Metal Oxide Nanoparticles Supported on Macro-Mesoporous Aluminosilicates for Catalytic Steam Gasification of Heavy Oil Fractions for On-Site Upgrading

Abstract
:1. Introduction
2. Results and Discussion
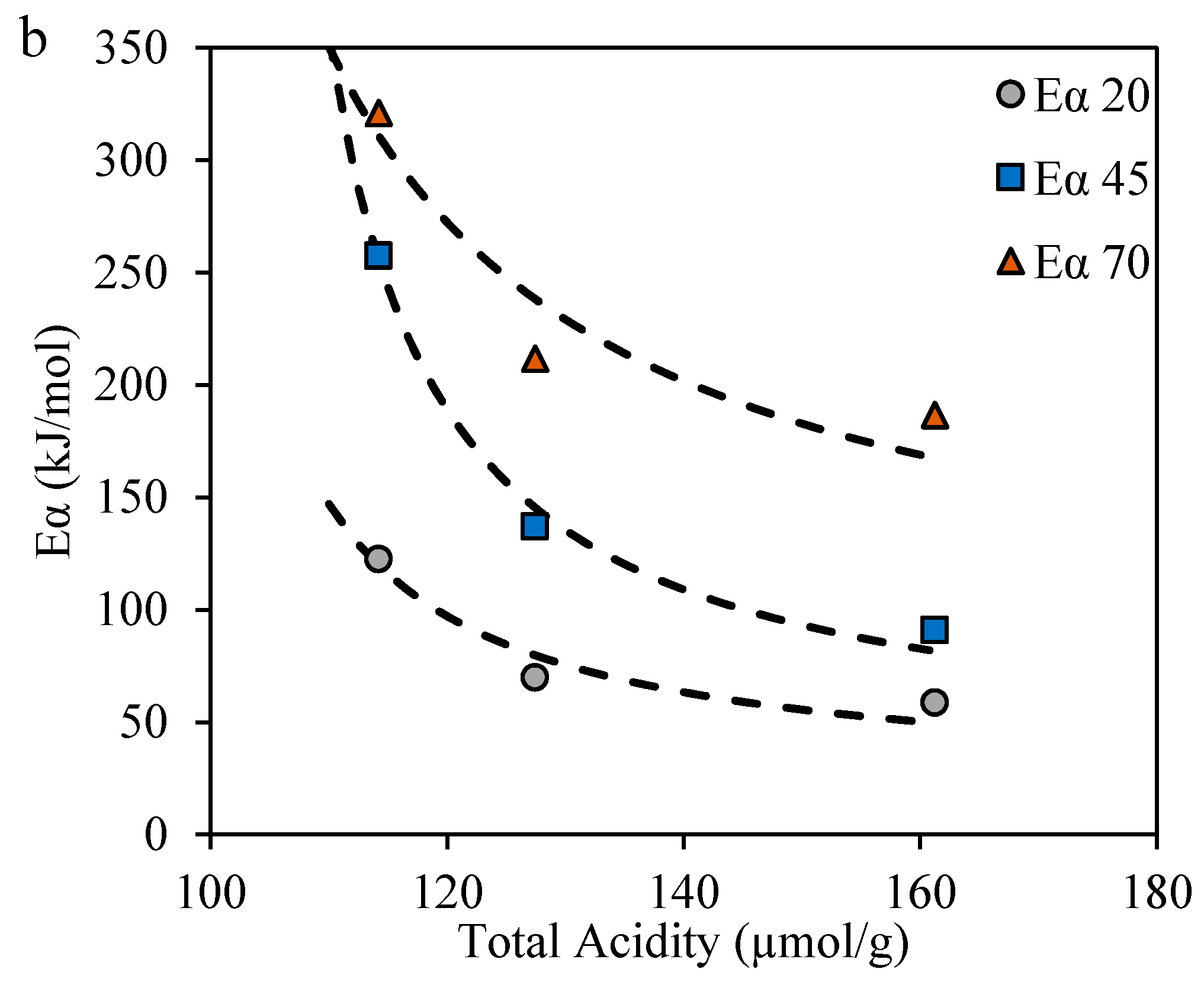
2.1. Catalyst Characterization
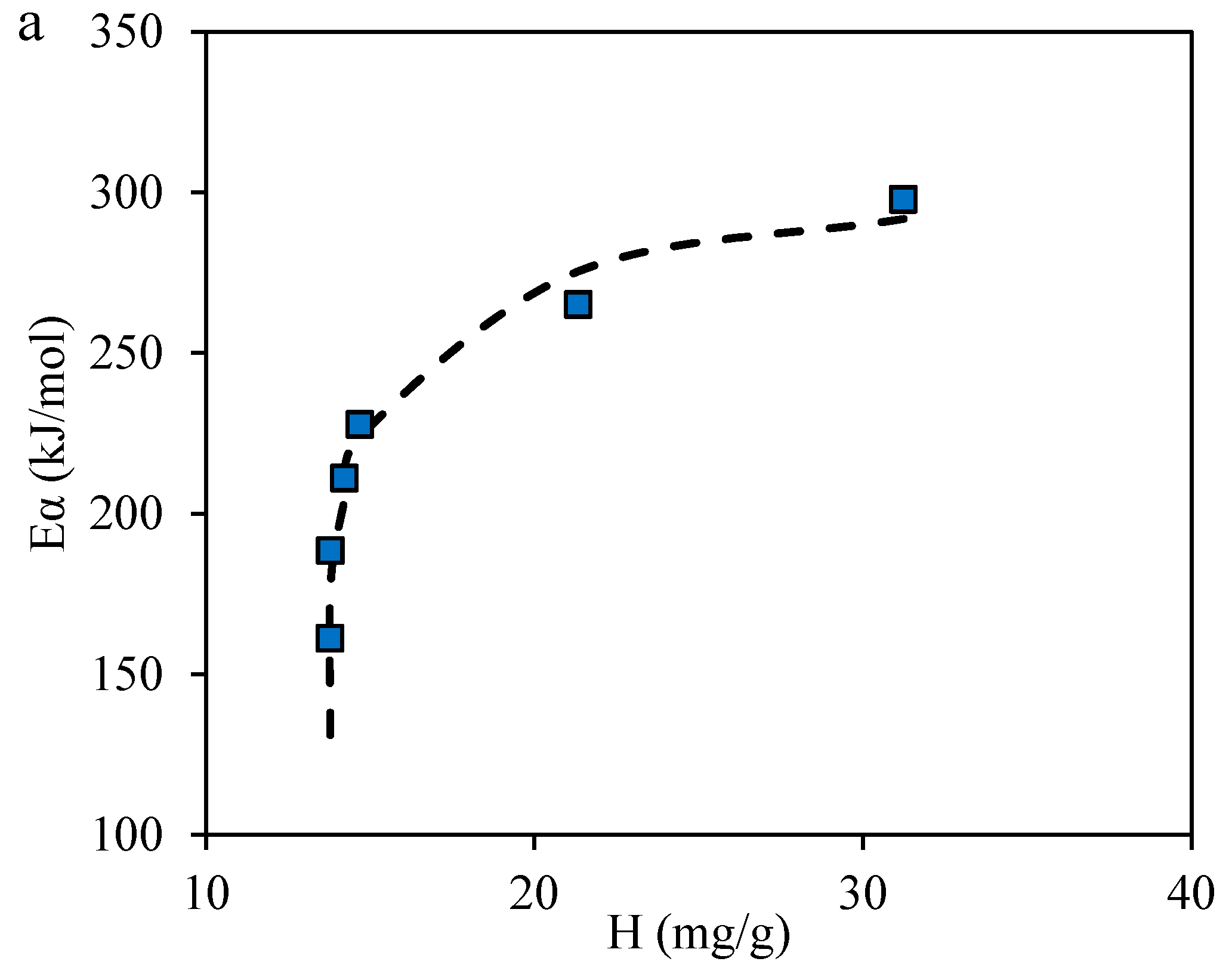
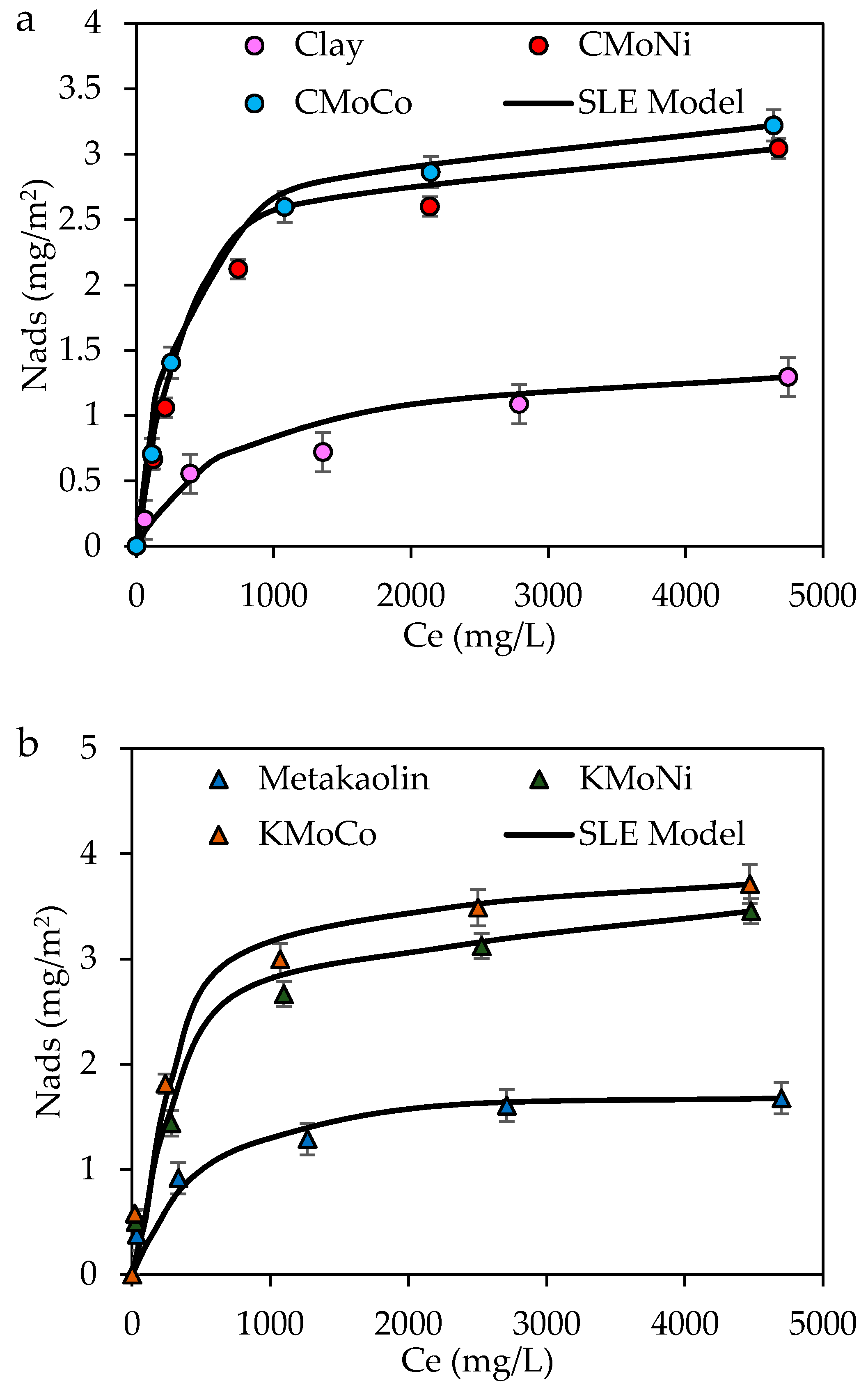
2.2. Adsorption Isotherms of Resins and Asphaltenes
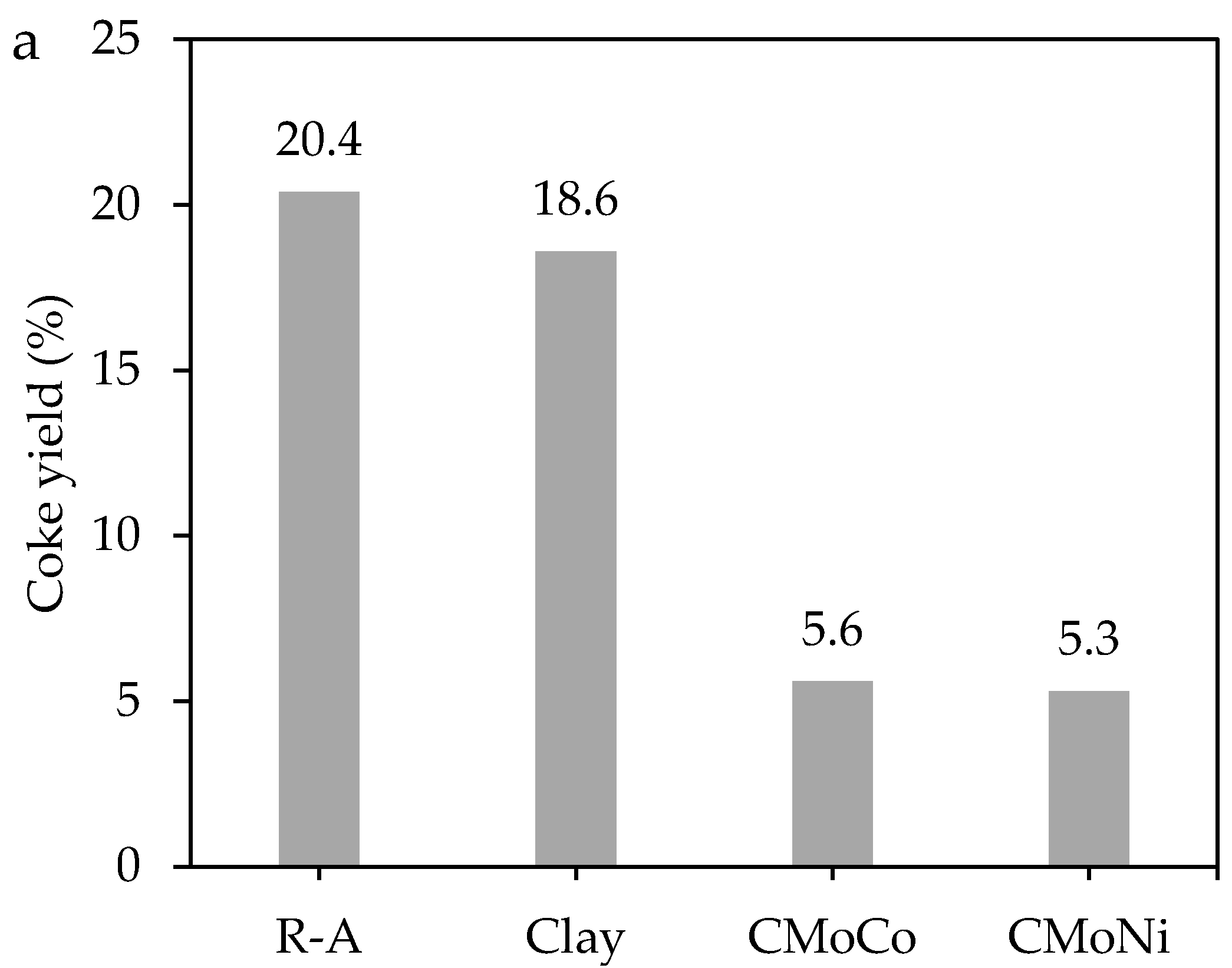
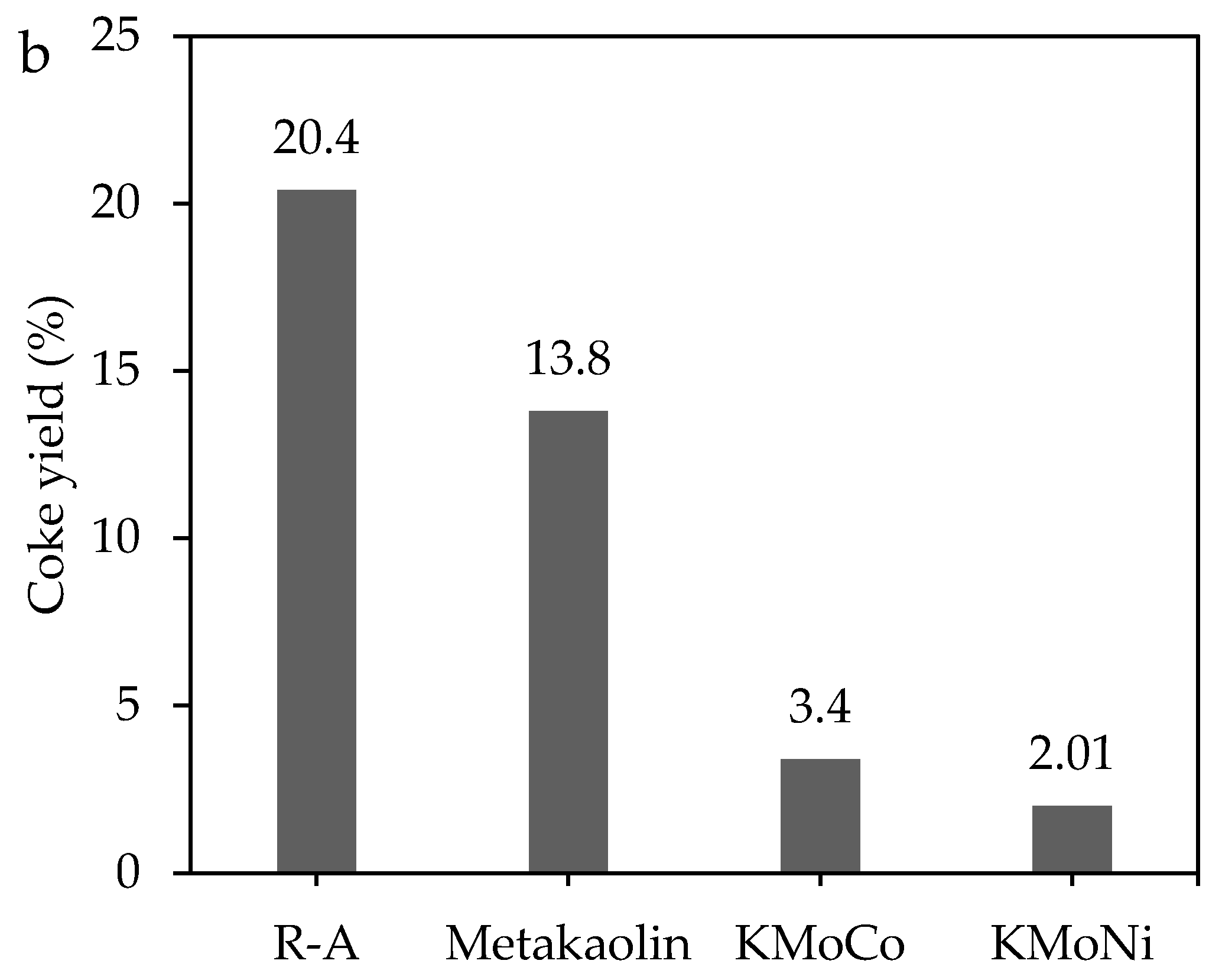
2.3. Catalytic Steam Gasification of Heavy Fractions
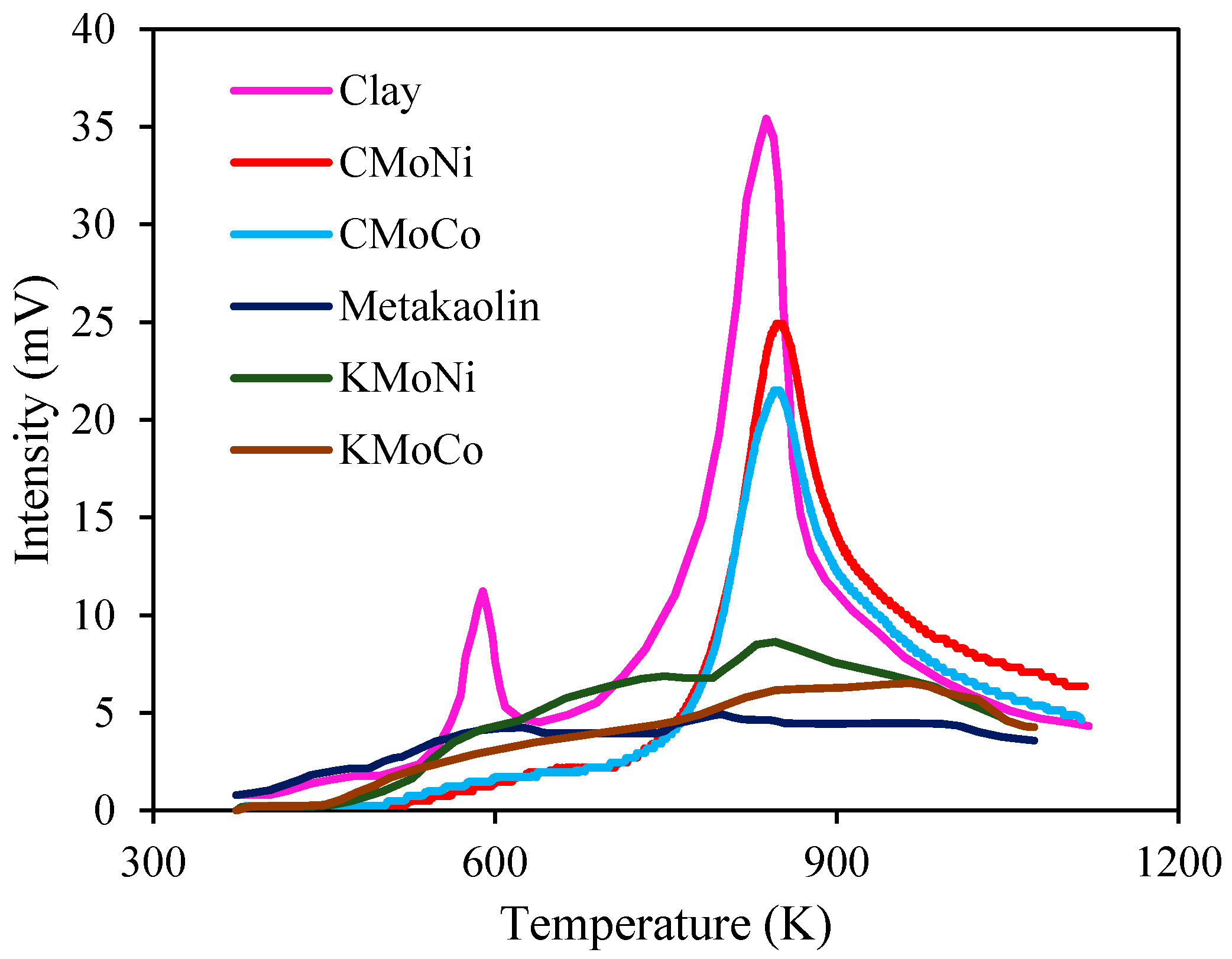
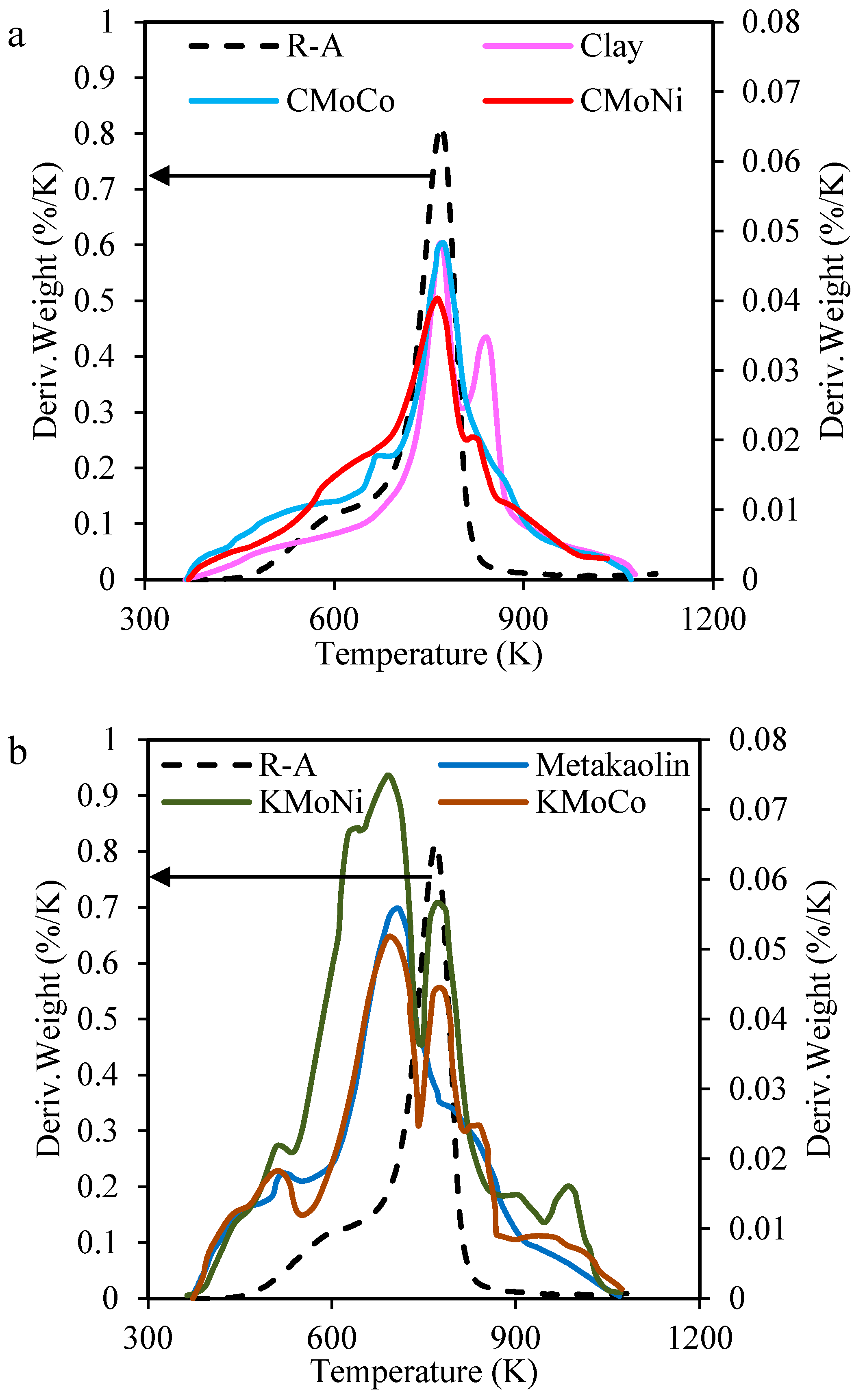
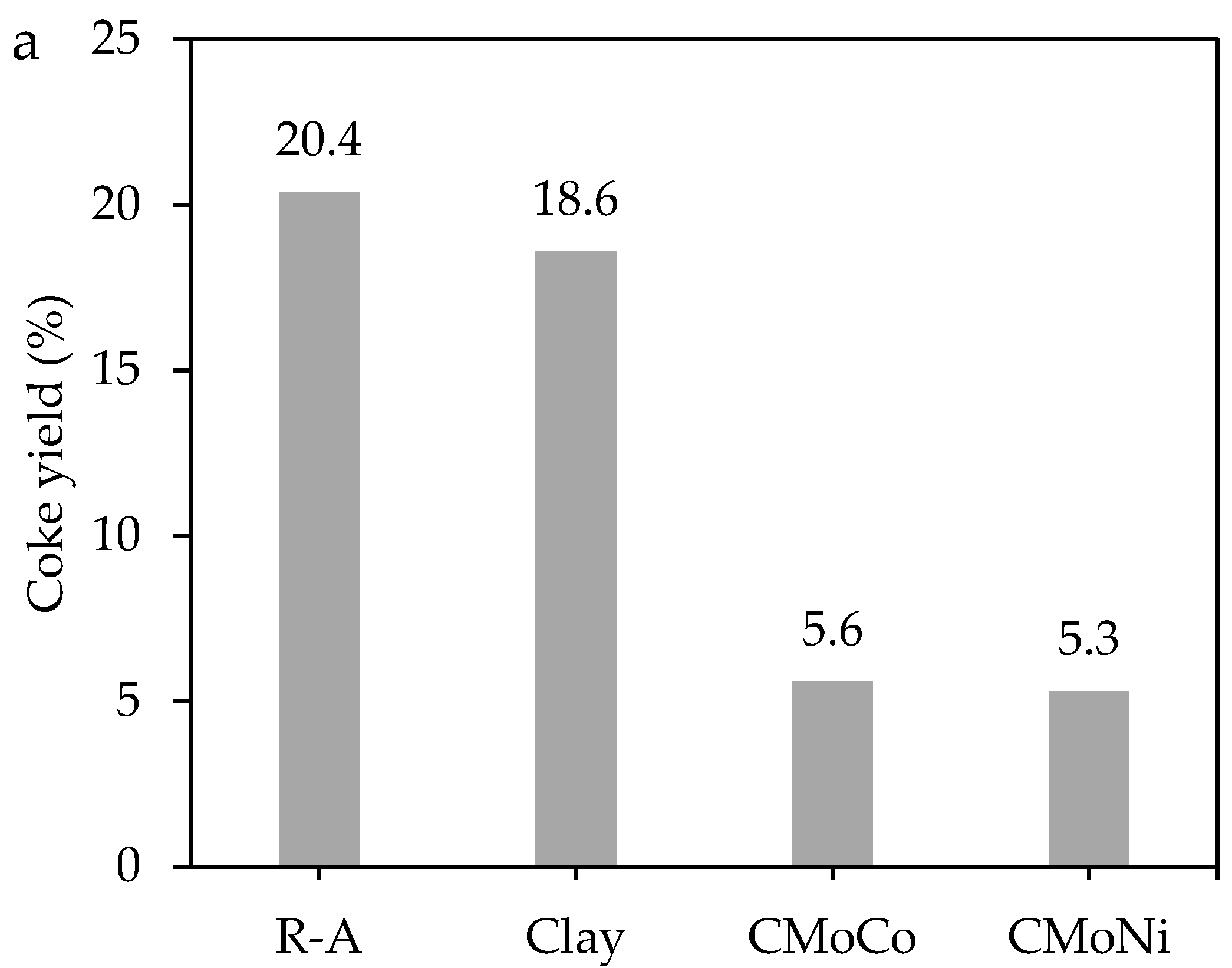
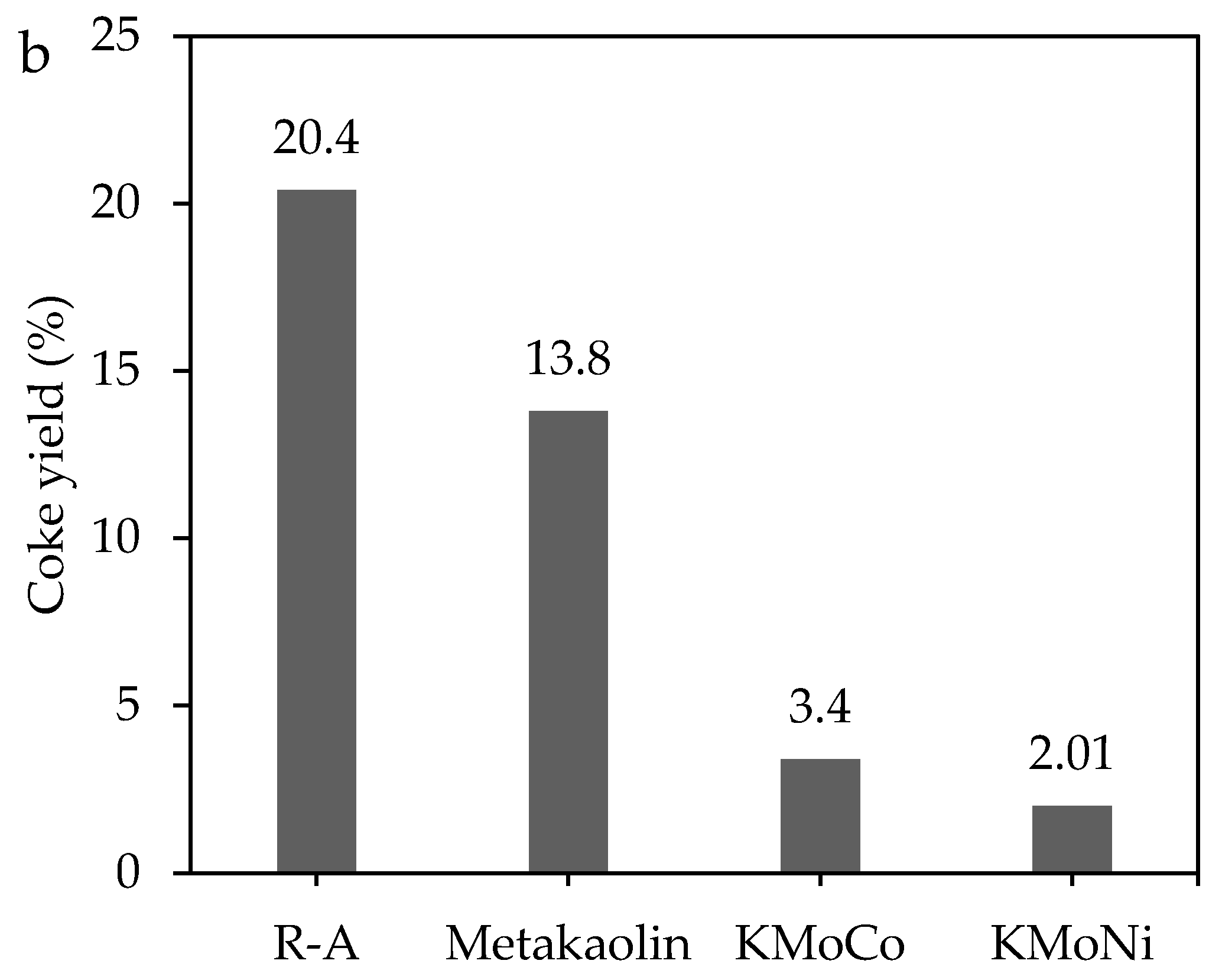
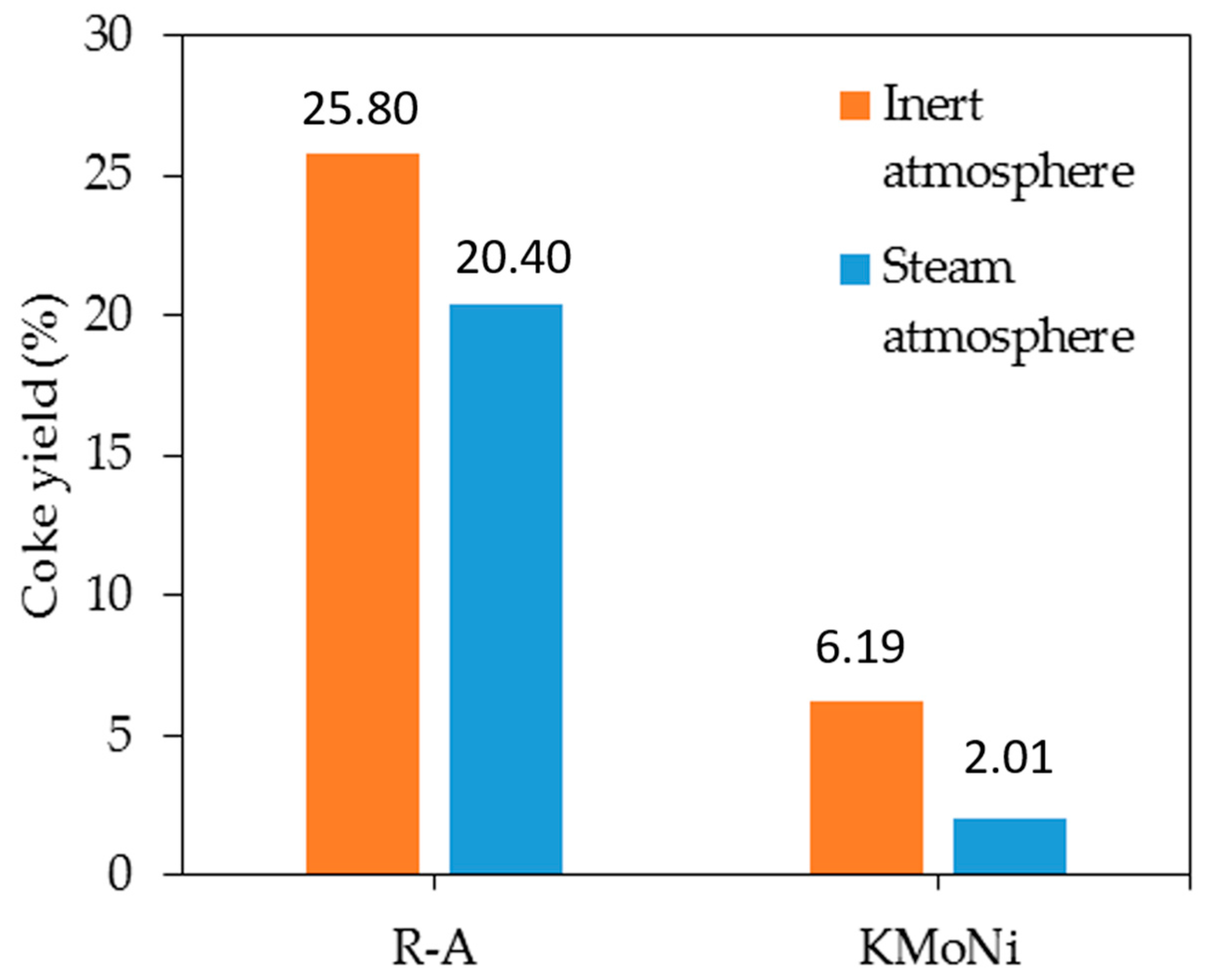
2.3.1. Mass Loss Analysis
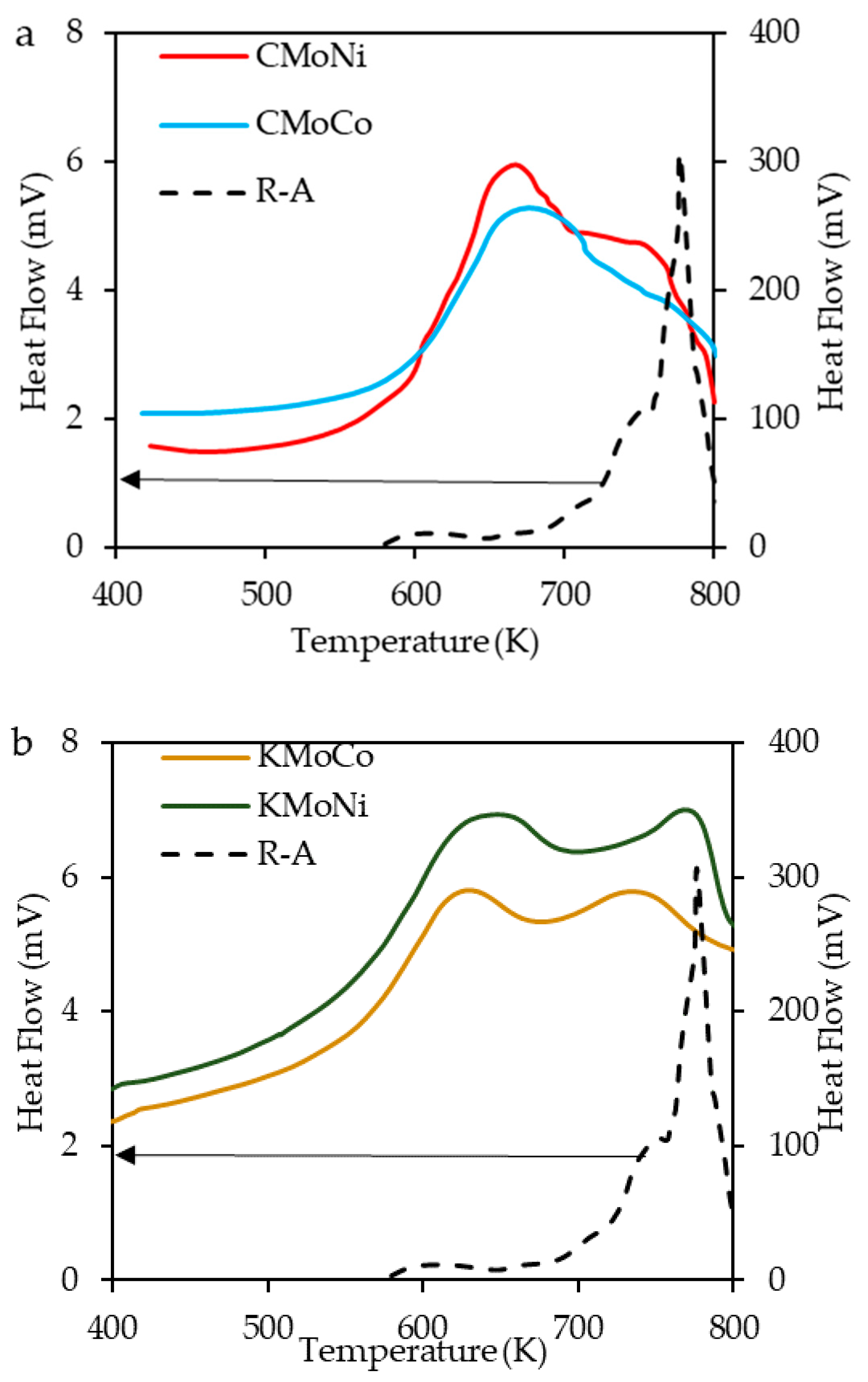
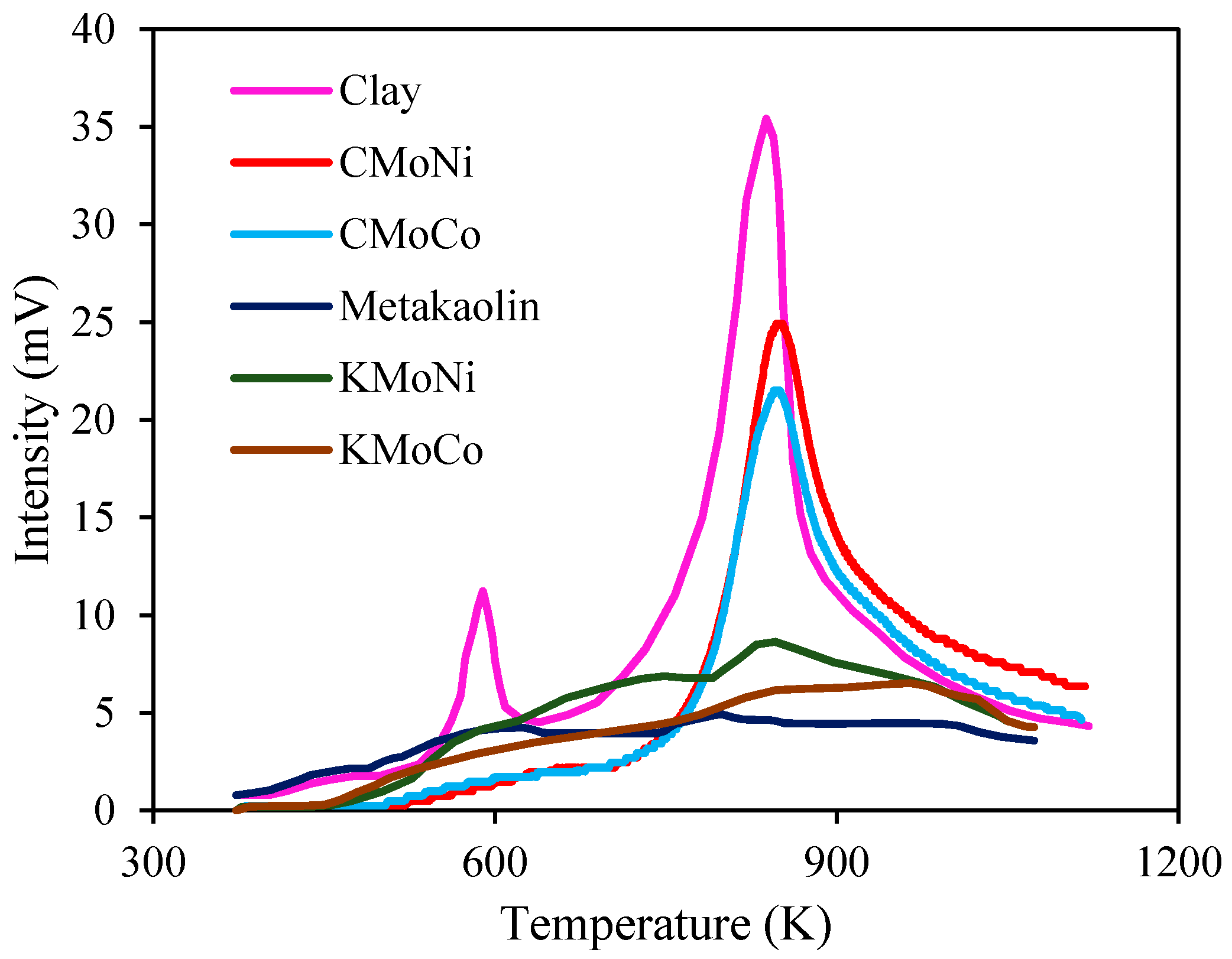
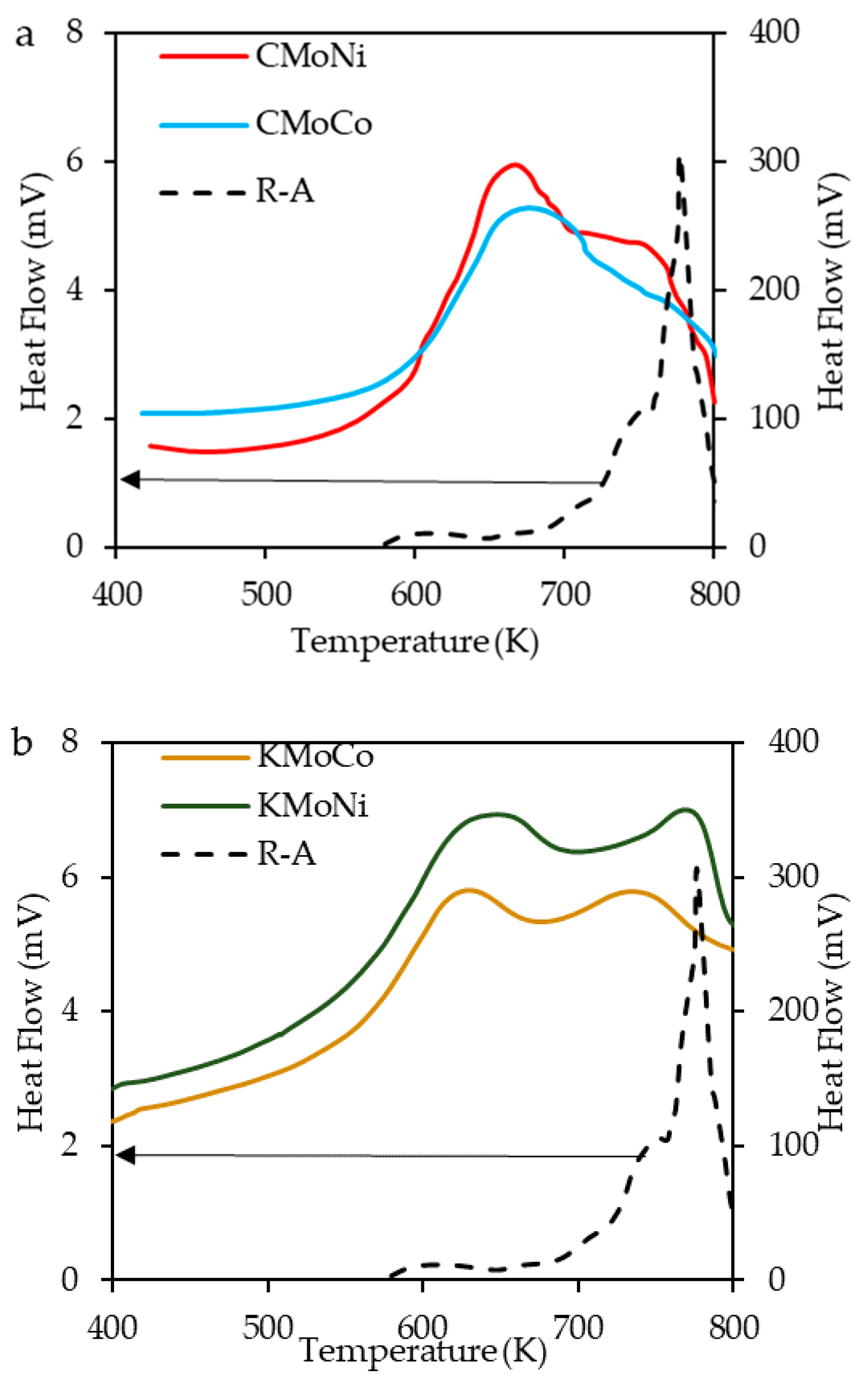
2.3.2. Differential Scanning Calorimetry (DSC) Analysis
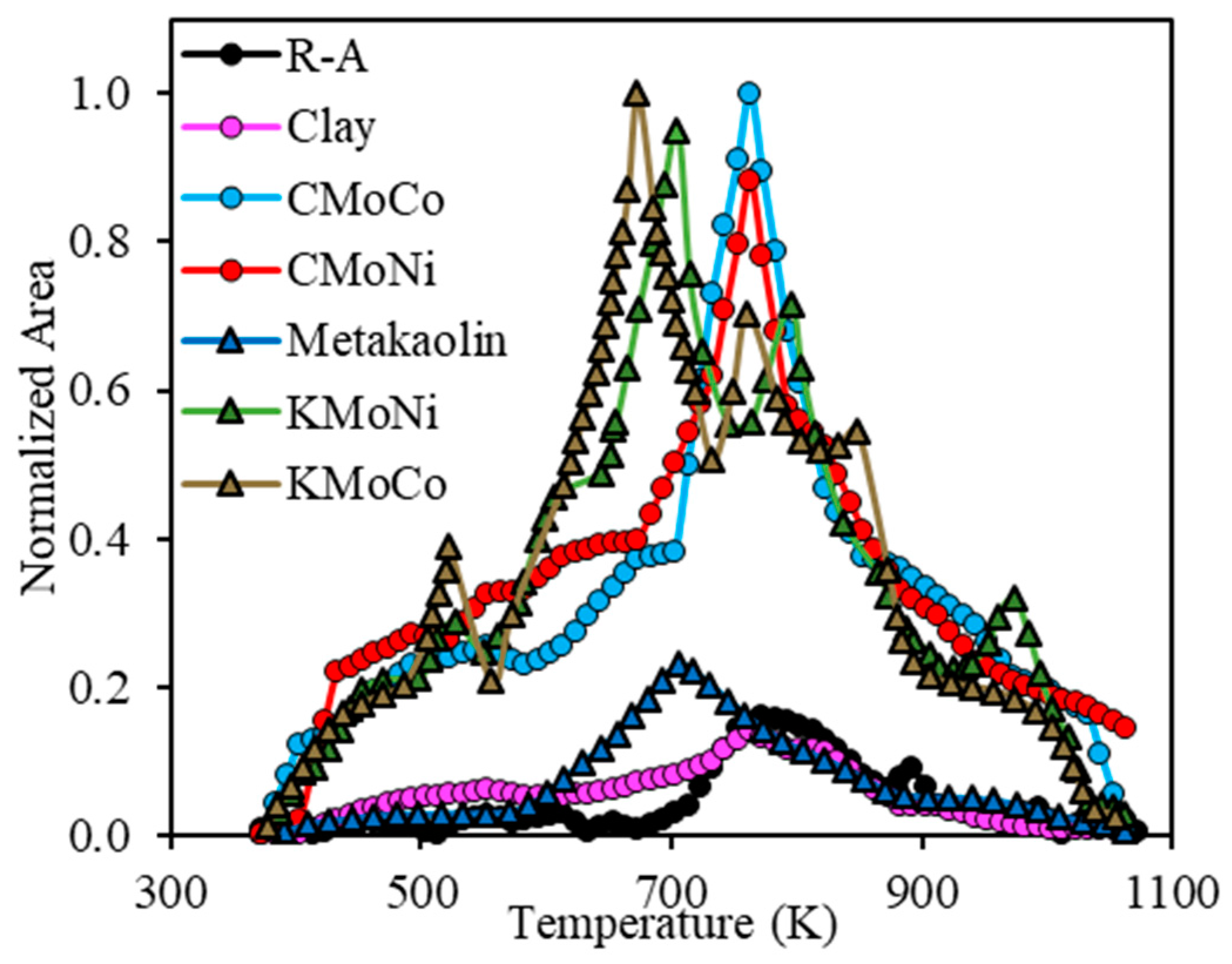
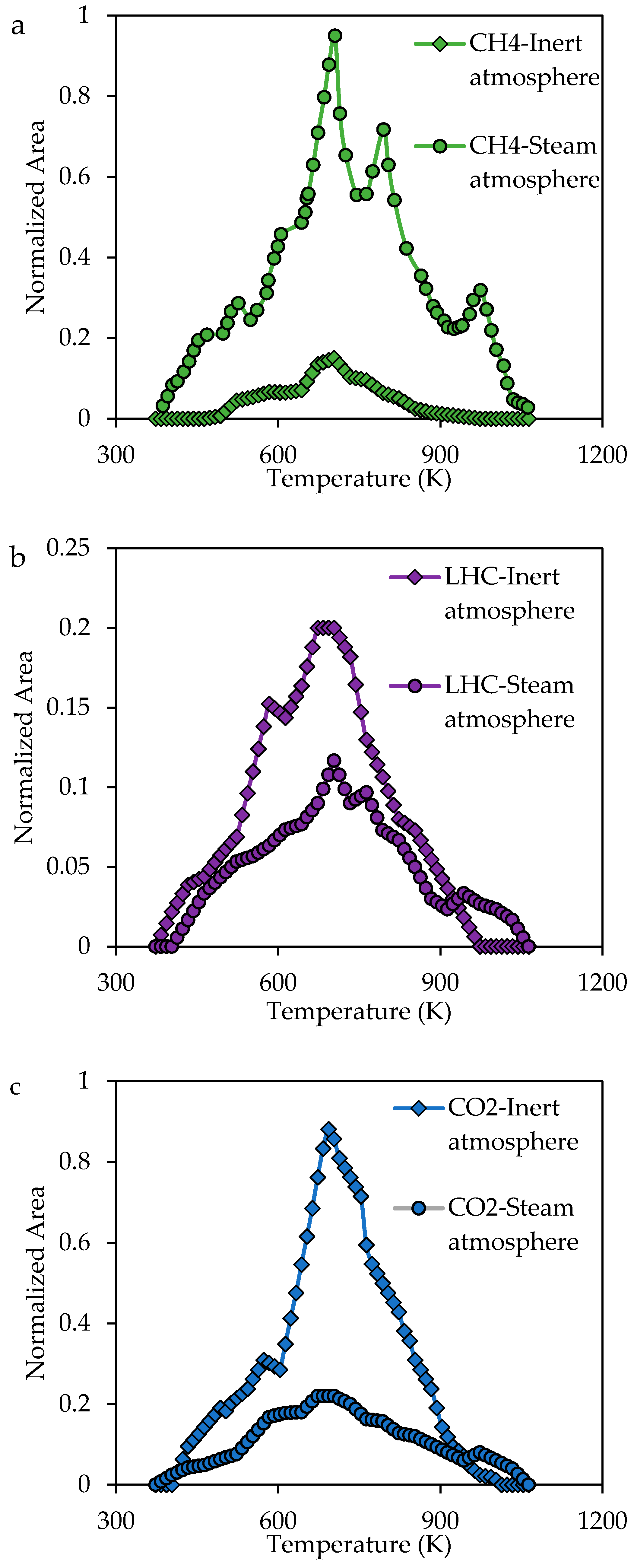
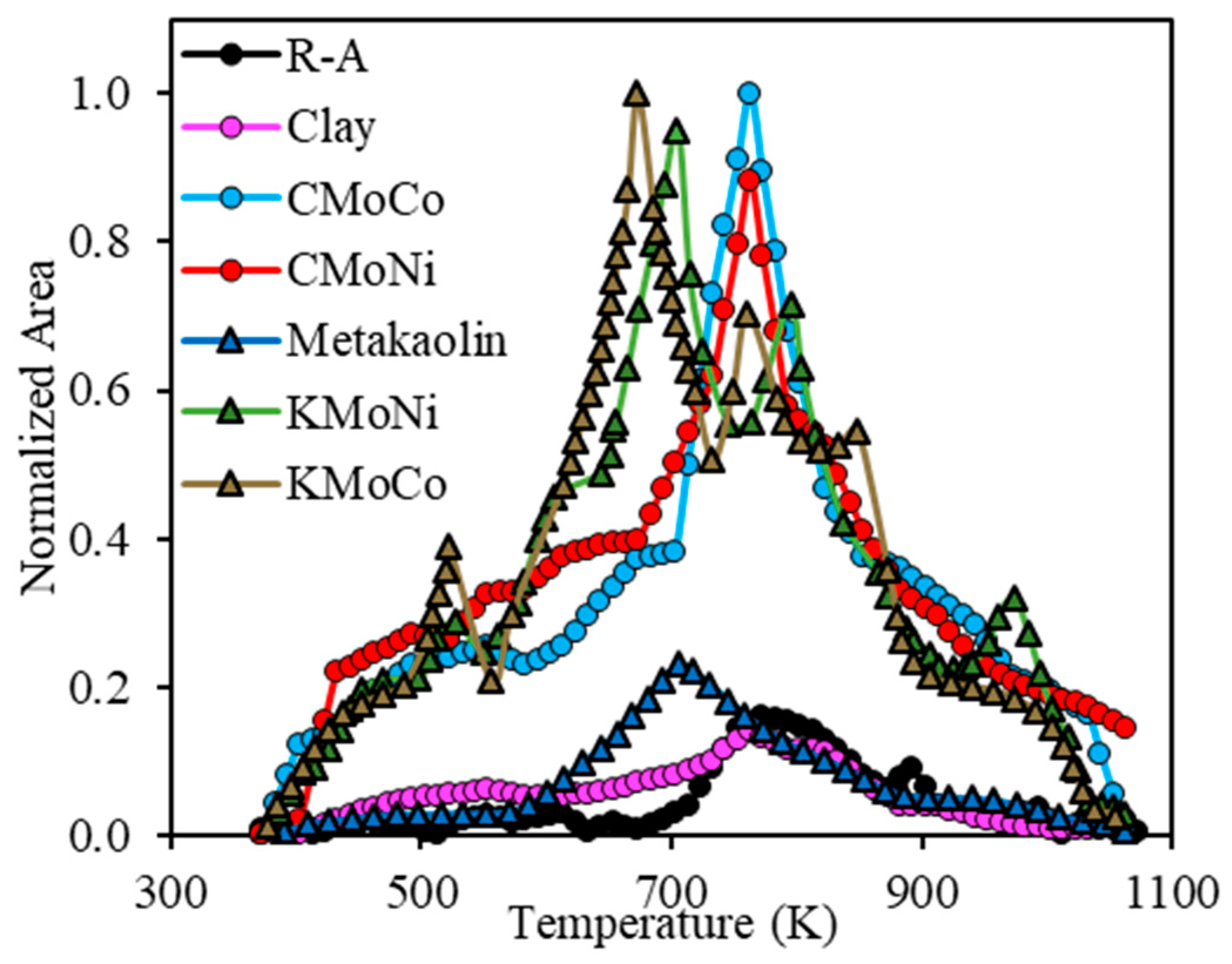
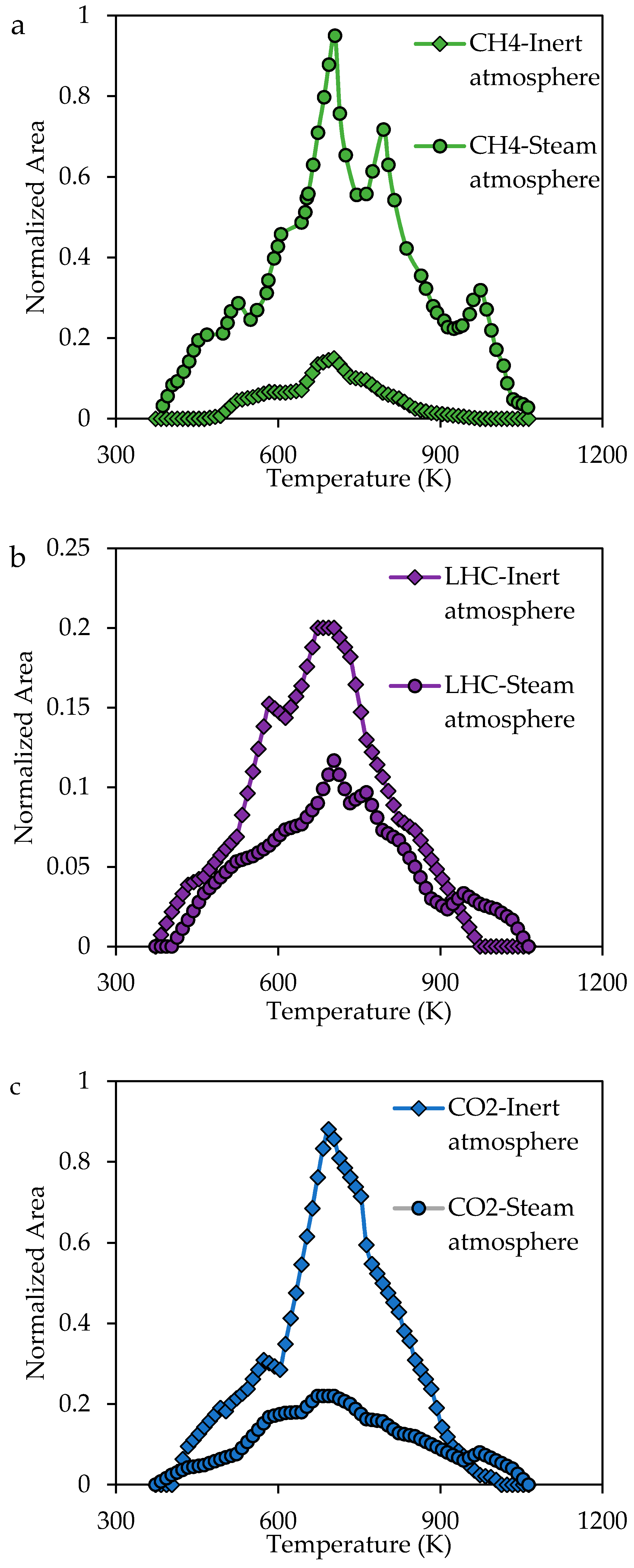
2.3.3. Analysis of the Evolution of the Gaseous Product during the Steam Gasification Process
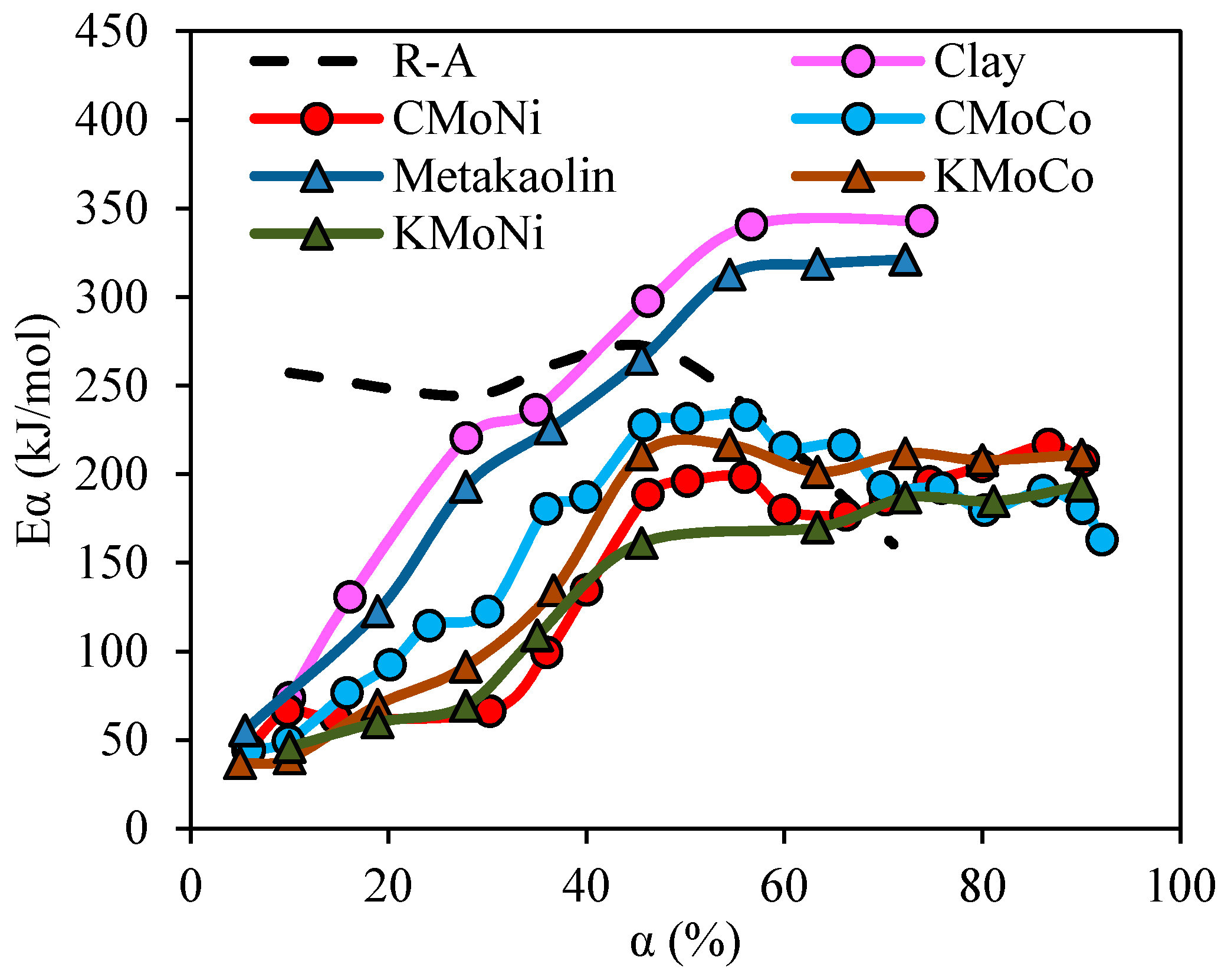
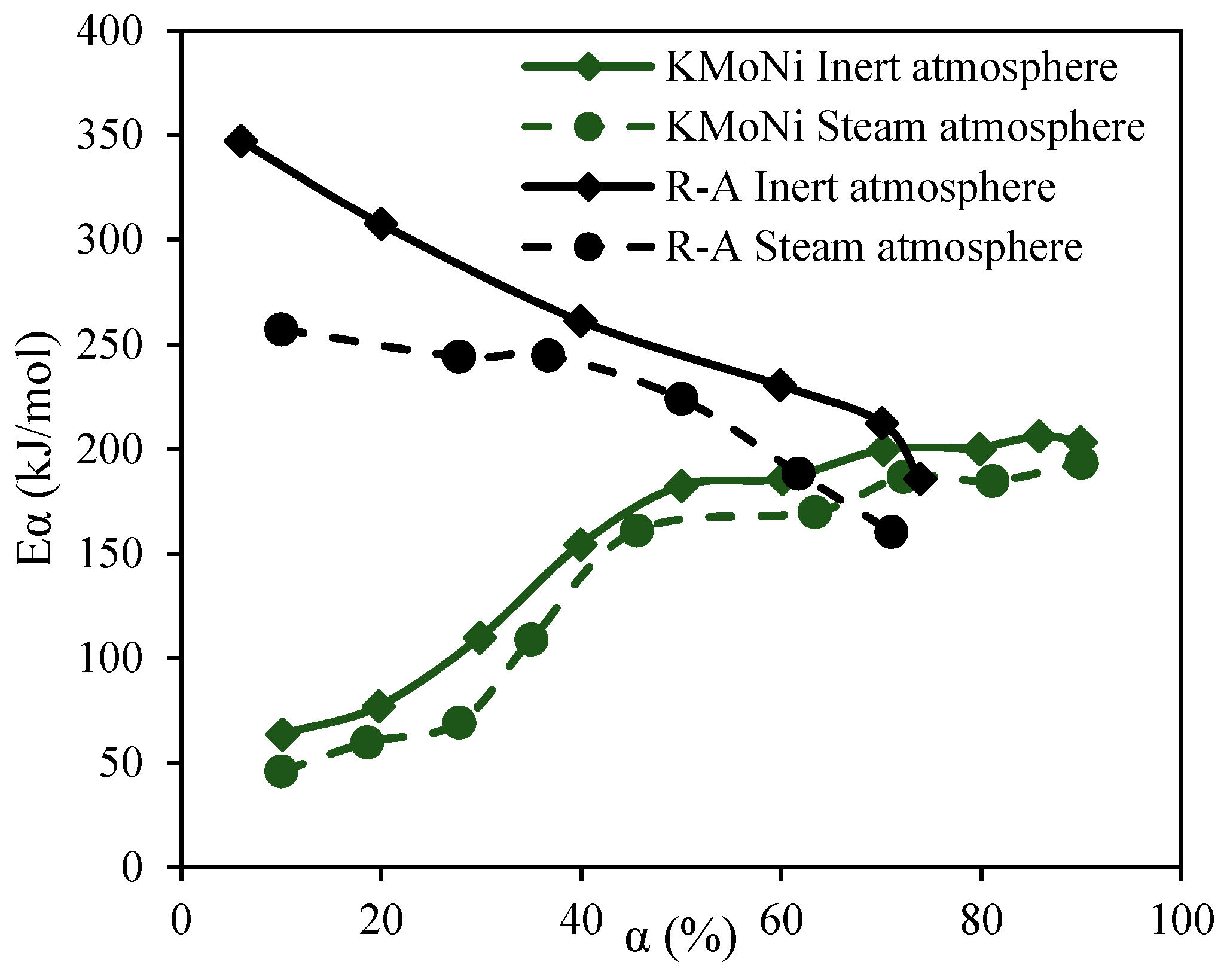
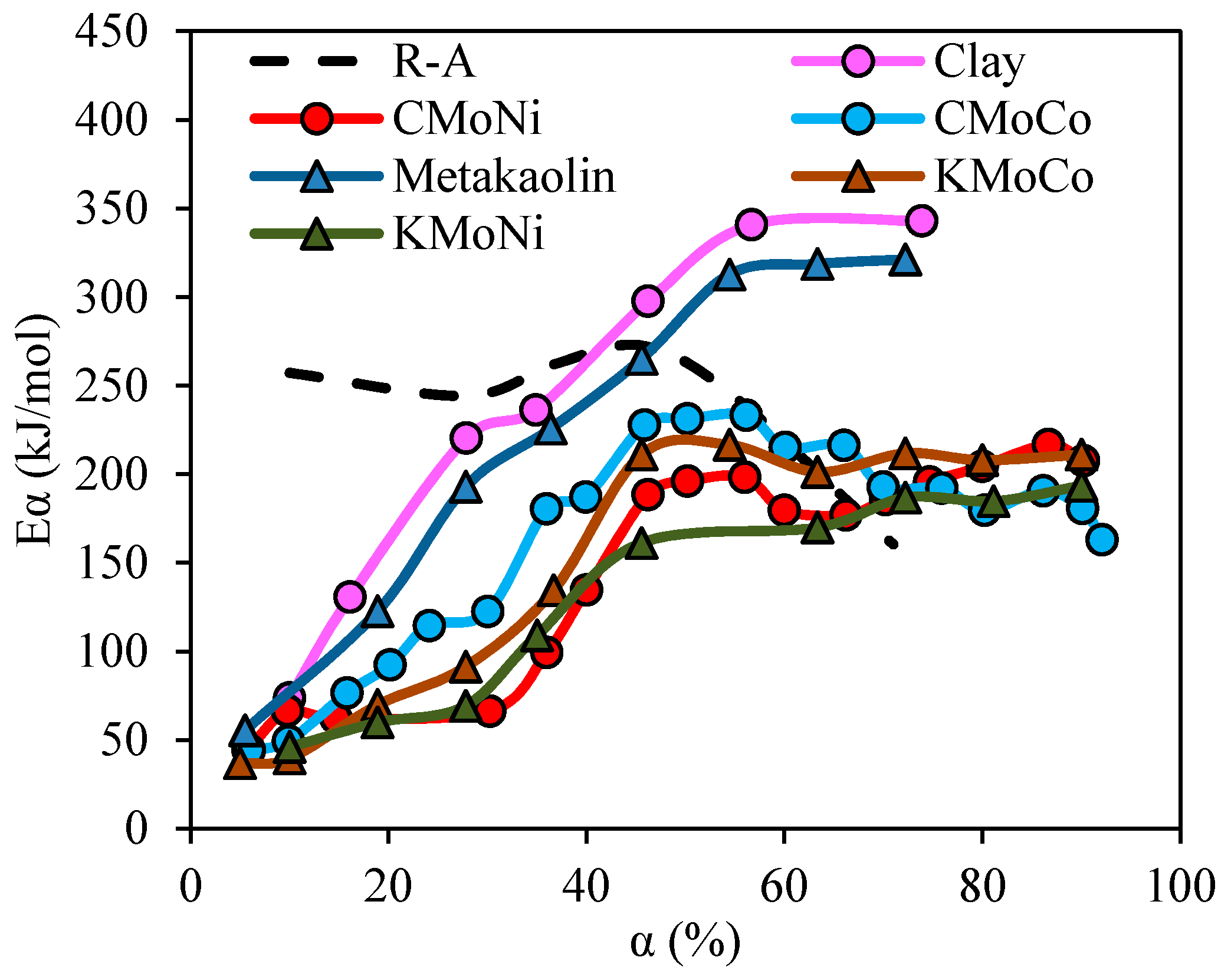
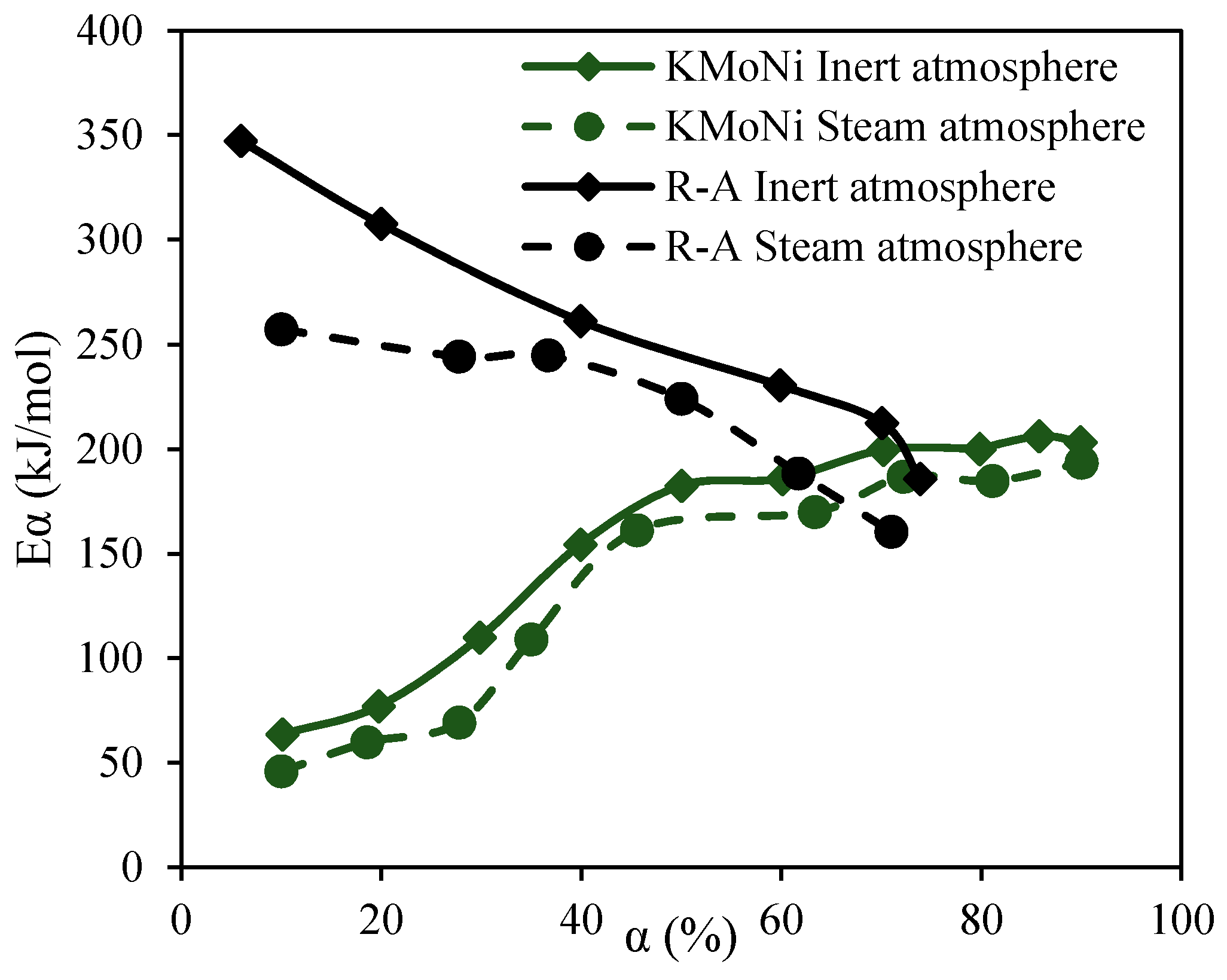
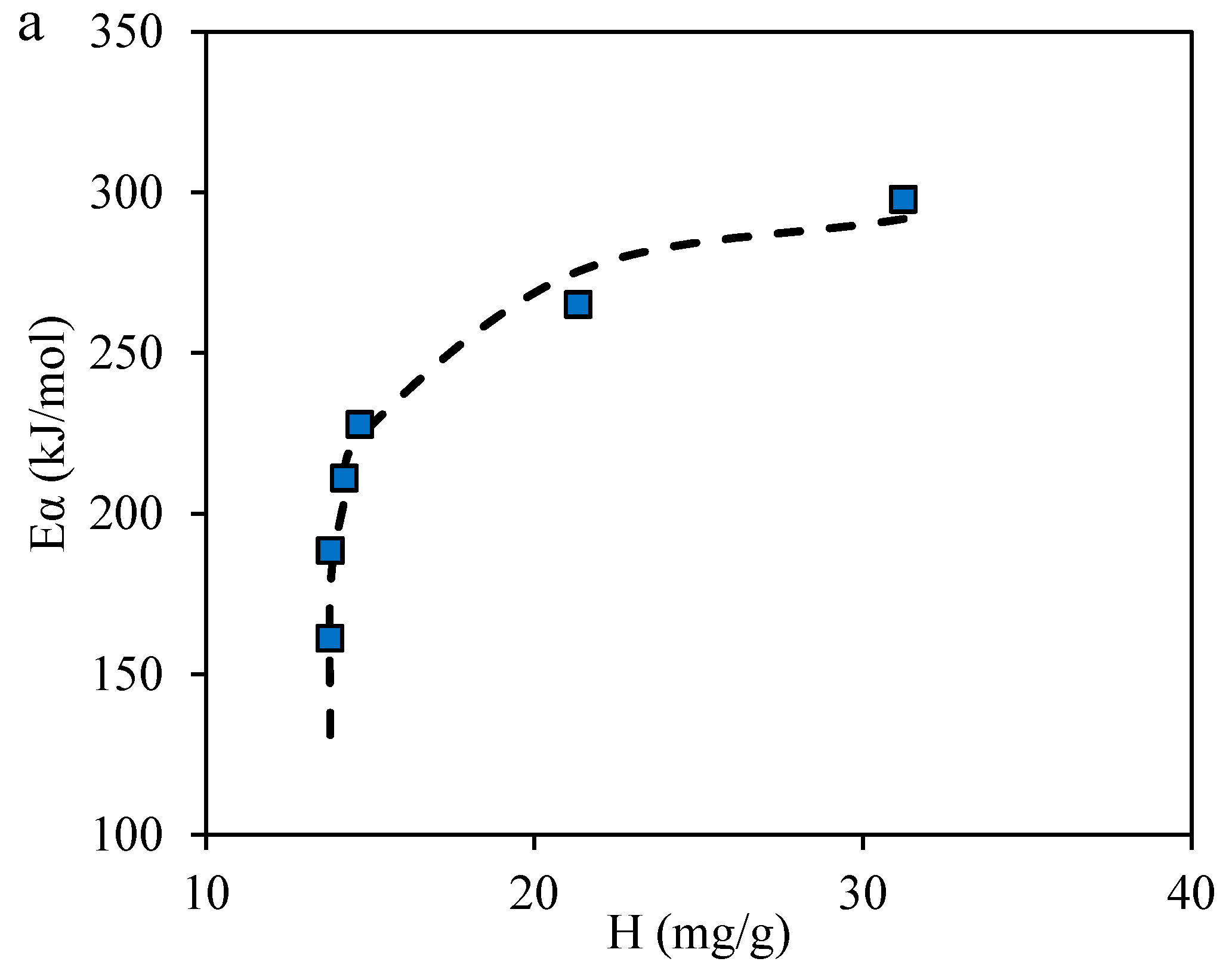
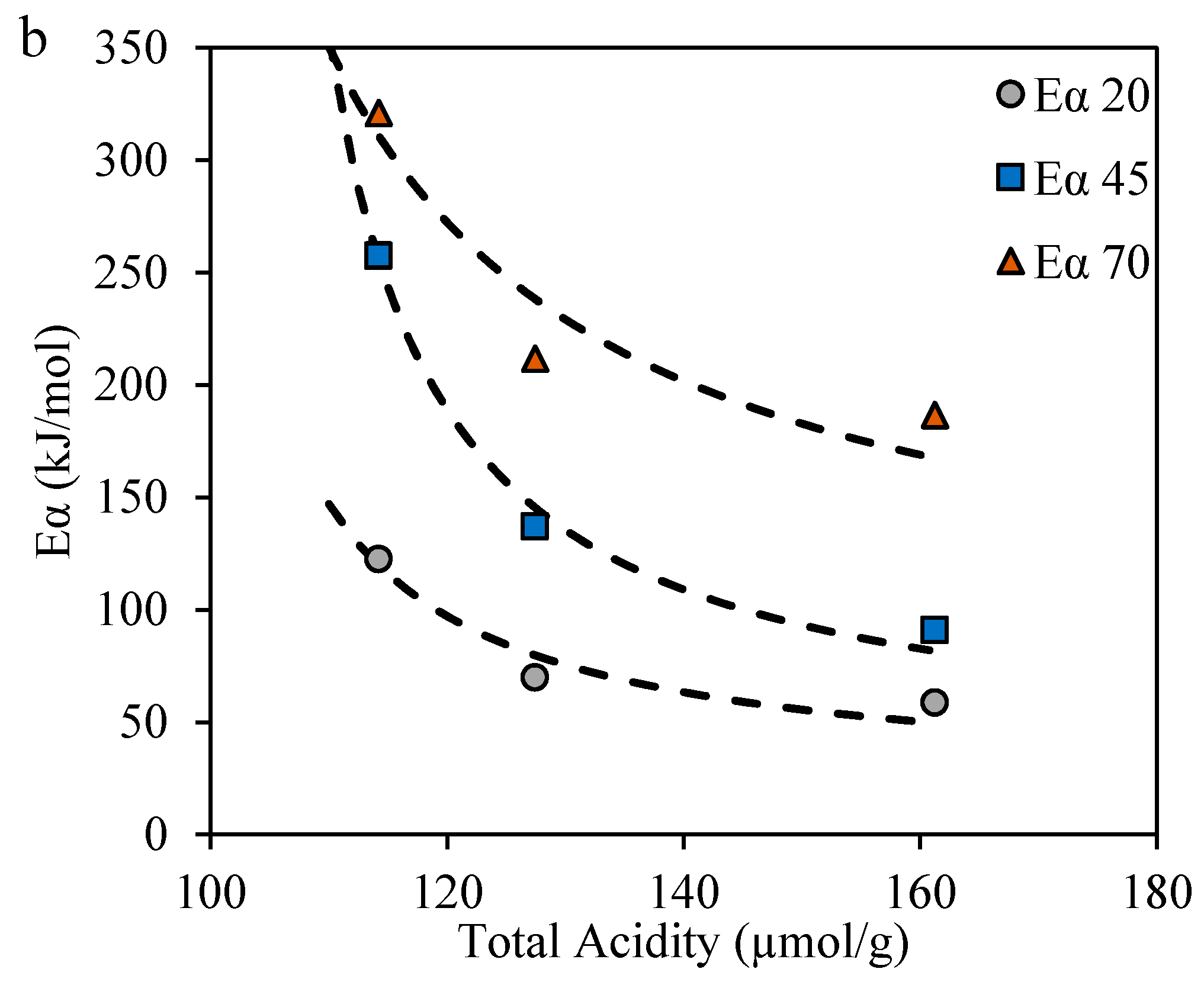
2.3.4. Effective Activation Energies
3. Materials and Methods
3.1. Materials and Chemical
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Batch Adsorption Experiments
3.5. Catalytic Steam Gasification of Asphaltenes and Resins
4. Modeling
4.1. Solid–Liquid Equilibrium (SLE) Model
4.2. Estimation of the Effective Activation Energy for R–A Decomposition
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chew, K.J. The future of oil: Unconventional fossil fuels. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2014, 372, 20120324. [Google Scholar] [CrossRef] [PubMed]
- Shokrlu, Y.H.; Babadagli, T. Viscosity reduction of heavy oil/bitumen using micro-and nano-metal particles during aqueous and non-aqueous thermal applications. J. Pet. Sci. Eng. 2014, 119, 210–220. [Google Scholar] [CrossRef]
- Ashrafizadeh, S.; Motaee, E.; Hoshyargar, V. Emulsification of heavy crude oil in water by natural surfactants. J. Pet. Sci. Eng. 2012, 86, 137–143. [Google Scholar] [CrossRef]
- Tedeschi, M. Reserves and Production Of Heavy Crude Oil and Natural Bitumen. In Proceedings of the 13th World Petroleum Congress, Buenos Aires, Argentina, 20–25 October 1991. [Google Scholar]
- Salameh, M.G. The potential of unconventional oil resources: Between expediency & reality. Int. Assoc. Energy Econ. 2012, Fourth Quarter, 17–20. [Google Scholar]
- Barrero, R.; Afanador, L.E.; Leal, G.; Grosso, J.L.; Parra, M.; Cuadrado, C.E.; Vidales, H.; Guzman, E.; Rodriguez, L. Method For the Well-Head Treatment of Heavy and Extra-Heavy Crudes in Order to Improve the Transport Conditions Thereof. U.S. Patents 8257579 B2, 4 September 2012. [Google Scholar]
- Franco, C.A.; Nassar, N.N.; Montoya, T.; Cortés, F.B.; Ambrosio, J. NiO and PdO Supported on Fumed Silica Nanoparticles for Adsorption and Catalytic Steam Gasification of Colombian C7 Asphaltenes. In Handbook on Oil Production Research; Nova Science Publishers: Hauppauge, NY, USA, 2014; pp. 101–145. [Google Scholar]
- U.S. E.I. Administration. Country Analysis Brief: Colombia; EIA: Washington, DC, USA, 2016. [Google Scholar]
- Martínez-Palou, R.; de Lourdes Mosqueira, M.; Zapata-Rendón, B.; Mar-Juárez, E.; Bernal-Huicochea, C.; de la Cruz Clavel-López, J.; Aburto, J. Transportation of heavy and extra-heavy crude oil by pipeline: A review. J. Pet. Sci. Eng. 2011, 75, 274–282. [Google Scholar] [CrossRef]
- Hashemi, R.; Nassar, N.N.; Almao, P.P. Nanoparticle technology for heavy oil in-situ upgrading and recovery enhancement: Opportunities and challenges. Appl. Energy 2014, 133, 374–387. [Google Scholar] [CrossRef]
- Shah, A.; Fishwick, R.; Wood, J.; Leeke, G.; Rigby, S.; Greaves, M. A review of novel techniques for heavy oil and bitumen extraction and upgrading. Energy Environ. Sci. 2010, 3, 700–714. [Google Scholar] [CrossRef]
- Ghanavati, M.; Shojaei, M.-J.; SA, R.S. Effects of asphaltene content and temperature on viscosity of Iranian heavy crude oil: Experimental and modeling study. Energy Fuels 2013, 27, 7217–7232. [Google Scholar] [CrossRef]
- Hosseinpour, N.; Mortazavi, Y.; Bahramian, A.; Khodatars, L.; Khodadadi, A.A. Enhanced pyrolysis and oxidation of asphaltenes adsorbed onto transition metal oxides nanoparticles towards advanced in-situ combustion EOR processes by nanotechnology. Appl. Catal. A Gen. 2014, 477, 159–171. [Google Scholar] [CrossRef]
- Ancheyta, J.; Centeno, G.; Trejo, F.; Marroquin, G. Changes in asphaltene properties during hydrotreating of heavy crudes. Energy Fuels 2003, 17, 1233–1238. [Google Scholar] [CrossRef]
- Rana, M.S.; Samano, V.; Ancheyta, J.; Diaz, J. A review of recent advances on process technologies for upgrading of heavy oils and residua. Fuel 2007, 86, 1216–1231. [Google Scholar] [CrossRef]
- Saniere, A.; Hénaut, I.; Argillier, J. Pipeline transportation of heavy oils, a strategic, economic and technological challenge. Oil Gas Sci. Technol. 2004, 59, 455–466. [Google Scholar] [CrossRef]
- Argillier, J.; Henaut, I.; Heraud, J.-P.; Glenat, P. Heavy Oil Dilution. In Proceedings of the SPE International Thermal Operations and Heavy Oil Symposium, Calgary, AB, Canada, 1–3 November 2005. [Google Scholar]
- Almao, P.P.; Trujillo, G.L.; Peluso, E.; Galarraga, C.; Sosa, C.; Algara, C.S.; Lopez-Linares, F.; Ortega, L.A.C.; Reques, N.G.Z. Systems and Methods for Catalytic Steam Cracking of Non-Asphaltene Containing Heavy Hydrocarbons. U.S. Patent 9562199 B2, 7 February 2012. [Google Scholar]
- Hasan, S.W.; Ghannam, M.T.; Esmail, N. Heavy crude oil viscosity reduction and rheology for pipeline transportation. Fuel 2010, 89, 1095–1100. [Google Scholar] [CrossRef]
- Gao, Y.; Li, K. New models for calculating the viscosity of mixed oil. Fuel 2012, 95, 431–437. [Google Scholar] [CrossRef]
- Fukuyama, H.; Nakamura, T.T.; Ikeda, A. Partial Upgrading of Bitumen at Sagd Wellsite. In Proceedings of the Canadian Unconventional Resources and International Petroleum Conference, Calgary, AB, Canada, 19–21 October 2010. [Google Scholar]
- Chanda, D.; Sarmah, A.; Borthakur, A.; Rao, K.; Subrahmanyam, B.; Das, H. Combined effect of asphaltenes and flow improvers on the rheological behaviour of Indian waxy crude oil. Fuel 1998, 77, 1163–1167. [Google Scholar] [CrossRef]
- Ribeiro, F.S.; Mendes, P.R.S.; Braga, S.L. Obstruction of pipelines due to paraffin deposition during the flow of crude oils. Int. J. Heat Mass Transf. 1997, 40, 4319–4328. [Google Scholar] [CrossRef]
- Shigemoto, N.; Al-Maamari, R.S.; Jibril, B.Y.; Hirayama, A. A study of the effect of gas condensate on the viscosity and storage stability of Omani heavy crude oil. Energy Fuels 2006, 20, 2504–2508. [Google Scholar] [CrossRef]
- Speight, J. Petroleum Asphaltenes-Part 1: Asphaltenes, resins and the structure of petroleum. Oil Gas Sci. Technol. 2004, 59, 467–477. [Google Scholar] [CrossRef]
- Kerr, R.; Birdgeneau, J.; Batt, B.; Yang, P.; Nieuwenburg, G.; Rettger, P.; Arnold, J.; Bronicki, Y. The Long Lake Project-The First Field Integration Of Sagd And Upgrading. In Proceedings of the SPE International Thermal Operations and Heavy Oil Symposium and International Horizontal Well Technology Conference, Calgary, AB, Canada, 4–7 November 2002. [Google Scholar]
- Wiley, J. FOCUS: Latin America’s “Big Four” struggle to raise production. Oil Energy Trends 2014, 39, 3–6. [Google Scholar]
- Pilehvari, A.; Saadevandi, B.; Halvaci, M.; Clark, D. Pipeline Transportation of Heavy Crude as Emulsions. In Proceedings of the Third International Symposium on Liquid Solid Flows. ASME 1988, 75, 161. [Google Scholar]
- Ashrafizadeh, S.; Kamran, M. Emulsification of heavy crude oil in water for pipeline transportation. J. Pet. Sci. Eng. 2010, 71, 205–211. [Google Scholar] [CrossRef]
- Zaki, N.N. Surfactant stabilized crude oil-in-water emulsions for pipeline transportation of viscous crude oils. Coll. Surf. A Phys. Eng. Asp. 1997, 125, 19–25. [Google Scholar] [CrossRef]
- Yaghi, B.M.; Al-Bemani, A. Heavy crude oil viscosity reduction for pipeline transportation. Energy Sources 2002, 24, 93–102. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Nassar, A.M.; Zaki, N.N.; Gharieb, H.K. Formation of fluid heavy oil-in-water emulsions for pipeline transportation. Fuel 1999, 78, 593–600. [Google Scholar] [CrossRef]
- Shan, Z.F.; Guang, W.J. Advances in Chemical Viscosity-Reducing Methods and Techniques for Viscous Crude Oils. Oilfield Chem. 2001, 3, 24. [Google Scholar]
- Storm, D.A. Method for Reducing the Pipeline Drag of Heavy Oil and compositions Useful Therein. U.S. Patent 6,178,980, 30 January 2001. [Google Scholar]
- Manfield, P.D.; Lawrence, C.J.; Hewitt, G.F. Drag Reduction with Additives in Multiphase Flow: A Literature Survey. Multiph. Sci. Technol. 1999, 11, 197–221. [Google Scholar] [CrossRef]
- Castañeda, L.; Muñoz, J.; Ancheyta, J. Combined process schemes for upgrading of heavy petroleum. Fuel 2012, 100, 110–127. [Google Scholar] [CrossRef]
- Rankel, L.A. Pipelineable Syncrude (Synthetic Crude) from Heavy Oil. U.S. Patent No. 4,933,067, 12 June 1990. [Google Scholar]
- Lokhandwala, T.; Barrufet, M. Post-Production Heavy Oil Operations: A Case for Partial Upgrading. In Proceedings of the IPTC 2014: International Petroleum Technology Conference, Kuala Lumpur, Malaysia, 10–12 December 2014. [Google Scholar]
- Veith, E.J. Performance of a Heavy to Light Crude Oil Upgrading Process. In Proceedings of the International Oil Conference and Exhibition in Mexico, Veracruz, Mexico, 27–30 June 2007. [Google Scholar]
- Fumoto, E.; Matsumura, A.; Sato, S.; Takanohashi, T. Recovery of Lighter Fuels by Cracking Heavy Oil with Zirconia−Alumina−Iron Oxide Catalysts in a Steam Atmosphere†. Energy Fuels 2009, 23, 1338–1341. [Google Scholar] [CrossRef]
- Funai, S.; Fumoto, E.; Tago, T.; Masuda, T. Recovery of useful lighter fuels from petroleum residual oil by oxidative cracking with steam using iron oxide catalyst. Chem. Eng. Sci. 2010, 65, 60–65. [Google Scholar] [CrossRef]
- Fengya, T.; Qinghe, Y.; Dadong, L.; Lishun, D.; Zhonghuo, D. Residue Upgrading in Slurry Phase over Ultra-fine NiMo/γ-Al2O3 Catalyst. China Pet. Proc. Petrochem. Technol. 2015, 3, 1. [Google Scholar]
- Hassan, A.; Lopez-Linares, F.; Nassar, N.N.; Carbognani-Arambarri, L.; Pereira-Almao, P. Development of a support for a NiO catalyst for selective adsorption and post-adsorption catalytic steam gasification of thermally converted asphaltenes. Catal. Today 2013, 207, 112–118. [Google Scholar] [CrossRef]
- Carrazza, J.; Pereira, P.; Martinez, N. Process and Catalyst for Upgrading Heavy Hydrocarbon. U.S. Patent 5688395 A, 18 November 1997. [Google Scholar]
- Janssen, M.J.; Ou, J.D.; Heeter, G.A.; van Oorschot, C.W. Removal of Asphaltene Contaminants from Hydrocarbon Streams Using Carbon Based Adsorbents. U.S. Patent 9,321,971, 26 April 2009. [Google Scholar]
- Jada, A.; Debih, H.; Khodja, M. Montmorillonite surface properties modifications by asphaltenes adsorption. J. Pet. Sci. Eng. 2006, 52, 305–316. [Google Scholar] [CrossRef]
- Pernyeszi, T.; Patzko, A.; Berkesi, O.; Dékány, I. Asphaltene adsorption on clays and crude oil reservoir rocks. Coll. Surf. A Physicochem. Eng. Asp. 1998, 137, 373–384. [Google Scholar] [CrossRef]
- Dean, K.R.; McAtee, J.L. Asphaltene adsorption on clay. Appl. Clay Sci. 1986, 1, 313–319. [Google Scholar] [CrossRef]
- Bantignies, J.-L.; dit Moulin, C.C.; Dexpert, H. Asphaltene adsorption on kaolinite characterized by infrared and X-ray absorption spectroscopies. J. Pet. Sci. Eng. 1998, 20, 233–237. [Google Scholar] [CrossRef]
- Acevedo, S.; Ranaudo, M.A.; García, C.; Castillo, J.; Fernández, A. Adsorption of asphaltenes at the toluene-silica interface: A kinetic study. Energy Fuels 2003, 17, 257–261. [Google Scholar] [CrossRef]
- Marchal, C.; Abdessalem, E.; Tayakout-Fayolle, M.; Uzio, D. Asphaltene diffusion and adsorption in modified NiMo alumina catalysts followed by ultraviolet (UV) spectroscopy. Energy Fuels 2010, 24, 4290–4300. [Google Scholar] [CrossRef]
- López-Linares, F.; Carbognani, L.; Sosa-Stull, C.; Pereira-Almao, P.; Spencer, R.J. Adsorption of virgin and visbroken residue asphaltenes over solid surfaces. 1. Kaolin, smectite clay minerals, and athabasca siltstone. Energy Fuels 2009, 23, 1901–1908. [Google Scholar] [CrossRef]
- Marriott, T.; Gonzalez, M.; Carbognani, L.; Zurita, M.P.; Lopez-Linares, F.; Husein, M.; Moore, G.; Pereira, P. Visbreaking Based Integrated Process for Bitumen Upgrading and Hydrogen Production. In Proceedings of the Canadian International Petroleum Conference, Calgary, Alberta, 13–15 June 2006. [Google Scholar]
- Carbognani, L.; González, M.F.; Lopez-Linares, F.; Stull, C.S.; Pereira-Almao, P. Selective adsorption of thermal cracked heavy molecules. Energy Fuels 2008, 22, 1739–1746. [Google Scholar] [CrossRef]
- Ali, M.; Tatsumi, T.; Masuda, T. Development of heavy oil hydrocracking catalysts using amorphous silica-alumina and zeolites as catalyst supports. Appl. Catal. A Gen. 2002, 233, 77–90. [Google Scholar] [CrossRef]
- Chianelli, R.R.; Ho, T.C.; Jacobson, A.J.; Young, A.R. Supported Chromium-Molybdenum and Tungsten Sulfide Catalysts. U.S. Patent 4,748,142, 31 May 1988. [Google Scholar]
- Lopez-Linares, F.; Carbognani, L.; Hassan, A.; Pereira-Almao, P.; Rogel, E.; Ovalles, C.; Pradhan, A.; Zintsmaster, J. Adsorption of Athabasca vacuum residues and their visbroken products over macroporous solids: Influence of their molecular characteristics. Energy Fuels 2011, 25, 4049–4054. [Google Scholar] [CrossRef]
- Ancheyta, J.; Betancourt, G.; Marroquın, G.; Centeno, G.; Castañeda, L.; Alonso, F.; Muñoz, J.A.; Gómez, M.T.; Rayo, P. Hydroprocessing of Maya heavy crude oil in two reaction stages. Appl. Catal. A Gen. 2002, 233, 159–170. [Google Scholar] [CrossRef]
- Carbognani, L. Upgrading of a Visbroken Vacuum Residue by Adsorption and Catalytic Steam Gasification of the Adsorbed Components. Master’s Thesis, University of Calgary, Calgary, AB, Canada, 2014. [Google Scholar]
- Sosa, C. Adsorption of Heavy Hydrocarbons for the Purpose of Hydrogen Production. MSc Thesis, University of Calgary, Calgary, AB, Canada, 2007. [Google Scholar]
- Hassan, A.; Carbognani-Arambarri, L.; Nassar, N.N.; Vitale, G.; Lopez-Linares, F.; Pereira-Almao, P. Catalytic steam gasification of n-C5 asphaltenes by kaolin-based catalysts in a fixed-bed reactor. Appl. Catal. A Gen. 2015, 507, 149–161. [Google Scholar] [CrossRef]
- Carbognani, L. Effects of iron compounds on the retention of oil polar hydrocarbons over solid sorbents. Pet. Sci. Technol. 2000, 18, 335–360. [Google Scholar] [CrossRef]
- Nassar, N.N. Asphaltene adsorption onto alumina nanoparticles: Kinetics and thermodynamic studies. Energy Fuels 2010, 24, 4116–4122. [Google Scholar] [CrossRef]
- Zimmer, A.K.; Becker, C.; Chambliss, C.K. Exploiting Metal Oxide Nanoparticle Selectivity in Asphaltenes for Identification of Pyridyl-Containing Molecules. Energy Fuels 2013, 27, 4574–4580. [Google Scholar] [CrossRef]
- Franco, C.A.; Montoya, T.; Nassar, N.N.; Pereira-Almao, P.; Cortés, F.B. Adsorption and subsequent oxidation of colombian asphaltenes onto Nickel and/or Palladium oxide supported on fumed silica nanoparticles. Energy Fuels 2013, 27, 7336–7347. [Google Scholar] [CrossRef]
- Nassar, N.N.; Hassan, A.; Carbognani, L.; Lopez-Linares, F.; Pereira-Almao, P. Iron oxide nanoparticles for rapid adsorption and enhanced catalytic oxidation of thermally cracked asphaltenes. Fuel 2012, 95, 257–262. [Google Scholar] [CrossRef]
- Nassar, N.N.; Hassan, A.; Pereira-Almao, P. Effect of the particle size on asphaltene adsorption and catalytic oxidation onto alumina particles. Energy Fuels 2011, 25, 3961–3965. [Google Scholar] [CrossRef]
- Franco, C.A.; Guzmán, J.D.; Cortés, F.B. Adsorption and catalytic oxidation of asphaltenes in fumed silica nanoparticles: Effect of the surface acidity. J. Fac. Minas Univ. Nac. Colomb.-Medellin Campus 2016, 83, 171. [Google Scholar]
- Nassar, N.N.; Hassan, A.; Pereira-Almao, P. Metal oxide nanoparticles for asphaltene adsorption and oxidation. Energy Fuels 2011, 25, 1017–1023. [Google Scholar] [CrossRef]
- Cortés, F.B.; Mejía, J.M.; Ruiz, M.A.; Benjumea, P.; Riffel, D.B. Sorption of asphaltenes onto nanoparticles of nickel oxide supported on nanoparticulated silica gel. Energy Fuels 2012, 26, 1725–1730. [Google Scholar] [CrossRef]
- Tarboush, B.J.A.; Husein, M.M. Adsorption of asphaltenes from heavy oil onto in situ prepared NiO nanoparticles. J. Coll. Interf. Sci. 2012, 378, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Nassar, N.N.; Franco, C.A.; Montoya, T.; Cortés, F.B.; Hassan, A. Effect of oxide support on Ni–Pd bimetallic nanocatalysts for steam gasification of nC 7 asphaltenes. Fuel 2015, 156, 110–120. [Google Scholar] [CrossRef]
- Lozano, M.M.; Franco, C.A.; Acevedo, S.A.; Nassar, N.N.; Cortés, F.B. Effects of resin I on the catalytic oxidation of n-C 7 asphaltenes in the presence of silica-based nanoparticles. RSC Adv. 2016, 6, 74630–74642. [Google Scholar] [CrossRef]
- Van Duin, A.C.; Larter, S.R. Molecular dynamics investigation into the adsorption of organic compounds on kaolinite surfaces. Org. Geochem. 2001, 32, 143–150. [Google Scholar] [CrossRef]
- Lopez-Linares, F.; Sosa, C.; Gonzalez, M.; Pereira-Almao, P. The adsorption of asphaltenes compared to model heavy molecules over macroporous solids. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2005; p. U1706. [Google Scholar]
- Fumoto, E.; Tago, T.; Tsuji, T.; Masuda, T. Recovery of useful hydrocarbons from petroleum residual oil by catalytic cracking with steam over zirconia-supporting iron oxide catalyst. Energy Fuels 2004, 18, 1770–1774. [Google Scholar] [CrossRef]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore texture of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Martins, G.; Berlier, G.; Bisio, C.; Coluccia, S.; Pastore, H.; Marchese, L. Quantification of Brønsted acid sites in microporous catalysts by a combined FTIR and NH3-TPD study. J. Phys. Chem.C 2008, 112, 7193–7200. [Google Scholar] [CrossRef]
- Hunger, B.; Heuchel, M.; Clark, L.A.; Snurr, R.Q. Characterization of acidic OH groups in zeolites of different types: An interpretation of NH3-TPD results in the light of confinement effects. J. Phys. Chem. B 2002, 106, 3882–3889. [Google Scholar] [CrossRef]
- AlSawalha, M.; Roessner, F.; Novikova, L.; Bel’chinskaya, L. Acidity of different Jordanian Clays characterized by TPD-NH3 and MBOH Conversion. World Acad. Sci. Eng. Technol. 2011, 5, 7–29. [Google Scholar]
- Caillot, M.; Chaumonnot, A.; Digne, M.; van Bokhoven, J.A. The variety of Brønsted acid sites in amorphous aluminosilicates and zeolites. J. Catal. 2014, 316, 47–56. [Google Scholar] [CrossRef]
- Pena, D.A.; Uphade, B.S.; Smirniotis, P.G. TiO2-supported metal oxide catalysts for low-temperature selective catalytic reduction of NO with NH 3: I. Evaluation and characterization of first row transition metals. J. Catal. 2004, 221, 421–431. [Google Scholar] [CrossRef]
- Diao, W.; Tengco, J.M.M.; Regalbuto, J.R.; Monnier, J.R. Preparation and Characterization of Pt–Ru Bimetallic Catalysts Synthesized by Electroless Deposition Methods. ACS Catal. 2015, 5, 5123–5134. [Google Scholar] [CrossRef]
- Shastri, A.; Schwank, J. Metal dispersion of bimetallic catalysts via stepwise chemisorption and surface titration: I. Ru AuSiO2. J. Catal. 1985, 95, 271–283. [Google Scholar] [CrossRef]
- Sinfelt, J.H. Catalysis by alloys and bimetallic clusters. Acc. Chem. Res. 1977, 10, 15–20. [Google Scholar] [CrossRef]
- Contreras, J.L.; Fuentes, G.A. ChemInform Abstract: Sintering of Supported Metal Catalysts. ChemInform 2013, 44. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- León, O.; Contreras, E.; Rogel, E.; Dambakli, G.; Acevedo, S.; Carbognani, L. Adsorption of native resins on asphaltene particles: A correlation between adsorption and activity. Langmuir 2002, 18, 5106–5112. [Google Scholar] [CrossRef]
- Franco, C.A.; Lozano, M.M.; Acevedo, S.; Nassar, N.N.; Cortes, F.B. Effects of Resin I on Asphaltene Adsorption onto Nanoparticles: A Novel Method for Obtaining Asphaltenes/Resin Isotherms. Energy Fuels 2015, 30, 264–272. [Google Scholar] [CrossRef]
- Montoya, T.; Coral, D.; Franco, C.A.; Nassar, N.N.; Cortés, F.B. A novel solid–liquid equilibrium model for describing the adsorption of associating asphaltene molecules onto solid surfaces based on the “chemical theory”. Energy Fuels 2014, 28, 4963–4975. [Google Scholar] [CrossRef]
- Clementz, D.M. Interaction of petroleum heavy ends with montmorillonite. Clays Clay Miner. 1976, 24, 312–319. [Google Scholar] [CrossRef]
- Adams, J.J. Asphaltene adsorption, a literature review. Energy Fuels 2014, 28, 2831–2856. [Google Scholar] [CrossRef]
- Kaneko, K. Determination of pore size and pore size distribution: 1. Adsorbents and catalysts. J. Membr. Sci. 1994, 96, 59–89. [Google Scholar] [CrossRef]
- Bardon, C.; Barre, L.; Espinat, D.; Guille, V.; Li, M.H.; Lambard, J. The colloidal structure of crude oils and suspensions of asphaltenes and resins. Fuel Sci. Technol. Int. 1996, 14, 203–242. [Google Scholar] [CrossRef]
- Andersen, S.I.; Birdi, K.S. Aggregation of asphaltenes as determined by calorimetry. J. Coll. Interf. Sci. 1991, 142, 497–502. [Google Scholar] [CrossRef]
- Rogel, E. Molecular thermodynamic approach to the formation of mixed asphaltene-resin aggregates. Energy Fuels 2008, 22, 3922–3929. [Google Scholar] [CrossRef]
- Spiecker, P.M.; Gawrys, K.L.; Trail, C.B.; Kilpatrick, P.K. Effects of petroleum resins on asphaltene aggregation and water-in-oil emulsion formation. Coll. Surf. A Physicochem. Eng. Asp. 2003, 220, 9–27. [Google Scholar] [CrossRef]
- Murgich, J.; Rodríguez, J.; Aray, Y. Molecular recognition and molecular mechanics of micelles of some model asphaltenes and resins. Energy Fuels 1996, 10, 68–76. [Google Scholar] [CrossRef]
- Murugan, P.; Mahinpey, N.; Mani, T. Thermal cracking and combustion kinetics of asphaltenes derived from Fosterton oil. Fuel Proc. Technol. 2009, 90, 1286–1291. [Google Scholar] [CrossRef]
- Montoya, T.; Argel, B.L.; Nassar, N.N.; Franco, C.A.; Cortés, F.B. Kinetics and mechanisms of the catalytic thermal cracking of asphaltenes adsorbed on supported nanoparticles. Pet. Sci. 2016, 13, 561–571. [Google Scholar] [CrossRef]
- Nassar, N.N.; Hassan, A.; Pereira-Almao, P. Application of nanotechnology for heavy oil upgrading: catalytic steam gasification/cracking of asphaltenes. Energy Fuels 2011, 25, 1566–1570. [Google Scholar] [CrossRef]
- Trujillo-Ferrer, G. Thermal and Catalytic Steam Reactivity Evaluation of Athabasca Vacuum Gasoil. Master’s Thesis, University of Calgary, Calgary, AB, Canada, 2008. [Google Scholar]
- Franco, C.A.; Montoya, T.; Nassar, N.N.; Cortés, F.B. NiO and PdO Supported on Fumed Silica Nanoparticles for Adsorption and Catalytic Steam Gasification of Colombian C7 Asphaltenes. In Handbook on Oil Production Research; Nova Science Publishers: Hauppauge, NY, USA, 2014; pp. 101–145. [Google Scholar]
- Vélez, J.F.; Chejne, F.; Valdés, C.F.; Emery, E.J.; Londoño, C.A. Co-gasification of Colombian coal and biomass in fluidized bed: An experimental study. Fuel 2009, 88, 424–430. [Google Scholar] [CrossRef]
- Ertl, G.; KnÃ, H.; Weitkamp, J. Preparation of Solid Catalysts; John Wiley & Sons: Weinheim, Germany, 2008. [Google Scholar]
- Nguyen-Huy, C.; Shin, E.W. Hierarchical macro-mesoporous Al2O3-supported NiK catalyst for steam catalytic cracking of vacuum residue. Fuel 2016, 169, 1–6. [Google Scholar] [CrossRef]
- Franco, C.; Patiño, E.; Benjumea, P.; Ruiz, M.A.; Cortés, F.B. Kinetic and thermodynamic equilibrium of asphaltenes sorption onto nanoparticles of nickel oxide supported on nanoparticulated alumina. Fuel 2013, 105, 408–414. [Google Scholar] [CrossRef]
- Navarro, L.; Álvarez, M.; Grosso, J.-L.; Navarro, U. Separación y caracterización de resinas y asfaltenos provenientes del crudo Castilla. Evaluación de su interacción molecular. CT&F-Ciencia Tecnología y Futuro 2004, 2, 53–67. [Google Scholar]
- Carnahan, N.F.; Salager, J.-L.; Antón, R.; Dávila, A. Properties of resins extracted from Boscan crude oil and their effect on the stability of asphaltenes in Boscan and Hamaca crude oils. Energy Fuels 1999, 13, 309–314. [Google Scholar] [CrossRef]
- Shan, S.; Yuan, P.; Han, W.; Shi, G.; Bao, X. Supported NiW catalysts with tunable size and morphology of active phases for highly selective hydrodesulfurization of fluid catalytic cracking naphtha. J. Catal. 2015, 330, 288–301. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Kuśtrowski, P.; Chmielarz, L.; Bożek, E.; Sawalha, M.; Roessner, F. Acidity and basicity of hydrotalcite derived mixed Mg–Al oxides studied by test reaction of MBOH conversion and temperature programmed desorption of NH3 and CO2. Mater. Res. Bull. 2004, 39, 263–281. [Google Scholar] [CrossRef]
- Guzmán, J.D.; Betancur, S.; Carrasco-Marín, F.; Franco, C.A.; Nassar, N.N.; Cortés, F.B. Importance of the Adsorption Method Used for Obtaining the Nanoparticle Dosage for Asphaltene-Related Treatments. Energy Fuels 2016, 30, 2052–2059. [Google Scholar] [CrossRef]
- Franco, C.A.; Zabala, R.D.; Zapata, J.; Mora, E.; Botero, O.; Candela, C. Inhibited Gas Stimulation To Mitigate Condensate Banking and Maximize Recovery in Cupiagua Field. SPE Prod. Oper. 2013, 28, 154–167. [Google Scholar] [CrossRef]
- Talu, O.; Meunier, F. Adsorption of associating molecules in micropores and application to water on carbon. AIChE J. 1996, 42, 809–819. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. A quick, direct method for the determination of activation energy from thermogravimetric data. J. Polym. Sci. Part B Polym. Lett. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Ozawa, T. A new method of analyzing thermogravimetric data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Doyle, C.D. Series approximations to the equation of thermogravimetric data. Nature 1965, 207, 290–291. [Google Scholar] [CrossRef]
- Šimon, P. Isoconversional methods. J. Therm. Anal. Calorim. 2004, 76, 123–132. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Oxides (wt %) | ||||
|---|---|---|---|---|---|
| SiO2 | Al2O3 | Fe2O3 | TiO2 | K2O | |
| Clay | 60.9 | 25.4 | 0.9 | 1.6 | 3.2 |
| Metakaolin | 56.6 | 37.3 | 1.3 | 0.5 | 0.4 |
| Sample | SBET ± 0.1 m2/g | Total Acidity ± 0.02 µmol/g | Mean Particle Size of Active Phase ± 0.1 nm | Dispersion (%) |
|---|---|---|---|---|
| Clay | 19.6 | 376.02 | - | - |
| CMoNi | 12.3 | 231.82 | 4.5 | 9.9 |
| CMoCo | 11.2 | 227.31 | 4.4 | 9.8 |
| Metakaolin | 18.0 | 114.00 | - | - |
| KMoNi | 15.1 | 161.24 | 8.9 | 20 |
| KMoCo | 14.3 | 127.41 | 13.5 | 15 |
| Material | H (mg/g) | K (g/g) × 104 | Nads (mg/m2) | R2 | RSME% |
|---|---|---|---|---|---|
| Clay | 31.23 | 19.87 | 1.6 | 0.96 | 9.82 |
| CMoNi | 13.79 | 33.90 | 3.21 | 0.95 | 9.73 |
| CMoCo | 14.69 | 34.01 | 3.32 | 0.99 | 8.27 |
| Metakaolin | 21.34 | 22.38 | 2.12 | 0.99 | 8.65 |
| KMoNi | 13.78 | 36.23 | 3.76 | 0.99 | 7.91 |
| KMoCo | 14.21 | 35.19 | 3.41 | 0.99 | 7.23 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, D.; Giraldo, L.J.; Salazar, J.P.; Zapata, D.M.; Ortega, D.C.; Franco, C.A.; Cortés, F.B. Metal Oxide Nanoparticles Supported on Macro-Mesoporous Aluminosilicates for Catalytic Steam Gasification of Heavy Oil Fractions for On-Site Upgrading. Catalysts 2017, 7, 319. https://doi.org/10.3390/catal7110319
López D, Giraldo LJ, Salazar JP, Zapata DM, Ortega DC, Franco CA, Cortés FB. Metal Oxide Nanoparticles Supported on Macro-Mesoporous Aluminosilicates for Catalytic Steam Gasification of Heavy Oil Fractions for On-Site Upgrading. Catalysts. 2017; 7(11):319. https://doi.org/10.3390/catal7110319
Chicago/Turabian StyleLópez, Daniel, Lady J. Giraldo, Juan P. Salazar, Dioni M. Zapata, Diana C. Ortega, Camilo A. Franco, and Farid B. Cortés. 2017. "Metal Oxide Nanoparticles Supported on Macro-Mesoporous Aluminosilicates for Catalytic Steam Gasification of Heavy Oil Fractions for On-Site Upgrading" Catalysts 7, no. 11: 319. https://doi.org/10.3390/catal7110319