,189, 18 October 1955. [Google Scholar]
- Truett, W.L.; Johnson, D.R.; Robinson, I.M.; Montague, B.A. Polynorbornene by coordination polymerization. J. Am. Chem. Soc. 1960, 82, 2337–2340. [Google Scholar] [CrossRef]
- Banks, R.L.; Bailey, G.C. Olefin disproportionation. A new catalytic process. Ind. Eng. Chem. Prod. Res. Dev. 1964, 3, 170–173. [Google Scholar] [CrossRef]
- Calderon, N. Olefin metathesis reaction. Acc. Chem. Res. 1972, 5, 127–132. [Google Scholar] [CrossRef]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef] [PubMed]
- Rouen, M.; Queval, P.; Borré, E.; Falivene, L.; Poater, A.; Berthod, M.; Hugues, F.; Cavallo, L.; Baslé, O.; Olivier-Bourbigou, H.; et al. Selective metathesis of α-olefins from bio-sourced Fischer–Tropsch feeds. ACS Catal. 2016, 6, 7970–7976. [Google Scholar] [CrossRef]
- Pozgan, F.; Dixneuf, P.H. Metathesis chemistry. In Nanostructure Design to Synthesis of Advanced Materials; Springer: Dordrecht, The Netherlands, 2007; Volume 243, pp. 195–222. [Google Scholar]
- Hérisson, J.L.; Chauvin, Y. Catalyse de transformation des olefins par les complexes du tungstène. Makromol. Chem. 1971, 141, 161–176. [Google Scholar] [CrossRef]
- Katz, T.J.; McGinnis, J. Mechanism of the olefin metathesis reaction. J. Am. Chem. Soc. 1975, 97, 1592–1594. [Google Scholar] [CrossRef]
- Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Probing the relevance of NHC ligand conformations in the Ru-catalysed ring-closing metathesis reaction. Chem. Eur. J. 2013, 19, 10492–10496. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M.S.; Ulman, M.; Grubbs, R.H. New insights into the mechanism of ruthenium-catalyzed olefin metathesis reactions. J. Am. Chem. Soc. 2001, 123, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. A computational perspective of olefins metathesis catalyzed by N-heterocyclic carbene ruthenium (pre) catalysts. Catal. Sci. Technol. 2011, 1, 1287–1297. [Google Scholar] [CrossRef]
- Weskamp, T.; Schattenmann, W.C.; Spiegler, M.; Herrmann, W.A. A novel class of ruthenium catalysts for olefin metathesis. Angew. Chem. Int. Ed. 1998, 37, 2490–2493. [Google Scholar] [CrossRef]
- Huang, J.; Stevens, E.D.; Petersen, J.L.; Nolan, S.P. Olefin metathesis-active ruthenium complexes bearing a nucleophilic carbene ligand. J. Am. Chem. Soc. 1999, 121, 2674–2678. [Google Scholar] [CrossRef]
- Veldhuizen, J.J.V.; Garber, S.B.; Kingsbury, J.S.; Hoveyda, A.H. A recyclable chiral Ru catalyst for enantioselective olefin metathesis. Efficient catalytic asymmetric ring-opening/cross metathesis in air. J. Am. Chem. Soc. 2002, 124, 4954–4955. [Google Scholar] [CrossRef] [PubMed]
- Samojłowicz, C.; Bieniek, M.; Grela, K. Ruthenium-based olefin metathesis catalysts bearing N-heterocyclic carbene ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.J.; Manzini, S.; Urbina-Blanco, C.A.; Nolan, S.P. Key processes in ruthenium-catalysed olefin metathesis. Chem. Commun. 2014, 50, 10355–10375. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Bahri-Laleh, N.; Cavallo, L. Rationalizing current strategies to protect N-heterocyclic carbene-based ruthenium catalysts active in olefin metathesis from C-H (de)activation. Chem. Commun. 2011, 47, 6674–6676. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Credendino, R.; Slugovc, C.; Cavallo, L. Exploring new generations of ruthenium olefin metathesis catalysts: The reactivity of a bis-ylidene ruthenium complex by DFT. Dalton Trans. 2013, 42, 7271–7275. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.P.; Vummaleti, S.V.C.; Falivene, L.; Nolan, S.P.; Cavallo, L.; Solà, M.; Poater, A. In silico olefin metathesis with Ru-based catalysts containing N-heterocyclic carbenes bearing C60 fullerenes. Chem. Eur. J. 2016, 22, 6617–6623. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Vummaleti, S.V.C.; Pump, E.; Cavallo, L. Comparing Ru and Fe-catalyzed olefin metathesis. Dalton Trans. 2014, 43, 11216–11220. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Pump, E.; Vummaleti, S.V.C.; Cavallo, L. The activation mechanism of Fe-based olefin metathesis catalysts. Chem. Phys. Lett. 2014, 610–611, 29–32. [Google Scholar] [CrossRef]
- De Brito Sá, E.; Rodríguez-Santiago, L.; Sodupe, M.; Solans-Monfort, X. Toward olefin metathesis with iron carbene complexes: Benefits of tridentate σ-donating ligands. Organometallics 2016, 35, 3914–3923. [Google Scholar]
- Poater, A.; Solans-Monfort, X.; Clot, E.; Copéret, C.; Eisenstein, O. DFT calculations of d0 M(NR)(CHtBu)(X)(Y) (M = Mo, W; R = CPh3, 2,6-iPr–C6H3; X and Y = CH2tBu, OtBu, OSi(OtBu)3) olefin metathesis catalysts: Structural, spectroscopic and electronic properties. Dalton Trans. 2006. [Google Scholar] [CrossRef] [PubMed]
- Poater, A. Moving from classical Ru-NHC to neutral or charged Rh-NHC based catalysts in olefin metathesis. Molecules 2016, 21, 177. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H. Phosphorus ylides in the coordination sphere of transition metals: An inventory. Angew. Chem. Int. Ed. 1983, 22, 907–927. [Google Scholar] [CrossRef]
- Vicente, J.; Chicote, M. The ‘acac method’ for the synthesis of coordination and organometallic compounds: Synthesis of gold complexes. Trans. Coord. Chem. Rev. 1999, 193–195, 1143–1161. [Google Scholar] [CrossRef]
- Vignolle, J.; Donnadieu, B.; Bourissou, D.; Soleilhavoup, M.; Bertrand, G. Cyclic C-amino phosphorus ylides as a source of bidentate heteroditopic ligands (phosphine/aminocarbene) for transition metals. J. Am. Chem. Soc. 2006, 128, 14810–14811. [Google Scholar] [CrossRef] [PubMed]
- Transition Metal Complexes of Neutral η1-Carbon Ligands; Chauvin, R.; Canac, Y. (Eds.) Springer: Berlin, Germany, 2010; pp. 1–256.
- Canac, Y.; Chauvin, R. Atropochiral C,X- and C,C-chelating carbon ligands. Eur. J. Inorg. Chem. 2010, 16, 2325–2335. [Google Scholar] [CrossRef]
- Canac, Y.; Lepetit, C.; Abdalilah, M.; Duhayon, C.; Chauvin, R. Diaminocarbene and phosphonium ylide ligands: A systematic comparison of their donor character. J. Am. Chem. Soc. 2008, 130, 8406–8413. [Google Scholar] [CrossRef] [PubMed]
- Lepetit, C.; Maraval, V.; Canac, Y.; Chauvin, R. On the nature of the dative bond: Coordination to metals and beyond. The carbon case. Coord. Chem. Rev. 2016, 308, 59–75. [Google Scholar] [CrossRef]
- Chauvin, R. Zwitterionic organometallates. Eur. J. Inorg. Chem. 2000, 4, 577–591. [Google Scholar] [CrossRef]
- Leitgeb, A.; Abbas, M.; Fischer, R.C.; Poater, A.; Cavallo, L.; Slugovc, C. Latent ruthenium based olefin metathesis catalyst with a sterically demanding NHC ligand (pre)catalysts. Catal. Sci. Technol. 2012, 2, 1640–1643. [Google Scholar] [CrossRef]
- Viau, L.; Lepetit, C.; Commenges, G.; Chauvin, R. Chiral phosphine-phosphonium ylide rhodium complexes. Organometallics 2001, 20, 808–810. [Google Scholar] [CrossRef]
- Zurawinski, R.; Donnadieu, B.; Mikolajczyk, M.; Chauvin, R. Chiral phosphino(sulfinylmethyl)triarylphosphonium ylide ligands: Rhodium complexes and catalytic properties. Organometallics 2003, 22, 4810–4817. [Google Scholar] [CrossRef]
- Viau, L.; Chauvin, R. ‘Diopium’, a chiral phosphoniophosphine derived from Kagan’s diop: Rhodium complexes and reducing catalytic properties. J. Organomet. Chem. 2002, 654, 180–186. [Google Scholar] [CrossRef]
- Ohta, T.; Sasayama, H.; Nakajima, O.; Kurahashi, N.; Fujii, T.; Furukawa, I. Asymmetric allylic substitution catalyzed by palladium Yliphos complex. Tetrahedron Asymmetry 2003, 14, 537–542. [Google Scholar] [CrossRef]
- Canac, Y.; Duhayon, C.; Chauvin, R. A Diaminocarbene-phosphonium ylide: Direct access to C,C-chelating ligands. Angew. Chem. Int. Ed. 2007, 46, 6313–6315. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Heyndrickx, W.; Occhipinti, G.; Jensen, V.R. An evolutionary algorithm for de novo optimization of functional transition metal compounds. J. Am. Chem. Soc. 2012, 134, 8885–8895. [Google Scholar] [CrossRef] [PubMed]
- Rissner, F.; Ma, M.Y.; Hofmann, O.T.; Slugovc, C.; Shuai, Z.; Zojer, E. Radical self-assembled monolayers on Au(111) formed by the adsorption of closed-shell molecules. J. Mater. Chem. 2012, 22, 4269–4272. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Correa, A.; Cavallo, L. Comparison of different ruthenium-alkylidene bonds in the activation step with N-heterocyclic carbene Ru-catalysts for olefins metathesis. Dalton Trans. 2011, 40, 11066–11069. [Google Scholar] [CrossRef] [PubMed]
- Bantreil, X.; Citadelle, C.A.; Slawin, A.M.Z.; Cazin, C.S.J. Phosphites as ligands in ruthenium-benzylidene catalysts for olefin metathesis. Chem. Commun. 2011, 47, 7060–7062. [Google Scholar]
- Romero, P.E.; Piers, W.E. Mechanistic studies on 14-electron ruthenacyclobutanes: Degenerate exchange with free ethylene. J. Am. Chem. Soc. 2007, 129, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Correa, A.; Pump, E.; Cavallo, L. Cis/trans coordination in olefin metathesis by static and molecular dynamic DFT calculations. Chem. Heter. Comp. 2014, 3, 424–430. [Google Scholar] [CrossRef]
- Benitez, D.; Tkatchouk, E.; Goddard, W.A. Relevance of cis- and trans-dichloride Ru intermediates in Grubbs-II olefin metathesis catalysis (H2IMesCl2Ru=CHR). Chem. Commun. 2008, 46, 6194–6196. [Google Scholar] [CrossRef] [PubMed]
- Vidavsky, Y.; Anaby, A.; Lemcoff, N.G. Chelating alkylidene ligands as pacifiers for ruthenium catalysed olefin metathesis. Dalton Trans. 2012, 41, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Cavallo, L. The elusive mechanism of olefin metathesis promoted by (NHC)Ru-based catalysts: A trade between steric, electronic, and solvent effects. J. Am. Chem. Soc. 2006, 128, 13352–13353. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.; Goddard, W.A., III. The isomerization equilibrium between cis and trans chloride ruthenium olefin metathesis catalysts from quantum mechanics calculations. J. Am. Chem. Soc. 2005, 127, 12218–12219. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Cavallo, L. A comprehensive study of olefin metathesis catalyzed by Ru-based catalysts. Beilstein J. Org. Chem. 2015, 11, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Urbina-Blanco, C.A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A.M.Z.; Cavallo, L.; Nolan, S.P. The activation mechanism of Ru−indenylidene complexes in olefin metathesis. J. Am. Chem. Soc. 2013, 135, 7073–7079. [Google Scholar] [CrossRef] [PubMed]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A web tool for analyzing catalytic pockets with topographic steric maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Understanding the M–(NHC) (NHC = N-heterocyclic carbene) bond. Coord. Chem. Rev. 2009, 253, 687–703. [Google Scholar] [CrossRef]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A web application for the calculation of buried volumes of N-heterocyclic carbene ligands. Eur. J. Inorg. Chem. 2009, 1759–1766. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Poater, A.; Childers, M.I.; Widger, P.C.B.; LaPointe, A.M.; Lobkovsky, E.B.; Coates, G.W.; Cavallo, L. Enantioselective polymerization of epoxides using biaryl-linked bimetallic cobalt catalysts: A mechanistic study. J. Am. Chem. Soc. 2013, 135, 18901–18911. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Falivene, L.; Urbina-Blanco, C.A.; Manzini, S.; Nolan, S.P.; Cavallo, L. How does the addition of steric hindrance to a typical N-heterocyclic carbene ligand affect catalytic activity in olefin metathesis? Dalton Trans. 2013, 42, 7433–7439. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; von Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Haeusermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Accuracy of energy-adjusted quasirelativistic ab initio pseudopotentials. Mol. Phys. 1993, 78, 1211–1224. [Google Scholar] [CrossRef]
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Martin, R.L.; Hay, P.J.; Pratt, L.R. Hydrolysis of ferric ion in water and conformational equilibrium. J. Phys. Chem. A 1998, 102, 3565–3573. [Google Scholar] [CrossRef] [Green Version]
- Poater, A.; Pump, E.; Vummaleti, S.V.C.; Cavallo, L. The right computational recipe for olefin metathesis with Ru-based catalysts: The whole mechanism of ring-closing olefin metathesis. J. Chem. Theory Comput. 2014, 10, 4442–4448. [Google Scholar] [CrossRef] [PubMed]
- García-Melchor, M.; Pacheco, M.C.; Nájera, C.; Lledós, A.; Ujaque, G. Mechanistic exploration of the Pd-catalyzed copper-free Sonogashira reaction. ACS Catal. 2012, 2, 135–144. [Google Scholar] [CrossRef]
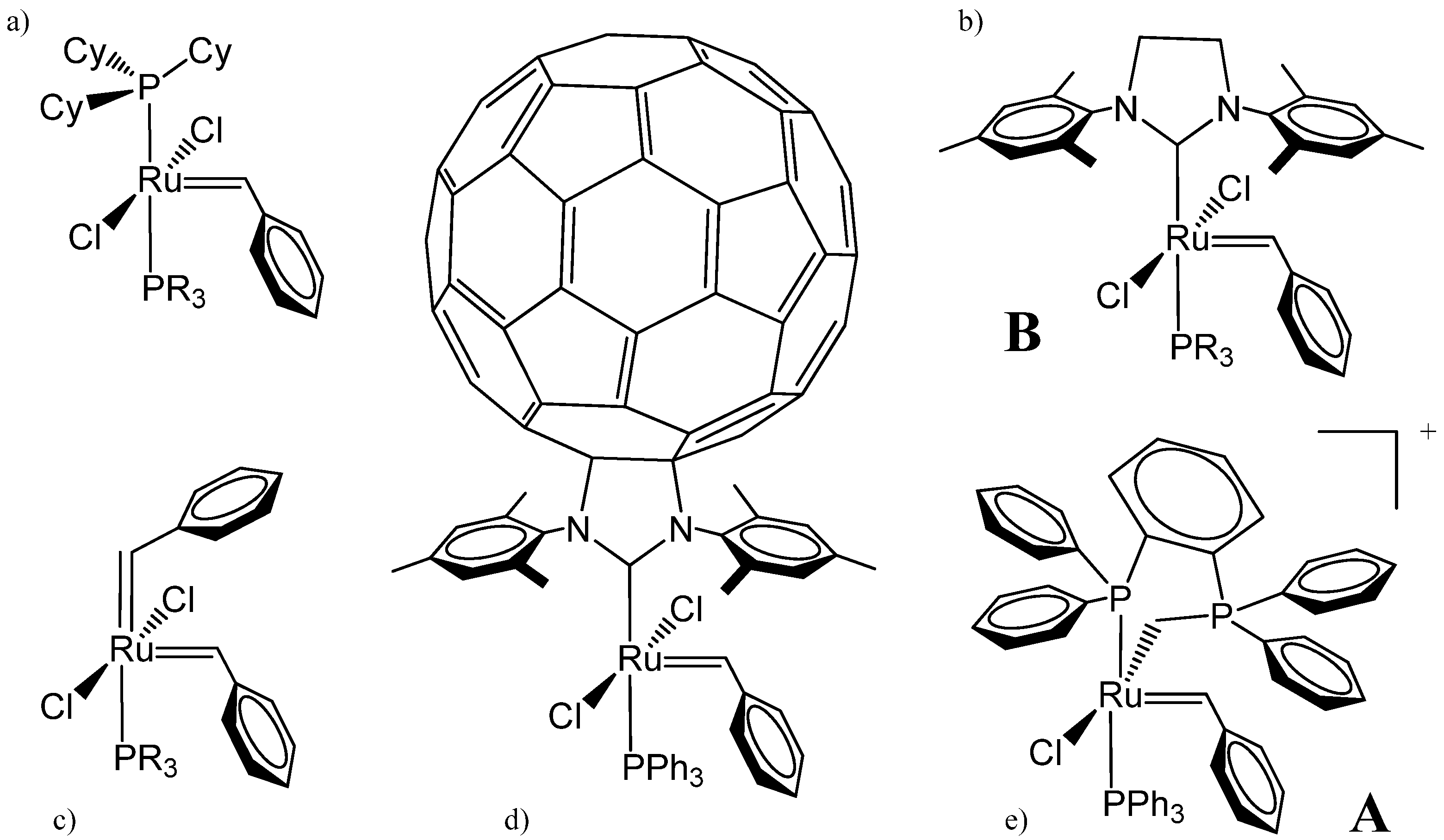
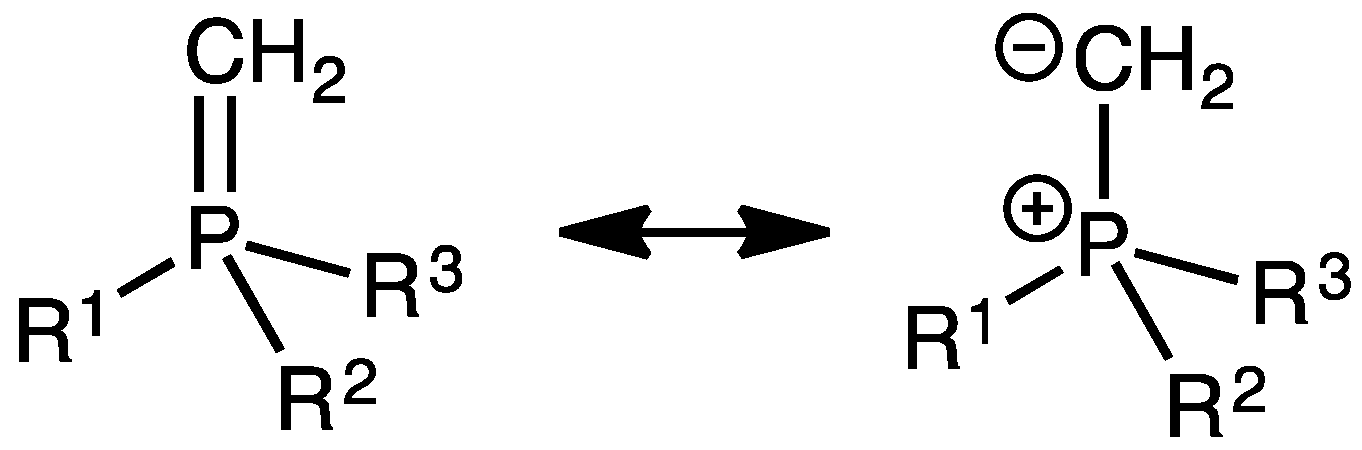
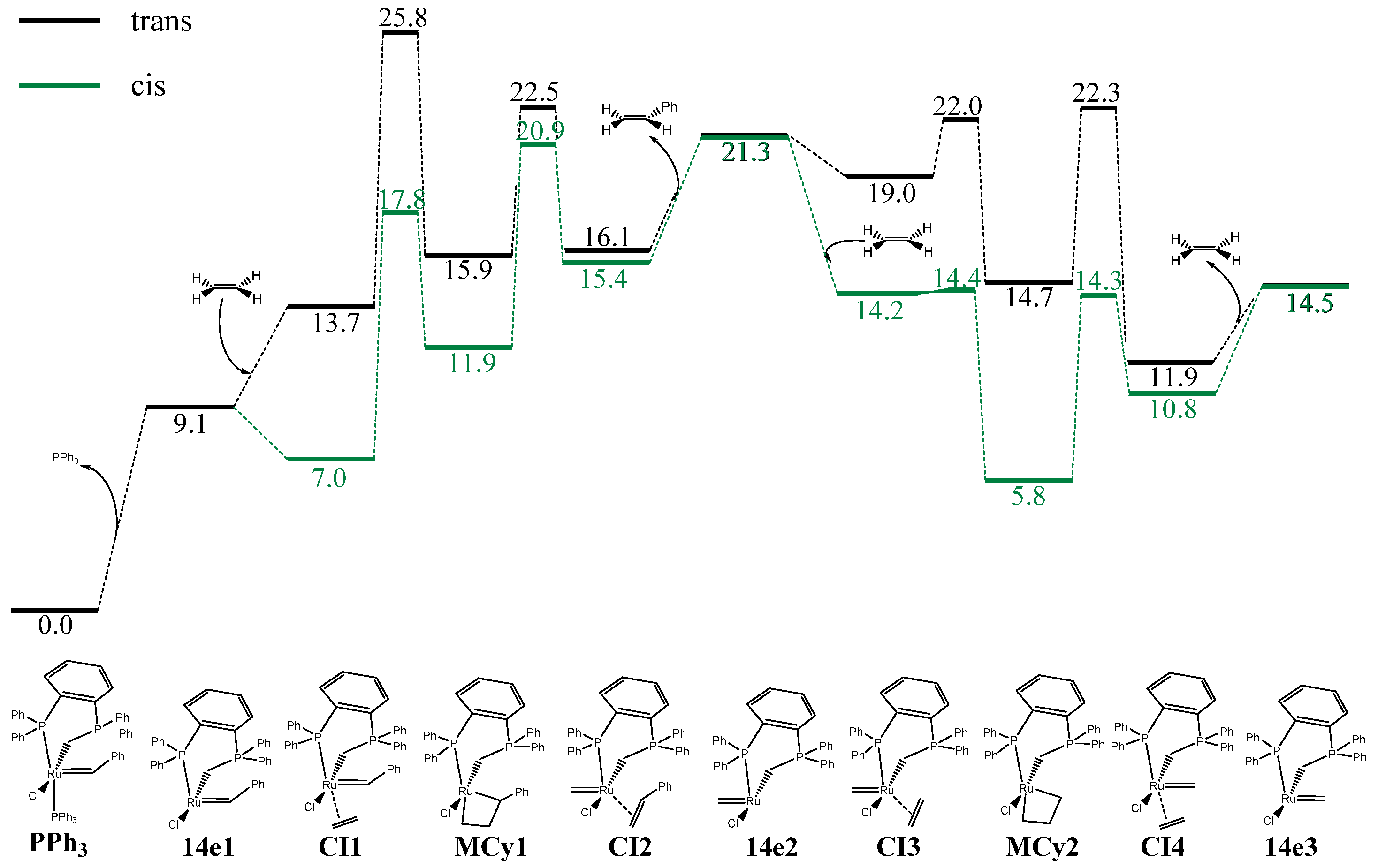
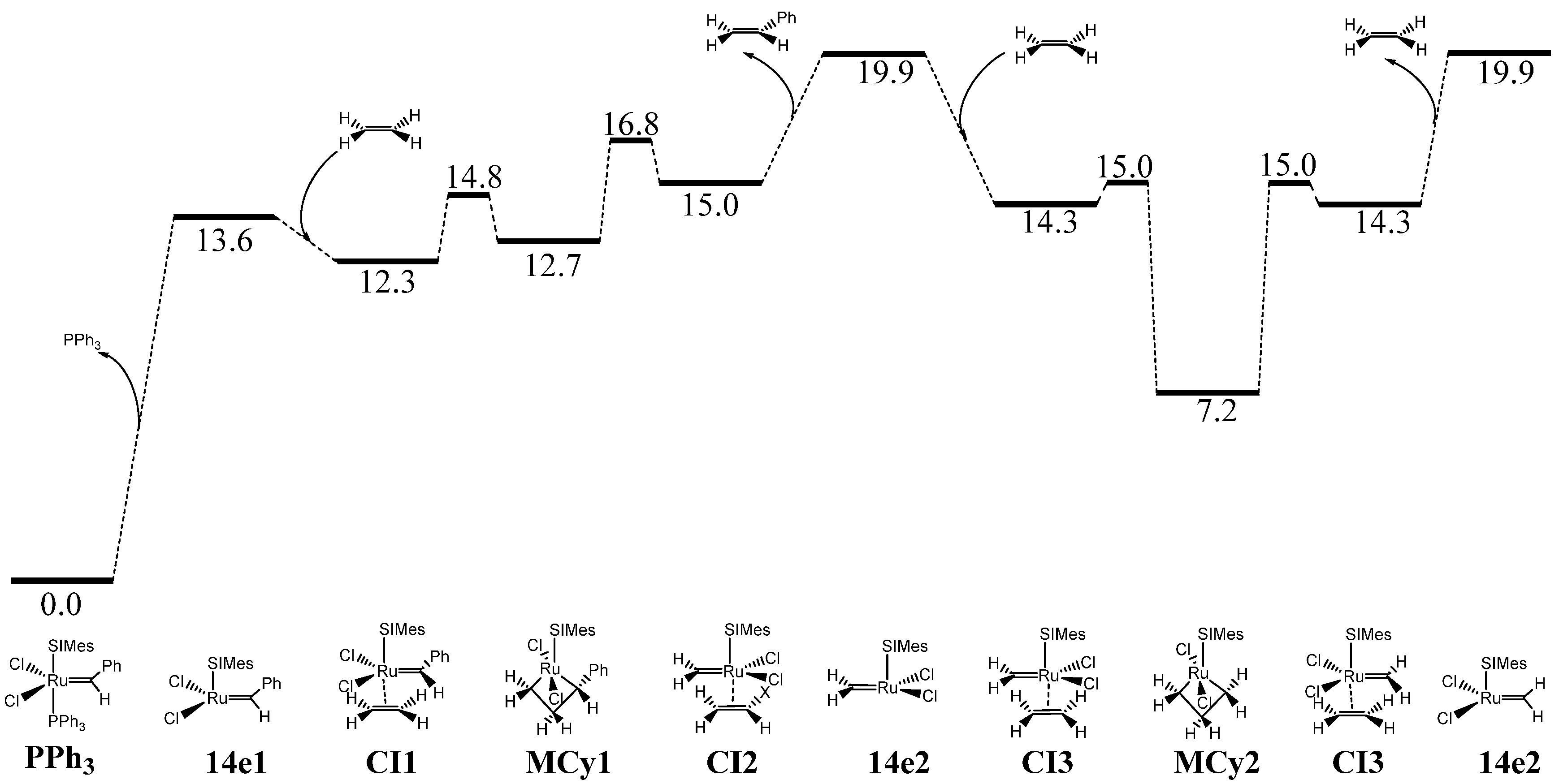
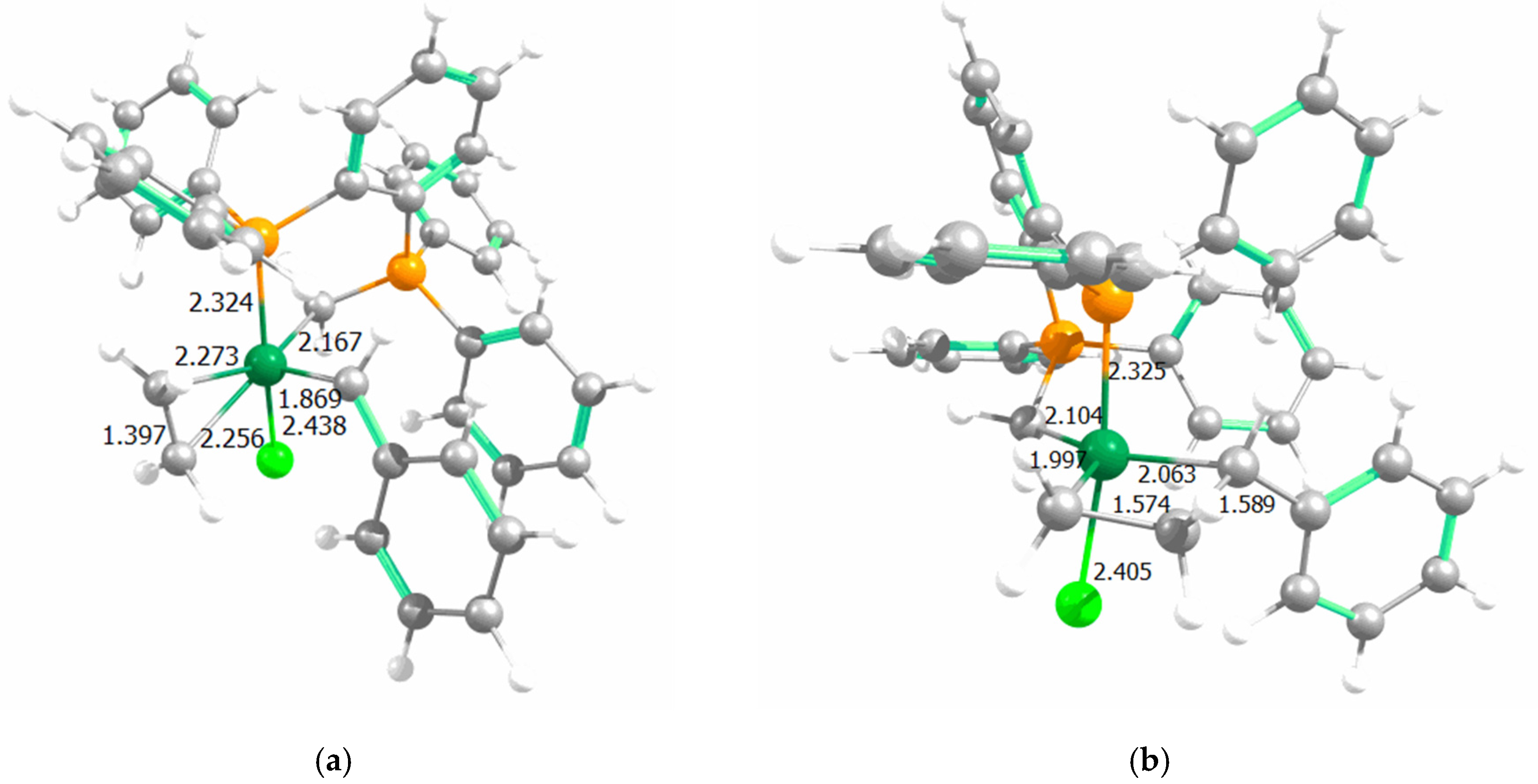

{kind=link}
{kind=link}
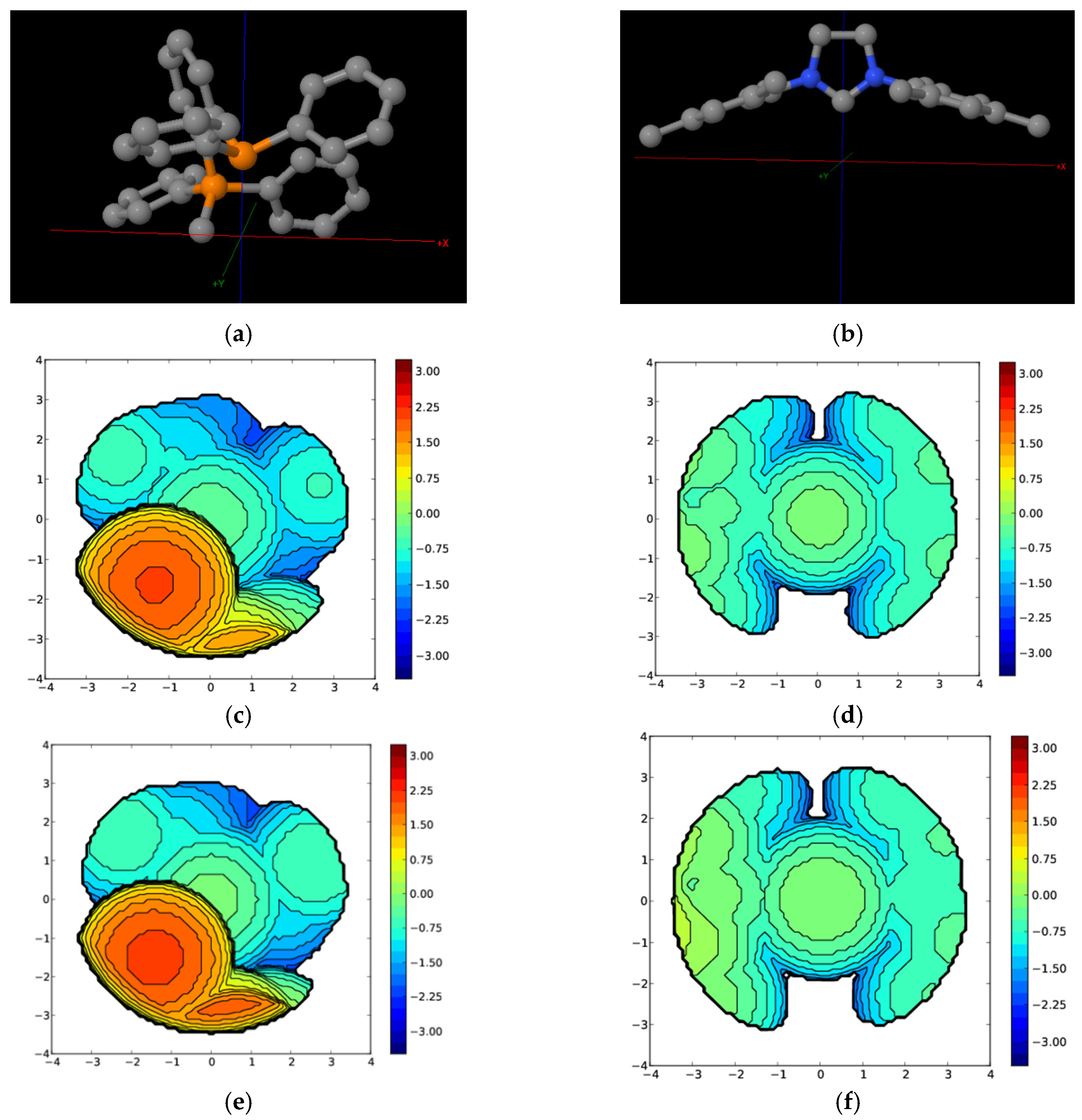
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | %VBur | |
|---|---|---|
| A | B | |
| PPh3 | 45.8 (77.6/33.1/28.2/44.3) | 33.4 (32.3/35.3/35.1/30.7) |
| 14e1 | 48.5 (80.6/35.8/31.8/45.6) | 36.4 (38.4/38.4/36.8/31.9) |
| Complex | Species | Ru–Cl | Ru–Pphosphine | Ru=Cmethylenic | Ru=Cylidene | Ru–P(Ph3) |
|---|---|---|---|---|---|---|
| A | PPh3 | 2.427 | 2.338 | 2.109 | 1.857 | 2.429 |
| 14e1 | 2.307 | 2.200 | 2.093 | 1.850 | - | |
| CI1 | 2.403 | 2.376 | 2.129 | 1.864 | - | |
| MCy1 | 2.432 | 2.345 | 2.144 | 2.067 | - | |
| CI2 | 2.397 | 2.324 | 2.137 | 2.456 | - | |
| 14e2 | 2.306 | 2.235 | 2.111 | - | - | |
| CI3 | 2.387 | 2.407 | 2.145 | 1.829 | - | |
| MCy2 | 2.408 | 2.339 | 2.149 | 1.994 | - | |
| CI4 | 2.465 | 2.397 | 2.170 | 1.819 | - | |
| - | Ru–Cl | Ru–CNHC | Ru–Clbis | Ru=Cylidene | Ru–P(Ph3) | |
| B | PPh3 | 2.442 | 2.039 | 2.421 | 1.850 | 2.400 |
| 14e1 | 2.323 | 1.921 | 2.331 | 1.845 | - | |
| CI1 | 2.403 | 1.998 | 2.416 | 1.870 | - | |
| MCy1 | 2.409 | 1.997 | 2.418 | 2.001 | - |
| Complex | Species | Ru–Cl | Ru–Pphosphine | Ru=Cmethylenic | Ru=Cylidene | Ru–P(Ph3) |
|---|---|---|---|---|---|---|
| A | PPh3 | 1.047 | 0.662 | 0.867 | 1.709 | 0.533 |
| 14e1 | 1.150 | 1.073 | 0.843 | 1.671 | - | |
| CI1 | 1.071 | 0.619 | 0.834 | 1.677 | - | |
| MCy1 | 1.199 | 0.678 | 0.841 | 0.935 | - | |
| B | - | Ru–Cl | Ru–CNHC | Ru–Clbis | Ru=Cylidene | Ru–P(Ph3) |
| PPh3 | 0.894 | 1.712 | 1.068 | 0.887 | 0.543 | |
| 14e1 | 1.111 | 1.648 | 1.114 | 1.249 | - | |
| CI1 | 1.021 | 1.569 | 1.153 | 0.978 | - | |
| MCy1 | 1.029 | 1.009 | 1.099 | 0.945 | - |
| Complex | Species | Ru–Cl | Ru–Pphosphine | Ru=Cylidene | Ru=Cylidene | Ru–P(Ph3) |
|---|---|---|---|---|---|---|
| A | PPh3 | −0.265 | 1.168 | −0.947 | 0.006 | 1.129 |
| 14e1 | −0.199 | 1.330 | −0.938 | −0.066 | - | |
| CI1 | −0.240 | 1.172 | −0.924 | 0.017 | - | |
| MCy1 | −0.312 | 1.203 | −0.945 | −0.155 | −0.315 | |
| - | Ru–Cl | Ru–CNHC | Ru–Cl | Ru=Cylidene | Ru–P(Ph3) | |
| B | PPh3 | −0.311 | −0.012 | −0.316 | 0.429 | 1.131 |
| 14e1 | −0.255 | −0.026 | −0.249 | 0.505 | - | |
| CI1 | −0.296 | 0.033 | −0.299 | 0.462 | - | |
| MCy1 | −0.307 | −0.151 | −0.294 | 0.482 | −0.328 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnedo, L.; Chauvin, R.; Poater, A. Olefin Metathesis with Ru-Based Catalysts Exchanging the Typical N-Heterocyclic Carbenes by a Phosphine–Phosphonium Ylide. Catalysts 2017, 7, 85. https://doi.org/10.3390/catal7030085
Arnedo L, Chauvin R, Poater A. Olefin Metathesis with Ru-Based Catalysts Exchanging the Typical N-Heterocyclic Carbenes by a Phosphine–Phosphonium Ylide. Catalysts. 2017; 7(3):85. https://doi.org/10.3390/catal7030085
Chicago/Turabian StyleArnedo, Laia, Remi Chauvin, and Albert Poater. 2017. "Olefin Metathesis with Ru-Based Catalysts Exchanging the Typical N-Heterocyclic Carbenes by a Phosphine–Phosphonium Ylide" Catalysts 7, no. 3: 85. https://doi.org/10.3390/catal7030085