
Synthesis and Electrochemical Evaluation of Carbon Supported Pt-Co Bimetallic Catalysts Prepared by Electroless Deposition and Modified Charge Enhanced Dry Impregnation

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
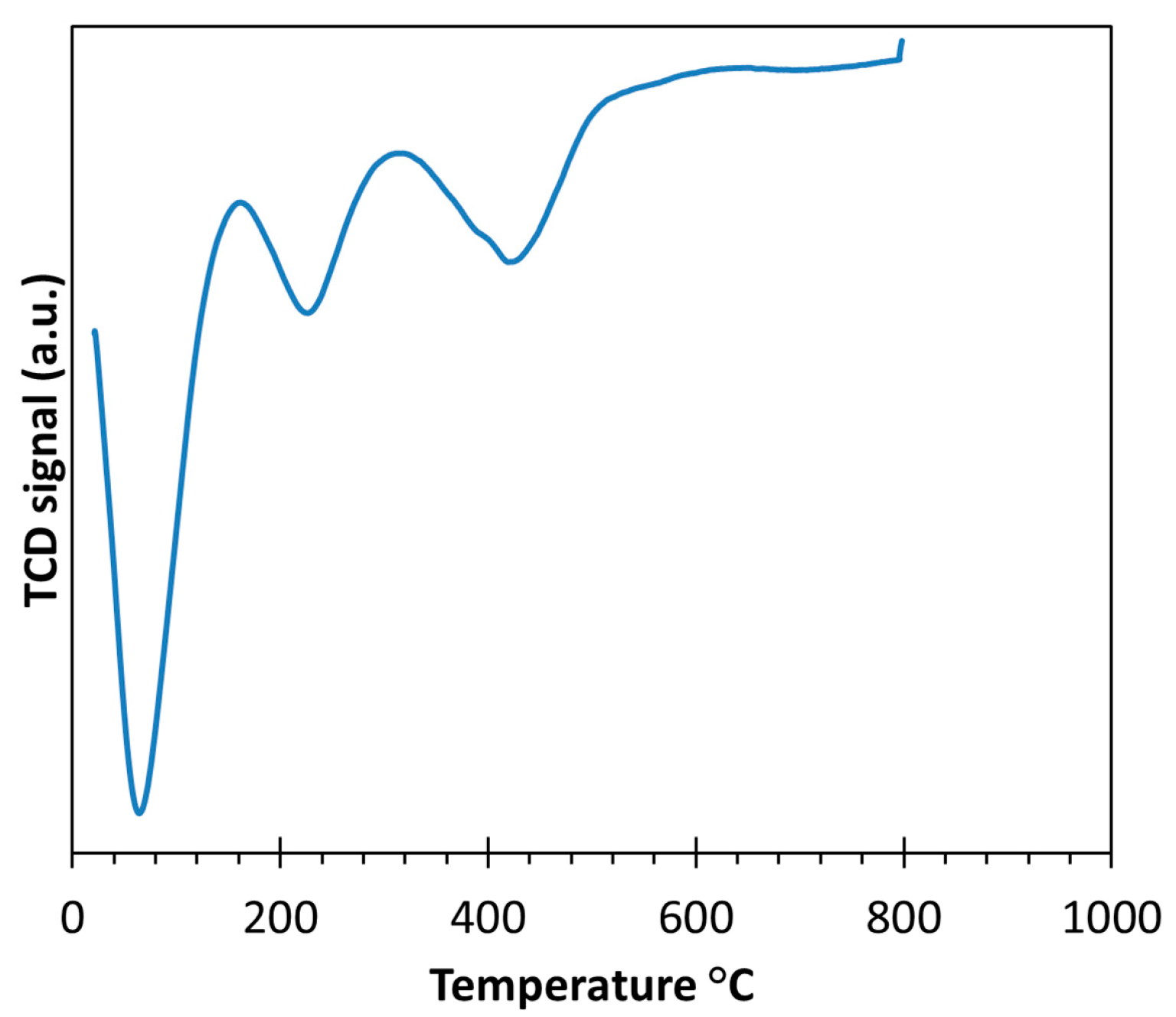
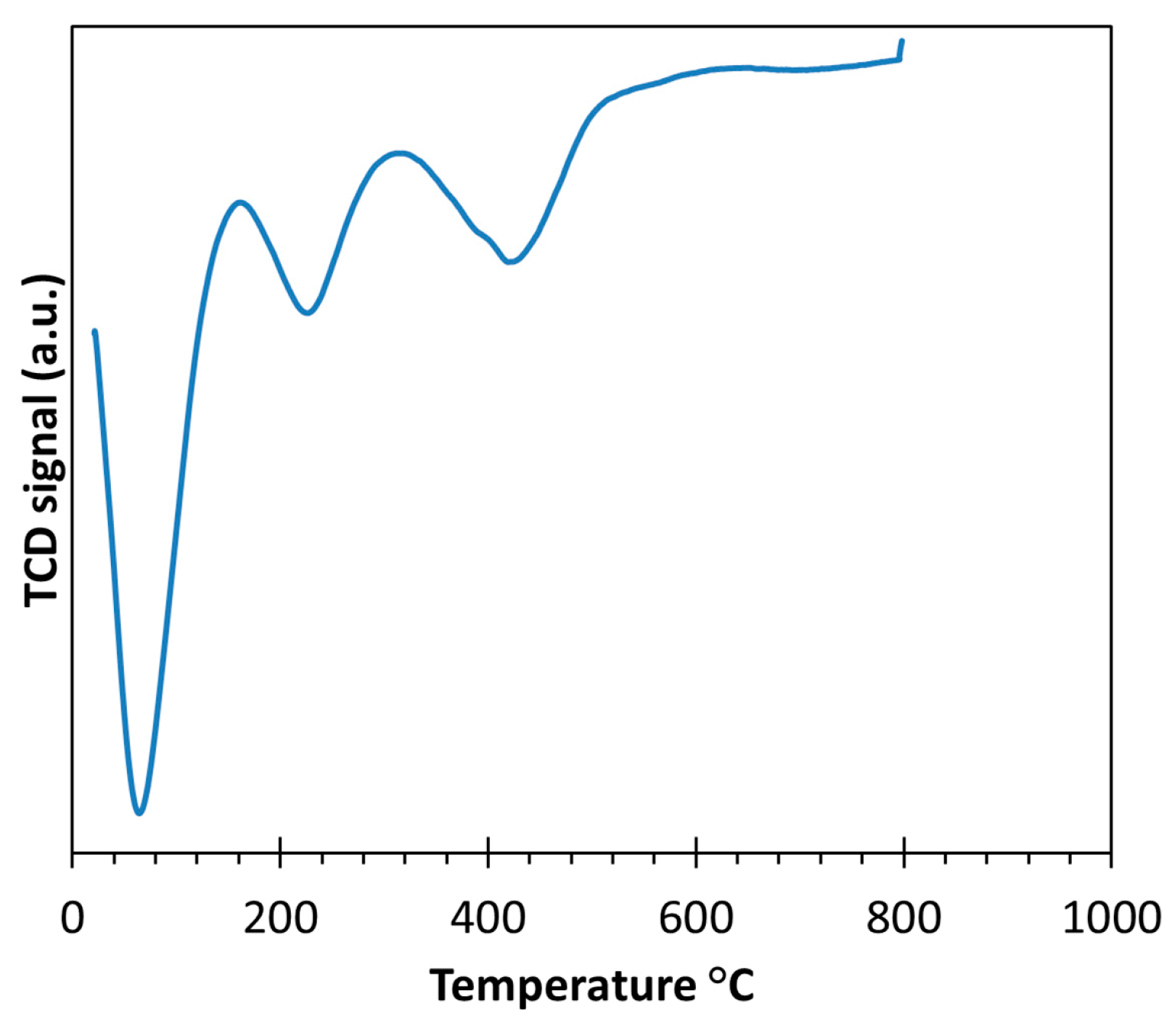
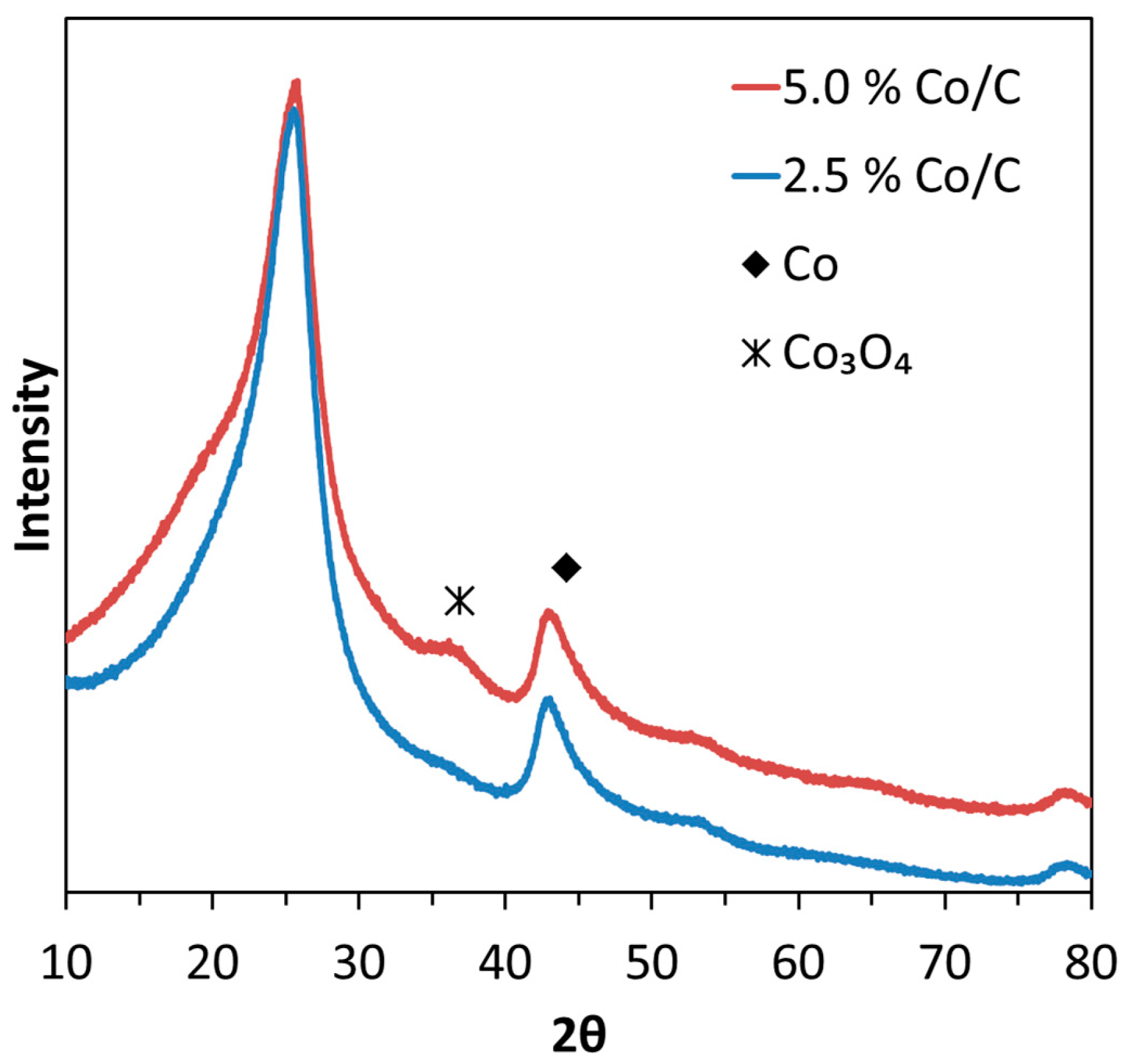
2.1. Thermal Treatment and Dispersion of Co/C Base Catalyst
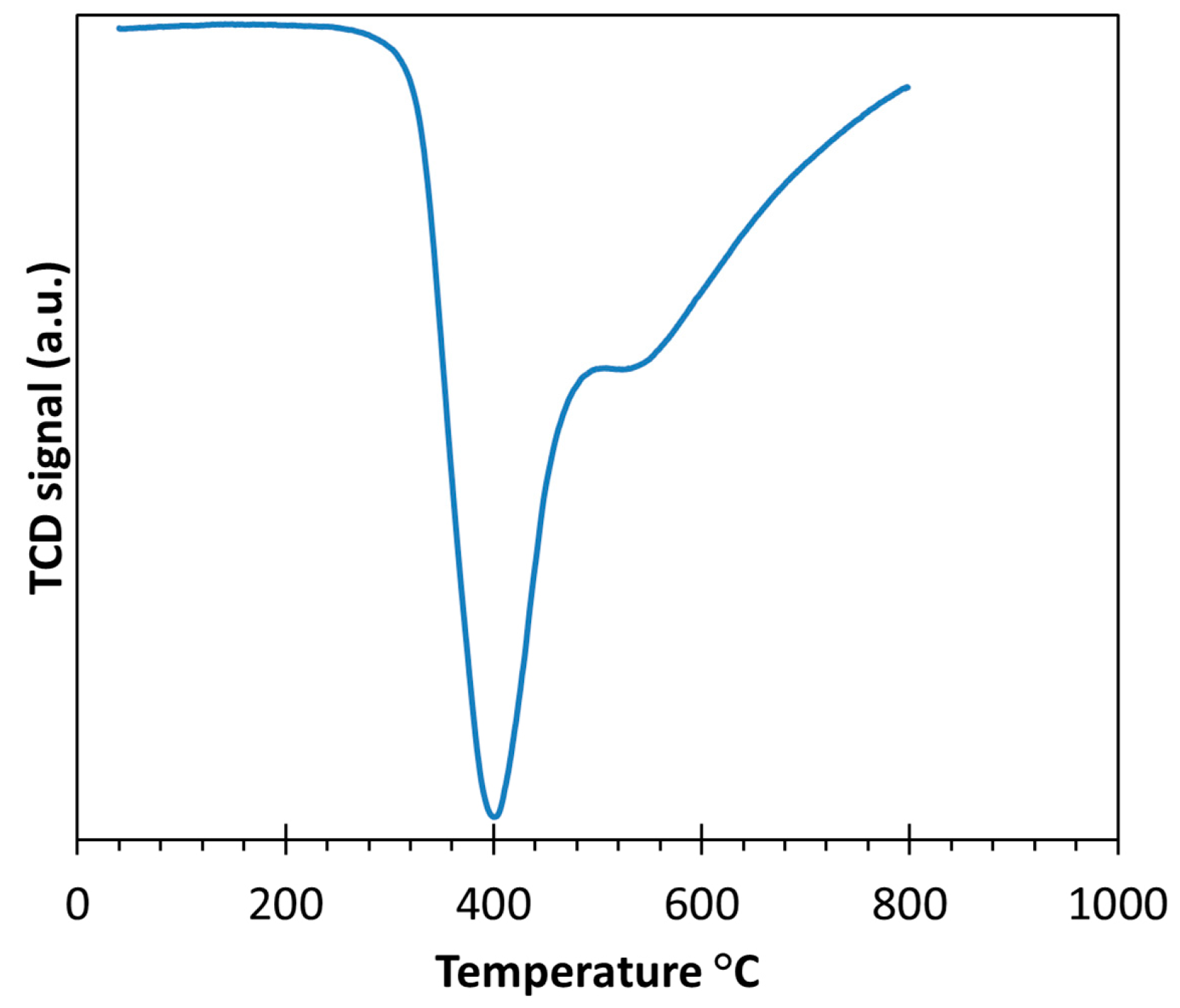
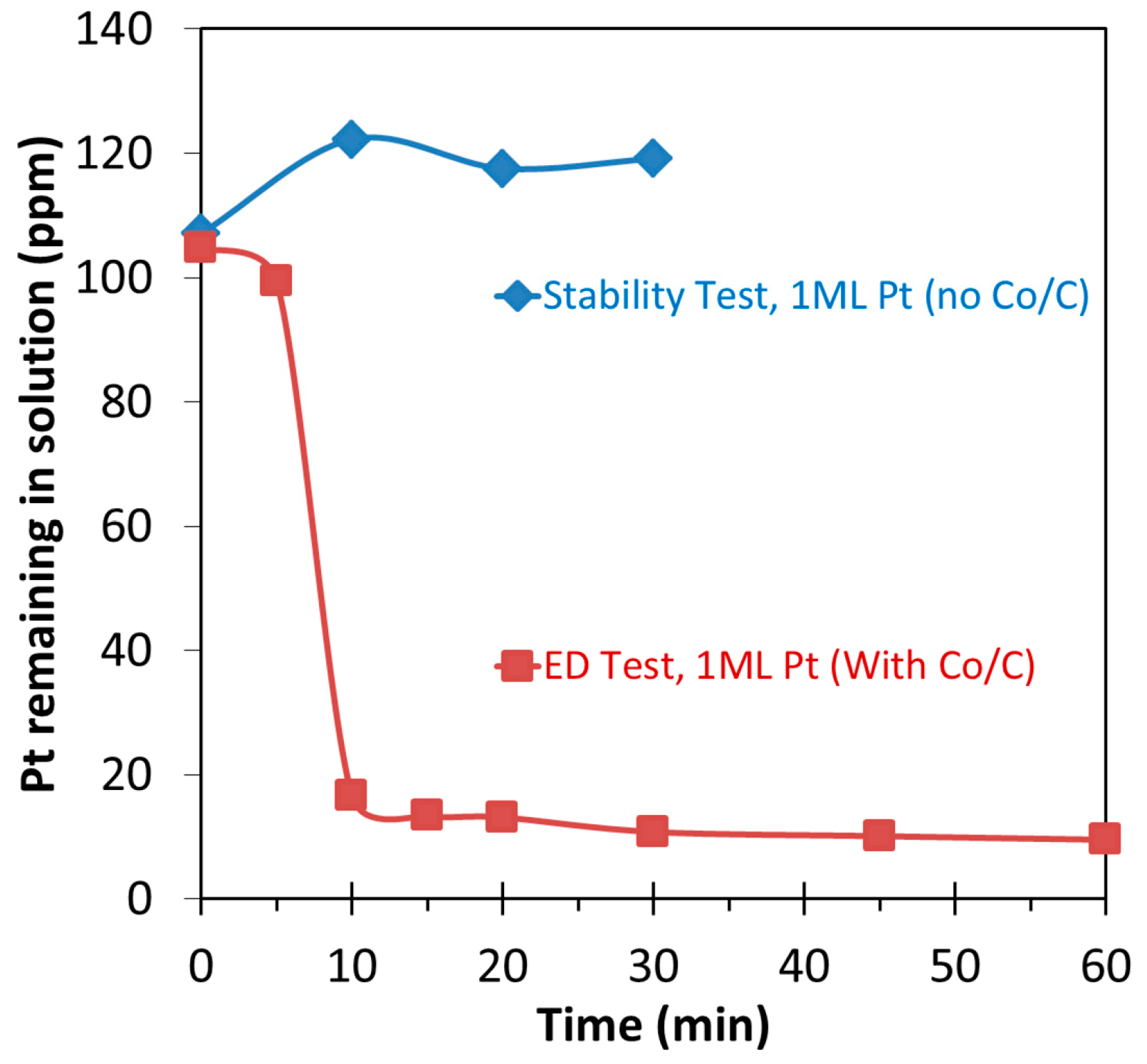
2.2. Bimetallic Pt-Co/C Catalysts from Electroless Deposition Experiments
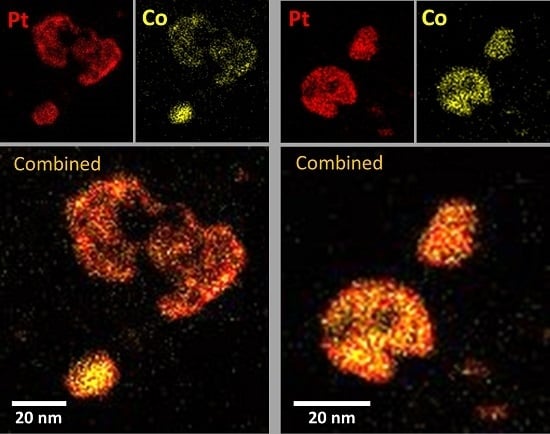
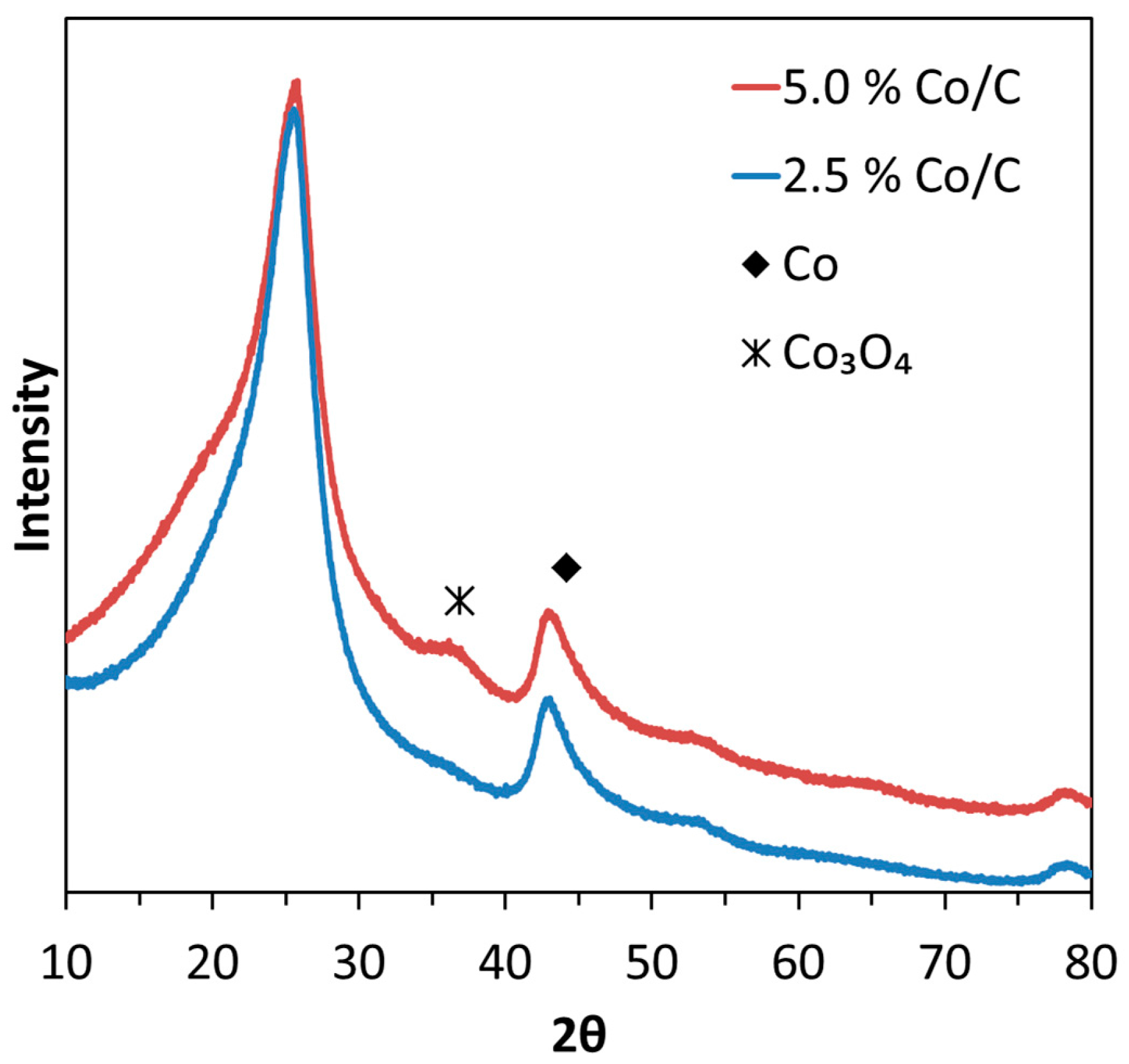
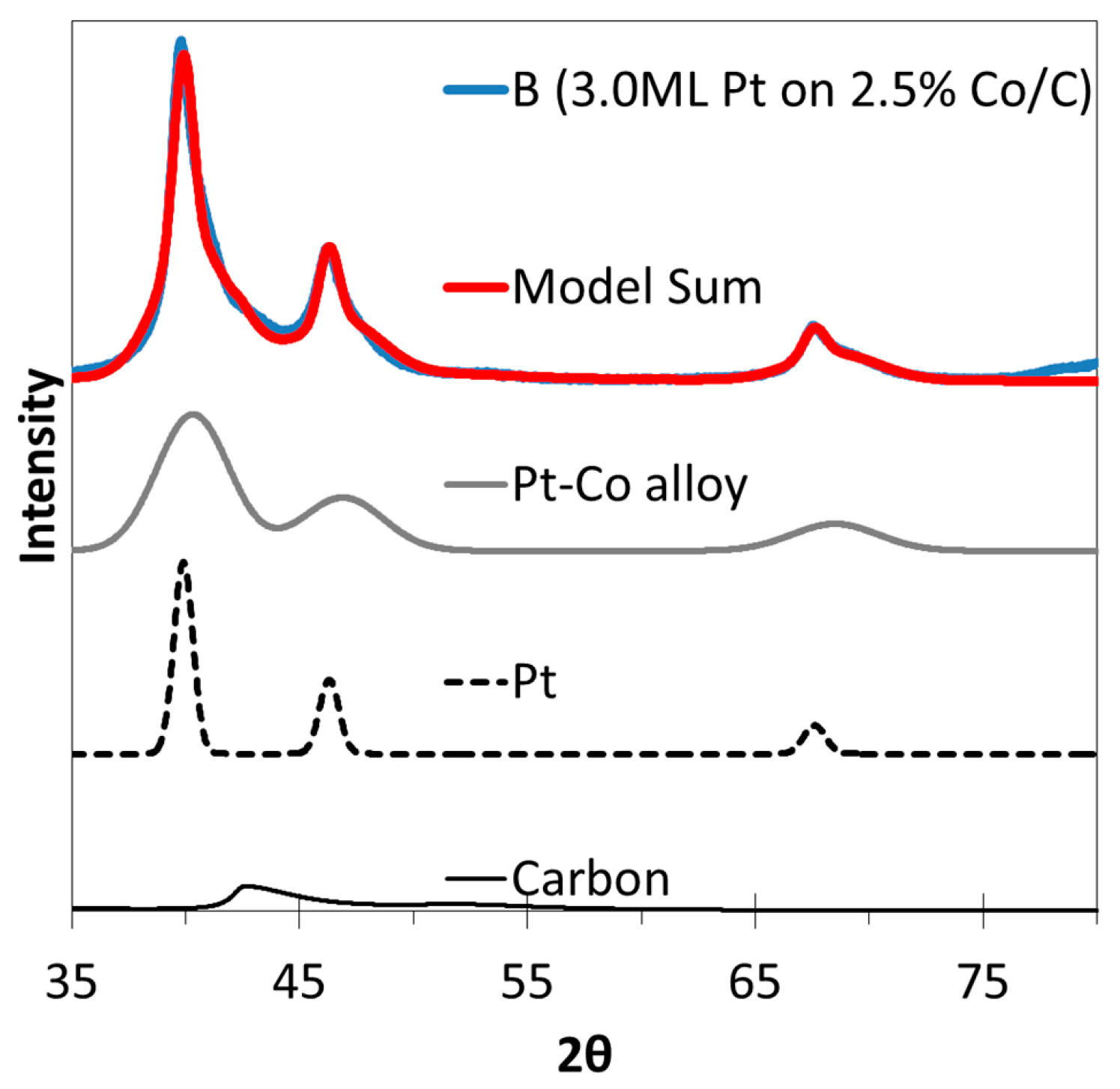
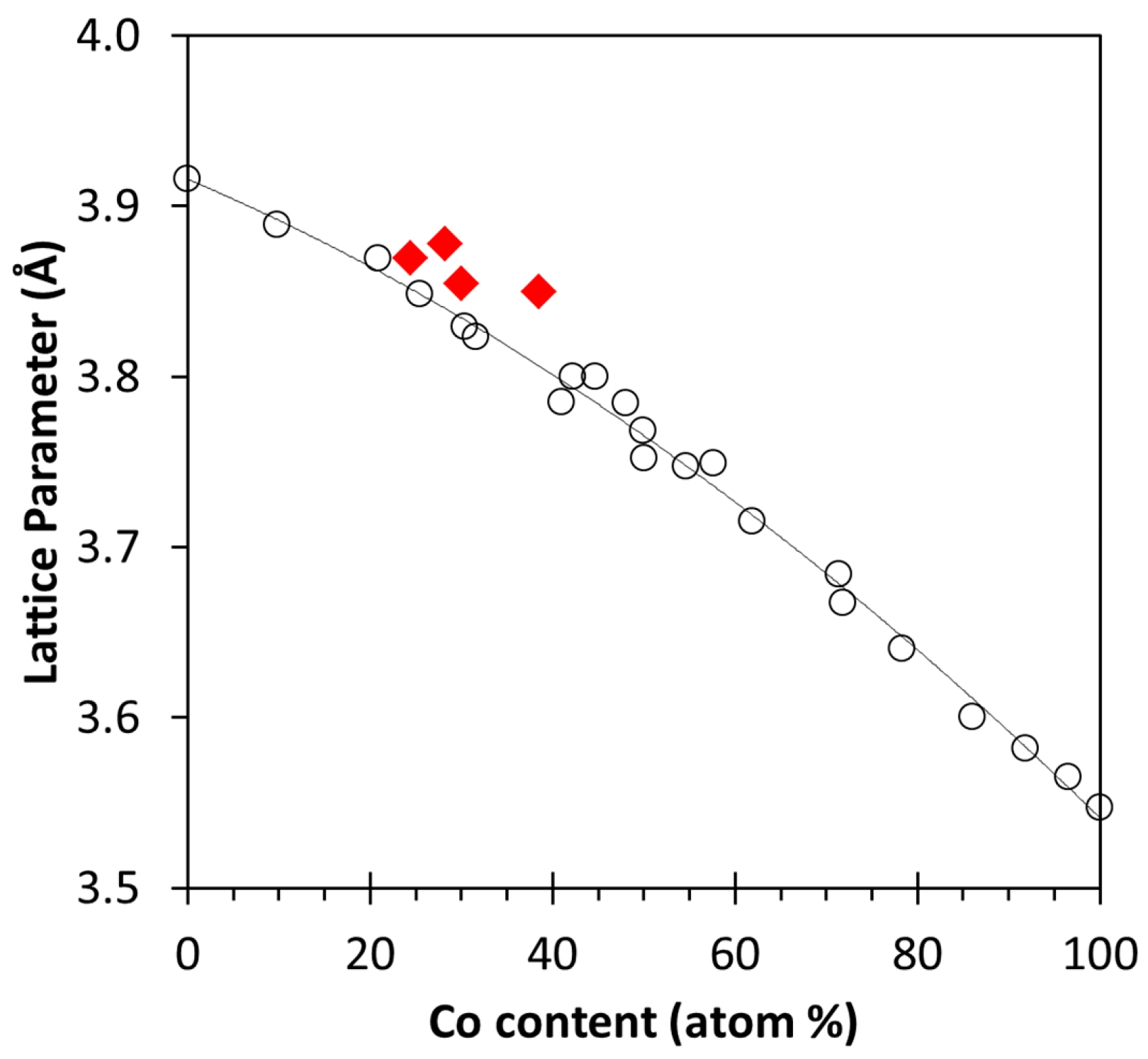
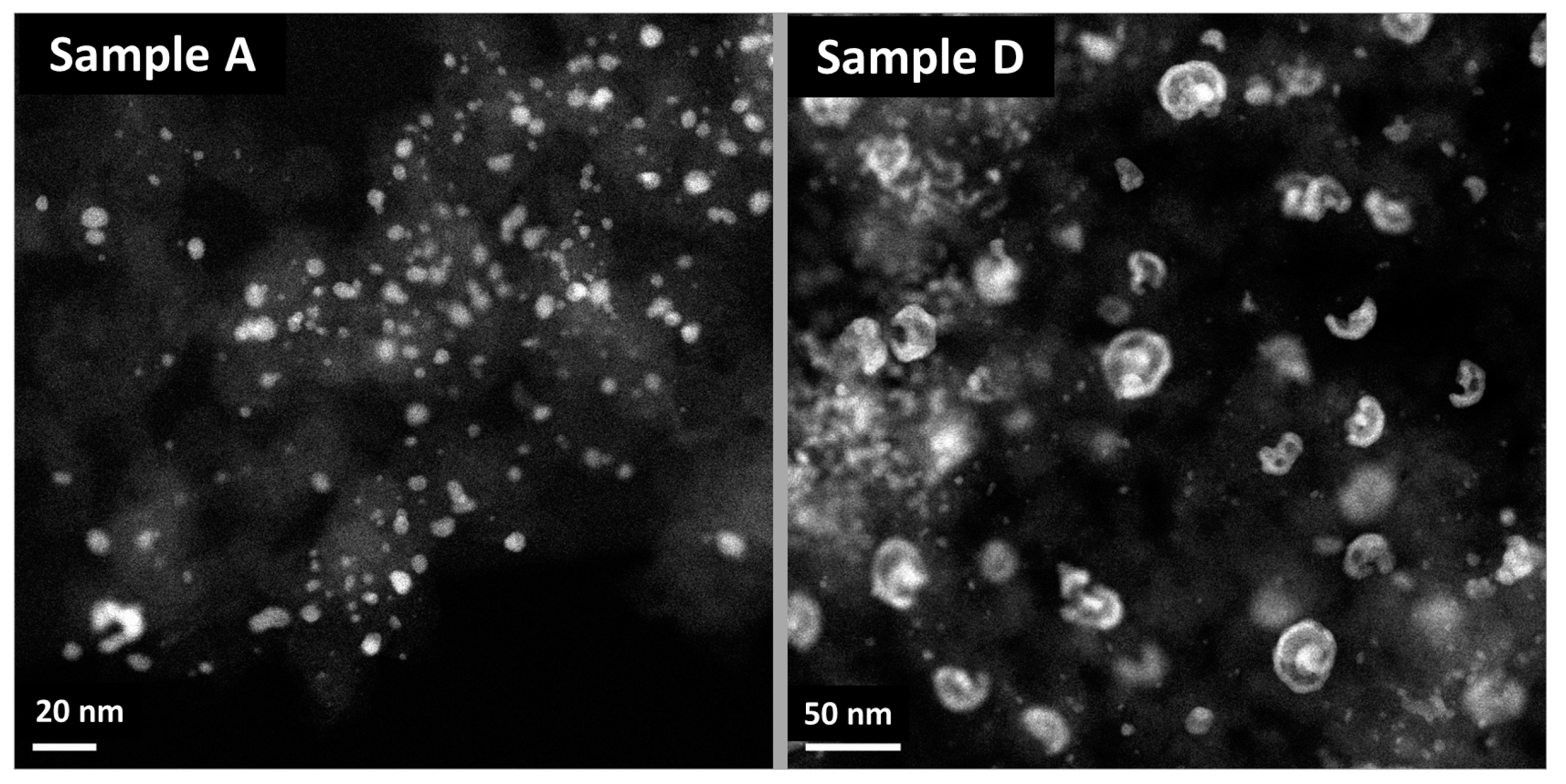
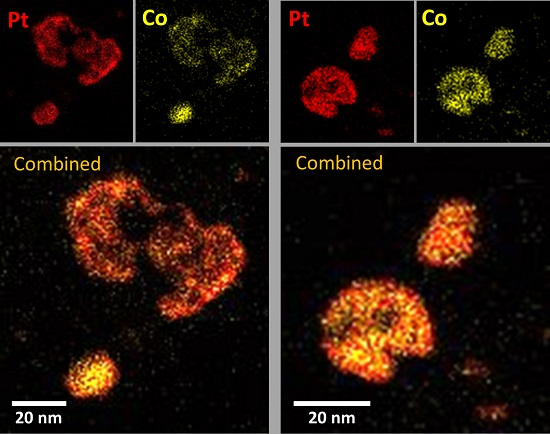
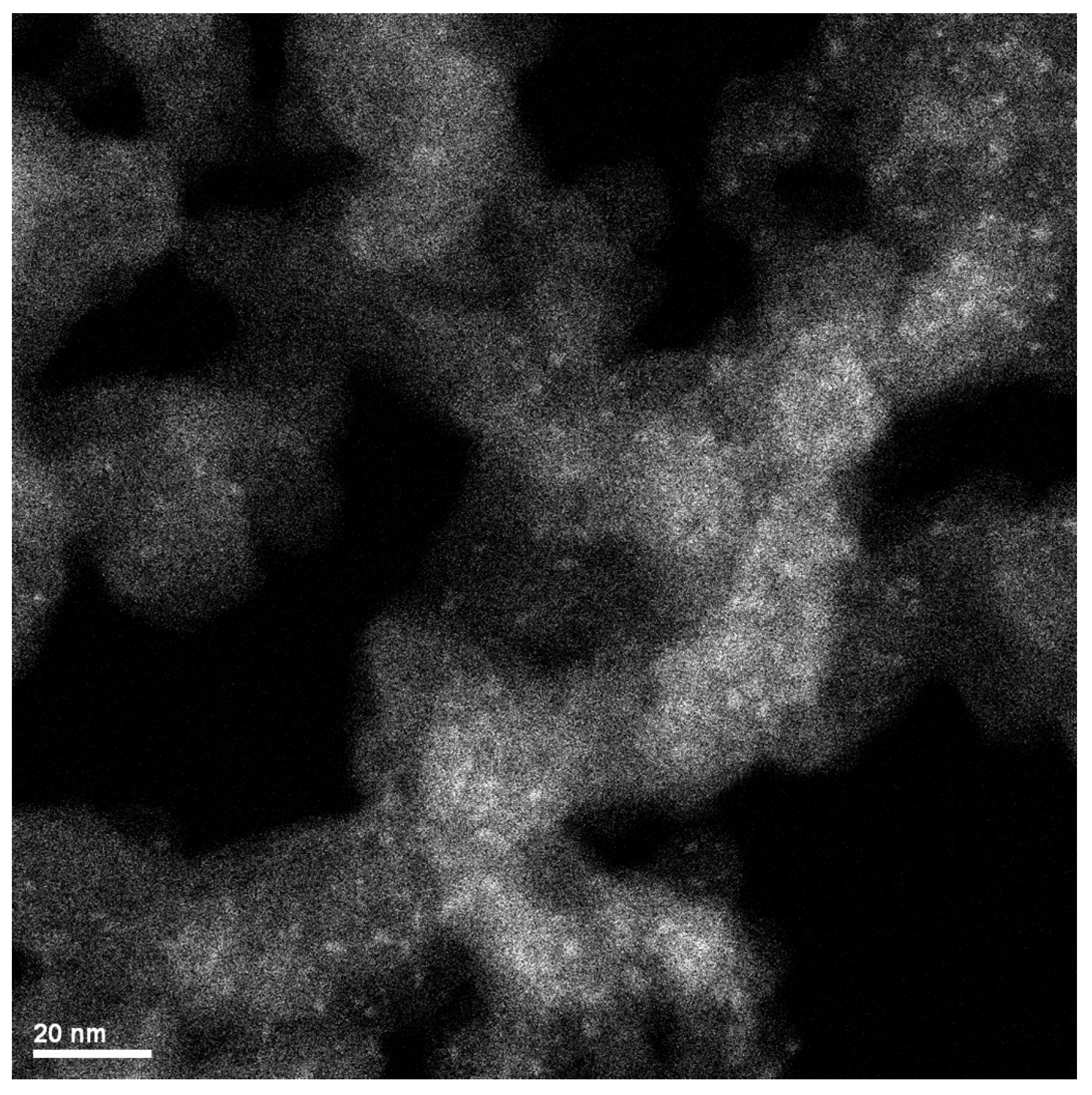
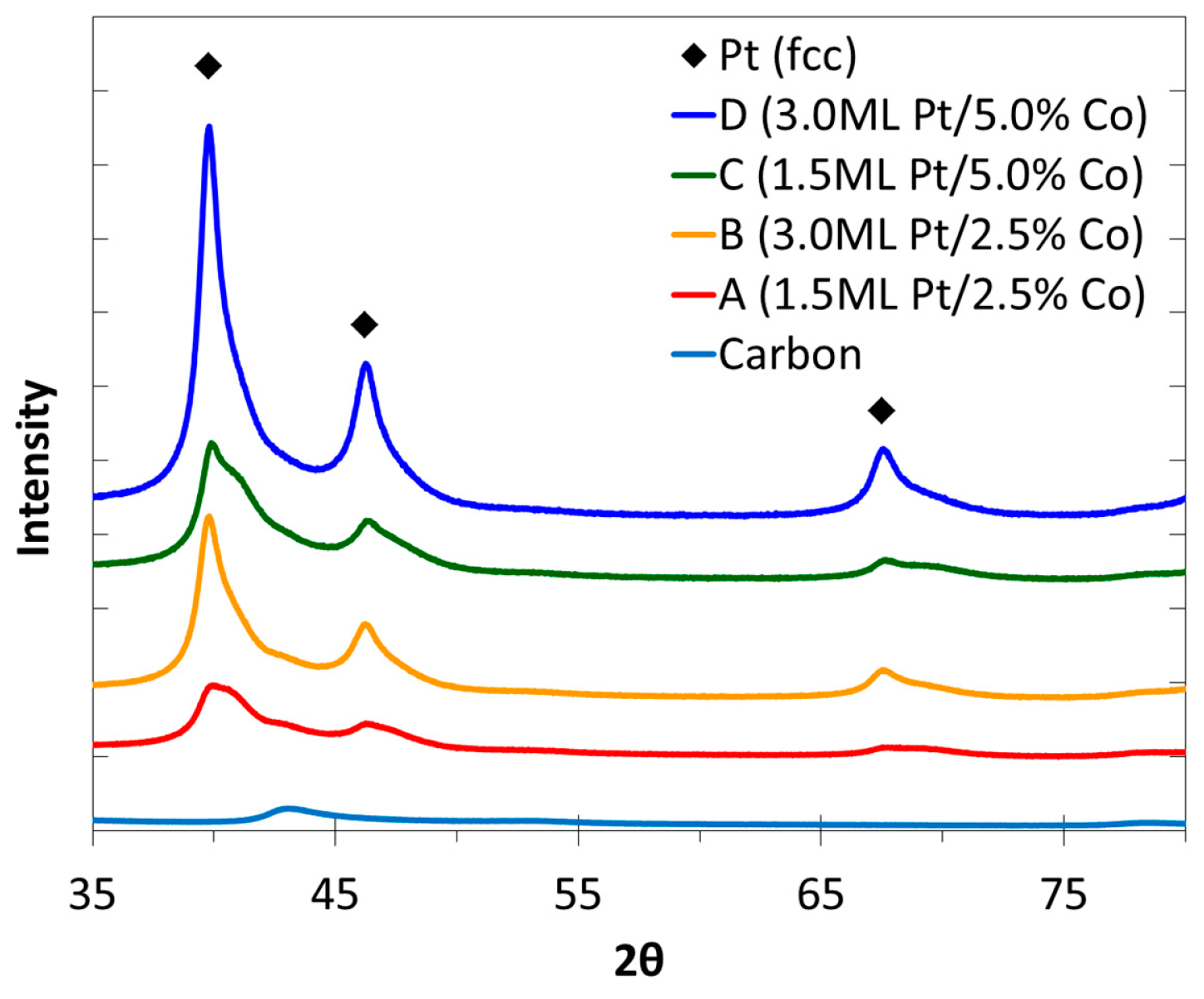
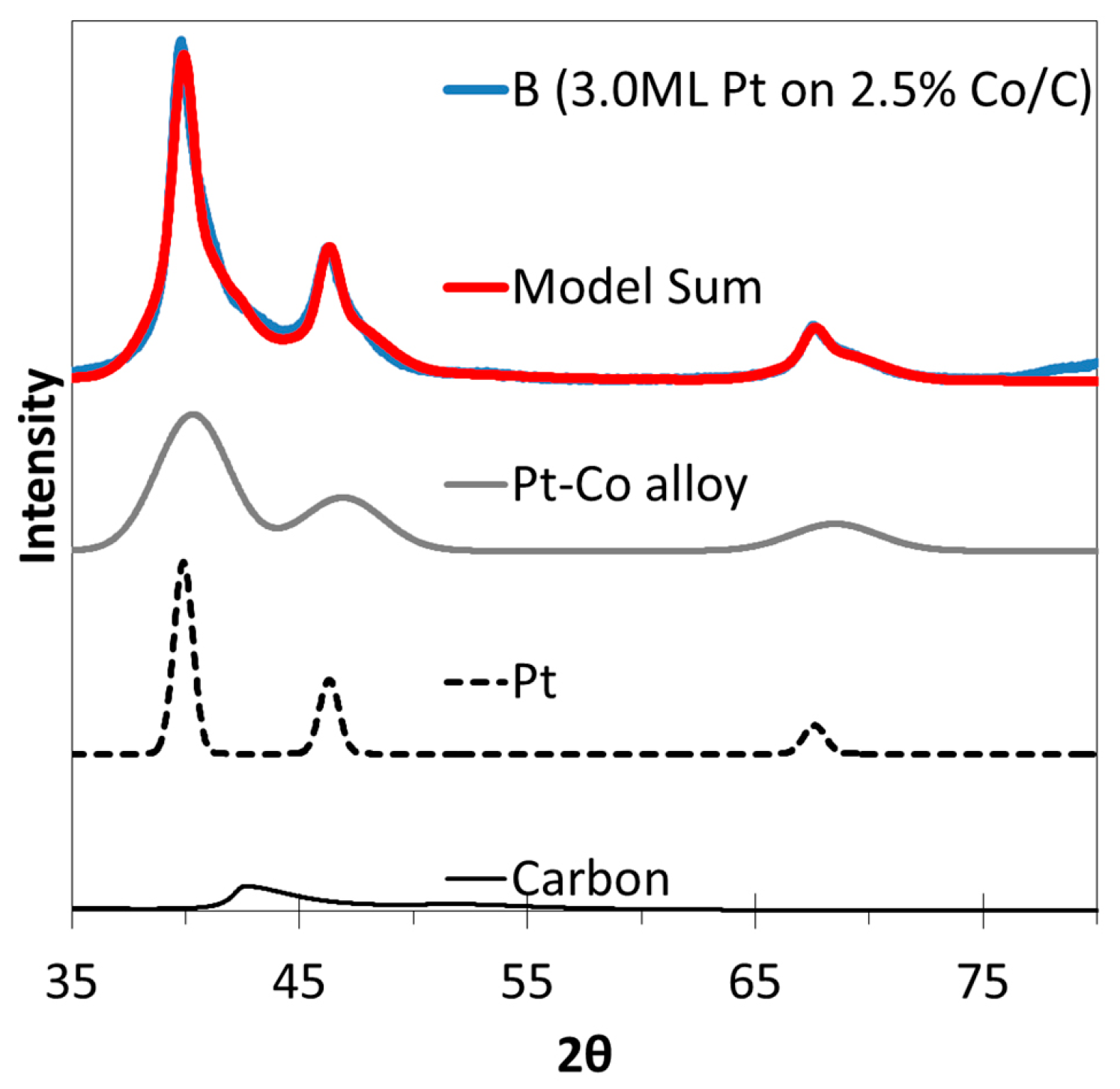
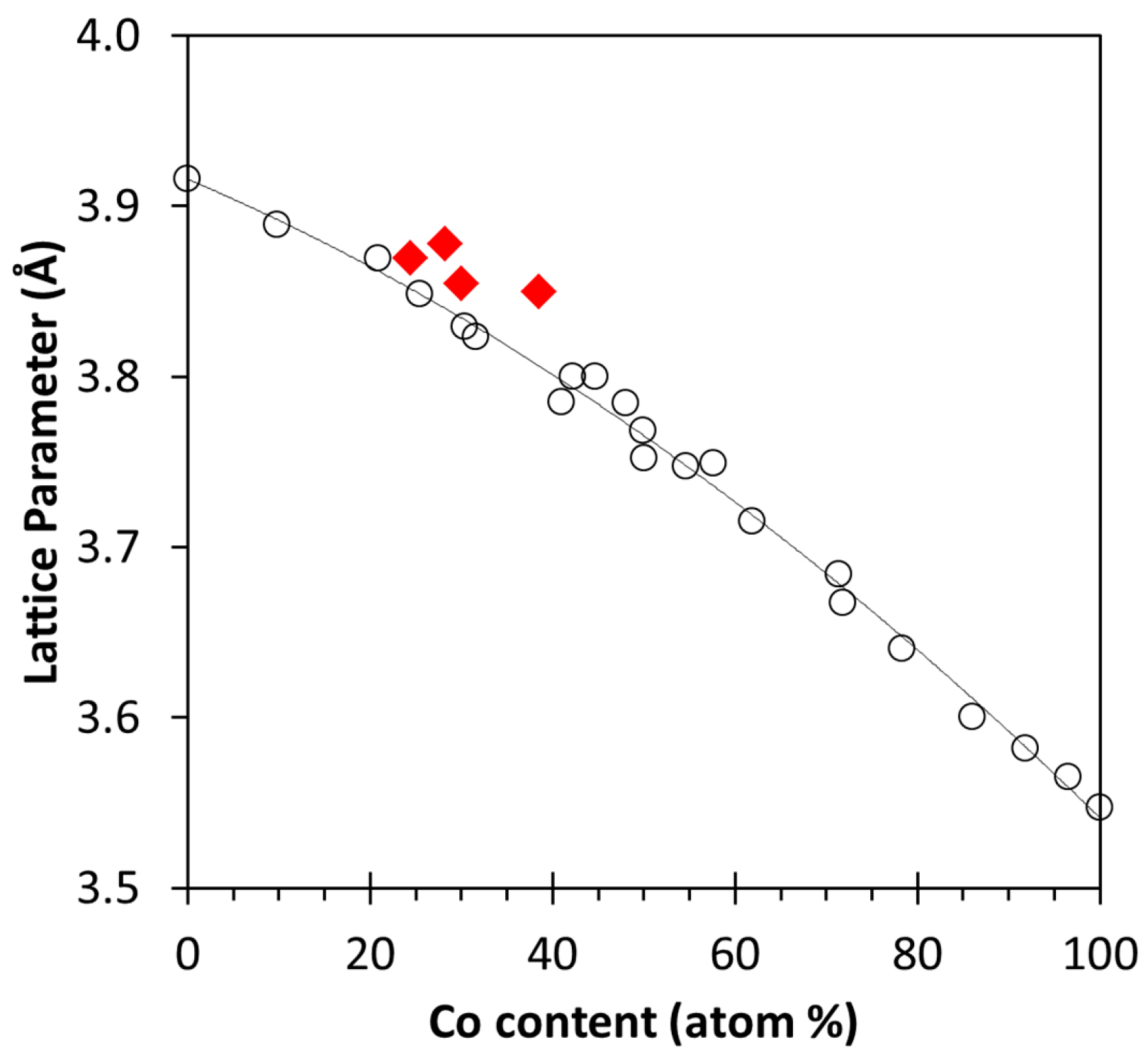
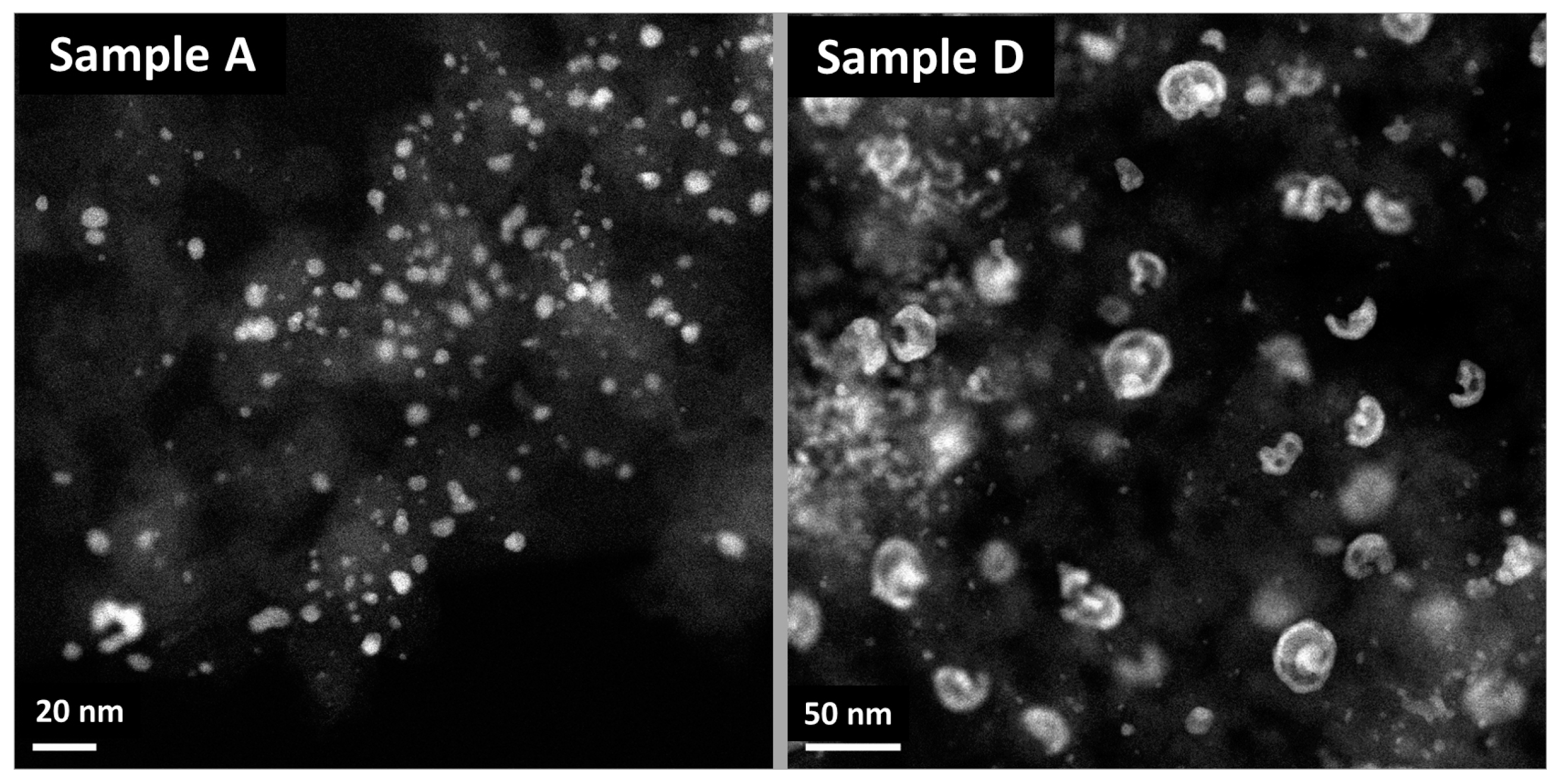
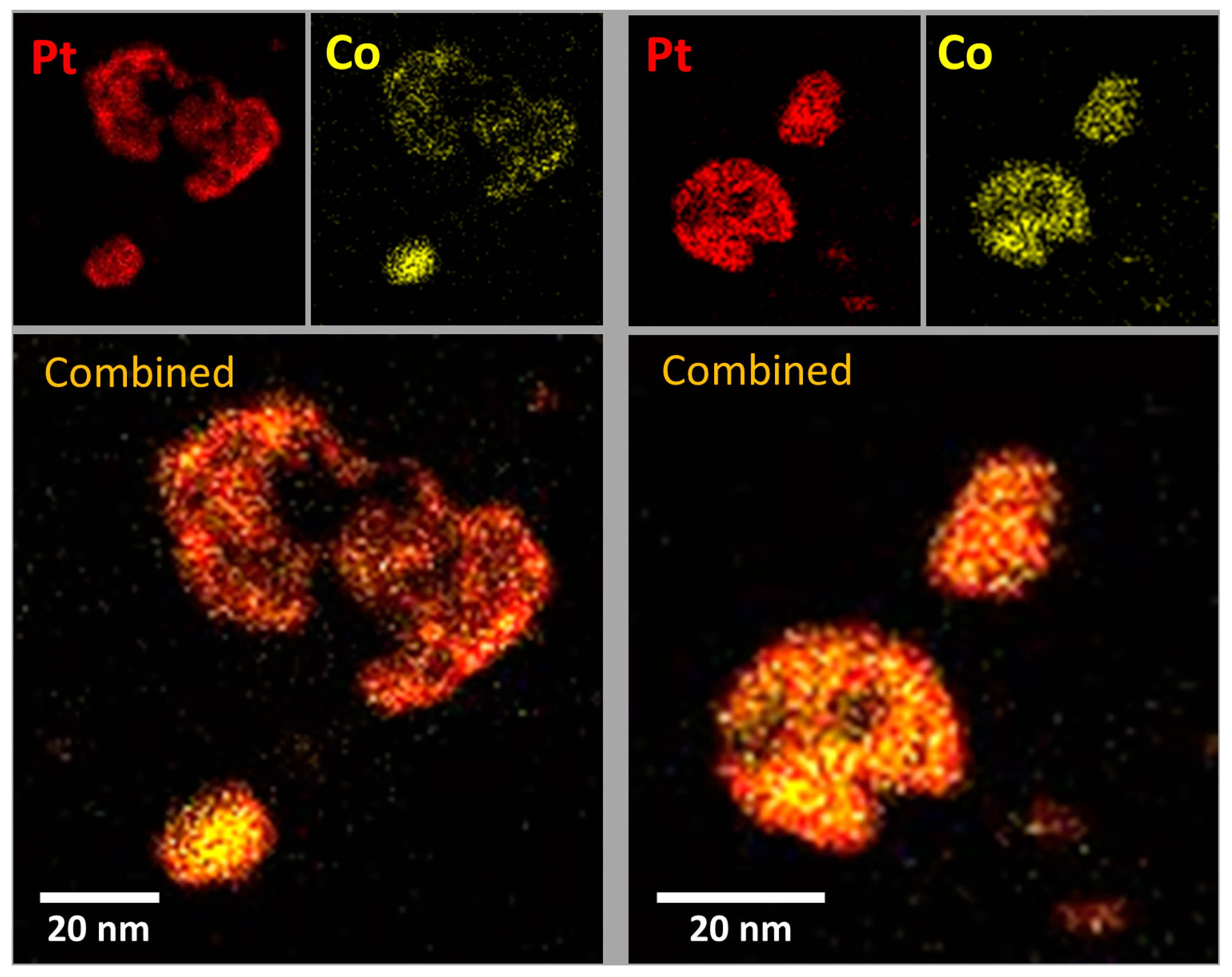
2.3. XRD, HAADF-STEM and XEDS Characterization of Pt-Co/C Catalysts
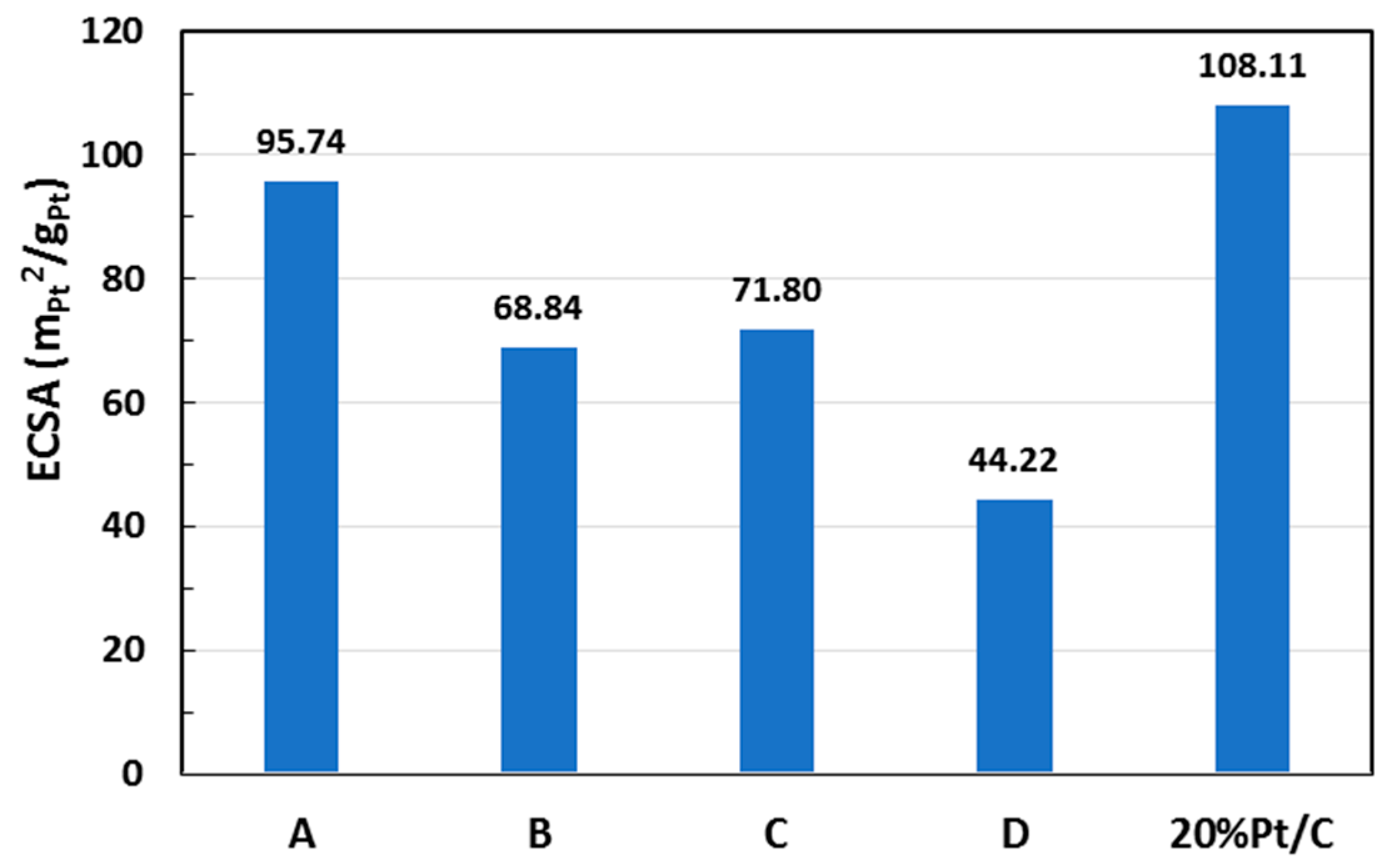
2.4. Catalyst Electrochemical Active Surface Area
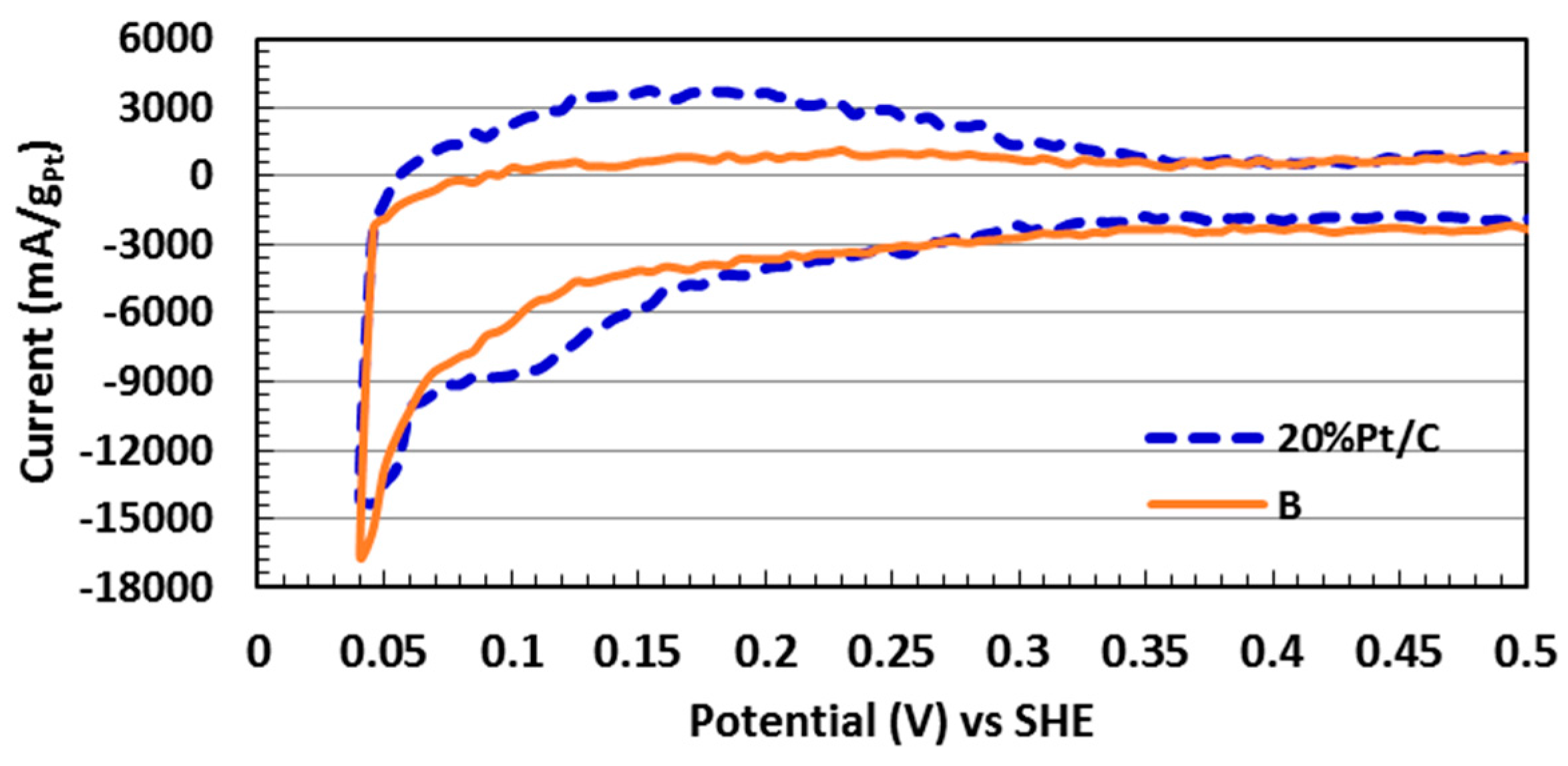
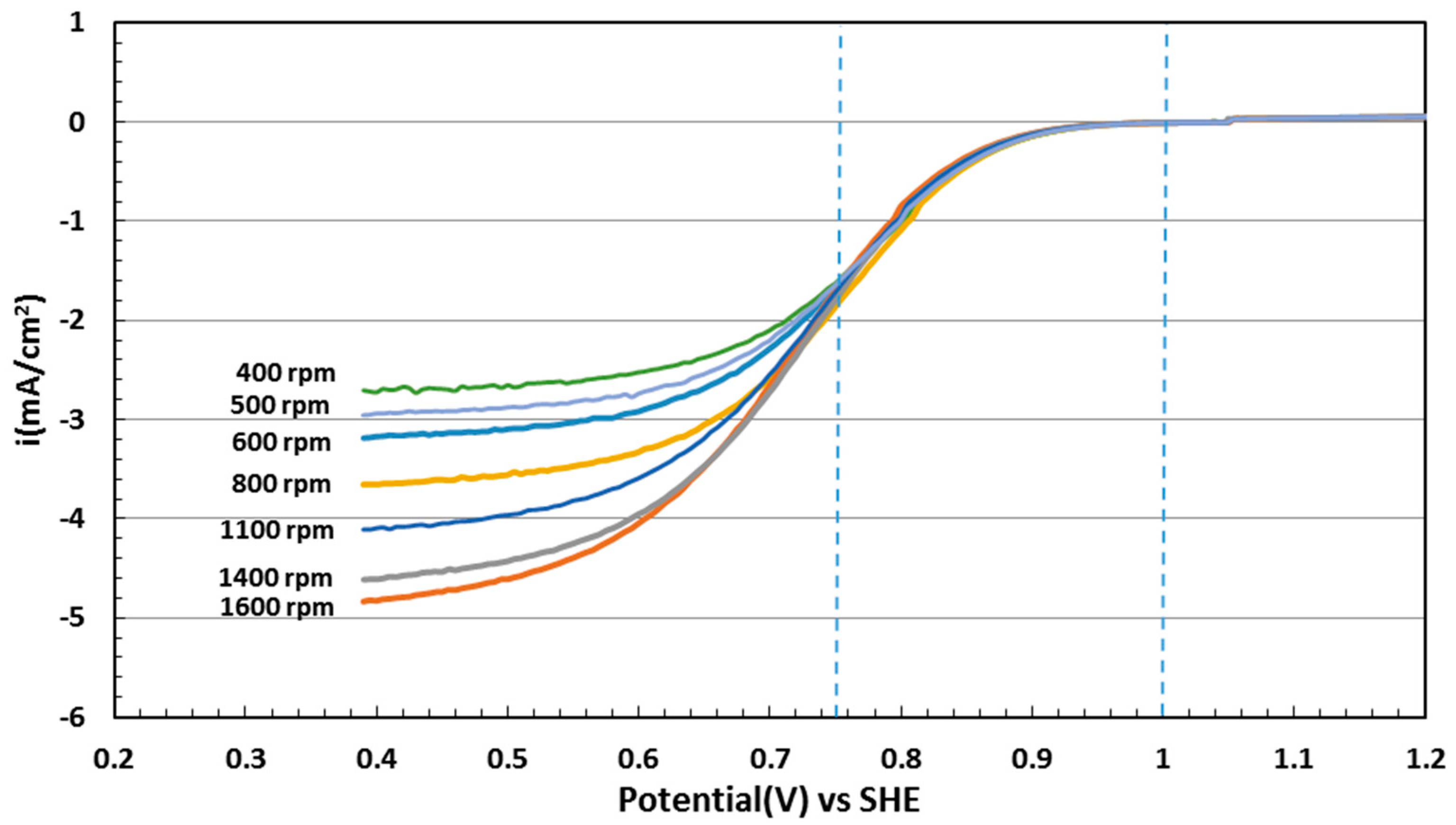
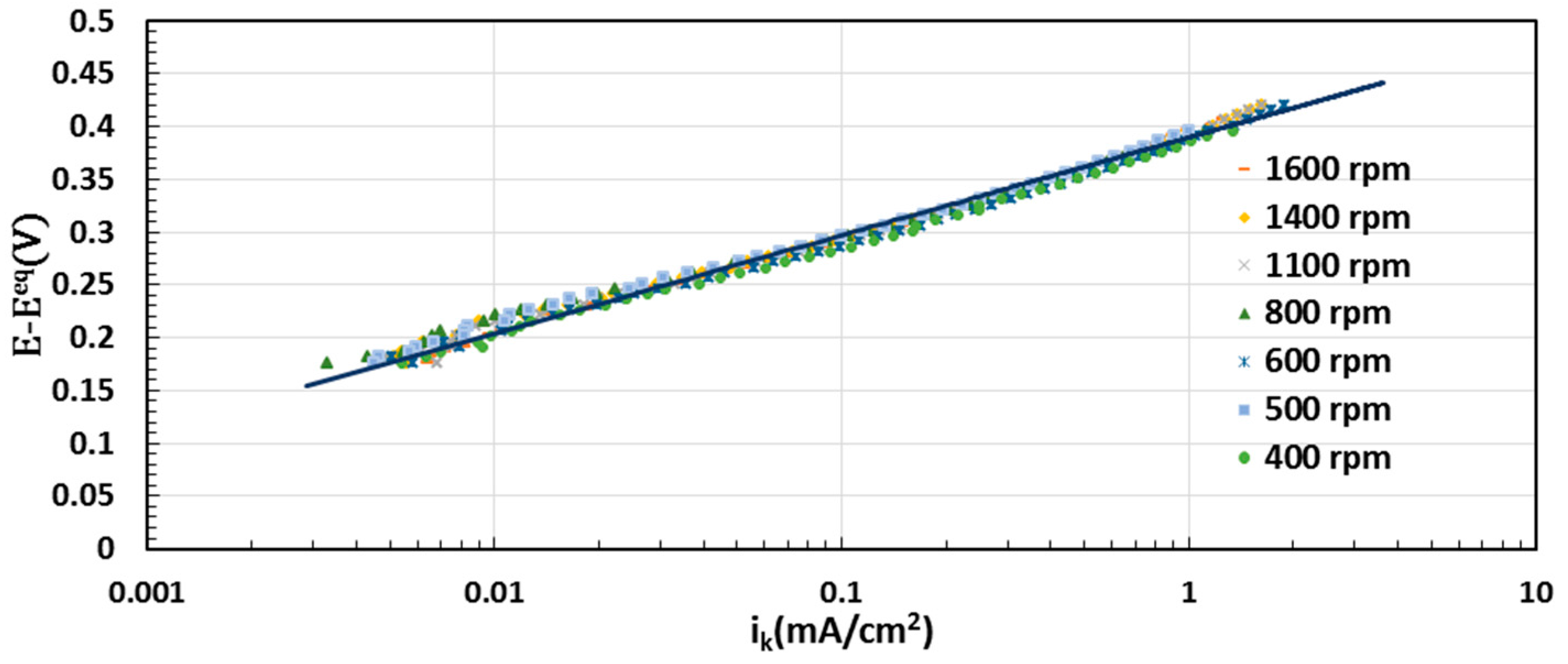
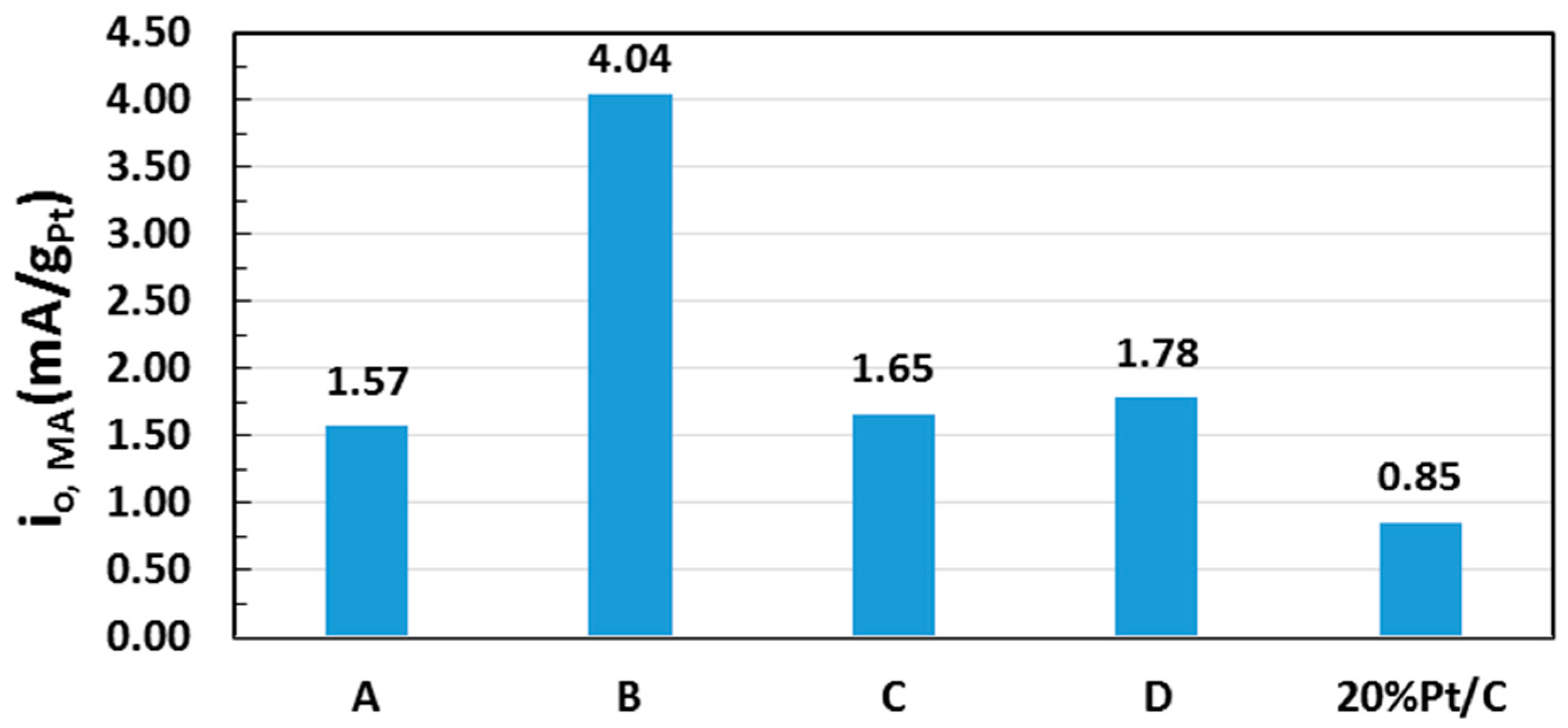
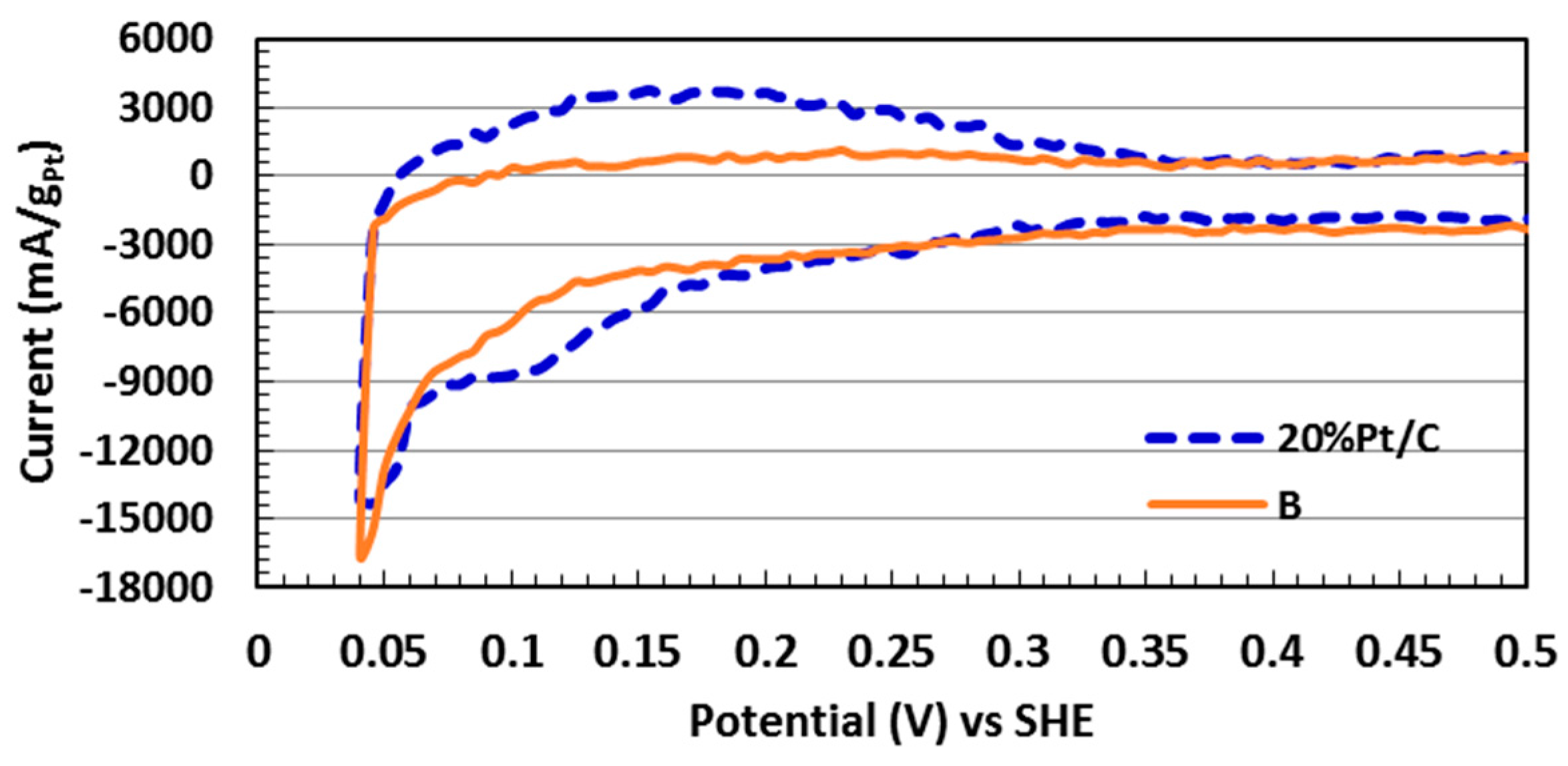
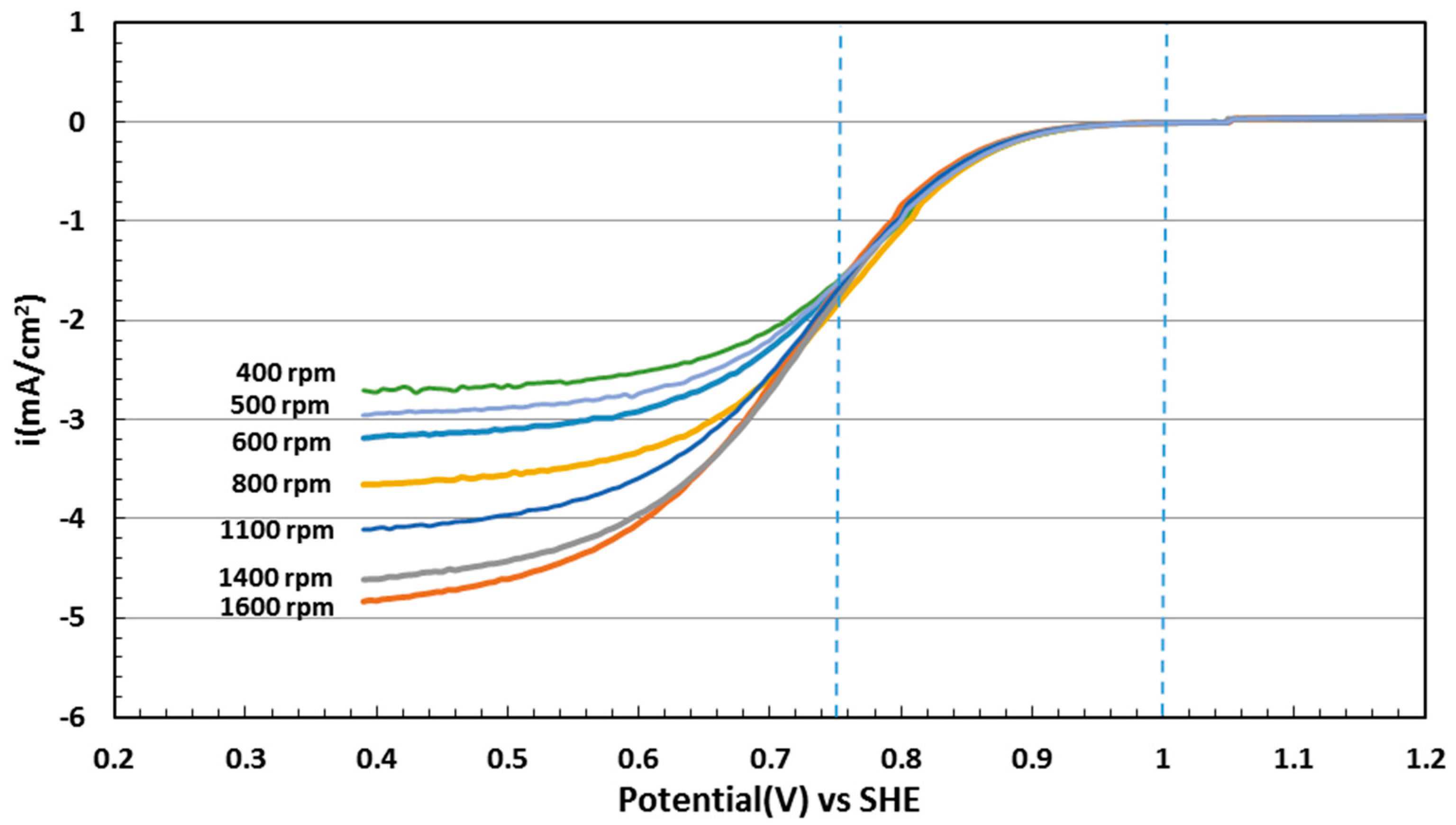
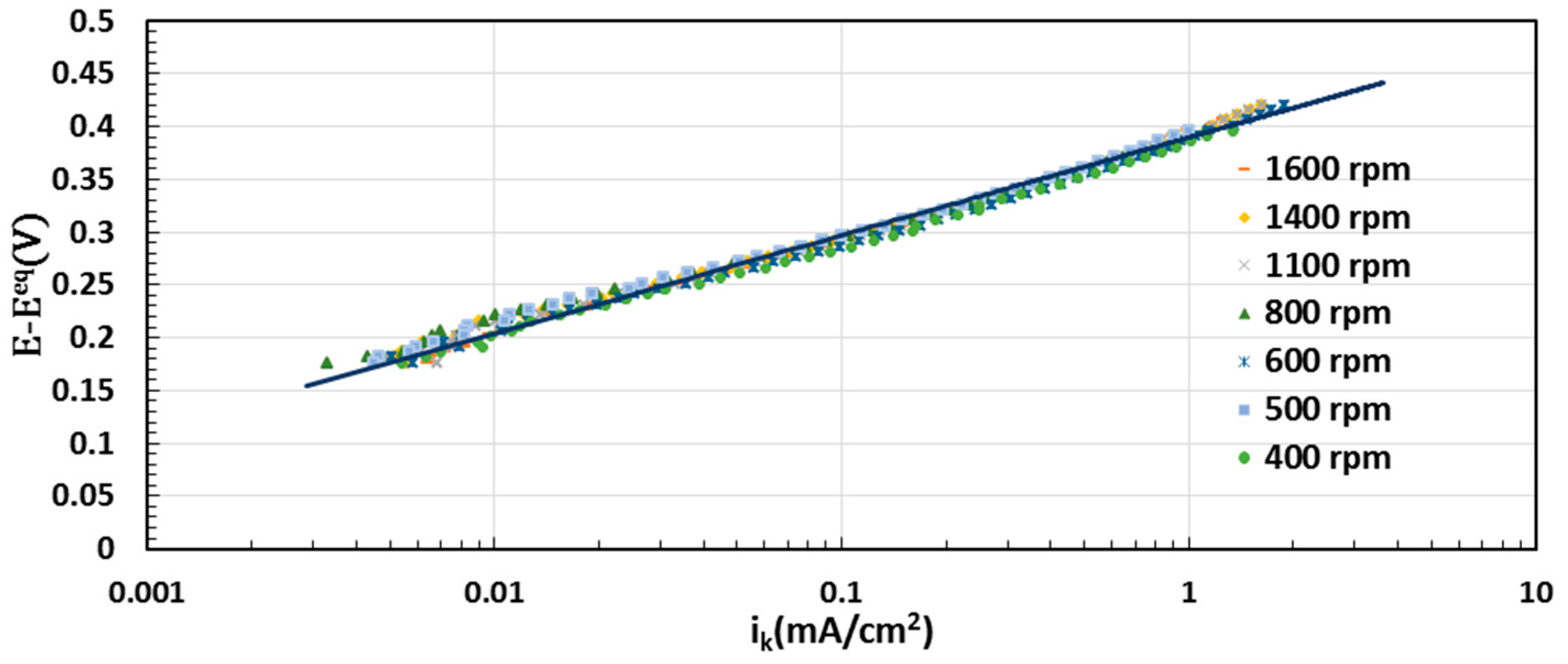
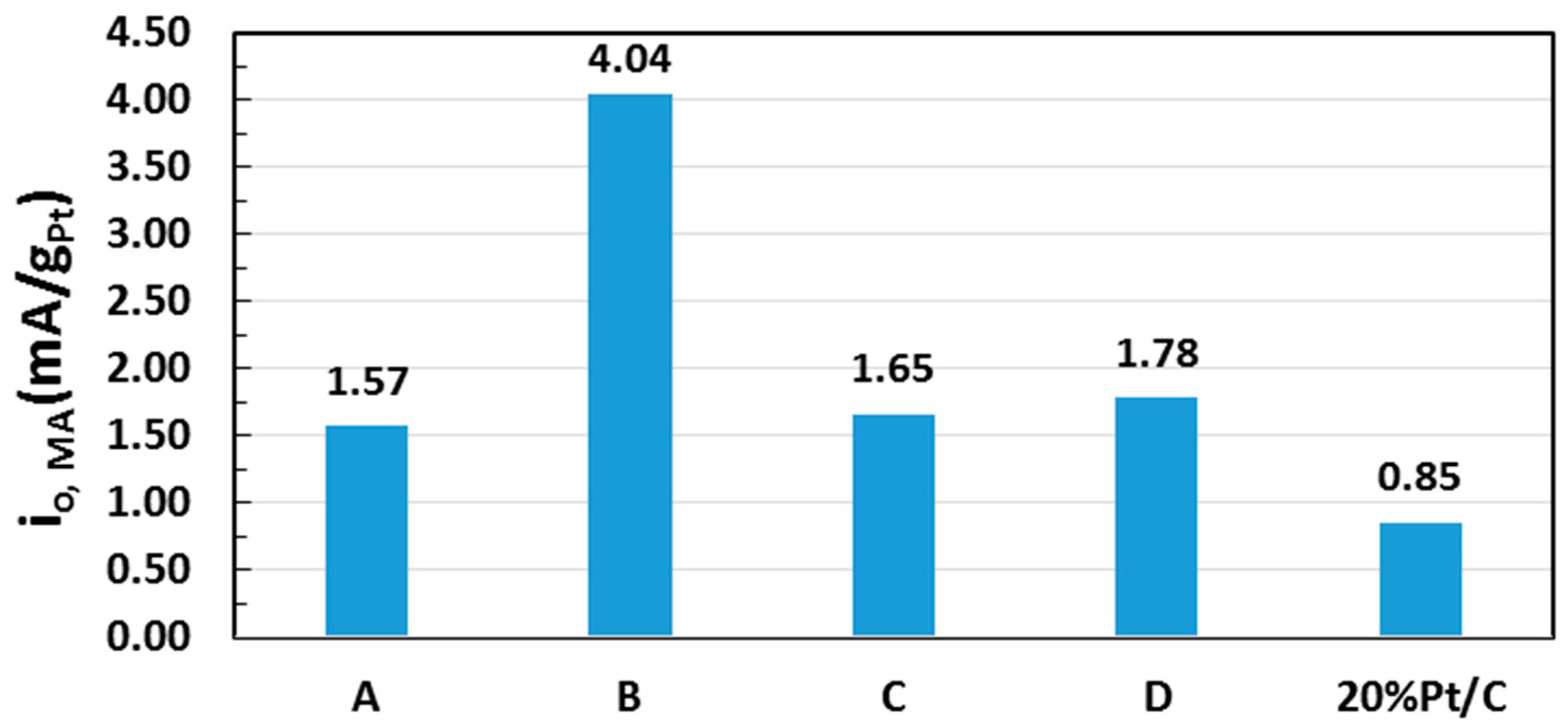
2.5. Electrocatalytic Activity toward ORR
3. Materials and Methods
3.1. Carbon Supported Cobalt (Co/C) Base Catalyst
3.2. Electroless Deposition of Pt
3.3. Catalyst Characterization
3.4. Electrochemical Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Redmond, E.L.; Setzler, B.P.; Juhas, P.; Billinge, S.J.L.; Fuller, T.F. In-Situ Monitoring of Particle Growth at PEMFC Cathode under Accelerated Cycling Conditions. Electrochem. Solid State Lett. 2012, 15, B72–B74. [Google Scholar] [CrossRef]
- Takeuchi, N.; Fuller, T.F. Modeling and Investigation of Carbon Loss on the Cathode Electrode during PEMFC Operation. J. Electrochem. Soc. 2010, 157, B135–B140. [Google Scholar] [CrossRef]
- Kinoshita, K. Particle Size Effects for Oxygen Reduction on Highly Dispersed Platinum in Acid Electrolytes. J. Electrochem. Soc. 1990, 137, 845–848. [Google Scholar] [CrossRef]
- Kabbabi, A.; Gloaguen, F.; Andolfatto, F.; Durand, R. Particle size effect for oxygen reduction and methanol oxidation on Pt/C inside a proton exchange membrane. J. Electroanal. Chem. 1994, 373, 251–254. [Google Scholar] [CrossRef]
- Wikander, K.; Ekström, H.; Palmqvist, A.E.C.; Lindbergh, G. On the influence of Pt particle size on the PEMFC cathode performance. Electrochim. Acta 2007, 52, 6848–6855. [Google Scholar] [CrossRef]
- Nikkuni, F.R.; Dubau, L.; Ticianelli, E.A.; Chatenet, M. Accelerated degradation of Pt3Co/C and Pt/C electrocatalysts studied by identical-location transmission electron microscopy in polymer electrolyte environment. Appl. Catal. B Environ. 2015, 176–177, 486–499. [Google Scholar] [CrossRef]
- Xu, Q.; Kreidler, E.; He, T. Performance and durability of PtCo alloy catalysts for oxygen electroreduction in acidic environments. Electrochim. Acta 2010, 55, 7551–7557. [Google Scholar] [CrossRef]
- Wang, D.L.; Xin, H.L.L.; Hovden, R.; Wang, H.S.; Yu, Y.C.; Muller, D.A.; DiSalvo, F.J.; Abruna, H.D. Structurally ordered intermetallic platinum-cobalt core-shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 2013, 12, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Habibi, B.; Ghaderi, S. Synthesis, characterization and electrocatalytic activity of Co@Pt nanoparticles supported on carbon-ceramic substrate for fuel cell applications. Int. J. Hydrog. Energy 2015, 40, 5115–5125. [Google Scholar] [CrossRef]
- Xin, H.; Holewinski, A.; Linic, S. Predictive Structure-Reactivity Models for Rapid Screening of Pt-Based Multimetallic Electrocatalysts for the Oxygen Reduction Reaction. ACS Catal. 2012, 2, 12–16. [Google Scholar] [CrossRef]
- Ferrin, P.; Nilekar, A.U.; Greeley, J.; Mavrikakis, M.; Rossmeisl, J. Reactivity descriptors for direct methanol fuel cell anode catalysts. Surf. Sci. 2008, 602, 3424–3431. [Google Scholar] [CrossRef]
- Herron, J.A.; Mavrikakis, M. On the composition of bimetallic near-surface alloys in the presence of oxygen and carbon monoxide. Catal. Commun. 2014, 52, 65–71. [Google Scholar] [CrossRef]
- Cao, C.; Yang, G.; Dubau, L.; Maillard, F.; Lambert, S.D.; Pirard, J.-P.; Job, N. Highly dispersed Pt/C catalysts prepared by the Charge Enhanced Dry Impregnation method. Appl. Catal. B Environ. 2014, 150–151, 101–106. [Google Scholar] [CrossRef]
- Zhu, X.; Cho, H.-R.; Pasupong, M.; Regalbuto, J.R. Charge-Enhanced Dry Impregnation: A Simple Way to Improve the Preparation of Supported Metal Catalysts. ACS Catal. 2013, 3, 625–630. [Google Scholar] [CrossRef]
- Regalbuto, J.R. Strong Electrostatic Adsorption of Metals onto Catalyst Supports. In Catalyst Preparation: Science and Engineering; Regalbuto, J.R., Ed.; Taylor and Francis/CRC Press: Boca Raton, FL, USA, 2007; pp. 297–318. [Google Scholar]
- Rodriguez, A.A.; Williams, C.T.; Monnier, J.R. Selective liquid-phase oxidation of glycerol over Au-Pd/C bimetallic catalysts prepared by electroless deposition. Appl. Catal. A Gen. 2014, 475, 161–168. [Google Scholar] [CrossRef]
- Diao, W.; Tengco, J.M.M.; Regalbuto, J.R.; Monnier, J.R. Preparation and Characterization of Pt-Ru Bimetallic Catalysts Synthesized by Electroless Deposition Methods. ACS Catal. 2015, 5, 5123–5134. [Google Scholar] [CrossRef]
- Zhang, Y.; Diao, W.; Williams, C.T.; Monnier, J.R. Selective hydrogenation of acetylene in excess ethylene using Ag- and Au-Pd/SiO2 bimetallic catalysts prepared by electroless deposition. Appl. Catal. A Gen. 2014, 469, 419–426. [Google Scholar] [CrossRef]
- Schaal, M.T.; Pickerell, A.C.; Williams, C.T.; Monnier, J.R. Characterization and evaluation of Ag-Pt/SiO2 catalysts prepared by electroless deposition. J. Catal. 2008, 254, 131–143. [Google Scholar] [CrossRef]
- Beard, K.D.; Borrelli, D.; Cramer, A.M.; Blom, D.; van Zee, J.W.; Monnier, J.R. Preparation and Structural Analysis of Carbon-Supported Co Core/Pt Shell Electrocatalysts Using Electroless Deposition Methods. ACS Nano 2009, 3, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Beard, K.D.; Schaal, M.T.; van Zee, J.W.; Monnier, J.R. Preparation of highly dispersed PEM fuel cell catalysts using electroless deposition methods. Appl. Catal. B Environ. 2007, 72, 262–271. [Google Scholar] [CrossRef]
- Beard, K.D.; van Zee, J.W.; Monnier, J.R. Preparation of carbon-supported Pt-Pd electrocatalysts with improved physical properties using electroless deposition methods. Appl. Catal. B Environ. 2009, 88, 185–193. [Google Scholar] [CrossRef]
- Wongkaew, A.; Zhang, Y.; Tengco, J.M.M.; Blom, D.A.; Sivasubramanian, P.; Fanson, P.T.; Regalbuto, J.R.; Monnier, J.R. Characterization and evaluation of Pt-Pd electrocatalysts prepared by electroless deposition. Appl. Catal. B Environ. 2016, 188, 367–375. [Google Scholar] [CrossRef]
- Garrick, T.R.; Diao, W.; Tengco, J.M.; Stach, E.A.; Senanayake, S.D.; Chen, D.A.; Monnier, J.R.; Weidner, J.W. The Effect of the Surface Composition of Ru-Pt Bimetallic Catalysts for Methanol Oxidation. Electrochim. Acta 2016, 195, 106–111. [Google Scholar] [CrossRef]
- Bezemer, G.L.; Radstake, P.B.; Koot, V.; van Dillen, A.J.; Geus, J.W.; de Jong, K.P. Preparation of Fischer-Tropsch cobalt catalysts supported on carbon nanofibers and silica using homogeneous deposition-precipitation. J. Catal. 2006, 237, 291–302. [Google Scholar] [CrossRef]
- Yang, Y.; Jia, L.; Meng, Y.; Hou, B.; Li, D.; Sun, Y. Fischer-Tropsch Synthesis over Ordered Mesoporous Carbon Supported Cobalt Catalysts: The Role of Amount of Carbon Precursor in Catalytic Performance. Catal. Lett. 2011, 142, 195–204. [Google Scholar] [CrossRef]
- Lu, J.; Huang, C.; Bai, S.; Jiang, Y.; Li, Z. Thermal decomposition and cobalt species transformation of carbon nanotubes supported cobalt catalyst for Fischer-Tropsch synthesis. J. Nat. Gas Chem. 2012, 21, 37–42. [Google Scholar] [CrossRef]
- Bychko, I.; Kalishyn, Y.; Strizhak, P. TPR Study of Core-Shell Fe@Fe3O4 Nanoparticles Supported on Activated Carbon and Carbon Nanotubes. Adv. Mater. Phys. Chem. 2012, 2, 17–22. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63–77. [Google Scholar] [CrossRef]
- Bergwerff, J.A.; Lysova, A.A.; Espinosa-Alonso, L.; Koptyug, I.V.; Weckhuysen, B.M. Monitoring Transport Phenomena of Paramagnetic Metal-Ion Complexes Inside Catalyst Bodies with Magnetic Resonance Imaging. Chem. A Eur. J. 2008, 14, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Hara, T.; Koizumi, N.; Yamada, M. Novel preparation method of highly active Co/SiO2 catalyst for Fischer-Tropsch synthesis with chelating agents. Catal. Lett. 2007, 113, 165–169. [Google Scholar] [CrossRef]
- Miulović Snežana, M.; Nikolić Vladimir, M.; Laušević Petar, Z.; Aćimović Danka, D.; Tasić Gvozden, S.; Marčeta-Kaninski Milica, P. Electrochemistry of cobalt ethylenediamine complexes at high pH. J. Serbian Chem. Soc. 2015, 80, 1515–1527. [Google Scholar] [CrossRef]
- Lee, C.-L.; Tseng, C.-M. Ag-Pt Nanoplates: Galvanic Displacement Preparation and Their Applications As Electrocatalysts. J. Phys. Chem. C 2008, 112, 13342–13345. [Google Scholar] [CrossRef]
- Lee, C.-L.; Tseng, C.-M.; Wu, R.-B.; Wu, C.-C.; Syu, S.-C. Catalytic characterization of hollow silver/palladium nanoparticles synthesized by a displacement reaction. Electrochim. Acta 2009, 54, 5544–5547. [Google Scholar] [CrossRef]
- Micheaud, C.; Marécot, P.; Guérin, M.; Barbier, J. Preparation of alumina supported palladium-platinum catalysts by surface redox reactions. Activity for complete hydrocarbon oxidation. Appl. Catal. A Gen. 1998, 171, 229–239. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Zhumaev, U.E.; Maksimov, Y.M. Galvanic displacement of copper adatoms on platinum in solutions. J. Electroanal. Chem. 2011, 651, 30–37. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Darling, A.S. Cobalt-platinum alloys. Platin. Met. Rev. 1963, 7, 96–104. [Google Scholar]
- Ohashi, M.; Beard, K.D.; Ma, S.; Blom, D.A.; St-Pierre, J.; van Zee, J.W.; Monnier, J.R. Electrochemical and structural characterization of carbon-supported Pt-Pd bimetallic electrocatalysts prepared by electroless deposition. Electrochim. Acta 2010, 55, 7376–7384. [Google Scholar] [CrossRef]
- Ohno, I. Electrochemistry of electroless plating. Mater. Sci. Eng. A 1991, 146, 33–49. [Google Scholar] [CrossRef]
- Ohno, I.; Wakabayashi, O.; Haruyama, S. Anodic Oxidation of Reductants in Electroless Plating. J. Electrochem. Soc. 1985, 132, 2323–2330. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, C.; Zhu, Y.; Si, R.; Cai, Y.; Wang, J.X.; Adzic, R.R. Hollow core supported Pt monolayer catalysts for oxygen reduction. Catal. Today 2013, 202, 50–54. [Google Scholar] [CrossRef]
- Zhou, W.-P.; Vukmirovic, M.; Sasaki, K.; Adzic, R. Oxygen Reduction Reaction on a Pt Monolayer on Pd2Co(111) Crystals. Meet. Abstr. 2008, 13, 23–28. [Google Scholar]
- Tegou, A.; Papadimitriou, S.; Mintsouli, I.; Armyanov, S.; Valova, E.; Kokkinidis, G.; Sotiropoulos, S. Rotating disc electrode studies of borohydride oxidation at Pt and bimetallic Pt-Ni and Pt-Co electrodes. Catal. Today 2011, 170, 126–133. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Armyanov, S.; Valova, E.; Hubin, A.; Steenhaut, O.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Methanol Oxidation at Pt-Cu, Pt-Ni, and Pt-Co Electrode Coatings Prepared by a Galvanic Replacement Process. J. Phys. Chem. C 2010, 114, 5217–5223. [Google Scholar] [CrossRef]
- Djokić, S.S. Electroless Deposition of Metals and Alloys. In Modern Aspects of Electrochemistry; Conway, B.E., White, R.E., Eds.; Springer: Boston, MA, USA, 2002; pp. 51–133. [Google Scholar]
- Haruyama, S.; Ohno, I. Catalytic Aspects in Electroless Deposition; Paunovic, M., Ohno, I., Eds.; Electrochemical Society: Pennington, NJ, USA, 1988; pp. 20–36. [Google Scholar]
- Cooper, K.R. In Situ PEM Fuel Cell Electrochemical Surface Area and Catalyst Utilization Measurement. Fuel Cell Mag. 2009, 9. [Google Scholar]
- Huang, H.; Sun, D.; Wang, X. PtCo alloy nanoparticles supported on graphene nanosheets with high performance for methanol oxidation. Chin. Sci. Bull. 2012, 57, 3071–3079. [Google Scholar] [CrossRef]
- Jiang, R.; Tran, D.T.; McClure, J.P.; Chu, D. Increasing the electrochemically available active sites for heat-treated hemin catalysts supported on carbon black. Electrochim. Acta 2012, 75, 185–190. [Google Scholar] [CrossRef]
- Dong, Q.; Santhanagopalan, S.; White, R.E. Simulation of the Oxygen Reduction Reaction at an RDE in 0.5 M H2SO4 Including an Adsorption Mechanism. J. Electrochem. Soc. 2007, 154, A888–A899. [Google Scholar] [CrossRef]
- Paulus, U.A.; Wokaun, A.; Scherer, G.G.; Schmidt, T.J.; Stamenkovic, V.; Radmilovic, V.; Markovic, N.M.; Ross, P.N. Oxygen Reduction on Carbon-Supported Pt-Ni and Pt-Co Alloy Catalysts. J. Phys. Chem. B 2002, 106, 4181–4191. [Google Scholar] [CrossRef]
- Ishiguro, N.; Saida, T.; Uruga, T.; Nagamatsu, S.-I.; Sekizawa, O.; Nitta, K.; Yamamoto, T.; Ohkoshi, S.-I.; Iwasawa, Y.; Yokoyama, T.; et al. Operando Time-Resolved X-ray Absorption Fine Structure Study for Surface Events on a Pt3Co/C Cathode Catalyst in a Polymer Electrolyte Fuel Cell during Voltage-Operating Processes. ACS Catal. 2012, 2, 1319–1330. [Google Scholar] [CrossRef]
- Zhang, J.; Lima, F.H.B.; Shao, M.H.; Sasaki, K.; Wang, J.X.; Hanson, J.; Adzic, R.R. Platinum Monolayer on Nonnoble Metal-Noble Metal Core-Shell Nanoparticle Electrocatalysts for O2 Reduction. J. Phys. Chem. B 2005, 109, 22701–22704. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.; Regalbuto, J.R. High Sensitivity Silicon Slit Detectors for 1 nm Powder XRD Size Detection Limit. Catal. Lett. 2015, 145, 777–783. [Google Scholar] [CrossRef]
- Fuentes, R.E.; García, B.L.; Weidner, J.W. Effect of Titanium Dioxide Supports on the Activity of Pt-Ru toward Electrochemical Oxidation of Methanol. J. Electrochem. Soc. 2011, 158, B461–B466. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Pt Coverage θPt (Theoretical) | Co Loading before ED wt % (Nominal) | Final Pt Loading | Final Co Loading | ||
|---|---|---|---|---|---|---|
| wt % | Atomic% (Metal Basis) | wt % | Atomic% (Metal Basis) | |||
| A | 1.5 | 2.5 | 6.7 | 70.1 | 0.9 | 29.9 |
| B | 3.0 | 2.5 | 12.2 | 75.6 | 1.2 | 24.4 |
| C | 1.5 | 5.0 | 12.6 | 61.5 | 2.4 | 38.5 |
| D | 3.0 | 5.0 | 22.2 | 71.9 | 2.6 | 28.1 |
| Catalyst | Theoretical Monodisperse Coverage of Pt (ML—monolayer) | Initial Co Loading (wt %) | XRD Sizes | ||
|---|---|---|---|---|---|
| Alloy (Pt-Co) Phase | Non-Alloyed Pt Size (nm) | ||||
| Lattice Parameter (Å) | Size (nm) | ||||
| A | 1.5 | 2.5 | 3.85 | 2.6 | 11.8 |
| B | 3.0 | 2.5 | 3.87 | 2.3 | 8.9 |
| C | 1.5 | 5.0 | 3.85 | 2.3 | 11.0 |
| D | 3.0 | 5.0 | 3.88 | 2.3 | 10.6 |
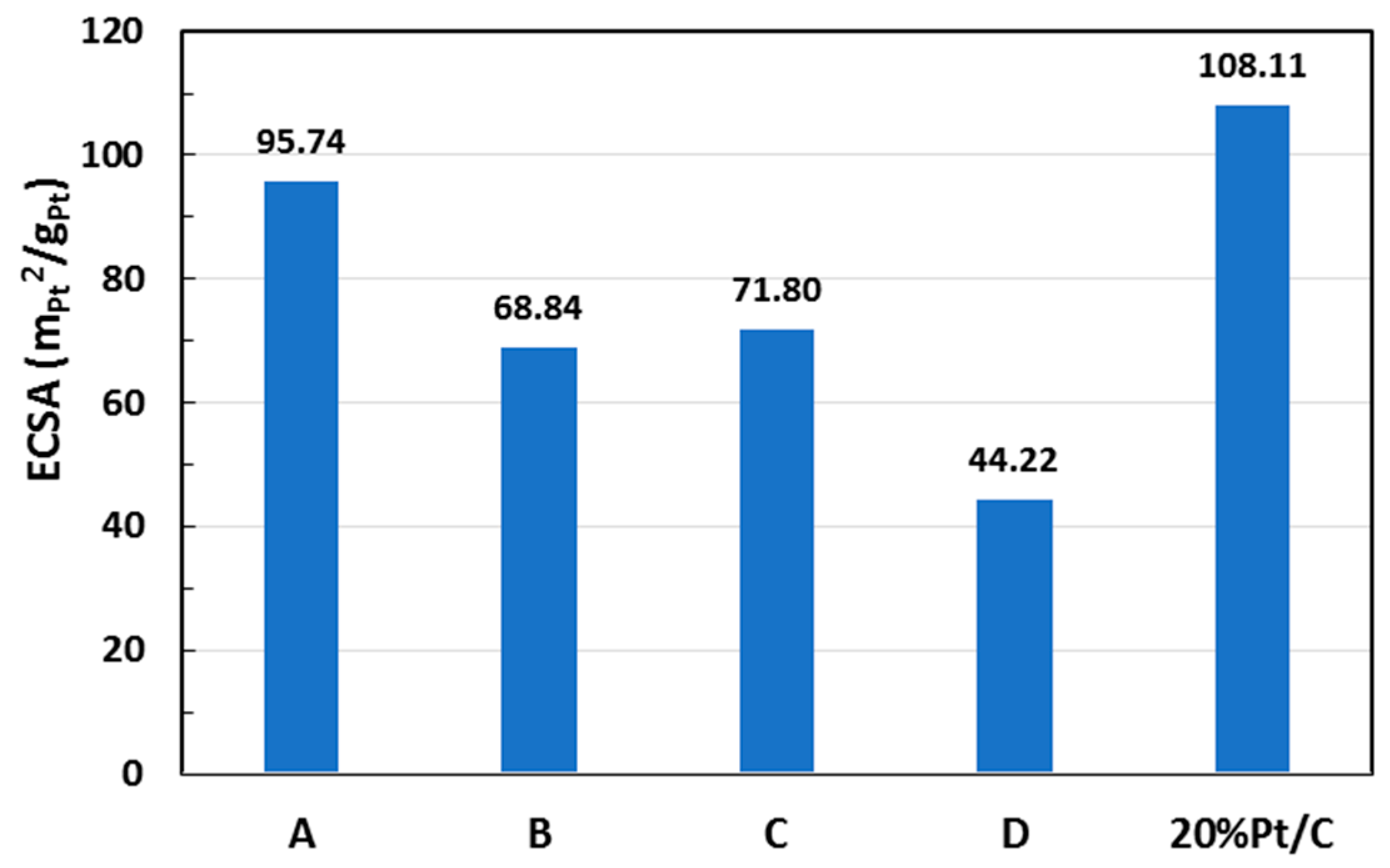
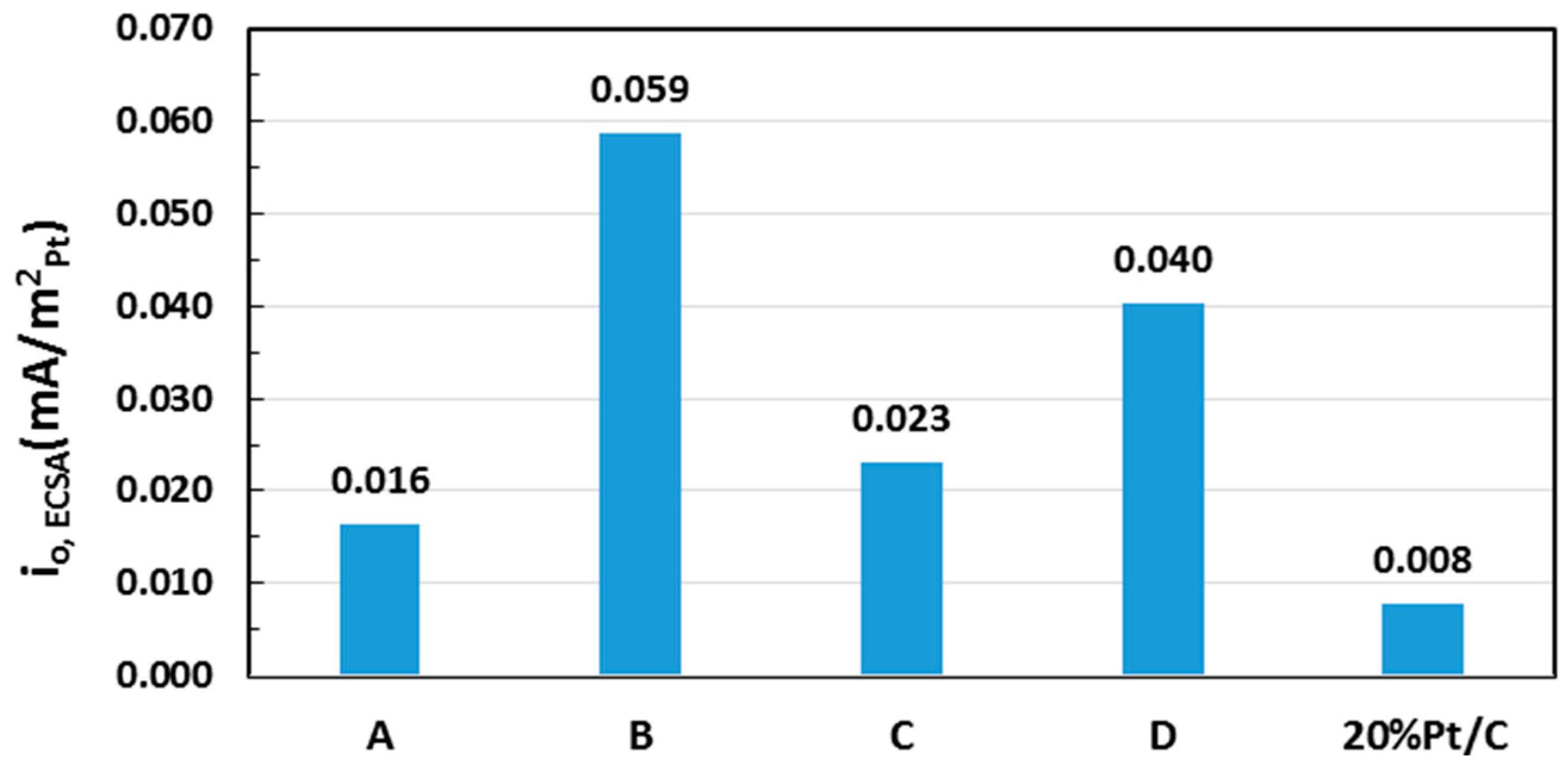
| Sample | Pt (% Loading) | ECSA (m2/gPt) | i0, ECSA (mA/m2Pt) |
|---|---|---|---|
| A | 6.71 | 95.7 | 0.016 |
| B | 12.2 | 68.8 | 0.059 |
| C | 12.6 | 71.8 | 0.023 |
| D | 22.2 | 44.2 | 0.040 |
| 20 wt % Pt/C * | 20.0 | 108.1 | 0.008 |
| Catalyst | Target Pt Coverage (ML) | Total Co/C Amount Used (g) | Total Bath Volume (mL) |
|---|---|---|---|
| 2.5% Co/CB1 | 1.5 | 1.0 | 600 |
| 3.0 | 0.5 | 600 | |
| 5.0% Co/CB1 | 1.5 | 1.0 | 1200 |
| 3.0 | 0.5 | 1200 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tengco, J.M.M.; Tavakoli Mehrabadi, B.A.; Zhang, Y.; Wongkaew, A.; Regalbuto, J.R.; Weidner, J.W.; Monnier, J.R. Synthesis and Electrochemical Evaluation of Carbon Supported Pt-Co Bimetallic Catalysts Prepared by Electroless Deposition and Modified Charge Enhanced Dry Impregnation. Catalysts 2016, 6, 83. https://doi.org/10.3390/catal6060083
Tengco JMM, Tavakoli Mehrabadi BA, Zhang Y, Wongkaew A, Regalbuto JR, Weidner JW, Monnier JR. Synthesis and Electrochemical Evaluation of Carbon Supported Pt-Co Bimetallic Catalysts Prepared by Electroless Deposition and Modified Charge Enhanced Dry Impregnation. Catalysts. 2016; 6(6):83. https://doi.org/10.3390/catal6060083
Chicago/Turabian StyleTengco, John Meynard M., Bahareh Alsadat Tavakoli Mehrabadi, Yunya Zhang, Akkarat Wongkaew, John R. Regalbuto, John W. Weidner, and John R. Monnier. 2016. "Synthesis and Electrochemical Evaluation of Carbon Supported Pt-Co Bimetallic Catalysts Prepared by Electroless Deposition and Modified Charge Enhanced Dry Impregnation" Catalysts 6, no. 6: 83. https://doi.org/10.3390/catal6060083