
Composition, Structural Evolution and the Related Property Variations in Preparation of Mixed Cesium/Ammonium Acidic Salts of Heteropolyacids

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Elemental Differences between the Initial Precipitates and the Evaporation Residues
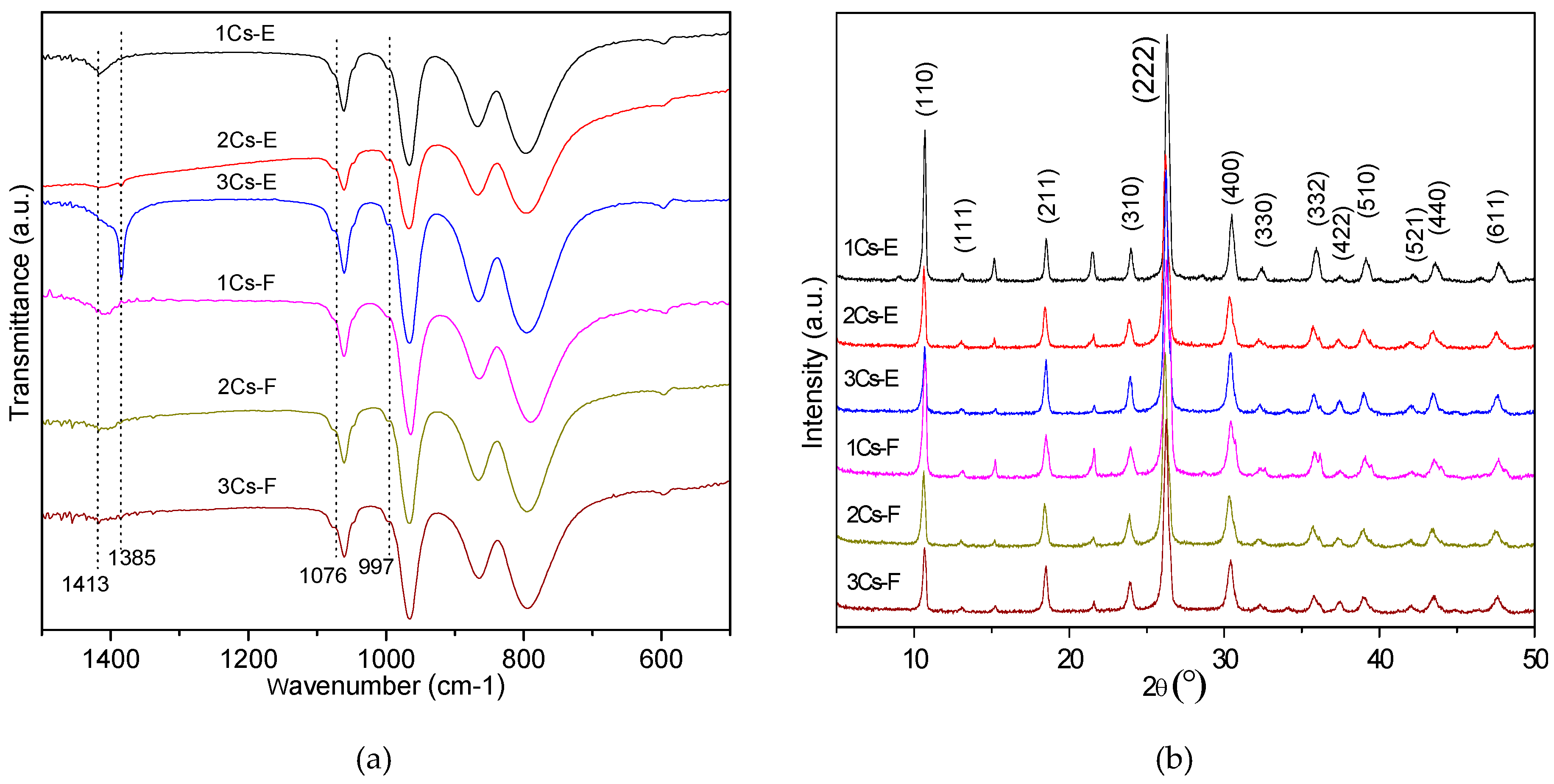
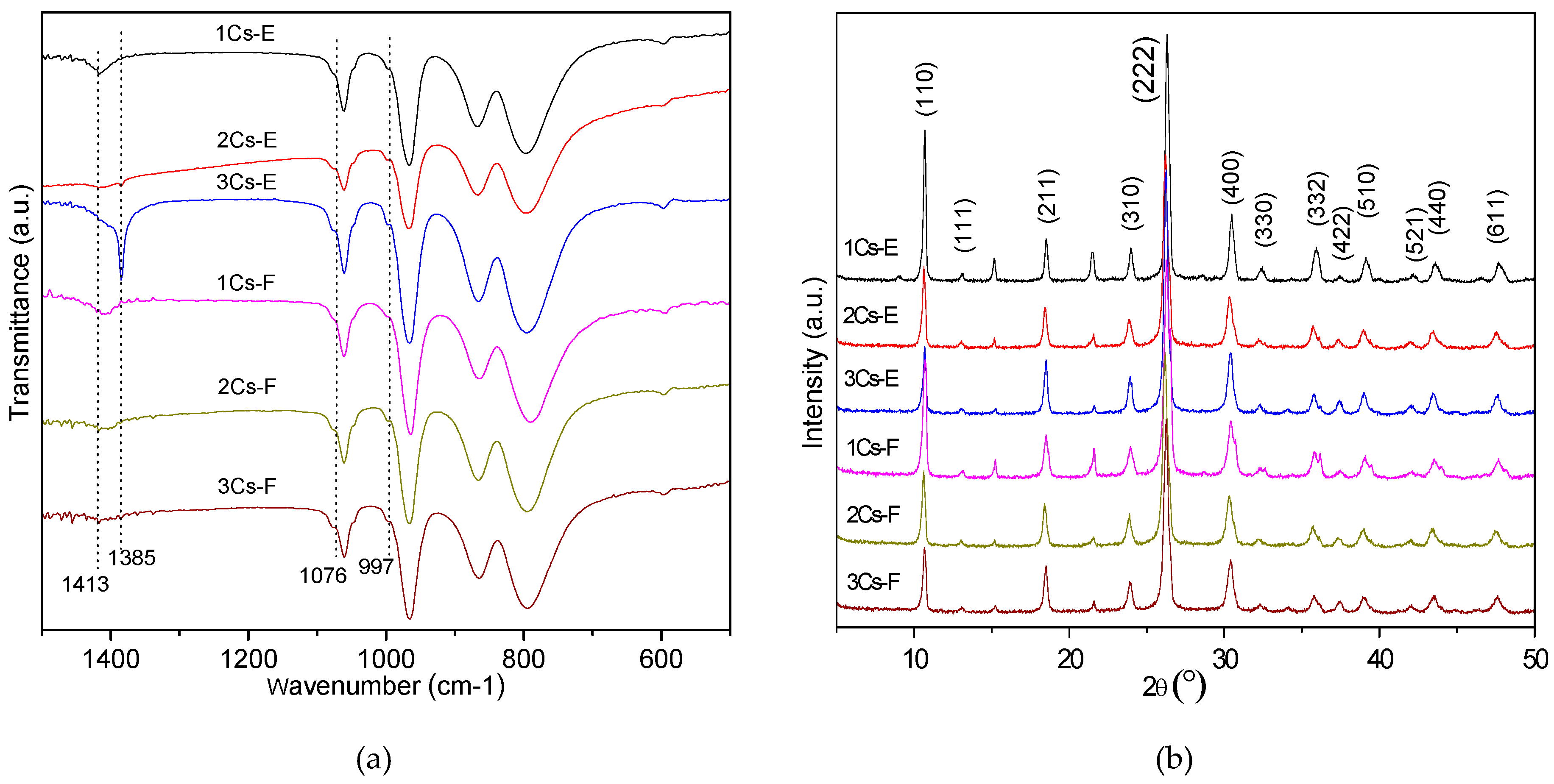
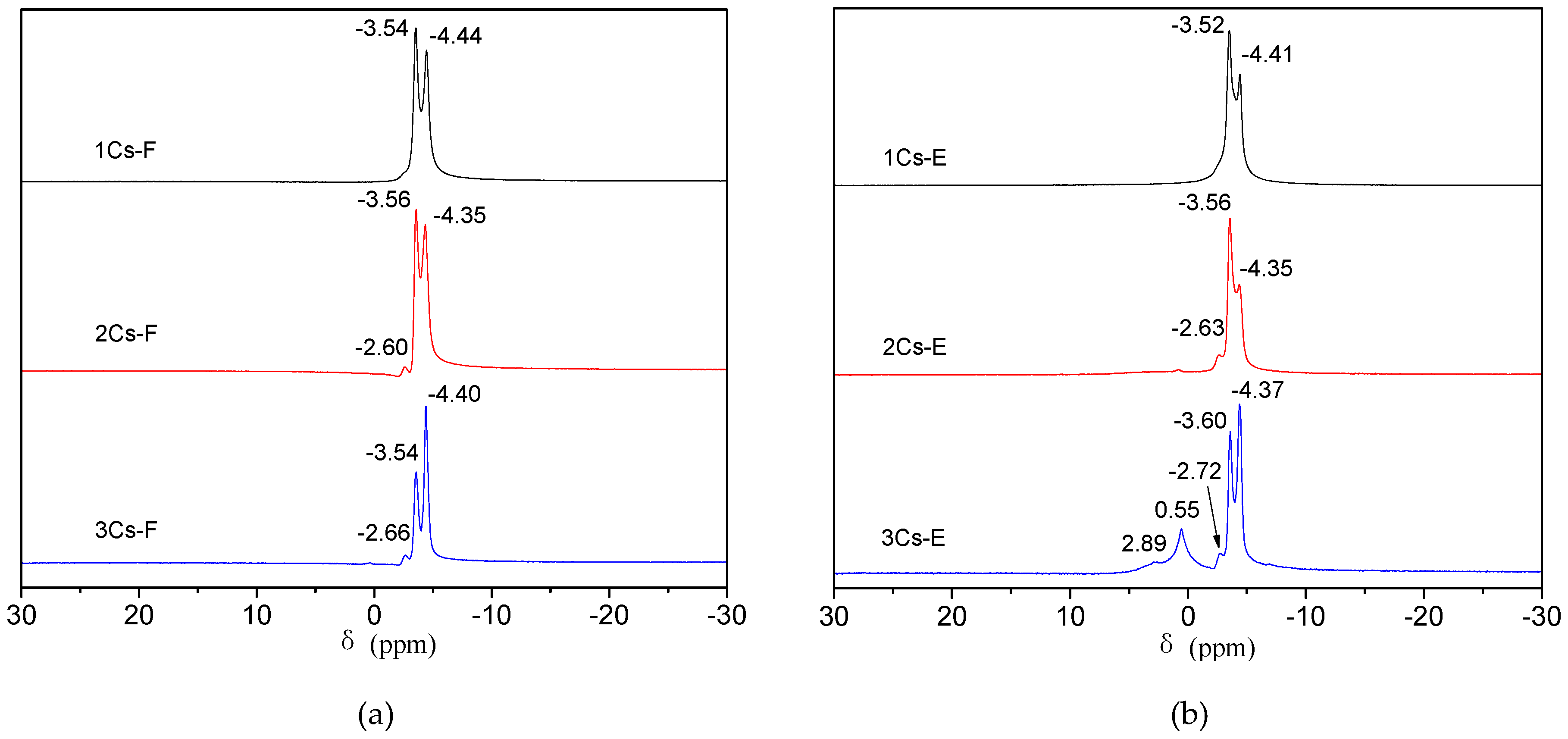
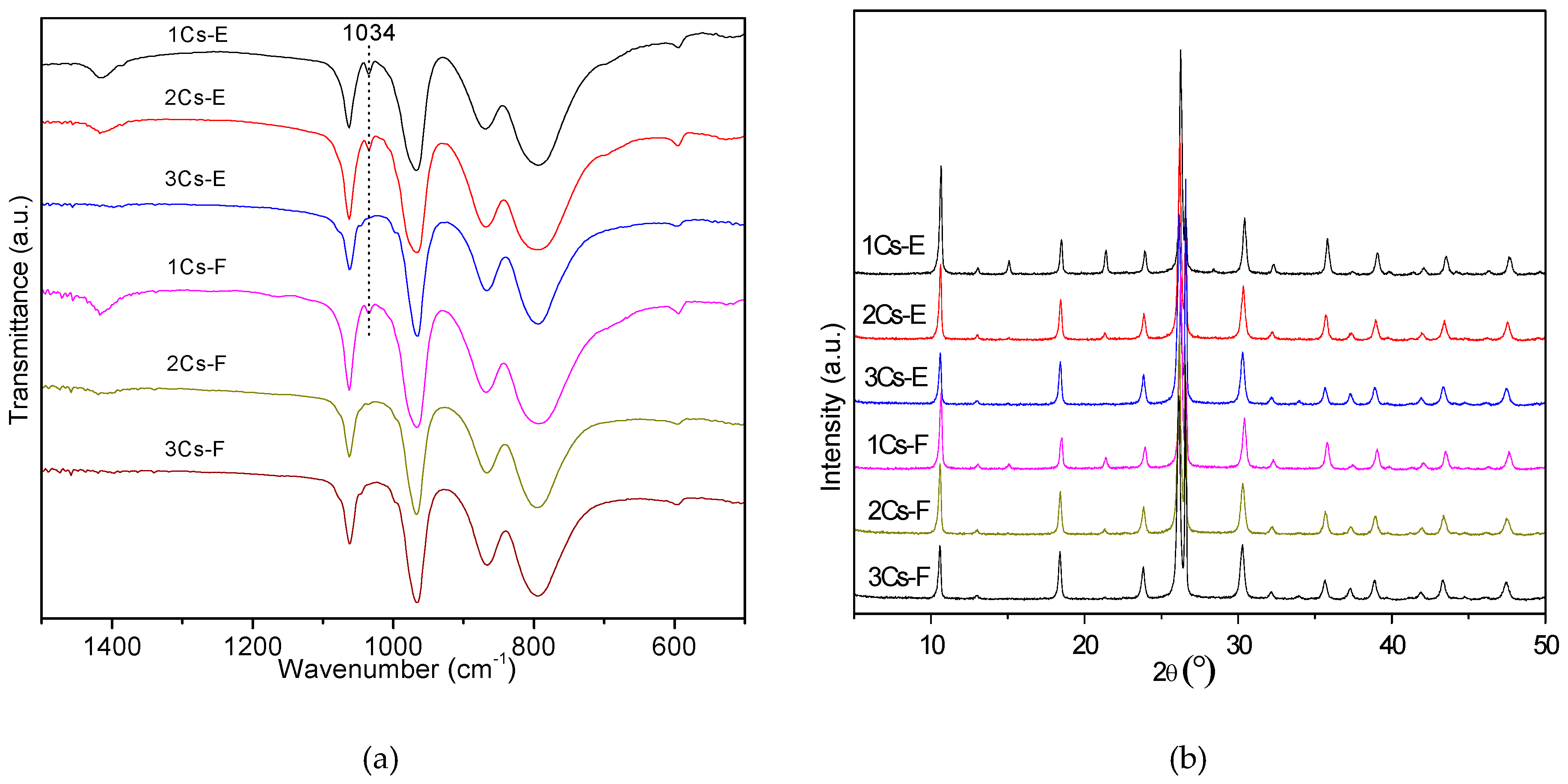
2.2. Structure Analysis of the Initial Precipitates and the Evaporation Residues
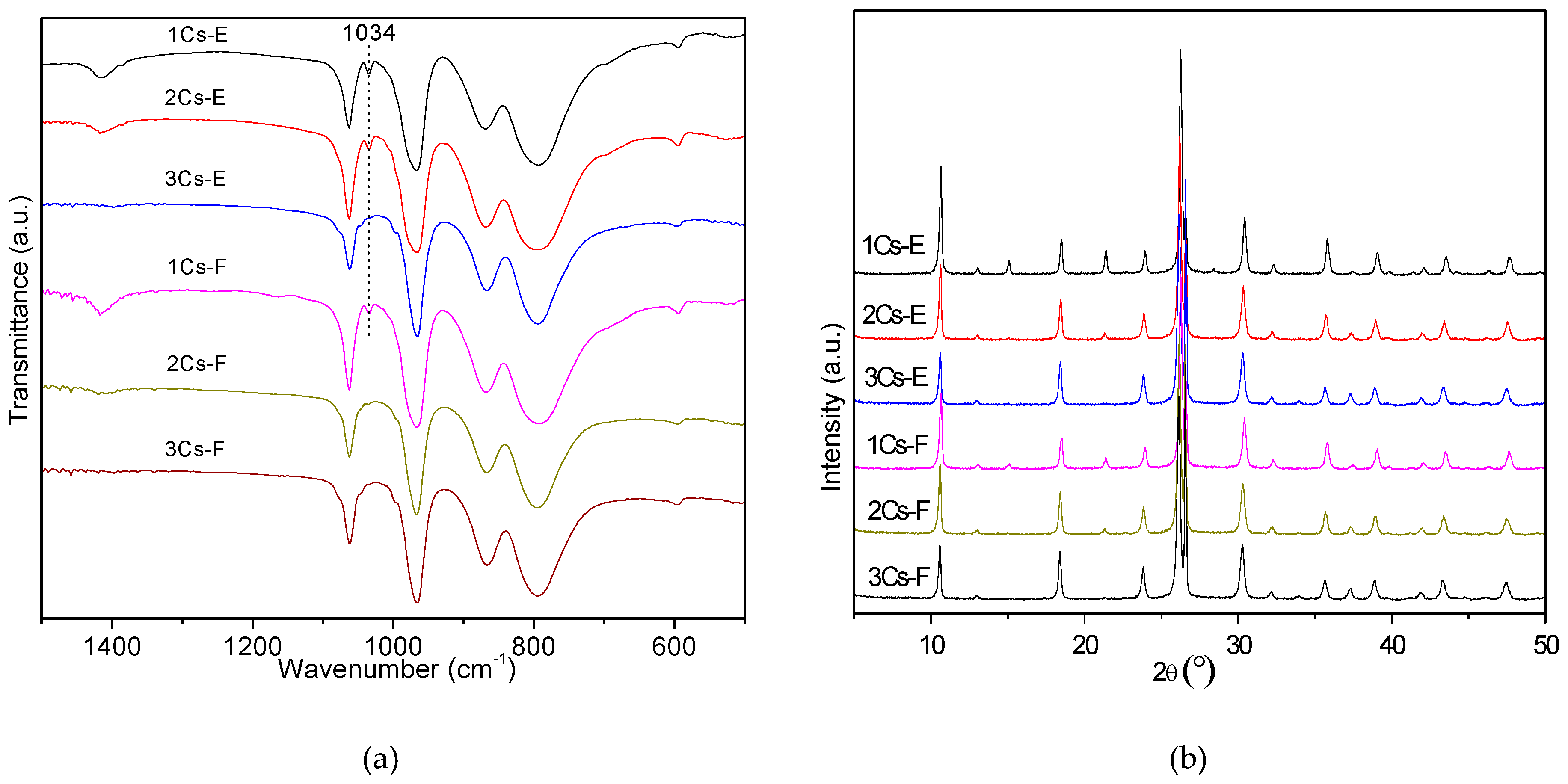
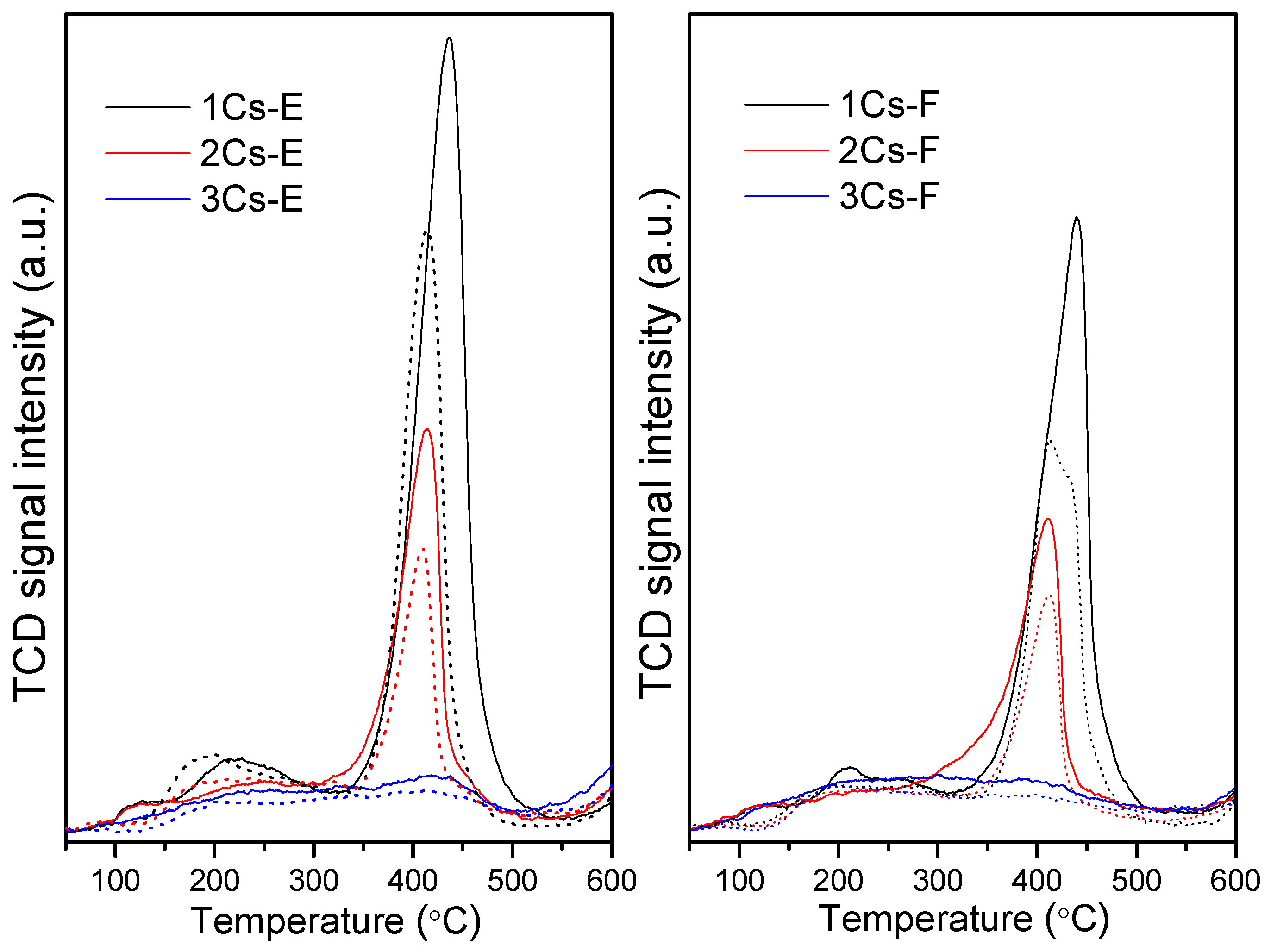
2.3. Structural Evolution during the Calcination Process
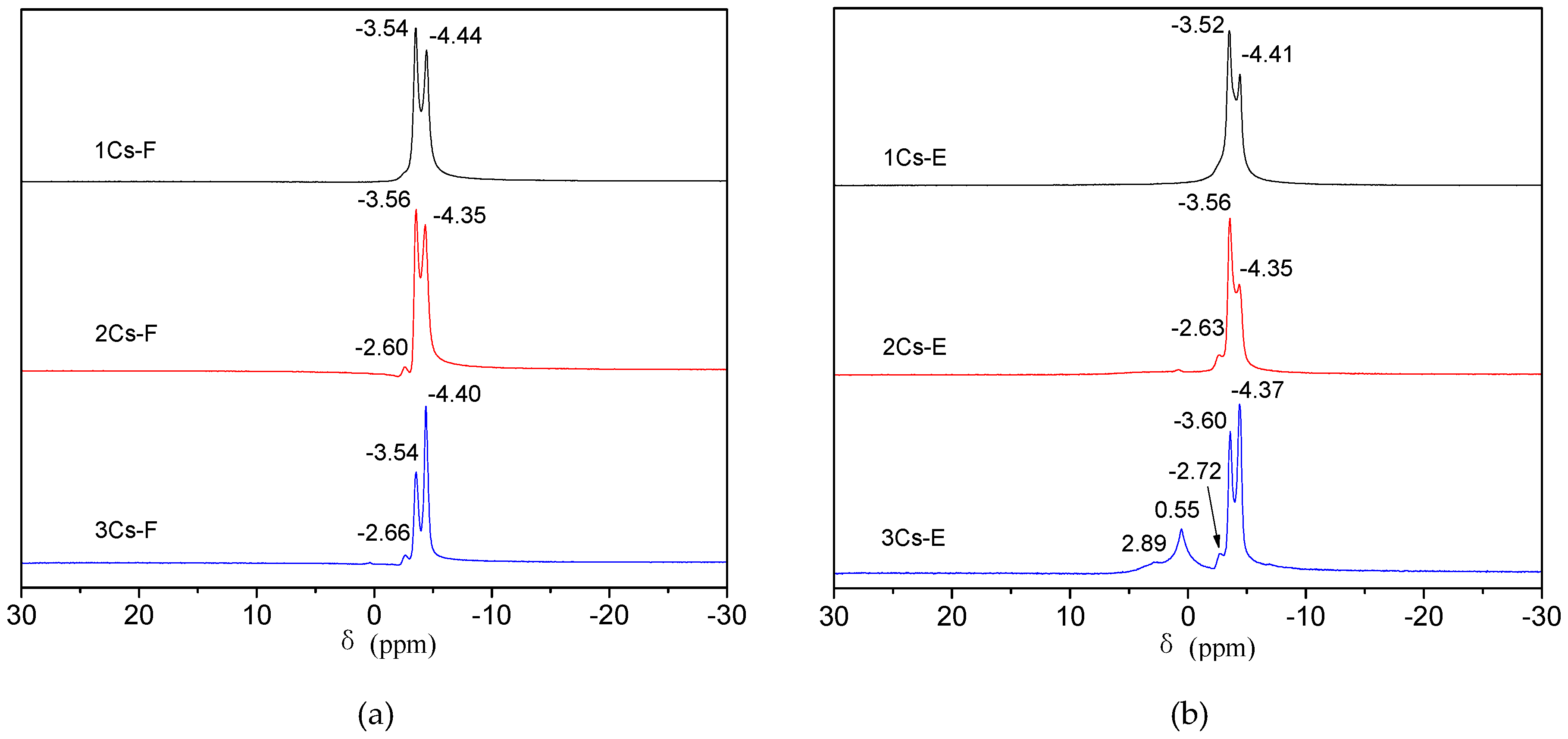
2.4. Determination of the Relative Content of Counterions in Calcined Compounds
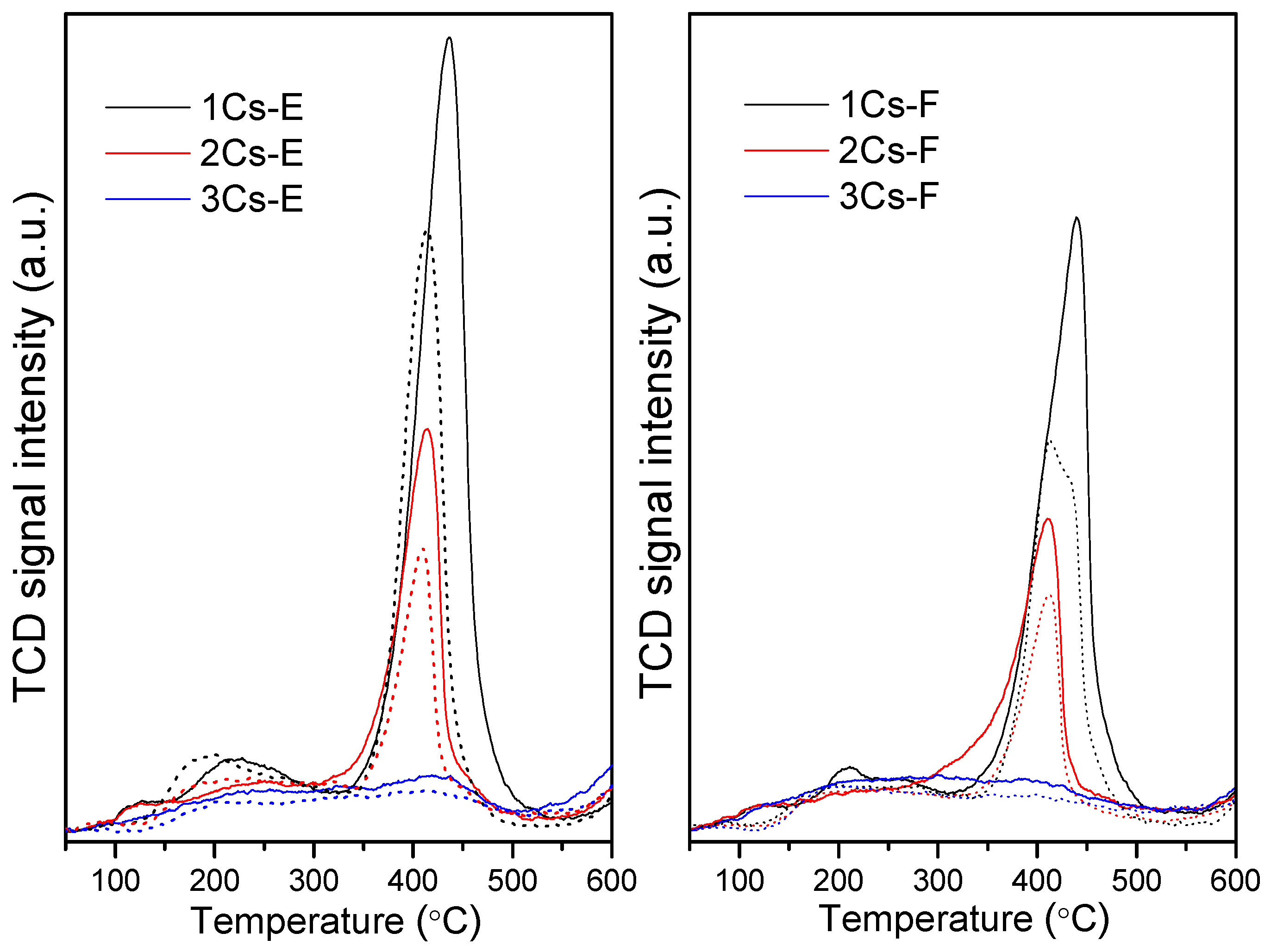
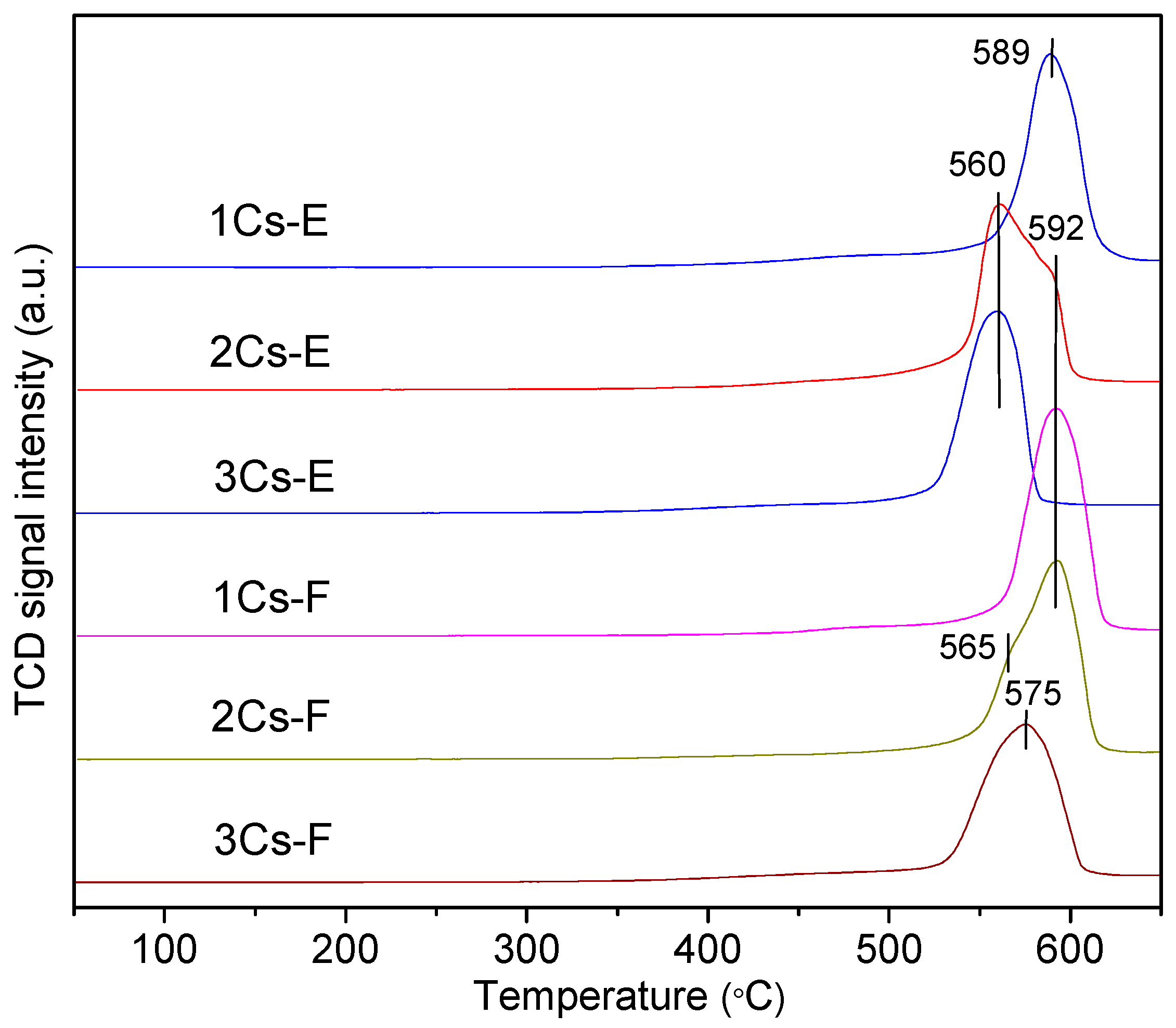
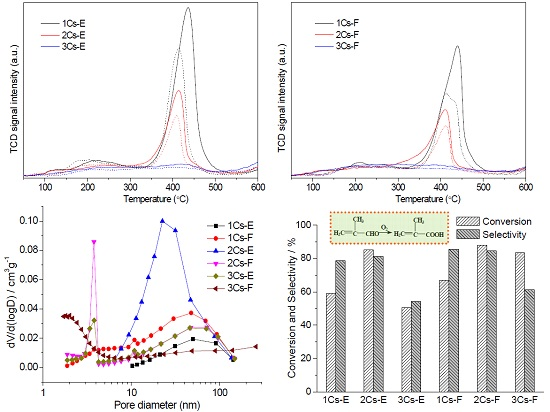
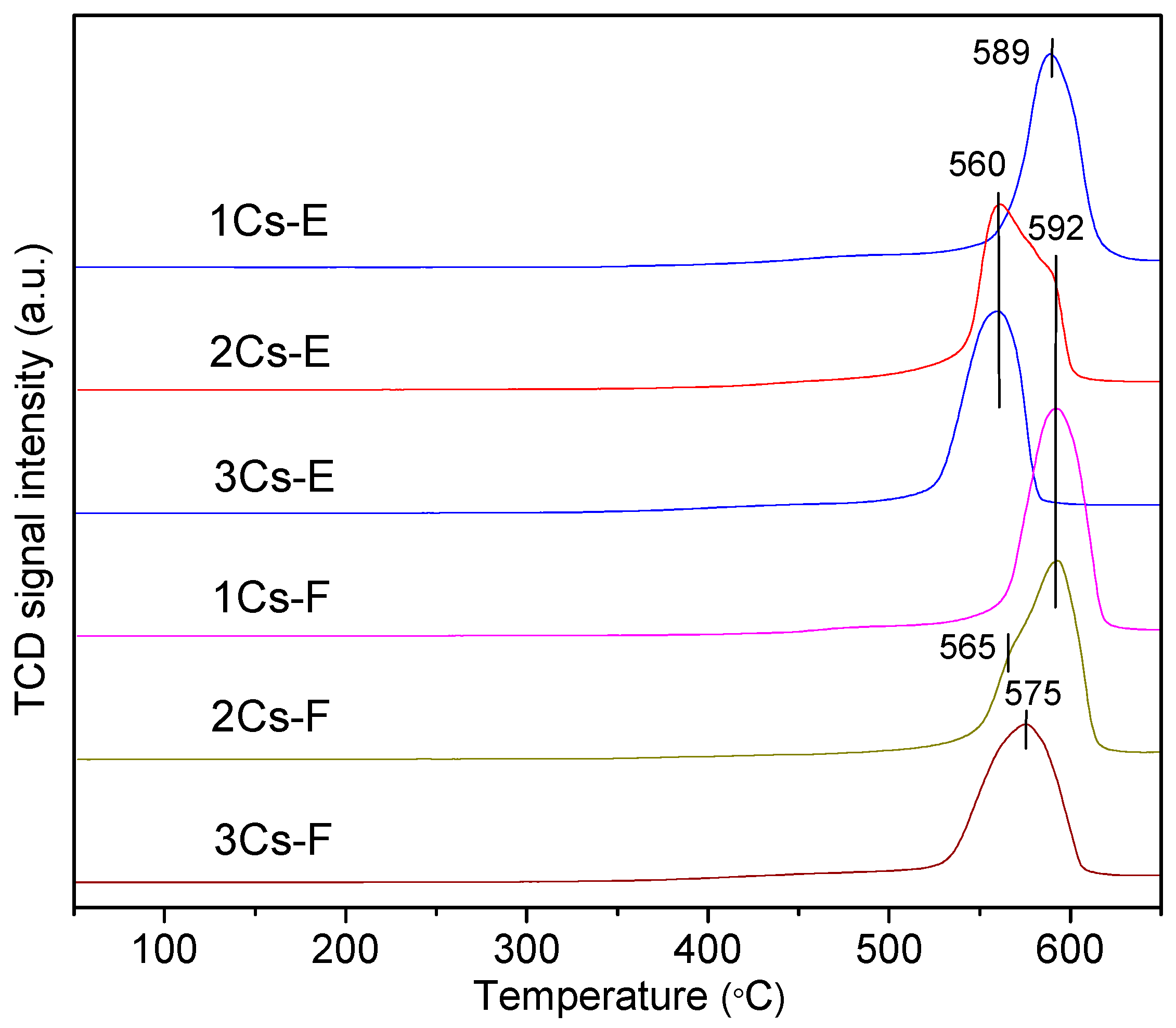
2.5. Redox Property of the Calcined Compounds
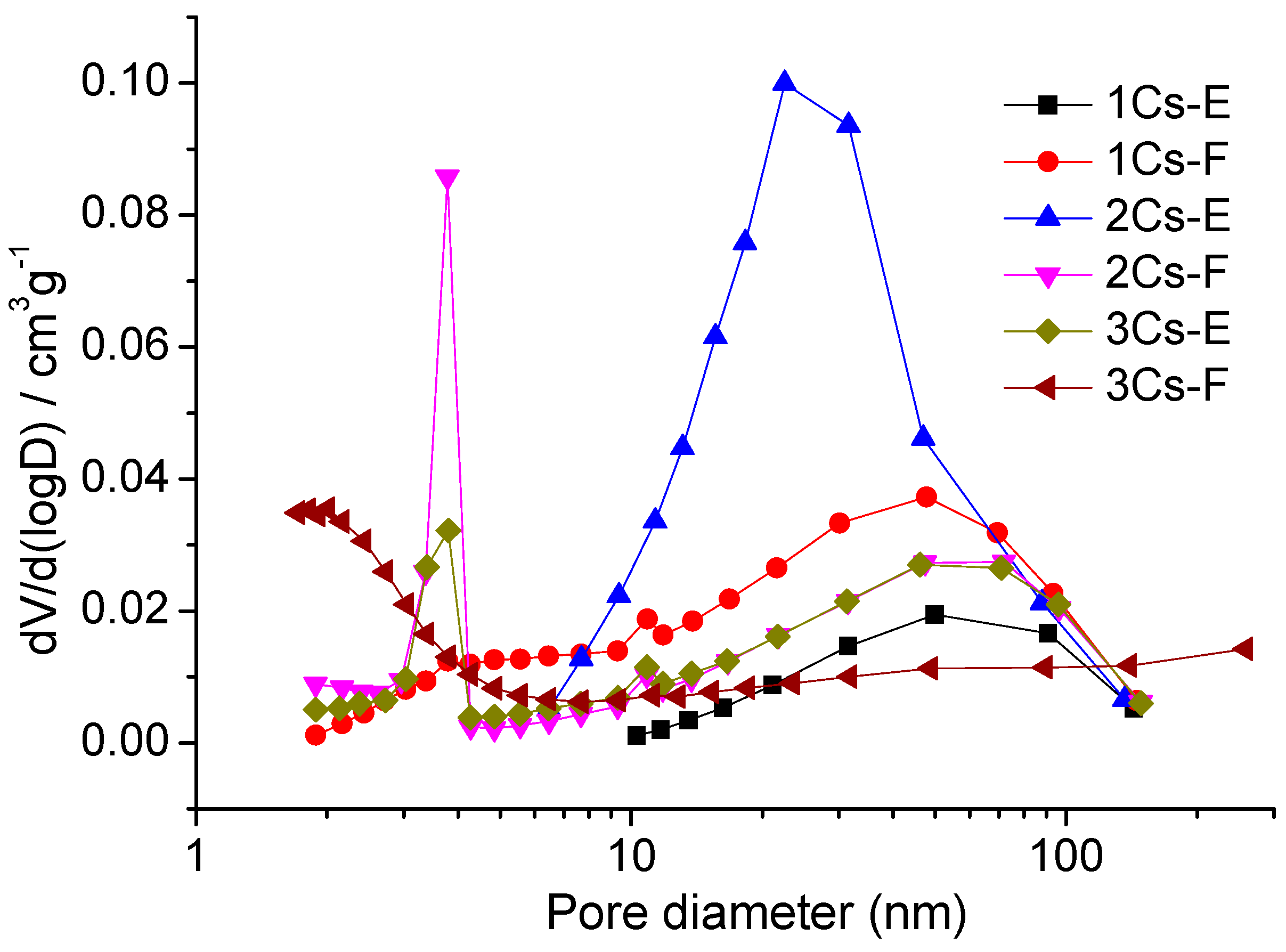
2.6. Textural Properties of the Calcined Compounds
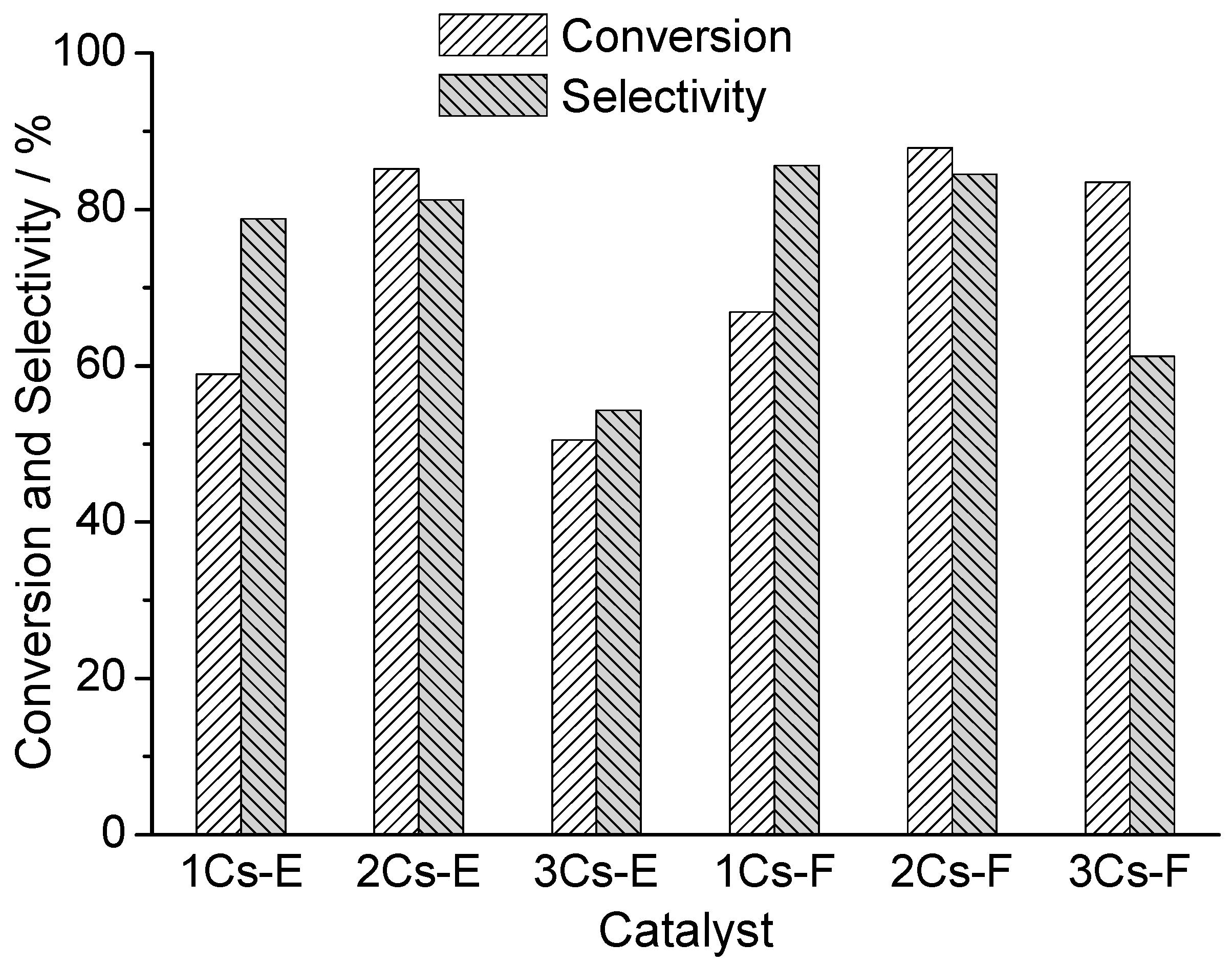
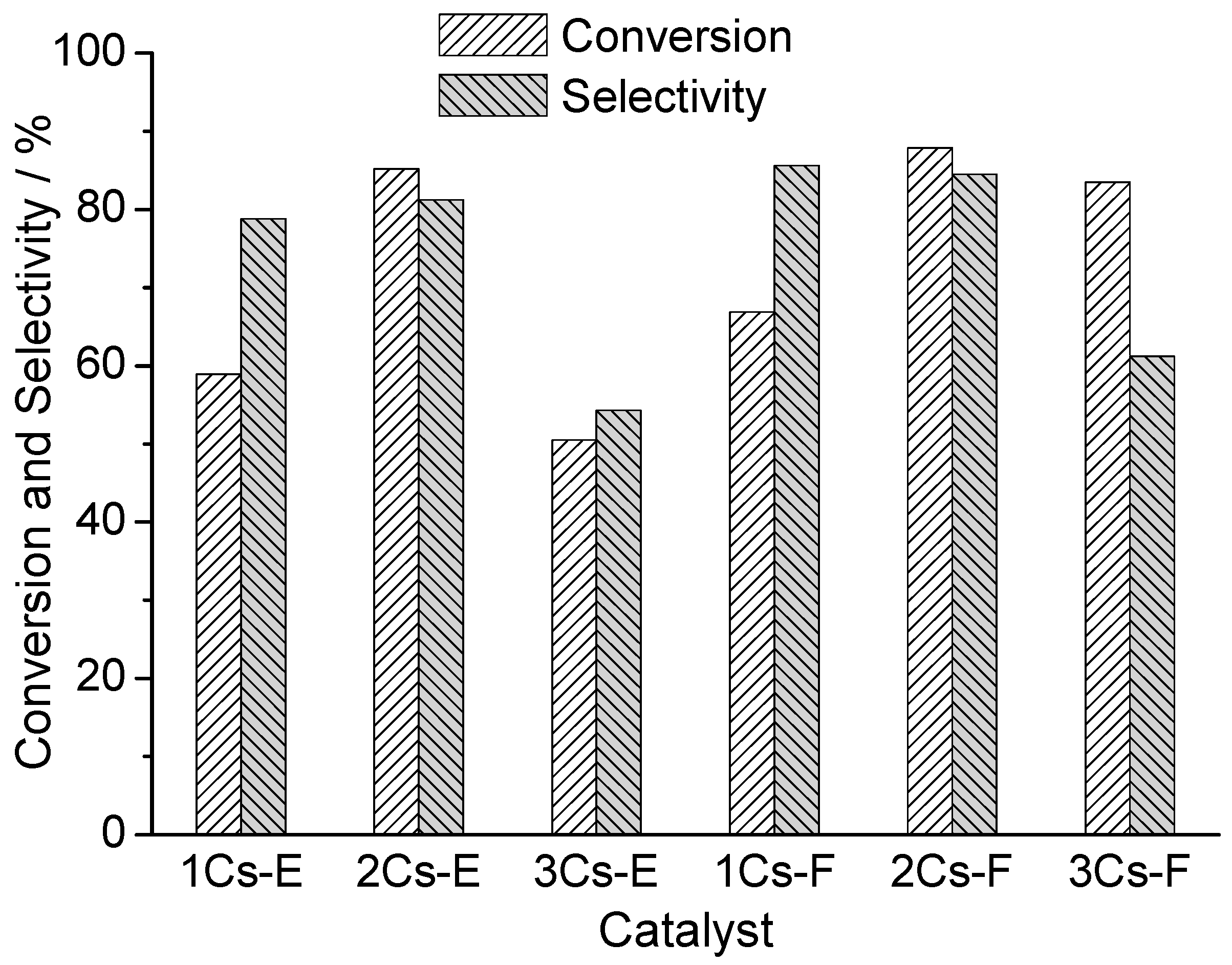
2.7. Catalytic Performance for the Oxidation of MAL to MAA
3. Materials and Methods
3.1. Synthesis
3.2. Characterization
3.3. Catalytic Activity Measurement
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tungatarova, S.A.; Abdukhalykov, D.B.; Baizhumanova, T.S.; Komashko, L.V.; Grigorieva, V.P.; Chanysheva, I.S. Oxidation of alkanes into olefins on the polyoxide catalysts. Catal. Today 2015, 256, 276–286. [Google Scholar] [CrossRef]
- Kuznetsova, N.I.; Popova, G.Y.; Kuznetsova, L.I.; Zaikovskii, V.I.; Koscheev, S.V.; Andrushkeich, T.V.; Lisitsyn, A.S.; Likholobov, V.A.; Han, S. Improving the performance of Pt-H3PMo12O40 catalysts in the selective dehydrogenation of propane with O2 and H2. Catal. Today 2015, 245, 179–185. [Google Scholar] [CrossRef]
- Mizuno, N.; Tateishi, M.; Iwamoto, M. Pronounced catalytic activity of Fe0.08Cs2.5H1.26PVMo11O40 for direct oxidation of propane into acrylic acid. Appl. Catal. A 1995, 128, L165–L170. [Google Scholar] [CrossRef]
- Liu, L.; Ye, X.P.; Bozell, J.J. A comparative review of petroleum-based and bio-based acrolein production. ChemSusChem 2012, 5, 1162–1180. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, L.; Wang, G.; Liu, J.; Gao, Y.; Li, C.; Shan, H. Effects of Cs-substitution and partial reduction on catalytic performance of Keggin-type phosphomolybdic polyoxometalates for selective oxidation of isobutene. J. Energy Chem. 2016, 25, 85–92. [Google Scholar] [CrossRef]
- Nagai, K. New developments in the production of methyl methacrylate. Appl. Catal. A 2001, 221, 367–377. [Google Scholar] [CrossRef]
- Li, X.K.; Zhang, Y.G. Oxidative dehydration of glycerol to acrylic acid over vanadium-substituted cesium salts of Keggin-type heteropolyacids. ACS Catal. 2016, 6, 2785–2791. [Google Scholar] [CrossRef]
- Srinivas, M.; Raveendra, G.; Parameswaram, G.; Sai Prasad, P.S.; Lingaiah, N. Cesium exchanged tungstophosphoric acid supported on tin oxide: An efficient solid acid catalyst for etherification of glycerol with tert-butanol to synthesize biofuel additives. J. Mol. Catal. A 2016, 413, 7–14. [Google Scholar] [CrossRef]
- Costa, V.V.; da Silva Rocha, K.A.; Oliveira, L.C.A.; Kozhevnikova, E.F.; Kozhevnikov, I.V.; Gusevskaya, E.V. Heteropoly acid catalysts for the synthesis of fragrance compounds from bio-renewables: Acetylation of nopol and terpenic alcohols. RSC Adv. 2016, 6, 43217–43222. [Google Scholar] [CrossRef]
- Trautwein, G.; El Bakkali, B.; Alcaniz-Monge, J.; Artetxe, B.; Reinoso, S.; Gutierrez-Zorrilla, J.M. Dimeric assemblies of lanthanide-stabilised dilacunary Keggin tungstogermanates: A new class of catalysts for the selective oxidation of aniline. J. Catal. 2015, 331, 110–117. [Google Scholar] [CrossRef]
- Mizuno, N.; Misono, M. Heterogeneous catalysis. Chem. Rev. 1998, 98, 199–217. [Google Scholar] [CrossRef] [PubMed]
- Stytsenko, V.D.; Lee, W.H.; Lee, J.W. Catalyst design for methacrolein oxidation to methacrylic acid. Kinet. Catal. 2001, 42, 212–216. [Google Scholar] [CrossRef]
- Bayer, R.; Marchal, C.; Liu, F.X.; Tézé, A.; Hervé, G. Catalysis of the oxidation of isobutyric acid by vanadyl, copper and mixed vanadyl-copper salts of H3[PMo12O40] and H4[PMo11VO40]. J. Mol. Catal. A 1996, 114, 277–286. [Google Scholar] [CrossRef]
- Zhou, L.L.; Wang, L.; Zhang, S.J.; Yan, R.Y.; Diao, Y.Y. Effect of vanadyl species in Keggin-type heteropoly catalysts in selective oxidation of methacrolein to methacrylic acid. J. Catal. 2015, 329, 431–440. [Google Scholar] [CrossRef]
- Kendell, S.M.; Alston, A.S.; Ballam, N.J.; Brown, T.C.; Burns, R.C. Structural and activity investigation into Al3+, La3+ and Ce3+ addition to the phosphomolybdate heteropolyanion for isobutane selective oxidation. Catal. Lett. 2011, 141, 374–390. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, R.Y.; Yang, L.; Diao, Y.Y.; Wang, L.; Zhang, S.J. Investigation of Cu- and Fe-doped CsH3PMo11VO40 heteropoly compounds for the selective oxidation of methacrolein to methacrylic acid. Ind. Eng. Chem. Res. 2013, 52, 4484–4490. [Google Scholar] [CrossRef]
- Zheng, Y.X.; Zhang, H.; Wang, L.; Zhang, S.J.; Wang, S.J. Transition metal-doped heteropoly catalysts for the selective oxidation of methacrolein to methacrylic acid. Front. Chem. Sci. Eng. 2016, 10, 139–146. [Google Scholar] [CrossRef]
- Lee, K.Y.; Oishi, S.; Igarashi, H.; Misono, M. Acidic cesium salts of molybdovanadophosphoric acids as efficient catalysts for oxidative dehydrogenation of isobutyric acid. Catal. Today 1997, 33, 183–189. [Google Scholar] [CrossRef]
- Jing, F.; Katryniok, B.; Dumeignil, F.; Bordes-Richard, E.; Paul, S. Catalytic selective oxidation of isobutane over Csx(NH4)3−xHPMo11VO40 mixed salts. Catal. Sci. Technol. 2014, 4, 2938–2945. [Google Scholar] [CrossRef]
- Santos, J.S.; Dias, J.A.; Dias, S.C.L.; Garcia, F.A.C.; Macedo, J.L.; Sousa, F.S.G.; Almeida, L.S. Mixed salts of cesium and ammonium derivatives of 12-tungstophosphoric acid: Synthesis and structural characterization. Appl. Catal. A 2011, 394, 138–148. [Google Scholar] [CrossRef]
- Langpape, M.; Millet, J.M.M.; Ozkan, U.S.; Boudeulle, M. Study of cesium or cesium-transition metal-substituted Keggin-type phosphomolybdic acid as isobutane oxidation catalysts: I. Structural characterization. J. Catal. 1999, 181, 80–90. [Google Scholar] [CrossRef]
- Villabrille, P.; Romanelli, G.; Vázquez, P.; Cáceres, C. Vanadium-substituted Keggin heteropolycompounds as catalysts for ecofriendly liquid phase oxidation of 2,6-dimethylphenol to 2,6-dimethyl-1, 4-benzoquinone. Appl. Catal. A 2004, 270, 101–111. [Google Scholar] [CrossRef]
- Mizuno, N.; Suh, D.; Han, W.; Kudo, T. Catalytic performance of Cs2.5Fe0.08H1.26PVMo11O40 for direct oxidation of lower alkanes. J. Mol. Catal. A 1996, 114, 309–317. [Google Scholar] [CrossRef]
- Ilkenhans, T.; Herzog, B.; Braun, T.; Schlögl, R. The nature of the active phase in the heteropolyacid catalyst H4PVMo11O40·32H2O used for the selective oxidation of isobutyric acid. J. Catal. 1995, 153, 275–292. [Google Scholar] [CrossRef]
- Marosi, L.; Platero, E.E.; Cifre, J.; Areán, C.O. Thermal dehydration of H3+xPVxM12−xO40·yH2O Keggin type heteropolyacids; formation, thermal stability and structure of the anhydrous acids H3PM12O40, of the corresponding anhydrides PM12O38.5 and of a novel trihydrate H3PW12O40·3H2O. J. Mater. Chem. 2000, 10, 1949–1955. [Google Scholar] [CrossRef]
- Dimitratos, N.; Védrine, J.C. Study of Ga modified Cs2.5H1.5PV1Mo11O40 heteropolyoxometallates for propane selective oxidation. J. Mol. Catal. A 2006, 255, 184–192. [Google Scholar] [CrossRef]
- Okuhara, T.; Noritaka, N.; Misono, M. Catalytic chemistry of heteropoly compounds. Adv. Catal. 1996, 41, 113–252. [Google Scholar]
- Dimitratos, N.; Védrine, J.C. Properties of Cs2.5 salts of transition metal M substituted Keggin-type M1−xPV1MxMo11−xO40 heteropolyoxometallates in propane oxidation. Appl. Catal. A 2003, 256, 251–263. [Google Scholar] [CrossRef]
- Spojakina, A.A.; Kostova, N.G.; Sow, B.; Stamenova, M.W.; Jiratova, K. Thiophene conversion and ethanol oxidation on SiO2-supported 12-PMoV-mixed heteropoly compounds. Catal. Today 2001, 65, 315–321. [Google Scholar] [CrossRef]
- Molinari, J.E.; Nakka, L.; Kim, T.; Wachs, I.E. Dynamic surface structures and reactivity of vanadium-containing molybdophosphoric acid (H3+xPMo12–xVxO40) Keggin catalysts during methanol oxidation and dehydration. ACS Catal. 2011, 1, 1536–1548. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, J.; Cao, C.; Zhang, Q.; Wang, Y.; Wan, H. Significant effect of acidity on catalytic behaviors of Cs-substituted polyoxometalates for oxidative dehydrogenation of propane. Appl. Catal. A 2008, 349, 212–221. [Google Scholar] [CrossRef]
- Damyanova, S.; Spojakina, A.; Jiratova, K. Effect of mixed titania-alumina supports on the phase composition of NiMo/TiO2–Al2O3 catalysts. Appl. Catal. A 2007, 125, 257–269. [Google Scholar] [CrossRef]
- Damyanova, S.; Cubeiro, M.L.; Fierro, J.L.G. Acid-redox properties of titania-supported 12-molybdophosphates for methanol oxidation. J. Mol. Catal. A 1999, 142, 85–100. [Google Scholar] [CrossRef]
- Misono, M.; Nojiri, N. Recent progress in catalytic technology in Japan. Appl. Catal. 1990, 64, 1–30. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Atomic Ratio | ||
|---|---|---|---|
| Mo/P | V/P | Cs/P | |
| 1Cs-E | 11.0 | 1.0 | 1.0 |
| 2Cs-E | 10.9 | 1.1 | 1.9 |
| 3Cs-E | 11.1 | 1.1 | 3.0 |
| 1Cs-F | 11.5 | 0.7 | 1.7 |
| 2Cs-F | 11.3 | 0.9 | 2.4 |
| 3Cs-F | 10.9 | 1.0 | 2.9 |
| Sample | NH4+ (mmol·g−1) | H+ (mmol·g−1) | Calculated Number of NH4+ and H+ Per Molecule of the Mixed Heteropoly Salts | |
|---|---|---|---|---|
| NH4+ | H+ | |||
| 1Cs-E | 0.571 | 0.342 | 1.07 | 0.67 |
| 2Cs-E | 0.214 | 0.161 | 0.44 | 0.33 |
| 3Cs-E | 0.0235 | 0.0163 | 0.052 | 0.036 |
| 1Cs-F | 0.444 | 0.237 | 0.90 | 0.48 |
| 2Cs-F | 0.184 | 0.141 | 0.39 | 0.30 |
| 3Cs-F | 0.0153 | 0.00075 | 0.033 | 0.002 |
| Sample | SBET (m2·g−1) | Pore Volume (cm3·g−1) | Average Pore Size (nm) |
|---|---|---|---|
| 1Cs-E | 3.54 | 0.016 | 42.8 |
| 1Cs-F | 8.94 | 0.041 | 15.6 |
| 2Cs-E | 13.9 | 0.067 | 23.6 |
| 2Cs-F | 21.1 | 0.033 | 9.84 |
| 3Cs-E | 14.6 | 0.031 | 12.4 |
| 3Cs-F | 34.0 | 0.029 | 5.85 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Wang, T.; Ma, X.; Zhu, W. Composition, Structural Evolution and the Related Property Variations in Preparation of Mixed Cesium/Ammonium Acidic Salts of Heteropolyacids. Catalysts 2016, 6, 187. https://doi.org/10.3390/catal6120187
Zhang H, Wang T, Ma X, Zhu W. Composition, Structural Evolution and the Related Property Variations in Preparation of Mixed Cesium/Ammonium Acidic Salts of Heteropolyacids. Catalysts. 2016; 6(12):187. https://doi.org/10.3390/catal6120187
Chicago/Turabian StyleZhang, Heng, Tingting Wang, Xinxin Ma, and Wancheng Zhu. 2016. "Composition, Structural Evolution and the Related Property Variations in Preparation of Mixed Cesium/Ammonium Acidic Salts of Heteropolyacids" Catalysts 6, no. 12: 187. https://doi.org/10.3390/catal6120187