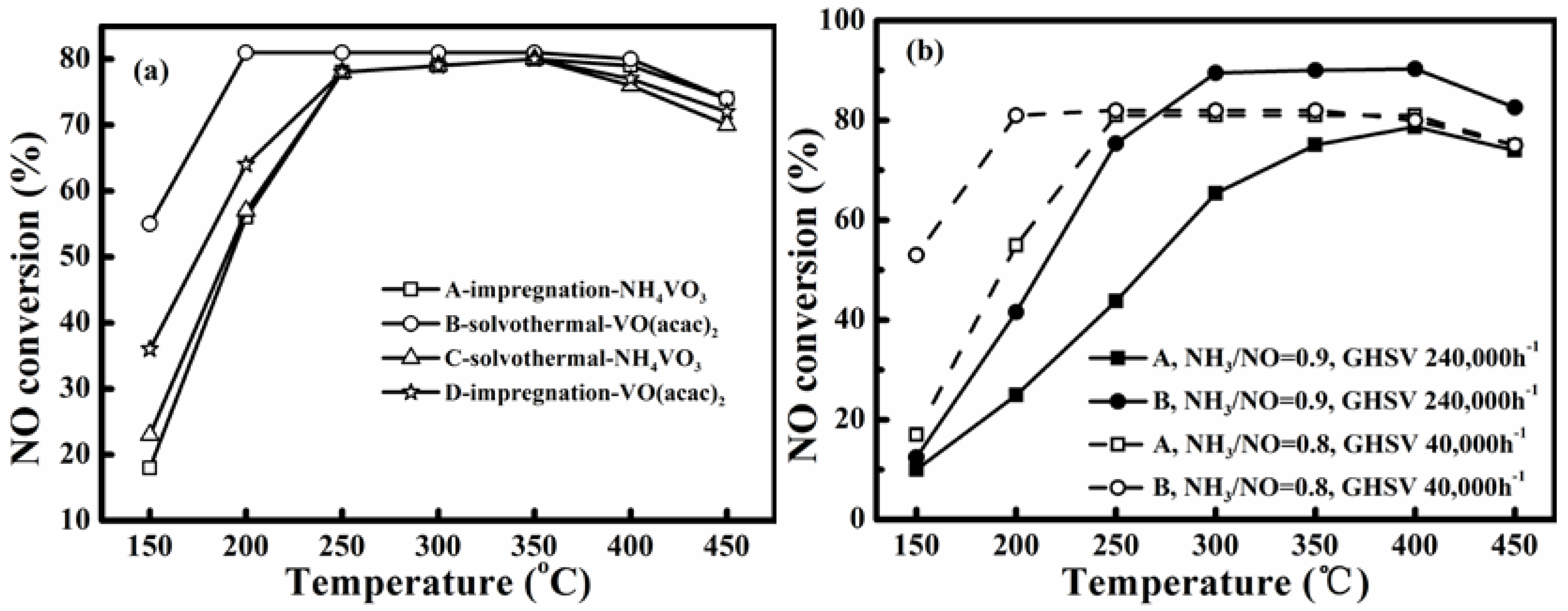
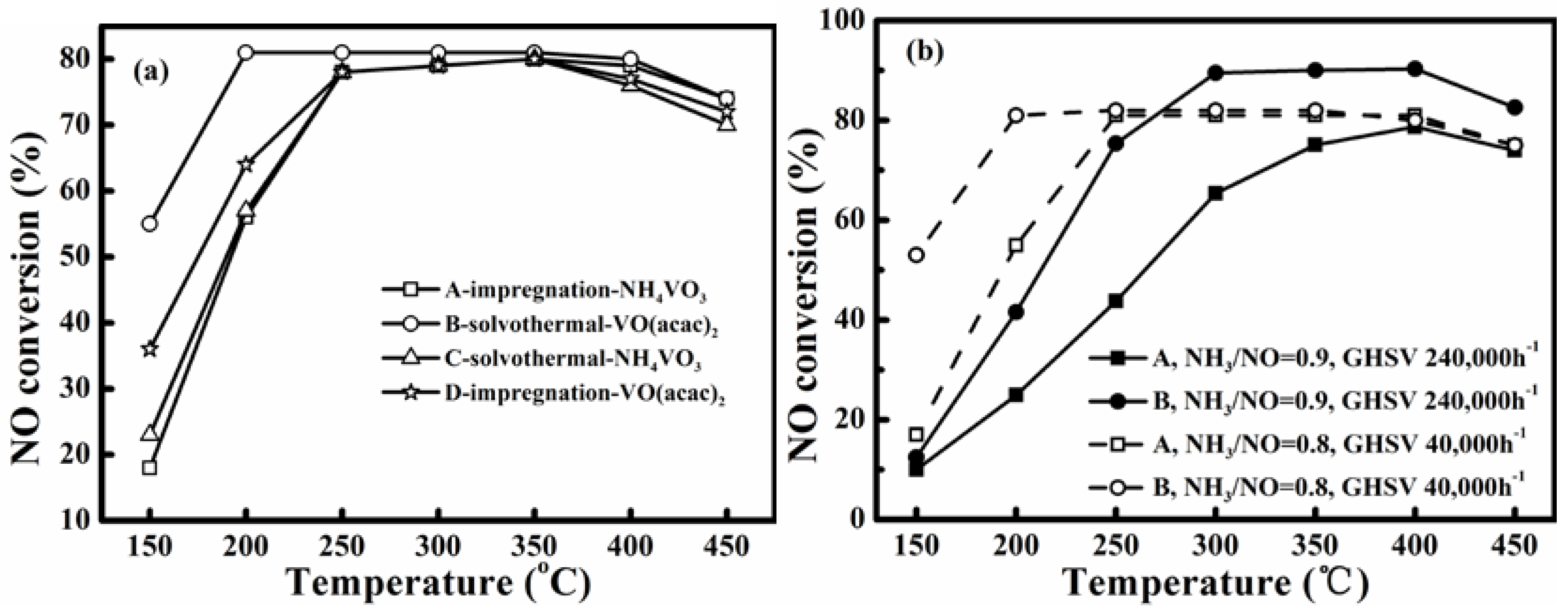
Figure 1a compares the NO conversions realized over the catalysts A to D under similar conditions shown in the experimental section for simulated flue gas without SO
2 and water vapor. Comparing the catalysts A and C, which had the same vanadium precursor NH
4VO
3 but different preparation methods, one can see that there is almost no obvious difference in the realized NO conversion that actually represents the de-NO
x activity of a catalyst. For the catalysts B and D using VO(acac)
2 as the vanadium precursor, however, their realized de-NO
x activities are higher than the catalysts A and C based on NH
4VO
3 precursor. Furthermore, the catalyst B manifested the best activity at low temperatures. Therefore, for analyzing the potential improvement on low-temperature activity and its mechanism by using different precursors, all tests herein were conducted only for the catalysts A and B.
Figure 1.
(a) NO conversions of four prepared catalysts A to D for simulated flue gas without SO2 and water vapor; (b) NO conversion of catalyst A and B under different GHSV: NH3/NO = 0.8, GHSV 40,000 h−1, and NH3/NO = 0.9, GHSV 240,000 h−1. (Gas compositions in Experimental section).
2.1. Physical Characterization
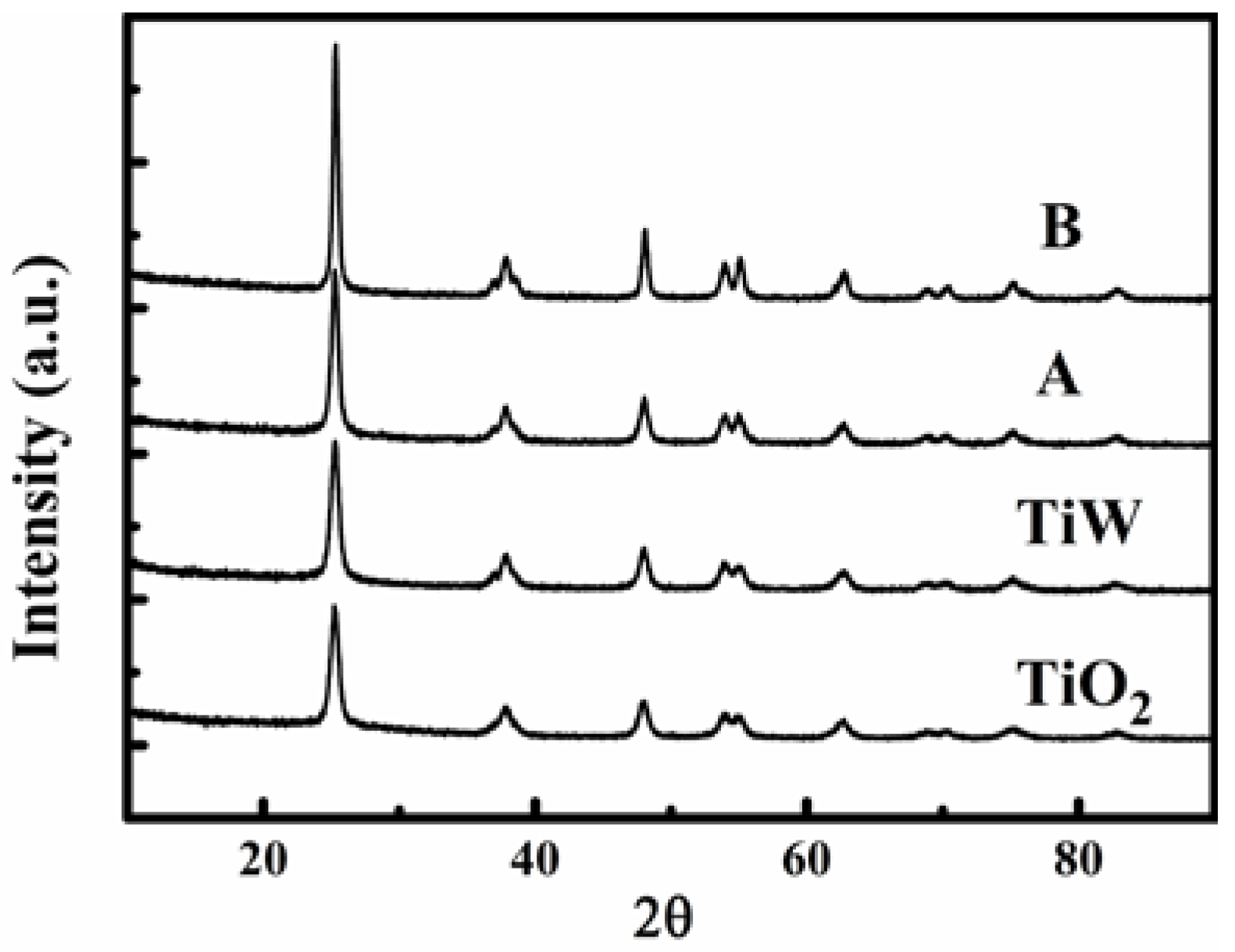
Figure 2 shows the XRD patterns of the catalysts A and B as well as of the TiO
2 support and WO
3/TiO
2 material (denoted as TiW). All of them were made in this study. Among the TiO
2 support, TiW, catalysts A, B, C, and D (the XRD profiles of catalysts C and D were not given here), there are no differences in the shape and position of the diffraction peaks except for the strength. The obtained diffraction patterns for all the samples comply with those of the TiO
2 anatase phase. No visible tungsten and vanadium oxides can be observed. Because the concentrations of tungsten and vanadium oxides were too low or well dispersed, for example, as monolayer species which are undetectable by XRD on the surface of TiO
2 [
35]. The full width at half-maximum (FWHM) of the characteristic peak (with the highest intensity) for the catalyst B (0.22) is smaller than that of the catalyst A (0.47), meaning that the grain size of anatase TiO
2 crystal particles on catalyst B were larger than on catalyst A.
Figure 2.
X-ray diffraction (XRD) profiles of catalysts A and B and major materials adopted in this study.
Figure 2.
X-ray diffraction (XRD) profiles of catalysts A and B and major materials adopted in this study.
Table 1 summarizes the BET surface areas, pore volumes, and pore sizes of the catalysts A and B, TiW and TiO
2 support. A significant loss of surface area occurred upon the addition of V
2O
5 and WO
3 into the catalyst samples, possible due to dispersion of such added species into the pores of TiO
2 [
36]. In
Table 1, the specific surface area was 108 m
2·g
−1 for TiO
2 support. After doping with tungsten and vanadium, it decreased to 85 m
2·g
−1 and 75 m
2·g
−1 for the catalysts A and B, respectively. Both tungsten and vanadium oxides could be dispersed onto the support to occupy some channels of TiO
2 crystallite. Meanwhile, the second calcination after impregnation of the W and V species during the catalyst preparation might also increase the grain size of TiO
2 (see
Table 1). In addition, the solvothermal method also led to a bigger crystallite size of TiO
2. All of these caused lowered surface areas and smaller pore sizes of the catalysts than of the original support.
Table 1.
Texture properties of the catalysts A and B and their materials.
Table 1.
Texture properties of the catalysts A and B and their materials.
| Samples | BET Surface Area (m2·g−1) | Pore Volume (cm3·g−1) | Average Pore Diameter (nm) | TiO2 Crystallite Size a (nm) |
|---|
| TiO2 | 108 | 0.41 | 14.3 | 14.6 |
| TiW | 95 | 0.39 | 16.1 | 16.1 |
| A | 85 | 0.27 | 13.0 | 17.8 |
| B | 75 | 0.31 | 13.4 | 38.3 |
Table 1 clarifies that the pore volume and average pore diameter of the catalyst B were both larger than those of the catalyst A, indicating that there are more meso, or even macro pores in the catalyst B. Especially, the catalyst B even had a pore diameter bigger than that of the original TiO
2 support. Combining with the lowest surface area and relatively higher pore volume of this catalyst, we can suggest that in the preparation of the catalyst B both V and W oxides would mainly exist on micro channels causing more meso and macro pores to remain, indicating deep dispersion of such active metal species onto the TiO
2 support. Compared to this, the dispersion of V and W species for the catalyst A would be more onto the meso or macro pores of the TiO
2 support, having few meso and macro pores in this catalyst. Essentially, the results demonstrate the great influence of the catalyst preparation method on the catalyst pore and surface characteristics due to, for example, the difference in solvent and precursors. The method for making the catalyst B, which was newly devised in this study and uses toluene as solvent and VO(acac)
2 precursor, delivered a better dispersion of the V and W species on TiO
2 than the conventional aqueous impregnation method using ammonium metavanadate precursor.
Figure 3.
Transmission electron microscopy (TEM) images at different magnifications of catalysts (a) A; and (b) B.
Figure 3.
Transmission electron microscopy (TEM) images at different magnifications of catalysts (a) A; and (b) B.
Figure 3 shows the TEM images for the catalysts A and B at different magnifications. The surface features in
Figure 3a1,b1 reveal that the catalysts A and B had obviously spherical primary particles with average particle sizes of 18 nm and 38 nm, respectively. The high-resolution images (HRTEM) in
Figure 3a2,b2 further shows that the catalysts were single crystals with an average space of 0.2 nm between the aligned crystal lattice. This shows also the anatase phase of TiO
2, as identified by XRD analysis in
Figure 2. It suggests that for the catalysts A and B the oxides of tungsten and vanadium, which are the active metallic oxides of the catalysts, were highly dispersed into the lattice structure of TiO
2 to closely interact with the TiO
2 support.
2.2. Performance for SCR of NO
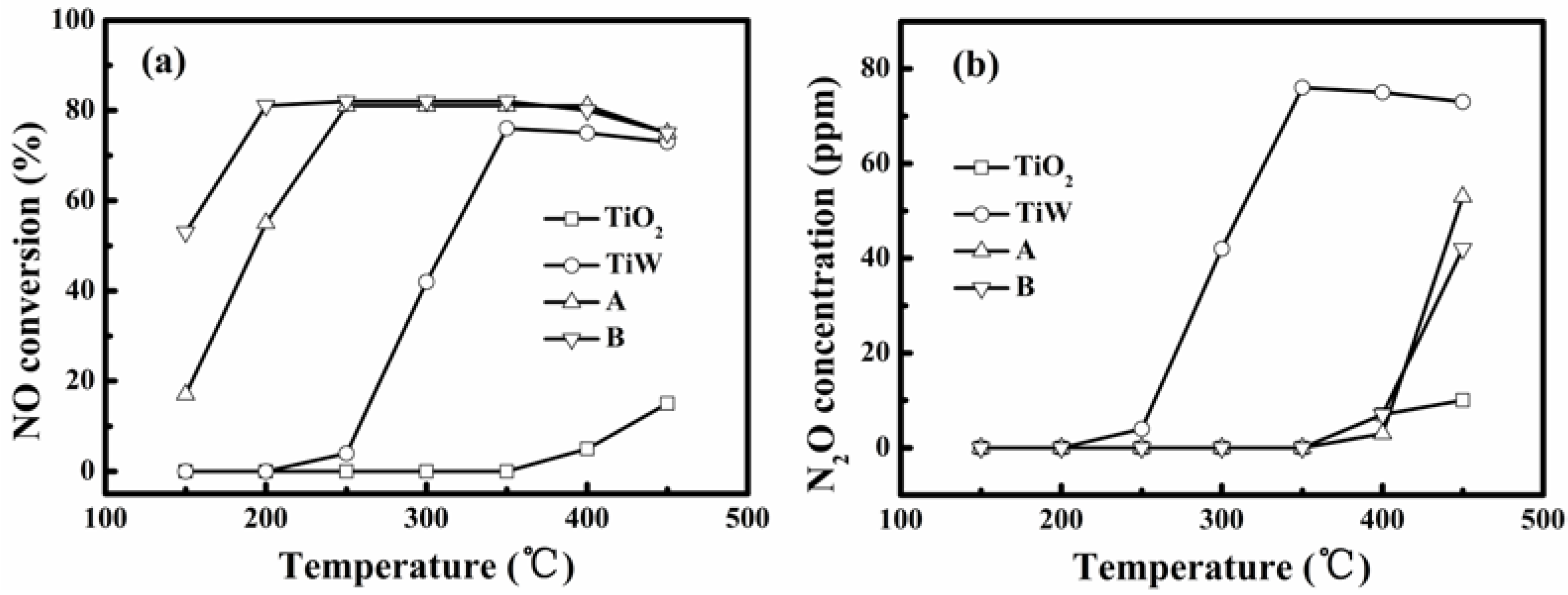
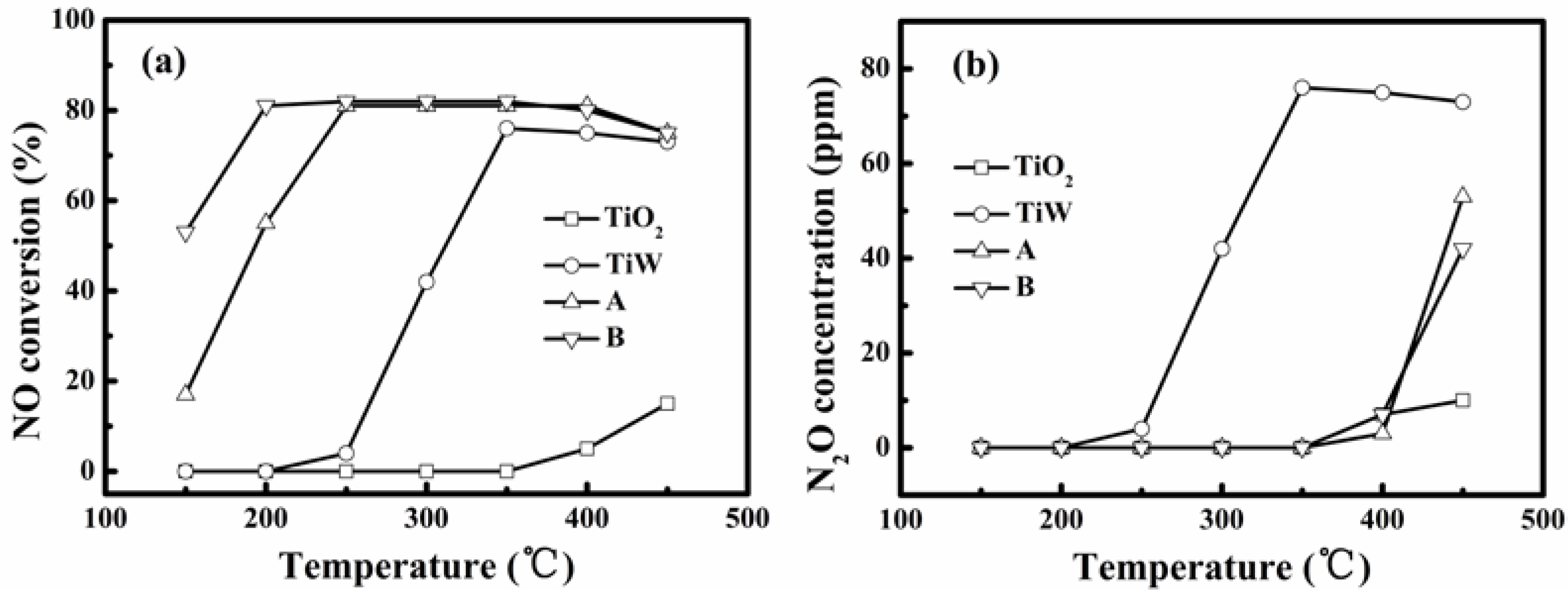
Figure 4a shows the realized NO conversions for simulated flue gas without SO
2 and water vapor at different reaction temperatures over the self-made catalysts A and B, TiO
2 and WO
3/TiO
2 (denoted as TiW). On raising the temperature the NO conversion first increased and then remained constant until finally turning to decrease. This variation tendency is common for SCR catalysts for denitration, but we can see that the temperature window enabling the steady NO conversion was 250–400 °C and 200–400 °C for the catalysts A and B, respectively. This means that the catalyst B had the better low-temperature activity. There was almost no NO reduction over pure TiO
2. The NO conversion over TiW was about 75% when the temperature was higher than 350 °C. However, the N
2O concentration in the effluent gas shown in
Figure 4b reached 73 ppm at such a temperature for TiW indicating its high oxidation to NH
3 or bad N
2 selectivity for SCR of NO. Consequently, the dispersion of V onto WO
3/TiO
2 is critical to achieve the activity of the catalyst for SCR of NO.
Figure 4.
Evaluation for selective catalytic reduction (SCR) of NO at different temperatures over different catalysts: (a) NO conversion and (b) N2O concentration in reacted gas (without SO2 and water vapor in simulated flue gas and gas conditions in Experimental section).
Figure 4.
Evaluation for selective catalytic reduction (SCR) of NO at different temperatures over different catalysts: (a) NO conversion and (b) N2O concentration in reacted gas (without SO2 and water vapor in simulated flue gas and gas conditions in Experimental section).
The steady NO conversion for the catalysts A and B were similar and reached a value almost equal to the NH
3/NO molar ratio of 0.8. At 150 °C and 200 °C, the NO conversions over catalyst B were 55% and 81%, respectively. These were obviously higher than those of 18% and 56%, realized by catalyst A.
Figure 4b shows further that below 400 °C there was no obvious formation of N
2O for both the catalysts. All of this shows that the prepared catalysts A and B both allowed good selectivity in SCR of NO with NH
3 to form N
2 at temperatures below 400 °C. The suitable activity temperature windows are respectively 250–400 °C and 200–400 °C for the catalysts A and B. This proves that the method of liquid-phase impregnation adopted in this study can lead to good low-temperature activity. It is related to the high-degree of dispersion of V and W oxides on the TiO
2 support realized by the liquid-phase impregnation. Furthermore, the result proves that the vanadium precursors also affect low-temperature activity. The use of VO(acac)
2 via impregnation in toluene enabled the lowest activity temperature of 200 °C for the flue gas without presence of SO
2 and water vapor.
Below 300 °C, the poisoning of SO
2 and water vapor in the SCR of NO is usually serious because of their adsorption on the catalytic sites in competition with NH
3 and NO and the easy formation of NH
4HSO
4 that hardly decomposes at such temperatures [
37].
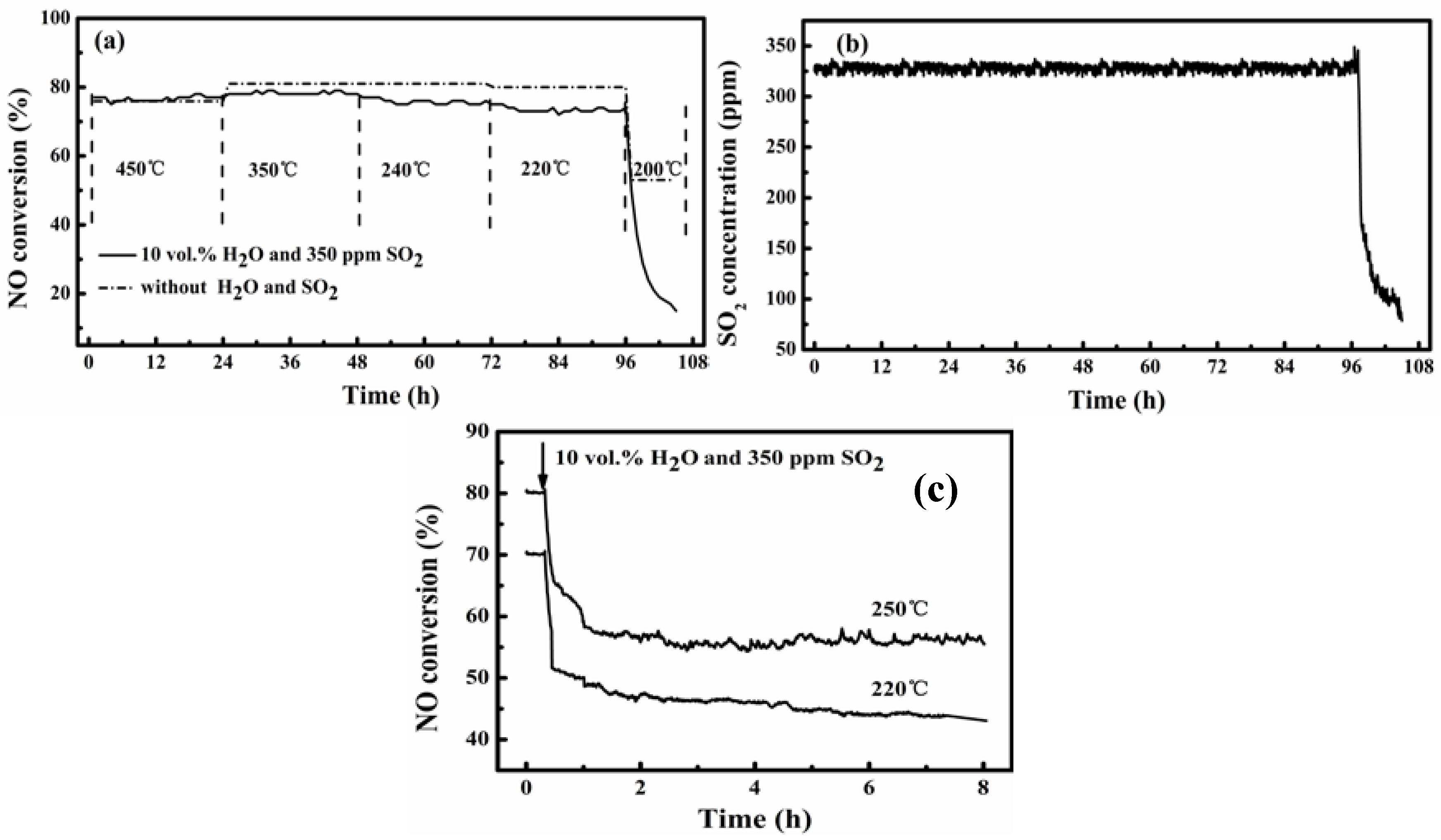
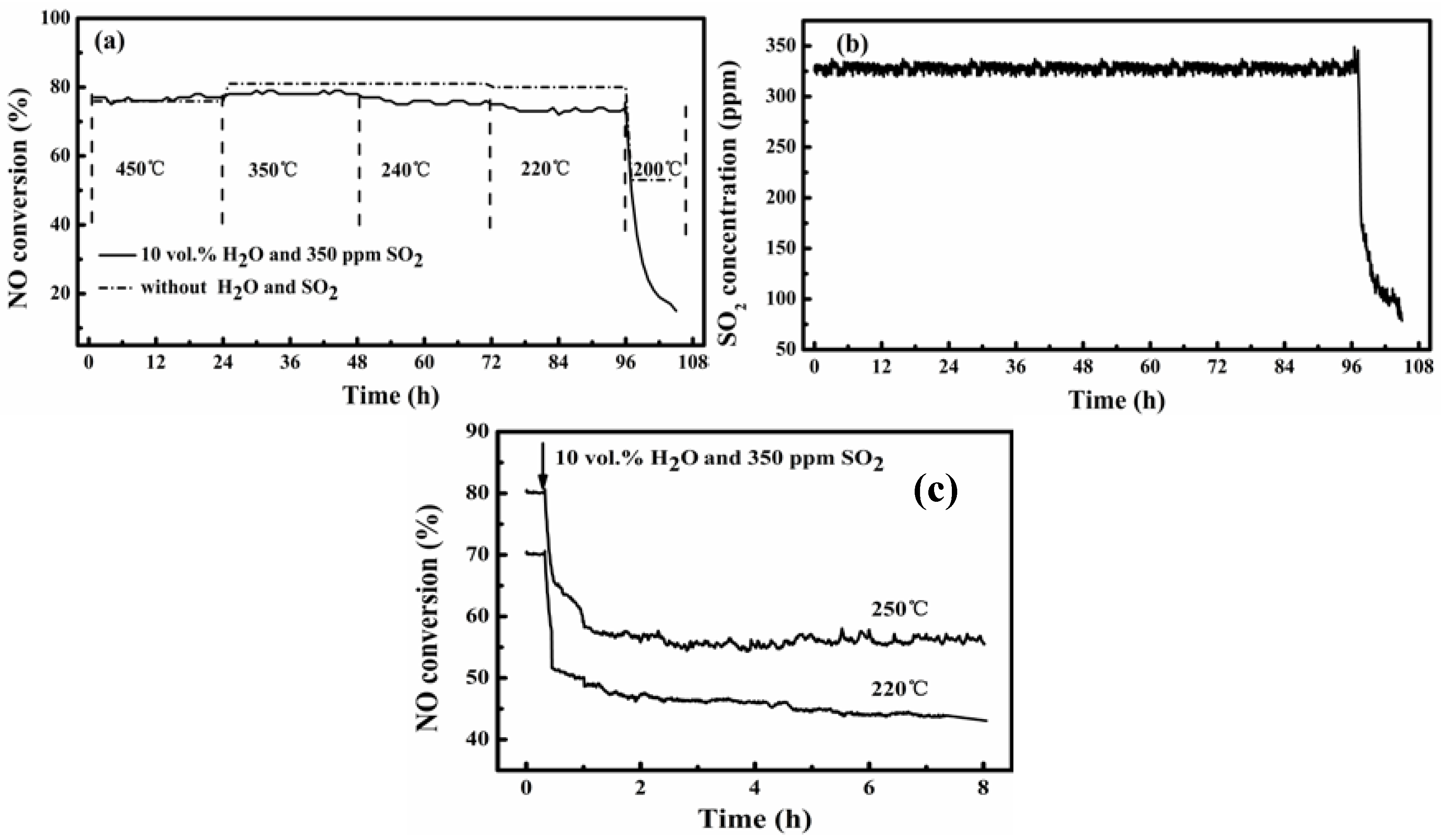
Figure 5 shows the results from testing the activity stability of the catalysts A and B at 200–450 °C for SCR of NO from flue gas containing 350 ppm SO
2 and 10 vol. % water vapor. For comparison, a test over catalyst B was also conducted for the simulated flue gas without SO
2 and water vapor, as is shown by the data of the dotted line in
Figure 5a. For all the tests, the GHSV was kept at 40,000 h
−1 and NH
3/NO was at 0.8 to indicate that the results shown below are for such specified conditions.
Figure 5.
Stability of catalysts A and B for SCR of NO at 200–450 °C in flue gas with SO2 and water vapor: (a) NO conversion over catalyst B in 108-h test at varied temperatures; (b) SO2 concentration at reactor exit corresponding to (a); (c) NO conversion over catalyst A at 220 °C and 250 °C (see gas conditions in the Experimental section).
Figure 5.
Stability of catalysts A and B for SCR of NO at 200–450 °C in flue gas with SO2 and water vapor: (a) NO conversion over catalyst B in 108-h test at varied temperatures; (b) SO2 concentration at reactor exit corresponding to (a); (c) NO conversion over catalyst A at 220 °C and 250 °C (see gas conditions in the Experimental section).
Over the catalyst B,
Figure 5a shows that its activity was stable at temperatures of 220–450 °C for both the tested gas conditions (with and without SO
2 and H
2O). Except for the test at 450 °C where the NO conversion was about 76% for both gases, the presence of SO
2 and H
2O in the flue gas slightly decreased the realized NO conversion. The decrease was below 10%, but it was more obvious at the lower reaction temperature. Decreasing the temperature from 450 °C to 220 °C only slightly lowered the NO conversion, from about 80% to about 72% even for the case with SO
2 and water vapor in the flue gas. Corresponding to this, the simultaneously monitored SO
2 concentration of the effluent gas from the reactor was constantly about 330 ppm at 220–450 °C but quickly decreased with time for the test at 200 °C (shown in
Figure 5b). The latter just corresponded to a rapid decrease in the NO conversion with the progress of the reaction.
Figure 5a shows further that without SO
2 and H
2O in the flue gas there was a final steady NO conversion of about 52%, whereas the presence of SO
2 and H
2O caused the NO conversion to sharply decrease to below 20% in 6 h to indicate complete deactivation of the catalyst. The corresponding decrease of effluent SO
2 concentration was from 330 ppm to below 100 ppm in 6 h.
The preceding results justify the potential application of the catalyst B for denitration from flue gases having temperatures above 220 °C. Meanwhile,
Figure 5c shows that the catalyst A prepared in this work is also difficult to function at 220 °C and 250 °C for flue gases containing SO
2 and H
2O. While it cannot give a steady NO removal at 220 °C, the possibly steady NO removal shown for 250 °C is about 25 percentage points lower (80% to 55%) than that for the case without SO
2 and water vapor in the flue gas. This shows that the V impregnation through VO(acac)
2 in toluene really granted the catalyst a better low-temperature activity and stability.
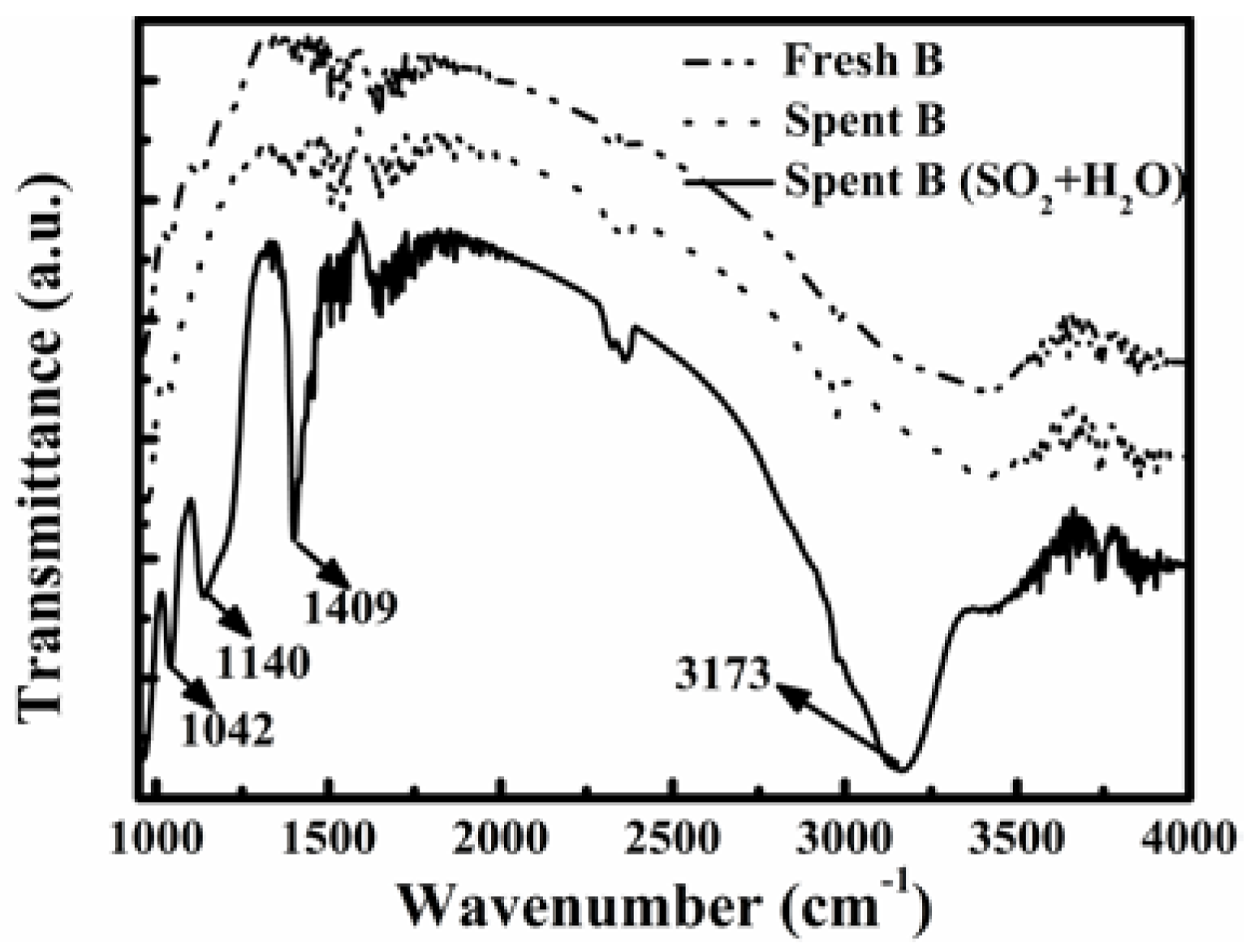
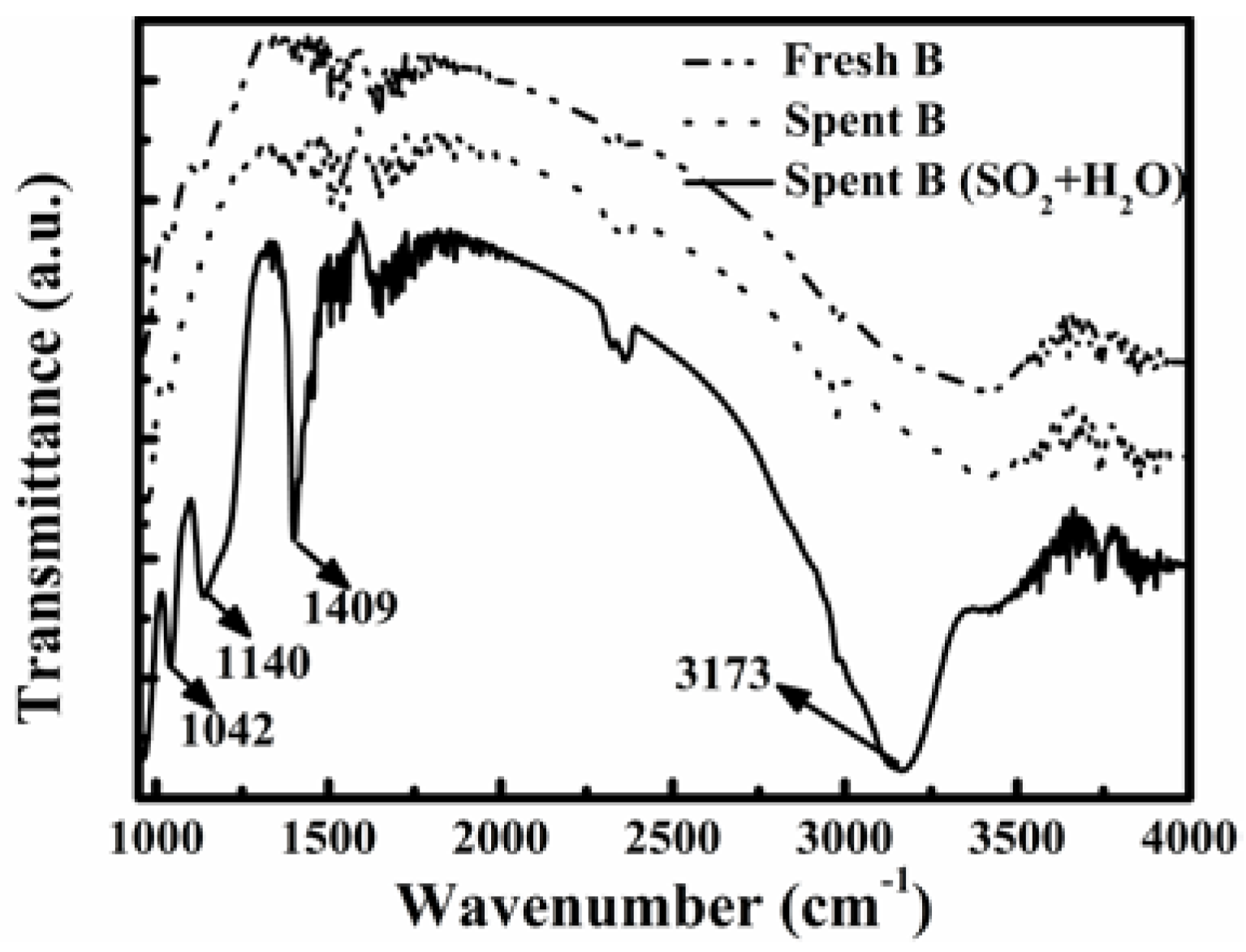
Figure 6 shows the FT-IR spectra of fresh and spent catalyst B after the stability tests shown in
Figure 5a. There was no obvious difference in the spectra for the fresh and spent catalyst B after 110-h reaction in flue gas without SO
2 and water vapor (dotted line in
Figure 5a). The presence of SO
2 and H
2O in the flue gas caused the FT-IR spectrum of the spent catalyst to have some new bands at 1042 cm
−1, 1140 cm
−1, 1409 cm
−1, and 3173 cm
−1, respectively. The bands at 1042 cm
−1 and 1140 cm
−1 are associated with SO
42− species, and those at 1409 cm
−1 and 3173 cm
−1 are due to NH
4+ species. Furthermore, the band at 1409 cm
−1 represents the asymmetric bending vibrations of NH
4+ [
38], while that at 3173 cm
−1 indicates the NH stretching [
39].
Figure 6.
FT-IR spectra of fresh and spent catalyst B after tests in different simulated flue gases.
Figure 6.
FT-IR spectra of fresh and spent catalyst B after tests in different simulated flue gases.
Table 2 shows the weight losses at 200–600 °C from TG analysis of spent catalyst after testing SCR of NO at 220 °C for 24 and 48 h in flue gas containing SO
2 and water vapor (conditions being the same as stated in the Experimental section). The weight loss of the spent catalyst A tested for 24 h was 2.7% and it increased on prolonging the testing time to reach 5.8% after a 48-h test. In comparison, the catalyst B had much lower weight losses of 1.3% for 24-h test and 1.5% for 48-h test, respectively. All of these justify the formation of (NH
4)
2SO
4 or NH
4HSO
4 in the NH
3-SCR reactions for flue gas containing SO
2 and water vapor (H
2O), which in turn deposit on the active sites to deactivate the catalyst for SCR of NO [
10]. Nonetheless, at high temperatures such as over 300 °C, in the SCR of NO over commercial SCR catalysts the formed ammonium sulfites or sulfates can be decomposed to remove their inhibition on catalytic activity. To have steady catalytic activity, there must be a dynamic balance between the formation and decomposition of ammonium sulfite or sulfate. Thus, for catalyst B, such a dynamic balance can be built at temperatures as low as 220 °C, meaning that this catalyst would facilitate the decomposition of ammonium sulfite or sulfate at the low temperatures of 220–300 °C so that it can work stably at such low temperatures, as is shown in
Figure 5a. Nonetheless, more fundamental studies, as worthwhile future work, are needed to justify the facilitated decomposition of ammonium sulfite or sulfate over catalyst B.
Table 2.
Weight loss of thermogravimetric (TG) analysis at 200–600 °C for spent catalysts after SCR of NO in gas containing SO2 and water vapor.
Table 2.
Weight loss of thermogravimetric (TG) analysis at 200–600 °C for spent catalysts after SCR of NO in gas containing SO2 and water vapor.
| Catalysts | Reacted for 24 h (wt. %) | Reacted for 48 h (wt. %) |
|---|
| A | 2.7 | 5.8 |
| B | 1.3 | 1.5 |
2.3. Further Justification
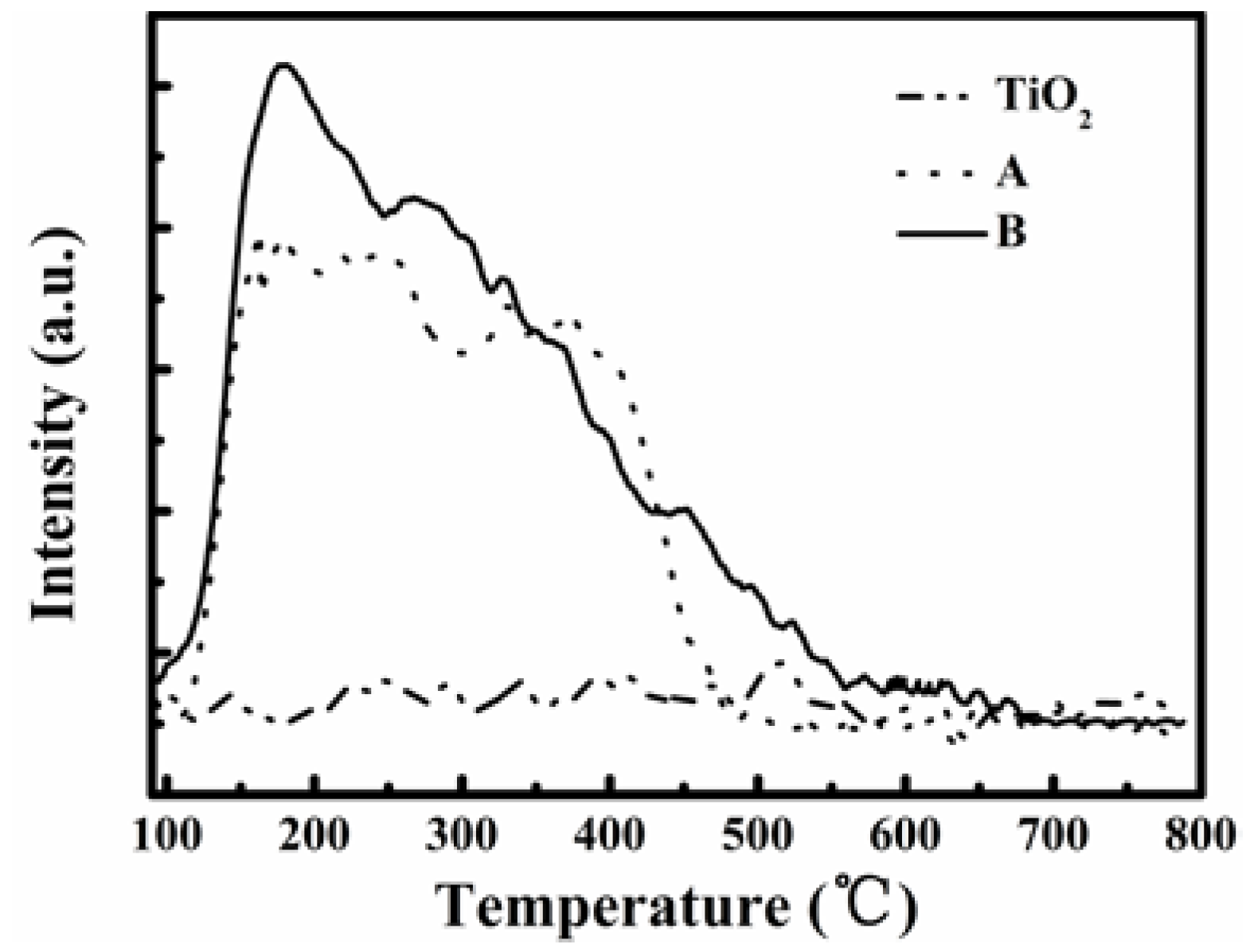
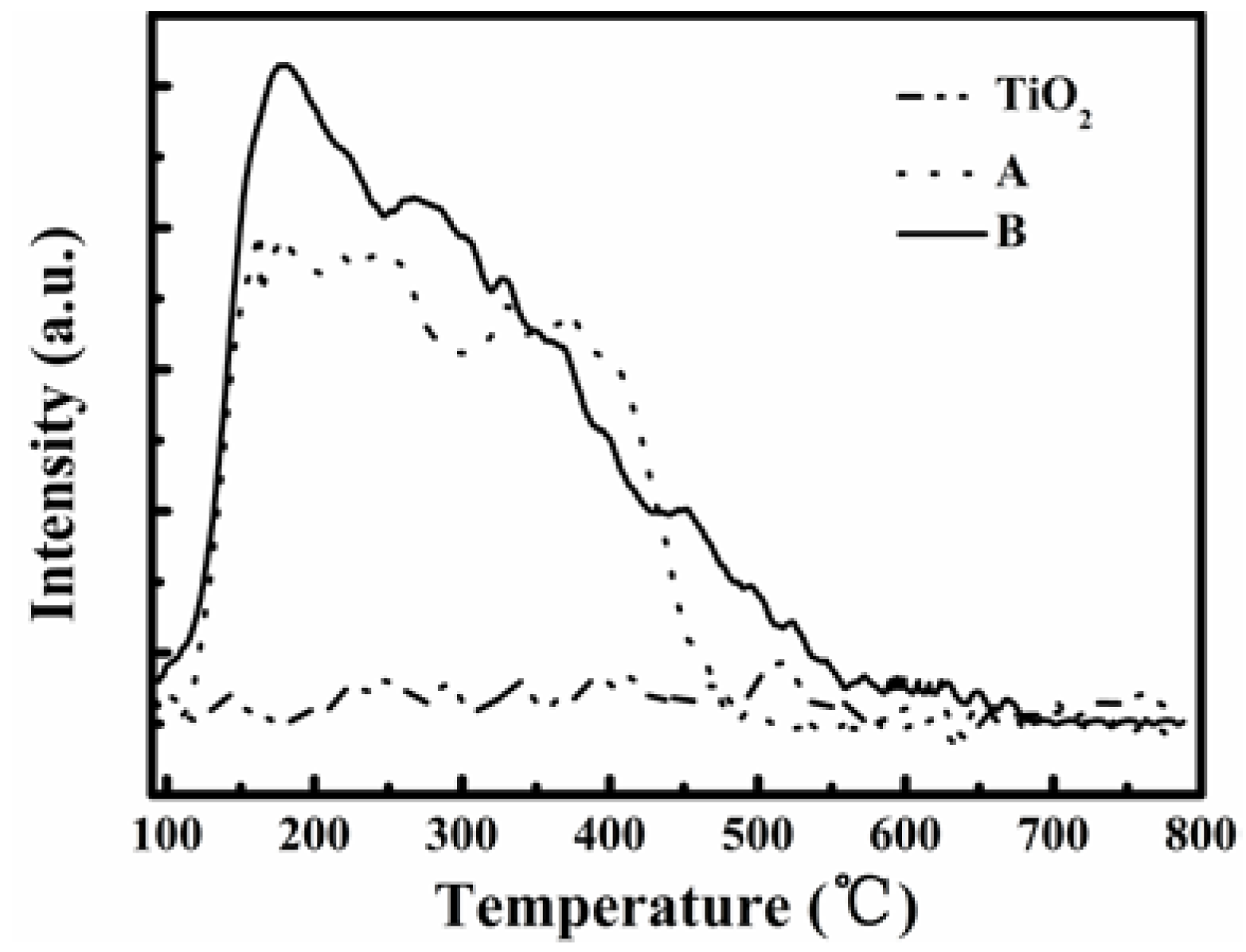
Figure 7 compares the NH
3-TPD profiles of the catalysts A and B. The NH
3 desorption occurred at temperatures above 100 °C for both catalysts but it was not obvious for TiO
2 until 800 °C, the highest tested temperature. Thus, there were very few acidic sites on TiO
2, although it had a high surface area of about 108 m
2·g
−1. There are two NH
3 desorption peaks demarcated at about 300 °C for catalyst A, while catalyst B showed one major NH
3 desorption peak at 175 °C and another shoulder peak starting from 250 °C. These peaks obviously show the existence of acidic sites on the catalysts, and catalyst B had more acidic sites at a low temperature of about 175 °C leading to higher NH
3 desorption. On further noting that over TiO
2 there was no obvious NO conversion until 400 °C and catalyst B enabled a stoichiometric NO removal of 81% at 200 °C, we can conclude that the activity of a catalyst for SCR of flue gas NO is closely related to the acidic sites on the catalyst surface, and more NH
3 adsorption could improve the SCR reaction rate.
Figure 7.
NH3-TPD profiles for catalysts A and B and TiO2 support.
Figure 7.
NH3-TPD profiles for catalysts A and B and TiO2 support.
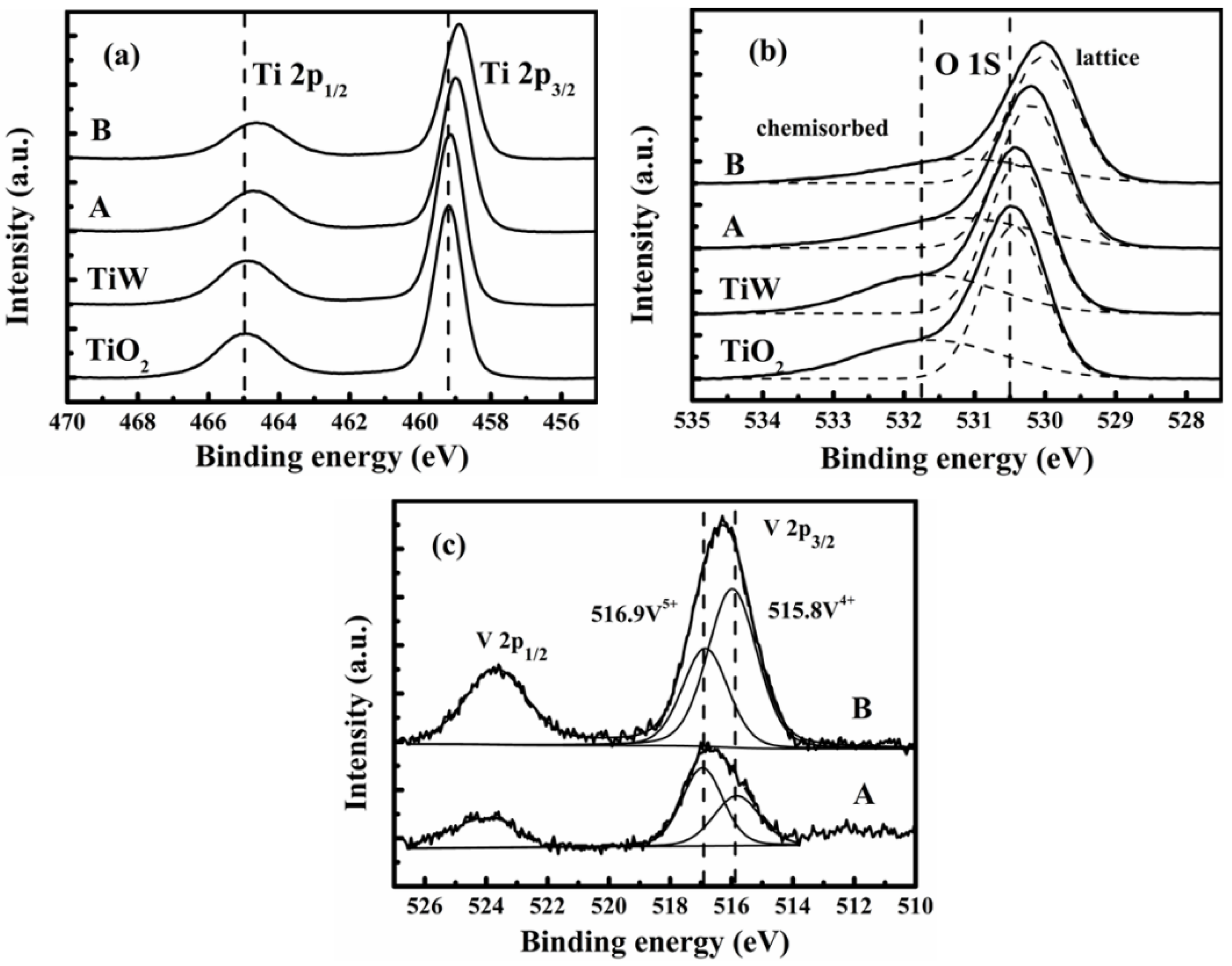
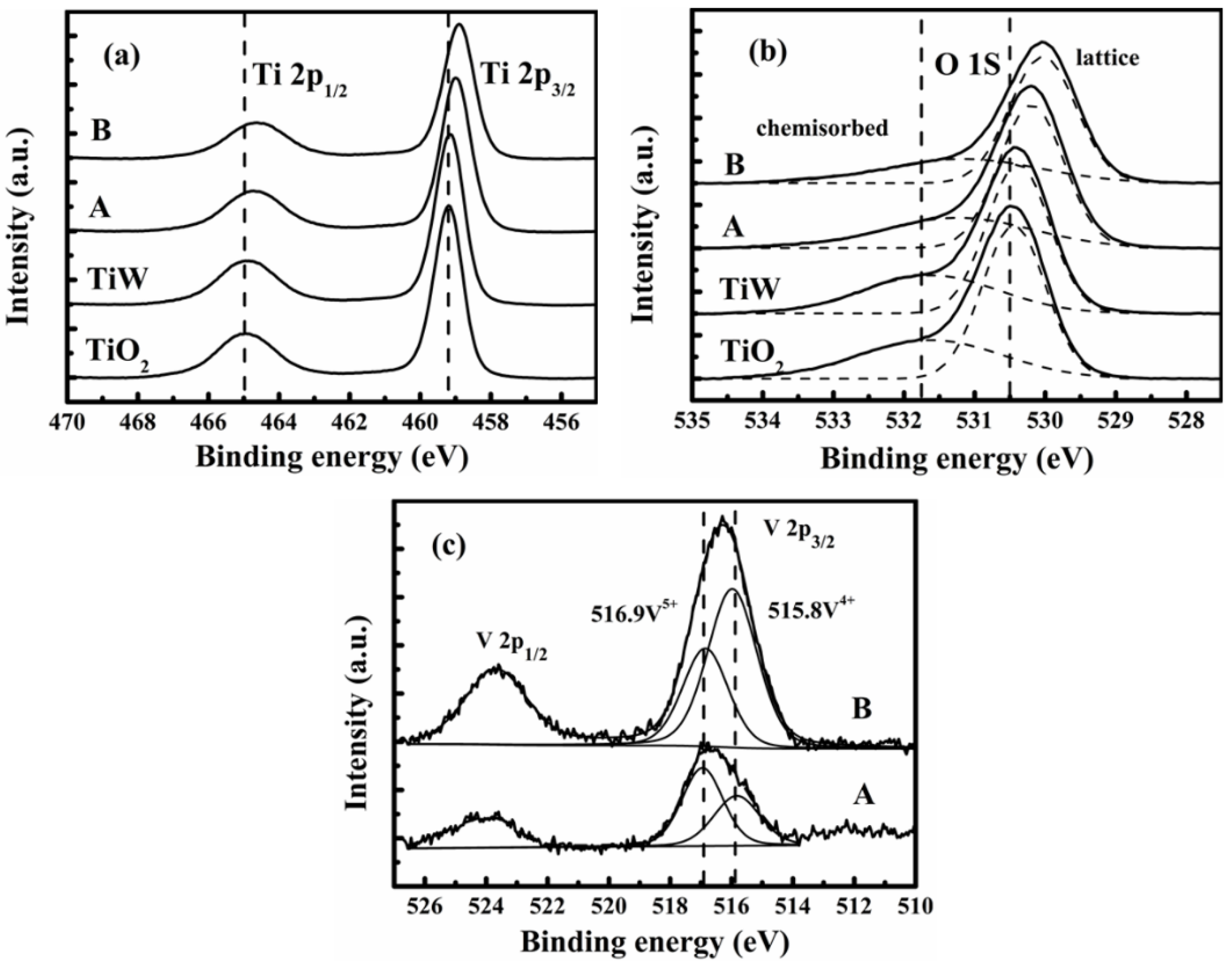
Figure 8 presents the XPS spectra of V, O, and Ti given by XPS analyses for the catalysts A and B and also the self-made TiO
2 and WO
3/TiO
2 (TiW). A few ratios of surface atomic concentrations are summarized in
Table 2 for elements V, O, and Ti. The Ti 2p XPS spectra in
Figure 8a consists of double peaks (Ti 2p1/2 and 2p3/2). For TiO
2, the binding energies of these peaks were 464.5 and 458.6 eV, respectively. This indicates that Ti existed in the Ti
4+ state on the TiO
2 support. With the presence of V and W, the binding energies for such peaks moved to the right side with slightly lower values. Thus, the impregnated V and W would have strong interactions with the TiO
2 support. Especially, such a kind of interaction on catalyst B is stronger than on catalyst A because the binding energies of the two peaks are lowest for catalyst B.
The O 1s peaks in
Figure 8b could be fitted into two peaks referring to the lattice oxygen (denoted as O
α) at 529.8–530.4 eV and the chemisorbed oxygen (denoted as O
β) at 531.1–531.9 eV, as shown by the two dotted fitting curves. The peaks of O 1s are shifted to the side with lower binding energy values, and the concentrations of chemisorbed oxygen O
β gradually increased according to the order of TiO2, TiW, catalyst A, and catalyst B. The ratio of O
β/(O
α + O
β) calculated from the areas of the two fractionate peaks was 0.31 for the catalyst B, bigger than those for the others. Jing
et al. [
40] reported that chemisorbed oxygen O
β is the most active oxygen and plays an important role in NH3-SCR of NO. Because the chemisorbed oxygen is more active in oxidation, the high O
β ratio in catalyst B would accelerate the oxidation of NO during SCR reactions.
Figure 8c displays the XPS spectra for V 2p of the catalysts A and B. There are two cross peaks to represent the V 2P
1/2 and V 2P
3/2, respectively. Each of them can be further separated into two fractionating peaks to analyze the valence interchange between V
4+ and V
5+. A similar result should be obtained by treating the peak of either V 2P
1/2 or V 2P
3/2, and here we take the latter. The fractionate peaks appearing at 515.8 and 516.9 eV can be ascribed to V
4+ 2p
3/2 and V
5+ 2p
3/2, respectively. Thus, the V species were in V
4+ and V
5+ states on the catalysts A and B. The surface atomic ratio of V
4+/(V
4+ + V
5+) was 0.64 for catalyst B (the area ratio of the two fractionated peaks), higher than that for catalyst A (0.41). Zhang
et al. [
41] reported that the reduction species V
4+ and V
3+ were the active sites for the formation of superoxide ions that enhance activity at low temperatures. The transformation between V
4+ and V
5+ contributes to the catalytic activity for NH
3-SCR reactions [
42]. Nonetheless, the catalysts A and B had very different ratios of peak areas between V and Ti, as shown in
Table 3 via the V/Ti ratio; 0.03 and 0.09 for catalysts A and B, respectively. Both a high ratio of V
4+/(V
4+ + V
5+) and a high atomic concentration of vanadium (
i.e., a high V/Ti ratio) on the catalyst surface are critical for the catalytic activity of a catalyst [
41,
42,
43]. Furthermore, the higher surface atomic concentration of vanadium indicates more active sites on the catalyst surface, which in turn increases the activity and stability of the catalyst.
Figure 8.
X-ray photoelectron spectrometry (XPS) spectra of (a) Ti 2p; (b) O 1s; and (c) V 2p for two catalysts and self-made TiO2 support and WO3/TiO2.
Figure 8.
X-ray photoelectron spectrometry (XPS) spectra of (a) Ti 2p; (b) O 1s; and (c) V 2p for two catalysts and self-made TiO2 support and WO3/TiO2.
Table 3.
Surface atomic concentration from XPS spectra in
Figure 7.
Table 3.
Surface atomic concentration from XPS spectra in Figure 7.
| Samples | Surface Atomic Concentration |
|---|
| V/Ti | Oα/(Oα + Oβ) | V4+/(V4+ + V5+) |
|---|
| TiO2 | - | 0.37 | - |
| A | 0.03 | 0.33 | 0.41 |
| B | 0.09 | 0.31 | 0.64 |
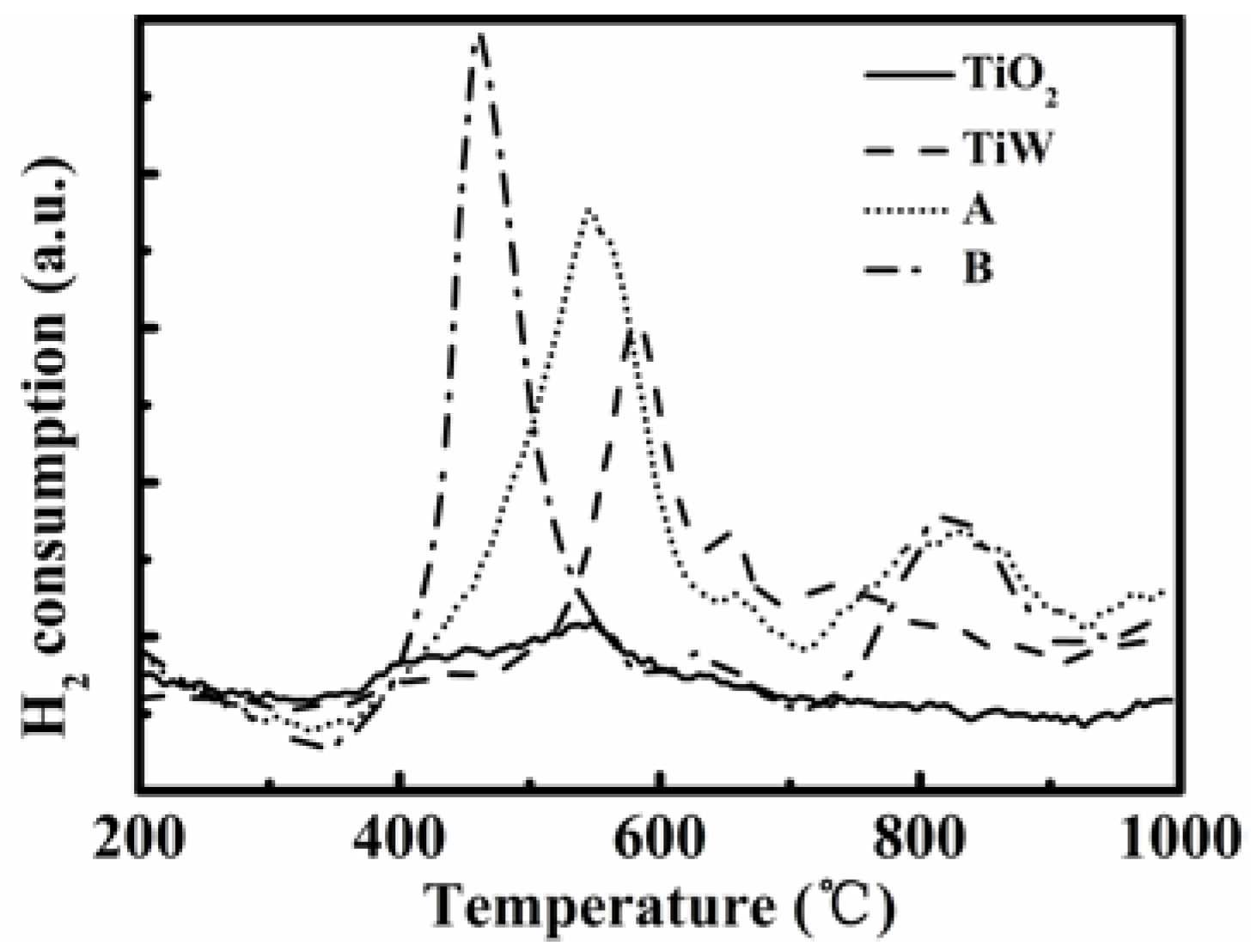
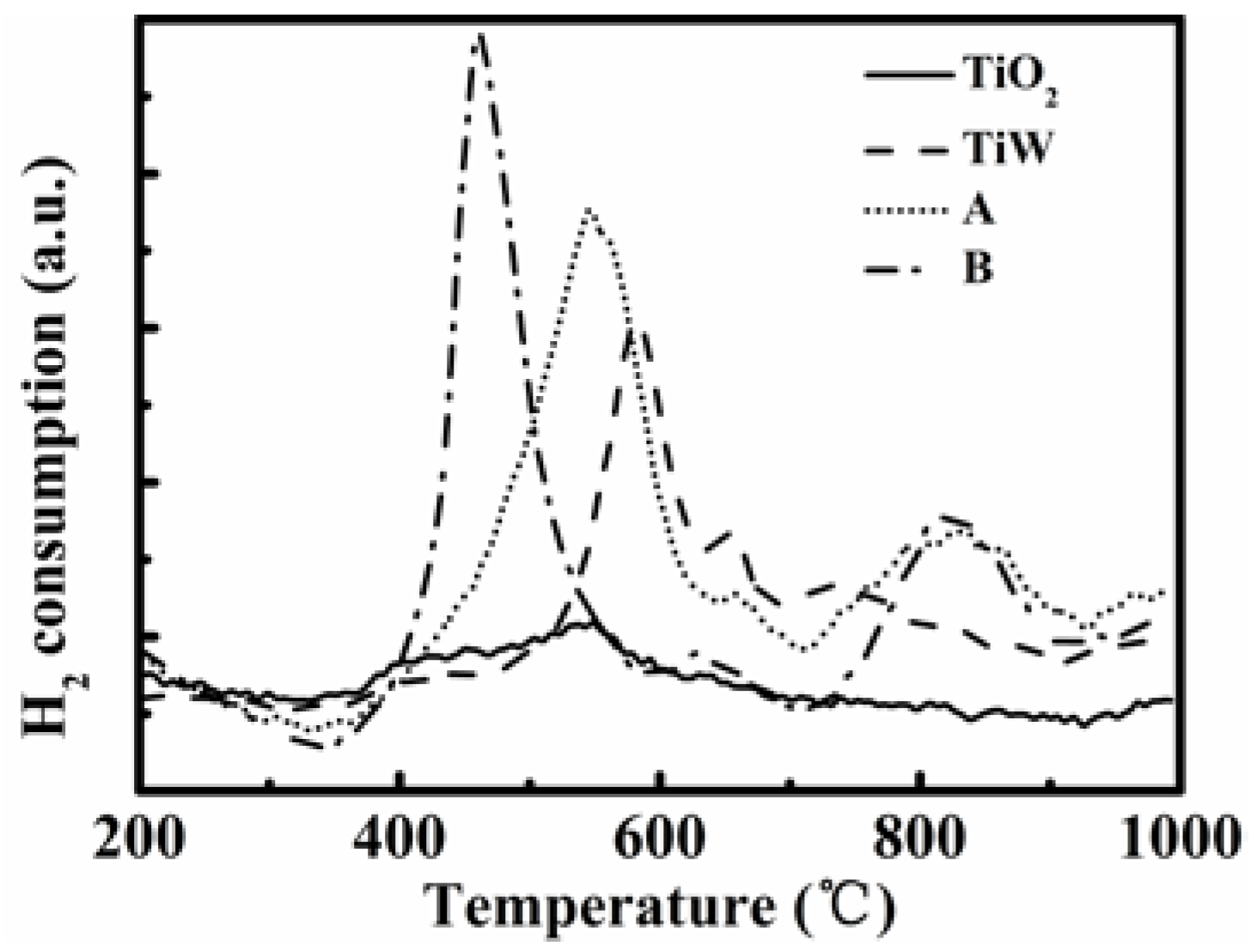
Figure 9 shows the H
2-TPR profiles for all the tested samples. For TiO
2 there was almost no reduction peak. Dispersing WO
3 onto a TiO
2 support caused a strong peak at 583 °C and two weak peaks centered around 660 °C and 743 °C for WO
3/TiO
2 (TiW). Reiche
et al. [
44] reported two peaks around 467 °C and 782 °C, which were attributed to reduction of W
6+ species. Here we also consider that all three peaks of TiW are attributed to the reduction of W
6+ species. When W and V were further introduced into the TiO
2 support, three peaks at 544 °C, 660 °C, and 824 °C were found for catalyst A. According to literature studies [
4,
44,
45], the peak at 544 °C is indicative of the well-dispersed V species, while the others are suggested to be reduction of W
6+ species. For catalyst B, three peaks appeared at about 460 °C, 632 °C, and 825 °C, respectively. The peak at 460 °C, which moved to a much lower temperature compared to 544 °C for the catalyst A, was attributed to the reduction of V species. Comparing with TiW, one can see that the H
2 consumption profile due to the V species is shown by a broad peak with a similar starting temperature of about 350 °C but different finishing temperatures for catalysts A and B. While the peak was complete at about 620 °C for catalyst A, the temperature was about 580 °C for catalyst B. The latter indicates that the vanadium species on catalyst B are easier to reduce, implying a better dispersion of the V species and thus a better low-temperature activity.
Figure 9.
H2-TPR (temperature-programmed reduction) curves of the tested two catalysts A and B, TiO2 support and WO3/TiO2.
Figure 9.
H2-TPR (temperature-programmed reduction) curves of the tested two catalysts A and B, TiO2 support and WO3/TiO2.
The lower reduction temperature of the V species for catalyst B shown in
Figure 9 suggests that its V species is easier to be reduced by H
2. This indicates that the V species have stronger interaction with the TiO
2 support and better dispersion over the surface of the catalyst when using VO(acac)
2 as the vanadium precursor. Corresponding to the lower reduction temperature of the V species, catalyst B enabled a higher SCR activity as shown above. Literature studies have widely recognized the higher reducibility of the SCR catalyst, that the higher catalyst activity is for NH
3-SCR at the reduction temperature [
46].
As a consequence, the illustrations from
Figure 7 to
Figure 9, not only justify the better low-temperature activity and stability of catalyst B prepared by following a newly proposed method using VO(acac)
2 precursor in toluene solvent, but verify also that the redox of the active component or the interaction between the active component and TiO
2 support plays an important role on the activity of a catalyst for NH
3-SCR of NO.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}